

Abstract
Drug discovery and efficacy in cancer treatments are limited by the inability of pre-clinical models to predict successful outcomes in humans. Limitations remain partly due to their lack of a physiologic tumor microenvironment (TME), which plays a considerable role in drug delivery and tumor response to therapy. Chemotherapeutics and immunotherapies rely on transport through the vasculature, via the smallest capillaries and stroma to the tumor, where passive and active transport processes are at play. Here, a 3D vascularized tumor on-chip is used to examine drug delivery in a relevant TME within a large bed of perfusable vasculature. This system demonstrates highly localized pathophysiological effects of two tumor spheroids (Skov3 and A549) which cause significant changes in vessel density and barrier function. Paclitaxel (Taxol) uptake is examined through diffusivity measurements, functional efflux assays and accumulation of the fluorescent-conjugated drug within the TME. Due to vascular and stromal contributions, differences in the response of vascularized tumors to Taxol (shrinkage and CD44 expression) are apparent compared with simpler models. This model specifically allows for examination of spatially resolved tumor-associated endothelial dysfunction, likely improving the representation of in vivo drug distribution, and has potential for development into a more predictable model of drug delivery.
Keywords: vascularized tumor, drug transport, engineered vessels, multi-drug resistance, P-gp
Graphical Abstract
A vascularized human tumor on-chip examines chemotherapeutic drug delivery in a more relevant tumor microenvironment. Tumors dramatically alter the local vascular and stromal landscape and can contribute to the negative impacts of chemotherapeutic treatment on vascular transport. This model has the potential for development into a relevant pre-clinical model, where tumor-endothelial and tumor-stromal interactions are considered in drug-delivery.
1. Introduction
Successful cancer treatment strategies rely heavily on the phenotypic and targeted drug development process, where, unfortunately, a very limited number of drugs pass clinical trials .[1] The low predictive ability of current pre-clinical models remains a huge challenge to overcome.[2] Species and disease-relevant differences often contribute to inaccuracies in predicting toxicity and efficacy of chemotherapeutic compounds in humans. For this reason, research into the development of physiologic and human-relevant pre-clinical models has surged. Yet, adoption and standardized use of complex pre-clinical models is non-existent in the drug development pipeline, as challenges regarding their relevance and validation remain. In light of this, and in contrast to costly and inefficient animal model development (xenografts, for example), microfluidics and other in vitro techniques (reviewed in [3, 4]) have become important tools for rapidly generating more predictive human models.
Development of complex 3D in vitro systems incorporating spheroids and organoids for large compound screens on-chip are on the rise. Spheroids are particularly useful for high-throughput screening due to their reproducibility and ease of generation. These 3D tumor models have been used typically with cancer cells alone or in combination with other cell types (typically fibroblasts or endothelial cells). For instance, co-culture of endothelial and tumor cells has been used in an attempt to pre-vascularize spheroids and to recapitulate metastatic-like behaviour, seen by SW620 colon cancer cells intravasating into angiogenic vessels.[5] Similarly, Nashimoto et al. generated tri-culture tumor spheroids incorporating vessels and fibroblasts, to demonstrate the impact of fluid flow on tumor cell proliferation and drug effectiveness.[6] However, successful intra-tumoral angiogenesis could only be demonstrated in one cancer cell line (MCF-7) with the co-culture of fibroblasts in their model. As an alternative to using cancer cell-lines, patient-derived organoids, typically grown from processed biopsies in matrigel, may offer a more heterogeneous (phenotype and genotype) and arguably more relevant tumor model. Organoids self-assemble into complex structures and retain their genetic alterations, as shown with patient-derived lung organoids, that responded to targeted therapy similarly to the patient.[7] These systems have not been accepted as a drug screening tool due in part to limitations in patient and control organoid development. While both 3D tumor-forming strategies provide increased tumor-like behaviours, and attempts have been made to integrate vasculature intra-tumorally,[8] they have not been integrated with the larger extra-tumoral microenvironment.
Emphasis has recently shifted to understanding the tumor within its microenvironment,[9] given that the TME can play a role in cancer progression, drug uptake and/or resistance (reviewed in [10]). For instance, tumor microvasculature is dysfunctional within the TME, and is a critical determinant of drug delivery, as shown with intravital microscopy in animal models.[11] How tumors influence microvascular remodelling and function, particularly in the context of drug response, is relatively poorly understood. This lack of knowledge regarding tumor-endothelial interactions in the TME leads to poor predictive features of existing human tumor models. With an increasing number of tools (including sacrificial molds and microfluidics) developed to generate in vitro microvessels (reviewed in [12]), the number of studies investigating the role of vasculature in the progression of tumor development and metastases is also increasing.[13, 14] Patient-derived samples have also been employed in vitro, where localization and migration of tumor cells were examined in the presence of microvessels on-chip.[15] Other models have focused on paracrine signalling between tumor cells and vasculature in an effort to study mass transfer,[8] cancer-cell intravasation,[16, 17] extravasation,[18] or directed angiogenesis and lymphangiogenesis.[19] While many of the aforementioned models generate more physiologic TMEs than simple 2D tumor cultures, none have integrated tumors with vasculature to examine drug delivery. Considering that many therapeutic compounds target tumor proliferation and viability, it is necessary to consider their effect on the TME, since unintended consequences like microvascular dysfunction can reduce treatment efficacy.[20]
In an effort to develop a system capable of predicting drug delivery within a more physiologic tumor microenvironment, 3D tumor spheroids are embedded within a perfusable microvascular network. For the first time, tumor spheroids are cultured within our previously developed vessel system, [21, 22] in order to explore drug delivery within a more physiologic tumor microenvironment, and to specifically investigate the impact on, and role of, the endothelium. A comparatively large field of microvessels are grown in order to examine localized regions near and far from a tumor. Vessels are significantly, and importantly, locally impacted (within millimetres) by two representative tumors (ovarian and lung carcinomas). Morphologic and permeability changes in tumor-adjacent vasculature contribute to differences in drug delivery, which are apparent between our complex TME and simpler spheroids (examined here), demonstrating the need to use more relevant TMEs for drug testing. Drug delivery is also examined in the presence of clinically employed drug efflux pump inhibitors in an attempt to improve chemotherapeutic uptake, which is demonstrated to be in-part regulated by the endothelium. Besides highlighting the role of the vasculature in drug delivery, further development of this culture method will be useful for investigating a variety of targeted and physiologic compounds, as well as for examining immune interactions and primary or acquired drug resistance – features that require the presence of a 3D vascularized microenvironment and so cannot be accurately examined using simplistic models.
2. Results
2.1. Development of a vascularized tumor model
In an effort to enhance the predictive capacity of cancer drug screens, an in vitro vascularized tumor model was developed as an improved pathophysiologic platform. This model exploits two 3D in vitro technologies – spheroids as simplified tumor models, and the use of hydrogel-based microfluidics for generating a relevant vascularized tumor microenvironment (TME). Embedding tumor spheroids (TSs) within our device allows for tumor growth/death to be probed in the presence of perfusable microvessels (Figure 1). This system allows for direct examination of the influence of the TSs on the endothelium, and importantly drug transport through the vessels to the tumor. First, tumor cells are cultured in non-adherent 96-well plates for one week allowing for TS stabilization (Figure S1a–b, Supplementary Information). Herein, two cancerous human cell lines were used to examine the utility of the platform; an ovarian carcinoma (Skov3) and a lung adenocarcinoma (A549). Both cancer cells result in dense aggregate TSs (Figure 1b) which are then co-cultured with human umbilical vein endothelial cells (HUVEC) and normal human lung fibroblasts (nhLF) in a fibrin gel. Fibrin has been proposed to have a direct role in modulating tumor growth and metastasis, and is often found adjacent to a variety of tumor types.[23] As described in our previous work,[22] microvessels develop in a process mimicking vasculogenesis over the course of approximately one week. Briefly, individual cells extend, interconnect, and form into a vessel network during the first 72 hours, followed by lumen formation by day 5, in the presence of a TS. During co-culture, TS growth is measured in the device, and is shown to increase in diameter (Figure 1c–d). Despite similar initial TS diameters, Skov3 grew significantly larger than A549 during five days of co-culture with microvascular networks, demonstrating the capacity for this method to capture distinct phenotypes of different cancer cell types. In some cases following extended (two weeks) co-culture, vessels were seen within the tumor spheroid, and on very rare occasions tumor cells could be observed partially embedded intravascularly (Figure S1d, Supplementary Information).
Figure 1.

3D vascularized tumor model for cancer-specific characterization and drug dissemination. A) Schematic of the protocol used to generate tumor spheroids within microvascular networks (TS-MVNs). Gel-cell mixture was inserted into the middle gel port of 3mm width, 0.5mm height, 15mm length, and media added to the two adjacent channels following polymerization. B) Histology image (H&E stain) of TS within gel and microvascular network after 7 days in co-culture. C) Epi-fluorescent images showing a region of interest centered on the tumor within the TS-MVN chips. TS diameter increases over time in culture with MVNs. TSs (red), HUVEC (green). White dashed line shows approximate diameter at day 1 overlaid on each subsequent time-point. Scale bar is 200 microns. D) Effective diameters (2D) measurements were performed for n=6 spheroids each. Significance is indicated by * for P < 0.0001 with paired t-tests between subsequent days (example between days 1 and 2). Two sample t-tests between tumor types is significant (P < 0.0001) between days 2-5.
2.2. Tumor-specific impacts on vasculature formation
Besides tumor characterization, this culture method is useful for investigating tumor-associated changes in the surrounding vasculature. Following seven days in culture, striking differences are apparent in local regions of vasculature near the two types of TSs (Figure 2). For both tumor types, vessel density (vessel/total area) in the most adjacent (proximal) region to the TS was reduced in relation to a more distal radial region (Figure 2a–b). However, Skov3 tumors result in a significant reduction in vessel density in comparison to those of A549. Correspondingly, Skov3 induce a localized region of apoptosis in the vascular networks proximal to the TS, as opposed to more homogeneous distribution near A549, seen by the introduction of a fluorescent caspase 3/7 probe (Figure 2c). We postulated that differences in vessel density and apoptosis result from either an altered migration pattern or cytokine expression between the two tumor types.
Figure 2.

Tumor-associated changes are present in the vasculature as tumor cells infiltrate the TME. A) Confocal image demonstrating local changes in vessel density near the TS at day 7. HUVEC are green and Skov3 are red. Scale bar is 200 microns. B) Vessel density measured proximally and distally from the TS. Regions of interest are indicated in (A), where concentric regions of interest 150 microns from the outer TS were used to generate proximal and distal regions. C) Apoptosis is seen in the endothelium adjacent to the TS. Stitched, maximum projection confocal images (left) show highly local apoptosis in ECs adjacent to Skov3-TS. A549-TS results in more homogeneous distributions of EC apoptosis, more clearly demonstrated by heat maps (right). White dashed line indicates the TS. D) Cell migration in number (top) and distance (bottom), as measured from the TS exterior are shown for both Skov3 and A549. Single cell migration was measured at day 7 from stitched confocal images. E) A cytokine array is shown to qualitatively compare inflammatory cytokine expression between microvessels with and without tumor spheroids. Two scales are shown (normalized to highest expression levels) since the top 5 are much higher in signal than those shown separately below. Significance in (B) and (D) is *P<0.05, ††P<E−7, †††††P<E−11, ††††††P<E−12 using a t-test, as measured across 2 independent experiments with 6 samples each.
Therefore, individual tumor cell migration was tracked from the TSs at day 7 (Figure 2d). While a significantly larger population of Skov3 migrate from the TS surface, they do so at a significantly closer distance from the TS than A549 (see Figure S1d for an example of Skov3 migration). This migratory response can contribute to the differences observed in vessel death/density near the TS, but may also alter local pro-inflammatory marker concentrations (as well as other excreted danger-associated molecular pattern (DAMP)) proteins, metabolites, and growth factors, or exosomes containing RNA or proteins, all worth future exploration with this culture method). Therefore, a human cytokine antibody array was used to investigate these differences (Figure 2e). Supernatants were collected and pooled from n=5 devices each at day 7. In all cases, including control vessels (without TS), there was high expression of inflammatory markers (IL-6, IL-8 and MCP-1, top panel in Figure 2e), similarly shown by vessel supernatant in our previous work [21]. A549 TSs result in higher expression of angiogenic factors including angiogenin, which could partially explain the increased vessel density adjacent to these TSs compared to Skov3 TSs. However, only subtle differences in cytokine expression were observed between tumors and even microvessels alone. This is in part due to the global measurement of cytokines collected from the devices. Considering the highly localized changes in both vessel remodelling and permeability, it is likely that local changes in soluble factors within the TME, particularly near the TS (~1-3 millimeters, of a >1cm long vascularized chip), will not be detected and is a limitation of this model.
2.3. Tumor-induced changes in vascular permeability
It is well-known that cancer cells induce increases in vascular permeability, largely due to signalling molecules such as VEGF.[24] While tumor-associated vascular leakiness has been shown in simpler models,[9] our system is primed for investigating this phenomenon with high spatial resolution near and distant from the 3D tumors. To do so, TS were co-cultured with nascent vascular networks until day 4, when monolayers were seeded into the media channels (to limit diffusion through the gel from the media channels, as in [22]), and finally microvessels were perfused with fluorescent dextran (40 kDa) at day 7. We and others have shown that size [25] (amongst other parameters like charge and concentration) of solute molecules will impact vessel permeability. Importantly, we have shown that dextrans are transported paracellularly and provide a facile means for measuring an effective permeability.[22] The size of dextran (40kDa) was chosen to limit diffusion through the gel. A series of confocal z-stacks, spanning from the TS to a ~4mm contiguous distance, were acquired over 12 min intervals (Figure 3a). Microvascular permeability was computed from leakage of fluorescent tracer from the intra- to extravascular spaces, as we have done previously.[22] Microvessel permeability is within the range expected in vivo for normal vessels (~10−7 cm/s), perfused with similarly sized tracer molecules (66kDa and 40kDa).[26, 27] However, permeability in the regions adjacent to the TS is significantly increased in comparison to distal regions and in comparison to control vessels without co-cultured TSs (shown as the grey area in Figure 3b). Skov3 have a more pronounced impact on vessel permeability adjacent to the TS, as well as at a greater distance away, in comparison to A549. These results may be due to changes in tumor-associated signalling. The expression of TNFα was examined due to its well-known role in vascular barrier dysfunction.[28] Supernatants collected from devices at day 7 were measured by ELISA and demonstrated only marginal increases in TNFα concentrations in TS-MVN cultures (Figure 3c). Considering the highly localized effect on vascular permeability, it is likely that any increase in TNFα (or any other low concentration inflammatory cytokine) is localized to regions near the TS, again limiting detection in this system. Multiple pro-inflammatory cytokines, or other locally secreted factors, could contribute to the impaired barrier function observed, but will require further investigation. Importantly, the local effects of tumors on adjacent vessel remodelling and permeability will undoubtedly affect drug delivery to the tumor.
Figure 3.

Endothelial permeability is locally affected by the presence of a tumor. A) A stitched image demonstrating a TS-MVN system perfused with 40kDa fluorescein labelled dextran. B) Corresponding permeability measurements were made in consecutive regions next to the TS. Separate measurements were made for n=4 devices for TS-MVN samples containing either Skov3 or an A549 TS at day 7. Orange dots represent average across samples, and the grey shaded region shows the range of measured values for control samples (without a TS). C) ELISA measurements of TNFα concentrations are shown for control microvessels and those containing TSs. Shown is mean ± SEM measured from n=8 independent samples each across 2 experiments.
2.4. Vascular transport of chemotherapeutics in 3D
To test the efficacy of drug delivery in this system, we treated both microvessels and spheroids with a common chemotherapeutic reagent, Paclitaxel (also known as Taxol). A member of the taxane family, Taxol arrests cancer cells in the cell cycle, and has been used for decades in the treatment of both ovarian and lung cancers in vivo.[29] First, to determine concentrations of Taxol which would cause significant effects on the viability of all cell types (HUVEC, Skov3, and A549), CCK-8 and Calcein-AM fluorescent indicator assays were employed (Figure S2a–b, Supplementary Information). For tumor cells alone in 2D culture, a 5μM treatment for 1hr resulted in initial cell death, followed by observed recovery after 72hrs (Figure S2c, Supplementary Information). Thus, a treatment regime of 1hr, followed by complete media change, was carried out at day 7 of culture in our 3D devices (Figure 4a) at concentrations relevant to those seen in solid tumors in vivo. [30].
Figure 4.

Taxol treatment affects the endothelium and tumor. A) Time-line for drug treatment. B) Permeability measurements to 40kDa dextran made in n=6 independent control MVN samples in response to drug treatment. Box plots are shown with outer bars as SD and inner limits of SEM overlaid over individual data points. C) Tumor spheroids are shown following Taxol treatment and incubation with a caspase 3/7 indicator. Triton-X (TX) was incubated with several spheroids to demonstrate increased apoptosis. D) Stitched confocal images demonstrate caspase 3/7 expression in the TS-MVN system 72hrs following treatment with 5μM Taxol. Corresponding heat maps (right) are shown. E) Increasing concentrations of Taxol result in increased apoptosis of Skov3 and A549 TSs, shown by normalized increases in caspase 3/7. Spheroids were treated directly in 96-well plates and imaged 72hrs later. F) Normalized caspase 3/7 intensity is shown for TS-MVN samples perfused and imaged at day 10 (72hrs after treatment). G) Integrated density measurements made in extra-tumoral region of interest. Intensities were normalized to the mean across each experiment. Shown are results from 3 separate experiments with n≥2 samples. H) Relative changes in tumor diameter were measured for TS-MVN and TSs alone at 24 (no color), 48 (color), and 72hrs (color + hashed lines) after treatment. For E-G) mean ± SEM is shown. Significance is indicated for t-tests, with *P<0.05, **P<0.01. ***P<0.001, and ****P<0.0001.
Considering that microvascular viability and function is often neglected in the study of chemotherapeutic delivery, we first examined the effect of Taxol on the barrier property of our perfusable MVNs in the absence of tumor cells. Permeability of MVNs to 40kDa dextran was significantly increased following just 1hr of treatment with increasing concentrations of Taxol (Figure 4b), as expected from the increased cell death in the MVNs with increasing Taxol concentration. Indeed, doses higher than 5μM resulted in total microvascular death 72hrs later (data not shown), corresponding with previous effects seen for HUVEC.[31] This finding highlights that MVN functionality, even in healthy vessels, is altered during and following successive rounds of treatment, which can recapitulate the adverse effects of chemotherapy often inferred,[32] and directly shown recently in ex vivo microvasculature.[33]
Next, we compared drug delivery between a simple spheroid model and our system. Taxol was used to treat TSs alone and within vascular networks (TS-MVN) (Figure 4c–g). Cell death in response to treatment was measured using a fluorescent indicator of caspase 3/7 as shown in Figure 4c for TS alone and Figure 4d for TS-MVNs. As seen in TS-MVNs, significant cell death in tumors and particularly in surrounding microvessels is also shown 72hrs following 5μM Taxol treatment (Figure 4d). As expected, for treatment of spheroids alone (TS), increasing concentrations of Taxol resulted in a significantly higher caspase 3/7 signal for both tumor cell lines (Figure 4e). For 1μM and 5μM Taxol respectively, there was a 1.8x and 2.3x increase for Skov3, and a 1.6x and 2.8x increase in cell death for A549, on average. Next, the same experiment was performed using the TS-MVN system, which was quantified by measuring intensity (integrated density) across only the tumor (Figure S3a, Supplementary Information). Caspase 3/7 intensity increased with Taxol treatment (Figure 4f), similar to that of TS alone. TS-MVNs result in marginal increases in apoptosis (on average, 1.2x for Skov3 and 1.4x for A549). Relative measurements of fluorescence intensity (i.e. drug/control ratios) can be compared between the TS and TS-MVN systems (unlike absolute measurements, limited by different imaging modalities for the different systems), and lower ratios for TS-MVNs suggest that the existence of a TME has an impact on drug uptake and apoptosis. It is possible that the tumor embedded within an ECM containing stromal cells and a perfusable vasculature limits the chemotherapeutic concentration at the site of the TS (in comparison to TS alone), by redirection of the drug spatially and by cellular uptake of non-cancerous cells (examined for ECs in the following section). Observed apoptosis in surrounding vessels (as seen in Figure 4d) could mitigate the effect of increasing concentrations of Taxol in the TS-MVN model. While there was no difference in caspase intensity (normalized to controls) between Skov3 and A549 TSs, the extra-tumoral region (see Figure S3a) adjacent to the TS (including vessels, fibroblasts, and any migrated cells) resulted in significantly increased apoptosis in comparison to A549 TSs (Figure 4g).
The difference between the TS and TS-MVN systems prompted the use of an additional method to compare drug efficacy. In addition to measuring apoptosis, we measured tumor size (diameter) 24, 48, and 72hrs following 5μM Taxol treatment (Figure 4h). Surprisingly, A549 TSs continued to increase in overall diameter following treatment - an effect also demonstrated by A549 TS-MVNs. Taxol treatment in these cases did result in significantly reduced increase in growth in comparison to control (Figure S3b, Supplementary Information). In contrast, Skov3 TSs alone shrank in diameter; however, this was not the case for TS-MVNs (which grew overall), and is potentially explained by a reduction in Taxol delivery due to dysfunctional adjacent vasculature, highlighting the difference in drug efficacy between TSs and our more physiologically relevant 3D TMEs.
2.5. Drug resistance in a relevant TME
Another major barrier to drug delivery is tumor-acquired drug resistance. Following exposure to taxanes (Taxol), tumors have often been shown to respond via increased expression of efflux pumps, resulting in multi-drug resistance (MDR) by actively pumping out chemotherapeutic agents from the cell cytoplasm, which can also be influenced by the TME. [34] Since P-glycoprotein (P-gp) is a common efflux pump associated with MDR, and one for which many inhibitors have been developed and used clinically to promote bioavailability of chemotherapeutics, including Taxol,[35] we examined the use of P-gp inhibitors and Taxol treatment in the 3D TS-MVN system. First, using 2D cultures we confirmed expression of P-gp (also known as MDR1) by western blot and flow cytometry in all cell types (Figure 5a–b), including HUVEC (as observed by others)[36]. Following exposure to 5μM Taxol (1hr treatment), mean P-gp expression (72hrs later) was reduced in A549 and Skov3, but was significantly (P=0.007) higher in HUVEC (Figure 5a). Nevertheless, Taxol pre-treated tumor cells resulted in decreased cleaved caspase-3 (apoptosis marker), while increased caspase-3 was observed for Taxol pre-treated HUVEC, as expected comparing to those that were untreated. Considering that HUVEC express lower levels of P-gp (prior to Taxol treatment), it is unsurprising that uptake of 488-Taxol was higher when compared to A549 and Skov3, as measured by flow cytometry (Figure 5c). It is possible that the decrease in expression of P-gp in the tumor cells corresponded with increased expression of other drug efflux proteins to aid in drug resistance.
Figure 5.

Intracellular uptake of Taxol is cell and efflux-dependent. A) WB data are shown for n=2 separate experiments targeting P-gp expression in response to control or Taxol-treated cells. Mean intensity (normalized to β-actin) ± SEM is shown. B) Flow cytometry data for P-gp expression using standard and recombinant (rP-gp) form of antibody. C) Mean fluorescent intensity (MFI) of intracellular 488-Taxol, as measured by flow cytometry. Mean intensity ± SEM is shown. D) Normalized MFI of intracellular 488-Taxol is shown for control (488-Tax) and cells pre-treated with inhibitors (Verapamil and Elacridar). Shown are box-plots (with SE and SD outer bars) overlaid on data from n=3 samples. E) Schematic of pre-treatment with P-gp inhibitor in the TS-MVN system. F) P-gp inhibition has no effect on diffusive permeability measurements made using 40kDa dextran in the MVN system (without TS). Shown are box-plots overlaid onto raw data. G) Normalized intra-tumor (TS) 488-Taxol intensity for control and P-gp-inhibited TS-MVN samples. Significance is indicated for t-tests, with *P<0.05, **P<0.01.
Next, to further understand the role of P-gp, an efflux experiment was performed using 488-conjugated Taxol and two commonly employed inhibitors. Verapamil (an L-type calcium channel blocker used to inhibit P-glycoprotein) and Elacridar (a dual inhibitor of P-gp and breast cancer resistance protein, BCRP) were used to inhibit P-gp activity in HUVEC, A549 and Skov3. Briefly, cells were incubated with 488-Taxol for 30 min prior to subsequent 30 min incubation with either inhibitor or vehicle control. As mentioned, uptake of 488-Taxol was significantly higher in HUVEC in comparison to Skov3 and A549. Verapamil only had a significant effect on Skov3, as demonstrated by 488-Taxol being retained within these cells. Interestingly, Elacridar had a counter-intuitive effect, contrary to other reports of successful P-gp inhibition,[37] and resulted in decreased 488-Taxol retention within all cells. Indeed, intensity was significantly decreased (488-Taxol was depleted) using this inhibitor for HUVEC and A549, further hinting at compounding effects of other drug resistance mechanisms. Efflux inhibition was additionally examined using calcein - a common substrate for P-gp,[38] and these assays showed similar results (Figure S4, Supplementary Information).
Finally, P-gp function was examined in our 3D model (Figure 5e). Given the reduction in Taxol uptake of HUVECs seen in the efflux experiments upon treatment with Elacridar, we first examined the potential of this treatment to affect microvessel permeability. MVNs were cultured for 7 days, and then perfused (via convective gradient applied across the gel and through the vessels) with media containing Elacridar or a vehicle control. Elacridar treatment had no effect on the permeability of vessels to 40kDa dextran (Figure 5f). Next, we examined the same treatment in the TS-MVNs prior to addition of 488-Taxol. After 1hr incubation, devices were washed with PBS and fixed in order to preserve the 488-Taxol distribution within the vessels and tumor spheroid. Integrated fluorescent intensity distributions were measured in the tumor spheroids using maximal projections (confocal images). Across multiple experiments (n≥3 samples each) pre-treatment with the inhibitor did not have any effect on the 488-Taxol distribution in the TS-MVNs, as opposed to the effects observed in 2D cell culture (Figure 5D). This result suggests that transport of both Taxol, and possibly P-gp inhibitors, are hindered by the presence of the MVNs, as opposed to in their absence (with 2D assays or spheroids alone). For this reason P-gp-inhibitors (as well as possibly other small molecule inhibitors) ultimately have little or no effect on chemotherapeutic (488-Taxol) accumulation in the tumor at the concentration tested, further attesting to the importance of employing a relevant pathophysiological 3D vascularized tumor model.
2.6. TME and diffusion-limited drug delivery
Apart from possible obstruction of luminal transport through the vasculature, barriers to drug transport in the TME persist. Inclusion of vessels and stromal cells allows for the measurement of transport through a more physiologic TME in the TS-MVN system. For instance, density of cells and extracellular matrix proteins in the surrounding microenvironment can impact diffusion-limited transport of small molecules to the tumor.[39] In the TS-MVN system, fibroblasts strongly associate with the tumor spheroids (Figure 6a). The cells occupy volume otherwise available for diffusion, and may also deposit large amounts of extra-cellular matrix (ECM). In fact, increased expression of matrix proteins such as Collagen III are associated with drug resistance,[40] and are strongly expressed in the vicinity of our tumors (Figure 6b). Here, to compare molecular diffusivity within a relevant TME between the two cancer cell types and in the absence of binding events, the TS-MVN system was perfused with fluorescein-labelled dextran (Figure 6c–d). Following 1hr incubation, media was refreshed and fluorescence recovery after photo-bleaching (FRAP) measurements were performed. FRAP was performed in the matrix (distal to the TS), TME stroma (adjacent to the TS), and within the TS, for each cancer type. As expected, for both cancer cell types there was a significant decrease in diffusivity in the immediate TME stroma directly adjacent to the TS and furthermore within the TS itself (Figure 6c). While there was no difference in diffusivity in the matrix or TME stroma between the two cell types, the tumors themselves were significantly different. A549 particularly limited molecular diffusion within the TS in comparison to Skov3, as is also shown by fluorescence intensity measurements across the TSs (Figure 6d).
Figure 6.

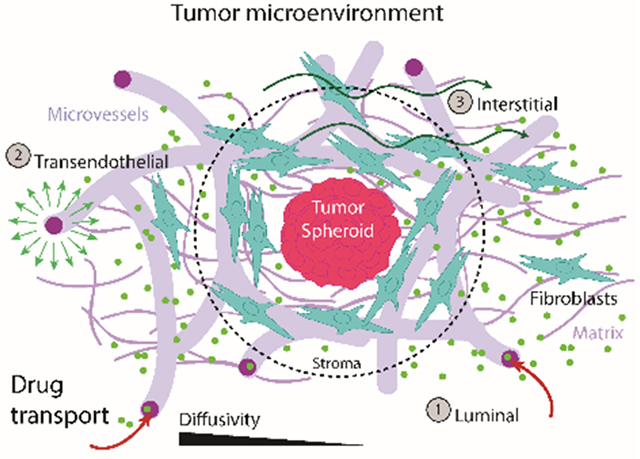
Drug transport is affected by the tumor microenvironment. A) Fixed image of Skov3 (TS-MVN) at day 7 following co-culture. Mid-TS planar confocal image is shown demonstrating HUVEC-RFP and Skov3 (H2B-mcherry), and arrow indicating ring of fibroblasts (+Phalloidin, −RFP) surrounding TS. B) Max-projection image of A549 (TS-MVN) fixed at day 7 and stained for EpCAM and Collagen III (left). Right shows only Col III stain. C) Diffusivities measured in matrix, stroma, and within the TS at day 7 for both cancer cell lines (n=10 samples each). Shown are box-plots (inner SEM, outer bars SD) overlaid on individual samples. Significance is indicated for paired t-tests, with *P<0.05, **P<0.01, and †P<E−6, ††††P<E−10, †††††P<E−11. D) A representative (normalized) intensity of 488-Taxol accumulation within each TS – from outer TS to mid-point. The graph above demonstrates measurements made across the TS. E) A graphical summary of diffusive drug transport through the vasculature and TME, demonstrating the 3 transport routes in our model system. All co-cultured components dictate the efficacy of drug transport in the TS-MVN system.
Tumor-associated changes in the endothelial and stromal components of the TME will considerably contribute to the effectiveness of transport of therapeutic reagents over time. As examined in our model, and summarized in Figure 6e, diffusive mechanisms of transport through the circulatory system to the tumor are quite important, particularly given that tumors can promote vessel dysregulation in their vicinity (causing leakiness or complete vessel regression with decreased perfusability). The model tumors examined herein demonstrated significantly altered effects on the surrounding vasculature and TME. Moreover, the tumors themselves demonstrated differences in diffusivity, which might explain why A549 do not respond to 5μM Taxol in the same manner as Skov3 (see Figure 4g and Figure 5d). Considering that other adaptive mechanisms such as increased cancer stem cell (CSC) phenotype may also be at play, we investigated one particular CSC marker following Taxol treatment. Increased CD44 expression (a well-known CSC marker [41, 42]) was seen in both tumor cell types (significantly for A549) following treatment with Taxol in 2D (Figure S5a–b, Supplementary Information). Interestingly, this result was not repeated when measured by flow cytometry in tumor cells extracted from 3D TS-MVNs (Figure S5c). This contrasting result again highlights the need for examining drug transport in a more complex TME – where tumor-associated remodelling of the TME can hinder drug dissemination.
3. Discussion
Despite the frequent use of in vitro and in vivo tumor models for investigating drug efficacy, many candidate compounds continue to fail, both in early and in the later stages of clinical trials.[43] This short-coming is partially due to the inability of pre-clinical models to accurately represent the human tumor microenvironment. While no model will perfectly recreate the complexities and heterogeneity of patient-specific TMEs, in vitro systems that closely resemble the in vivo TME have the potential to bridge the gap left by interspecies differences in current models. The predictability of treatment outcomes in vitro models still requires careful correlation with animal and clinical studies. Therefore, as a first step, we developed a model capable of recapitulating a more physiologic TME, inclusive of a 3D tumor spheroid embedded within a network of stromal cells (fibroblasts) and perfusable vasculature made entirely from human cells. This TME-relevant culture method using our previously employed vascular system [21, 22] allows for the direct observation and measurement of tumor-vessel and tumor-stromal interactions (Figure 1). Using two cancer cell lines as model tumors, we demonstrate highly localized changes in the TME over time in the TS-MVN model (Figure 2). Co-culture with these two tumor spheroids demonstrates changes in vessel density and cell apoptosis unique to each cancer cell type, which are captured with high spatial resolution using confocal imaging. Importantly, both tumors result in localized changes in endothelial barrier function, causing leakier vessels within ~1mm of the tumors (Figure 3). Key differences in the response of these two tumor types were also revealed: Skov3 are more dispersive and migrate into the surrounding TME, leading to increased intratumoral diffusivity as well as a drastic decrease in microvascular density in adjacent regions. On the other hand, A549 remain tightly packed and are thus less diffusive, and do not alter the surrounding vascular density as drastically (Figure 6). The choice of these cell lines was made due to their potential to form aggregates, and both of these cancer types are clinically treated by paclitaxel (ovarian cancer as a first-line treatment,[44] and lung cancer as an adjuvant therapy)[45] clinically relevant comparisons can be made. While cell lines are limited in their ability to capture the heterogeneity of patient samples, they are useful for establishing the model and show the sensitivity of our system to changes in tumor type. Differences in the response to treatment between these two tumors are investigated within the context of a more complex, yet more physiologic system. The endothelium is the main focus here, but stromal cells also impact the form and function of microvessels,[46] and have the potential to be investigated further in relation to the TME presented here. Overall, the TS-MVN system demonstrates the highly local effects of distinct tumors on the TME, which ultimately affects drug transport.
Successful drug transport relies on luminal, transendothelial and interstitial transport (within the surrounding TME and in the tumor itself) (Figure 6e). The first two mechanisms rely on functional microvessels, whereas the later mode depends on the cellular and ECM constituents present within the TME. An overview of the relations between diffusive and convective flux through the vasculature and interstitium is available in Stylianopoulos et al..[47] Here, by employing fluorescent dextran and a common taxane (Taxol) conjugated to a fluorophore, it was possible to examine diffusive transport mechanisms and characterize the drug’s distribution within the TME in the TS-MVN system. Importantly, Skov3 and A549 were shown to promote leaky vessels within several millimeters distance of the tumor mass. While tumors are well-known to alter barrier function,[48] this phenomenon had not been directly observed in other 3D in vitro studies. HUVEC and fibroblasts together allow for robust and perfusable culture of vessels; however, work is on-going to generate more tissue- and tumor-specific vasculature. Challenges will be faced generating hPSC-derived vessels, since they often represent early endothelial progenitors and can also lose endothelial phenotype in long-term culture, [49] so for this reason HUVEC (shown for us to express well-organized tight junctions and vessel morphology approximating in vivo microvasculature [22], and expression of an extensive repertoire of drug and nutrient transporters) were used as a representative model to assess tumor-dependent changes in vascular function. In this initial study, continuous luminal flow was not applied to the microvascular system, so that only diffusive measurements could be made. The impact of a dense tumor or surrounding stroma can greatly impact drug delivery time. Considering the maximum distance between blood vessels (L≈100μm), diffusion time can be roughly approximated by L2/D,[20] where D is diffusivity. For the TSs, A549 demonstrate a near 2-fold increase (~74s versus ~42s for FITC dextran) in diffusion time compared with Skov3. It is important to note that convection (which can be applied in this system)[50] will have a significant impact on permeability of the endothelium, interstitial fluid pressures, and ultimately drug transfer - investigations of which are on-going. Previous xenograft research has demonstrated that hydrostatic microvascular pressure drives the interstitial (hypertensive) tumor pressure.[51] Thus, hydrostatic conditions (as applied here) may still provide an appropriate indication of mostly diffusion-dependent drug delivery to the tumor.
While there are several opportunities for increased physiologic relevance, our TS-MVN system as used here already demonstrates clear differences between the response to Taxol between the TS-MVN model and the simpler TS without a TME (Figure 4e–h). In summary, while apoptosis markers increased as expected following Taxol treatment, tumor size was not necessarily correlated. Specifically, for A549 the TS (alone or within MVN) continued to increase in size, which could be attributed to transient enlargements, as seen during programmed cell death.[52] In contrast, Skov3 demonstrated an opposing effect in the TS versus TS-MVN systems, which could be attributed to changes in the local TME (decreased local chemo concentrations due to the lack of vessel delivery or changes due to cellular uptake in the surrounding stroma or nearby vasculature). Although Taxol is well-known to arrest cells in the mitotic phase, the subsequent outcome of this arrest is one of three fates: cell death, arrest, or continued cycling .[30] What determines this fate remains unknown, and we are still unable to predict accurately the sensitivity of cells to this drug, despite its widespread effectiveness and usage. Given the complexities in response to drugs like Taxol, our system provides a new avenue for further exploration of these differences between cancer types that can provide significant insight into its efficacy.
The significance of the endothelium is paramount in the drug delivery process, which is demonstrated by the TS-MVN model. Overall, our results show that Taxol treatment, as expected, has a significant impact on endothelial function and viability – with vessels becoming leakier with increasing concentrations and becoming non-perfusable at high concentrations (10μM). At concentrations (1-5μM) typically associated with intra-tumoral drug concentrations,[30] permeability to solutes is significantly increased, as is P-gp expression (in those cells that persist following 72hrs). Whether the expression level is increased or the population of cells already expressing P-gp are preferentially selected remains to be seen. Interestingly, intracellular uptake of Taxol was highest in endothelial cells, not in the tumor cells, suggesting that active transport mechanisms are at work. The effect of the efflux inhibitor Elacridar was also shown to not have a significant effect on uptake (or efflux) of 488-Taxol in the TS-MVN system – a result contradictory to what is expected,[53] and likely due to the hindered transport of the molecule in the system. Interestingly, the inhibitor did not have an effect on endothelial permeability, although the 40kDa (40000 g/mol) dextrans used were an order of magnitude larger than Taxol (~854 g/mol) (Figure 6), and might not be substrates for P-gp.
P-glycoprotein has been shown to limit uptake of chemotherapeutics like Taxol in vivo, and thus remains an attractive target for increasing drug delivery. In particular, the first line of treatment for ovarian cancer is often Taxol (in combination with other drugs), which is well-characterized as a substrate for P-gp.[54] Elacridar has been shown to be effective in Taxol-resistant ovarian cancer cell lines.[53] For this reason, we examined the utility of this P-gp inhibitor in our 3D TS-MVN system. A surprising effect was shown in HUVECs, A549, and Skov3 pre-treated with Elacridar, which all resulted in decreased short-term 488-Taxol retention in 2D. The result suggests that Elacridar either interferes with the uptake of 488-Taxol or causes increased efflux, possibly by compounding drug resistance mechanisms. While Elacridar is a specific but non-competitive target for P-gp, it has little effect on the other ATP-binding cassette (ABC) transporters, and so those other transporters could contribute to the observed effect and should be examined in the future. Cellular uptake of Elacridar could also be impeded, as it has been shown to be relatively insoluble.[55] No distinction was made here between passive and active transport of Taxol (either transcellular or paracellular), and we investigated only one active efflux mechanism in the 3D TS-MVN. Many other active transporters could be contributing to the efflux of Taxol in the tumor. Moreover, the 488-labelled version of the drug is larger and might interact differently with the inhibitors than non-labelled Taxol. Ultimately, examination of drug dissemination is complex; however the TS-MVN system provides an ideal platform to study drug delivery and tumor resistance in a pathophysiological representative TME.
Characterizing drug transport in the TS-MVN will be beneficial for targeting the correct tumor- and, importantly, the vascular response. For instance, drug transport through tumor-induced leaky vessels will be more effective in regions of dense vasculature close to the tumor, where drug transport is not reliant solely on diffusion (as is the case where vessel rarefaction occurs). These ~500 micron diameter tumors were easy to handle and provided sufficient cell number for flow cytometry measurements, but have only a few small vessels protruding into the TSs (Figure 2a and Figure S1d, Supplementary Information). In vivo, larger and more aggressive tumors undergoing rapid growth are typically associated with a necrotic core [56] and initiate pro-angiogenic signalling and/or vessel co-option. While not a focus herein, metabolic stress-induced tumor necrosis could be a focus of future work using this system, and it is possible that at the average size of 500 microns our TSs may indeed be necrotic. It is possible that over longer durations our TS-MVNs might be increasingly necrotic and more vascularized (as seen at day 14 in Figure S1d, Supplementary Information). Although mostly avascular tumors were examined here, a variant of this model could be used to culture larger vascularized spheroids or tumor organoids, where vessel normalization (by for example using anti-VEGF treatments like bevacizumab)[37] could be examined as a possible strategy to increase drug delivery into the tumor by improving the transvascular pressure gradient. The TS-MVN system could also be used to examine the underlying factors responsible for tumor-induced vascular dysfunction (growth factors, cytokines, MMPs, DAMPs, extracellular vesicles, etc.) in more detail.
With a more physiologic TME, our 3D TS-MVN model has the potential to test various treatment strategies by direct delivery through the vasculature. This system is capable of examining drug dissemination from diffusive (herein) and convective (on-going) transport mechanisms – which is an advantage over animal models, where the two modes cannot be decoupled. Moreover, this culture method is beneficial for modelling drug delivery to small developing tumors, as they transition from a dormant to a metastatic growth phase.[57] Considering that vessel density was significantly altered by both cancer cell types herein, this tool demonstrates its utility for demonstrating key differences in tumor-endothelial interactions, which will be important for detection of TME changes due to more complex and variant (patient) samples. Our previous in vitro models of cancer extravasation,[58, 59] and immune-mediated cancer cell migration assays,[60] have been validated against animal studies. Future work for this model necessitates correlation with clinical data and validation of our model in order for it to be proposed as a pre-clinical tool. Moreover, strategies that combine chemotherapeutics and immune cell interactions (as we previously showed with monocytes [61]) and immunotherapies (reviewed in [62]) could be examined in our TS-MVN on-chip. Importantly, the findings could be used to design novel vessel normalization therapies for better drug and immune cell (chimeric antigen receptor (CAR)T-cell) delivery.
4. Conclusion
Our human tumor on-chip model integrates perfusable microvessels directly with a tumor allowing for examination of key transport mechanisms. This model demonstrates the importance of an intact and functional vascular network for effective drug delivery, as tumor-induced remodelling significantly impacts the local TME and vasculature. Differences in response to Taxol treatment between our 3D model (dampened effects) and simpler systems (heightened effects), demonstrates the need for investigating drug delivery in a relevant TME. This model has potential for further development into a more accurate pre-clinical model to examine complex immune cell and targeted antibody interactions, alongside chemotherapeutics.
5. Experimental Section
Detailed materials and methods can be found in the Supporting Information. Briefly, tumor spheroids are first grown and then seeded with a combination of endothelial and stromal cells in a hydrogel-laden microfluidic device, allowing for the growth of tumors embedded within a perfusable microvascular network.
Supplementary Material
Acknowledgements
K.H. would like to acknowledge funding from an NSERC post-doctoral fellowship as well as partial funding from the National Science Foundation (CBET-0939511). G.O. is supported by an American-Italian Cancer Foundation Post-Doctoral Research Fellowship and would also like to acknowledge funding from Amgen, Incorporated. M.R.G. would like to acknowledge funding from a Canadian Institutes of Health Research post-doctoral fellowship. R.D.K. would like to also acknowledge funding from the National Institutes of Health (U01 CA214381) and the National Science Foundation (CBET-0939511).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library.
Contributor Information
Kristina Haase, Massachusetts Institute of Technology, Massachusetts, 02139, USA.
Giovanni S. Offeddu, Massachusetts Institute of Technology, Massachusetts, 02139, USA
Mark R. Gillrie, Massachusetts Institute of Technology, Massachusetts, 02139, USA; University of Calgary, Calgary, T2N 1N4, Canada
Roger D. Kamm, Massachusetts Institute of Technology, Massachusetts, 02139, USA
References
- [1].Swinney DC, Clinical pharmacology and therapeutics 2013, 93, 299. [DOI] [PubMed] [Google Scholar]
- [2].Mak IW, Evaniew N, Ghert M, American journal of translational research 2014, 6, 114. [PMC free article] [PubMed] [Google Scholar]
- [3].Rodenhizer D, Dean T, D’Arcangelo E, McGuigan AP, Advanced healthcare materials 2018, 7, e1701174. [DOI] [PubMed] [Google Scholar]
- [4].Tsai HF, Trubelja A, Shen AQ, Bao G, J R Soc Interface 2017, 14, 1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ehsan SM, Welch-Reardon KM, Waterman ML, Hughes CC, George SC, Integrative biology : quantitative biosciences from nano to macro 2014, 6, 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Nashimoto Y, Okada R, Hanada S, Arima Y, Nishiyama K, Miura T, Yokokawa R, Biomaterials 2019, 119547. [DOI] [PubMed] [Google Scholar]
- [7].Kim M, Mun H, Sung CO, Cho EJ, Jeon H-J, Chun S-M, Jung DJ, Shin TH, Jeong GS, Kim DK, Choi EK, Jeong S-Y, Taylor AM, Jain S, Meyerson M, Jang SJ, Nature Communications 2019, 10, 3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shirure VS, Bi Y, Curtis MB, Lezia A, Goedegebuure MM, Goedegebuure SP, Aft R, Fields RC, George SC, Lab on a chip 2018, 18, 3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tang Y, Soroush F, Sheffield JB, Wang B, Prabhakarpandian B, Kiani MF, Scientific reports 2017, 7, 9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Khawar IA, Kim JH, Kuh HJ, Journal of controlled release : official journal of the Controlled Release Society 2015, 201, 78. [DOI] [PubMed] [Google Scholar]
- [11].Fukumura D, Duda DG, Munn LL, Jain RK, Microcirculation 2010, 17, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Haase K, Kamm RD, Regen Med 2017, 12, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sobrino A, Phan DT, Datta R, Wang X, Hachey SJ, Romero-Lopez M, Gratton E, Lee AP, George SC, Hughes CC, Scientific reports 2016, 6, 31589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen MB, Whisler JA, Frose J, Yu C, Shin Y, Kamm RD, Nature protocols 2017, 12, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Xiao Y, Kim D, Dura B, Zhang K, Yan R, Li H, Han E, Ip J, Zou P, Liu J, Chen AT, Vortmeyer AO, Zhou J, Fan R, Adv Sci (Weinh) 2019, 6, 1801531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nagaraju S, Truong D, Mouneimne G, Nikkhah M, Advanced healthcare materials 2018, 7, e1701257. [DOI] [PubMed] [Google Scholar]
- [17].Zervantonakis IK, Hughes-Alford SK, Charest JL, Condeelis JS, Gertler FB, Kamm RD, Proceedings of the National Academy of Sciences of the United States of America 2012, 109, 13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jeon JS, Bersini S, Gilardi M, Dubini G, Charest JL, Moretti M, Kamm RD, Proceedings of the National Academy of Sciences of the United States of America 2015, 112, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chung M, Ahn J, Son K, Kim S, Jeon NL, Advanced healthcare materials 2017, 6. [DOI] [PubMed] [Google Scholar]
- [20].Dewhirst MW, Secomb TW, Nature reviews. Cancer 2017, 17, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Haase K, Gillrie MR, Hajal C, Kamm RD, Advanced Science 0, 1900878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Offeddu GS, Haase K, Gillrie MR, Li R, Morozova O, Hickman D, Knutson CG, Kamm RD, Biomaterials 2019, 212, 115. [DOI] [PubMed] [Google Scholar]
- [23].Kwaan HC, Lindholm PF, Seminars in thrombosis and hemostasis 2019, 45, 413. [DOI] [PubMed] [Google Scholar]
- [24].Azzi S, Hebda JK, Gavard J, Frontiers in oncology 2013, 3, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang HF, Ran R, Liu Y, Hui Y, Zeng B, Chen D, Weitz DA, Zhao CX, ACS nano 2018, 12, 11600. [DOI] [PubMed] [Google Scholar]
- [26].Brown EB, Campbell RB, Tsuzuki Y, Xu L, Carmeliet P, Fukumura D, Jain RK, Nature medicine 2001, 7, 864. [DOI] [PubMed] [Google Scholar]
- [27].Reyes-Aldasoro CC, Wilson I, Prise VE, Barber PR, Ameer-Beg M, Vojnovic B, Cunningham VJ, Tozer GM, Microcirculation 2008, 15, 65. [DOI] [PubMed] [Google Scholar]
- [28].Curnis F, Sacchi A, Corti A, The Journal of Clinical Investigation 2002, 110, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhu L, Chen L, Cellular & molecular biology letters 2019, 24, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Weaver BA, Molecular biology of the cell 2014, 25, 2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bocci G, Di Paolo A, Danesi R, Angiogenesis 2013, 16, 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Herrmann J, Yang EH, Iliescu CA, Cilingiroglu M, Charitakis K, Hakeem A, Toutouzas K, Leesar MA, Grines CL, Marmagkiolis K, Circulation 2016, 133, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hader SN, Zinkevich N, Norwood Toro LE, Kriegel AJ, Kong A, Freed JK, Gutterman DD, Beyer AM, American journal of physiology. Heart and circulatory physiology 2019, 317, H705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jeong SY, Lee JH, Shin Y, Chung S, Kuh HJ, PloS one 2016, 11, e0159013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Saneja A, Khare V, Alam N, Dubey RD, Gupta PN, Expert opinion on drug delivery 2014, 11, 121. [DOI] [PubMed] [Google Scholar]
- [36].Krawczenko A, Bielawska-Pohl A, Wojtowicz K, Jura R, Paprocka M, Wojdat E, Kozlowska U, Klimczak A, Grillon C, Kieda C, Dus D, PloS one 2017, 12, e0172371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Jain RK, Science 2005, 307, 58. [DOI] [PubMed] [Google Scholar]
- [38].Karaszi E, Jakab K, Homolya L, Szakacs G, Hollo Z, Telek B, Kiss A, Rejto L, Nahajevszky S, Sarkadi B, Kappelmayer J, British journal of haematology 2001, 112, 308. [DOI] [PubMed] [Google Scholar]
- [39].Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK, Cancer research 2000, 60, 2497. [PubMed] [Google Scholar]
- [40].R J, M S, K S, K W, M N, M Z, J Cancer 2016, 7, 1295.27390605 [Google Scholar]
- [41].Klemba A, Purzycka-Olewiecka JK, Wcislo G, Czarnecka AM, Lewicki S, Lesyng B, Szczylik C, Kieda C, Contemp Oncol (Pozn) 2018, 22, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen C, Zhao S, Karnad A, Freeman JW, Journal of hematology & oncology 2018, 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Dowden H, Munro J, Nature reviews. Drug discovery 2019, 18, 495. [DOI] [PubMed] [Google Scholar]
- [44].Ledermann JA, Therapeutic advances in medical oncology 2018, 10, 1758835918768232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ramalingam S, Belani CP, Expert opinion on pharmacotherapy 2004, 5, 1771. [DOI] [PubMed] [Google Scholar]
- [46].Whisler JA, Chen MB, Kamm RD, Tissue engineering. Part C, Methods 2014, 20, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Stylianopoulos T, Munn LL, Jain RK, Trends in cancer 2018, 4, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].McDonald DM, Baluk P, Cancer research 2002, 62, 5381. [PubMed] [Google Scholar]
- [49].Lee SJ, Kim KH, Yoon YS, Current cardiology reports 2018, 20, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Offeddu GS, Possenti L, Loessberg-Zahl JT, Zunino P, Roberts J, Han X, Hickman D, Knutson CG, Kamm RD, Small 2019, 15, e1902393. [DOI] [PubMed] [Google Scholar]
- [51].Goel S, Fukumura D, Jain RK, Proceedings of the National Academy of Sciences of the United States of America 2012, 109, E1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang Y, Chen X, Gueydan C, Han J, Cell research 2018, 28, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Vaidyanathan A, Sawers L, Gannon AL, Chakravarty P, Scott AL, Bray SE, Ferguson MJ, Smith G, British journal of cancer 2016, 115, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nanayakkara AK, Follit CA, Chen G, Williams NS, Vogel PD, Wise JG, Scientific reports 2018, 8, 967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sane R, Mittapalli RK, Elmquist WF, Journal of pharmaceutical sciences 2013, 102, 1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lee SY, Ju MK, Jeon HM, Jeong EK, Lee YJ, Kim CH, Park HG, Han SI, Kang HS, Oxidative medicine and cellular longevity 2018, 2018, 3537471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Pradhan S, Sperduto JL, Farino CJ, Slater JH, Journal of biological engineering 2018, 12, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Chen MB, Lamar JM, Li R, Hynes RO, Kamm RD, Cancer research 2016, 76, 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Chen MB, Hajal C, Benjamin DC, Yu C, Azizgolshani H, Hynes RO, Kamm RD, Proceedings of the National Academy of Sciences of the United States of America 2018, 115, 7022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Li R, Hebert JD, Lee TA, Xing H, Boussommier-Calleja A, Hynes RO, Lauffenburger DA, Kamm RD, Cancer research 2017, 77, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Boussommier-Calleja A, Atiyas Y, Haase K, Headley M, Lewis C, Kamm RD, Biomaterials 2019, 198, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Boussommier-Calleja A, Li R, Chen MB, Wong SC, Kamm RD, Trends in cancer 2016, 2, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Marupudi NI, Han JE, Li KW, Renard VM, Tyler BM, Brem H, Expert opinion on drug safety 2007, 6, 609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.