

Abstract
Excessive fear learning and generalized, extinction-resistant fear memories are core symptoms of anxiety and trauma-related disorders. Despite significant evidence from clinical studies reporting hyperactivity of the bed nucleus of stria terminalis (BNST) under these conditions, the role of BNST in fear learning and expression is still not clarified. Here, we tested how BNST modulates fear learning in male mice using a chemogenetic approach. Activation of GABAergic neurons of BNST during fear conditioning or memory consolidation resulted in enhanced cue-related fear recall. Importantly, BNST activation had no acute impact on fear expression during conditioning or recalls, but it enhanced cue-related fear recall subsequently, potentially via altered activity of downstream regions. Enhanced fear memory consolidation could be replicated by selectively activating somatostatin (SOM), but not corticotropin-releasing factor (CRF), neurons of the BNST, which was accompanied by increased fear generalization. Our findings suggest the significant modulation of fear memory strength by specific circuits of the BNST.
SIGNIFICANCE STATEMENT The bed nucleus of stria terminalis (BNST) mediates different defensive behaviors, and its connections implicate its integrative modulatory role in fear memory formation; however, the involvement of BNST in fear learning has yet to be elucidated in detail. Our data highlight that BNST stimulation enhances fear memory formation without direct effects on fear expression. Our study identified somatostatin (SOM) cells within the extended amygdala as specific neurons promoting fear memory formation. These data underline the importance of anxiety circuits in maladaptive fear memory formation, indicating elevated BNST activity as a potential vulnerability factor to anxiety and trauma-related disorders.
Keywords: BNST, DREADD, extended amygdala, fear, somatostatin.
Introduction
Forming memories of negative events are essential for the survival of the individual, however, excessive, generalized, and extinction-resistant memories results in maladaptive, inflexible phenotype, or symptoms present in anxiety and trauma-related disorders, i.e., posttraumatic stress disorder (PTSD; Duits et al., 2015; Singewald et al., 2015). The extended amygdala, including the bed nucleus of stria terminalis (BNST), shows activation during acute threats and sustained anxiety-like states in healthy populations, which becomes elevated in patients with anxiety disorders (Alheid and Heimer, 1988; Avery et al., 2016; Brinkmann et al., 2017; Klumpers et al., 2017). Early studies investigating the rodent BNST observed functional division between the amygdala and the BNST, which was supported by human fMRI data. According to this model, the amygdala mediates imminent phasic “fear-like” states whereas the BNST mediates more diffuse unconditioned “anxiety-like” states (Davis et al., 2010; Daniel and Rainnie, 2016; Asok et al., 2018; Goode et al., 2019). Recently, accumulating evidence showed that both the amygdala and the BNST are recruited under both phasic and sustained (or conditioned and unconditioned) fear-like states in humans and primates (Gungor and Paré, 2016; Shackman and Fox, 2016). In vivo electrophysiological recordings in rodents also demonstrated that BNST neurons are recruited during fear acquisition and conditioned stimulus (CS)-dependent fear recall, when stimuli are not diffuse as a threatening context (Haufler et al., 2013; Jennings et al., 2013; Daldrup et al., 2016; Bjorni et al., 2020); however, involvement and the exact role of the BNST in fear learning and expression is still unclear. The functional heterogeneity and competing effects of local circuits have been also demonstrated in the extended amygdala (Jennings et al., 2013; Kim et al., 2013; Daniel and Rainnie, 2016; Gungor and Paré, 2016), pointing out the necessity of specific manipulations targeting subregions or neurochemically distinct neuronal populations. In the central amygdala, which shows remarkable similarities to the BNST, opposing functional impact of somatostatin (SOM) and corticotropin-releasing factor (CRF) neurons on fear expression has been documented (Fadok et al., 2017; Hartley et al., 2019). More specifically, SOM neurons drive passive, whereas CRF neurons drive active fear responses, which seem to be more universal in the light of similar effects in the prefrontal cortex (Cummings and Clem, 2020). The functional role of these two major neuronal populations of the BNST related to fear responses is yet to be clarified.
Our study investigated how BNST hyperactivation (as recognized in anxiety disorders) modulates fear learning characteristics in a Pavlovian fear conditioning paradigm that is suitable for translational studies by targeting conserved mechanisms (Deslauriers et al., 2018; Flandreau and Toth, 2018). We investigated different phases of fear learning, i.e., fear acquisition, consolidation, and recall evoked by contextual and conditioned stimuli (CSs), and their generalization to safe context and safety cues. Additionally, we aimed to dissect how SOM and CRF neurons contribute to these processes. Here, we show the significant activation of the BNST during fear acquisition, but not during CS-dependent fear recall. In accordance, chemogenetic activation of the major cell type of the BNST (i.e., vesicular GABA transporter positive neurons, BNSTvGAT) during fear conditioning resulted in enhanced CS-dependent fear recall, which was replicated when BNSTvGAT neurons were activated during fear memory consolidation. In contrast, chemogenetic activation during fear expression (recall) did not alter freezing levels. Finally, we show that activation of somatostatin-expressing (BNSTSOM), but not CRF-expressing (BNSTCRF), neurons mediate this effect with additional increase of fear generalization. These data suggest the modulatory role of the BNST in fear memory formation potentially via plasticity changes in the fear circuitry as indicated by altered neuronal activation in downstream regions to the BNST during fear memory consolidation.
Materials and Methods
Subjects
Adult (more than eight weeks old) male mice from the following strains were used in the present study: C57Bl/6J, vgat-ires-cre, crh-ires-cre and som-ires-cre mice (all strains from The Jackson Laboratory; Taniguchi et al., 2011; Vong et al., 2011). To visualize CRF and SOM neurons (by reporter fluorescent proteins) in the BNST, we crossed crh-ires-cre and som-ires-cre mice with Gt(ROSA)26Sor-CAG/LSL-ZsGreen1 mice (The Jackson Laboratory). All animals were group-housed (three to four mice/cage) in Plexiglas chambers at constant temperature (22 ± 1°C) and humidity (40–60%), under a reverse circadian light-dark cycle (lights-off at 7 A.M., lights-on at 7 P.M.). Behavioral experiments were performed during the first half of the active (dark) cycle. Mice were isolated 3 d before fear conditioning and kept single-housed during testing to prevent social buffering/modulatory effects. Regular laboratory chow (Sniff) and water was available ad libitum.
Stereotaxic surgery for viral gene transfer
Mice underwent stereotaxic surgery to bilaterally inject virus constructs into the BNST (anterior-posterior +0.8 mm, medio-lateral ±0.8 mm, dorso-ventral –4.2 mm to bregma; Paxinos and Franklin, 2001). Animals were anesthetized with a ketamine-xylazine solution (16.6 mg/ml ketamine and 0.6 mg/ml xylazine-hydrochloride in 0.9% saline, 10 ml/kg body weight, i.p.) and placed in a stereotaxic frame (David Kopf Instruments). Viral vectors (20–40 nl volume/hemisphere) were microinjected through a glass pipette (tip diameter: 20–30 μm) at a rate of 100 nl/min by using a Nanoject II precision microinjector pump (Drummond). The pipette was left in place for an additional 5 min to ensure diffusion before slow retraction. This volume provided an optimal, global designer receptors exclusively activated by designer drugs (DREADD) expression restricted to the BNST. After the surgeries, mice received buprenorphine injection (Bupaq; 0.1 mg/kg) subcutaneously as analgesic treatment. Behavioral experiments were conducted four to six weeks after virus injection to allow time for DREADD expression.
Virus vectors
Adeno-associated viruses (AAVs) carrying Cre-inducible (double-inverse orientation; DIO) transgenes were purchased from Addgene. We used stimulatory AAV8-hSyn::DIO-hM3Dq-mCherry (4.0e12 GC/ml titer, #44 361), inhibitory AAV8-hSyn::DIO-hM4Di-mCherry (1.9e13 GC/ml titer, #44 362) DREADD constructs, and inactive control fluorophore AAV8-hSyn::DIO-mCherry (4.1e12 GC/ml titer, #50 459).
Drugs
Designer receptor-ligand clozapine-N-oxide (CNO; Tocris Bioscience; 4936, CAS #34233-69-7) was dissolved in 0.9% saline solution at a concentration of 0.1 mg/ml and administered intraperitoneally at a dose of 1 mg/kg 40 min before testing (in case of fear acquisition or fear recall modulation), or immediately after fear conditioning (in case of modulation of fear memory consolidation).
Behavioral testing
Auditory fear conditioning and recall testing
Auditory fear conditioning was started four to six weeks after viral surgeries. On day 1, mice were placed into a clear Plexiglas chamber (25 × 25 × 30 cm) with an electrical grid floor (Coulbourn Instruments) used to deliver footshocks. The chamber was cleaned with 20% ethanol between the animals. Fear conditioning was performed with maximum light intensity in the test room (700 lux). This setting was considered as the conditioning context (CtxA: light on, ethanol odor, a specific box with grid floors). Following a 150-s baseline period, mice were presented with seven auditory stimuli as CSs (CS+: 80-dB pure tones at 7 kHz, SuperTech Instruments) of 30-s duration spaced with pseudorandom interstimulus intervals (ITIs, ranged between 60–90 s). All CS+ co-terminated with a 1 s scrambled footshock as an unconditioned stimulus (US; 0.7 mA in all experiments, except low-intensity training of crh-ires-cre mice when 0.4 mA was applied, Fig. 1A and Fig. 2E).
Figure 1.
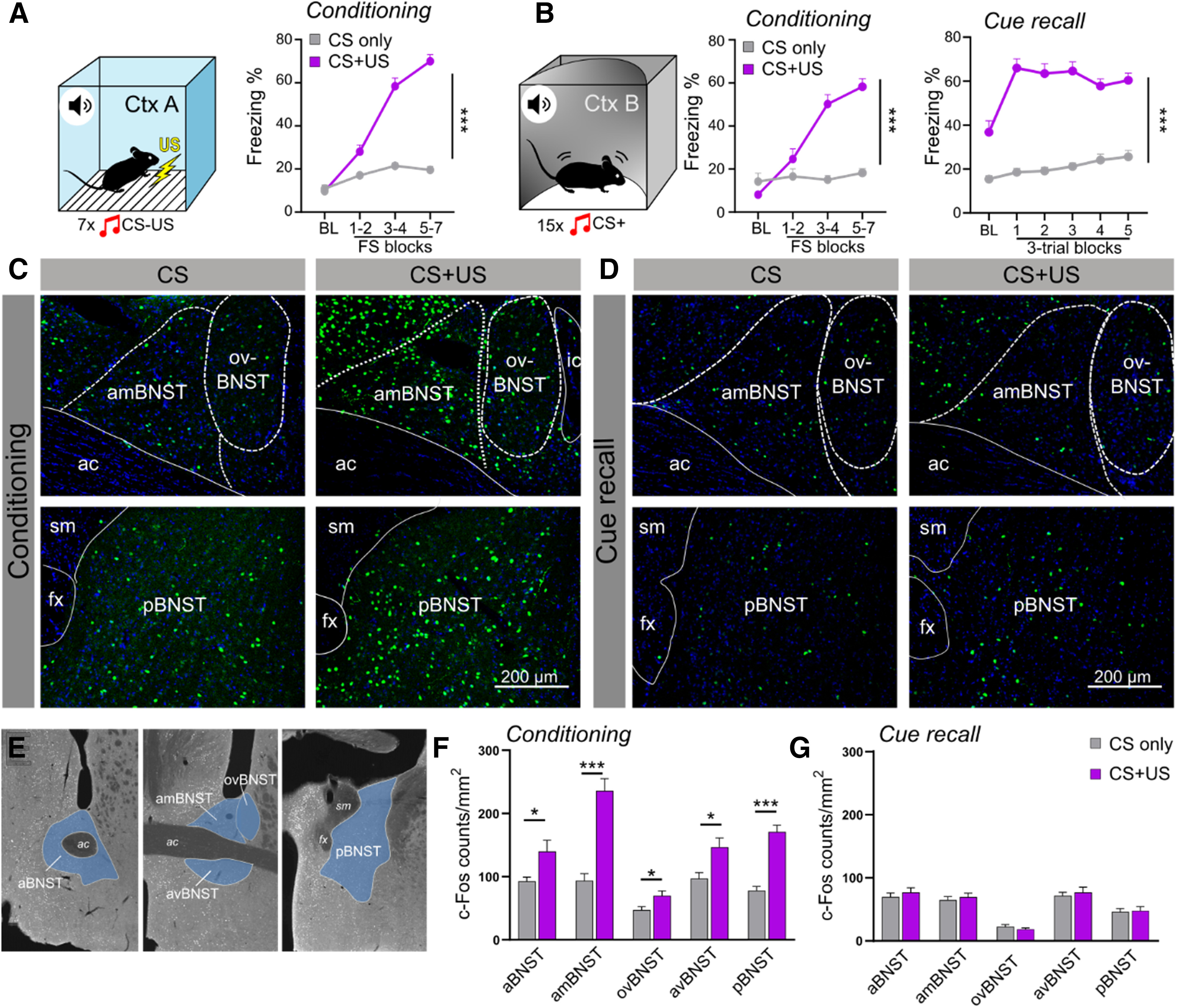
BNST neurons are activated during fear conditioning but not during CS-dependent fear recall. A, B, Freezing behavior during auditory fear conditioning and CS recall. Control mice were exposed to the auditory conditioned stimuli (CSs) without footshocks, whereas conditioned animals received seven pairings of shock-cue (CS+US) pairings. During auditory fear recall, mice were exposed to 15 CS+ in an altered context (illustrated in panel B). C, D, Representative fluorescent photomicrographs showing c-Fos immunostaining of BNST subregions during fear conditioning and auditory fear recall, respectively. E, Illustration of investigated BNST subregions for c-Fos quantification. F, Fear conditioning significantly increased c-Fos expression of all BNST subregions (n = 10/groups). G, In contrast, cued fear recall induced no changes in c-Fos expression (n = 10–11/groups). On freezing time curves, each major tick depicts two to three footshock (FS) blocks in case of conditioning, and a 180-s block, starting with a 150-s pretone baseline period (BL) in case of recall test. All data are represented as mean ± SEM. Asterisks represent main effect of ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001. aBNST, anterior BNST; ac, anterior commissure; amBNST, anteromedial BNST; avBNST, anteroventral BNST; fx, fornix; ic, internal capsule; ovBNST, oval nucleus of the BNST; pBNST, posterior BNST; sm, stria medullaris.
Figure 2.

Chemogenetic activation of the BNST facilitates fear learning, but not fear recall. A, Schematics of virus injections in vgat-ires-cre mice and representative photomicrograph of mCherry expression. Right panels show minimum (filled areas) and maximum (areas with colored outlines) extensions of mCherry expression in the BNST. B, Patch clamp recordings from BNST slices: representative photomicrograph depicting a vGAT+ neuron filled with biocytin. Right panel shows representative trace of a hM3Dq expressing neuron indicating CNO-mediated depolarization in the presence of TTX, which was absent in BNSTvGAT neurons expressing control fluorophore. C, CNO administration also elevated the frequency of APs evoked by depolarizing current steps and decreased the rheobase. D, Intraperitoneal injection of CNO under homecage condition induced significant c-Fos expression in hM3Dq-mCherry-expressing neurons (control: n = 9, hM3Dq: n = 9). E, Experimental design for auditory fear conditioning. F, I, Schematics of chemogenetic modulation of the BNST during fear conditioning or memory consolidation, respectively, and freezing behavior exhibited during conditioning. BNST stimulation did not affect acute fear response (freezing) during conditioning. G, BNST stimulation during conditioning did not affect contextual fear recall, but enhanced cued fear recall (H; control: n = 8, hM3Dq: n = 9). Similarly, chemogenetic activation of BNSTvGAT neurons during fear memory consolidation resulted in enhanced cued fear recall with additional fear generalization on day 2 (freezing during baseline; K), without altering contextual fear recall in context A (J). L, Experimental design for chemogenetic activation of the BNST during cued fear recall with conditioned (CS+) and safety cue (CS–) presentations. Freezing response of hM3Dq and control groups were similar during all testing phase, i.e., conditioning (M), short CS+ and CS– recall (N), CS+ and CS– induced fear recall (O; control: n = 8, hM3Dq: n = 13). On freezing time curves, each major tick depicts two to three footshock (FS) blocks in case of conditioning, and a 180-s block, starting with a 150-s pretone baseline period (BL) in case of recall test. All data are represented as mean ± SEM. Asterisks represent main effect of one-way, two-way, or repeated-measure ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; # represents significant difference in BL period p < 0.05.
On day 3, mice were exposed briefly to context A for 5 min without CS+ to study contextual fear recall without inducing fear extinction. On days 4 and 5, mice were subjected to sessions of auditory fear recall in an altered context B (CtxB). Context B was altered in all dimensions to differ from context A (i.e., experimenter, room, 20 lux red light, plastic floor instead of grids, plastic inserts changing the shape of the chamber, cleaning with soapy water with fruit odor). Both sessions started with a 150-s baseline period (no CS+ presented) to measure fear response in a safe context (conditioned vs safe context fear responses as an index of generalization). The baseline period was followed by 15 or 9 CS+ (30-s duration, spaced with 30 s ITIs) on days 4 and 5, respectively, to measure cue-dependent fear recall, as well as within-session and between-session fear extinction. In case of SOM-ires-cre mice, one cohort was exposed to context B without CS presentations (for the same time period, i.e., 17 min) to test contextual generalization and CS-dependent effects. Time spent with freezing was considered as an index of fear, which was analyzed using EthoVision XT 13 software. Software parameters and thresholds were set and optimized to reach R > 0.90 correlation with hand scoring by an experimenter blind to treatments.
Differential auditory fear conditioning and recall testing
Fear conditioning and recall testing took place in CtxA and CtxB, respectively (Fig. 2L). In this paradigm, we presented two types of auditory stimuli with 30-s duration, i.e., 7-kHz pure tone and white noise pips. They were used as CS and US (CS+ and CS–, randomized between animals, and counterbalanced between groups). On day 1, mice were habituated to auditory cues in CtxA, i.e., after a 150-s baseline period, four of each cue were delivered in alternating order with 30 s ITIs (not shown in Fig. 2). The next day, fear conditioning was performed in CtxA: after 150-s baseline period, seven CS+ and seven CS– (30-s duration, 30-s ITIs) were presented in alternating order. All CS+, but none of CS–, were co-terminated with 1 s scrambled footshock (0.5 mA). We used lower shock intensity to achieve better discriminative learning (CS+ vs CS–) in mice based on previous studies (Duvarci et al., 2009; Kim and Cho, 2017; Sanford et al., 2017). Two days later, cue-dependent fear recall and discrimination between CS+ and CS– were briefly tested by exposing subjects to four CS+ and four CS– in alternating order in CtxB (30-s duration, 30-s ISIs). The next day, cue-dependent fear recall (with CS+/ CS– discrimination) and fear extinction were tested following chemogenetic activation of the BNST (1 mg/kg, i.p., CNO; 40-min preinjection time) by exposing subjects to 15 CS+ and four CS– in alternating order in CtxB (after 150-s baseline period). We also tested extinction recall by exposing mice to nine CS+ in CtxB 24 h later. Analysis of freezing behavior was conducted as above.
Ex vivo electrophysiology and slice preparation
For validation of hM3Dq-mediated depolarization, current-clamp recordings were performed on brain slices obtained from vGAT-cre mice (postnatal days 90–110, matching experimental age) expressing hM3Dq or control fluorophore in BNST (n = 4/group; Fig. 2B,C). Animals were given four to six weeks to express transgenes after virus injection. After decapitation, the brain was removed rapidly and immersed in ice-cold low-sodium solution (205.0 mm saccharose, 2.5 mm KCl, 26.0 mm NaHCO3, 1.0 mm CaCl2, 5.0 mm MgCl2, 1.25 mm NaH2PO4, and 10 mm glucose) bubbled with a mixture of 95% O2 and 5% CO2. Coronal sections of the BNST were sliced at 250 μm on a VT-1000S Vibratome (Leica Microsystems) in the low-sodium solution. The slices were transferred into artificial CSF (aCSF; 130.0 mm NaCl, 3.5 mm KCl, 26.0 mm NaHCO3, 2.5 mm CaCl2, 1.2 mm MgSO4, 1.25 mm NaH2PO4, and 10 mm glucose) saturated with O2/CO2, and kept in it for 1 h to equilibrate. The initial temperature of aCSF was 33°C, which was left to cool to room temperature during equilibration. Electrophysiological recording, during which the brain slices were oxygenated by bubbling the aCSF with O2/CO2 gas, was performed at 33°C. An Axopatch 200B patch-clamp amplifier, a Digidata-1322A Data Acquisition System, and pCLAMP version 10.4 software (Molecular Devices) were used for recording.
Recordings were performed under visual guidance using a BX51WI IR-DIC microscope (Olympus) located on a S'Table antivibration table (Supertech Instruments) to detect mCherry-positive cells.
The patch electrodes (outer diameter, 1.5 mm, thin wall; Hilgenberg) were pulled with a Flaming-Brown P-97 puller (Sutter Instrument) and polished with an MF-830 Microforge (Narishige). The pipette solution contained the following: 130 mm K-gluconate, 10 mm NaCl, 10 mm KCl, 0.1 mm MgCl2, 10 mm HEPES, 1 mm EGTA, 4 mm Mg-ATP, and 0.3 mm Na-GTP (pH 7.3 with KOH). Osmolarity was adjusted to 295–300 mOsm with sorbitol. Neurons were recorded in whole-cell mode, the intrapipette solution contained 0.2% biocytin. To record CNO-induced depolarization of hM3Dq expressing neurons, 10 μm CNO was bath applied for 10 min after a 3-min baseline in the presence of 660 nm tetrodotoxin (TTX). The firing was induced by 30- or 60-pA current pulses (900 ms), respectively, 5 min after the onset of CNO application. Similar recordings were performed on control mice (expressing inactive control fluorophore DREADD). Recordings were stored and analyzed off-line using the Clampfit module of the PClamp version 10.4 software (Molecular Devices).
Immunohistochemistry and image analysis
Tissue processing
Mice were anesthetized with a ketamine-xylazine solution (16.6 and 0.6 mg/ml, respectively) and transcardially perfused with ice-cold PBS, followed by ice-cold paraformaldehyde (PFA; 4% in PBS). Brains were rapidly removed and postfixed in 4% PFA at 4°C, and cryoprotected in 30% sucrose solution in PBS before slicing; 30-μm coronal sections were collected on a sliding microtome and stored in a cryoprotectant solution (containing 20% glycerin, 30% ethylene glycol) at −20°C until immunohistochemical analysis.
Verification of virus extensions
We labeled mCherry by immunohistochemistry using a primary antibody against red fluorescent protein (RFP) to verify virus expression in the BNST. Briefly, after several rinses in PBS, sections (90 μm apart) were incubated in PBS containing 0.3% Triton X-100 (TxT; Sigma-Aldrich) and 0.3% H2O2 for 30 min followed by 2% bovine serum albumin (BSA; Sigma-Aldrich) diluted in PBS for 1 h. Primary antibody solution (1:4000 rabbit anti-RFP, #600-401-379, Rockland; diluted in PBS containing 2% BSA and 0.1% TxT) was left over on the slices for 2 d at 4°C. After several rinsing with PBS, slices were incubated in biotin-conjugated donkey anti-rabbit secondary antibody (1:1000 in 2% BSA and PBS, #711-065-152, Jackson ImmunoResearch) for 2 h. Labeling was amplified by avidin–biotin complex (1:1000; Vector Laboratories) by incubation for 1 h at room temperature. The peroxidase reaction was developed in the presence of diaminobenzidine tetrahydrochloride (0.2 mg/ml), nickel–ammonium sulfate (0.1%), and hydrogen peroxide (0.003%) dissolved in Tris buffer. Sections were mounted onto gelatin-coated slides, dehydrated, and coverslipped with DPX Mountant (Sigma-Aldrich/Merck). Regions of interest were digitalized by an Olympus DP70 Light Microscope and CCD camera system. All animals with virus extension outside of the BNST were excluded from the analysis. Generally, mCherry-positive cell bodies were observed along the whole rostro-caudal axis of the BNST, including anteroventral, anteromedial, and posterior regions of the BNST, with limited expression in the oval nucleus.
Viral tracing
We also used anti-RFP fluorescent immunolabeling and confocal microscopy to assess projections of BNSTCRF, BNSTSOM, and BNSTvGAT neurons. The protocol was slightly modified as above: non-specific binding sites were blocked by 10% normal goat serum (NGS; diluted in TBS, Jackson ImmunoResearch) for 1 h, and slices were incubated in the primary antibody solution for 3 d (monoclonal rabbit anti-RFP IgG 1:1000, #600-401-379, Rockland; 0.15 TxT in TBS). Several rinsing with TBS was followed by incubation in secondary antibody solution for 2 h (1:500 Cy3-conjugated goat anti-rabbit IgG, #134845, Jackson ImmunoResearch).
C-Fos immunohistochemistry
We used c-Fos immunohistochemistry to assess neuronal activity in BNST and its downstream regions at different time points of fear learning. Mice were anesthetized (with ketamine-xylazine mixture) and transcardially perfused 90 min after either testing or CNO injection (in case of homecage condition to verify chemogenetic activation of BNST in vivo) as described above. We used fluorescent immunolabeling against c-Fos and RFP as described above (1:2000 guinea-pig polyclonal anti-c-Fos IgG, #226004, Synaptic Systems with monoclonal rabbit anti-RFP IgG 1:1000, #600-401-379, Rockland), which were detected by fluorescent-conjugated antibodies (1:500 Cy3-conjugated donkey anti-rabbit, #134845, Jackson ImmunoResearch, and 1:500 Alexa Fluor 488-conjugated donkey anti-guinea-pig, #S32354, ThermoFisher Scientific).
SOM- and CRF-Zsgreen assessment
To quantify the distribution of SOM and CRF neurons in BNST subregions, neuronal somata were visualized by NeuN immunolabeling (1:1000, monoclonal mouse anti-NeuN IgG, #MAB377, Merck) in crh-ires-cre: and som-ires-cre::Gt(ROSA)26Sor-CAG/LSL-ZsGreen1 crossed mouse lines. The primary antibody was incubated overnight in 2% normal donkey serum and 0.1% TxT). The next day, after PBS rinses, the primary antibody was detected by Cy3-conjugated donkey anti-mouse IgG (1:1000, #715-165-151, Jackson ImmunoResearch). After several rinsing in PBS, sections were mounted on glass slides and coverslipped using Mowiol4-88 (Merck).
Microscopy and quantification of labeling
All imaging and quantification were performed by experimenters blind to treatments. As mentioned above Ni-DAB-stained sections were digitalized by an Olympus DP70 Light Microscope and CCD camera system. Fluorescent c-Fos labeling was imaged using C2 Confocal Laser-Scanning Microscope (Nikon CFI Plan Apo VC60X/NA 1.40 Oil objective, z step size: 0.13 μm, xy: 0.08 μm/pixel and CFI Plan Apo VC20X/N.A. 0.75, xy: 0.62 μm/pixel, Nikon Europe), whereas projections of the BNST (anti-RFP) were imaged using a Panoramic Digital Slide Scanner (Zeiss, Plan-Apochromat 10×/NA 0.45, xy: 0.65 μm/pixel, Pannoramic MIDI II; 3DHISTECH) equipped with LED (Lumencor, SPECTRA X light engine). To assess c-Fos counts and ZsGreen-positive BNSTSOM/BNSTCRF neurons, we delineated regions of interest or used fixed rectangular/oval frames on fluorescent pictures using CaseViewer 2.3 software (3DHISTECH). C-Fos signal was counted bilaterally in two or three sections that were 180 µm apart, covering the whole antero-posterior extension of the actual region/subregion. For statistical analyses and figures, average counts were considered. Section planes were standardized according to the atlas of Paxinos and Franklin (2001). All c-Fos results are presented as c-Fos counts normalized for mm2, which were counted manually using standardized settings (contrast, intensity) across subjects and regions.
Statistics
Data are expressed as mean ± SEM. Differences between groups were evaluated by one-way, two-way, or repeated-measure ANOVA (time/block as within-subject factor), followed by Tukey's post hoc analyses using Statistica software (Tibco). Ex vivo electrophysiological data were also analyzed by Student's one-sample t test. The significance level was set at p < 0.05 throughout, all p values are indicated with exact numbers.
Results
The BNST is activated during cued fear acquisition but not during recall
To interrogate the role of the BNST in different phases of conditioned fear response, we mapped neuronal activity during fear acquisition (i.e., conditioning) and CS-induced fear recall in adult male C57Bl/6J mice through analysis of c-Fos expression. During fear conditioning, mice were presented with seven 30 s pure tones (7 kHz), each co-terminating with a 1-s footshock (0.7 mA). Control mice were exposed to the same (contextual and auditory) stimuli without footshocks. Mice were killed for c-Fos staining 90 min after fear conditioning. As expected, shocked mice showed significantly higher freezing levels than controls during conditioning (F(1,18) = 153.944 and p < 0.0001; Fig. 1A). Fear conditioning induced significant c-Fos expression in all BNST subregions (Fig. 1E), particularly in the anteromedial and posterior regions (all regions: F(1,18) > 5.55, p = 0.029; Fig. 1C,F), whereas c-Fos expression was similar in conditioned and control mice during cue-dependent fear recall (all regions: F(1,19) > 1.008, p > 0.327; Fig. 1D,G) despite a high level of freezing exhibited by conditioned mice (F(1,19) = 9.777 and p < 0.0001; Fig. 1B). These data suggest that the BNST is recruited during cued fear conditioning, but not when conditioned fear is recalled. Accordingly, we aimed to test how enhanced BNST activity contributes to fear learning.
Chemogenetic activation of the BNST facilitates fear learning but not fear expression
We expressed stimulatory hM3Dq DREADD in the BNST of adult male vgat-ires-cre mice to target the major neuronal population of the BNST, i.e., GABAergic neurons. By using standard injection method and four weeks incubation time, we were able to induce widespread expression of DREADD receptors in the BNST, but infection consistently avoided the oval nucleus (Fig. 2A). Control animals were injected with a viral vector carrying only mCherry fluorophore protein without active hM3Dq receptor. Consistently with previous studies (Mazzone et al., 2018), whole-cell patch-clamp recordings confirmed that CNO activated hM3Dq receptors indicated by depolarized resting membrane potential (4.18 ± 0.60 vs −0.54 ± 1.11 mV in controls; F(1,7) = 17.319, p = 0.004; t = 9.286, p < 0.001 compared with baseline) and increased firing rate of hM3Dq-expressing BNSTvGAT cells [30-pA pulse: from 0.9 ± 0.23 action potentials (APs) to 4.4 ± 0.37 APs, F(1,9) = 169.615, p < 0.001; 60-pA pulse: from 3.2 ± 0.29 APs to 16.5 ± 0.80 APs, F(1,9) = 214.846, p < 0.001; Fig. 2B,C]. C-Fos immunohistochemistry also confirmed that intraperitoneal injection of CNO in 1 mg/kg dose resulted in marked activation of BNSTvGAT neurons under baseline (homecage) condition (F(1,16) = 73.123, p < 0.001; Fig. 2D).
To test how BNST activation modulates fear acquisition, we chemogenetically activated BNSTvGAT neurons during fear conditioning (Fig. 2F). Interestingly, it did not affect fear acquisition acutely, i.e., freezing levels during fear conditioning (F(1,15) = 0.041, p = 0.841; Fig. 2F), but it resulted in elevated CS-induced fear recall (F(1,15) = 6.774, p = 0.019), which effect diminished by the next extinction session 1 d later (i.e., extinction recall: F(1,15) = 1.275, p = 0.276; Fig. 2H). Contextual fear recall and freezing in a safe context during baseline period (i.e., before CS presentation as an index of contextual fear generalization) did not reveal any alterations (F(1,15) = 0.025, p = 0.875; F(1,15) = 0.961, p = 0.342, respectively; Fig. 2G,H).
Since chemogenetic activation has been reported to last for several hours (Roth, 2016), chemogenetic stimulation could potentially modulate both fear acquisition and consolidation phases of fear memory formation. To dissect the temporal dynamics of our effect, in the next experiment CNO was injected immediately after fear conditioning to activate BNSTvGAT neurons specifically during fear memory consolidation (Fig. 2I). Consolidation-specific stimulation replicated the enhancement of CS-induced fear recall (F(1,15) = 5.320, p = 0.035; Fig. 2K), with a trend for extinction deficit on day 1 (block × group interaction: F(1,75) = 1.897, p = 0.105). CS-induced fear recall showed a non-significant trend during the next session (day 2: F(1,16) = 2.863, p = 0.109) with more apparent contextual generalization indicated by significant difference during baseline period without CS (F(1,16) = 5.763, p = 0.028; Fig. 2K, right panel). CS-specific freezing analysis (based on subtraction of baseline period freezing from CS-induced freezing) confirmed that enhanced freezing response was CS-dependent on day 1 (F(1,15) = 5.146, p = 0.038), but it was rather because of a general increase in the safe context independent from CS-presentation on day 2 (F(1,16) = 0.006, p = 0.937), suggesting a potential development of contextual generalization in BNST-stimulated mice. In contrast, contextual recall in context A showed no difference (F(1,17) = 1.560, p = 0.228; Fig. 2J), and fear acquisition was similar between groups as indicated by freezing (F(1,17) = 0.311, p = 0.584; Fig. 2I).
Finally, we tested how BNSTvGAT neurons modulate CS-induced fear recall, or its generalization to safety cues using a differential auditory fear conditioning paradigm (Fig. 2L). Again, fear acquisition was similar between groups (F(1,19) = 0.001, p = 0.987; Fig. 2M), and groups could similarly differentiate between CS+ and CS– during a brief recall test (CS–/CS+: F(1,18) = 0.451, p = 0.510; stimulus: F(1,36) = 9.603, p = 0.003; stimulus × group interaction: F(1,36) = 0.008, p = 0.927; Fig. 2N). In line with unaltered c-Fos activity, we did not observe changes in CS-induced freezing levels and CS+/CS– discrimination, when BNSTvGAT neurons were activated during fear recall (CS+-induced recalls: F(1,19) = 0.303, p = 0.588; F(1,19) = 0.040, p = 0.843; CS–/CS+: F(1,19) = 18.595, p < 0.001; Fig. 2O). Together, our findings suggested that BNST is actively recruited during fear acquisition and enhancement of this activity results in enhanced fear memory formation via consolidatory mechanisms. Next, we aimed to identify potential downstream targets, which could mediate these effects.
Chemogenetic stimulation of BNSTvGAT neurons alters c-Fos activity in downstream regions involved in fear regulation
To point out potential downstream targets mediating our effects, we mapped c-Fos activity 6 h after fear conditioning, i.e., consolidation phase (Fig. 3A). First, we confirmed marked activation of hM3Dq-expressing neurons in the BNST at 6 h (86.36% vs 6.09% in controls; F(1,8) = 1117.353, p < 0.001; Fig. 3C,D). Second, we assessed c-Fos expression in densely innervated regions of the fear circuitry (Fig. 3B,C), where significant BNST projections could be observed (indicated by mCherry labeling), i.e., in the nucleus accumbens (NAc) shell, dorsal midline thalamus (DMT), central amygdala (CeA, medial nucleus-CeM), lateral hypothalamus (LH), paraventricular nucleus of the hypothalamus (PVN), substantia nigra pars compacta (SNc), ventral tegmental area (VTA), dorsal raphe (DR), ventrolateral periaqueductal gray (vlPAG). Noteworthy, this pattern described above was in high accordance with previous reports in rats and mice (Dong et al., 2001; Dong and Swanson, 2004, 2006; Kodani et al., 2017). Among these regions, DMT, VTA (interfascicular part-ifVTA), and vlPAG exhibited increased c-Fos expression (F(1,13) = 7.516, p = 0.016; F(1,13) = 8.191, p = 0.013; F(1,14) = 18.919, p < 0.001, respectively; Fig. 3C,F), whereas PVN exhibited reduced c-Fos expression in hM3Dq mice (F(1,14) = 5.208, p = 0.038; Fig. 3C,E). DR also exhibited a trend for increased activity (F(1,14) = 4.013, p = 0.064).
Figure 3.

Postsynaptic activity following chemogenetic stimulation of the BNST during memory consolidation. A, Schematics of virus injections and representative photomicrograph of mCherry expression in the BNST. Right panel shows experimental design: c-Fos expression was assesses 6 h after fear conditioning combined with subsequent chemogenetic activation (i.e., in the consolidation phase). B, Representative wide-field fluorescence photomicrographs depicting major projection areas of BNSTvGAT neurons. C, Representative single-plane confocal photomicrographs showing altered c-Fos expression during consolidation in the BNST and downstream regions, where white arrows indicate activated hM3Dq-expressing BNSTvGAT neurons (mCherry+c-Fos). D, C-Fos activity was significantly increased in the BNST, and DMT, vlPAG, ifVTA downstream regions (F), with additional decrease in PVN (E). ac, anterior commissure; amBNST, anteromedial BNST; Aq, cerebral aqueduct; BLA, basolateral amygdala; CeL/CeC, central amygdala, lateral/capsular part; CeM, central amygdala, medial part; cpd, cerebral peduncle; D3V, dorsal part of the third ventricle; DMT, dorsal midline thalamus; DR, dorsal raphe; fx, fornix; ic, internal capsule; LHA, lateral hypothalamic area; MD, mediodorsal thalamus; MHb, medial habenula; MM, medial mammillary nucleus; NAc, nucleus accumbens; opt, optic tract; PVN, paraventricular hypothalamic nucleus; SNc, substantia nigra, pars compacta; st, stria terminalis; vlPAG/lPAG, periaqueductal gray, ventrolateral/lateral part; ifVTA, ventral tegmental area, interfascicular nucleus. Asterisks represent main effect of one-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001.
BNSTSOM, but not BNSTCRF, neurons facilitate fear memory consolidation
To interrogate the involvement of specific cell types of the BNST in fear memory consolidation, we selectively modulated BNSTSOM and BNSTCRF neurons, which constitute two major GABAergic cell types in the BNST (Nguyen et al., 2016; Dedic et al., 2018; Ye and Veinante, 2019). First, we quantified the intra-BNST distribution of CRF and SOM positive neurons using cre-dependent reporter mouse lines expressing Zsgreen fluorescent protein (Gt(ROSA)26Sor-CAG/LSL-ZsGreen1). We observed similar expression levels in the anterior and medial divisions (∼15–20%), and significant dominance of SOM neurons in the oval nucleus and posterior regions compared with CRF (Figs. 4A, 5A). Noteworthy, in contrast to this distribution we detected very limited expression of DREADDs in our experiments (indicated by mCherry labeling) in the oval nucleus (Figs. 2A, 3A, 4A, 5A), which should be taken into account when interpreting our findings, particularly when one appreciates the heterogeneity and competing effects across BNST subregions, and anxiogenic effects mediated by the oval nucleus (Choi et al., 2007; Kim et al., 2013; Daniel and Rainnie, 2016).
Figure 4.
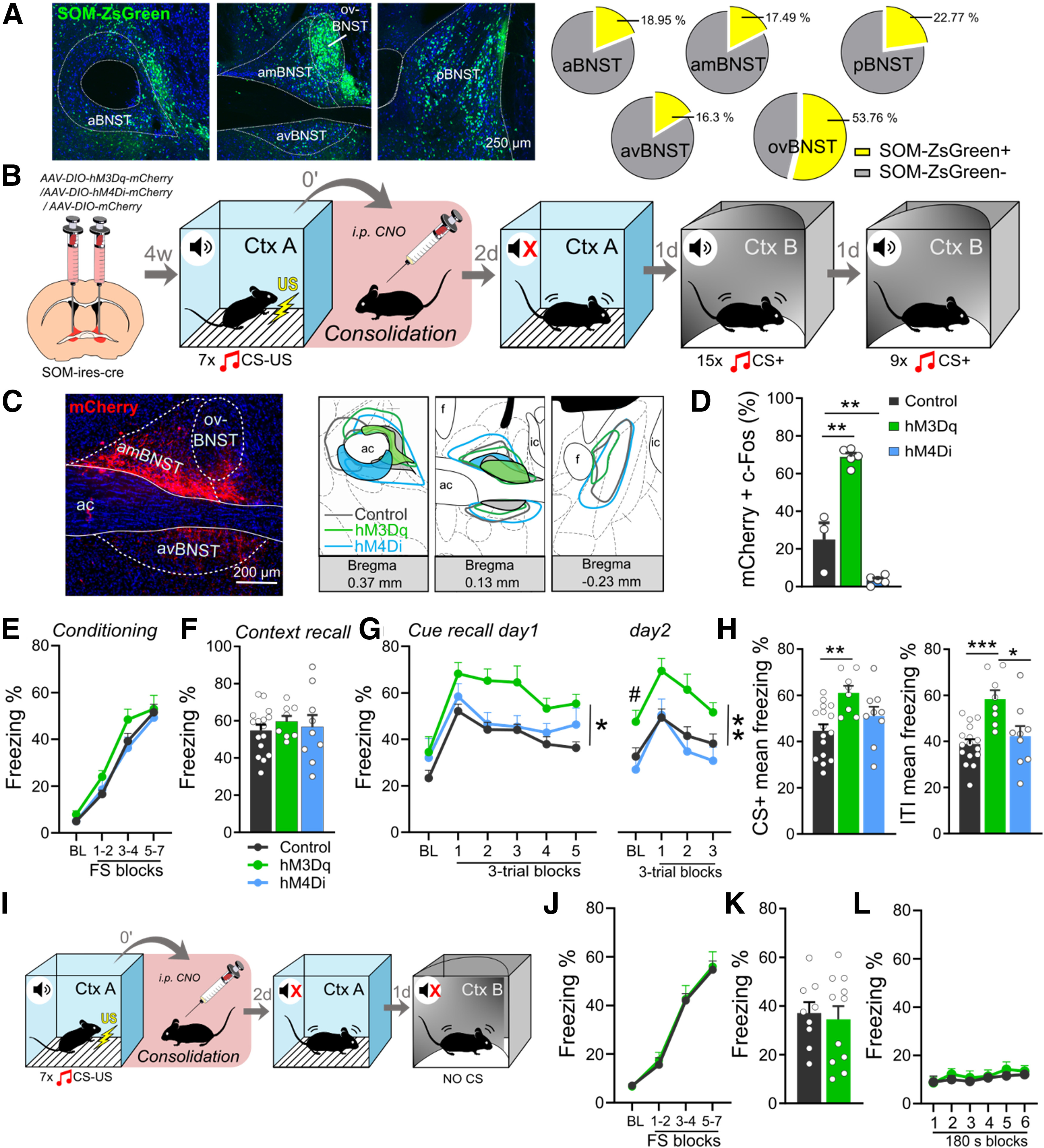
Chemogenetic activation of BNSTSOM neurons during fear memory consolidation enhances CS-induced fear recall. A, Distribution of BNSTSOM neurons illustrated by representative single-plane confocal photomicrographs from reporter ZsGreen fluorescent protein-expressing mouse lines, and their proportional quantification (% of all neurons, NeuN+) across subregions. B, Experimental design for chemogenetic modulation of BNSTSOM neurons during fear memory consolidation. C, Photomicrograph showing mCherry expression in BNSTSOM neurons and illustration of minimum (filled areas) and maximum (areas with colored outlines) extensions of mCherry expression. D, Intraperitoneal injection of CNO (1 mg/kg) under baseline (homecage) condition significantly increased c-Fos expression in hM3Dq-mCherry-expressing BNSTSOM neurons, while hM4Di-expressing BNSTSOM neurons showed reduced c-Fos expression (control: n = 3, hM3Dq: n = 5, hM4Di: n = 5). E, Freezing behavior during fear conditioning of som-ires-cre mice. Chemogenetic activation of BNSTSOM neurons during fear memory consolidation resulted in enhanced cued fear recall (G, left panel) with additional contextual fear generalization during baseline period 1 d later (G, right panel) and disrupted discrimination between CS and ITI (H). Contextual fear recall in context A was similar between groups (F; control: n = 8, hM3Dq: n = 7). In contrast to stimulation, chemogenetic inhibition of BNSTSOM neurons had no impact on fear recalls (F–H, hM4Di: n = 9). I, Experimental design to test CS-independent contextual fear generalization following chemogenetic stimulation of BNSTSOM neurons. J–L, Chemogenetic stimulation of BNSTSOM neurons did not affect contextual fear recall, either in context A or B (control: n = 9, hM3Dq: n = 11). On freezing time curves, each major tick depicts two to three footshock (FS) blocks in case of conditioning, and a 180-s block, starting with a 150-s pretone baseline period (BL) in case of recall test. All data are represented as mean ± SEM. Asterisks represent main effect of repeated-measure ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001; Hash sign represents significant difference during BL period: #p < 0.05. ac, anterior commissure; aBNST, anterior BNST; amBNST, anteromedial BNST; avBNST, anteroventral BNST; ovBNST, oval nucleus of the BNST; pBNST, posterior BNST.
Figure 5.
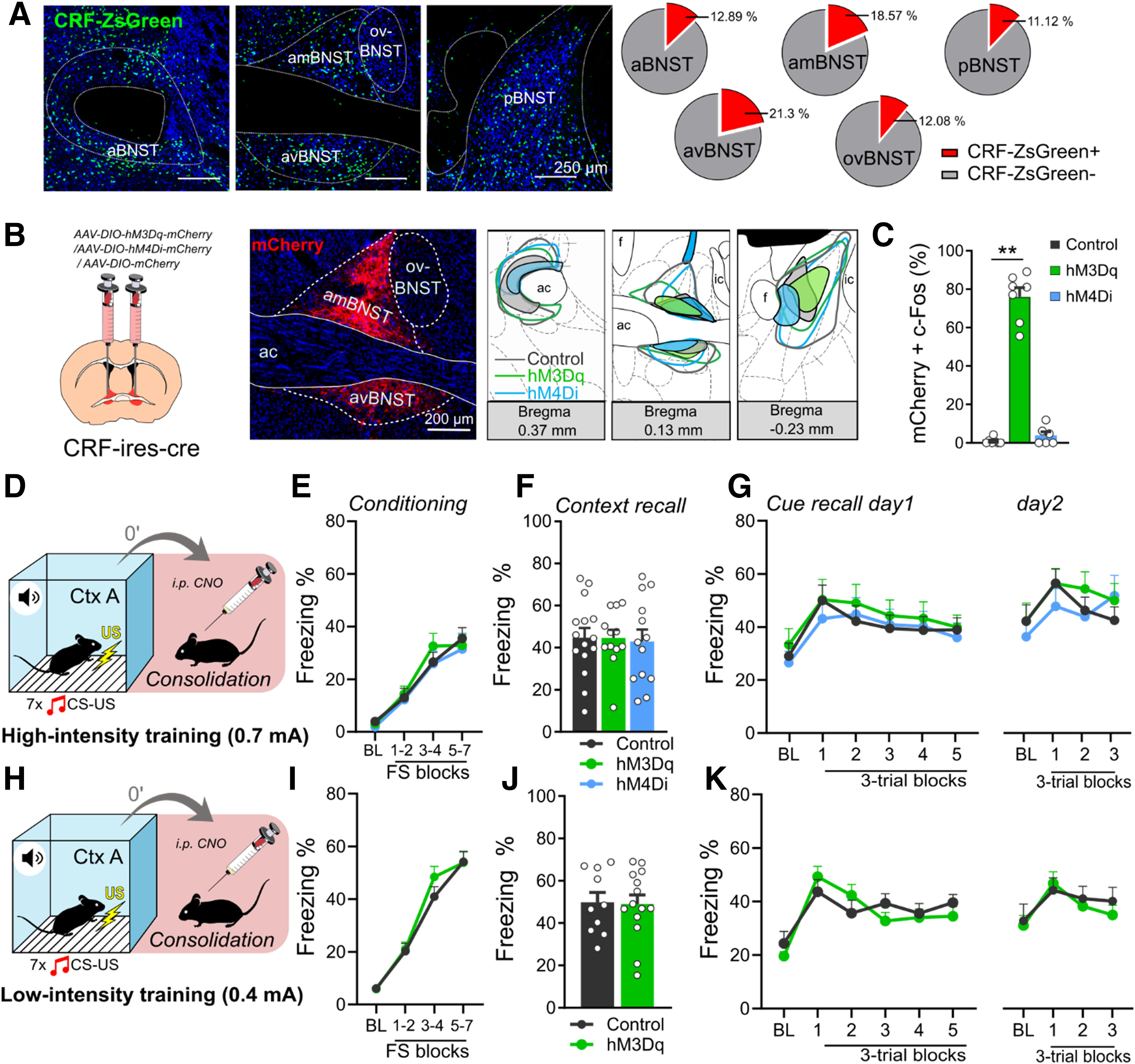
Chemogenetic modulation of BNSTCRF neurons during fear memory consolidation does not affect fear recalls. A, Distribution of BNSTCRF neurons illustrated by representative single-plane confocal photomicrographs from reporter ZsGreen fluorescent protein-expressing mouse lines and their proportional quantification (% of all neurons, NeuN+) across subregions. B, Representative photomicrographs of mCherry expression in BNSTCRF neurons and illustration of minimum (filled areas) and maximum (areas with colored outlines) extensions of mCherry expression. C, Intraperitoneal injection of CNO (1 mg/kg) under baseline (homecage) condition significantly increased c-Fos expression in hM3Dq-mCherry-expressing BNSTCRF neurons (control: n = 7, hM3Dq: n = 7, hM4Di n = 6). D and H, Illustrations depicting high (0.7-mA footshocks) and low-intensity (0.4-mA footshocks) fear conditioning in crh-ires-cre mice with CNO administration after conditioning. Fear recalls were independent of BNST manipulation following both high-intensity (F–G) and low-intensity (J–K) trainings. E and I, Freezing during high- and low-intensity trainings, respectively (control: n = 15 and n = 10, hM3Dq: n = 12 and n = 14 for low-intensity and high-intensity trainings, respectively; hM4Di: n = 13). Asterisks represent main effect of one-way ANOVA: **n < 0.01.
Since SOM and CRF neurons have been shown to drive fear expression in opposite ways in the CeA (Fadok et al., 2017), we applied SOM and CRF-specific chemogenetic activation and inhibition during the consolidation phase in som-ires-cre and crh-ires-cre mice. First, we validated that intraperitoneal CNO injection can reliably modulate neuronal activity (Figs. 4D, 5C): hM3Dq expressing SOM and CRF neurons showed significantly higher co-expression with c-Fos compared with mCherry-expressing controls [SOM: F(2,10) = 93.361, p < 0.001 (Fig. 4D); Tukey's post hoc test p < 0.001; CRF: F(2,17) = 199.509, p < 0.001, Tukey's post hoc test p < 0.001 (Fig. 5C)]. hM4Di mediated decrease in c-Fos expression was also observed in SOM neurons (Tukey's post hoc test p = 0.008), but could not be detected in CRF neurons because of floor effect (minimal baseline activity in control mice, Tukey's post hoc test p = 0.786).
Chemogenetic activation of BNSTSOM neurons during fear consolidation (Fig. 4B) could partially replicate our BNSTvGAT findings by enhancing fear recall in the safe context, even with more pronounced and persistent effects (cue recall day 1: F(2,30) = 8.067, p = 0.001, Tukey's post hoc test p = 0.001 and p = 0.391 control vs hM3Dq and hM4Di, respectively; day 2/extinction recall: F(2,29) = 5.547, p = 0.009, Tukey's post hoc test p = 0.021 and p = 0.773 control vs hM3Dq and hM4Di, respectively; Fig. 4G). Importantly, this effect became non-significant when CS-specific freezing levels were calculated (baseline subtracted from CS-induced freezing; Cue recall day 1: F(2,30) = 1.025, p = 0.370; day 2: F(2,30) = 0.116, p = 0.890), implying that enhanced freezing was more because of contextual generalization in the safe context. Similarly to BNSTvGAT stimulation, BNSTSOM stimulation enhanced freezing during baseline period on day 2 (BL freezing for day 1: F(2,31) = 0.769, p = 0.471 and day 2: F(2,29) = 4.225, p = 0.024; Fig. 4G), which again indicated enhanced contextual generalization. Moreover, higher freezing levels in the hM3Dq group were not only observed during CS-presentations but during ITIs, as well (CS: F(2,30) = 5.975, p = 0.006, Tukey's post hoc test p = 0.004 vs controls; ITIs: F(2,30) = 9.324, p < 0.001, Tukey's post hoc test p < 0.001 vs controls; Fig. 4H). Latter also appeared in the lack of CS/ITI discrimination in the hM3Dq group (1.06 ± 0.05; t = 1.080, p = 0.316), although CS/ITI discrimination ratio was also weak in the control and hM4Di groups (1.15 ± 0.04, t = 4.037, p = 0.001; 1.24 ± 0.05, t = 4.400, p = 0.002, respectively). Similar to previous experiments, contextual fear recall in context A was not changed (F(2,31) = 0.160, p = 0.852; Fig. 4F), and fear conditioning was similar between groups (F(2,29) = 1512, p = 0.237; Fig. 4E). Chemogenetic inhibition did not affect any forms of fear recall, despite its potential to lower neuronal activity indicated by c-Fos (p = 0.008; Fig. 4D).
Based on the above generalization tendencies, we aimed to test whether our effect represents contextual generalization independent of CS. In a separate cohort of mice, we tested the impact of chemogenetic stimulation of BNSTSOM neurons during consolidation (using the same experimental design) on fear recall without CS-presentation in the safe context (Fig. 4I). Freezing response showed no difference in the safe context without CS presentation (F(1,18) < 0.275, p > 0.606; Fig. 4L), suggesting that the memory-enhancing effect of BNSTSOM stimulation was dependent on, but not limited to, CS–presentation (i.e., during CS blocks).
In BNSTCRF neurons, neither chemogenetic activation, nor inhibition altered contextual (F(2,37) = 0.041, p = 0.959; Fig. 5F) or CS-induced fear recalls (cue recalls: F < 0.270, p > 0.765; Fig. 5G), or fear generalization (CtxB BLs: F < 0.416, p > 0.662). Fear acquisition was similar between groups as indicated by freezing (F(2,37) = 0.205, p = 0.815; Fig. 5E). Considering recruitment of CeACRF neurons during weak threats, i.e., fear conditioning with low-intensity footshock as shown before (Sanford et al., 2017), one could hypothesize that stimulus intensity determines BNSTCRF neurons recruitment, which could contribute to our negative findings. To test this hypothesis, we performed an experiment with identical experimental design but using 0.4-mA footshocks as US (Fig. 5H). Similar to high-intensity conditioning, we did not observe alteration in fear memory consolidation indicated by similar contextual or CS-dependent fear recalls between groups (F(1,22) = 0.045, p = 0.833 and F(1,22) = 0.117, p = 0.734, respectively; Fig. 5J,K). Latter findings suggest that BNSTCRF neurons are not, or minimally involved in the memory-enhancing effect of the BNST, although lack of DREADD expression in the oval nucleus could be a significant limitation in our experiment contributing to negative findings.
Discussion
Our study demonstrates that BNST neurons are activated during fear acquisition and chemogenetic stimulation of BNSTvGAT neurons during fear conditioning or consolidation enhances CS-related fear memory formation. In contrast, the BNST seems to be minimally involved in conditioned fear expression indicated by unaltered freezing during fear acquisition and CS-dependent fear recall when the same neurons were chemogenetically stimulated. Stimulation of BNSTSOM neurons during the consolidation phase recapitulated these effects by resulting in strong enhancement of CS-related fear, but it also increased contextual fear generalization. Although CS-presentation was necessary to detect significant enhancement of fear recall in the safe context, it was not limited to CS periods, suggesting that CS may represent an ambiguous stimulus in a safe environment manifesting fear recall alterations regulated by the BNST. Accordingly, our data support the importance of the BNST in fear memory formation related to threat predicting cues.
It is now well accepted that the BNST does not only regulate anxiety responses, but it is also capable to modulate short-term conditioned fear responses (Goode and Maren, 2017). Although the involvement of the BNST in CS-related fear conditioning is still debated, in vivo recordings from BNST neurons revealed that a substantial population of recorded BNST cells is excited by CS during fear learning, especially in the anteromedial BNST (Bjorni et al., 2020), which showed robust activity during acquisition in our experiments. Most studies investigating the role of the BNST in cued conditioned fear inhibited the BNST during fear retrieval. The majority of these studies showed that BNST inactivation disrupted only conditioned fear to ambiguous cues (Sullivan et al., 2004; Lange et al., 2017; Goode et al., 2019), without any effect on temporally predictable CS-induced fear. In line with these observations, in our experiments, BNST stimulation did not affect acute fear responses (during conditioning or recall), supporting that predictable CS-induced fear expression is not dependent on BNST activity.
In contrast to fear expression, increased BNST activity had a significant impact on fear memory formation that subsequently manifested in enhanced fear. It is important to note that few studies investigated the BNST with gain of function approaches or with cell type-specific modulations. Here, we repeatedly showed that stimulation of BNST neurons facilitate fear learning, however, the exact circuitries mediating this effect need to be determined. To identify potential mediators of our effect, we mapped c-Fos activity in BNST-projecting areas. We confirmed that BNST neurons project to several brain regions involved in learned fear responses including NAc, DMT, LH, CeA, VTA, DR, vlPAG. Elevated activity in DMT indicates enhanced aversive learning as previous reports showed the involvement of DMT in fear memory consolidation (Li et al., 2013; Penzo et al., 2014, 2015; Do-Monte et al., 2015; Gao et al., 2020). Similarly, enhanced vlPAG activity suggests an alteration in the amygdalar output initiating fear responses (Dejean et al., 2015), which region has also been recently shown to encode danger-cue associations and its probability (Ozawa et al., 2017; Wright and McDannald, 2019; Wright et al., 2019). Moreover, the avBNST-vlPAG pathway was recently shown to modulate fear memory consolidation in the active avoidance paradigm (Lingg et al., 2020). Accordingly, the BNST (as extension of CeA-DMT and CeA-vlPAG circuitry) could be a significant modulator of fear memory strength (Do-Monte et al., 2015; Penzo et al., 2015; Ozawa et al., 2017). Altered activity in monoaminergic regions (ifVTA, DR) and PVN are also potential mediators based on their significant impact on fear memory formation (Dedic et al., 2018; Groessl et al., 2018; Sengupta and Holmes, 2019). Although reduced PVN activity is in line with previous studies documenting the mediatory/inhibitory role of the BNST on hypothalamic-pituitary-adrenal axis (HPA) axis, it contradicts the significant memory-enhancing effect of glucocorticoids (de Quervain et al., 2009; Radley and Sawchenko, 2011; Lingg et al., 2020). Considering the limitation of our c-Fos mapping, we cannot conclude on HPA-axis activity without CRF-specific neuronal activity assessment, particularly when one considers opposing impact of BNST subregions on HPA-axis activity (Choi et al., 2007), or the importance of temporal dynamics, i.e., activity changes during 6 h of consolidation. Finally, it is more realistic to assume that enhanced fear response following BNST stimulation was a result of complex circuitry changes with multiple nodes, where potential competing effects exist. Future studies targeting specific efferent pathways of BNST need to clarify how these specific circuit elements contribute to fear learning.
Projection areas of SOM, CRF, and vGAT neurons of the BNST described here showed minimal differences, which also correspond highly with previous descriptions (Dabrowska et al., 2016; Dedic et al., 2018 (Fig. 6)). The distribution of these projections also highly overlaps with reported CeA projections (Hartley et al., 2019; Li, 2019), suggesting coordinated functioning between the amygdala and the BNST. In respect of our contrasting findings on BNSTCRF and BNSTSOM stimulations, the high distributional overlap between SOM and CRF neurons suggests that they target functionally distinct postsynaptic partners in the same projection areas. Interpreting our BNSTSOM findings in the light of previous reports, it seems that SOM neurons in the central extended amygdala have a rather uniform impact on fear-like responses, shifting it toward passive “coping” forms, i.e., freezing versus escape (Li et al., 2013; Penzo et al., 2014, 2015; Yu et al., 2016; Fadok et al., 2017; Hartley et al., 2019). Moreover, CeASOM neurons enhance fear learning via connections with DMT and vlPAG, regions where we detected marked elongated c-Fos hyperactivity after chemogenetic stimulation of the BNST in the consolidation phase (Li et al., 2013; Penzo et al., 2014, 2015; Li, 2019). However, whereas CeASOM neurons drive passive fear response directly, BNSTSOM neurons modulates fear responses more indirectly via modulating the fear circuitry likely via plastic changes (i.e., potentiation). BNSTSOM neurons also shifted fear expression toward more generalized forms, although this effect was highly dependent on, but not limited to, CS-presentation. Likely CS in a safe context represent an ambiguous stimulus for the animal, which recruits BNST-related circuitry as shown before (Daldrup et al., 2016; Goode et al., 2019; Naaz et al., 2019).
Figure 6.
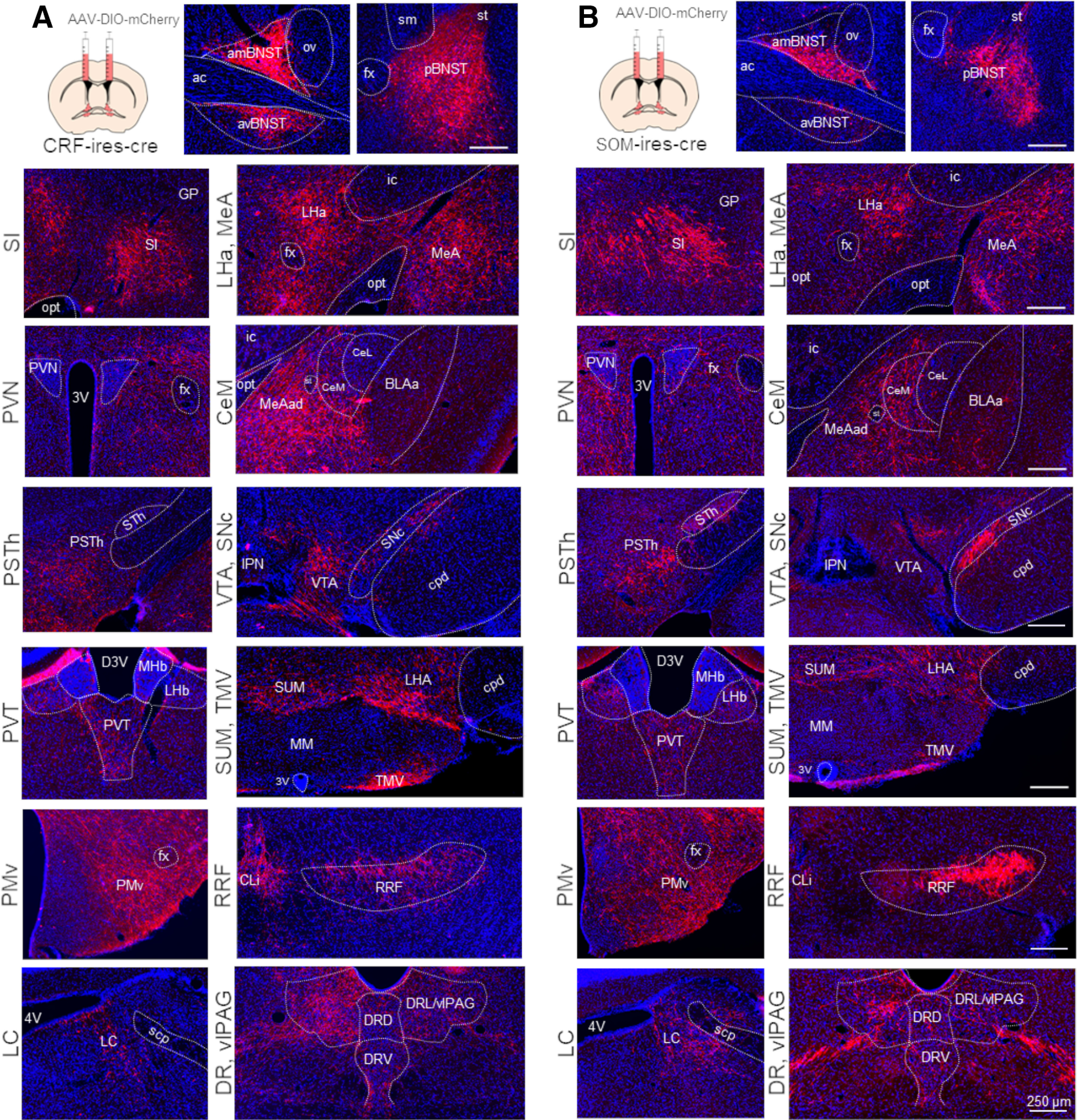
AAV based mapping of major projection areas of BNSTCRF (A) and BNSTSOM (B) neurons. Representative wide-field fluorescence photomicrographs show a highly similar distribution of BNSTCRF and BNSTSOM projections. 3V, third ventricle; 4V, fourth ventricle; ac, anterior commissure; amBNST, anteromedial BNST; avBNST, anteroventral BNST; BLAa, basolateral amygdala, anterior part; CeL, central amygdala, lateral part; CeM, central amygdala, medial part; CLi, central linear nucleus raphe; cpd, cerebral peduncle; D3V, dorsal part of the third ventricle; DRD, dorsal raphe, dorsal part; DRL, dorsal raphe, lateral part; DRV, dorsal raphe, ventral part; fx, fornix; GP, globus pallidus; ic, internal capsule; IPN, interpeduncular nucleus; LC, locus coeruleus; LHa, lateral hypothalamic area; LHb, lateral habenula; MeA, medial amygdala; MeAad, medial amygdala nucleus, anterodorsal part; MHb, medial habenula; MM, medial mammillary nucleus; opt, optic tract; ov, oval nucleus of the BNST; pBNST, posterior BNST; PMv, ventral premammillary nucleus; PSTh, parasubthalamic nucleus; PVN, paraventricular hypothalamic nucleus; PVT, paraventricular thalamic nucleus; RRF, retrorubral field; scp, superior cerebellar peduncles; SI, substantia innominate; sm, stria medullaris; SNc, substantia nigra, pars compacta; st, stria terminalis; STh, subthalamic nucleus; SUM, supramammillary nucleus; TMV, tuberomammillary nucleus, ventral part; vlPAG, ventrolateral periaqueductal gray; VTA, ventral tegmental area.
It is important to note that BNSTSOM neurons may not be inevitable for intact fear learning as chemogenetic inhibition showed no effect. It is possible that these neurons only exert a modulatory effect on fear memory formation and canonical BLA-dependent fear learning could be intact with BNST inhibition. A previous study indicated that BNST and BLA-dependent circuits may represent alternative pathways for fear learning and could compensate each other, i.e., rats were able to acquire conditioned fear with BLA lesion, which was diminished with combined BLA and BNST lesion (Poulos et al., 2010). The lack of bidirectional effects with BNST modulation is not unusual: involvement of extended amygdala neurons in innate anxiety is well documented; however, some studies pointed out inhibition often fails to modulate baseline anxiety-like behavior without prior stress (Regev et al., 2011; Pomrenze et al., 2019). Accordingly, enhanced BNST activity may represent a vulnerability-like factor for exaggerated fear learning: although canonical amygdalar pathways are major mediators of acute fear responses and conditioning, enhanced BNST present in high-anxiety individuals may potentiate fear learning. Unfortunately, our chemogenetic approach made it unfeasible for us to define behavioral extremes (e.g., low-high anxiety quartiles) in the present study, which is to be tested by future studies. Another important question for future studies how these mechanisms appear in females, particularly considering the sexually dimorphic nature of the BNST.
Finally, negative findings on BNSTCRF activation is in accordance with a previous report using BNSTCRF manipulation (Marcinkiewcz et al., 2016). Interestingly, lowered intensity training could not reveal any difference as it was shown in the amygdalar CRF circuit (Sanford et al., 2017), suggesting that stimulus intensity was not the issue in our study. However, we cannot exclude that (1) our paradigm was not sufficient to reveal “active-defensive” phenotype, i.e., escape behavior, which is driven by CRF neurons in multiple brain regions (Fadok et al., 2017; Daviu et al., 2020); and (2) DREADD expression consistently avoided the oval nucleus, which has been shown to mediate anxiogenic effects or responses to aversive stimuli (Kim et al., 2013; Pelrine et al., 2016; Giardino et al., 2018). Despite these limitations, it is important to note that SOM and CRF expression in the mouse BNST is much less restricted to the oval nucleus and exhibit more widespread distribution (Nguyen et al., 2016), in accordance with our observations in Zsgreen reporter lines, and significant DREADD expression across multiple BNST subregions.
Together, our study points out a specific role of the BNST in the facilitation of fear learning. Particularly, BNSTvGAT neurons enhance fear memory strength (likely via plastic changes in several regions of the fear circuitry) without affecting acute fear reactivity and expression. Moreover, BNSTSOM neurons promote fear generalization, although this effect is dependent on threat-predictive cues (CS), providing further data how the amygdala and the BNST exert complementing functions in the fear circuitry. BNST hyperactivity may also translate into maladaptive fear memories and generalization apparent in anxiety disorders (Avery et al., 2016; Brinkmann et al., 2018).
Footnotes
This work was supported by National Research, Development and Innovation Office Grants (Hungary) #FK129296 and #PD116589 (to M.T.) and the Hungarian Brain Research Program Grant #2017-1.2.1-NKP-2017-00002 (to E.M.). This work was also supported by New National Excellence Program of the Ministry for Innovation and Technology Grants #UNKP-18-3-III-SE-22, #UNKP-19-2-I-ELTE-574, and #UNKP-20-5-SE (to L.B., H.S., and M.T.) from the source of the National Research, Development and Innovation Fund and by the Bolyai Janos Research Fellowship (M.T.). We thank all the core facilities of our institute for their supportive help: the Behavioral Studies Unit for help with behavioral testing (Dr. Kornél Demeter), the Nikon Microscopy Center for help with microscopy (Dr. László Barna and Dr. Csaba Pongor), and the Medical Gene Technology Unit for help with mouse lines. We also thank Beata Barsvari and Nikolett Bakos for technical assistance.
The authors declare no competing financial interests.
References
- Alheid GF, Heimer L (1988) New perspectives in basal forebrain organization of special relevance for neuropsychiatric disorders: the striatopallidal, amygdaloid, and corticopetal components of substantia innominata. Neuroscience 27:1–39. 10.1016/0306-4522(88)90217-5 [DOI] [PubMed] [Google Scholar]
- Asok A, Draper A, Hoffman AF, Schulkin J, Lupica CR, Rosen JB (2018) Optogenetic silencing of a corticotropin-releasing factor pathway from the central amygdala to the bed nucleus of the stria terminalis disrupts sustained fear. Mol Psychiatry 23:914–922. 10.1038/mp.2017.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery SN, Clauss JA, Blackford JU (2016) The human BNST: functional role in anxiety and addiction. Neuropsychopharmacology 41:126–141. 10.1038/npp.2015.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorni M, Rovero NG, Yang ER, Holmes A, Halladay LR (2020) Phasic signaling in the bed nucleus of the stria terminalis during fear learning predicts within- and across-session cued fear expression. Learn Mem 27:83–90. 10.1101/lm.050807.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann L, Buff C, Feldker K, Tupak SV, Becker MPI, Herrmann MJ, Straube T (2017) Distinct phasic and sustained brain responses and connectivity of amygdala and bed nucleus of the stria terminalis during threat anticipation in panic disorder. Psychol Med 47:2675–2688. 10.1017/S0033291717001192 [DOI] [PubMed] [Google Scholar]
- Brinkmann L, Buff C, Feldker K, Neumeister P, Heitmann CY, Hofmann D, Bruchmann M, Herrmann MJ, Straube T (2018) Inter-individual differences in trait anxiety shape the functional connectivity between the bed nucleus of the stria terminalis and the amygdala during brief threat processing. Neuroimage 166:110–116. 10.1016/j.neuroimage.2017.10.054 [DOI] [PubMed] [Google Scholar]
- Choi DC, Furay AR, Evanson NK, Ostrander MM, Ulrich-Lai YM, Herman JP (2007) Bed nucleus of the stria terminalis subregions differentially regulate hypothalamic-pituitary-adrenal axis activity: implications for the integration of limbic inputs. J Neurosci 27:2025–2034. 10.1523/JNEUROSCI.4301-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings KA, Clem RL (2020) Prefrontal somatostatin interneurons encode fear memory. Nat Neurosci 23:61–74. 10.1038/s41593-019-0552-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowska J, Martinon D, Moaddab M, Rainnie DG (2016) Targeting corticotropin-releasing factor projections from the oval nucleus of the bed nucleus of the stria terminalis using cell-type specific neuronal tracing studies in mouse and rat brain. J Neuroendocrinol. Advance online publication. Retrieved November 2, 2019. doi: 10.1111/jne.12442. 10.1111/jne.12442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daldrup T, Lesting J, Meuth P, Seidenbecher T, Pape HC (2016) Neuronal correlates of sustained fear in the anterolateral part of the bed nucleus of stria terminalis. Neurobiol Learn Mem 131:137–146. 10.1016/j.nlm.2016.03.020 [DOI] [PubMed] [Google Scholar]
- Daniel SE, Rainnie DG (2016) Stress modulation of opposing circuits in the bed nucleus of the stria terminalis. Neuropsychopharmacology 41:103–125. 10.1038/npp.2015.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M, Walker DL, Miles L, Grillon C (2010) Phasic vs sustained fear in rats and humans: role of the extended amygdala in fear vs anxiety. Neuropsychopharmacology 35:105–135. 10.1038/npp.2009.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daviu N, Füzesi T, Rosenegger DG, Rasiah NP, Sterley TL, Peringod G, Bains JS (2020) Paraventricular nucleus CRH neurons encode stress controllability and regulate defensive behavior selection. Nat Neurosci 23:398–410. 10.1038/s41593-020-0591-0 [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Aerni A, Schelling G, Roozendaal B (2009) Glucocorticoids and the regulation of memory in health and disease. Front Neuroendocrinol 30:358–370. 10.1016/j.yfrne.2009.03.002 [DOI] [PubMed] [Google Scholar]
- Dedic N, Kühne C, Jakovcevski M, Hartmann J, Genewsky AJ, Gomes KS, Anderzhanova E, Pöhlmann ML, Chang S, Kolarz A, Vogl AM, Dine J, Metzger MW, Schmid B, Almada RC, Ressler KJ, Wotjak CT, Grinevich V, Chen A, Schmidt MV, et al. (2018) Chronic CRH depletion from GABAergic, long-range projection neurons in the extended amygdala reduces dopamine release and increases anxiety. Nat Neurosci 21:803–807. 10.1038/s41593-018-0151-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejean C, Courtin J, Rozeske RR, Bonnet MC, Dousset V, Michelet T, Herry C (2015) Neuronal circuits for fear expression and recovery: recent advances and potential therapeutic strategies. Biol Psychiatry 78:298–306. 10.1016/j.biopsych.2015.03.017 [DOI] [PubMed] [Google Scholar]
- Deslauriers J, Toth M, Der-Avakian A, Risbrough VB (2018) Current status of animal models of posttraumatic stress disorder: behavioral and biological phenotypes, and future challenges in improving translation. Biol Psychiatry 83:895–907. 10.1016/j.biopsych.2017.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do-Monte FH, Quiñones-Laracuente K, Quirk GJ (2015) A temporal shift in the circuits mediating retrieval of fear memory. Nature 519:460–463. 10.1038/nature14030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Swanson LW (2004) Organization of axonal projections from the anterolateral area of the bed nuclei of the stria terminalis. J Comp Neurol 468:277–298. 10.1002/cne.10949 [DOI] [PubMed] [Google Scholar]
- Dong HW, Swanson LW (2006) Projections from bed nuclei of the stria terminalis, anteromedial area: cerebral hemisphere integration of neuroendocrine, autonomic, and behavioral aspects of energy balance. J Comp Neurol 494:142–178. 10.1002/cne.20788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Watts AG, Swanson LW (2001) Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol 436:430–455. 10.1002/cne.1079 [DOI] [PubMed] [Google Scholar]
- Duits P, Cath DC, Lissek S, Hox JJ, Hamm AO, Engelhard IM, van den Hout MA, Baas JM (2015) Updated meta-analysis of classical fear conditioning in the anxiety disorders. Depress Anxiety 32:239–253. 10.1002/da.22353 [DOI] [PubMed] [Google Scholar]
- Duvarci S, Bauer EP, Paré D (2009) The bed nucleus of the stria terminalis mediates inter-individual variations in anxiety and fear. J Neurosci 29:10357–10361. 10.1523/JNEUROSCI.2119-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok JP, Krabbe S, Markovic M, Courtin J, Xu C, Massi L, Botta P, Bylund K, Müller C, Kovacevic A, Tovote P, Lüthi A (2017) A competitive inhibitory circuit for selection of active and passive fear responses. Nature 542:96–100. 10.1038/nature21047 [DOI] [PubMed] [Google Scholar]
- Flandreau EI, Toth M (2018) Animal models of PTSD: a critical review. Curr Top Behav Neurosci 38:47–68. 10.1007/7854_2016_65 [DOI] [PubMed] [Google Scholar]
- Gao C, Leng Y, Ma J, Rooke V, Rodriguez-Gonzalez S, Ramakrishnan C, Deisseroth K, Penzo MA (2020) Two genetically, anatomically and functionally distinct cell types segregate across anteroposterior axis of paraventricular thalamus. Nat Neurosci 23:217–228. 10.1038/s41593-019-0572-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardino WJ, Eban-Rothschild A, Christoffel DJ, Li SB, Malenka RC, de Lecea L (2018) Parallel circuits from the bed nuclei of stria terminalis to the lateral hypothalamus drive opposing emotional states. Nat Neurosci 21:1084–1095. 10.1038/s41593-018-0198-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode TD, Maren S (2017) Role of the bed nucleus of the stria terminalis in aversive learning and memory. Learn Mem 24:480–491. 10.1101/lm.044206.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode TD, Ressler RL, Acca GM, Miles OW, Maren S (2019) Bed nucleus of the stria terminalis regulates fear to unpredictable threat signals. Elife 8:e46525. 10.7554/eLife.46525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groessl F, Munsch T, Meis S, Griessner J, Kaczanowska J, Pliota P, Kargl D, Badurek S, Kraitsy K, Rassoulpour A, Zuber J, Lessmann V, Haubensak W (2018) Dorsal tegmental dopamine neurons gate associative learning of fear. Nat Neurosci 21:952–962. 10.1038/s41593-018-0174-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gungor NZ, Paré D (2016) Functional heterogeneity in the bed nucleus of the stria terminalis. J Neurosci 36:8038–8049. 10.1523/JNEUROSCI.0856-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley ND, Gaulden AD, Báldi R, Winters ND, Salimando GJ, Rosas-Vidal LE, Jameson A, Winder DG, Patel S (2019) Dynamic remodeling of a basolateral-to-central amygdala glutamatergic circuit across fear states. Nat Neurosci 22:2000–2012. 10.1038/s41593-019-0528-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haufler D, Nagy FZ, Pare D (2013) Neuronal correlates of fear conditioning in the bed nucleus of the stria terminalis. Learn Mem 20:633–641. 10.1101/lm.031799.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, Stuber GD (2013) Distinct extended amygdala circuits for divergent motivational states. Nature 496:224–228. 10.1038/nature12041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Adhikari A, Lee SY, Marshel JH, Kim CK, Mallory CS, Lo M, Pak S, Mattis J, Lim BK, Malenka RC, Warden MR, Neve R, Tye KM, Deisseroth K (2013) Diverging neural pathways assemble a behavioural state from separable features in anxiety. Nature 496:219–223. 10.1038/nature12018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WB, Cho JH (2017) Encoding of discriminative fear memory by input-specific LTP in the amygdala. Neuron 95:1129–1146.e5. 10.1016/j.neuron.2017.08.004 [DOI] [PubMed] [Google Scholar]
- Klumpers F, Kroes MCW, Baas JMP, Fernández G (2017) How human amygdala and bed nucleus of the stria terminalis may drive distinct defensive responses. J Neurosci 37:9645–9656. 10.1523/JNEUROSCI.3830-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodani S, Soya S, Sakurai T (2017) Excitation of GABAergic neurons in the bed nucleus of the stria terminalis triggers immediate transition from non-rapid eye movement sleep to wakefulness in mice. J Neurosci 37:7164–7176. 10.1523/JNEUROSCI.0245-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange MD, Daldrup T, Remmers F, Szkudlarek HJ, Lesting J, Guggenhuber S, Ruehle S, Jüngling K, Seidenbecher T, Lutz B, Pape HC (2017) Cannabinoid CB1 receptors in distinct circuits of the extended amygdala determine fear responsiveness to unpredictable threat. Mol Psychiatry 22:1422–1430. 10.1038/mp.2016.156 [DOI] [PubMed] [Google Scholar]
- Li B (2019) Central amygdala cells for learning and expressing aversive emotional memories. Curr Opin Behav Sci 26:40–45. 10.1016/j.cobeha.2018.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Penzo MA, Taniguchi H, Kopec CD, Huang ZJ, Li B (2013) Experience-dependent modification of a central amygdala fear circuit. Nat Neurosci 16:332–339. 10.1038/nn.3322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingg RT, Johnson SB, Emmons EB, Anderson RM, Romig-Martin SA, Narayanan NS, McGaugh JL, LaLumiere RT, Radley JJ (2020) Bed nuclei of the stria terminalis modulate memory consolidation via glucocorticoid-dependent and -independent circuits. Proc Natl Acad Sci USA 117:8104–8114. 10.1073/pnas.1915501117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewcz CA, Mazzone CM, D'Agostino G, Halladay LR, Hardaway JA, DiBerto JF, Navarro M, Burnham N, Cristiano C, Dorrier CE, Tipton GJ, Ramakrishnan C, Kozicz T, Deisseroth K, Thiele TE, McElligott ZA, Holmes A, Heisler LK, Kash TL (2016) Serotonin engages an anxiety and fear-promoting circuit in the extended amygdala. Nature 537:97–101. 10.1038/nature19318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzone CM, Pati D, Michaelides M, DiBerto J, Fox JH, Tipton G, Anderson C, Duffy K, McKlveen JM, Hardaway JA, Magness ST, Falls WA, Hammack SE, McElligott ZA, Hurd YL, Kash TL (2018) Acute engagement of G(q)-mediated signaling in the bed nucleus of the stria terminalis induces anxiety-like behavior. Mol Psychiatry 23:143–153. 10.1038/mp.2016.218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naaz F, Knight LK, Depue BE (2019) Explicit and ambiguous threat processing: functionally dissociable roles of the amygdala and bed nucleus of the stria terminalis. J Cogn Neurosci 31:543–559. 10.1162/jocn_a_01369 [DOI] [PubMed] [Google Scholar]
- Nguyen AQ, Dela Cruz JA, Sun Y, Holmes TC, Xu X (2016) Genetic cell targeting uncovers specific neuronal types and distinct subregions in the bed nucleus of the stria terminalis. J Comp Neurol 524:2379–2399. 10.1002/cne.23954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Ycu EA, Kumar A, Yeh LF, Ahmed T, Koivumaa J, Johansen JP (2017) A feedback neural circuit for calibrating aversive memory strength. Nat Neurosci 20:90–97. 10.1038/nn.4439 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, Ed 2. San Diego: Academic Press. [Google Scholar]
- Pelrine E, Pasik SD, Bayat L, Goldschmiedt D, Bauer EP (2016) 5-HT2C receptors in the BNST are necessary for the enhancement of fear learning by selective serotonin reuptake inhibitors. Neurobiol Learn Mem 136:189–195. 10.1016/j.nlm.2016.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo MA, Robert V, Li B (2014) Fear conditioning potentiates synaptic transmission onto long-range projection neurons in the lateral subdivision of central amygdala. J Neurosci 34:2432–2437. 10.1523/JNEUROSCI.4166-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo MA, Robert V, Tucciarone J, De Bundel D, Wang M, Van Aelst L, Darvas M, Parada LF, Palmiter RD, He M, Huang ZJ, Li B (2015) The paraventricular thalamus controls a central amygdala fear circuit. Nature 519:455–459. 10.1038/nature13978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomrenze MB, Tovar-Diaz J, Blasio A, Maiya R, Giovanetti SM, Lei K, Morikawa H, Hopf FW, Messing RO (2019) A corticotropin releasing factor network in the extended amygdala for anxiety. J Neurosci 39:1030–1043. 10.1523/JNEUROSCI.2143-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulos AM, Ponnusamy R, Dong HW, Fanselow MS (2010) Compensation in the neural circuitry of fear conditioning awakens learning circuits in the bed nuclei of the stria terminalis. Proc Natl Acad Sci USA 107:14881–14886. 10.1073/pnas.1005754107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radley JJ, Sawchenko PE (2011) A common substrate for prefrontal and hippocampal inhibition of the neuroendocrine stress response. J Neurosci 31:9683–9695. 10.1523/JNEUROSCI.6040-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regev L, Neufeld-Cohen A, Tsoory M, Kuperman Y, Getselter D, Gil S, Chen A (2011) Prolonged and site-specific over-expression of corticotropin-releasing factor reveals differential roles for extended amygdala nuclei in emotional regulation. Mol Psychiatry 16:714–728. 10.1038/mp.2010.64 [DOI] [PubMed] [Google Scholar]
- Roth BL (2016) DREADDs for neuroscientists. Neuron 89:683–694. 10.1016/j.neuron.2016.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanford CA, Soden ME, Baird MA, Miller SM, Schulkin J, Palmiter RD, Clark M, Zweifel LS (2017) A central amygdala CRF circuit facilitates learning about weak threats. Neuron 93:164–178. 10.1016/j.neuron.2016.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta A, Holmes A (2019) A discrete dorsal raphe to basal amygdala 5-HT circuit calibrates aversive memory. Neuron 103:489–505.e7. 10.1016/j.neuron.2019.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shackman AJ, Fox AS (2016) Contributions of the central extended amygdala to fear and anxiety. J Neurosci 36:8050–8063. 10.1523/JNEUROSCI.0982-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ (2015) Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol Ther 149:150–190. 10.1016/j.pharmthera.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan GM, Apergis J, Bush DE, Johnson LR, Hou M, Ledoux JE (2004) Lesions in the bed nucleus of the stria terminalis disrupt corticosterone and freezing responses elicited by a contextual but not by a specific cue-conditioned fear stimulus. Neuroscience 128:7–14. 10.1016/j.neuroscience.2004.06.015 [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Kvitsani D, Fu Y, Lu J, Lin Y, Miyoshi G, Shima Y, Fishell G, Nelson SB, Huang ZJ (2011) A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71:995–1013. 10.1016/j.neuron.2011.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S,Jr, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154. 10.1016/j.neuron.2011.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KM, McDannald MA (2019) Ventrolateral periaqueductal gray neurons prioritize threat probability over fear output. Elife 8:e45013. 10.7554/eLife.45013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KM, Jhou TC, Pimpinelli D, McDannald MA (2019) Cue-inhibited ventrolateral periaqueductal gray neurons signal fear output and threat probability in male rats. Elife 8:e50054. 10.7554/eLife.50054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Veinante P (2019) Cell-type specific parallel circuits in the bed nucleus of the stria terminalis and the central nucleus of the amygdala of the mouse. Brain Struct Funct 224:1067–1095. 10.1007/s00429-018-01825-1 [DOI] [PubMed] [Google Scholar]
- Yu K, Garcia da Silva P, Albeanu DF, Li B (2016) Central amygdala somatostatin neurons gate passive and active defensive behaviors. J Neurosci 36:6488–6496. 10.1523/JNEUROSCI.4419-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]