

Abstract
Microsatellites are short, repetitive segments of DNA, which are dysregulated in mismatch repair-deficient (MMRd) tumors resulting in microsatellite instability (MSI). MSI has been identified in many human cancer types with varying incidence, and microsatellite instability-high (MSI-H) tumors often exhibit increased sensitivity to immune-enhancing therapies such as PD-1/PD-L1 inhibition. Next-generation sequencing (NGS) has permitted advancements in MSI detection, and recent computational advances have enabled characterization of tumor heterogeneity via NGS. However, the evolution and heterogeneity of microsatellite changes in MSI-positive tumors remains poorly described. We determined MSI status in six patients using our previously published algorithm, MANTIS, and inferred subclonal composition and phylogeny with Canopy and SuperFreq. We developed a simulated annealing-based method to characterize microsatellite length distributions in specific subclones and assessed the evolution of MSI in the context of tumor heterogeneity. We identified three to eight tumor subclones per patient, and each subclone exhibited MMRd-associated base substitution signatures. We noted that microsatellites tend to shorten over time, and that MMRd fosters heterogeneity by introducing novel mutations throughout the disease course. Some microsatellites are altered among all subclones in a patient, whereas other loci are only altered in particular subclones corresponding to subclonal phylogenetic relationships. Overall, our results indicate that MMRd is a substantial driver of heterogeneity, leading to both MSI and subclonal divergence.
Keywords: microsatellite instability, mismatch repair, tumor heterogeneity, cancer genomics, hypermutation
Introduction
Microsatellite instability (MSI) arises from defects in the mismatch repair (MMR) system, which includes MSH2, MSH6, MLH1 and PMS2 [1]. Defects in MMR may occur sporadically, for instance via MLH1 promoter hypermethylation [2], or be inherited, such as in Lynch syndrome [3]. MSI-H (microsatellite instability-high) is known to occur frequently in colorectal and endometrial cancer [4], and has been described in multiple other cancer types [5]. During DNA replication, DNA polymerases are prone to slippage in microsatellite regions, which are short (10–60 bp), repetitive segments of DNA found throughout the human genome. This slippage erroneously adds or removes repeat units, which if uncorrected by MMR leads to MSI, or variable microsatellite lengths among affected cells.
Cancers have been increasingly recognized to comprise collections of heterogeneous malignant cells of divergent clonal ancestries, which ultimately arose from a single neoplastic ancestor [6]. Thus, genetically distinct tumor cell populations, or subclones, may develop at different times in different regions of the body. Understanding tumor heterogeneity is important for modeling the evolution and diversification of tumor cells over time while under the influences of carcinogenesis and selective pressures. For instance, heterogeneity may result in tumor subclones with variable metastatic potential [7] or treatment resistance [8]. The presence of tumor heterogeneity also complicates diagnostic testing, as prognostic and predictive biomarkers may be found in some tumor cells but not others [9], and tumor biopsies represent only a small portion of a single tumor region [10]. Since tumor mutations serve as potential neoantigen targets for the immune system, subclonal mutations may contribute to differential sensitivity to immunotherapies [11].
Multiple computational methods have been developed to detect MSI-H utilizing next-generation sequencing (NGS) data, such as mSINGS [12] and MANTIS [13]. Many of these sequencing-based methods have demonstrated superiority over the traditional methods of MSI-PCR [14], which assays the lengths of 5–7 microsatellite loci, and MMR immunohistochemistry (IHC), which measures the expression of the MSH2, MSH6, MLH1, and PMS2 proteins. In addition, modern NGS permits detailed analysis of tumor heterogeneity. Whole exome sequencing (WES) of multiple tumors from the same patient enables characterization of subclonal tumor populations and inference of tumor evolution [15–17] through utilization of software tools such as THetA [18], SuperFreq [19] and Canopy [20].
However, despite these recent NGS-powered break-throughs in both MSI-H testing of individual tumor biopsies and in analysis of tumor heterogeneity using WES of multiple tumor samples, the subclonal genomic heterogeneity in MSI-H tumors remains uncharacterized. Furthermore, the success of immunotherapy in MMR deficient cancers has attracted particular interest in the biology of MSI-H tumors, as recent studies have demonstrated that MSI-H tumors exhibit increased sensitivity and durable response rates to immune checkpoint inhibitors [21]. A better understanding of clonality in MSI-H tumors is important for improved diagnostic testing, tumor sampling, identification and selection of tumor antigen targets for therapy, and prediction of which patients are likely to respond to immunotherapy. In this study, we examine subclonal tumor evolution with respect to mutations and microsatellites in six patients with MSI-H malignancies. We show that MMRd generates a substantial degree of tumor heterogeneity, which is reflected both in subclonal microsatellites and single-nucleotide variants, and that microsatellite instability follows subclonal evolution.
Materials and Methods
Sample acquisition and sequencing
Written informed consent was obtained from all six patients for participation in an IRB-approved study for high-throughput sequencing of tumor and normal specimens (OSU-13053, NCT02090530) at the James Cancer Hospital and The Ohio State University. This study was conducted in accordance with the Declaration of Helsinki. Frozen tumor specimens were obtained from tumor biopsies performed at The Ohio State University, formalin-fixed paraffin-embedded (FFPE) biopsy and surgical specimens were obtained from pathology archives at The Ohio State University and the Mayo Clinic, and matched normal blood samples were collected from each patient. Tumor samples were reviewed for tumor content by a board-certified pathologist. Whole exome sequencing, alignment, and variant calling was performed as previously described [16, 17] (see Supplemental Methods, Sequencing and alignment).
Microsatellite instability analysis
Per-sample microsatellite length distributions and microsatellite instability were called using MANTIS [13] version 1.0.4 run with recommended whole exome settings; min read quality 20, min locus quality 25, and min read length 35. 2551 microsatellite loci (Supplemental File S1) were assessed in each sample; the union of 2539 loci originally used by Salipante et al [12] and 99 loci targeted by our in-house MSI probe panel.
Subclonal inference
Subclonal cancer cell populations were identified using Canopy [20] as previously described [16]. Briefly, the set of somatic single-nucleotide variants (SNVs) in each patient was filtered for ultra-high-confidence SNVs (Supplemental Figure S1, Supplemental Methods, Variant and copy number calling). In addition, rather than utilizing the Frobenius norm of the εM and εm matrices from FALCON [22], missing values in these matrices were imputed using missForest with default settings [23]. Canopy was run with SNVs only for patients 1–5, as no CNVs in those patients passed curation. For all six patients, the maximum permitted simulation runs was increased to at least 100,000. This procedure resulted in phylogenetic trees of tumor subclones, along with the estimated prevalence of each subclone in each tumor sample. We attempted to remove the hysterectomy sample from patient 5 due to its poor tumor purity (10–20%), however Canopy failed to find a tree without it, likely due to lack of clonal diversity among the other patient 5 samples. Note that subclones were not renumbered, therefore subclone numbers do not necessarily correspond to evolutionary relationships. After trees were generated, we performed post hoc assignment of the remaining SNVs (which were not used by Canopy for tree-building) and indels as well as estimating temporal ordering of mutations in each patient as previously described [17].
Analysis of subclonal microsatellite instability
Within a single patient, define K as the number of subclones + 1 (germline), with k = 1 corresponding to germline, and define N as the number of samples. Additionally, define R as the set of microsatellite loci meeting MANTIS coverage thresholds in all samples from this patient, and for any locus r ∈ R define L as the set of observed microsatellite lengths in any k ∈ K. We applied a simulated annealing [24] algorithm (with 50 chains and 50,000 iterations) to learn a matrix for each locus, corresponding to the per-subclone microsatellite length distributions most consistent with per-sample microsatellite distributions and subclonal composition, and its first column fixed to the germline microsatellite distribution (see Supplemental Methods, Subclonal microsatellite estimation).
We benchmarked this approach (see Supplemental Methods, Benchmarking) by computationally mixing sequencing reads from five separate MSI-H tumor samples from different patients and a normal blood sample (Supplemental File S2). With each tumor sample used as a pure subclone, we created five virtual patients with seven virtual samples (mixes of real samples) each, and applied this algorithm to recover the original samples’ microsatellite distributions (Supplemental Figure S2). A locus was considered as recovered in a subclone if its estimated subclonal microsatellite length distribution had a Pearson correlation with the corresponding tumor sample’s microsatellite length distribution yielding P < 0.001 (Supplemental File S3). Our subclonal microsatellite inference method achieved an average recovery rate of 96.2% (s.d. 1.8%). Code for microsatellite inference and benchmarking is available at Code Ocean: https://codeocean.com/capsule/7080731/.
After determining per-subclone microsatellite distributions, we computed MANTIS-equivalent microsatellite instability scores. Although their average in a subclone indicates a relative degree of microsatellite instability, we avoid assigning MSI status to subclones on this basis since MANTIS was originally calibrated with tumor tissue. We classified each locus r in subclone k with score dr(k) as unstable if 1.0 < dr(k) ≤ 2.0 or stable if dr(k) ≤ 1.0 (Supplemental Figure S3). Furthermore, loci unstable in all subclones are termed “ubiquitously unstable”, in more than one but not all subclones “shared unstable”, in one subclone only “private unstable”, and in no subclones “stable”. We estimate expected values (E) for the number of ubiquitous, shared, private, and stable loci through simulating 1000 random distributions of unstable loci in each subclone, and use chi-square tests to compute P values. For significance of overlapping ubiquitously unstable loci across patients, P values were calculated by comparison versus 10,000 random distributions of ubiquitous loci.
We computed normalized most recent common ancestor distances (nMRCA) as follows. Within any subclone k ∈ 2‥K within a patient, we have a set of variants Vk. For any pair of subclones k1, k2 ∈ 2‥K, we have:
| (1) |
We computed the Pearson correlation coefficient between d(k1) and d(k2) of per-locus stepwise differences, and utilized linear regression of these correlations versus nMRCA to quantify the relationship between common clonal ancestry and similarity of microsatellite instability.
The statistical significance of this regression was computed via a bootstrapping scheme. We generated 1000 random P matrices for each of the six patients with the same dimensions as the original matrices, and applied our simulated annealing approach to estimate subclonal microsatellite distributions. For computational considerations, only one chain was run for 10,000 iterations for each set of matrices. Regression between nMRCA and pairwise microsatellite distance correlation was performed as above to generate a set of correlations under this random null.
Results
Whole exome sequencing demonstrates mismatch repair-deficient hypermutation
We performed whole exome sequencing on 31 tumor samples and six matched normal blood samples from six patients (average coverage 218x, Supplemental File S2) with MSI-H metastatic cancers (Figure 1). Clinical histories for all patients are available in the Supplemental Materials. Except for the hysterectomy sample from patient 5, all tumor samples were hypermutated (Figure 2), with an average tumor mutational burden (TMB) of 53.0 mutations/Mb in patient 1 (Lynch syndrome gastric cancer), 23.0 in patient 2 (Lynch colon), 58.1 in patient 3 (somatic MMRd colon), 19.7 in patient 4 (somatic duodenal), 79.3 in patient 5 (somatic endometrial), and 47.7 in patient 6 (somatic prostate). There were 1396 to 5601 total unique mutations (SNVs and indels) with a minimum variant allele fraction (VAF) of 6% detected in each patient (Supplemental Figure S1, Supplemental File S4). Except for the patient 5 hysterectomy sample, all samples were estimated to contain at least 30% tumor cells. We also identified six discrete CNVs in patient 6 (Supplemental File S4). None of the other patients had CNVs passing thresholds.
Figure 1:
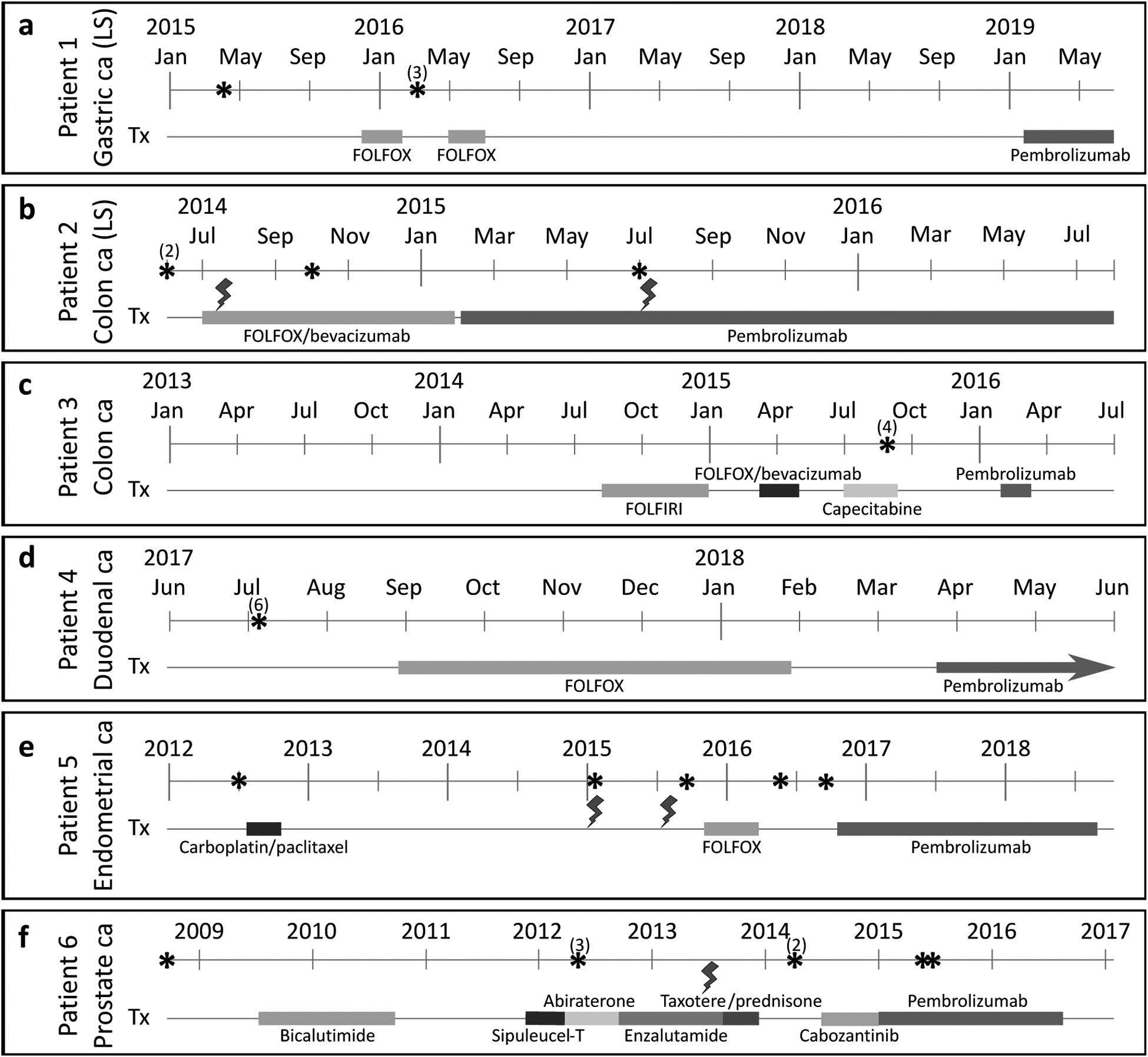
Clinical courses of six patients with MSI-H metastatic cancers. (a): 53-year old female with gastric cancer. (b): 31-year old female with colon cancer. (c): 77-year old female with colon cancer. (d): 67-year old male with duodenal cancer. (e): 63-year old female with endometrial cancer. (f): 85-year old male with prostate cancer. Colored lines represent the duration of each treatment modality. Asterisks delineate time points when samples were acquired, and numbers in parentheses indicate acquisition of multiple samples from the same time point. Lightning symbols denote radiotherapy. FOLFOX: folinic acid, 5-fluorouracil, oxaliplatin. FOLFIRI: folinic acid, 5-fluorouracil, irinotecan. LS: Lynch syndrome.
Figure 2:
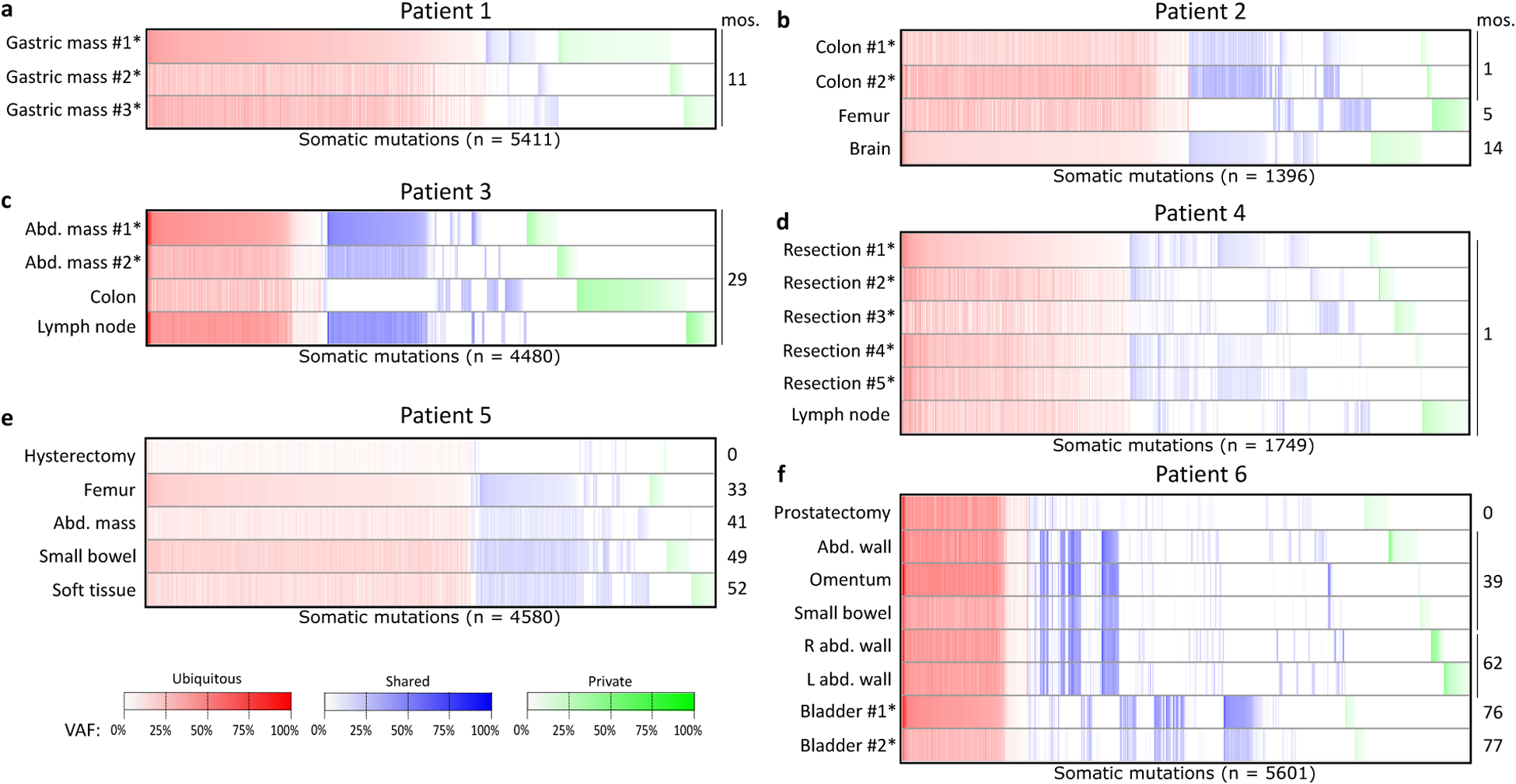
Somatic mutations (SNVs and indels) called in at least one tumor sample from six patients with MSI-H cancers (a–f). The colon polyp sample from patient 1 was excluded due to suspicion of arising from a separate primary tumor than the gastric mass samples. Note that in patient 6, bladder #1 and bladder #2 were acquired from separate surgeries one week apart. Within each heatmap, columns in red represent mutations with at least one alternate-supporting read in all tumor samples (whether or not the variant was called in all samples), columns in blue represent mutations with alternate-supporting reads in one but not all tumor samples, and columns in green represent mutations with alternate-supporting reads in only one tumor sample. Samples marked with asterisks were acquired from a single tumor mass in that patient. mos: months post-diagnosis. Abd: abdominal. VAF: variant allele fraction.
All samples from each patient were called MSI-H by MANTIS (Supplemental File S1) and bore mutational signatures characteristic of MMRd-induced hypermutation (any of signatures 6, 15, 20, and 26) (Supplemental File S4). Patients 1–4 and 6 possessed signature 6 in all samples, and all patient 5 samples contained signature 20. Patient 5 was also notable for signature 14, which in conjunction with signature 20 is associated with combined polymerase proofreading and MMRd [25]. We identified mutations in MMR genes in five of six patients. Patients 1 and 2 had the germline mutations MSH2 p.Q493X and MLH1 p.K618del respectively. All patient 3 and 5 samples contained MMR stop-gain mutations (MLH1 p.Q346X and MSH2 p.Q324X, respectively), and a region of chromo-some 2p containing MSH6 was deleted in all samples from patient 6. For patient 4, an epigenetic modification such as MLH1 hypermethylation is a likely possibility, however IHC was not available to identify specific MMR protein loss. In addition, a POLD1 p.E318K mutation was found in three samples from patient 5.
Next, we assessed the overlap of mutations among tumor samples within each patient (Figure 2, Supplemental Table S1). Groupings of mutations were observed by organ sites, for instance among the patient 2 colon samples and patient 6 abdominal vs. bladder samples. Fewer than 40% of mutations were ubiquitous (present in all tumor samples with at least 6% VAF) in any patient (Supplemental Figure S1), which suggests the presence of substantial heterogeneity among these tumors. Patients 1 and 5 had very low proportions of ubiquitous mutations, instead containing an abundance of private (present in one tumor sample) and shared (neither ubiquitous nor private) mutations. Within patient 1, the colon polyp sample contained almost exclusively private mutations, with only 50 of its 2407 mutations (2.1%) found in any of the three gastric mass samples. In contrast, the three gastric masses were genetically more similar, with 1489 of the 5411 total mutations (27.5%) in this patient present in every gastric sample. Of the 2836 mutations called in at least one gastric mass sample but not the colon polyp, only 42 (1.5%) had at least five alternate-supporting reads in the colon polyp. As 47 (1.7%) of those mutations had at least five alternate-supporting reads in patient 1’s abdominal mass, and in conjunction with the neighbor-joining tree of these samples (Supplemental Figure S4), the colon polyp likely represents a separate primary and was therefore excluded from further analysis, which focuses on her gastric cancer. In patient 5 (Supplemental Figure S1), 4283 of the 4580 total mutations were not called in the hysterectomy sample at 6% VAF. This is most likely due to its low tumor purity, as 1179 of these 4283 mutations (27.5%) had at least five alternate-supporting reads in the hysterectomy sample (Figure 2). Based on this substantial overlap, we conclude that the 2015 and 2016 samples represent recurrence of the 2012 endometrial cancer, contrary to the clinical diagnosis of separate primaries.
Quantification and phylogenetic classification of tumor subclones
We applied Canopy [20], post hoc tree assignment of mutations, and relative mutational ordering (see Methods, Subclonal inference) within each patient to identify tumor subclones and determine their phylogeny (Figure 3, Supplemental File S5), along with SuperFreq [19] as an alternate method (see Supplemental Methods). For Canopy, the number of subclones yielding the lowest BIC (Bayesian Information Criterion) score was selected for patients 1–5, however for patient 6 an eight-subclone model was manually chosen due to its comparable BIC with the optimal nine-subclone model (Supplemental Figure S5). For brevity, we refer to Canopy subclones in patients as P#C#; e.g. P2C3 refers to patient 2 subclone 3.
Figure 3:
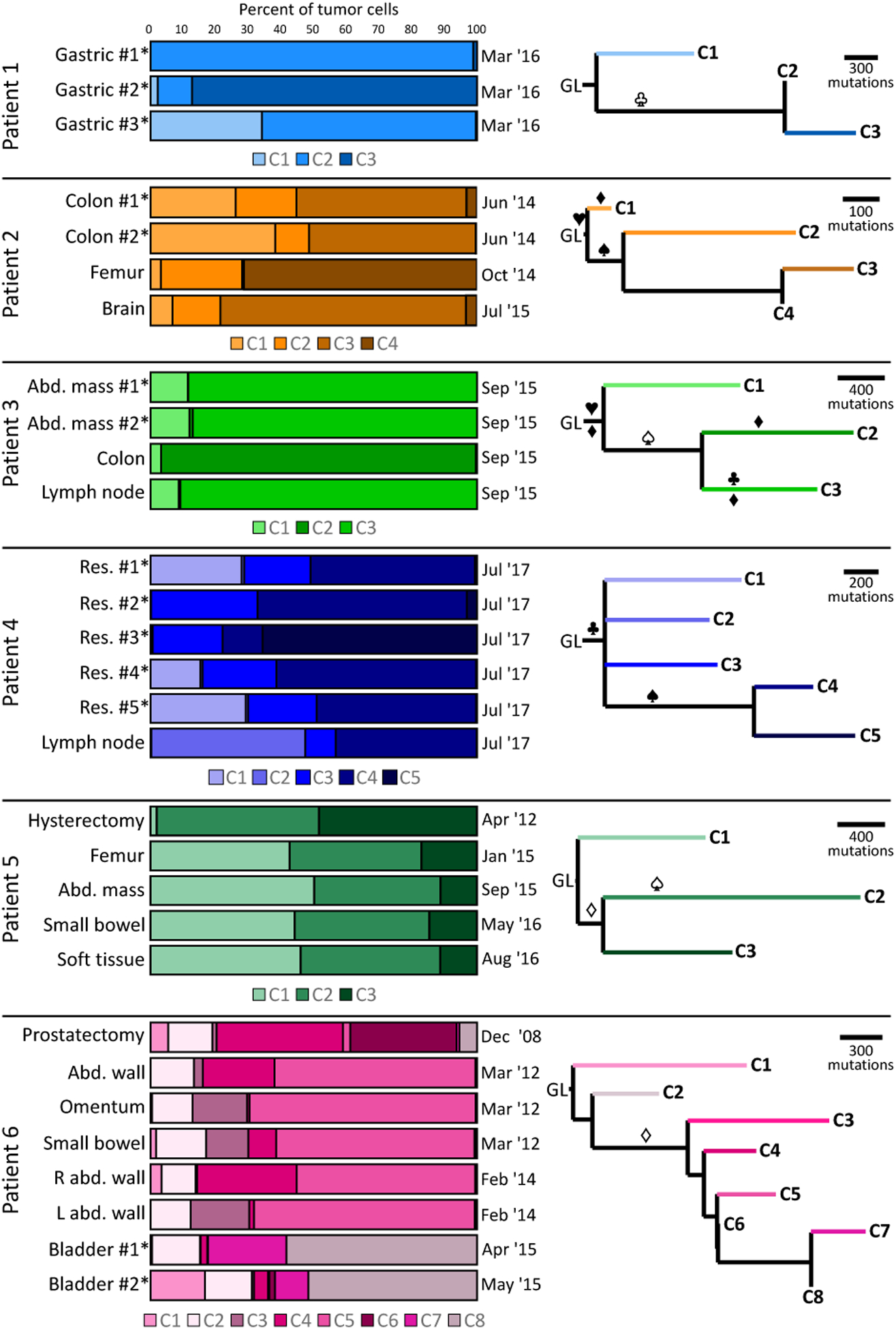
Fractions of tumor subclones and estimated subclonal phylogenetic trees within six patients with MSI-H cancers. Three subclones were identified in patients 1, 3 and 5, four in patient 2, five in patient 4, and eight in patient 6. Listed fractions represent the estimated proportion of tumor cells in each sample derived from each subclone. Sample acquisition dates are listed above. For patients 1–4, sequentially numbered samples were obtained from the same surgeries. Note that in patient 6, bladder #1 and bladder #2 were acquired from separate surgeries one week apart. For each tree, horizontal distance corresponds with increased number of mutations. Samples marked with asterisks were acquired from a single tumor mass in that patient. Symbols on the tree correspond to mutations present in a branch, with unfilled symbols indicating potentially ambiguous mutation positions based on relative statistical likelihood of assignment to other branches, and filled symbols indicating more certain position. ♦: MMR gene (MLH1, MSH2, MSH6, or PMS2). ♥: CTNNB1. ♠: KRAS. ♣: TP53. Res: resection. Abd: abdominal. GL: germline.
Varying numbers of subclones were identified by Canopy in these six patients, from three subclones in patients 1, 3 and 5, to eight subclones in patient 6. Intratumor heterogeneity was particularly pronounced in patients 2, 4, 5 and 6, while samples in patients 1 and 3 tended to consist of pure subclones (with the exception of patient 1 gastric mass #3). In contrast, some level of intertumor heterogeneity was evident in all patients. At least one clonally distinct sample was present in each patient, for instance subclones P1C1, P1C3, P2C4, P3C2, P4C5, and P6C6 were only prominent in one sample each, and patient 5’s hysterectomy sample uniquely lacked P5C1. In addition, P2C2, P3C1, P4C3, and P6C2 were found at low levels in all samples from these patients. Consistent with the presence of substantial clonal diversity, SuperFreq identified three to sixteen clones in each patient (Supplemental Figure S6, Supplemental File S6).
Patients 5 and 6 demonstrated clonal shifts with time. P5C1 was essentially absent in the hysterectomy sample, and emerged in the recurrent tumors sampled in 2015 and 2016. In patient 6, P6C6 was only present in the earliest sample (prostatectomy, from 2008), and absent at all later time points. The prostatectomy contained substantial P6C4 as well, which was also seen in abdominal wall samples from 2012 and 2014, but not in either 2015 sample. P6C5 was predominant in all of the 2012 and 2014 samples from the abdomen, and P6C3 was seen in two 2012 samples (omentum and small bowel) and one 2014 sample (left abdominal wall). P6C8 was predominant in both 2015 bladder samples, which also uniquely contained P6C7.
All of the phylogenetic trees inferred by Canopy for these six patients were notable for very short trunks (Figure 3) containing less than 4% of mutations (Supplemental File S6). Short trunks were also found by SuperFreq, with less than 12% of mutations truncal in all patients except patient 1 (Supplemental Figure S6). Despite these short trunks, potentially driver truncal mutations were identified in five patients. Patients 2 and 3 had truncal CTNNB1 mutations (p.T41A and p.S45F respectively), both known driver mutations in colorectal cancer [26], and patient 4 harbored a truncal TP53 p.R273C mutation. Patient 1 possessed a TP53 p.R175H mutation in clones 2–3, however its log-likelihood of assignment to the trunk was similar, suggesting it may have been a truncal driver mutation misplaced by Canopy (Supplemental Table S2). The truncal branch of patient 6 was notable for LYST p.P3167S, predicted as damaging by DANN with a score of 0.999 [27]. LYST mutations have previously been described as likely drivers in chordoma [28].
Known and candidate pathogenic mutations were also found in specific subclones, most notably KRAS p.G12D in P2C2–C4 and P5C2, p.G13D in P3C2–C3, and p.G12V in P4C4–C5. However, the KRAS assignments in patients 3 and 5 were ambiguous due to similar log-likelihoods (Supplemental Table S3), leaving open the possibility that this is a truncal driver mutation. This likely occurred because of P3C1’s low prevalence in all samples, and the poor hysterectomy tumor purity along with low intertumor heterogeneity among the other patient 5 samples. Other notable mutations included FBXW7 p.R385H, APC p.P2712L and GNAS p.R186H in P1C2-P1C3, ERBB2 p.V842I in P3C2–C3, and TP53 p.P278L and PIK3CA p.C378R in P3C3. Patient 6 possessed six separate coding mutations in the androgen receptor gene AR: p.V904A in P6C1, p.C570W and p.S889G in P6C3, p.W742C and p.T878A in P6C5, and a stop-loss mutation c.*1232T>A in subclones P6C5–C8. These mutations, previously implicated in resistance to bicalutamide, enzalutamide and abiraterone [29], demonstrates emergence of diverse subclonal anti-androgen resistance mechanisms, likely in response to the three different anti-androgen therapies received by this patient.
Most Canopy-defined subclones contained hundreds to thousands of mutations, characteristic of hypermutation (Figure 3). MMRd-associated signatures (6, 15, 20, or 26) were identified in all subclones in all patients (Supplemental File S6), along with all branches but one with at least 50 SNVs. All four subclones in patient 2 were also notable for signature 12 (currently unknown etiology), which was found in at least one subclone in all other patients. Patient 3 had a truncal MLH1 p.Q346X mutation along with MSH6 p.A1055T in P3C2 and PMS2 p.S517N in P3C3. Patient 5 contained a MSH2 p.Q324X mutation, assigned to P5C2–C3 but with similar log-likelihood to a truncal assignment (Supplemental Table S4), and called by SuperFreq as truncal (Supplemental File S6). Also of interest was a MCM9 p.G302X mutation in P5C2, affecting a gene previously associated with MMRd [30]. In patient 6, two somatic frameshift mutations in MSH6, p.F1088fs*2/c.3254del and p.F1104fs*11/c.3306del, along with a single copy loss of MSH6, were found in subclones P6C3–C8, however, their assignment to this branch was ambiguous (Supplemental Table S5). The p.F1088fs*2/c.3254del mutation was a deletion in a (C)8 repeat, and the p.F1104fs*11/c.3306del mutation was a deletion in a (T)7 repeat, suggesting that these were secondary to pre-existing MMRd not identified in the phylogenetic tree [31]. As these mutations were early in the branch leading to P6C3–C8 (15th and 18th, respectively), a MMR deficiency mechanism preceding them would likely have been present in P6C2 as well.
Assessment of microsatellite loci within subclones
In order to interrogate microsatellites within each subclone identified by Canopy, we applied machine learning to estimate microsatellite length distributions within individual subclones (Supplemental File S7, Supplemental Figure S7). The hysterectomy sample from patient 5 was excluded from this analysis due to its low tumor purity (10–20%). We computed MANTIS-equivalent measures of per-subclonal aggregate instability (Figure 4), and found that all subclones in all patients except for P2C1 exhibited substantially increased instability compared to those patients’ tumor samples. Patients 1, 2, and 6 contained direct ancestor to descendant subclonal relationships, providing a microcosm of linear evolution within otherwise branched trees. MANTIS score correspondingly increased along the P1C2–C3 and P6C6–C8–C7 lineages. Patient 2 did not follow this pattern, as P2C4 had a higher MANTIS score compared to its direct descendant P2C3. This may be explained by the fact that subclone 3 was identified in the 2015 brain sample as well as 2014 samples (Figure 3), and subclone 4 was only found in a mid-2014 sample, therefore subclone 3 reflects an increased length of time for microsatellites to diverge. This may also be due to the relatively low average coverage of microsatellites by subclone 4 (29.3x) compared to subclone 3 (82.5x). Increased time for microsatellite divergence could also contribute to the increase in instability from P6C6 to P6C8.
Figure 4:
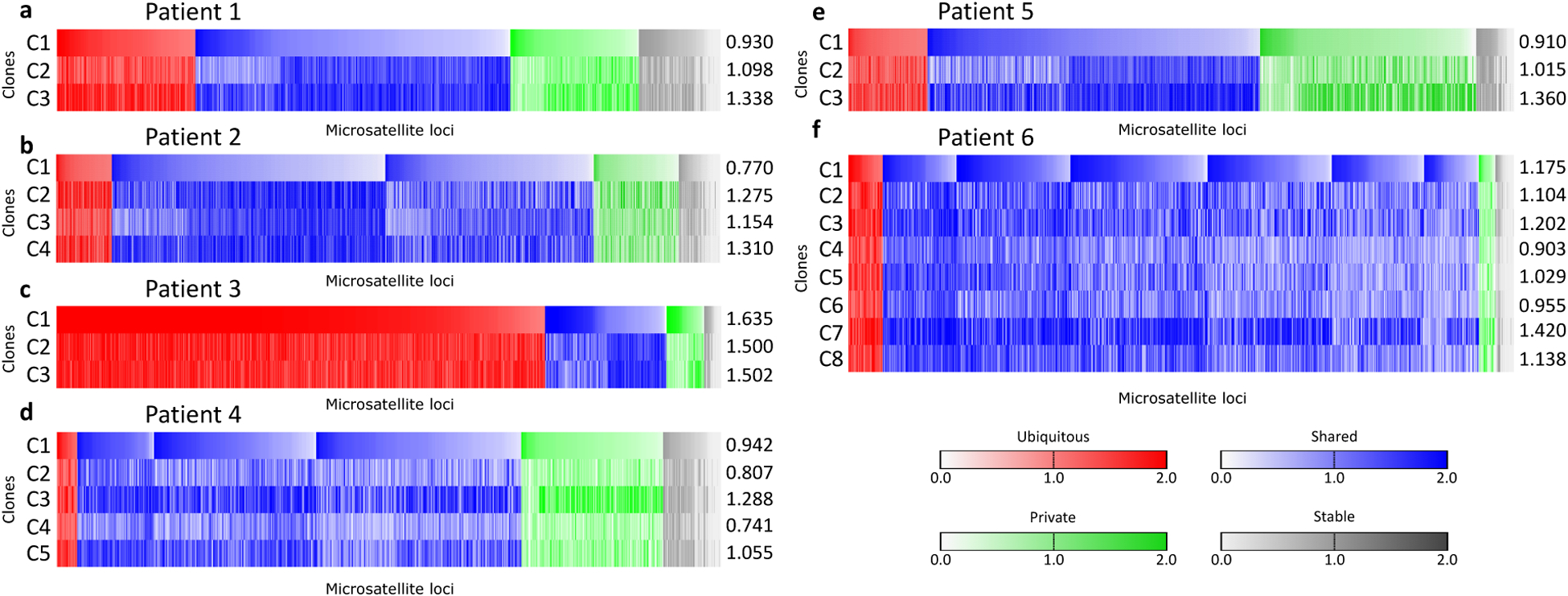
MANTIS scores of 714 microsatellite loci from 3 subclones in patient 1 (a), 1098 loci from 4 subclones in patient 2 (b), 1203 loci from 3 subclones in patient 3 (c), 655 loci from 5 subclones in patient 4 (d), 663 loci from 3 subclones in patient 5 (e), and 756 loci from 8 subclones in patient 6 (f). Loci with MANTIS score ≥ 1.0 were classified as unstable. Numbers to the right of each plot indicate the MANTIS-equivalent scores of each subclone, computed by averaging across all loci.
We next investigated subclonal microsatellite changes in individual loci, and found a pattern of unstable microsatellites corresponding to subclonal phylogeny. Per-locus subclonal MANTIS scores were stratified into unstable and stable groups (see Methods, Analysis of subclonal microsatellite instability). Loci were classified across subclones as “ubiquitous” if unstable in all subclones, “shared” if unstable in more than one but not all clones, “private” if unstable in only one subclone, or “stable” if not unstable in any subclone (Figure 4). 397 loci were unstable in P1C2–C3 (E ≈ 336.6, P < 0.001), consistent with their common ancestor, along with 420 loci unstable in subclones P2C2–C4 (irrespective of P2C1) (E ≈ 348.5, P ≈ 0.002). This trend was not seen for P5C2–C3, which had 267 unstable loci in common (E ≈ 264.6, n.s.), possibly due to their lesser degree of common ancestry vs. P2C3–C4 and P3C2–C3. The overall distributions of ubiquitous/shared/private/stable loci were statistically different than expected for patients 1–4 and 6 (P < 0.001 for each), but not for patient 5 (P ≈ 0.09) (Figure 5a). Patients 1–4 and 6 also possessed significantly more stable loci than expected, consistent with inheritance of unstable microsatellites rather than uniform selection of unstable loci among subclones. In addition, we note that all subclones from all patients demonstrated a negative shift of median microsatellite length (average median change −0.89, s.d. 0.59), indicating that microsatellite loci tend to shorten in MMRd cells (Supplemental Figure S8). This shift was less pronounced in P1C1, P2C1, P4C4 and P5C1, reflecting their lower level of microsatellite instability than other subclones. Similarly, this shift was particularly evident in patient 3’s highly unstable subclones.
Figure 5:
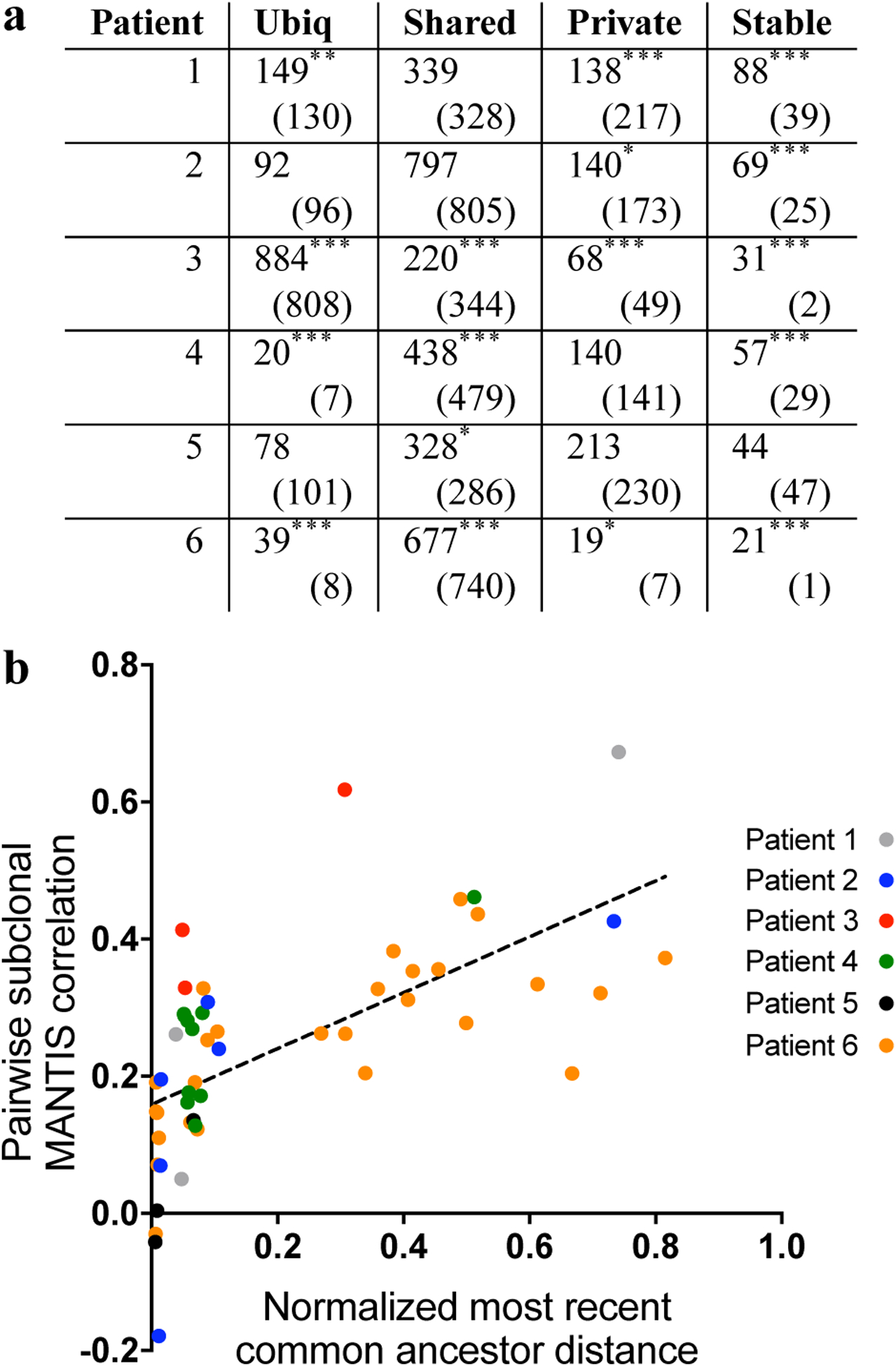
(a): Unstable microsatellites are more commonly shared among clones than expected. Ubiquitous loci are defined as unstable in all clones, shared loci unstable in more than one but not all clones, private loci unstable in one subclone, and stable loci as not unstable in any subclone. Numbers in parentheses represent the expected amount of loci in each category from each patient. Ubiq: ubiquitous. *: P < 0.05. **: P < 0.01. ***: P < 0.001. (b): Subclonal MANTIS score correlates with common clonal ancestry. Normalized most recent common ancestor distance refers to the proportion of mutations in common between two clones versus the most mutated subclone (see Equation 1).
Assessing loci across patients, we found that no loci were ubiquitously unstable in all six patients. Two loci were ubiquitously unstable in five patients; chr8:100287518–100287535, upstream of exon 19 of VPS13B, a gene previously implicated in small cell lung cancer [32], along with chr11:102080326–102080340, downstream of exon 6 of YAP1. We next sought to further investigate the relationship of subclonal microsatellite instability with tumor phylogeny, and found that development of instability in microsatellites follows subclonal evolution. Pooling subclones from all six patients, pairwise normalized common ancestor distance (see Equation 1) moderately correlated with pairwise subclonal microsatellite MANTIS scores (r = 0.638, P < 10−5 of regression) (Figure 5b). This was highly statistically significant (P < 10−3, one-sided test) versus a null hypothesis of no clonal relation. This correlation rose when limited to the three patients (2, 5, 6) with samples available from multiple time points (r = 0.712, P < 10−5 of regression). As our approach for subclonal microsatellite inference does not utilize phylogenetic information, this indicates its ability to recapitulate features of subclonal evolution via analysis of microsatellites independent of the SNV and CNV-based evolutionary trees from Canopy.
Discussion
Tumor heterogeneity can impact diagnostic testing, therapy selection and efficacy, and tumor antigen selection [8]. For instance, heterogeneity has been shown to affect metastasis of colorectal cancer [33] and clinical outcomes in renal cell carcinoma [34]. Previous studies have demonstrated histologic heterogeneity within different regions of MSI-H tumors [35] and that heterogeneity impacts the accuracy of MSI-PCR and IHC [36, 37]. In this study, we utilized multi-sample sequencing to characterize temporal and spatial evolution of microsatellites and subclonal heterogeneity within six patients with MSI-H malignancies, modeling both acquisition of discrete mutations (SNVs/indels/CNVs) and changes in microsatellite loci. We demonstrate that microsatellite instability follows subclonal evolution, while instability at specific loci appears to be a fundamentally stochastic process (Figure 6).
Figure 6:
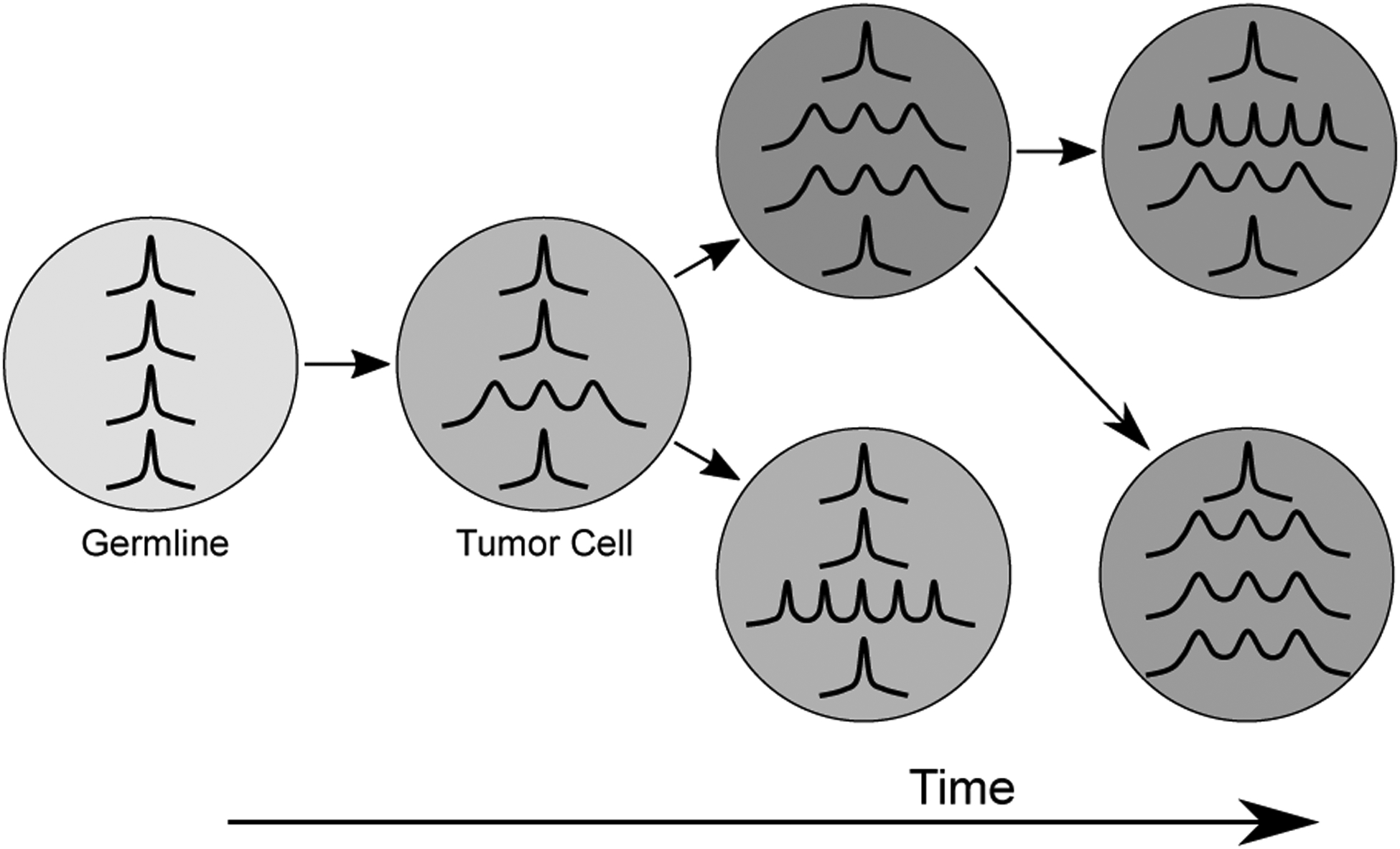
Hypothesized model of clonal microsatellite instability. In this model, mismatch repair deficiency leading to MSI develops early in tumor evolution, and is present in the founding tumor cell and all descendent subclones. As MMRd is stochastic, it introduces insertions and deletions at different loci in different cells, which fosters tumor heterogeneity. MMRd continues to cause MSI throughout the development of the cancer, which increases the level of microsatellite divergence within subclones over time. Instability at specific loci is preserved and expanded within the progeny of affected cells.
Through the evaluation of 31 tumor samples in six patients, three of these patients having samples from multiple time points, we observed branching evolution, dynamic accumulation of mutations, and unique subclones. We noted a substantial level of heterogeneity in patients 1–4 and 6 (Figure 3), with analysis in patient 5 likely limited by poor sample quality. Patients 2–3 and 4–6 demonstrated a branched pattern of evolution [38] (patient 1 having insufficient branches to classify), consistent with continued accumulation of mutations throughout the disease course and selection for subclonal driver mutations. All six patients responded to immunotherapy, and five of them displayed branching rather than neutral evolution. We speculate that the emergence of subclones containing particularly potent neoantigens was selected against by the host immune system, thus necessitating PD-1 axis inhibition to enable immunity against the subclones we detected. Branching evolution can also be driven by subclonal acquisition of mutations conferring selective advantages (such as immune evasion), enabling them to outcompete other subclones. Though five of the six patients received FOLFOX chemotherapy at some point, MMRd signatures were prominent throughout all phylogenetic trees. Taken together, these evolutionary patterns show intrinsic MMRd as a persistent generator of mutations leading to heterogeneity in MSI-H cancers. Contrary to Sveen et al [39], which found high proportions of truncal mutations in MSI-H tumors (median of 85%), we made a striking observation that less than 4% of mutations were truncal in any of these six patients. This is likely due to usage of multiple tumor samples in this study, as subclones present at similar frequencies in a single sample are difficult to separate without diversity conferred by other samples [40]. We identified multiple examples of subclones unique to particular time points and organ sites in our study, representing diversity which cannot be captured by a single sample.
We observed intra-patient tumor heterogeneity in patients with MSI-H cancers. For instance, we identified a non-hypermutated subclone P2C1 in patient 2 (TMB 2.1 mutations/Mb), which possessed a mutation in CTNNB1 but not the KRAS mutation found in P2C2–C4. We suspect that P2C1 may represent remnants of a pre-malignant population of cells derived from the polyp, with lower levels of MSI, which eventually gave rise to cancer in this patient. P2C1 was essentially unique to the colon samples (which included the right colon primary site), and this fits with well-described genetic events for the transformation of colorectal adenoma to become carcinoma [41], in which Wnt pathway (CTNNB1) alterations precede KRAS mutations. This would also be consistent with recent observations of increased MSI in endometrial carcinoma versus paired pre-malignant tissue [42]. In patient 6, we detected six AR mutations in four different phylogenetic branches, potentially accounting for resistance to multiple antiandrogen therapies during his long disease course. Multiple co-existing AR mutations have been previously documented in MSS prostate cancer in both individual tissue samples [43] and cell-free DNA (cfDNA) [29]. Patients 2, 3, 4 and 6 contain genetic features typical of MSS malignancies of each type, indicating that MMRd can coexist with or potentially even account for other oncogenic pathways.
We found MSI in all subclones in all six patients, with differing levels of MSI corresponding to subclonal timing and phylogeny. Admixture of different subclones has the potential to complicate NGS-based diagnostic tests for MSI, as higher tumor purity would be necessary to detect MSI in less unstable subclones. This is consistent with tumor and cfDNA testing for MSI, in which limit of detection and sensitivity decrease in samples with lower tumor content. We found that the time points of samples containing subclones substantially influenced the level of microsatellite instability. For instance, P2C1 was exclusive to earlier time points than P2C2–C4, which provides an alternative explanation for the lower level of instability in P2C1. Subclone P6C7 exhibited substantially higher microsatellite instability than P6C1–C6, likely because it was unique to the 2015 bladder biopsies and a descendant of P6C8, while other clones were observed at earlier time points. Consistent with multiple previous studies [44, 45], we noted that unstable microsatellites preferentially shorten in length rather than expand. Notably, we found that locus-specific instability corresponded to subclonal evolution and that microsatellite analysis recapitulated phylogenetic relationships. With further refinement, this finding may enable usage of microsatellites to “fingerprint” tumor subclones in patients during treatment.
This study is subject to multiple limitations, primarily stemming from next-generation sequencing and clonality. Our analysis of MSI most depends on accurate estimation of subclonal prevalence. Accurate subclone detection requires precise VAFs, which in turn depend on sufficient coverage and error rates. Although we enforce a minimum coverage of 100x in all samples for tree-building mutations, the remaining mutations retroactively assigned to the tree can suffer from the granularity of discrete reads at low coverage. Sequencing error can also confound our deconvolution of subclonal microsatellite distributions despite the error filters provided by MANTIS, as microsatellite regions are difficult to sequence with current technology [46]. Our analysis was limited by the low quantity of tumor samples available, especially in patient 1, and in patient 5 by poor tumor cell content. Improved coverage and increased quality and quantity of tumor specimens can mitigate these issues.
The results from this study lend themselves to several avenues of further investigation. Inclusion of more samples from more patients would permit further investigation of the trends identified in this study, both by increasing the power to resolve subclones and by providing more exemplars of subclonal microsatellites. Furthermore, a larger sample size may permit cohort studies to identify differences in microsatellite evolution due to Lynch status, cancer type, specific MMR alterations, or other covariates. Research autopsy, as performed by our group [16] and others, may provide an excellent resource for high-quality tumor samples. Of clinical importance, inclusion of patients who do not respond to immunotherapies or who relapse may yield additional insights into their subclonal effects on microsatellites, especially in conjunction with emerging methods of neoantigen prediction [47] and findings of microsatellite alleles relevant to host CD8+ T cell response [48]. An expanded mathematical model including time points could enable joint tracking of temporal and spatial evolution of microsatellite alleles, with the potential to guide therapy selection in early and late disease. Such an integrated model, building on the model developed in this study, may provide direct clinical benefit through forecasting of checkpoint inhibitor sensitivity.
In conclusion, we utilized multi-sample sequencing from multiple time points to infer subclonal phylogeny within six patients with MSI-H malignancies, identifying genetically distinct tumor subclones and branching patterns of subclonal evolution which reflect ongoing selective pressure. Improved understanding of tumor heterogeneity in MSI-H cancers has implications for improved diagnostic testing, overcoming resistance to immunotherapy, anti-tumor vaccine development, and treatment decisions over time.
Supplementary Material
Statement of Implication:
We leveraged subclonal inference to assess clonal evolution based on somatic mutations and microsatellites, which provides insight into MMRd as a dynamic mutagenic process in MSI-H malignancies.
Acknowledgements
SR is supported by an American Cancer Society grant MRSG-12-194-01-TBG, the Prostate Cancer Foundation, NCI UH2CA202971, NCI UH2CA216432, American Lung Association, and Pelotonia. RB is supported by NIGMS T32GM068412 and a Pelotonia Graduate Research Fellowship. AP is supported by a Pelotonia Post-Doctoral Research Fellowship. MAK is supported by NCATS TL1TR002735. HZC is supported by NCI K08CA241309, a Pelotonia Post-Doctoral Research Fellowship, and an American Society of Clinical Oncology Young Investigator Award.
We are grateful for administrative support from Jenny Badillo, the Comprehensive Cancer Center at The Ohio State University, computational resources from the Ohio Supercomputer Center (OSC), community support from Pelotonia, technical assistance with SuperFreq from Gregory Wheeler, PhD (Nationwide Children’s Hospital, Columbus, OH), and most importantly the patients included in this study and their families. Sequencing reads from all six patients in this cohort have been deposited in dbGaP (https://ncbi.nlm.nih.gov/gap) under accession number phs001925.v1.p1.
Footnotes
Competing interests
SR participated in Advisory Boards for Incyte Corporation (2017), AbbVie, Inc. (2017), and QED Therapeutics (2018, 2019). SR received honoraria from IDT Integrated DNA Technologies (2017) and Illumina (2018). SR received consulting fees from QED Therapeutics (2018). SR received travel reimbursement (less than $999 USD) from Incyte Corporation (2019).
References
- 1.Strand M, Prolla TA, Liskay RM & Petes TD Destabilization of Tracts of Simple Repetitive DNA in Yeast by Mutations Affecting DNA Mismatch Repair. Nature 365, 274–276 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Kane MF et al. Methylation of the hMLH1 Promoter Correlates with Lack of Expression of hMLH1 in Sporadic Colon Tumors and Mismatch Repair-defective Human Tumor Cell Lines. Cancer Research 57, 808–811. ISSN: 0008–5472. eprint: http://cancerres.aacrjournals.org/content/57/5/808.full.pdf. http://cancerres.aacrjournals.org/content/57/5/808 (1997). [PubMed] [Google Scholar]
- 3.Lynch HT, Shaw MW, Magnuson CW, Larsen AL & Krush AJ Hereditary Factors in Cancer: Study of Two Large Midwestern Kindreds. Archives of Internal Medicine 117, 206–212. ISSN: 0003–9926. eprint: https://jamanetwork.com/journals/jamainternalmedicine/articlepdf/572426/archinte_117_2_009.pdf. 10.1001/archinte.1966.03870080050009 (February. 1966). [DOI] [PubMed] [Google Scholar]
- 4.Watson P & Lynch HT The Tumor Spectrum in HN-PCC. Anticancer research 14, 1635–1639. ISSN: 0250–7005. http://europepmc.org/abstract/MED/7979199 (1994). [PubMed] [Google Scholar]
- 5.Bonneville R et al. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precision Oncology 1, 1–15. 10.1200/PO.17.00073 (October. 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gerlinger M et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. New England Journal of Medicine 366. 883–892. 10.1056/NEJMoa1113205 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong WS, Shpak M & Townsend JP Inferring the Origin of Metastases from Cancer Phylogenies. Cancer Research 75, 4021–4025. ISSN: 0008–5472. eprint: http://cancerres.aacrjournals.org/content/75/19/4021.full.pdf. http://cancerres.aacrjournals.org/content/75/19/4021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGranahan N & Swanton C Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 168, 613–628. ISSN: 0092–8674. http://www.sciencedirect.com/science/article/pii/S0092867417300661 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Gerashchenko TS et al. Intratumor Heterogeneity: Nature and Biological Significance. Biochemistry (Moscow) 78, 1201–1215. ISSN: 1608–3040 (November. 2013). [DOI] [PubMed] [Google Scholar]
- 10.Stanta G, Jahn SW, Bonin S & Hoefler G Tumour Heterogeneity: Principles and Practical Consequences. Virchows Archiv 469, 371–384. ISSN: 1432–2307 (October. 2016). [DOI] [PubMed] [Google Scholar]
- 11.Lu Y & Robbins PF Cancer Immunotherapy Targeting Neoantigens. Seminars in Immunology 28. T cell therapies for cancer, 22–27. ISSN: 1044–5323. http://www.sciencedirect.com/science/article/pii/S1044532315000731 (February. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salipante SJ, Scroggins SM, Hampel HL, Turner EH & Pritchard CC Microsatellite Instability Detection by Next Generation Sequencing. Clinical Chemistry 65. ISSN: 0009–9147. eprint: http://clinchem.aaccjnls.org/content/early/2014/06/23/clinchem.2014.223677.full.pdf. http://clinchem.aaccjnls.org/content/early/2014/06/23/clinchem.2014.223677 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Kautto E et al. Performance Evaluation for Rapid Detection of Pan-Cancer Microsatellite Instability With MANTIS. Oncotarget 8, 7452–7463 (January. 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boland CR et al. A National Cancer Institute Work-shop on Microsatellite Instability for Cancer Detection and Familial Predisposition: Development of International Criteria for the Determination of Microsatellite Instability in Colorectal Cancer. Cancer Research 58, 5248–5257. ISSN: 0008–5472. eprint: http://cancerres.aacrjournals.org/content/58/22/5248.full.pdf. http://cancerres.aacrjournals.org/content/58/22/5248 (1998). [PubMed] [Google Scholar]
- 15.Savas P et al. The Subclonal Architecture of Metastatic Breast Cancer: Results from a Prospective Community-Based Rapid Autopsy Program “CASCADE”. PLOS Medicine 13, 1–25. 10.1371/journal.pmed.1002204 (December. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen H-Z et al. Genomic Characterization of Metastatic Ultra-Hypermutated Interdigitating Dendritic Cell Sarcoma through Rapid Research Autopsy. Oncotarget 10, 277–288 (January. 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krook MA et al. Tumor Heterogeneity and Acquired Drug Resistance in FGFR2-Fusion-Positive Cholangiocarcinoma through Rapid Research Autopsy. Molecular Case Studies 5, a004002. eprint: http://molecularcasestudies.cshlp.org/content/5/4/a004002.full.pdf+html. http://molecularcasestudies.cshlp.org/content/5/4/a004002.abstract (August. 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oesper L, Mahmoody A & Raphael BJ THetA: Inferring Intra-Tumor Heterogeneity from High-Throughput DNA Sequencing Data. Genome Biology 14, R80. ISSN: 1474–760X. 10.1186/gb-2013-14-7-r80 (July 2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flensburg C, Sargeant T, Oshlack A & Majewski IJ SuperFreq: Integrated Mutation Detection and Clonal Tracking in Cancer. PLOS Computational Biology 16, 1–21. 10.1371/journal.pcbi.1007603 (February. 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang Y, Qiu Y, Minn AJ & Zhang NR Assessing Intratumor Heterogeneity and Tracking Longitudinal and Spatial Clonal Evolutionary History by Next-Generation Sequencing. Proc. Natl. Acad. Sci. U.S.A 113, E5528–5537 (September. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le DT et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. New England Journal of Medicine 372. 2509–2520. 10.1056/NEJMoa1500596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Bell JM, Zavala NA, Ji HP & Zhang NR Allele-Specific Copy Number Profiling by Next-Generation DNA Sequencing. Nucleic Acids Res. 43, e23 (February. 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stekhoven DJ & Bühlmann P MissForest—Non-Parametric Missing Value Imputation for Mixed-Type Data. Bioinformatics 28, 112–118. ISSN: 1367–4803. eprint: http://oup.prod.sis.lan/bioinformatics/article-pdf/28/1/112/583703/btr597.pdf. 10.1093/bioinformatics/btr597 (October. 2011). [DOI] [PubMed] [Google Scholar]
- 24.Kirkpatrick S, Gelatt CD & Vecchi MP Optimization by Simulated Annealing. Science 220, 671–680. ISSN: 0036–8075. eprint: http://science.sciencemag.org/content/220/4598/671.full.pdf. http://science.sciencemag.org/content/220/4598/671 (1983). [DOI] [PubMed] [Google Scholar]
- 25.Haradhvala NJ et al. Distinct Mutational Signatures Characterize Concurrent Loss of Polymerase Proofreading and Mismatch Repair. Nature Communications (May 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anwar M et al. Frequent Activation of the β-Catenin Gene in Sporadic Colorectal Carcinomas: A Mutational & Expression Analysis. Molecular Carcinogenesis 55, 1627–1638. https://onlinelibrary.wiley.com/doi/abs/10.1002/mc.22414 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Quang D, Chen Y & Xie X DANN: a Deep Learning Approach for Annotating the Pathogenicity of Genetic Variants. Bioinformatics 31, 761–763 (March. 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tarpey PS et al. The Driver Landscape of Sporadic Chordoma. Nat Commun 8, 890 (October. 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lallous N et al. Functional Analysis of Androgen Receptor Mutations that Confer Anti-Androgen Resistance Identified in Circulating Cell-Free DNA from Prostate Cancer Patients. Genome Biology 17, 10. ISSN: 1474–760X. 10.1186/s13059-015-0864-1 (January. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Traver S et al. MCM9 Is Required for Mammalian DNA Mismatch Repair. Molecular Cell 59, 831–839. ISSN: 1097–2765. http://www.sciencedirect.com/science/article/pii/S1097276515005687 (August. 2015). [DOI] [PubMed] [Google Scholar]
- 31.Shia J et al. Secondary Mutation in a Coding Mononucleotide Tract in MSH6 causes Loss of Immunoexpression of MSH6 in Colorectal Carcinomas with MLH1/PMS2 Deficiency. Modern Pathology 26. Original Article, 131–138. 10.1038/modpathol.2012.138 (August. 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwakawa R et al. Expression and Clinical Significance of Genes Frequently Mutated in Small Cell Lung Cancers Defined by Whole Exome/RNA Sequencing. Carcinogenesis 36, 616–621. ISSN: 0143–3334. eprint: http://oup.prod.sis.lan/carcin/article-pdf/36/6/616/774342/bgv026.pdf. 10.1093/carcin/bgv026 (April. 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Joung J-G et al. Tumor Heterogeneity Predicts Metastatic Potential in Colorectal Cancer. Clinical Cancer Research 23, 7209–7216. ISSN: 1078–0432. eprint: http://clincancerres.aacrjournals.org/content/23/23/7209.full.pdf. http://clincancerres.aacrjournals.org/content/23/23/7209 (December. 2017). [DOI] [PubMed] [Google Scholar]
- 34.Huang Y et al. Clonal Architectures Predict Clinical Outcome in Clear Cell Renal Cell Carcinoma. Nature Communications 10, 1245 (March. 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Smedt L et al. Microsatellite Instable vs Stable Colon Carcinomas: Analysis of Tumour Heterogeneity, Inflammation and Angiogenesis. British Journal of Cancer 113, 500–509 (July 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chapusot C et al. Microsatellite Instability and Intratumoural Heterogeneity in 100 Right-Sided Sporadic Colon Carcinomas. British Journal of Cancer 87, 400–404 (August. 2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi YJ, Kim MS, An CH, Yoo NJ & Lee SH Regional Bias of Intratumoral Genetic Heterogeneity of Nucleotide Repeats in Colon Cancers with Microsatellite Instability. Pathology & Oncology Research 20, 965–971. ISSN: 1532–2807. 10.1007/s12253-014-9781-y (October. 2014). [DOI] [PubMed] [Google Scholar]
- 38.Davis A, Gao R & Navin N Tumor Evolution: Linear, Branching, Neutral or Punctuated? Biochimica et Biophysica Acta (BBA) - Reviews on Cancer 1867. Evolutionary principles - heterogeneity in cancer?, 151–161. ISSN: 0304–419X. http://www.sciencedirect.com/science/article/pii/S0304419X17300197 (April. 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sveen A et al. Multilevel Genomics of Colorectal Cancers with Microsatellite Instability—Clinical Impact of JAK1 Mutations and Consensus Molecular Subtype 1. Genome Medicine 9, 46. ISSN: 1756–994X. 10.1186/s13073-017-0434-0 (May 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abécassis J et al. Assessing Reliability of Intra-Tumor Heterogeneity Estimates from Single Sample Whole Exome Sequencing Data. PLOS ONE 14, 1–22. 10.1371/journal.pone.0224143 (November. 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armaghany T, Wilson JD, Chu Q & Mills G Genetic Alterations in Colorectal Cancer. Gastrointest Cancer Res 5, 19–27 (January. 2012). [PMC free article] [PubMed] [Google Scholar]
- 42.Chapel DB et al. Quantitative Next-Generation Sequencing-based Analysis Indicates Progressive Accumulation of Microsatellite Instability between Atypical Hyperplasia/Endometrial Intraepithelial Neoplasia and Paired Endometrioid Endometrial Carcinoma. Modern Pathology, 1 (June 2019). [DOI] [PubMed] [Google Scholar]
- 43.Robinson D et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 161, 1215–1228. ISSN: 0092–8674. http://www.sciencedirect.com/science/article/pii/S0092867415005486 (May 2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim T-M, Laird PW & Park PJ The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell 155, 858–868 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Currey N, Daniel JJ, Mladenova DN, Dahlstrom JE & Kohonen-Corish MRJ Microsatellite Instability in Mouse Models of Colorectal Cancer. Canadian Journal of Gastroenterology and Hepatology 2018, 6152928 (March. 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zavodna M, Bagshaw A, Brauning R & Gemmell NJ The Accuracy, Feasibility and Challenges of Sequencing Short Tandem Repeats Using Next-Generation Sequencing Platforms. PLOS ONE 9, 1–14. 10.1371/journal.pone.0113862 (December. 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roudko V, Greenbaum B & Bhardwaj N Computational Prediction and Validation of Tumor-Associated Neoantigens. Frontiers in Immunology 11, 27. ISSN: 1664–3224. https://www.frontiersin.org/article/10.3389/fimmu.2020.00027 (January. 2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maby P, Galon J & Latouche J-B Frameshift Mutations, Neoantigens and Tumor-Specific CD8+ T Cells in Microsatellite Unstable Colorectal Cancers. OncoImmunology 5, e1115943. 10.1080/2162402X.2015.1115943 (April. 2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.