

Abstract
Biomaterial carriers offer modular features to control the delivery and presentation of vaccines and immunotherapies. This tunability is a distinct capability of biomaterials. Understanding how tunable material features impact immune responses is important to improve vaccine and immunotherapy design, as well as clinical translation. Here we discuss the modularity of biomaterial properties as a means of controlling encounters with immune signals across scales – tissue, cell, molecular, and time – and ultimately, to direct stimulation or regulation of immune function. We highlight these advances using illustrations from recent literature across infectious disease, cancer, and autoimmunity. As the immune engineering field matures, informed design criteria could support more rational biomaterial carriers for vaccination and immunotherapy.
Keywords: vaccine, immunotherapy, autoimmunity, cancer, nanoparticle and microparticle
The physicochemical properties of biomaterials can be harnessed to improve vaccines and immunotherapies. Biomaterials exhibit tunable properties which can facilitate their trafficking throughout the host, as well as their interactions with immune cells. The physicochemical properties of biomaterials offer control over several key determinants of immune processing to facilitate activation of specific immune responses.
Graphical Abstract
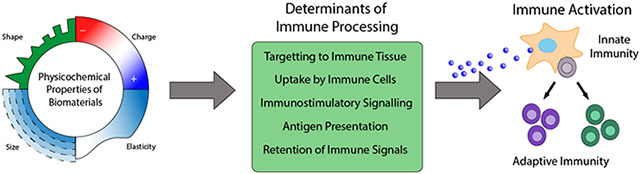
1. Biomaterials offer modularity that can be exploited for vehicles to improve vaccines and immunotherapies
Vaccines and immunotherapies are unique in their ability to exert specific and long-lasting effects to combat infection and disease. Advances in our understanding of the underlying cellular processes that govern these responses have paved the way for new vaccines and immunotherapies, but putting this new insight into practice is a work in progress.[1–4] One ongoing hurdle facing new strategies is the evolving nature of pathogens and cancerous cells that constantly mutate to evade immune recognition. Likewise, coaxing the immune system to recognize a particular fragment of a particular pathogen to mount a response – and determining which fragments to focus on – represent other key challenges. Additionally, emerging pathogens such as Zika and SARS-CoV-2 – the cause of novel COVID-19 – highlight the challenges of quickly identifying targetable antigens – molecular fragments of pathogens – without raising safety concerns.[5–7] Because the immune system is a complex amplification system, changes in immune activity can lead to broad effects, which underscores the constant need for safety considerations as new technologies are developed for the clinic. These challenges are true not only in vaccines for infectious disease, but also cancer immunotherapies aimed at enabling selective immune responses to destroy tumors. Likewise, similar hurdles are faced in designing better immunotherapeutics to tackle aberrant immune recognition and excess inflammation that may occur in autoimmune disease (i.e. multiple sclerosis, type 1 diabetes), transplantation, and allergies. Current treatments for these areas seek to control excess inflammation and dysfunctional immune attack, but often lead to side effects. For example, immunotherapies for autoimmune diseases – in which immune cells mistakenly identify self-molecules as foreign – often leave patients immunocompromised and are non-curative, requiring frequent and life-long treatment.[8] As such, new strategies that offer safer and more controlled modulation of the immune system are critical for new options across infectious disease vaccines and cancer immunotherapy, as well as for therapeutics that maintain immunological tolerance in autoimmune or inflammatory settings.
Many strategies are exploring biomaterials as carriers for immune signals for the generation, enhancement, inhibition, or other selective direction of immune responses.[9–12] A defining feature motivating this interest is the modularity that biomaterials offer. The immune system integrates and responds to multi-dimensional cues including physiological location, the combination and relative concentration of signals present, the types of immune cells involved, the molecular conformation of the signals, as well as the kinetics at which these processes progress. These factors influence the generation, maintenance, and resolution of immune responses. Thus, the unique tunability of engineered materials enable the design of vaccines and immunotherapies that can specifically interact with the immune system to achieve these requirements.
The immune engineering field has rapidly blossomed by drawing on the drug delivery space to explore carriers spanning organic and inorganic compounds; all of these offer facile opportunities to modify physiochemical properties. For the purposes of this progress report, we restrict our discussion to scaffolds and particles designed as biomaterial carriers for vaccination and immunotherapy. Within these areas, we explore organic biomaterial carriers comprised of natural and synthetic polymers, including peptides and nucleic acids, or even cells. Polymers such as chitosan, poly(lactic-glycolic acid) (PLGA), agarose, and hyaluronic acid (HA) are well-studied in this class and have been widely explored due to their biocompatibility and biodegradability. Additionally, many organic materials allow for customizable architectures or properties that mimic natural aspects of natural tissue, or for tunable degradation rates. Our scope also includes inorganic material carriers (e.g. gold, silicon, carbon, aluminum), which while often non-biodegradable, have also been explored as important classes of biomaterials.[13] Inorganic material carriers – such as quantum dots and metal nanoparticles (NPs) – are also often easily-functionalized to alter physicochemical properties, providing stable templates and precision synthesis. Many also offer unique optical or electrical properties. While there are other large bodies of work involving biomaterials interacting with the immune system, such as tissue engineering and host response to implants, these are beyond the specific scope of this report, which focuses on biomaterial carriers with a primary focus of manipulating and delivering immune function.
Excitingly, the potential of biomaterials to improve immune outcomes is also already being explored in a number of clinical trials. For example, the potential for multivalent virus-like particles as improved vectors against HPV (NCT00943722[14]) and influenza (NCT04120194[15]) are being explored in Phase III clinical trials by Merck and Novavax, respectively. Combination vaccines delivered in liposomes are being assessed to prevent HIV infection (NCT03961438[16]), a disease that lacks an effective vaccine. A poly(lactic-glycolic) (PLG) scaffold vaccine against melanoma is being investigated in a phase I clinical trial (NCT01753089[17]), with parts of this technology now licensed by Novartis, suggesting interest in the pharmaceutical industry for the adoption of these approaches. Antigen loaded microparticles are being explored to shift the balance of immune cell types to combat celiac disease (NCT03738475[18]). Gold nanoparticles with surface coupled antigen are being explored in the treatment of Type I diabetes (NCT02837094[19]), which currently lacks a cure. While not exhaustive, these examples highlight the therapeutic potential that biomaterials enable in the vaccine and immunotherapy space.
Over the past decade, biomaterial systems for vaccines and immunotherapies have been intensly studied [20–23]. As alluded to above, distilling the driving tenants of this body of work, a unique feature is the ability to create tunable materials platforms that control how the immune system interacts with vaccine and immunotherapy components. For example, surface functionalization through absorbed proteins, intrinsic topographical materials features, and carrier geometries that mimic bacteria or promote certain cell-biomaterial interactions, are all routes explored along these lines. Likewise, carrier/scaffold size, shape, charge, hydrophobicity, and mechanical properties are now documented as playing important roles in determining immune cell function and differentiation.[21,24–27] While it is clear that the physicochemical properties of materials can impact immune responses, there remains a lack of comparative measures for the gestalt of emerging immune engineering platforms. Thus, another important need in the field is systematic studies and standardized approaches to benchmark trade-offs in design approaches and connect materials properties to modulating immune responses.[28,29]
In this progress report we use studies from the most recent three years to highlight centrals ways in which the modularity of biomaterials can be leveraged to direct immune outcomes. In particular, we connect tuning of material properties – such as size, charge, shape, elasticity, topography, and stability – to manipulating immune processes at several scales, including tissue, cell, molecular, and time. For example, altering design parameters can be used to control biodistribution and improve targeting to LNs or other important immune tissues (Figure 1A). Once contacting antigen presenting cells (APCs), many of these same properties can be exploited to promote or limit uptake and activation of immune cells (Figure 1B). Ultimately, the ability to control the distribution in immune tissues and cells, along with control over the context in which specific immune signals are received, plays a major role in programming APCs and subsequently the types of T and B cells responses that occur (Figure 1C). Additionally, carefully engineered designs can further improve the quality of generated immune responses by tuning the kinetics with which immune signals are encountered or displayed (Figure 1D). In the next section – Section 2 – we provide concise immunological background to introduce some of the key steps in immune response that biomaterial carriers are commonly designed to interact with. Moving to recent literature, we begin with Section 3, which focuses on improving targeting at the immune tissue scale by tuning size, charge, shape, and stiffness. Section 4 probes a shorter length scale – trafficking within immune cells – by highlighting how altering material designs can impact uptake and localization of immune signals to specific intracellular compartments. Having considered targeting at the tissue and cell level, in Section 5, we narrow in on manipulating material design for delivery of specific immune signals classes, beginning with the delivery of adjuvants to prime early, non-specific functions of innate immune cells. Maintaining the theme of molecular encounter, in Section 6 we focus on using biomaterials to control the context with which antigen is delivered to impact the slower, but highly specific functions of adaptive immune response. We conclude in Section 7 by discussing how biomaterial properties are being engineered to control the kinetics and persistence of immune signals. Examples of the recent pre-clinical approaches that modulate these areas of immune signal delivery, which we discuss in this progress report, are summarized in Table 1.
Figure 1.
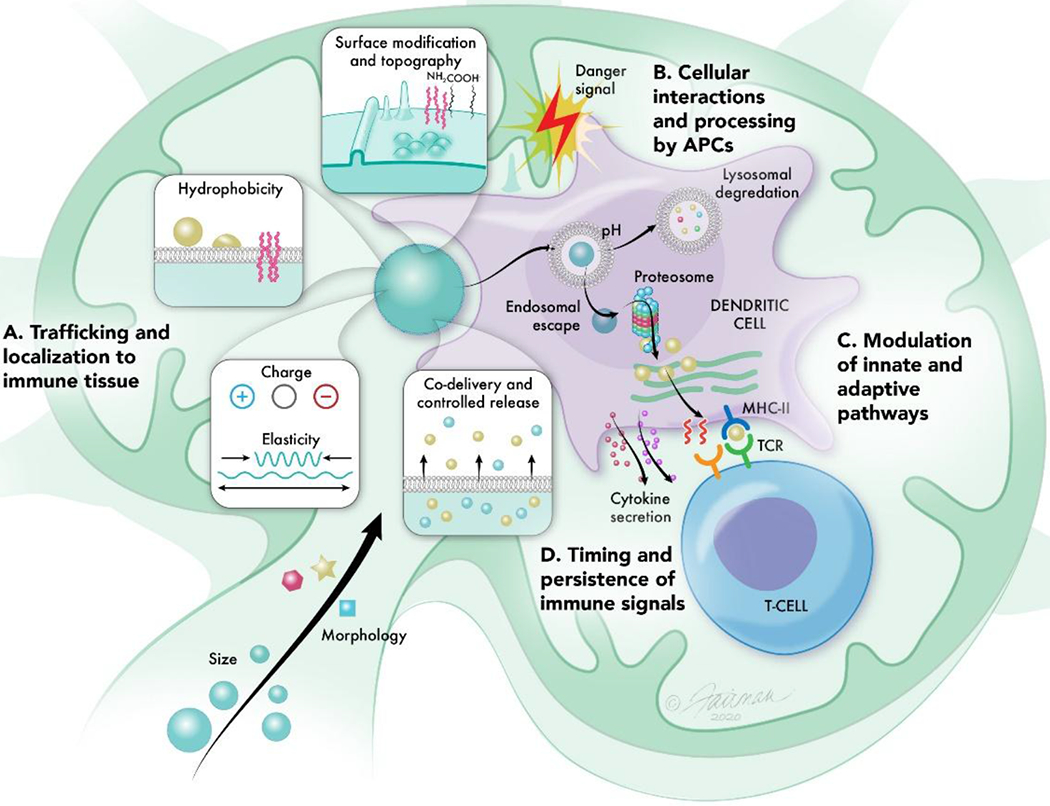
The physicochemical properties of biomaterials can be tuned to program immune responses. Properties such as size and morphology can A) promote trafficking of materials to immune tissues such as LNs (Section 3). These same properties in addition to surface modifications, hydrophobicity, charge, elasticity, and controlled release can B) impact the interactions of biomaterial carriers with APCs, altering their uptake and processing by immune cells (Section 4). Because APCs are key initiators of adaptive immune responses, molecular control over the delivery of immunostimulatory cues (Section 5) and antigen (Section 6) by biomaterial carriers can C) modulate innate and downstream adaptive immune pathways to generate potent specific immune responses. The modularity of biomaterials further allows for tuning over D) the lengthscales in which immune signals persist to control the quality and maintenance of responses (Section 7).
Table 1.
Key Examples of Control Over Immune Processing by Tuning Material Properties
| Immunological Process to be Controlled |
Biomaterial Parameter | Technique/Approach | Biological Outcome | Ref |
|---|---|---|---|---|
| Targeting to lymphoid organs | Size | Altering organic:water volumetric ratio during flash nanoprecipatiation | 20nm NPs rapidly drain to LNs, but 100nm NPs show minimal accumulation | [44] |
| Shape | Different NP seeding protocols from aqueous solution | Sphere and star-like particles accumulate in spleen | [47] | |
| Charge | Addition of cationic or anionic amino acids to the end of displayed antigen peptide sequence | Zwitterionic micelles promotes a combination of LN accumulation and cellular interactions | [49] | |
| Surface functionalization | PEGylation | Increased active transport of antigen to LNs, decreased ECM interactions with increasing MW of PEG |
[50,51] |
|
| Conjugating antigen to targetting moieties | Cell-mediated trafficking of antigen to LNs, enhanced LN accumulation | [56,58,59] | ||
| Elasticity | Electrostatic layer-by-layer assembly, followed by removal of template core to form hollow capsules | Hollow particles can pass through pores up to 4x smaller in size to faciliate LN trafficking | [55] | |
| Targeting APCs | Elasticity | Pickering emulsions | Highly deformable NPs due to raspeberry-like structure of pickering emulsions improves particle interactions with DCs | [67] |
| Varying polymer content (i.e. PLGA:PEG ratio) | Stiffer nanodiscs improve uptake by macrophages by increasing material-cell interaction times | [69] | ||
| Charge | Anionic modification of self-assembling polymer chains with different chain lengths of carboxyl group substitution | Intermediate levels of negative charge displays highest level of uptake; highly negatively charged particles are taken less eficiently by cells. | [72] | |
| Formation of amine containing hydrogels using Particle Replication In Non-wetting Templates (PRINT), followed by protonation/deprotonation of amine groups | In lung, cationic NPs are preferentially taken up by DCs, while anionic NPs are preferentially taken up by macrophages | [74] | ||
| Surface functionalization | Conjugating bacterial sugars or mimics to polymer NP surface | Increased intracellular accumulation targetting endoplasmic reticulum | [75] | |
| Hydrophobicity | Preparation of dendritic mesoporous organosilica and pure silica NPs | Hydrophobic particles facilitate lysosomal escape for delivery into the cytosol | [78] | |
| Charge | Altering number of basic amino acid arms on dendrimer | Positively charge NPs can rupture lysosomes to enter cytosol and improve inflammazome activation | [80] | |
| Functionalization with quarternary ammonium groups to polymer backbone | Positively charged hydrophobic microgels improve membrane disrupting potential to promote cytosolic delivery | [82] | ||
| Controlled Release | Materials selection: polymers with different degradation profiles | Faster release under acidic conditions enhances antigen presentation | [83] | |
| Delivery of Immunostimulatory Cues | Shape | Computationally designed nucleic acid sequences that self-assemble into 2D and 3D structures | Inflammatory cytokine secretion can be tuned based on dimensions (i.e. 2D vs. 3D), and the number of sides on polygonal structures | [91,95] |
| Conjugating poorly immunogenic RNA adjuvant to gold NP | Nanorods improve adjuvanticity | [109] | ||
| Controlled Release | Materials selection: polymers degradable by hydrolysis or degraded under acidic conditions | Products of polymer degradation can modulate immune activity | [96–98] | |
| Alter binding affinity of polymer to TLRa by using softer chained polymers or varying polymer:TLRA ratio | Reduction in adjuvanticity of TLRa with increased interaction strength of polymer carrier | [113,114] | ||
| Surface functionalization (ligand density) | Covalent linking of TLRa at different densities to polymer backbone allowing for chemically defined controlled loading | Higher density induces particle formation and improved activation of DCs and macrophages | [105] | |
| Topography | Hydrothermal assembly of titanium oxide nanostructural bundles to form nanospikes | Mechanical stress induced by nanospikes activates inflammasome pathway | [107] | |
| Antigen Presentation | Size | Covalent linkage of TLRa to polymers with different chain architectures with distinct hydrodynamic characteristics | Induction of CD8+ T cell responses increases with increasing polymer hydrodynamic radius | [115] |
| NPs of different sizes coated with pMHC and anti-CD28 to form aAPCs | Smaller aAPCs require saturated doses of pMHC or artificial magnetic clustering to activate T cells at similar levels compared to larger particles. | [126] | ||
| Controlled Release | Conjugation of antigen to adjuvant using a pH sensitive reversible linker | Release of unmodified antigen improves expansion of T cells | [120] | |
| Antigen localization | Surface conjugation of antigen and encapsulation of antigen onto polymers | Encapsulated antigens preferentially promote antigen presentation on MHC-I to enhance CD4+ resposnes. Surface conjugated antigens promote antigen presentation on MHC-II, enhancing CD8+ responses | [122–124] | |
| Surface functionalization | Biotinylated liposomes coated onto mesoprous silica microrods, followed by attachment of anti-CD8 and anti-CD3 antibodies | Fluidity of lipid bilayers allows for robust expnasion of T cells even with lower density of stimulatory cues | [127] | |
| Site specific binding of antigen to alum via multivalent phosphorylated serine groups that bind hydroxyl groups on alum. | Stable binding of antigen to alum offers conformational control over antigen presentation, allowing for tuning of B cell specificity towards specific antigen epitopes | [134] | ||
| Ligand density | Iron oxide NPs conjugated with pMHC | pMHC must exceed a threshold density for T cell activation to occur | [142] | |
| Antigen adsorbed to quantum dots | Controlling antigen display to APCs alters T cell responses | [144] | ||
| Immune Signal Retention | Controlled Release | Altering linker chemistry (e.g. thioether vs. dssulfide linker) between antigen and polymer | Slower release of antigen prolongs antigen release and antigen presentation over time leading to improved immune response | [147] |
| Altering MW and varying degree of cyclic acetal groups on acetylated-dextra MPs | Faster degrading MPs promote strong humoral and cellular responses at earlier timepoints. Slow-degrading MPs drive stronger responses at later timepoints | [150] | ||
| Size | Antigen conjugated to different sized NPs using carbodiimide-mediate coupling to control antigen dose | Larger particles prolong antigen presentation by APCs, resulting in improved antibody production | [148] | |
| Different gold NP seeding protocols | Retention of 50-100nm NPs on dendrites of FDCs in LNs | [149] | ||
| Surface Functionalization | Peptide conjugated to nucleic acid adjuvant and adsorbed to liposome via hydrophobic anchoring group (cholesterol) | Co-delivery of antigen and adjuvant allows for synchronized peptide presentation and expression of costimulatory markers, improving generation of memory T cells | [152] |
2. Immune responses arise from complex interactions between immune cell populations across distinct tissues and time scales
Immune responses are categorized into two major classes, innate and adaptive. Innate responses provide rapid defense against infection by non-specifically removing pathogens, infected cells, and damaged tissue. In contrast, adaptive immune responses develop more slowly, but are highly specific responses that are initiated within lymphoid organs such as the spleen and lymph nodes (LNs). In these tissues, resident B and T cells can differentiate into long-lived memory cells that provide rapid protection upon re-exposure to a pathogen. This section describes the key features of these two systems and how they interact.
2.1. The innate immune system is a rapid first line of defense, but lacks specificity
Innate immunity offers a quick-acting but less-specific defense mechanism comprised of both molecular and cellular components able to recognize general patterns common in frequently encountered pathogens. Biomaterial carriers can trigger innate immunity through surface engineering of molecules found on the cell walls of bacteria or through fabrication of particles with similar size scales and topographical features to pathogens. When encountered, cells express surface proteins and secrete cytokines – the protein signals of the immune system that play key roles in the activation and polarization of most immune cells. Additionally, a variety of innate immune cells survey the body for pathogens such as natural killer (NK) cells and APCs. APCs are specialized innate immune cells that are important in the detection and processing of pathogens and include macrophages and dendritic cells (DCs). Because they efficiently phagocytose or internalize fragments of pathogens – termed antigens, these cells can quickly generate non-specific inflammatory responses against pathogens. Equally important, APCs bridge the innate and adaptive immune system, providing signaling cues to initiate more specific responses.
Recognition and activation of APCs is dependent on surface receptor interactions and soluble signals (i.e. cytokines) that can be used to sense pathogens. APCs can recognize molecular motifs commonly found on pathogens but absent in healthy host cells, termed pathogen associated molecular patterns (PAMPs). PAMP recognition results in upregulation of co-stimulatory signals that help initiate immune response. Pattern recognition receptors (PRRs) on APCs can sense PAMPS and help identify a diverse range of these “warning signals”, making them a key target of interest in engineering immune responses. One major class of PRRs are Toll-like receptors (TLRs), a family of membrane bound receptors that recognize ligands on pathogens, leading to activation of inflammatory responses. Signaling may occur on the APC outer membrane surface through receptors evolved to detect extracellular pathogens. In contrast, many of the TLRs exist within specific intra-cellular domains such as endosomes to detect pathogens taken up by endocytosis or intracellular components exposed after pathogen degradation, such as viral RNA. Other PRRs, such as the inflammasome, can detect PAMPs within the cytosol. The inflammasome is a complex of proteins which ultimately triggers secretion of IL-1β, a key cytokine involved in initiating inflammatory processes. Of particular relevance, biomaterial carriers can promote internalization of immune signals to facilitate activation of these pathways.
Concurrent with PRR activation, internalization of antigens triggers processing and loading onto major histocompatibility complex (MHC) by APCs. Antigens are loaded onto either MHC-I or MHC-II, depending on the intracellular processing mechanisms that the antigen undergoes. Endocytosed materials are degraded in endosomal/lysosomal compartments and presented in MHC-II. On the other hand, MHC-I predominantly presents antigens localized within the cytosol. Importantly, endocytosed materials can also enter the cytosol and be presented via MHC-I through various mechanisms reviewed by others [30–33] such as lysosomal escape, through a process called “cross-presentation.” Biomaterial carriers offer features for improved delivery of immune cargo (i.e. co-delivery of signals, efficient internalization, tunable kinetics, cargo protection), which can better direct these outcomes. This is important because together, internalization of antigen and engagement of PRRs results in DC maturation and migration to spleen or LNs where these APCs can prime T cells. As such, APC activation and antigen presentation are not only important for the elimination of pathogens, but also provide signaling cues that can greatly influence the adaptive immune responses. The interactions of APCs with T and B cells within lymphoid organs are highly dependent on the appropriate signals being presented by APCs. Additionally, T cells recognize the antigen they are specific for – the “cognate” antigen, only when loaded within an MHC complex displayed by a DC or other APCs. As highlighted in several comprehensive reviews, these cells are thus frequent targets for vaccines and immunotherapies.[34–36] Current vaccination strategies employ adjuvants, molecules that mimic immune warning signals to trigger activation through co-stimulation and other mechanisms. Importantly, the immune pathways activated by pathogens and other foreign molecules can also sometimes be triggered by biomaterials. The particulate nature of many biomaterial carriers (e.g., NPs) also facilitates uptake by APCs. These features, along with the prominent role of APCs in initiating adaptive immunity, have made APC populations a major target for materials-based strategies.
2.2. Adaptive immune responses develop more slowly but are high specialized and highly specific
Generation of specific, long-lasting immunity involves the activation of adaptive lymphocytes that respond to a particular antigen in LNs and spleens. Thus, one way in which nanocarriers and microcarriers facilitate adaptive immune responses is through targeting immune signals to these sites. Adaptive immunity is a highly selective response that is initiated by interactions between APCs and lymphocytes, such as T and B cells. While T and B cells are responsible for carrying out processes to combat and remove pathogens, APCs are responsible for activating these cells. As discussed above, APCs take up, process, and display antigen peptide fragments to molecularly-specific receptors on lymphocytes by loading antigens into MHC. These complexes can then be presented in combination with costimulatory molecules to, for example, T cell receptors (TCRs) on the T cells, leading to activation of T cells specific to the presented antigen. The activated T cells then leave LNs or spleen and return to sites of infection or disease to selectively combat the pathogen the cell is now armed against. A small number of activated cells can become long-lasting memory cells that persist in the body to quickly mount protective immune responses if the pathogens these cells are armed against are re-encountered in the years or decades to come.
Antigen presentation, costimulatory molecules, and cytokines are all cues that initiate and maintain immune response against pathogens, tumors, or other targets. The intricate balance between these signals is also important in maintaining immunological tolerance that prevents host tissue from being attacked. For example, T cell activation results when these costimulatory signals occur in tandem with presentation of an antigen that the T cell engaging the APC is specific for. Conversely, the absence of costimulatory signals during antigen presentation can give rise to different outcomes, such as the generation of regulatory T cells (TREGs) that can help regulate immune response and combat autoimmunity, conditions in which immune cells mistaken recognize self-antigen as foreign. Synthetic microparticles (MPs) present another opportunity for biomaterials to engage with immune responses, through the engineering of APC mimics that can directly interacting with T cells to regulate their differentiation and function.
Depending on the signals that T cells receive, a number of different T cell subsets can arise. For example, recognition of peptide antigens in MHC are restricted to particular T cell types such that CD8+ T cells recognize cognate antigen displayed in MHC I and CD4+ T cells recognize antigen displayed in MHC II. CD8+ T cells, become cytotoxic T lymphocytes (CTLs) upon activation. These cells directly target and destroy diseased host cells – such as those infected with intracellular pathogens (e.g., viruses) – to prevent spread of infection; these cells are also sometimes able to destroy cancerous cells. Activated CD4+ T cells, on the other hand, become T helper (TH) cells. TH cells exhibit specific phenotypes, such as TH1 and TH2 function, that provide support to other immune cells through secretion of signaling molecules called cytokines.
B cells are also important components of adaptive immunity. These cells share some of the same features of T cell activation, including the ability to generate memory cells. B cells become activated following recognition of a cognate antigen. Importantly, B cell activation requires cross-linking of the B cell receptors displayed on the surface of these cells by the antigen. However, maturation and long-live antibody-production requires additional activation from TH cells. Development of B cell memory and long-live antibody-production requires that B and T cells organize into specialized domains that form in lymph nodes called germinal centers (GCs); the resulting activated B cells produce more potent strongly-binding antibodies.[37,38] This brief description demonstrates an important point, that generation of strong antibody responses requires activation of both B and T cell subsets. This is one more example of the inter-connected nature of the immune system, motivating the need to understand how biomaterial properties can be tuned to control the cues governing adaptive immunity. Along these lines, in the following section we focus on how materials can be engineered to improve targeting to immune tissue.
3. Biomaterial properties can be engineered to target or enrich immune signals in lymphoid tissues.
As discussed in Section 2, lymphoid organs such as the spleen and LNs are the sites where APCs present antigen and co-stimulatory signals to drive differentiation and proliferation of the T and B cells residing in these sites.[39] For this reason, LNs and spleen are the tissue target of many vaccines and other immune signal delivery applications.[21,22,40] Reaching these sites is important in quickly generating strong and selective immune responses, using doses that minimize toxicity or off-target effects. Efficient delivery of materials to LNs is also important to minimize toxicity of the carriers themselves, as the biocompatibility of candidate biomaterial carriers can be highly dependent on microenvironment, further underscoring the importance of directed targeting to lymphoid organs.[41] A number of approaches now exist that enable vaccines and immunotherapies – typically administered peripherally in muscle or under the skin – to accumulate at high levels in LNs.[42,43] We begin by discussing targeting and accumulation of biomaterial carriers to lymphoid organs by tuning biomaterials, focusing on design parameters that promote passive targeting, strategies to overcome barriers that impede entry, and active targeting approaches.
3.1. Design parameters can be tailored to promote passive targeting to spleen and LNs
While conventional non-biomaterial vaccines rely on free drainage to LNs or trafficking by APCs that encounter the vaccine, biomaterials offer additional properties to leverage in directing the trafficking and targeting of vaccines and immunotherapies after injection. For example, the ability to control size has been pivotal in improving LN drainage following injection. Howard et. al investigated the “size gate” for effective LN drainage by synthesizing PLGA-b-PEG NPs with average diameters of 20,40 and 100-nm. Following subcutaneous (s.c.) administration in mice, 20-nm NPs were found to drain rapidly across proximal and distal LNs and displayed improved retention compared to NPs with an average diameter of 40-nm. The drainage of 100-nm NPs was negligible.[44] These results support seminal studies that polypropylene sulfide NPs larger than 100 nm do not passively diffuse to LNs, relying instead on uptake and trafficking by APCs at the site of injection.[45] Another opportunity that biomaterials offer is the ability to control size to assist in the delivery of small molecules. As one example, mellitin is a small molecule adjuvant that preferentially enters the blood. Encapsulation of the mellitin, a small molecule adjuvant, into nanolipids 10-20 nm in size has been shown to promote LN accumulation of melittin, but not other organs.[46]
Manipulation of multiple parameters offers an additional layer of control. The size and shape of gold NPs (AuNP) has been leveraged to impact the biodistribution and trafficking to spleen.[47] In particular, this study revealed AuNPs of 50 nm diameter accumulated more in the spleen than AuNPs of 10 nm. When using AuNPs of similar size but with different shapes – spheres, star-like, and rod shaped – only spheres and star-like particles accumulated in the spleen. In contrast, rod-shaped particles displayed a poor ability to penetrate organs and were rapidly cleared. In another example, the icosahedral-shaped cowpea mosaic virus has been found to display superior transport and retention to LNs compared to the filamentous-shaped potato virus (Figure 2A).[48]
Figure 2.
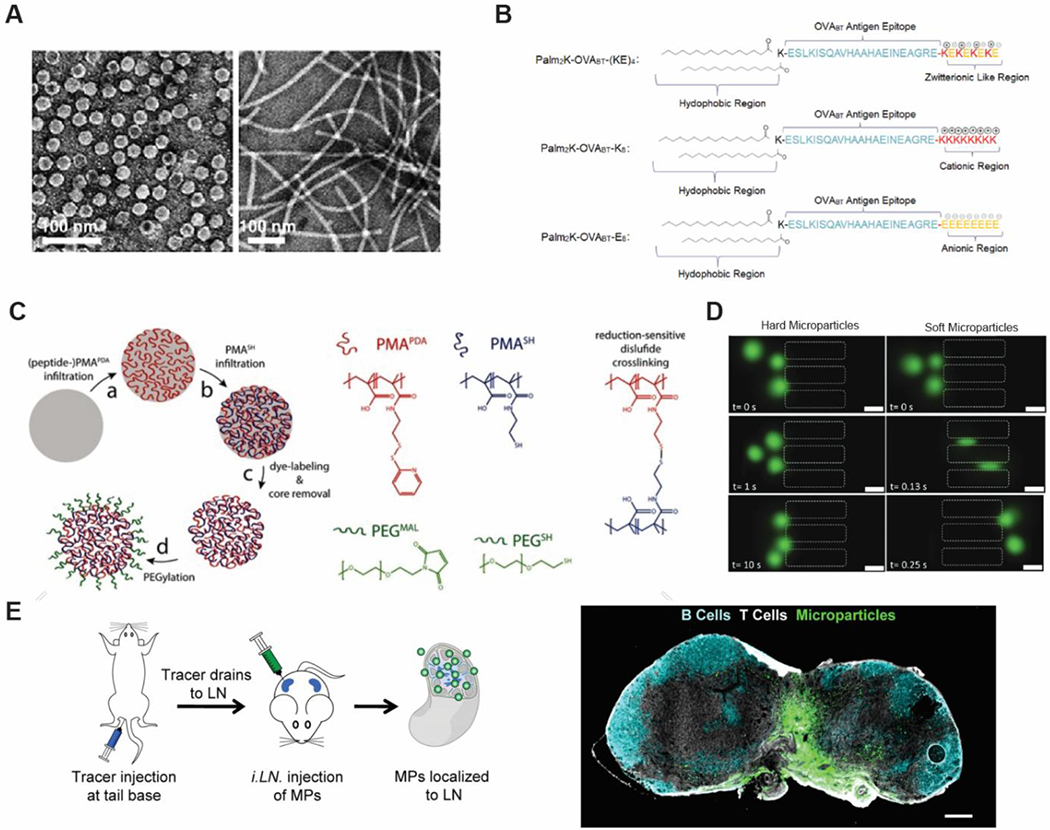
Engineering biomaterial properties to target LNs. A) Morphology of viral nanoparticles improves transport and retention within LNs. Reproduced with permission.[48] Copyright 2017 , Elsevier. B) Engineering of PAM charge influences lymph node accumulation and cell association, with cationic PAMS exhibiting less accumulation in LN than zwitterionic or anionic PAMs. Macrophages incubated with anionic PAMs display reduced uptake compared with other tested formulations. Highly charged PAMS induce lower antibody responses, compared to zwitterionic particles. Reproduced with permission.[49] Copyright 2018, American Chemical Society. C) PEGylation of PMA particles improves particle stability, blocking unfavorable interactions of PMA side chains with the ECM that would otherwise hinder particle mobility and transport through the lymphatics. Reproduced with permission.[51] Copyright 2015, Wiley-VCH. D) Deformability supports the passage of soft MPs through confined microchannels, whereas hard MPs exhibit poor migration (scale bar= 30um). Reproduced with permission.[54] Copyright Wiley-VCH 2018. E) Direct LN injection to locally deposits vaccine depots into LNs. A tracer dye is injected subcutaneously at the tail base which then drains to inguinal LNs allowing for visualization of LNs through the skin. MP depots can then be injected into the skin. MPs are retained within LN 28 days following injection of LN with fluorescent depots (scale bar= 200um). Reproduced according to the terms of the Creative Commons Attribution 4.0 International License.[62] Copyright 2016, Springer US.
In addition to size and shape, charge is another property that can be leveraged to promote LN targeting. For example, the charge on peptide amphiphile micelles (PAMs) can be readily modified through the addition of positively charge lysine or negatively charge glutamic acid residues, while maintaining similar shape and size, ranging 60-70nm (Figure 2B).[49] Entry of these PAMs and their subsequent interactions with APCs in the LNs has been shown to be maximized when the PAM surface was zwitterionic. Anionic surfaces allowed accumulation in LNs but failed to interact with APCs, eliminating immune activation. Cationic surfaces on the other hand, had significantly lower accumulation in the LN than both zwitterionic and anionic, but were able to interact with APCs in circulation and peripheral tissues; this binding however was relatively non-specific, as the cationic PAMs also bound non-phagocytic cells. Only zwitterionic PAMs provided the appropriate combination of LN accumulation and LN interactions. Collectively, these results demonstrate another biomaterial property lever – charge – to promote or diminish access and interaction with immune cells and tissues.
Thus, a range of biomaterials properties – size, shape, and charge– each have significant roles in enhancing trafficking to LNs and spleen. This is important as lymphoid organs control many aspects of immune function; this targeting also provides dose-sparing which can minimize systemic toxicity. Although the trends examined here may not apply across all platforms or biomaterials, they highlight different opportunities to improve targeting to lymphoid organs by altering biomaterial design parameters. It is important to note, however, that the examples presented here reflect only carriers within size ranges below 100 nm, which, as discussed, favor efficient LN drainage. While size remains a critical component to effective LN drainage, a number of biomaterial-based approaches vary by orders of magnitude between 100-1000 nm, yet still display efficient LN targeting. Similarly, some positively charged NPs have been observed to accumulate within LNs. Critically, biomaterials modulate immune function through an interplay of multiple design parameters, which enables additional modalities to overcome barriers to LN trafficking; this topic is the focus of the next subsection.
3.2. Rational design enables biomaterials carriers to overcome barriers to entry into LNs
Although the examples in the previous section highlight how the tunability of biomaterial carriers allows for their accumulation in lymphoid organs, there are many physiological barriers that limit efficient delivery of vaccines and immunotherapies from peripheral injections sites to LNs. While entry into the lymphatics offers a direct route to LNs, biomaterial-based vaccines and immunotherapies that enter systemic circulation – passively from the injection site or through direct intravenous (i.v.) injection – require design strategies to promote prolonged circulation. As previously mentioned, most traditional vaccines are administered into muscle or under the skin. At the site of injection, administered agents must navigate a collection of extravascular fluid, solute, extracellular matrix (ECM), and cellular environment. The pore size and highly negatively charged moieties within the ECM present another obstacle for larger positively charged carriers. Thus, biomaterials must also be tuned to overcome these hurdles.
Work by the Collier lab has shown that for sublingual delivery of vaccines, in which the network of mucin presents similar obstacles to those found in ECM, nanofiber interactions with mucin were found to decrease as a function of increasing MW of PEG.[50] Increasing MW of PEG elicitted larger antibody responses, suggesting a role for improving immune responses by making biomaterial carriers more inert against extracellular environments. Utilizing a similar principle, De Koker et al. has approached this design need by PEGylating 200 nm PMA hydrogel particles (Figure 2C).[51] These studies revealed that the addition of PEG increased lymphatic draining and the active transport of antigen to the LN. This observation was attributed to the blocking of redox-sensitive groups on the PMA particles upon addition of PEG, which may have increased mobility through the ECM and the circulation half-life. The association of the particles to immune cells also increased with PEGylation. A similar outcome has been observed by conjugating peptide antigens to 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-PEG (DSPE-PEG) to enhance lymphatic drainage.[52] After conjugation these peptides traveled to nearby and distant LNs. These findings build on established drug delivery approaches based on PEGylation of liposomes or small molecular drugs to increase circulation.[53]
In an alternative approach to PEGylation, Hasani-Sadrabadi et al. described polymeric gel alginate particles with elastic moduli that mimic naive and activated helper T cells (CD4+).[54] Naïve and activated T cells are able to pass though capillaries of much smaller size than their diameters, thus, the investigators created a microfluidic chip model using capillary channels with diameters of 5 µm to study the traversal of 12 µm diameter particles. Only softer particles (e.g., modulus of 3. 3 kPa) were able to pass through the pores, then regain their shape (Figure 2D). Even when chemotaxis – movement of cells towards a chemical attractant – was simulated using magnetic fields, stiffer particles (e.g., modulus of 11.1 kPa) were unable to traverse the microfluidic channels. This result highlights the importance of deformability for larger particles to navigate smaller pore sizes. In addition to simulating CD4+ T cell movement across barriers, the particles could also be readily loaded for immune signaling molecules, interleukin-2 (IL-2) and interferon γ (IFN-γ), demonstrating the potential of softer particles to improve delivery of immune signals.
Supporting the importance of deformability to facilitate entry into LNs, the Moon lab has developed nanocapsules 220nm in size with a hollow core and shell composed of microbial polysaccharides to mimic the structural and immunological properties of bacterial cell walls.[55] The nanocapsules were developed using a layer-by-layer assembly technique onto a rigid silica NP core template, followed by removal of the silica template. Hollow nanocapsules administered s.c. were found to efficiently drained to inguinal LNs compared to NPs which maintained the rigid silica core. These differences were attributed to the hollow design which allowed for high deformability while NPs with the rigid cores maintained their structure. Additional studies revealed that hollow particles easily passed through 100nm pores and 30% of the hollow particles were still recovered when hollow particles were flowed through a 50nm pore membrane. On the other hand, NPs that maintained the rigid silica core failed to pass through even a 200nm sized pore membrane, supporting design of elastic materials to enable improved delivery of NPs larger than 100nm.
While the above examples highlight ways in which biomaterials have been designed to improve passive drainage to LNs, active targeting approaches are also being developed for efficient delivery to these immune tissues. One common target is albumin, a protein that regularly filters through LN. In these approaches, the natural shuttling ability of albumin to LNs is exploited by conjugating peptide antigens or vaccine adjuvants to albumin-binding structures to enhance LN trafficking.[56,57] Alternatively, biomaterials can target mannose receptors on DCs to promote LN accumulation through cell-mediated trafficking.[58] While the above strategies describe targeting of LNs through the lymphatics, systemic circulation offers an alternative transport route to LNs. In one study, NPs were conjugated to an antibody that efficiently targeted high endothelial venules – the vasculature structure through which lymphocytes enter LNs from circulation – to enhance LN accumulation.[59] This is important as systemic administration has been shown to be the superior route of administration for some vaccines.[60,61] We have developed an alternate idea based on direct delivery of biomaterials depots directly to LNs (Figure 2E). These particles are synthesized to be too large to freely drain from the sites and are thus mechanically restricted. Instead, the depots slowly degrade, releasing stimulatory or regulatory cues to reprogram the local LN microenvironment. For example, this strategy can be used to deliver antigens or immunostimulatory adjuvants,[62–64] as well as signals to promote immune tolerance and combat autoimmune disease.[65]
4. Biomaterial properties can impact single cell interactions, uptake and processing
While in Section 3 we discussed how biomaterial properties are being manipulated to target delivery to immune tissues across the body, we now turn to a much shorter length scale, focusing on how the properties of biomaterials can influence their interactions with and within immune cells. We discuss how biomaterial carriers can be engineered to influence binding, internalization, and immune signal processing by APCs within these cells. In the context of immune engineering, the studies highlighted in this section suggest that strategies to enhance the immunogenicity of biomaterial-enabled vaccines and immunotherapies can be achieved through more direct targeting to LNs.
4.1. Biomaterial stiffness and charge alters interactions with immune cells to promote uptake
In Section 3 we discussed how physicochemical properties are important for tissue-level biodistribution, but some of these same parameters impact how biomaterials interact and are processed by immune cells. Internalization of materials requires direct and effective immune cell-material interactions. Recent studies have generated increasing evidence that physical properties – including size, shape, charge and stiffness – play significant roles in the internalization of particles by APCs, by favorably associating with cell membranes allowing for improved uptake. However, it remains difficult to define how each parameter affects particle fate and function, and some inconsistencies exist across the literature findings. For example, in studies using micron-sized polymer particles, the effect of stiffness on uptake has been found to be shape-dependent, such that only softer variants of rods display increased uptake, while spheres displayed no enhancement in uptake as stiffness was varied.[66] However, in these studies, the effects of shape and stiffness were eclipsed by the effect of size, whereby larger particles (6µm) resulted in poor uptake. These larger sized particles may exhibit limited uptake due to the higher membrane deformation energy required for cells to engulf these particles, highlighting the importance of particle-cell interactions in the uptake of biomaterials.
Recognizing that internalization of particles requires increased contact areas and multivalent interactions with APCs, the Ma lab designed PLGA NP-stabilized Pickering emulsions (PPAS) (Figure 3A).[67] In this design, NPs form a fluid raspberry-like structure to stabilize a hydrophobic core, resulting in particles that can readily deform under mechanical stress. PPAS exhibited significantly improved uptake compared to traditional PLGA NPs stabilized with surfactant. Confocal microscopy revealed that deformability of particles during uptake improved the ability of DCs to wrap around NPs and allowed for increased contact area with the DC, and thus more multivalent interactions; together, these features facilitated phagocytosis. In contrast, PLGA NPs with smoother surfaces appeared to hinder the ability for cells to interact with NPs, resulting in sterically blocked DC interactions. Thus, in addition to improving LN accumulation, softer particles can also improve immune cell interactions.
Figure 3.
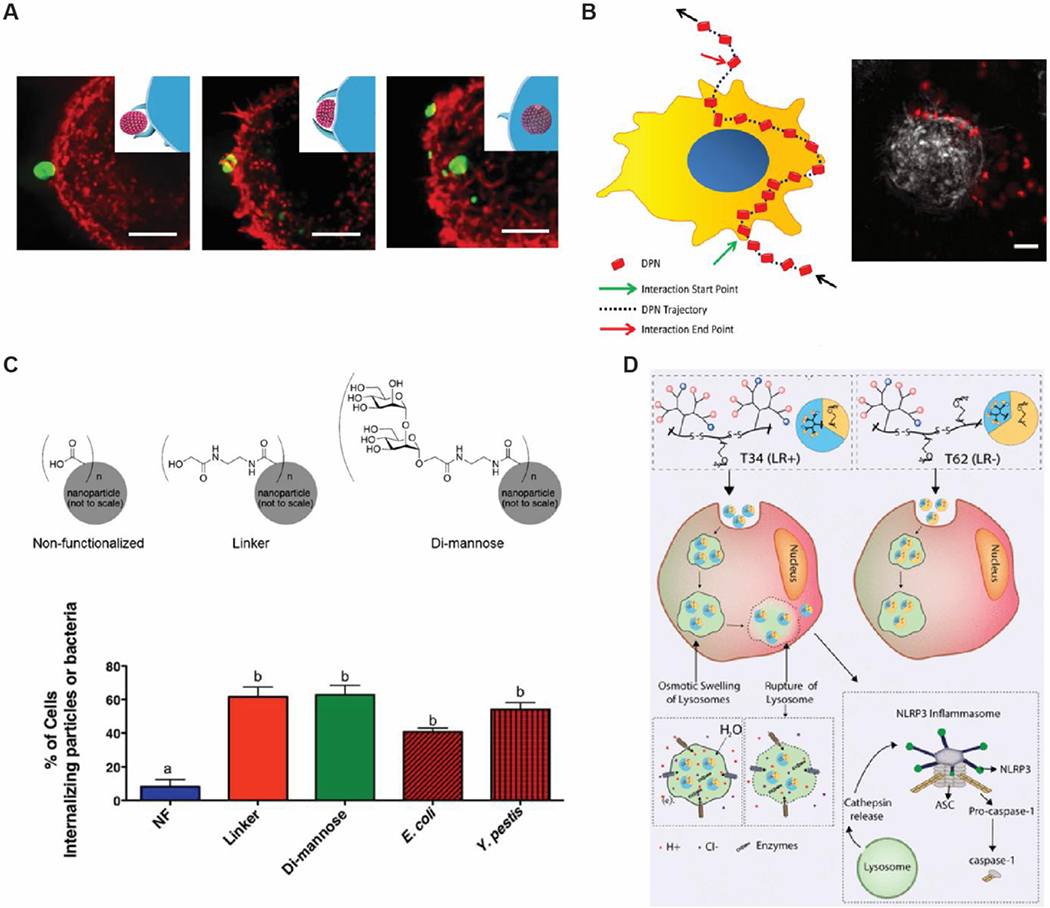
Biomaterials can be designed to improve uptake and processing by APCs. A) Pickering emulsions allow for improved deformability, which facilitates uptake of NPs by APCs. Reproduced with permission.[67] Copyright 2018, Nature Publishing Group. B) Time-lapse microscopy analysis of NPs to observe cell-particle interactions. Rigid constructs have prolonged interactions with cells compared to soft constructs. Reproduced with permission.[69] C) Functionalization of polyanhydride NPs with di-mannose via a glycolic acid linker to mimic bacterial surfaces promotes internalization of NPs. Enhanced uptake is observed even with the glycolic acid alone. Copyright 2018, American Chemical Society. Reproduced with permission.[75] D) Modification of a single amino acid can alter intracellular processing. Reproduced with permission.[80] Copyright 2018, American Chemical Society.
Contrary to these findings, soft silica nanocapsules have been reported to have 3 times less uptake than their stiffer counterparts.[68] In this study, functionalization with folic acid to improve uptake by macrophages was shown to only affect stiffer particles. In a similar vein, Palomba et. al developed polymeric nanodiscs of defined shapes and size.[69] The stiffness of each particle was readily tunable over several orders of magnitude by varying the relative ratio of PLGA and PEG. Regardless of shape and size, softer nanoconstructs were taken up less efficiently compared to rigid constructs, although it was noted that soft elliptical particles were also readily internalized. Live cell microscopy indicated that soft nanodiscs experienced short-lived interactions, diminishing their likelihood of recognition and internalization by macrophages (Figure 3B). Further analysis identified the bending stiffness of nanodiscs as a discriminating factor for uptake: nanodiscs with bending stiffnesses much higher or lower than cells facilitated internalization, while a bending stiffness similar to cells opposed internalization. Although the data set is not yet complete, these studies highlight that stiffness is a key parameter for modulating interactions with the immune system to improve cellular uptake. Importantly, the role of stiffness in facilitating uptake may be dependent on the type of APC. While softer particles were found to improve uptake in DCs, as highlight by the Ma lab, the above studies suggest that stiffer particles facilitate uptake by macrophages. These differences suggest that stiffness can also be leveraged to target specific APCs.
Charge is another parameter that can be tuned to improve uptake. In several studies, positively charged particles have been found promote interactions with cells through electrostatic interactions with the cell membrane, which is often negatively charged [70,71] This interaction can translate to improved uptake that affects processing of biomaterials and immune cargo by the APC. On the other hand, negatively charged materials or assemblies have been found to hinder uptake by APCs, impeding signal processing and the ultimate downstream T cell and antibody responses. These findings, however, are not universally true. In one study, negatively charged polysaccharide nanogels modified with varying levels of carboxyl groups showed preferential uptake by APCs in LNs over unmodified nanogels.[72] Importantly, however, the level of uptake was greatest in nanogels that displayed intermediate levels of charge; highly negatively charged particles were less efficiently taken up by cells. One possibility is that highly negatively charged particles offered improved trafficking to LNs (as discussed in Section 3) however were taken up less efficiently by APCs, once within LNs. Nanogels that displayed intermediate levels of negative charges, on the other hand, could still be taken up efficiently, while offering improved LN targeting. These findings illustrate a critical design dilemma that can arise in designing vehicles that can efficiently be internalized by APCs: material properties that promote lymph node accumulation can hinder internalization by APCs. As such, in the development of carriers that can efficiently deliver immune signals to cells and tissue, systematic studies of material design parameters are important to balance opposing design needs. This task that is readily accomplished through the tunability of materials.
In addition to improving material-cell interactions to promote uptake, biomaterials can also be targeted to specific populations of APCs. For example, while the underlying mechanisms are unknown, ferritin NPs demonstrate intrinsic preferential capture by specific APC subsets both in vivo and in vitro.[73] Studies in pulmonary antigen delivery to the lungs suggest that particle charge can also differentially affect uptake by specific cell types.[74] In one report, cationic NPs were found to be preferentially taken up by DCs, the targeted APC in this study. Conversely, anionic NPs were preferentially taken up by alveolar macrophages, whose primary function is in maintenance and clearance of air spaces from foreign particulates. Thus, uptake by specific APCs can be manipulated by surface charge, presenting another important variable through which biomaterials can regulate immunogenicity.
Functionalization of biomaterial carriers to alter the surface chemistry of particles is another important strategy, offering two layers of control over cell interaction and uptake: i) an additional method to influence interactions with immune cells and ii) the mode of uptake. As one example, polyanhydride NPs were modified with a glycolic acid linker conjugated to dimannose, a sugar found on bacteria, or with glycolic acid linker alone (Figure 3C).[75] Both linkers exhibited 8-fold higher uptake by DC in vitro compared to unfunctionalized polyanhydride, which was attributed in part to the positive charge of the NPs after modifications. Interestingly, d-mannose-functionalized NPs caused increased intracellular accumulation compared to NPs functionalized with glycolic acid linker alone. Both linker types also resulted in markedly different levels of cytokine secretion and activation marker expression, suggesting that uptake may occur through different pathways depending on surface modification. Additionally, in contrast with non-functionalized particles, a fraction of the linker-functionalized NPs was found to co-localize to the endoplasmic reticulum, further supporting different mechanisms of uptake between NPs. These findings highlight that surface functionalization may play a role not just in mediating the initial material-cell association and the level of uptake, but also in the mechanisms by which biomaterials are internalized. This is important because the mechanism of uptake affects intracellular processing and control antigen presentation by APCs, as discussed in the follow section.
4.2. Hydrophobicity and charge can regulate intracellular processing pathways
In addition to efficient uptake, the ability to control compartmentalization of biomaterials within particular intracellular locations is critical for proper antigen processing, detection of danger signals, and initiation of specific immune responses. A key concern for the delivery of antigens and adjuvants is the array of cellular compartments that can be targeted. For example, immune recognition of PAMPs – described in Section 2 - can occur in either intracellular structures (e.g., endosomes) or within the cytosol. Nucleic acid based PAMPs such as viral RNA or bacterial DNA are recognized within endosomal membranes, while intracellular danger sensors such as the inflammasome reside in the cytosol. As such, activation of certain pathways requires that immune signals are able to escape endosomal membranes following uptake to deliver immune signals to the cytosol. Furthermore, activation of specific adaptive immune responses is dependent on antigen internalization by APCs and processing. Antigen that ultimately reaches the cytosol is presented on MHC-I, while antigen that remains in the endosome is presented on MHC-II, resulting in engagement of different subsets of T cells. Thus, the intracellular fate of immune signals (i.e., degradation, localization to specific cellular compartments, agglomeration) within APCs shapes both the innate response and downstream adaptive immune responses. As such, materials must not only be designed for efficient internalization of immune signals, but also for delivery to the appropriate cellular compartments for a particular signal or application.[76,77]
Hydrophobicity and charge emerge as key parameters to control how biomaterials interact with membranes to deliver immune signals to the cytosol. This is largely due to membranes being comprised of negatively charged lipids. For example, in one study, hydrophobic mesoporous organosilica NPs have been shown to better facilitate lysosomal escape into the cytosol compared to hydrophilic silica particles.[78] In another example, aluminum oxyhydroxide nanorods (ALNRs) were functionalized with either -NH2 or -SO3H to alter surface charge and assessed for cellular uptake.[79] Although all ALNRs were taken up by immune cells with similar efficiencies, -NH2 functionalized ALNRs exhibited higher levels of lysosomal damage and activation than -SO3H functionalized ALNRs and unfunctionalized ALNRs.
In addition to facilitating membrane penetration by physical interactions of materials with the membrane, charge also plays a role due to its effect on the capacity of materials to buffer pH. Following initial uptake of materials by cells into endosomes, cells naturally lower the intravessicular pH as endosomes mature into lysosomes to support degradation into resources cells can use. The reductive environment and relative low pH of lysosomes create opportunities for materials that exhibited altered properties or triggered response when these changing environmental cues occur. For example, positively charged polymer particles containing pH buffering units can induce an osmotic pressure buildup leading to lysosomal disruption for cytosol delivery. In one study, increasing the number of histidine residues on side arms of dendrimers offers more protonation sites under acidic endosomal conditions, altering the osmolarity in the intracellular compartment, leading to increased lysosomal disruption.[80] Importantly, tuning the ability of particles to rupture lysosomes and enter the cytosol has been shown to modulate activation the inflammasome (Figure 3D). Following a similar mechanism, adsorption of cationic PEI to mesoporous silicon microrods also allows for lysosomal rupture following uptake.[81] These results exemplify that positively charged particles can facilitate lysosomal escape.
The relative roles of charge and hydrophobicity has been investigated by the Su group.[82] In these studies, chitosan microgels were evaluated for their uptake efficiency and ability to activate bone marrow derived DCs (BMDCs) depending of the extent of functionalization of positively-charged quaternary ammonium groups to a chitosan backbone; this level was termed the “quartenization”. Importantly, quartenization was a critical factor in dictating the microgel hydrophobicity and charge. While low quartenization microgels possess high hydrophobicity and lower surface charge, higher quartenization results in lower hydrophobicity and higher surface charge. Only moderate quartenization microgels display both high, positive surface charge and high hydrophobicity. Although lower quartenization exhibited increased uptake, microgels with moderate quartenization elicited the strongest immune responses, displaying improved stimulation of BMDCs in vitro; this was true even at lower antigen doses. These findings were attributed to the improved ability of highly charged hydrophobic microgels to disrupt membranes. Thus, the membrane disrupting potential of materials is impacted by the combination of hydrophobic and electrostatic interactions. These differences in uptake are particularly important in biomaterials designed to deliver antigen, because the mode of uptake affects which molecular machinery in immune cells encounter the antigen, which in turn determines how antigen is processed.
While the examples above present ways in which biomaterials can be tuned to promote immune signal localization to specific compartments within cells, it is equally important that immune signals are then released from biomaterial carriers in a manner that will enable processing by their target pathways (e.g. MHC, TLRs, inflammasome). The ability for carriers to quickly degrade and release cargo within the endosome is important for targeting immune receptors localized within this compartment. Biomaterials can be engineered to quickly release antigen under acidic conditions within endosomes to facilitate antigen processing and presentation. For example, poly(orthoester)s that rapidly degraded at pH 5.0 were found to enhance antigen presentation over PLGA NPs, suggesting that faster release kinetics improved antigen processing.[83] Additionally, the accumulation of particles within immune cells can pose problems. Accumulation of particles can lead to endosomal dysfunction, leading to blockage of other key cellular functions. For example, increased localization of smaller NPs in endosomes was associated with slowed antigen degradation into peptides in endosomes, a critical step for intracellular processing of antigen onto MHC.[84] Thus, controlled release and polymer degradation characteristics offer an avenue to facilitate the pacing of immune signal processing by cells. Control over the timing of immune signal delivery and availability will be addressed in further detail in Section 7.
5. Material Properties Regulate the Delivery of Immunostimulatory Cues to Control Immune Activation
In the previous two sections we focused on general targeting at the immune cell and immune tissue scales. Here, we focus on how the molecular control provided by biomaterials can direct delivery of immune signals, beginning with immunostimulatory cues to immune cells. As reviewed in Section 2, innate responses serve as the first line of defense against pathogens and allow for rapid protection by activating APCs and stimulating the release of inflammatory cytokines. Thus – owing to some of the same design capabilities explained in Sections 3 and 4 – biomaterial carriers can directly or indirectly (i.e. delivery of payload) drive generalized inflammation and concentrate immune signals to activate innate and subsequently adaptive immune cells. This is of particular interest from a design perspective due to the innate immune system’s role in initiating cellular and humoral immune responses needed in vaccines and immunotherapies. As such, APCs remain a frequent target in immune engineering.[33,36,85–87]
5.1. Biomaterials exhibit intrinsic immunogenic features that can trigger innate immune function
Many biomaterials are inherently immunogenic, in other words, the material itself can trigger or modulate immune function. While this activity can create problems – for example, in the context of autoimmune disease where carrier-induced inflammation could exacerbate the activity of the dysfunctional immune cells – the intrinsic immunogenicity of materials can also be harnessed to promote immune function for vaccines and immunotherapies against infectious disease and cancer. As one illustration, arginine rich polymers have been shown to activate the complement system, an innate immune pathway for the clearance of pathogens.[88] Different forms of the same material can also trigger different immune responses. Gold nanoparticles have been reported to preferentially activate different innate pathways depending on size.[89] Likewise, Chen et. al offer insight using thiolated poly(methacrylic acid) polymer capsules consisting of spheres and rods that demonstrate similar levels of uptake by macrophages.[90] In these studies, short rod-shaped capsules were found to promote a larger increase in inflammatory TNF and IL-8 cytokine secretion. Neither intracellular fate nor capsule size and volume appeared to play a role in the observed differences in cytokine secretion, suggesting that inflammatory cytokine secretion is dependent on shape.
The dependence on shape in altering intrinsic immune activity and innate immune cell interactions is not confined to polymer-based biomaterials. Nucleic acids offer a facile way to study size and morphology due to the relative ease of forming structures with well-defined configurations and have been studied by several labs.[91–94] As one example, the Afonin lab constructed a library of RNA and DNA based NPs of different sizes and shapes with the inclusion of fibrous and globular structures, in addition to planar structures with multiple facets (Figure 4A).[95] These NPs only stimulate immune responses when complexed to a polymer carrier; nucleic acid structures or carrier alone displayed no immunogenicity. This finding illustrates one way in which biomaterial structure can directly impact immunogenicity. DNA NPs were found to be overall less immunostimulatory than RNA based NPs. Again, shape and structure were found to influence immunogenicity with globular structures being more immunogenic than fibers, which were more immunogenic than planar structures.
Figure 4.
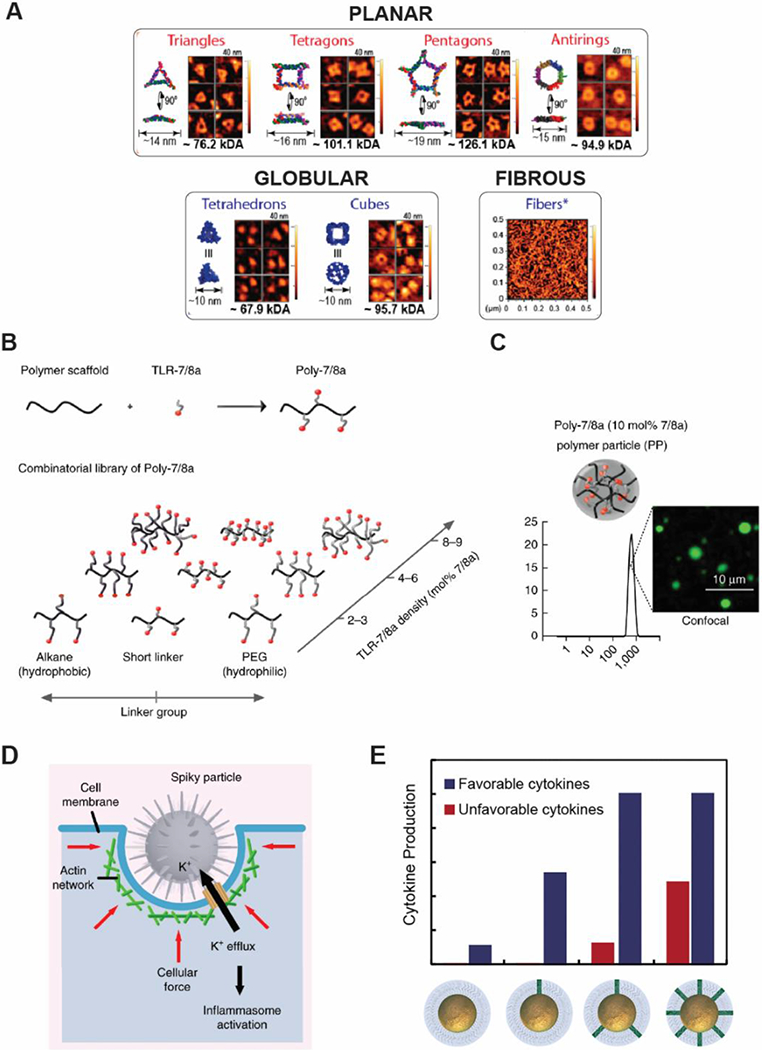
Shape and surface characteristics can be tailored to improve delivery of immunostimulatory signals to the immune system. A) A library of self-assembled nucleic acid structures offers tunable activation of innate immune responses. Reproduced with permission. [95] Copyright 2017, American Chemical Society. B) TLR7/8 is conjugated to a polymer scaffold at different densities differentially activate immune cells C) Higher densities of TLR7/8 form polymer particles, while lower density remain as random coils. Reproduced with permission.[105] Copyright 2015, Nature Publishing Group D) Nanospikes on nanoparticles exert mechanical stress on cells, leading to potassium efflux and inflammasome activation to enhance immune activation by “danger signals”. Reproduced with permission.[107] Copyright 2018, Springer Nature. E) Increased loading of CpG improves production of cytokines that promote anti-tumor responses. However, at higher concentrations cytokines that promote tumor growth and survival are produced. Reproduced with permission.[112] Copyright 2019, American Chemical Society.
Furthering these findings, the Guo lab has developed a library of RNA based NPs comprised of triangle, square, pentagon, and tetrahedron shapes of different sizes. For each shape, extended nucleic acid sequences were attached at each vertice to form an additional set of RNA structures with “arms” [91] Only NPs with extended sequences stimulated production of TNF and IL-6 by macrophages; and immunogenicity was also sequence dependent. When size was kept constant, inflammatory cytokine secretion levels correlated with the number of sides on polygons such that triangles exhibited the lowest secretion levels. This finding may have been influenced by the presence of additional extended sequences on higher ordered polygons. Additionally, the tetrahedron structure exhibited the highest level of cytokine secretion of all NPs, again, suggesting a role of dimensionality (i.e. planar vs. three-dimensional structure) in stimulating inflammatory responses. Importantly these results offer the potential of tunable immunogenicity to produce either a minimal immune response allowing for NPs that can serve as immunologically-inert therapeutic vectors, or a strong adjuvant immune response, such as those useful in vaccines and cancer immunotherapies.
In addition to shape, controlled release offered by degradable polymers can also lead to activation of immune responses by biomaterials themselves. Given that the properties of polymers are altered during degradation, our lab has explored how degradation of biodegradable polymeric carriers impacts immunogenicity over time. NPs comprised of poly(beta-amino ester) (PBAEs), a class of rapidly degradable polymers, exhibit changes in charge, size, and molecular weight as the polymers degrade.[96] Notably, PBAEs were observed to be immunogenic only in particulate form, with free polymer failing to activate DCs. Activation of DCs by polymer alone was dependent on the extent of degradation. Further studies have revealed that regardless of the starting molecular weight of the polymer, the immunogenicity was identified to be greatest when the molecular weight of degrading PBAEs decreased to a range of 1.5–3kDa, below which immunogenicity was eventually lost.[97] These studies demonstrate that the intrinsic immunogenicity of polymers evolves with degradation, highlighting another important consideration in biomaterial design in the control of APCs and other immune cells. This is particularly important in controlled release to prolong immune signal retention, which will be addressed in Section 7.
Similarly, PLGA, one of the most investigated biomaterials for particulate based immunoengineering, has also been shown to exhibit degradation-dependent immunomodulation of APCs. Allen et. al investigated the immunomodulatory properties of PLGA across multiple molecular weights over time.[98] In this studies, empty PLGA MPs with differing MW (10kDa, 22kDa, and 90kDa) were prepared. Notably, variations in the molecular weight and composition used influenced the degradation of PLGA. Treatment of DCs with PLGA MPs alone resulted in a time-dependent decreased expression of stimulatory markers MHC-II, CD80, and CD86 in the maturation level of cells. Even when challenged with LPS to stimulate DC activation, MP-treated cells resulted in a marked decreased expression of stimulatory markers and decreased inflammatory IL-12 secretion compared to treatment with LPS alone. The observed immune inhibition was correlated to increased lactic acid, both intracellularly and extracellularly. Importantly, lactic acid is a product of PLGA degradation and has been shown to be immunosuppressive in the tumor environment.[99,100] As such, accumulation of lactic acid as phagocytosed PLGA MPs degraded creates another mechanism that impacts innate immunity. This idea was further supported by findings that immunosuppression was dependent on MW; slower degrading high MW polymers that produce lactic acid more slowly required longer incubation times to produce comparable dampening of DC activation. Together, these results demonstrate that PLGA degradation can lead to immunosuppression of DCs via the accumulation of lactic acid byproducts. These studies illustrate how degradation alters biomaterial modulation of the local immune environment, highlighting the need for further studies to better understand the evolving immunogenicity of materials.
5.2. Biomaterials modify adjuvant function to direct innate and adaptive immune responses
Moving from intrinsic properties of biomaterials as stimulatory cues that direct immunity, here we focus on biomaterials to deliver adjuvants and stimulatory signals that activate APCs or other innate cells. For example, Loftus et. al demonstrated that conjugating innate-activating antibody ligands to a graphene oxide template can be used to stimulate specialized innate immune cells – NK cells – much more effectively than the soluble antibody cues.[101] Additionally, some immunostimulants can cause serious adverse immune-toxicity effects if disseminated via systemic circulation. This constraint creates an additional challenge when multiple adjuvants are involved, which sometimes generate synergistic or self-amplifying responses.[102] Encapsulation of adjuvants can reduce systemic exposure, limiting delivery of immunostimulatory cues to targeted cells and tissues as described in Sections 3 and 4. Further, encapsulation can also enhance uptake by APCs, offering the ability to deliver multiple adjuvants to the same innate-immune cells and control adjuvant display density. These materials can also be used to mimic common physiological properties of particulate pathogens – such as size, shape, or stiffness. Biomaterial carriers can be tuned to deliver cues in a manner that mimics the pathogens immune cells are specialized to detect and internalize, allowing for improved potency relative to soluble adjuvants or signals that innate immune cells may not as easily sense.
The ability to carefully tune polymer chemistry presents another mode of modularity for biomaterials. For example, PBAE chemistry can be readily tuned for hydrophobicity and charge density. In recent studies, a large library of PBAEs NP encapsulating the adjuvant polyIC were used to identify formulations that enhanced the magnitude and duration of antibody responses following vaccination.[103] These studies offer a path to improve our understanding of the role of polymer chemical and structural features in the effective delivery of adjuvants to enable the rational design of biomaterials based vaccines and immunotherapies. Importantly, Sofias et. al have demonstrated that the chemistry by which surface ligands are attached can also have a large effect on immune responses [104], reflecting a need for careful consideration over how adjuvants are conjugated to biomaterials.
The Seder lab has investigated how optimal delivery of TLRs can enhance delivery of immunostimulatory cues to APCs and improve vaccine immunogenicity.[105] TLR7/8a was conjugated to a polymer scaffold to generate a library of adjuvant-linked polymers (Poly-7/8a) with different densities of TLR7/8a displayed on the polymer (Figure 4B). In aqueous conditions, increasing density of TLR7/8a resulted in assembly of the Poly-7/8a into structures, such that low to intermediate densities produced random coils arrangements, while higher densities promoted formation of particles (Figure 4C). Higher density particles increased cytokine secretion and activation of DCs and macrophages relative to lower density particles, despite the overall dose of TLR7/8a being constant across formulations. Thus, particle formation, increasing densities of TLR-7/8a on the polymers, or both, were critical in determining the potency of immune responses. In similar studies, CpG – a TLR9 agonist, was conjugated to poly (L-glutamic acid) (PGA) via disulfide bonds that could readily be reduced under the acidic environment within lysosomes to release CpG.[106] The elasticity and cargo loading of these PGA-CpG conjugate NPs could be readily be tuned by varying crosslinking density. During in vitro experiments, activation levels of DCs could be readily varied by altering crosslinking density. Higher crosslinking density resulted in higher loading capacity, which led to increased DC activation. Together these studies highlight how biomaterials can alter adjuvant delivery to improve innate responses.
As alluded to above using the example of particular versus soluble signals, biomaterial can also improve innate immune cell activation and subsequent downstream responses by mimicking other features not present on soluble adjuvants. Wang et. al designed titanium oxide (TiO2) MPs decorated with nanospikes.[107] These spiky NPs were shown to activate and amplify innate immune responses. Bone marrow macrophages were incubated with spiky or rough particles or nanorods. Following priming with TLR4 agonist, lipopolysaccharide (LPS), spiky particles triggered inflammatory IL-1β secretion (Figure 4D). Additional studies revealed that mechanical stress exerted on the cell membrane during uptake of spiky TiO2 MPs by macrophages stimulated potassium efflux, resulting in inflammasome activation and increased IL-1β. Importantly, these findings were LPS-dependent; treatment with TiO2 MPs alone resulted in no significant changes. As such, while activation of innate immunity requires the presence of danger signals, morphology can play a role in potentiating the response.
From the above study, is it clear that co-administration of biomaterials with adjuvants can alter how APCs respond to these immunostimulatory signals. The Fahmy lab explored the effect that NPs may have in these skewing responses.[108] Silica NPs were coated with different poly(amino acid)s to form a library of NPs of different size, charge, and hydrophobicity. When DCs were treated with NPs in conjunction with TLR3/4 agonists, IL-1β secretion was dependent on size and hydrophobicity. Charge, on the other hand, did not have a significant effect on the generation of innate immune responses. However, cationic NPs were found to improve proliferation of T cells. These findings highlight that biomaterials can be used to enhance or alter the immune system’s response to an adjuvant. Intrinsic properties of biomaterials can also alter cytokine secretion profiles, which regulate and direct immune responses. Thus, material properties that exhibit intrinsic immunogenic effects as described in Section 5.1 can also enhance weakly immunogenic adjuvants. For instance, Tazaki et. al conjugated a safer (i.e., less toxic), but poorly immunogenic RNA adjuvant to gold NPs. In these studies, it was observed that nanorods, but not spheres, enhanced the adjuvanticity improving suppression of influenza infection in mice immunized intranasally.[109] Thus material properties can also be harnessed to improve adjuvanticity and improve safety.
Despite the need for more potent adjuvants, the ability to modulate and carefully manipulate the type and magnitude of response remains an important goal. For example, clinical translation of TLR-based adjuvants requires balancing the induction effective responses with safety concerns sometimes related to generation of excessive systemic inflammatory responses.[110,111]
The modularity of biomaterials can address this need by allowing for tunable loading of adjuvants to optimize immune activation in the absence of undesirable side effects. In one study, CpG was conjugated to gold NPs to improve macrophage activation to promote anti-tumor responses.[112] Maximal immunostimulation was achieved when CpG comprised as little as 5% of total oligonucleotides. NPs with higher compositions of CpG achieved similar levels of immunostimulation and production of inflammatory cytokines that promote anti-tumor immunity (i.e. TNF). However, higher levels of CpG was also associated with elevated levels of cytokines that have been linked to tumor growth (Figure 4E). As such, it is important to be able to precisely control the magnitude of the response as well as the profile of cytokines produced to stimulate desired responses.
While we previously discussed strategies that employ polymers as scaffolds to increase loading capacity of adjuvants, biomaterial carriers can also alter the immunostimulatory potential by limiting the accessibility of adjuvants. CpG complexes formed through electrostatic assembly with an arginine-based poly(ester amide) have been observed to elicit different immune responses in macrophages based on polymer chain stiffness.[113] CpG complexes lowered immune responses compared to soluble CpG alone, with softer chained polymers exhibiting the greatest reduction in the immune response. The reduction in immune responses was hypothesized to be the result of polymer binding to CpG, with softer chain polymers exhibiting more favorable binding interactions. In similar studies, cationic polymers such as poly(beta-amino esters) (PBAE), which are designed to bind nucleic acid, have also been found to tightly bind CpG to form NP complexes.[114] As such, at higher w/w ratio of PBAE:CpG, CpG remains tightly bound to PBAEs, rendering it inaccessible to activate TLR. Interestingly, however, additional studies revealed that higher w/w ratio of PBAE:CpG facilitated improved CpG uptake over soluble CpG. Thus, although higher interaction strength of polymers for adjuvants can decrease the activating potential, the ability to bind CpG to form NPs is important for promoting uptake. These findings present an example of design considerations that must be balanced for engineering effective vaccines and immunotherapies. From these studies, it is clear that the physicochemical properties of materials can play a key role in altering how innate immune cell receive immune signals. In the next section, we discuss how materials properties can be leveraged to manipulate downstream adaptive responses by controlling antigen encounter.
6. Biomaterials Control the Context in Which Antigen is Presented to Tune Adaptive Immune Responses
In this section, we highlight how the physicochemical properties of biomaterials can be used to influence adaptive immune response. We first discuss how controlled APC activation and antigen presentation by biomaterial carriers alters the immune microenvironment (e.g., LNs) in which T cells are activated. Next, beginning in Section 6.3, we explore how antigen presentation by materials are modulated to directly interact with T cells to activate and polarize their responses, followed by examples in which materials initiate B cell responses. Finally, we highlight examples of how biomaterials can be engineered to promote antigen-specific immune tolerance that could be useful in treating autoimmune disease, inflammatory disease, and for transplantation.
6.1. Biomaterials can be engineered to alter the microenvironment in which T cell responses are generated
The modulation of innate immune responses by biomaterials defines the conditions (i.e. immune signal presentation, cytokine milieu) under which induction of antigen specific adaptive immune responses occurs. Immune signal trafficking throughout the host and subsequent intracellular processing of these signals by innate cells all contribute to the types of adaptive immune response that result. Demonstrating that inducing a local inflammatory LN environment can enhance T cell responses, Lynn et. al synthesized polymer-TLR7/8a conjugates with different chemical compositions and chain architectures.[115] These conjugates exhibited distinct molecular conformation and size (e.g., random coil, polymer micelle and particles) to evaluate how these parameters impact the potency of the adjuvant for inducing CD8+ T cell response in mouse models. Cytokine production in LNs and the number of CD8+ T cells induced against antigen increased with increasing polymer-TLR-7/8a hydrodynamic radius, such that particles induced the highest magnitude responses, followed by micelles, then random coils. The ability of the particle to induce greater T cell responses was attributed to increased particle uptake by macrophages and monocytes within LNs, leading to increased activation of APCs and production of inflammatory cytokines, such as IL-12. Thus, molecular conformation and size of polymers laden with TLR-7/8a influences the local LN environment to improve T cell responses. Follow-up studies demonstrated that the physical form of peptide plays a similar role.[61] Synthetic peptides comprised of an antigen epitope conjugated to 30 amino acid long peptide sequence display different hydrodynamic behaviors depending on the hydrophobicity of peptide sequences. Hydrophobic sequences form particulates that result in 20-fold higher T cell responses compared to hydrophilic sequences that remain soluble in aqueous solution. This is due in part to particle peptides being retained longer in LNs, allowing for prolonged antigen presentation.
In addition to driving the activation of T cell responses, biomaterials can also be tuned to skew the specific features of adaptive responses. For example, the choice of carrier can impact the balance between inflammatory T cell subsets. As one illustration, antigen loaded onto calcium phosphate templates and aluminum hydroxide induced both TH1 and TH2 responses, which stimulate other T cells and B cells, respectively.[116] Chitosan templates, however, only induced TH1 responses. In the context of allergies and autoimmune diseases, the ability to inhibit infiltration of activated APCs at sites of disease can skew T cell responses towards tolerance. One unique approach that has been explored employs drug-free biodegradable NPs lacking any targeting ligands, but composed of different polymers to inhibit specific inflammatory cells from entering sites of disease.[117] NPs with higher MW polymers and higher hydrophilicity associated with inflammatory cells to redirect their trafficking. In another study, the Shea lab investigated the effects of poly (lactide-co-glycolide) (PLG) and poly (lactide) (PLA) NPs in delivering antigen.[118] Compared to PLG NPs, treatment with PLA NPs markedly ameliorated disease in a mouse model of multiple sclerosis (MS), an autoimmune disease in which immune cells mistakenly attack the myelin insulating neurons (Figure 5A). The addition of a methyl group in lactide makes PLA more hydrophobic than PLG, which was found to facilitate association with APCs and inhibit expression of costimulatory markers. This translated to reduced numbers of CD4+ T cells and B cells in the CNS. These observations illustrate how engineering to curtail inflammatory cues to T cells can limit immune responses and highlight the importance of modulating the microenvironment that T cells are exposed to.
Figure 5.
Biomaterials modulate the context of antigen delivery to regulate immunity for both immune activation and to promote tolerance. A) NP-cell interactions depend on the type of polymer used. Association of PLA particles with APC inhibit expression of costimulatory molecules to induce tolerance, resulting in lower disease severity. Reproduced with permission.[118] Copyright 2019, Elsevier. B) Reversible chemistry of OVA-p(Man-TLR7) linker allows for release of unmodified antigen, allowing for improved antigen presentation and downstream proliferation of both CD4+ and CD8+ T cells. Reproduced with permission.[120] Copyright 2019, Nature Publishing. C) TEM and schematic illustrations of different deliery methods of antigen that evoke different intracellular processing mechanisms by APCs. NPs with antigen presented on the surface promote antigen presentation and activation of CD4+ while NPs that encapsulate antigen, promote CD8+ T cell responses. Incorporation of antigen at both locales promotes expansion of both CD4+ and CD8+ T cells. Reproduced with permission.[124] Copyright 2016, Elsevier. D) Antigen presentation density plays an important role in activating T cells. Small aAPCs poorly expand T cells at lower concentrations compared to larger antigen coated particles. When the concentration is raised to improve clustered binding of antigen coated NPs, small aAPCs can expand T cells as well as larger aAPCs. Reproduced with permission.[126] Copyright 2017, American Chemical Society. E) Presentation of stimulatory T cell cues (e.g. anti-CD3, anti-CD28) on lipid bilayers mimcs the dynamic process of surface cue presentation, allowing for robust expansion of T cells even at lower presentation density. Reproduced with permission.[127] Copyright 2018, Nature Publishing. F) Antigen conjugated to alum using a linker composed of repeating phosphoserine units offers conformational control. Single particle electron microscopy analysis of antibodies in treated rabbits revealed that the ability to orient antigen display allowed for targetting of a larger repetoire of antigen epitopes. Reproduced with permission.[134] Copyright 2020, Nature Publishing. G) Antigen density also plays an important role in vaccines and immunotherapies for autoimmune diseases. Quantum dots enable the tuning of antigen display density. H) At constant overall antigen dose, treatment with more particles displaying antigen at a lower density can more effectively reduce clinical scores in a mouse model of multiple sclerosis (EAE). Reproduced according to the terms of the Creative Commons Attribution 4.0 International License.[144] Copyright 2017, Wiley-VCH.
6.2. Biomaterials can alter antigen presentation by APCs to activate CD4+ versus CD8 T+ cells
The spatial organization of cell surface proteins at immune interfaces is a central aspect of immune cell signaling. While Section 5 discussed antigen processing by APCs, we now turn our focus to antigen presentation. As the bridge between innate and adaptive immunity, APC presentation of antigen is critical and can alter subsequent priming of specific adaptive immune responses. For instance, increasing antigen presentation on APCs can increase the interactions of APCs with T cells. Optimizing the size of PLGA particles to improve uptake of antigen loaded particles can lead to increased peptide presentation in MHC and induce inflammatory cytokines.[119] Equally important is the ability to maintain the structural integrity of the presented antigen, because T cells can only recognize specific peptide sequences under precise conformations. This is a particularly important consideration for antigens conjugated to materials, which must be cleaved before they can be processed into peptides and loaded onto MHC. One strategy is the use of pH sensitive linkers to control release. In collaboration, the Swartz and Hubbell labs developed a pH sensitive self-immolative linker to conjugate antigen to a glyco-adjuvant conjugate.[120] Importantly, the self-immolative linker used reversible chemistry, allowing for the release of conjugated antigen without additional modifications, unlike other commonly used linkers which often chemically tag the antigen upon release. The ability to release unmodified antigen was revealed to augment antigen presentation to T cells, resulting in improved proliferation of both CD4+ and CD8+ T cells compared to a non-self-immolative linker (Figure 5B). This study highlights a role for controlled release of antigen to improve T cell activation.
Improving antigen presentation to increase the magnitude of T cell responses, however, is only one dimension to consider. To drive specific T cell subsets, antigens must be also be presented in the correct MHC. We previously discussed the ability of cationic particles to facilitate lysosomal escape (Section 3.2) to localize immune signals within the cytosol. While this is a key step in cross-presentation, excessive exposure to the endolysosomal environment can also lead to degradation of peptides, hindering its proper presentation following escape into the cytosol. This is evident in a comparison between anionic and cationic liposomes. Cationic, but not anionic liposomes have been found to increase cross-presentation of extracellular antigens.[121] This is due in part to elevation of lysosomal pH by cationic liposomes, which reduces the endolysosomal degradation of antigens. In contrast, anionic liposomes do not affect lysosomal pH. However, a critical limitation is that cationic liposomes can have increased cytotoxicity at higher concentrations, while anionic liposomes typically exhibit no cytotoxicity even at much higher doses. Thus, alternative biomaterial approaches to promote antigen presentation in the correct MHC are being explored.
In one approach, work by Zupancic et. al demonstrates that the nature of protein association to biomaterial carriers (i.e. adsorbed vs. entrapped) affects how these antigens are processed and displayed on DCs.[122] These studies support the need for antigen protection to promote cross-presentation. Immunization with antigen-adsorbed NPs upregulated MHC-II, while antigen-entrapped NPs upregulated MHC-I. This is significant because these results suggest that antigen-loaded NPs may be more efficient for cross-presentation, perhaps due to cargo protection offered by encapsulation. As previously mentioned, the priming of specific adaptive immune responses requires the presentation of antigen within specific classes of MHC. Thus, the ability to tune the presentation of antigen on MHC-I vs. MHC-II remains a key area of interest. This is exemplified in recent studies by Restrepo et. al, which examined how changes in the mode of antigen delivery – encapsulated antigen vs. antigen decorated on surfaces – could control activation of CD4+ and CD8+ T cell responses.[123] The model antigen OVA was delivered using either NPs or polymersomes (PSs), each composed of hydrophobic polymer poly(propylene sulfide) (PPS) and hydrophilic PEG; this design allowed considerable variation in interactions between the carriers and APCs. The NPs were comprised of a hydrophobic core of PPS and a corona of PEG onto which OVA was readily conjugated using disulfide bonds. In contrast, PSs consist of an aqueous core where antigen is loaded, surrounded by a polymer bilayer of PEG-PPS. These differences in material structure promoted unique T cell subsets. In particular, NPs promoted CD8+ responses, while PS preferentially enhanced CD4+ responses. While both carriers activated DCs to similar levels, they each displayed different intracellular processing of antigen. NPs were primarily found in early endosomes with uptake studies suggesting that the disulfide link is cleaved from the carrier to allow escape of OVA from the endosome to the cytosol. On the other hand, cargo protection by the polymer bilayer of PSs prevented early degradation, allowing for antigen retention within vesicles until these cellular structures acidified into lysosomes. These differences in processing contribute to differential antigen presentation on MHC-I and MHC-II by APCs, resulting in activation of CD8+ or CD4+ T cells, respectively.
Liu et. al suggest that the strongest responses occur by using a combination of the antigen displays described in the examples above (i.e. antigen adsorption and encapsulation).[124] In this study, nanoparticles with antigen both bound to the surface and encapsulated were compared to particles that localized antigen either on the surface or within particles (Figure 5C). Antigen loading was dose matched across all formulations. NPs that incorporated antigen through both encapsulation and adsorption were significantly more effectively, as this design offered not only adequate initial antigen exposure, but also long-term antigen persistence at the injection site due to cargo protection. More importantly, this design allowed for antigen presentation through both MHC-I and MHC-II. The ability of particles to simultaneous elicit CD4+ and CD8+ responses is critical for generating immunological memory. These results reveal the unique physical and chemical properties that result from carrier design can also generate distinct immune responses, even when the same building blocks are used. Collectively, these findings highlight how biomaterial design can control the context under which antigen is presented by APCs to ultimately help shape the resulting adaptive immune response.
6.3. Biomaterial mimics of APCs can present antigen directly to T cells
Biomaterials can also be engineered to directly interact with T cells to trigger differentiation and expansion into specific phenotypes. One opportunity created by such strategies is the ability to directly alter design parameters such as antigen display density, aspects ratio, or shape to impact T cell activation. Several labs have designed artificial APCs (aAPCs) to control immune signal display to APCs.[125] In one example, researchers in the Schneck lab have designed aAPCs composed of superparamagnetic iron oxide NPs coated with peptide-MHC (pMHC) and a costimulatory molecule, anti-CD28.[126] Magnetic NPs were used to control clustering of NPs. Size (i.e., 50nm, 300nm, 600nm) and stimulatory ligand density were then varied to determine whether these properties could improve the efficiency of T cell activation. In these studies, larger aAPCs more efficiently activated T cells (Figure 5D). Smaller aAPCs, on the other hand, required saturating doses of pMHC or artificial magnetic clustering of NPs to activate T cells at similar levels compared to the larger particles. These results suggest that T cell activation is dependent on the formation of TCR clusters. This was supported by an inverse relationship between aAPC size and the number of aAPCs needed to provide effective T cell signaling, whereby larger aAPCs required fewer aAPCs. Further, transmission electron microscopy studies revealed that while very few 50nm aAPCs attached to T cells, many more 300nm to 600nm aAPCs attached to T cells. This result suggests that ligand density and size affect the ability of aAPCs to interact with T cells. Collectively, these findings indicate that TCR activation requires multi-receptor ligation and formation of TCR nanoclusters.
Studies by the Mooney lab further elucidate the importance of TCR nanoclusters.[127] Under a different platform using mesoporous silica microrods coated with lipid bilayers to form APC mimetic scaffolds (APC-ms), similar findings were obtained (Figure 5E). Again, T cell expansion was observed to be dependent on the density of stimulatory cues and number of APC-ms. Strikingly, however, scaffolds with significantly lower density of stimulatory cues could still promote robust expansion of T cells. These findings were attributed to the presentation of stimuli on a fluid lipid membrane, which better emulated the dynamic process of surface cue presentation on APCs contrary to other synthetic aAPC systems which present immune signals on static surfaces. Additionally, formulations that presented higher amounts of T-cell stimuli (i.e. anti-CD3, anti-CD28, IL-2) skewed T cell expansion towards CD4+, while lower amounts of T-cell stimuli promoted a more balanced of CD4-to-CD8 T cells. These results suggest another role for ligand density in polarizing T cell responses. Supporting this idea, antigen density, in combination with surface area and particle size, has also been found to correlate with the subtype of immune responses generated. In one study, spherical NPs (193nm) with antigen conjugated to the surface produced a TH1-biased response, whereas large rod-shaped particles (1530 nm) produce a TH2-biased response.[128] Thus, the density of costimulatory signals and antigen displayed on biomaterials not only plays a critical role in their ability to interact with and trigger T cells, but can even alter the polarization of T cell responses. In the next section, we discuss how controlling antigen presentation on biomaterials can be harnessed to improve B cell maturation that controls antibody production.
6.4. Controlled antigen presentation on biomaterials enhances antibody responses
In addition to presenting antigen to T cells, biomaterials can also be engineered to promote B cell activation and antibody production. Many B cell activation processes begin with cross-linking of surface receptors by antigen, which implies that the conformation in which the antigen is displayed impacts these events, a criterion which biomaterials are well-suited to leverage. Several labs have demonstrated that particulate shape and size play a key role in eliciting higher levels of antibody titers on different substrates, such as gold [129] and hydroxyapatite.[130] Interestingly, conjugation of antigen to the outer surfaces of these particles typically elicits higher immune responses when conjugated to smaller sized particles with lower surface areas and therefore less antigen per particle. Similar trends have been noted when antigen is chemically grafted onto the shell of nanostructured lipid carriers, such that small anionic lipid particles elicit stronger antibody responses compared to larger, cationic lipid particles.[131] These findings are surprising because contrarily, positively charged particles favor uptake, suggesting that higher antibody responses are not solely dependent on internalization of antigen particles, but that other processing mechanisms may be involved. Because the overall antigen dose is maintained constant across particle types in such studies, a possible explanation is that antigen density may also play a role in eliciting strong humoral responses by offering more optimal antigen interaction with B cells. This possibility is corroborated by Marcandalli et. al, who explored the structure-based design of NP vaccines, using self-assembling proteins.[132] An antigen trimer protein was conjugated to NP subunit building blocks to form icosahedral assemblies that could present up to 20 copies of the trimer with tunable control of the antigen display density. Immunogens that could not assemble into NP complexes promoted much weaker antibody responses even with dose-matched antigen amounts. This result suggests the increase in antibody production was related to the structure of the NP guiding antigen display and interactions with the B cell. Additionally, antigen density on the NP exterior correlated with the magnitude of the response, with higher density eliciting higher levels of antibody production. These findings support the hypothesis that efficient BCR cross-linking by the dense array of antigen on the NP surface, at least in part, improves immunogenicity.
However, high antigen density does not always favor improved antibody responses. In another study, NPs displaying lower densities of viral protein antigens were found to more efficiently stimulated antigen-specific B cells than NPs displaying higher antigen densities.[133] NPs displaying a low density of antigen also increased the number of GC B cells in immunized mice, resulting in higher levels of antigen-specific antibodies. Taken together, these observations suggest that sparse antigen density on NPs allows improved GC reactions that, in turn, give rise to durable memory reservoirs and elevated, long-lived serum antibodies. In the case of more immunogenic antigens, it is possible that antigen density may influence antigen access and handling by innate immune cells localized at the site of immunization. In this scenario, a higher protein density could alter draining to LNs or improve uptake by DCs and macrophages, diminishing the number of NPs available to be efficiently captured and presented to B cells within the draining LN. Alternatively, by displaying antigen at a lower density, epitopes may become more accessible due to less steric hinderance, allowing for improved immunogenicity and activation of a larger percentage of the antigen-specific B cell repertoire.
Building on these findings, another approach to improving B cell responses leverages biomaterials to improve the conformational display of antigens on biomaterial carriers. For example, the Irvine lab conjugated phosphoserine linkers to antigen, allowing for the tunable binding of immunogens to aluminum hydroxide (alum), the gold standard for adjuvants that is used in many FDA approved human vaccines (Figure 5F).[134] Importantly, this system allowed for both the tuning of antigen orientation and density. Interestingly, B cells were also observed to take up alum-antigen conjugates. These findings suggest that when bound to alum via pSer linkages, antigens can behave as a multivalent, particulate vaccine that are internalized by B cells. While, the immobilization of antigens to alum allowed for control over the conformation of the antigen, the density of antigen binding to alum could be augmented by increasing the number of serine residues in the linker. Importantly, it was demonstrated that the directed orientation of immunogens with the pSer linker can alter the B cell specificity of the immune response, allowing for B cell specificity to be tuned towards specific epitopes. This is particularly relevant for universal protection against highly mutative viruses such as HIV and influenza, in which specific targeting to more conserved regions of the virus are needed. Because these vaccines are effective only against antigenically-matched viruses, new design strategies that can improve specificity towards conserved regions remains an important area of research. In another approach, Skwarczynski et. al, formed self-assembling amphiphilic particles comprised of antigen coupled to poly(amino acids) comprised of 10 repeat units of hydrophobic amino acids.[135] The hydrophobic properties and conformation were easily modified by changing the type and number of amino acids. These particles were found to be self-adjuvanting, with the most hydrophobic amphiphilic particles displaying the highest level antibody titers. However, only particles that maintained the helical conformation of the antigen could generate strong antibodies against multiple strains of group A Streptococcus. These findings highlight the importance of antigen conformation in promoting strong antibodies responses, which can be facilitated through the modularity of biomaterials .
6.5. Tunable loading of antigen onto biomaterials can promote tolerance to combat autoimmune diseases
While biomaterial based immunomodulation has largely focused on generating potent responses, much exciting work is exploiting biomaterials to promote tolerance during autoimmune disease.[136–138] As discussed in Section 5.1, biomaterials can exhibit intrinsic immunogenic feature, which are heavily influenced by the physicochemical properties of materials. Thus, it is important to understand how features of biomaterials such as size and charge, as well as formulation designs such as antigen loading contribute to immune polarization of immune responses towards tolerance. This insight could support rational design criteria for autoimmune therapies and anti-inflammatory materials.
Pearson et. al developed PLGA NPs with modular loading of one or multiple self-antigen types coupled to the NP surface to study the role of size and antigen loading on TREG induction.[139] Higher antigen loading induced more TREGs, but TREG induction was also dependent on size; 400nm NPs induced more TREGs than 80nm NPs at the same total antigen dose. Contrary to this finding, other studies have observed that smaller particles are more effective at inducing tolerogenic responses. For example, phosphatidylserine liposomes have been reported to induce hyporesponsiveness to an otherwise immunogenic antigen.[140] Investigation of the biophysical properties governing this result revealed that smaller liposomes reduced DC activation and increased secretion of anti-inflammatory TGF-β, polarizing DCs towards a more tolerogenic phenotype and reducing antibody production against the antigen. In other studies, peptides were conjugated onto a HA polymer backbone to form different size antigen arrays.[141] Treatment with smaller sized arrays delayed disease onset and lowered disease incidence in a mouse model of MS. However, the impact of smaller size was only observed at earlier stages of disease. At later time points, antigen arrays displayed similar levels of efficacy across sizes, suggesting a role of smaller size in allowing for quicker drainage to LNs for faster polarization of T cells. These differences in outcomes highlight the need for further studies to elucidate the role of carrier size in inducing tolerance.
As previously described, TREGs can form during interactions with APCs presenting antigen to a T cell in the absence of costimulation. Similar to the principles discussed in Section 6.3, biomaterials that directly interact with T cells can be engineered to create more favorable interactions that promote the formation and expansion of TREGs. As one example, the Santamaria lab demonstrated that peptide presentation density can promote long term interactions with the TCR of T cells to promote TREG formation using a different type of aAPC.[142] In these studies, iron oxide NPs were conjugated with pMHC at different densities. pMHC density was found to play a key role in the activation of CD8+ T cell responses, such that 11 pMHCs per NP resulted in strikingly higher levels of IFN-γ secretion compared to NPs with only 8 pMHCs. This suggested that a clear threshold of pMHC density on NPs is required to activate T cells. Confirming this observation, larger particles required a higher number of pMHC per NP to activate T cells, suggesting that density, rather than the absolute number of pMHC molecules, drives these responses. Importantly, pMHC-NP dose and density were also observed to enhance TREG expansion, but in distinct ways. While density influenced the expression levels of a TREG marker (CD49b), resulting in its upregulation on cells, dose had a more minimal effect on expression levels. However, the proliferation of TREGs was found to be dose-dependent. Thus, it was determined that pMHC density regulated the efficiency of TREG formation, and dose controlled the magnitude of expansion. Because TCRs have been found to organize into nanoclusters with ligands to promote TCR cross-linking [143], it was hypothesized that higher density pMHC improved NP interaction with T cells. Further investigation revealed that binding of pMHC-NP with the TCR on T cells promoted the formation of TCR microclusters that increased in size with increasing pMHC density. However, below a threshold density, clusters were unable form. These findings suggest that pMHC density controls TREG conversion by promoting sustained assembly of TCR microclusters.
In other studies, Hess et. al have demonstrated that the degree of tolerance induced in a mouse model of MS (EAE) correlates with the density of self-antigen presented on quantum dots.[144] In these studies, myelin peptide (MOG), a self-antigen that is attacked in MS was displayed on quantum dots with tunable control over the density (Figure 5G). Following disease induction, mice were treated with quantum dots displaying antigen at one of three MOG densities, while maintaining a constant dose of MOG; thus, mice receiving lower ligand densities received a higher number of quantum dots (Figure 5H). Interestingly, MOG displayed at lower densities on a greater number of the particles exhibited the lowest clinical scores, lowest disease incidence, and healthiest body weights. These findings suggest that a higher number of tolerogenic particles displaying lower levels of self-antigen is more effective for inducing tolerance than fewer particles each displaying a higher density of peptide. Additional studies to elucidate potential mechanisms suggest that improved clinical scores are the result of increased TREG expansion and colocalization of quantum dots with macrophages and scavenger receptors involved in promoting tolerance. This observation is particularly significant because it highlights the importance of antigen density in controlling T cell responses, not only through direct interaction with T cells as described in previous paragraphs, but also by controlling antigen display to APCs that then interact with T cells.
7. Biomaterials can be engineered to alter the timescales over which immune signals are available
As alluded to already, the kinetics with which immune signals are encountered play an important role in initiating and directing adaptive immune response. Generation of strong adaptive immune responses often requires multi-step treatments, comprised of an initial “prime” vaccination, and an additional “booster” injection at later time points. Prolonged and targeted adjuvant or antigen uptake by APCs enables sustained DC activation, which contributes to the enhanced immune response seen with particulate vaccine delivery. Additionally, the ability to improve the pharmacokinetics of vaccines and immunotherapies can improve administration schedules, leading to improved safety and compliance.
7.1. Material properties can be tuned to prolong retention of immune signals within LNs
Temporally controlled antigen delivery may optimize immune responses for specific diseases of interest when different antigen-APC interaction kinetics lead to distinct downstream signals. In exciting studies, mini osmotic pumps implanted into mice allowed for continuous antigen release over the span of one or two weeks, providing a controlled system to study the importance of antigen persistence in immune response.[145] Continuous antigen exposure increased germinal center and serum antibody responses. These results suggested that regulating antigen kinetics may enable increased vaccine potency, further fueling ongoing interest in controlled release systems for improving vaccines and immunotherapies.
Biomaterials offer a platform to study and tune release kinetics. For instance, unique release rates for poly(lactic-co-glycolic-acid) (PLGA) microspheres can be obtained by varying MW and lactide:glycolide ratios.[146] Another strategy for extending antigen release is the use of different linker chemistries to alter release of antigen. Kapadia et. al varied antigen release rate and presentation time by conjugating a model antigen, SIINFEKL, to PEG-hydrogels via a disulfide or thioether linkage (Figure 6A).[147] Compared to the disulfide linkage, thioether linkage allowed for sustained release of peptide that prolonged antigen presentation over 72 hours. To further examine the mechanism of how NP-peptide formulations deliver antigenic peptide, BMDCs were treated with soluble antigen and antigen NPs, followed by washing with acidic citrate phosphate buffer to remove MHC-I peptide complexes from the surface of BMDCs. Cells were then incubated for an additional 24, 48 and 72 hours, enabling internalized antigens to be processed and re-presented onto the cell surface. Citrate-phosphate treatment completely removed SIINFEKL from p-MHC complexes for cells treated with soluble SIINFEKL but was unable to completely remove cell bound NP-peptide. At each time point, particle-conjugated SIINFEKL induced significantly higher antigen presentation in BMDCs as compare to soluble SIINFEKL. Slower release from the thioether bond resulted in prolonged antigen release and higher bioavailability, and therefore improved antigen presentation over time. During tumor studies, mice treated with NPs conjugated with either linkage exhibited significantly lower tumor growth compared to mice treated with soluble SIINFEKL and a TLR-based adjuvant, CpG (Figure 6B). However, due to the improved controlled release of SIINFEKL from thioether-linked NP formulations, tumor growth was delayed up to 14 days, compared to 7 days during treatment with the disulfide linked NP formulation.
Figure 6.
Tunable properties of biomaterials allow control over the persistence and pharmacokinetics to improve the quality of adaptive immune responses. A) Slower release of antigen from NPs conjugated to antigen via slower releasing thioether-linkages results in improved antigen presentation over time compared to NPs conjugated to fast releasing disulfide bonds B) Mice vaccinated with NPs conjugated to antigen by thioether linkages exhibit enhanced protection against tumor challenge, resulting in tumor growth inhibition up to 14 days post inoculation compared to 3 days and 7 days for soluble vaccine and NPs conjugated to antigen with disulfide bonds, respectively. Reproduced with permission.[147] Copyright 2017, Elsevier. C) FDCs internalize smaller NPs while larger NPs are retained on the surface, allowing for prolonged antigen availability. Reproduced with permission.[149] Copyright 2019, American Chemical Society. D) Spherical nucleic acids (SNAs) comprised of CpG and peptide antigen that are compositionally similar but vary in the position and/or conjugation chemistry of antigen peptides. Differences in antigen delivery result in different kinetics of peptide presentation and expressions of co-stimulatory markers. Reproduced with permission.[152] Copyright 2019, National Academy of Sciences.
Particle size can also impact the duration of peptide/MHC-II presentation.[148] DCs have been observed to display antigens conjugated to larger particles for longer periods of time than when they display antigens on smaller particles. In one study, antigen was conjugated onto polystyrene NPs of varying size by covalent linkages. The overall administered antigen dose, mass of particles, and antigen density was maintained constant across formulations, by altering the amount of antigen per particles and the number of particles administered to mice. For instance, larger particles contained more antigen per particle due to greater surface area, and as such, mice were given fewer particles per mouse, compared to mice that received smaller antigen particles. To assess kinetics of antigen presentation, a traceable antigen, EαGFP was covalently linked to NPs. Smaller NPs showed rapid uptake and presentation as early as 6 hours. Larger antigen NPs, on the other hand, required longer time intervals (within 24 hours) to achieve equivalent levels of uptake by APCs, however, antigen presentation was maintained beyond 72 hours. As such, although larger antigen NPs did not alter the magnitude of antigen presentation, they changed the dynamics of T cell/DC interactions, promoting stable, long-term interactions. The increased presentation duration ultimately promoted GC formation and antibody production; this sequence exemplifies the importance of sustained antigen presentation in eliciting robust immune responses.
Size and surface chemistry can also play a role in the prolonged retention of biomaterials within LNs. Studies by the Chan lab demonstrated that antigen-conjugated gold NPs 5–15 nm in size are rapidly cleared from LNs, while larger gold NPs 50–100 nm in size are retained for over 5 weeks.[149] This translated to an increase in GC B cell formation, and 5-fold increase in antigen specific antibody production compared to smaller NPs. Interestingly, retention of nanoparticles was found to be facilitated by gold nanoparticle interactions with follicular dendritic cells (FDCs), a specialized set of APCs that can retain antigen within LNs and serve as antigen depots for B cells. Small gold NPs were taken up by FDCs, resulting in their subsequent clearance (Figure 6C). Larger NPs on the hand were retained on FDC dendrites. Additional studies suggested that serum protein adsorption to NPs during circulation to LNs promoted NP binding to FDCs. Thus, the combination of surface chemistry and increased size, offer another method to promote antigen retention.
7.2. Biomaterials can tune the timescales over which multiple immune signals are received
Although biomaterials can be harnessed to mimic specific dosing schedules, temporal control over the delivery of vaccines and immunotherapies can be complicated by the need to deliver multiple signals (i.e. adjuvants and antigen). This is highlighted in studies by Chen et. al. In this work, acetalated dextran (Ace-DEX) MPs with distinct degradation profiles were used to deliver model antigen or adjuvant.[150] Encapsulated adjuvant generated stronger responses then soluble antigen, as indicated by faster-degrading MPs that promoted larger humoral and cellular responses in mice at earlier time points, while slow-degrading MPs drove stronger responses at later time points. MPs that degraded very quickly were associated with the lowest level of antibody responses. This result was likely due to faster clearance and therefore, less adjuvant exposure. When antigen was encapsulated within Ace-DEX MPs to test for controlled antigen delivery, fast-degrading MPs induced greater antibody and cytokine production throughout the length of the experiment. These findings suggest that faster-degrading Ace-DEX MPs may be beneficial to fighting diseases requiring a rapid antibody response or for individuals who need protection quickly, such post-exposure prophylaxis during accidental exposure to pathogens. Slower degrading MPs may be advantageous for conditions that would benefit from a more sustained immune response. It is also important to note that antibody responses had opposing relationships for the release kinetics of adjuvant versus antigen: delivery of adjuvant with fast-degrading MPs resulted in lower antibody response, but delivery of antigen with fast-degrading MPs induced greater antibody responses. This suggests a possible need for distinct control over adjuvant and antigen delivery.
Further supporting the need for well-designed dosing schedules, Tzeng et. al have demonstrated that effective combination cancer immunotherapy is highly dependent on the order of administration of each individual immunotherapy. [151] In these studies, a tumor specific antibody was combined with immunostimulatory cytokine to activate DCs, IFNα, to treat established tumors in a mouse melanoma model. Interestingly, improved survival rates were achieved only when IFNα was administered at a later time point (48 hours, 96 hours), following treatment with the tumor antibody. This finding suggests a requirement that DC maturation occur after generation of antigenic tumor debris. One hypothesis is that DC maturation results in the loss of the ability to phagocytose antigen. As such, administration of IFNα prior to treatment with tumor antibody to expose tumor antigens resulted in markedly worse therapeutic outcomes and lower survival rates.
In contrast, studies from the Mirkin lab suggest a need for similar timescales of antigen and adjuvant delivery. [152] Spherical nucleic acids (SNAs) comprised of CpG and peptide antigen that are compositionally similar but vary in antigen incorporation were used to study different mechanisms of co-delivery of antigen and immunostimulatory signals. In this work, superior anti-tumor immune responses were generated when antigen presentation and costimulatory markers were presented in tandem. These constructs were synthesized by absorbing CpG to the surface of liposomes and varying the position and/or conjugation chemistry of antigen peptides by i) encapsulating soluble antigen (SNA-E) ii) absorption of antigen to the liposomal surface (SNA-A) iii) hybridizing peptide to adsorbed CpG (SNA-H) (Figure 6D). These differences in antigen delivery resulted in different kinetics of peptide presentation, with SNA-A and SNA-H particles inducing peptide presentation at slower rates compared to SNA-E. These differences are likely attributed to the need for processing and dissociation of antigen adsorbed to the surface of SNAs. Importantly, the synchronization of peptide presentation and costimulatory marker expression was found to be important for generating cytotoxic and memory T cell phenotypes. Mice immunized with SNA-H particles exhibited the highest numbers of these antigen-specific CD8+ T cells cell types. Thus, not only can the modularity of biomaterials control release kinetics of immune signals, but they can also be tuned to control the kinetics of immune signal presentation by other cells.
8. Concluding Remarks
Despite increased insight into how biomaterials can be engineered to promote immune responses, the interplay between material properties remains a challenge in defining universal design criteria. For instance, changes in shape can alter size. Similarly, surface modifications can introduce changes in both charge and hydrophocity, making it difficult to isolate individual physicochemical parameters. Further, biomaterial carriers span a large collection of platforms each with their own set of optimal design parameters. A detailed understanding of the immune processes that biomaterials can influence and how they can achieve this control will pave the way towards more sophisticated biomaterial designs. Many of the studies discussed here offer new insight into how material properties can be tuned to affect the delivery of immune signals to program the immune system towards immunity or tolerance. Importantly, several biological processes and considerations such as targeting to immune tissue, uptake, controlled delivery of adjuvant and antigens (i.e. conformation, timing) can heavily influence immune responses, offering multiple avenues and targets through which biomaterials can alter innate and adaptive immune function. Studies that isolate specific design parameters (e.g. size, shape) or investigate the relative roles of multiple parameters will allow for greater understanding of these mechanisms and support design of future biomaterial-based vaccines and immunotherapies that serve as precision technologies.
Acknowledgements
This work was supported in part by the United States Department of Veterans Affairs (Award # 1I01BX003690) and the National Institutes of Health (Awards R01EB026896, R01EB027143, and R01AI144667). S. J. Tsai is a trainee of the NIH T32 Host-Pathogen Interaction Fellowship (# AI089621).
Biographies
Author Biographies

Shannon J. Tsai is a graduate fellow in the Fischell Department of Bioengineering at University of Maryland. Her research focuses on how biomaterials can be harnessed to improve the delivery of immune signals. Specifically, she is interested in understanding how biomaterial design parameters impact immune responses to help define rational design criteria for more effective immunotherapies for autoimmune diseases. Shannon is a NIH T32 Host-Pathogen Interaction fellow.

Sheneil K. Black is a research technician and faculty specialist in the Fischell Department of Bioengineering at the University of Maryland, College Park. She received her B.A. in Chemistry from Bard College and her M.S in Biomedical Science, specializing in Molecular and Cellular Pharmacology from State University of New York at Stony Brook.

Christopher M. Jewell is the Minta Martin Professor of Engineering in the Fischell Department of Bioengineering at the University of Maryland. Dr. Jewell is also a Research Biologist with the United States Department of Veterans Affairs at the VA Maryland Healthcare System. Dr. Jewell’s research spans engineering and immunology to study immune function for therapeutic vaccines targeting autoimmunity and cancer. He earned his PhD in Chemical Engineering from the University of Wisconsin, then worked as a consultant at the Boston Consulting Group in the Healthcare Practice. Dr. Jewell completed his postdoctoral training at MIT and Harvard.
Footnotes
Disclosures
C.M.J. is an employee of the VA Maryland Health Care System. The views reported in this paper do not reflect the views of the Department of Veterans Affairs or the United States Government. C.M.J. has equity positions in Avidea Technologies and Cellth Systems, LLC.
Contributor Information
Shannon J. Tsai, Fischell Department of Bioengineering, 8278 Paint Branch Drive, College Park, MD 20742, USA
Sheneil K. Black, Fischell Department of Bioengineering, 8278 Paint Branch Drive, College Park, MD 20742, USA
Christopher M. Jewell, Fischell Department of Bioengineering, 8278 Paint Branch Drive, College Park, MD 20742, USA; Robert E. Fischell Institute for Biomedical Devices, 8278 Paint Branch Drive, College Park, MD 20742, USA; United States Department of Veterans Affairs, VA Maryland Health Care System, 10. N Green Street, Baltimore, MD 21201, USA; United States Department of Veterans Affairs, VA Maryland Health Care System, 10. N Green Street, Baltimore, MD 21201, USA; Marlene and Stewart Greenebaum Cancer Center, 22 South Greene Street, Baltimore, MD 21201, USA
References
- [1].Barrat FJ, Elkon KB, Fitzgerald KA, Annu. Rev. Med 2015. [DOI] [PubMed] [Google Scholar]
- [2].Reis ES, Mastellos DC, Ricklin D, Mantovani A, Lambris JD, Nat. Rev. Immunol 2018, 18, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hancock REW, Haney EF, Gill EE, Nat. Rev. Immunol 2016, 16, 321. [DOI] [PubMed] [Google Scholar]
- [4].Sharma P, Allison JP, Cell 2015, 161, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Diamond MS, Ledgerwood JE, Pierson TC, Annu. Rev. Med 2019, 70, 121. [DOI] [PubMed] [Google Scholar]
- [6].Yang Y, Peng F, Wang R, Guan K, Jiang T, Xu G, Sun J, Chang C, The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun 2020, 109, 102434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Koopmans M, de Lamballerie X, Jaenisch T, Rosenberger KD, Morales I, Marques ETA, Viana IFT, Brasil P, Rabello R, Avelino-Silva VI, Segurado A, Alexander N, Mayaud P, Netto EM, Tami A, Bethencourt S, Consuelo Miranda M, Lozano A, Soria C, Salgado Cisneros SP, Gotuzzo E, Guzmán MG, Rodriguez PAM, Lopez-Gatell H, Hegewisch-Taylor J, Borja-Aburto VH, Gonzales Bonilla C, Hoen B, van Roode M, Rockx B, Familiar barriers still unresolved—a perspective on the Zika virus outbreak research response. Lancet Infect. Dis 2019, 19, e59–e62. [DOI] [PubMed] [Google Scholar]
- [8].Focosi D, Tuccori M, Maggi F, Rev. Med. Virol 2019, 29. [DOI] [PubMed] [Google Scholar]
- [9].Leach DG, Young S, Hartgerink JD, Advances in immunotherapy delivery from implantable and injectable biomaterials. Acta Biomater. 2019, 88, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sestito LF, Thomas SN, ACS Pharmacol. Transl. Sci 2019, 2, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhao Y, Guo Y, Tang L, Engineering cancer vaccines using stimuli-responsive biomaterials. Nano Res. 2018, 11, 5355–5371. [Google Scholar]
- [12].Vishwakarma A, Bhise NS, Evangelista MB, Rouwkema J, Dokmeci MR, Ghaemmaghami AM, Vrana NE, Khademhosseini A, Engineering Immunomodulatory Biomaterials To Tune the Inflammatory Response; 2016; Vol. 34, pp. 470–482. [DOI] [PubMed] [Google Scholar]
- [13].Hess KL, Medintz IL, Jewell CM, Nano Today 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].A Study of V503 (A Multivalent Human Papillomavirus [HPV] L1 Virus-Like Particle [VLP] Vaccine) in Preadolescents and Adolescents (V503–002) - Full Text View - ClinicalTrials.gov. [Google Scholar]
- [15].Phase 3 Pivotal Trial of NanoFluTM in Older Adults - Full Text View - ClinicalTrials.gov. [Google Scholar]
- [16].Amsterdam UMC Clinical Trial With a Native-like HIV-1 Envelope Vaccine - Full Text View - ClinicalTrials.gov. [Google Scholar]
- [17].Dendritic Cell Activating Scaffold in Melanoma - Full Text View - ClinicalTrials.gov. [Google Scholar]
- [18].Study of the Safety, Pharmacodynamics, Efficacy, and PK of TIMP-GLIA in Subjects With Celiac Disease - Full Text View - ClinicalTrials.gov. [Google Scholar]
- [19].Enhanced Epidermal Antigen Specific Immunotherapy Trial −1 - Full Text View - ClinicalTrials.gov. [Google Scholar]
- [20].Moon JJ, Huang B, Irvine DJ, Adv. Mater 2012, 24, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gause KT, Wheatley AK, Cui J, Yan Y, Kent SJ, Caruso F, ACS Nano 2017, 11, 54. [DOI] [PubMed] [Google Scholar]
- [22].Li X, Wang X, Ito A, Chem. Soc. Rev 2018, 47, 4954. [DOI] [PubMed] [Google Scholar]
- [23].Jasinski D, Haque F, Binzel DW, Guo P, ACS Nano 2017, 11, 1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bookstaver ML, Tsai SJ, Bromberg JS, Jewell CM, Trends Immunol. 2017, 39, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Toy R, Roy K, Bioeng. Transl. Med 2016, 1, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bracho-Sanchez E, Xia CQ, Clare-Salzler MJ, Keselowsky BG, Micro and Nano Material Carriers for Immunomodulation. Am. J. Transplant 2016, 16, 3362–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pati R, Shevtsov M, Sonawane A, Nanoparticle vaccines against infectious diseases. Front. Immunol 2018, 9, 2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nicolas J, Liu S, Zhao D, Caruso F, Reichmanis E, Buriak JM, Best Practices for New Polymers and Nanoparticulate Systems; American Chemical Society, 2018; Vol. 30, pp. 6587–6588. [Google Scholar]
- [29].Faria M, Björnmalm M, Thurecht KJ, Kent SJ, Parton RG, Kavallaris M, Johnston APR, Gooding JJ, Corrie SR, Boyd BJ, Thordarson P, Whittaker AK, Stevens MM, Prestidge CA, Porter CJH, Parak WJ, Davis TP, Crampin EJ, Caruso F, Nat. Nanotechnol 2018, 13, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Kreer C, Rauen J, Zehner M, Burgdorf S, Front. Immunol 2012, 2, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Joffre OP, Segura E, Savina A, Amigorena S, Nat. Rev. Immunol 2012, 12, 557. [DOI] [PubMed] [Google Scholar]
- [32].Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT, Nat. Immunol 2004, 5, 190. [DOI] [PubMed] [Google Scholar]
- [33].Oakes RS, Froimchuk E, Jewell CM, Adv. Ther 2019, 2, 1800157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Le Gall CM, Weiden J, Eggermont LJ, Figdor CG, Nat. Mater 2018, 17, 474. [DOI] [PubMed] [Google Scholar]
- [35].Garg AD, Coulie PG, Van den Eynde BJ, Agostinis P, Trends Immunol. 2017, 38, 577. [DOI] [PubMed] [Google Scholar]
- [36].Tostanoski LH, Gosselin EA, Jewell CM, Discov. Med 2016, 21, 403. [PubMed] [Google Scholar]
- [37].Mesin L, Ersching J, Victora GD, Immunity 2016, 45, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Stavnezer J, Guikema JEJ, Schrader CE, Annu. Rev. Immunol 2008, 26, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McCullough KC, Summerfield A, ILAR J 2005, 46, 230. [DOI] [PubMed] [Google Scholar]
- [40].Sahdev P, Ochyl LJ, Moon JJ, Pharm. Res 2014, 31, 2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dai Q, Guo J, Yan Y, Ang C-S, Bertleff-Zieschang N, Caruso F, 2017. [DOI] [PubMed] [Google Scholar]
- [42].Ke X, Howard GP, Tang H, Cheng B, Saung MT, Santos JL, Mao HQ, Physical and chemical profiles of nanoparticles for lymphatic targeting. Adv. Drug Deliv. Rev 2019, 151–152, 72–93. [DOI] [PubMed] [Google Scholar]
- [43].Schudel A, Francis DM, Thomas SN, Material design for lymph node drug delivery. Nat. Rev. Mater 2019, 4, 415–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Howard GP, Verma G, Ke X, Thayer WM, Hamerly T, Baxter VK, Lee JE, Dinglasan RR, Mao HQ, Nano Res. 2019, 12, 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Reddy ST, Rehor A, Schmoekel HG, Hubbell JA, Swartz MA, Control J. Release 2006, 112, 26. [DOI] [PubMed] [Google Scholar]
- [46].Yu X, Dai Y, Zhao Y, Qi S, Liu L, Lu L, Luo Q, Zhang Z, Nat. Commun 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Talamini L, Violatto MB, Cai Q, Monopoli MP, Kantner K, Krpetić Ž, Perez-Potti A, Cookman J, Garry D, Silveira CP, Boselli L, Pelaz B, Serchi T, Cambier S, Gutleb AC, Feliu N, Yan Y, Salmona M, Parak WJ, Dawson KA, Bigini P, ACS Nano 2017, 11, 5519. [DOI] [PubMed] [Google Scholar]
- [48].Shukla S, Myers JT, Woods SE, Gong X, Czapar AE, Commandeur U, Huang AY, Levine AD, Steinmetz NF, Biomaterials 2017, 121, 15. [DOI] [PubMed] [Google Scholar]
- [49].Zhang R, Smith JD, Allen BN, Kramer JS, Schauflinger M, Ulery BD, ACS Biomater. Sci. Eng 2018, 4, 2463. [DOI] [PubMed] [Google Scholar]
- [50].Kelly SH, Wu Y, Varadhan AK, Curvino EJ, Chong AS, Collier JH, Biomaterials 2020, 241, 119903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].De Koker S, Cui J, Vanparijs N, Albertazzi L, Grooten J, Caruso F, De Geest BG, Angew. Chemie Int. Ed 2016, 55, 1334. [DOI] [PubMed] [Google Scholar]
- [52].Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ, Nature 2014, 507, 519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Fenton OS, Olafson KN, Pillai PS, Mitchell MJ, Langer R, Adv. Mater 2018, 30, 1705328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hasani-Sadrabadi MM, Majedi FS, Bensinger SJ, Wu BM, Bouchard L-S, Weiss PS, Moshaverinia A, Adv. Mater 2018, 30, 1706780. [DOI] [PubMed] [Google Scholar]
- [55].Son S, Nam J, Zenkov I, Ochyl LJ, Xu Y, Scheetz L, Shi J, Farokhzad OC, Moon JJ, Nano Lett. 2020, 20, 1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Moynihan KD, Holden RL, Mehta NK, Wang C, Karver MR, Dinter J, Liang S, Abraham W, Melo MB, Zhang AQ, Li N, Le Gall S, Pentelute BL, Irvine DJ, Cancer Immunol. Res 2018, 6, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhu G, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang H, Ma Y, Zhang F, Tian R, Ni Q, Cheng S, Wang Z, Lu N, Yung BC, Wang Z, Lang L, Fu X, Jin A, Weiss ID, Vishwasrao H, Niu G, Shroff H, Klinman DM, Seder RA, Chen X, Nat. Commun 2017, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhu D, Hu C, Fan F, Qin Y, Huang C, Zhang Z, Lu L, Wang H, Sun H, Leng X, Wang C, Kong D, Zhang L, Biomaterials 2019, 206, 25. [DOI] [PubMed] [Google Scholar]
- [59].Bahmani B, Uehara M, Jiang L, Ordikhani F, Banouni N, Ichimura T, Solhjou Z, Furtmüller GJ, Brandacher G, Alvarez D, Von Andrian UH, Uchimura K, Xu Q, Vohra I, Yilmam OA, Haik Y, Azzi J, Kasinath V, Bromberg JS, McGrath MM, Abdi R, J. Clin. Invest 2018, 128, 4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Darrah PA, Zeppa JJ, Maiello P, Hackney JA, Wadsworth MH, Hughes TK, Pokkali S, Swanson PA, Grant NL, Rodgers MA, Kamath M, Causgrove CM, Laddy DJ, Bonavia A, Casimiro D, Lin PL, Klein E, White AG, Scanga CA, Shalek AK, Roederer M, Flynn JAL, Seder RA, Nature 2020, 577, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lynn GM, Sedlik C, Baharom F, Zhu Y, Ramirez-Valdez RA, Coble VL, Tobin K, Nichols SR, Itzkowitz Y, Zaidi N, Gammon JM, Blobel NJ, Denizeau J, de la Rochere P, Francica BJ, Decker B, Maciejewski M, Cheung J, Yamane H, Smelkinson MG, Francica JR, Laga R, Bernstock JD, Seymour LW, Drake CG, Jewell CM, Lantz O, Piaggio E, Ishizuka AS, Seder RA, Nat. Biotechnol 2020, 38, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Andorko JI, Gammon JM, Tostanoski LH, Zeng Q, Jewell CM, Cell. Mol. Bioeng 2016, 9, 418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Andorko JI, Tostanoski LH, Solano E, Mukhamedova M, Jewell CM, J. Vis. Exp 2014, e50984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Jewell CM, Bustamante López SC, Irvine DJ, Proc. Natl. Acad. Sci 2011, 108, 15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tostanoski LH, Chiu Y-C, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM, Cell Rep. 2016, 16, 2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Garapaty A, Champion JA, Bioeng. Transl. Med 2017, 2, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Xia Y, Wu J, Wei W, Du Y, Wan T, Ma X, An W, Guo A, Miao C, Yue H, Li S, Cao X, Su Z, Ma G, Nat. Mater 2018, 17, 187. [DOI] [PubMed] [Google Scholar]
- [68].Hui Y, Wibowo D, Liu Y, Ran R, Wang H-F, Seth A, Middelberg APJ, Zhao C-X, ACS Nano 2018, 12, 2846. [DOI] [PubMed] [Google Scholar]
- [69].Palomba R, Palange AL, Rizzuti IF, Ferreira M, Cervadoro A, Barbato MG, Canale C, Decuzzi P, ACS Nano 2018, 12, 1433. [DOI] [PubMed] [Google Scholar]
- [70].Pino P, Yang F, Pelaz B, Zhang Q, Kantner K, Hartmann R, Martinez de Baroja N, Gallego M, Möller M, Manshian BB, Soenen SJ, Riedel R, Hampp N, Parak WJ, Angew. Chemie Int. Ed 2016, 55, 5483. [DOI] [PubMed] [Google Scholar]
- [71].Wen Y, Waltman A, Han H, Collier JH, ACS Nano 2016, 10, 9274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Miura R, Sawada SI, Mukai SA, Sasaki Y, Akiyoshi K, Biomacromolecules 2020, 21, 621. [DOI] [PubMed] [Google Scholar]
- [73].Wang W, Zhou X, Bian Y, Wang S, Chai Q, Guo Z, Wang Z, Zhu P, Peng H, Yan X, Li W, Fu YX, Zhu M, Nat. Nanotechnol 2020, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Fromen CA, Rahhal TB, Robbins GR, Kai MP, Shen TW, Luft JC, DeSimone JM, Nanomedicine Nanotechnology, Biol. Med 2016, 12, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Phanse Y, Carrillo-Conde BR, Ramer-Tait AE, Roychoudhury R, Broderick S, Pohl N, Rajan K, Narasimhan B, Wannemuehler MJ, Bellaire BH, J. Biomed. Mater. Res. Part A 2017, 105, 2762. [DOI] [PubMed] [Google Scholar]
- [76].Tonigold M, Mailänder V, Nanomedicine 2016, 11, 2625. [DOI] [PubMed] [Google Scholar]
- [77].Gros M, Amigorena S, Regulation of antigen export to the cytosol during cross-presentation. Front. Immunol 2019, 10, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yang Y, Lu Y, Abbaraju PL, Zhang J, Zhang M, Xiang G, Yu C, Angew. Chemie Int. Ed 2017, 56, 8446. [DOI] [PubMed] [Google Scholar]
- [79].Sun B, Ji Z, Liao Y-P, Chang CH, Wang X, Ku J, Xue C, Mirshafiee V, Xia T, ACS Appl. Mater. Interfaces 2017, 9, 21697. [DOI] [PubMed] [Google Scholar]
- [80].Manna S, Howitz WJ, Oldenhuis NJ, Eldredge AC, Shen J, Nihesh FN, Lodoen MB, Guan Z, Esser-Kahn AP, ACS Cent. Sci 2018, 4, 982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Li AW, Sobral MC, Badrinath S, Choi Y, Graveline A, Stafford AG, Weaver JC, Dellacherie MO, Shih T-Y, Ali OA, Kim J, Wucherpfennig KW, Mooney DJ, Nat. Mater 2018, 17, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Wang Y-Q, Fan Q-Z, Liu Y, Yue H, Ma X-W, Wu J, Ma G-H, Su Z-G, Int. J. Pharm 2016, 515, 84. [DOI] [PubMed] [Google Scholar]
- [83].Xing Y, Xu Z, Liu T, Shi L, Kohane D, Guo S, Angew. Chemie Int. Ed 2020, anie. 202001169. [DOI] [PubMed] [Google Scholar]
- [84].Seydoux E, Rothen-Rutishauser B, Nita IM, Balog S, Gazdhar A, Stumbles PA, Petri-Fink A, Blank F, von Garnier C, Int. J. Nanomedicine 2014, 9, 3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Coffman RL, Sher A, Seder RA, Immunity 2010, 33, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Maisonneuve C, Bertholet S, Philpott DJ, De Gregorio E, Proc. Natl. Acad. Sci 2014, 111, 12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Jones KS, Biotechnol. Prog 2008, 24, 807. [DOI] [PubMed] [Google Scholar]
- [88].Peleteiro M, Presas E, González-Aramundiz JV, Sánchez-Correa B, Simón-Vázquez R, Csaba N, Alonso MJ, González-Fernández Á, Front. Immunol 2018, 9, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Zhu M, Du L, Zhao R, Wang HY, Zhao Y, Nie G, Wang RF, ACS Nano 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Chen X, Yan Y, Müllner M, Ping Y, Cui J, Kempe K, Cortez-Jugo C, Caruso F, Biomacromolecules 2016, 17, 1205. [DOI] [PubMed] [Google Scholar]
- [91].Guo S, Li H, Ma M, Fu J, Dong Y, Guo P, Mol. Ther. - Nucleic Acids 2017, 9, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Jasinski DL, Li H, Guo P, Mol. Ther 2018, 26, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Chandler M, Afonin KA, Chandler M, Afonin KA, Nanomaterials 2019, 9, 611. [Google Scholar]
- [94].Parlea L, Puri A, Kasprzak W, Bindewald E, Zakrevsky P, Satterwhite E, Joseph K, Afonin KA, Shapiro BA, ACS Comb. Sci 2016, 18, 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Hong E, Halman JR, Shah AB, Khisamutdinov EF, Dobrovolskaia MA, Afonin KA, Nano Lett. 2018, 18, 4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Andorko JI, Hess KL, Pineault KG, Jewell CM, Acta Biomater. 2016, 32, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Andorko JI, Pineault KG, Jewell CM, J. Biomed. Mater. Res. - Part A 2017, 105, 1219. [DOI] [PubMed] [Google Scholar]
- [98].Allen RP, Bolandparvaz A, Ma JA, Manickam VA, Lewis JS, ACS Biomater. Sci. Eng 2018, 4, 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Colegio OR, Chu N-Q, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, Cline GW, Phillips AJ, Medzhitov R, Nature 2014, 513, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Nasi A, Fekete T, Krishnamurthy A, Snowden S, Rajnavölgyi E, Catrina AI, Wheelock CE, Vivar N, Rethi B, Immunol J. 2013, 191, 3090. [DOI] [PubMed] [Google Scholar]
- [101].Loftus C, Saeed M, Davis DM, Dunlop IE, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ebrahimian M, Hashemi M, Maleki M, Hashemitabar G, Abnous K, Ramezani M, Haghparast A, Front. Immunol 2017, 8, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Gale EC, Roth GA, Smith AAA, Alcántara-Hernández M, Idoyaga J, Appel EA, Adv. Ther 2020, 3, 1900174. [Google Scholar]
- [104].Sofias AM, Andreassen T, Hak S, Mol. Pharm 2018, 15, 5754. [DOI] [PubMed] [Google Scholar]
- [105].Lynn GM, Laga R, Darrah PA, Ishizuka AS, Balaci AJ, Dulcey AE, Pechar M, Pola R, Gerner MY, Yamamoto A, Buechler CR, Quinn KM, Smelkinson MG, Vanek O, Cawood R, Hills T, Vasalatiy O, Kastenmüller K, Francica JR, Stutts L, Tom JK, Ryu KA, Esser-Kahn AP, Etrych T, Fisher KD, Seymour LW, Seder RA, Nat. Biotechnol 2015, 33, 1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Cui J, De Rose R, Best JP, Johnston APR, Alcantara S, Liang K, Such GK, Kent SJ, Caruso F, Adv. Mater 2013, 25, 3468. [DOI] [PubMed] [Google Scholar]
- [107].Wang J, Chen H-J, Hang T, Yu Y, Liu G, He G, Xiao S, Yang B, Yang C, Liu F, Tao J, Wu MX, Xie X, Nat. Nanotechnol 2018, 13, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Kakizawa Y, Lee JS, Bell B, Fahmy TM, Acta Biomater. 2017, 57, 136. [DOI] [PubMed] [Google Scholar]
- [109].Tazaki T, Tabata K, Ainai A, Ohara Y, Kobayashi S, Ninomiya T, Orba Y, Mitomo H, Nakano T, Hasegawa H, Ijiro K, Sawa H, Suzuki T, Niikura K, RSC Adv. 2018, 8, 16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Petrovsky N, Comparative Safety of Vaccine Adjuvants: A Summary of Current Evidence and Future Needs. Drug Saf. 2015, 38, 1059–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Batista-Duharte A, Martínez DT, Carlos IZ, Efficacy and safety of immunological adjuvants. Where is the cut-off? Biomed. Pharmacother. 2018, 105, 616–624. [DOI] [PubMed] [Google Scholar]
- [112].Pallares RM, Choo P, Cole LE, Mirkin CA, Lee A, Odom TW, Bioconjug. Chem 2019, 30, 2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].You X, Gu Z, Huang J, Kang Y, Chu C-C, Wu J, Acta Biomater. 2018, 74, 180. [DOI] [PubMed] [Google Scholar]
- [114].Tsai SJ, Andorko JI, Zeng X, Gammon JM, Jewell CM, Nano Res. 2018, 11, 5642. [Google Scholar]
- [115].Lynn GM, Chytil P, Francica JR, Lagová A, Kueberuwa G, Ishizuka AS, Zaidi N, Ramirez-Valdez RA, Blobel NJ, Baharom F, Leal J, Wang AQ, Gerner MY, Etrych T, Ulbrich K, Seymour LW, Seder RA, Laga R, Biomacromolecules 2019, 20, 854. [DOI] [PubMed] [Google Scholar]
- [116].Abkar M, Alamian S, Sattarahmady N, Immunol. Lett 2019, 207, 28. [DOI] [PubMed] [Google Scholar]
- [117].Saito E, Kuo R, Pearson RM, Gohel N, Cheung B, King NJC, Miller SD, Shea LD, Control J. Release 2019, 300, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Saito E, Kuo R, Kramer KR, Gohel N, Giles DA, Moore BB, Miller SD, Shea LD, Biomaterials 2019, 222, 119432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Jia J, Zhang W, Liu Q, Yang T, Wang L, Ma G, Mol. Pharm 2017, 14, 14. [DOI] [PubMed] [Google Scholar]
- [120].Wilson DS, Hirosue S, Raczy MM, Bonilla-Ramirez L, Jeanbart L, Wang R, Kwissa M, Franetich JF, Broggi MAS, Diaceri G, Quaglia-Thermes X, Mazier D, Swartz MA, Hubbell JA, Nat. Mater 2019, 18, 175. [DOI] [PubMed] [Google Scholar]
- [121].Gao J, Ochyl J, Yang E, Moon JJ, Int. J. Nanomedicine 2017, 12, 1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Zupančič E, Curato C, Paisana M, Rodrigues C, Porat Z, Viana AS, Afonso CAM, Pinto J, Gaspar R, Moreira JN, Satchi-Fainaro R, Jung S, Florindo HF, Control J. Release 2017, 258, 182. [DOI] [PubMed] [Google Scholar]
- [123].Rincon-Restrepo M, Mayer A, Hauert S, Bonner DK, Phelps EA, Hubbell JA, Swartz MA, Hirosue S, Biomaterials 2017, 132, 48. [DOI] [PubMed] [Google Scholar]
- [124].Liu L, Ma P, Wang H, Zhang C, Sun H, Wang C, Song C, Leng X, Kong D, Ma G, 2016. [Google Scholar]
- [125].Gosselin EA, Eppler HB, Bromberg JS, Jewell CM, Designing natural and synthetic immune tissues. Nat. Mater 2018, 17, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Hickey JW, Vicente FP, Howard GP, Mao H-Q, Schneck JP, Nano Lett. 2017, 17, 7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Cheung AS, Zhang DKY, Koshy ST, Mooney DJ, Nat. Biotechnol 2018, 36, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S, Control J. Release 2015, 220, 141. [DOI] [PubMed] [Google Scholar]
- [129].Dykman LA, Staroverov SA, Fomin AS, Khanadeev VA, Khlebtsov BN, Bogatyrev VA, Int. Immunopharmacol 2018, 54, 163. [DOI] [PubMed] [Google Scholar]
- [130].Hayashi M, Aoshi T, Kogai Y, Nomi D, Haseda Y, Kuroda E, Kobiyama K, Ishii KJ, Vaccine 2016, 34, 306. [DOI] [PubMed] [Google Scholar]
- [131].Courant T, Bayon E, Reynaud-Dougier HL, Villiers C, Menneteau M, Marche PN, Navarro FP, Biomaterials 2017, 136, 29. [DOI] [PubMed] [Google Scholar]
- [132].Marcandalli J, Fiala B, Ols S, Perotti M, Schueren W, Snijder J, Hodge E, Benhaim M, Ravichandran R, Carter L, Sheffler W, Brunner L, Lawrenz M, Dubois P, Lanzavecchia A, Sallusto F, Lee KK, Veesler D, Correnti CE, Stewart LJ, Baker D, Loré K, Perez L, King NP, Cell 2019, 176, 1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Brewer MG, DiPiazza A, Acklin J, Feng C, Sant AJ, Dewhurst S, Vaccine 2017, 35, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Moyer TJ, Kato Y, Abraham W, Chang JYH, Kulp DW, Watson N, Turner HL, Menis S, Abbott RK, Bhiman JN, Melo MB, Simon HA, Herrera-De la Mata S, Liang S, Seumois G, Agarwal Y, Li N, Burton DR, Ward AB, Schief WR, Crotty S, Irvine DJ, Nat. Med 2020, 26, 430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Skwarczynski M, Zhao G, Boer JC, Ozberk V, Azuar A, Cruz JG, Giddam AK, Khalil ZG, Pandey M, Shibu MA, Hussein WM, Nevagi RJ, Batzloff MR, Wells JW, Capon RJ, Plebanski M, Good MF, Toth I, Sci. Adv 2020, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Northrup L, Christopher MA, Sullivan BP, Berkland C, Adv. Drug Deliv. Rev 2016, 98, 86. [DOI] [PubMed] [Google Scholar]
- [137].Pearson RM, Casey LM, Hughes KR, Miller SD, Shea LD, Adv. Drug Deliv. Rev 2017, 114, 240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Gammon JM, Jewell CM, Adv. Healthc. Mater 2019, 8, 1801419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Pearson RM, Casey LM, Hughes KR, Wang LZ, North MG, Getts DR, Miller SD, Shea LD, Mol. Ther 2017, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Ramakrishnan R, V Balu-Iyer S, V Balu-Iyer S, Sathyamangalam F, Balasubramanian V, J. Pharm. Sci 2016, 105, 3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Sestak JO, Fakhari A, Badawi AH, Siahaan TJ, Berkland C, AAPS J 2014, 16, 1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Singha S, Shao K, Yang Y, Clemente-Casares X, Solé P, Clemente A, Blanco J, Dai Q, Song F, Wan Liu S, Yamanouchi J, Sokke Umeshappa C, Hebbandi Nanjundappa R, Detampel P, Amrein M, Fandos C, Tanguay R, Newbigging S, Serra P, Khadra A, W Chan WC, Santamaria P, Nat. Nanotechnol 2017, 12, 701. [DOI] [PubMed] [Google Scholar]
- [143].Martin-Blanco N, Blanco R, Alda-Catalinas C, Bovolenta ER, Oeste CL, Palmer E, Schamel WW, Lythe G, Molina-París C, Castro M, Alarcon B, Nat. Commun 2018, 9, 2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Hess KL, Oh E, Tostanoski LH, Andorko JI, Susumu K, Deschamps JR, Medintz IL, Jewell CM, Adv. Funct. Mater 2017, 27, 1700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, Hu JK, Kumari S, Crampton J, Baldeon AD, Sanders RW, Moore JP, Crotty S, Langer R, Anderson DG, Chakraborty AK, Irvine DJ, Proc. Natl. Acad. Sci 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Guarecuco R, Lu J, McHugh KJ, Norman JJ, Thapa LS, Lydon E, Langer R, Jaklenec A, Vaccine 2018, 36, 3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Kapadia CH, Tian S, Perry JL, Sailer D, Christopher Luft J, DeSimone JM, Control J. Release 2018, 269, 393. [DOI] [PubMed] [Google Scholar]
- [148].Benson RA, Macleod MK, Hale BG, Patakas A, Garside P, Brewer JM, Elife 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Zhang YN, Lazarovits J, Poon W, Ouyang B, Nguyen LNM, Kingston BR, Chan WCW, Nano Lett. 2019, 19, 7226. [DOI] [PubMed] [Google Scholar]
- [150].Chen N, Johnson MM, Collier MA, Gallovic MD, Bachelder EM, Ainslie KM, Control J. Release 2018, 273, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Tzeng A, Kauke MJ, Zhu EF, Moynihan KD, Opel CF, Yang NJ, Mehta N, Kelly RL, Szeto GL, Overwijk WW, Irvine DJ, Wittrup KD, Cell Rep. 2016, 17, 2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Wang S, Qin L, Yamankurt G, Skakuj K, Huang Z, Chen PC, Dominguez D, Lee A, Zhang B, Mirkin CA, Proc. Natl. Acad. Sci. U. S. A 2019, 116, 10473. [DOI] [PMC free article] [PubMed] [Google Scholar]