

Abstract
Cocaine experience generates AMPA receptor (AMPAR)-silent synapses in the nucleus accumbens (NAc), which are thought to be new synaptic contacts enriched in GluN2B-containing NMDA receptors (NMDARs). After drug withdrawal, some of these synapses mature by recruiting AMPARs, strengthening the newly established synaptic transmission. Silent synapse generation and maturation are two consecutive cellular steps through which NAc circuits are profoundly remodeled to promote cue-induced cocaine seeking after drug withdrawal. However, the basic cellular processes that mediate these two critical steps remains underexplored. Using a combination of electrophysiology, viral-mediated gene transfer, and confocal imaging in male rats as well as knock-in (KI) mice of both sexes, our current study characterized the dynamic roles played by AMPARs and NMDARs in generation and maturation of silent synapses on NAc medium spiny neurons after cocaine self-administration and withdrawal. We report that cocaine-induced generation of silent synapses not only required synaptic insertion of GluN2B-containing NMDARs, but also, counterintuitively, involved insertion of AMPARs, which subsequently internalized, resulting in the AMPAR-silent state on withdrawal day 1. Furthermore, GluN2B NMDARs functioned to maintain these cocaine-generated synapses in the AMPAR-silent state during drug withdrawal, until they were replaced by nonGluN2B NMDARs, a switch that allowed AMPAR recruitment and maturation of silent synapses. These results reveal dynamic interactions between AMPARs and NMDARs during the generation and maturation of silent synapses after cocaine experience and provide a mechanistic basis through which new synaptic contacts and possibly new neural network patterns created by these synapses can be manipulated for therapeutic benefit.
SIGNIFICANCE STATEMENT Studies over the past decade reveal a critical role of AMPA receptor-silent, NMDA receptor-containing synapses in forming cocaine-related memories that drive cocaine relapse. However, it remains incompletely understood how AMPA and NMDA receptors traffic at these synapses during their generation and maturation. The current study characterizes a two-step AMPA receptor trafficking cascade that contributes to the generation of silent synapses in response to cocaine experience, and a two-step NMDA receptor trafficking cascade that contributes to the maturation of these synapses after cocaine withdrawal. These results depict a highly regulated cellular procedure through which nascent glutamatergic synapses are generated in the adult brain after drug experience and provide significant insight into the roles of glutamate receptors in synapse formation and maturation.
Keywords: AMPA receptor, cocaine, NMDA receptor, nucleus accumbens, silent synapses, trafficking
Introduction
Glutamatergic synapses in the nucleus accumbens (NAc) are targeted by cocaine experience to remodel neural circuits, promoting addiction associated emotional and motivational responses (Wolf, 2016; Wright and Dong, 2020). A prominent form of cocaine-induced synaptic changes in the NAc shell (NAcSh) is the generation of AMPA receptor (AMPAR)-silent excitatory synapses (Huang et al., 2009; Brown et al., 2011). These synapses are thought to be generated de novo, and may serve as the initial connections in forming new network connectivity patterns encoding memory traces associated with cocaine (Wang et al., 2020; Wright et al., 2020). After withdrawal from cocaine, some of the cocaine-generated silent synapses mature by recruiting AMPARs, resulting in consolidation of these synapses and enhancement of cue-induced cocaine seeking (Lee et al., 2013; Ma et al., 2014, 2016; Wright et al., 2020). These published results suggest that cocaine-generated NAcSh silent synapses are key neuronal substrates that can be selectively manipulated to reduce cocaine seeking and relapse. However, several key cellular processes mediating the assembly and maturation of NAcSh silent synapses remain unclear, preventing precise manipulation of these cocaine-specific synapses. These processes can be outlined by two basic questions, which are elaborated below.
First, recent results show that cocaine self-administration uses a developmental synaptogenic mechanism to generate NAcSh silent synapses de novo (Wang et al., 2020). To generate such AMPAR-absent synapses, the most parsimonious cellular process would be a selective insertion of GluN2B-containing NMDA receptors (NMDARs) to new synaptic contacts as initially postulated (Huang et al., 2009, 2015; Dong and Nestler, 2014; Dong, 2016). This possibility is supported by observations that the overexpression of GluN2B-containing NMDARs or modified GluN2A with the C terminus of GluN2B is sufficient to generate new spines (Barria and Malinow, 2002, 2005; Gambrill and Barria, 2011), whereas deletion of GluN2B decreases spine densities in the developing hippocampus (Gray et al., 2011). However, other results suggest that AMPARs are also involved in developmental synaptogenesis. Simultaneous time-lapse imaging detects that AMPARs and NMDARs are both present in mobile transport packets moving toward postsynaptic membranes during developmental synaptogenesis in hippocampal cultures (Washbourne et al., 2002), and both AMPARs and NMDARs displayed similar clustering kinetics at postsynaptic membranes (Friedman et al., 2000). Furthermore, both AMPAR- and NMDAR-mediated postsynaptic currents are detected during early synaptogenesis periods, suggesting the existence of functional AMPARs at newborn synapses (Diabira et al., 1999; Verhage et al., 2000; Groc et al., 2002). Overexpression of GluA2 AMPAR subunits is sufficient to induce large-scale spinogenesis in hippocampal neuronal cultures, suggesting that AMPARs actively promote synaptogenesis (Passafaro et al., 2003). These results raise a possibility that, during development, nascent excitatory synapses are generated initially with both AMPARs and NMDARs, but AMPARs are highly labile and prone to be internalized on moderate synaptic activities, resulting in AMPAR-silent synapses shortly after synaptogenesis (Xiao et al., 2004; Hanse et al., 2009, 2013). Occurring in the adult brain with different synaptogenic cellular conditions, an outstanding question is what cellular routes are used for cocaine-induced generation of silent synapses in NAcSh.
Second, a well documented maturation route for developmental silent synapses is through long-term synaptic potentiation (LTP), by which AMPARs are recruited and stabilized in an experience-dependent manner in the hippocampus (Isaac et al., 1995; Liao et al., 1995; Durand et al., 1996). Although this process requires abrupt activation of NMDARs and NMDAR-coupled CaMKII signaling (Wu et al., 1996; Paoletti et al., 2013), basal activities of GluN2B-containing NMDARs may indeed provide a sustained suppression of AMPAR insertion thus maintaining the silent state of these synapses (Adesnik et al., 2008; Gray et al., 2011). Furthermore, in contrast to its role in synaptogenesis (Passafaro et al., 2003), homomeric GluA2 AMPARs are not involved in silent synapse maturation (Shi et al., 2001). Cocaine-generated silent synapses are initially enriched in GluN2B NMDARs, which are subsequently replaced by nonGluN2B (likely GluN2A) NMDARs after prolonged cocaine withdrawal (Huang et al., 2009; Brown et al., 2011; Wright et al., 2020). In parallel, a portion of these silent synapses mature after cocaine withdrawal by recruiting GluA2-lacking, calcium-permeable (CP) AMPARs, a type of AMPAR that is not typically expressed at high levels in NAcSh medium spiny neurons (MSNs; Lee et al., 2013; Ma et al., 2014, 2016; Shukla et al., 2017; Wright et al., 2020). A critical question is whether GluN2B NMDARs play a pivotal role in regulating AMPAR trafficking as well as the functional state of cocaine-generated synapses in these neurons. By addressing these questions, our current study characterized the basic cellular processes mediating the generation and maturation of silent synapses after cocaine self-administration.
Materials and Methods
Subjects
Male Sprague Dawley rats (age, postnatal day 35–40; body weight on arrival, 160–170 g; Charles River), as well as female and male wild-type and GluN2B knock-in mice (weight, ∼25 g; age, 9–16 weeks) were used. Animals were singly housed on a 12 h light/dark cycle (light on/off at 7:00 A.M./7:00 P.M.) with food and water available ad libitum. Rats were allowed to habituate to their housing cages for ∼1 week before catheter or intracranial surgeries. All animals were used in accordance with protocols approved by the Institutional Care and Use Committees at the University of Pittsburgh, the University Medical Center Göttingen, and the Icahn School of Medicine at Mount Sinai.
Catheter implantation
Self-administration surgery was performed as described previously (Lee et al., 2013; Neumann et al., 2016; Baldwin and Eroglu, 2017). Briefly, animals were anesthetized with a xylazine–ketamine mixture (5–10/50–100 mg/kg, i.p.). A silicone catheter constructed from SILASTIC tubing [for rats: length, ∼10 cm; inner diameter (i.d.), 0.02 inch; outer diameter (o.d.), 0.037 inch; for mice: length, ∼5 cm; i.d., 0.012 inch; o.d., 0.025 inch] was inserted into the jugular vein and passed subcutaneously to the midscapular region and connected to a vascular access button (Instech Laboratories) subcutaneously implanted under the skin. Animals were allowed to recover for 5–7 d after surgery. For rats, catheters were flushed with sterile saline containing gentamycin (5 mg/ml) and heparin (10 U/ml) every 24 h during the recovery and training periods. For mice, catheters were flushed with sterile saline containing cefazolin (3 mg/ml) and heparin (30 U/ml) every 24 h during recovery and training periods.
Self-administration apparatus
Behavioral experiments were conducted in operant-conditioning chambers enclosed within sound-attenuating cabinets (Med Associates). For rats, each chamber contains an active and an inactive nose poke hole, a food dispenser, a conditioned stimulus light in each nose poke hole, and a house light. For mice, each chamber contains an active and an inactive lever, a food dispenser, a conditioned stimulus light in each lever press hole and a house light. No food or water was provided in the chamber during training sessions.
Cocaine self-administration training began ∼5 d after surgery. On day 1, animals were placed in self-administration chambers for an overnight training session on a fixed ratio 1 reinforcement schedule. Nose poking in the active hole or pressing active lever resulted in a cocaine infusion (0.75 mg/kg over 3–6 s) and illumination of the conditioned stimulus light inside the active hole and switch off of the house light simultaneously. The conditioned stimulus light remained on for 20 s, whereas the house light remained off for 20 s, during which active nose pokes or lever presses were counted but resulted in no cocaine infusions. After the 20 s timeout period, the house light was turned on, and the next “active” nose poke or lever press resulted in a cocaine infusion. Nose pokes in the inactive hole or presses on the inactive lever had no reinforced consequences but were recorded. During this first-day overnight training, only animals that received at least 50 cocaine infusions were allowed into the subsequent 5 d cocaine self-administration regimen (<2% of animals failed to meet this criterion). On the fifth day of training, animals were placed back into their home cages and remained there during subsequent withdrawal days. Cocaine-trained animals that failed to meet self-administration criteria (≥15 infusions/session) were excluded from further experimentation and analysis.
GluA23Y peptide administration
The peptide GluA23Y was used to interfere with experience-dependent endocytosis of GluA2-containing AMPARs. GluA2scr, an inactive form of the 3Y peptide, was used as control. These peptides were conjugated with a trans-activating transcriptional activator (TAT) sequence (YGRKKRRQRRR) to facilitate intracellular delivery, resulting in the TAT-GluA23Y (YGRKKRRQRRR-YKEGYNVYG) or the TAT-GluA2scr scrambled (YGRKKRRQRRR-VYKYGGYNE) peptide. Peptides were dissolved in double-distilled H2O and administered intravenously (1.5 nmol/g) at the time points specified in the main text.
Viral vectors
Recombinant adeno-associated virus (AAV) vectors were used for in vivo viral-mediated gene expression. Briefly, the AAV2 genomic back bone expressing Venus tagged with the intended gene was pseudotyped with AAV1/2 or 8 capsid proteins. HEK293T cells were cotransfected with the recombinant AAV2 plasmid, the adenoviral helper plasmid F6, and the rep2/capsid composite plasmid pDP8rs (Sonntag et al., 2011; Löw et al., 2013), using linear polyethylenimine-assisted transfection (Huang et al., 2011; Suska et al., 2013). Cultures grown in DMEM (Biochrom) with 10% substituted FBS (catalog #S0115, Biochrom) were harvested from 15 × 15 cm dishes after 48 h. AAVs were harvested and purified as described previously (Huang et al., 2011; Suska et al., 2013). The GluN2B-RNAi (bu2Bd: GCTGGTGATAATCCTTCTGAA) was inserted into the miR30 backbone in the 3′ UTR of the expression cassette (Fellmann et al., 2013), driven from a CAG promoter in a recombinant AAV2 vector pseudotyped with AAV8 capsids, as previously described (Suska et al., 2013; Cahill et al., 2018). A small interfering peptide (siP) RRPPRSPDHKRYFRDKE compromises the Cdk5–GluN2B interaction motif (Plattner et al., 2014) was fused C terminally to enhanced yellow fluorescent protein (eYFP) with the linker peptide GGGS, and its expression was driven from a human Synapsin I promoter in an AAV2, which was pseudotyped with AAV1/2 capsids.
Intracranial viral infusion
We stereotaxically delivered viral vectors to specific brain regions. Briefly, rats were anesthetized with a xylazine/ketamine mixture (5–10/50–100 mg/kg, i.p.). Stainless steel needles (28 gauge) were implanted bilaterally into the NAcSh (anteroposterior, +1.60 mm; mediolateral, ±0.95 mm; dorsoventral, −7.40 mm). Concentrated viral solutions (0.5 µl/side) were infused through a pump at a flow rate of 0.1 µl/min. The infusion needle was left in place for at least 5 min to minimize backflow and then was slowly withdrawn. The rats were placed on a heating pad for ∼1 h for postsurgical recovery before being transferred to the home cages. Electrophysiological analyses were conducted 4 weeks (withdrawal day 0/1) or 5 weeks (withdrawal day 7) after viral injection.
Preparation of acute brain slices
Rats were decapitated following isoflurane anesthesia. Coronal slices (250–300 μm thick) containing the NAc were prepared on a vibratome (catalog #VT1200S, Leica) in 4°C cutting solution containing the following (in mm): 135 N-methyl-d-glutamine, 1 KCl, 1.2 KH2PO4, 0.5 CaCl2, 1.5 MgCl2, 20 choline-HCO3, and 11 glucose, saturated with 95%O2/5%CO2 and pH adjusted to 7.4 with HCl. Osmolality was adjusted to 305 mmol/kg. Slices were incubated in the artificial CSF (aCSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, with the osmolality adjusted to 280–290 mmol/kg. Slices were placed in the aCSF saturated with 95% O2/5% CO2 at 37°C for 30 min and then held at 20–22°C for at least 30 min before experimentation.
Electrophysiological recordings
All recordings were made from MSNs located in the medial-ventral NAcSh. During recordings, slices were perfused with aCSF containing 100 μm picrotoxin (catalog #P1675, Sigma). The perfusion aCSF was heated to 31–33°C by passing the solution through a feedback-controlled inline heater (Warner) before entering the chamber. In all EPSC recordings, electrodes (2–5 MΩ) were filled with a cesium-based internal solution containing the following (in mm): 140 CsMeSO3, 5 TEA-Cl, 0.4 EGTA (Cs), 20 HEPES, 2.5 Mg-ATP, 0.25 Na-GTP, and 1 QX-314 (Br), with pH adjusted to 7.2–7.3. To evoke EPSCs, presynaptic afferents were stimulated by a constant-current isolated stimulator (Digitimer), using a monopolar electrode (glass pipette filled with aCSF). Series resistance was 7–20 MΩ, uncompensated and was monitored continuously during recording. Cells with a change in series resistance >20% were excluded for subsequent data collection and analysis. Synaptic currents were recorded with a MultiClamp 700B amplifier, filtered at 3–10 kHz, amplified five times, and then digitized at 20 kHz. To measure NMDAR-mediated EPSCs, 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX; 5–10 μm) and picrotoxin (100 μm) were included in the bath. Recorded neurons were held at +40 mV to relieve Mg2+-mediated blockage of NMDARs. A total of 25–35 consecutive responses were recorded and then averaged to a single EPCS with relatively smooth inactivation slope. The decay kinetics of NMDAR EPSCs were assessed by the time for the EPSCs declined from the peak amplitude to one-half of the peak amplitude. We previously verified that, under our experimental conditions, this approximation strategy introduced fewer technical concerns compared with the weighted time constant strategy for NMDA EPSCs in the NAcSh (Huang et al., 2009).
The minimal stimulation assay was performed as previously described (Liao et al., 1995; Marie et al., 2005; Huang et al., 2009). Briefly, after obtaining small (∼50 pA) EPSCs at −70 mV, the stimulation intensity was reduced in small decrements to the point that failures versus successes of EPSCs could be clearly distinguished. The stimulation intensity and frequency (0.1−0.2 Hz) were then kept constant for the rest of the experiment except for the experiments in which LTD induction protocol was used to manipulate labile AMPARs. The amplitudes of both AMPAR and NMDAR ESPCs resulting from single vesicular release are relatively large in NAcSh MSNs (e.g., ∼15 pA for AMPAR mEPSCs at −70 mV), which facilitates the judgment of successes versus failures. For each cell, 50–100 traces were recorded at −70 mV, and 50–100 traces were recorded at +40 to +50 mV. Recordings were then repeated at −70 mV and +40 to +50 mV for another round or two. Only cells with relatively constant failure rates (changes, <15%) between rounds were used to assess the percentage of silent synapses. We visually detected failures versus successes at each holding potential over 50–100 trials to calculate the failure rate, as described previously (Liao et al., 1995; Marie et al., 2005; Huang et al., 2009). We performed this analysis in a blind manner such that a small number of ambiguous responses were categorized in a fully unbiased way.
To assess the percentage of silent synapses, we made the following two theoretical assumptions: (1) the presynaptic release sites were independent; and (2) the release probability across all synapses, including silent synapses, was identical. Thus, the percentage of silent synapses was calculated using the following equation: 1 – ln(F–70)/ln(F+40), in which F–70 was the failure rate at −70 mV and F+40 was the failure rate at +40 mV, as rationalized previously (Liao et al., 1995; Ma et al., 2014). Note that in this equation, the failure rate was the only variable that determined the percentage of silent synapses. The amplitudes of EPSCs were used to present failures or successes, but otherwise did not have analytical value. It is possible that the aforementioned theoretical assumptions were not always true. Nevertheless, the above equation was still used, as it is valid in predicting the changes of silent synapses qualitatively as previously rationalized (Ma et al., 2014). The amplitude of an EPSC was determined as the mean value of the EPSC over a 1 ms time window around the peak, which was typically 3–4 ms after the stimulation artifact. At +40 mV, successful synaptic responses are conceivably mediated by both AMPARs and NMDARs. Indeed, inhibiting AMPARs by NBQX (5 μm) modestly reduces the amplitudes of EPSCs (Graziane et al., 2016). Despite the effects of NBQX on the amplitudes, the failure rate of synaptic responses at +40 mV is not altered during AMPAR inhibition (Graziane et al., 2016). Thus, in the minimal stimulation assay assessing the percentage of silent synapses, the results are thought to not be affected by whether synaptic responses of +40 mV are mediated by NMDARs alone or by both AMPARs and NMDARs.
Dendritic spine labeling and imaging
To visualize dendritic spines, MSNs were filled with Alexa Fluor 594 using single-cell microinjections, and the labeled dendritic spines were quantified as described previously (Graziane et al., 2016; Wang et al., 2020; Wright et al., 2020). Briefly, the rats were transcardially perfused with ice-cold 1% PFA in 0.1 m phosphate buffer, followed by 4% PFA and 0.125% glutaraldehyde in 0.1 m phosphate buffer. Brains were removed and postfixed in 4% PFA and 0.125% glutaraldehyde in 0.1 m phosphate buffer for 12–14 h at 4°C. Following postfixing, the brains were transferred into 0.01 m PBS and sectioned into 250-µm-thick slices using a vibratome (model #VT1200S, Leica). Cells were filled shortly after slicing (within 4 h). Cells within the NAcSh were impaled with a fine micropipette containing 5 mm Invitrogen Alexa Fluor 594 hydrazide (Thermo Fisher Scientific) and injected with 1–10 nA of negative current until dendrites and spines were filled. After filling, the slices were mounted in Invitrogen ProLong Gold Antifade (Thermo Fisher Scientific) on slides for imaging. Spacers (240 µm thick) were placed along the edge of the slide before mounting to avoid compression of the slices by the overlying coverslip and to prevent deformation of spine morphologies.
Images were captured with a Leica TCS SP5 confocal microscope equipped with the Leica Application Suite software. Individually filled neurons were visualized with a 63× oil-immersion objective for final verification of their neuronal types. Individual dendritic segments were focused on and scanned at 0.3 µm intervals along the z-axis to obtain a z-stack. After capture, all images were deconvolved within the Leica Application Suite software. Analyses were performed on two-dimensional projection images using ImageJ (NIH). Secondary dendrites were sampled and analyzed for spine densities because of their significant cellular and behavioral correlates (Wright et al., 2020; Xia et al., 2020). For each neuron, one to four dendritic segments ∼20 µm in length were analyzed. For each group, 2–10 cells/rat were analyzed. Similar to our previous studies (Graziane et al., 2016; Wang et al., 2020; Wright et al., 2020), we operationally divided spines into the following three categories: (1) mushroom-like spines were dendritic protrusions with a head diameter of >0.5 µm or more than two times the spine neck diameter; (2) stubby spines were dendritic protrusions with no discernable heads and a length of ≤0.5 µm; and (3) thin/filopodia-like spines were dendritic protrusions with a length of >0.5 µm and head diameters of <0.5 µm or no discernable heads.
Western analysis of GluN2B knockdown
Primary neuron cultures were prepared from mice at postnatal day 0 and maintained on poly-d-lysine-coated 24-well plates with Neurobasal medium (Krüger et al., 2013). On day 3 in vitro, the cultures were transduced with the AAV vector expressing the shRNA against GluN2B. Ten days after transduction, the cultures were harvested and lysed with SDS-PAGE sample buffer (Krüger et al., 2013). Samples were separated by SDS-PAGE and transferred onto nitrocellulose membranes. GluN2B expression was probed with anti-GluN2B/NR2B glutamate receptor (clone N59/20, NeuroMab) and IRDye 800CW Goat anti-Mouse IgG Secondary Antibody (LI-COR). Mortalin (clone N52A/9, NeuroMab) expression was used as the equal loading control. The bands were visualized on a LI-COR Odyssey imaging system and quantified using Image Studio software. Signals of GluN2B from transduced samples were normalized to the GluN2B expression of nontransduced neurons from the same culture dish.
Drugs and reagents
Cocaine HCl (provided by the National Institute on Drug Abuse Drug Supply Program) was dissolved in 0.9% NaCl saline. Ketamine and xylazine were mixed for anesthesia (purchased from a Drug Enforcement Agency-designated vendor at the University of Pittsburgh). All chemicals were purchased from Sigma-Aldrich, Tocris Bioscience, J.T.Baker, Fluka, or Alomone Labs.
Data acquisition and statistics
All results are shown as the mean ± SEM. All experiments were replicated in 3–14 animals. All data collection was randomized. Specifically, animals were randomly assigned for saline versus cocaine groups. NAcSh MSNs were randomly chosen for electrophysiological recording. Secondary dendrites of MSNs were randomly chosen for spine analysis. All data were analyzed offline, and investigators were blinded to experimental conditions during the analyses.
A total of 411 rats and 96 mice were used for this study, among which 137 rats and 33 mice were excluded from the final data analysis and interpretation because of the following reasons: (1) 116 rats and 20 mice were excluded because of health issues after surgery (e.g., >20% drop in body weight in a day), failed catheterization, or failure to reach the self-administration criteria; (2) 5 rats were excluded because of off-target stereotaxic injections or poor viral expression; and (3) 16 rats and 13 mice were excluded because of experimental failures (e.g., unsuccessful slice preparations, failed recordings, or other experimental incidents). No animals were excluded after data acquisition was accomplished.
Repeated experiments for the same group were pooled together for statistical analysis. Sample sizes were based on our previous studies performing similar experiments. For electrophysiology and dendritic spine experiments, the sample sizes are presented as n/m, where n is the number of cells and m is the number of animals. Cell-based statistics were used for the analysis of all electrophysiology data. For results of dendritic spines, we averaged the spine densities of individual dendritic segments from each cell to obtain a cell or animal-based value for statistical analysis. Statistical significance was assessed using two-tailed unpaired t tests, one-way or two-way ANOVA followed by Bonferroni post-test, as specified in the related text. Statistical significance was set at p < 0.05 for all experiments. Statistical analyses were performed in GraphPad Prism (version 7).
Results
Role of GluN2B NMDARs in silent synapse generation
Based on the effects of cocaine on synaptic GluN2B-containing NMDARs and dendritic spines of NAcSh MSNs, a straightforward cellular model was hypothesized for cocaine-induced generation of silent synapses, namely, these synapses are generated by insertion of GluN2B NMDARs to new synaptic locations (Huang et al., 2009; Dong and Nestler, 2014). We tested this model and explored alternatives by examining GluN2B, dendritic spines, and silent synapses in NAcSh MSNs, all with the same cocaine procedure (Lee et al., 2013; Neumann et al., 2016; Wright et al., 2020). Briefly, we trained rats (age, 12 weeks) to self-administer cocaine, initially with an overnight (10–12 h) session, and then with a 5 d procedure comprising a 2 h daily self-administration session in the operant boxes and 22 h daily drug abstinence/withdrawal in the home cages (Fig. 1A,O–R). Saline-trained rats were used as controls. Rats exhibited a clear acquisition of cue-conditioned nose poke responding to cocaine and stable consumption of cocaine through this procedure.
Figure 1.
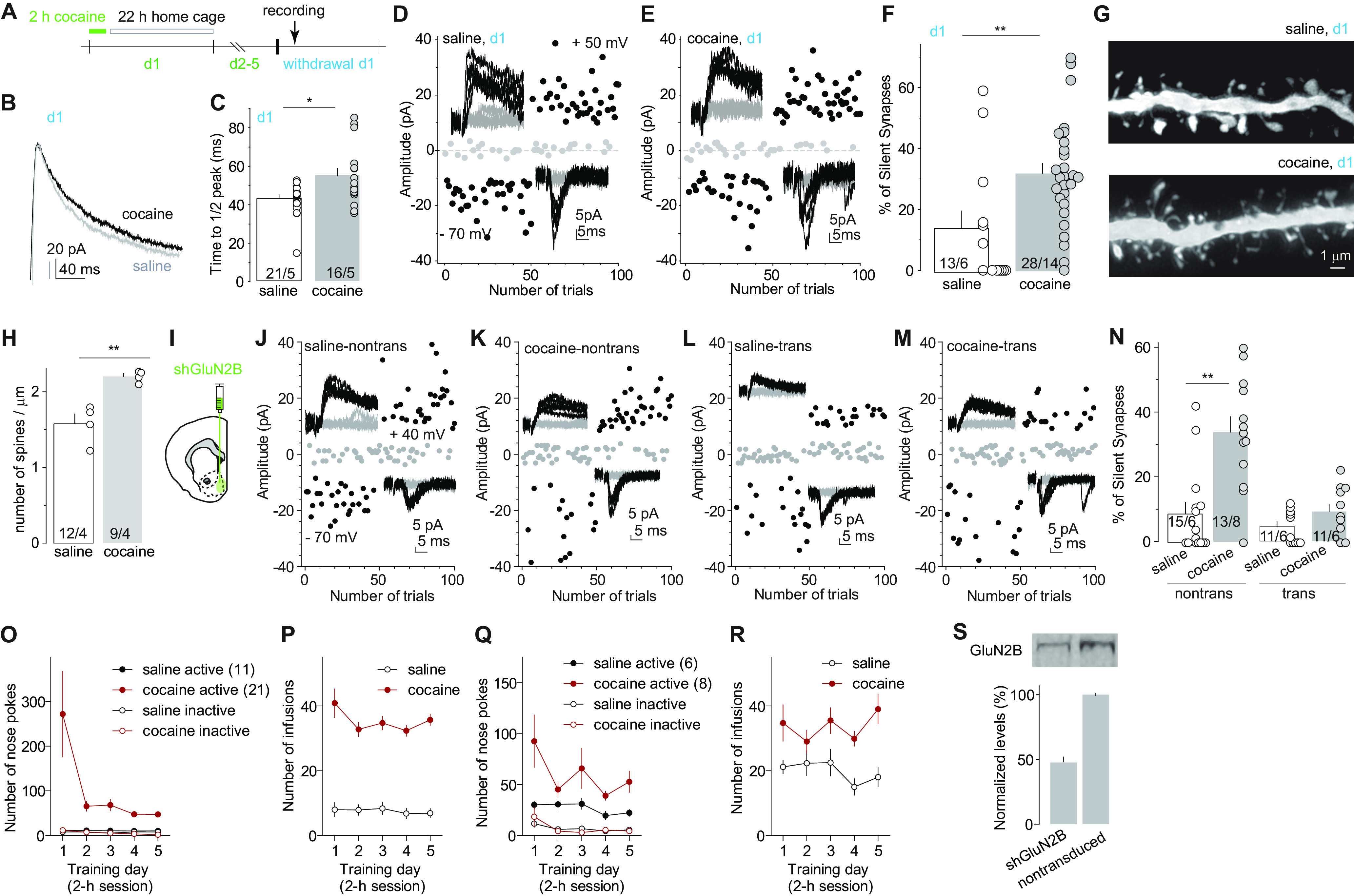
GluN2B NMDARs are essential for cocaine-induced generation of silent synapses. A, Diagram showing experimental timeline. B, C, Example traces (B) and summary (C) showing that on withdrawal day 1, the decay kinetics of NMDAR EPSCs in NAcSh MSNs from cocaine-trained rats were significantly slower compared with saline-trained rats. D, E, Example EPSCs evoked at −70 or +50 mV (insets) over 100 consecutive trials in the minimum stimulation assay from saline-trained (D) or cocaine-trained (E) rats 24 h after the last self-administration session. F, Summary showing that the percentage of silent synapses in NAcSh MSNs was increased on withdrawal day 1 after cocaine self-administration. G, Example dendrites of NAcSh MSNs from saline- and cocaine-trained rats on withdrawal day 1. H, Summary showing that the density of total dendritic spines was increased after 1 d withdrawal from cocaine. I, Diagram showing the injection of shGluN2B-expressing AAV into the NAcSh. J–M, Example EPSCs evoked at −70 or +40 mV (insets) over 100 consecutive trials in the minimum stimulation assay of transduced and nontransduced NAcSh MSNs in saline- or cocaine-trained rats after 1 d withdrawal. N, Summary showing that on withdrawal day 1, NAcSh MSNs with expression of shGluN2B in cocaine-trained rats exhibited a low percentage of silent synapses, comparable to that in saline-trained rats. O, P, Self-administration results of rats whose electrophysiological and morphologic results are presented in B–H. Q, R, Self-administration results of rats whose electrophysiological results are presented in J–N. S, Western assays verifying the efficacy of shGluN2B-expressing AAV in reducing protein levels of GluN2B in neuronal cultures. *p < 0.05; **p < 0.01; ns, no significance.
A signature property of cocaine-generated silent synapses is the enrichment of GluN2B-containing NMDARs, which can be detected by prolongation of the decay kinetics of NMDAR-mediated EPSCs (Huang et al., 2009; Brown et al., 2011; Dong and Nestler, 2014; Graziane et al., 2016; Wright et al., 2020). Twenty-four hours after the last training session (defined as withdrawal day 1), we observed a similar prolongation of the decay kinetics of NMDAR EPSCs in NAcSh MSNs, assessed by the elapsed time for the EPSC from the peak amplitude to one-half of the peak amplitude (Huang et al., 2009; Brown et al., 2011; Fig. 1B,C; see Materials and Methods for isolation of NMDAR EPSCs; saline: 43.4 ± 1.8 ms, n/m = 21/5; cocaine: 54.9 ± 3.9 ms, n/m = 16/5, t35 = 2.9, p < 0.01, two-tailed t test). In parallel, we performed the minimal stimulation assay, an established biophysical procedure assessing levels of silent synapses (Isaac et al., 1995; Liao et al., 1995; Marie et al., 2005; Lee et al., 2013; Ma et al., 2014; 2016; also see Materials and Methods). We observed an increased percentage of silent synapses in cocaine-trained rats compared with saline-trained rats (Fig. 1D–F; saline, 13.8 ± 5.7%, n/m =13/6; cocaine: 31.9 ± 3.3%, n/m = 28/14; t(39) = 3.0, p < 0.01, two-tailed t test). Using iontophoresis of Alexa Fluor 594 into fixed MSNs, we imaged spines on the secondary dendrites through confocal microscopy. The overall density of dendritic spines was higher in cocaine-trained than in saline-trained rats (Fig. 1G,H; saline: 1.6 ± 0.1, n/m = 12/4; cocaine: 2.2 ± 0.1, n/m = 9/4, t(19) = 4.8; p < 0.01, two-tailed t test), and this increase was primarily attributable to the increase in relatively immature, thin spines (saline: 0.50 ± 0.05, m/n = 12/4; cocaine: 1.19 ± 0.08, m/n = 9/4; t(19) = 7.9, p < 0.01) rather than mushroom-like spines (saline: 0.61 ± 0.05, m/n = 12/4; cocaine: 0.55 ± 0.06, m/n = 9/4; t(19) = 0.77, p = 0.45). These results correlate synaptic enrichment of GluN2B NMDARs to cocaine-induced spinogenesis and generation of silent synapses under the same experimental condition.
The above correlative alterations prompted us to test the role of GluN2B in cocaine-induced generation of silent synapses. We bilaterally injected the rats with an AAV8 vector that expresses an shRNA against GluN2B into the NAcSh, a previously established approach that decreases protein levels of GluN2B in neuronal cultures (control, 1.00 ± 0.01; shRNA, 0.49 ± 0.06; n = 4; Fig. 1I,S) and reduces synaptic transmission mediated by GluN2B NMDARs (Wang et al., 2018). For this and other electrophysiological experiments, we injected small volumes (∼0.5 µl) of viral solution such that a relatively small number of neurons were transduced, minimizing the potential behavioral consequences of this manipulation. After 4 weeks of viral expression, these rats were trained with the same cocaine procedure described above, which exhibited similar acquisition of operant responding and cocaine consumption (Fig. 1O–R). On withdrawal day 1, nontransduced NAcSh MSNs in cocaine-trained rats exhibited an increased percentage of silent synapses compared with saline-trained rats, but, in the same rats, this effect of cocaine on silent synapse generation was abolished in transduced MSNs in which GluN2B was knocked down (Fig. 1J–N; saline-nontransduced: 8.7 ± 3.4%, n/m = 15/6; cocaine-nontransduced: 33.8 ± 4.7%, n/m = 13/8; saline-transduced: 4.6 ± 1.5%, n/m = 11/6; cocaine-transduced: 9.4 ± 2.4%, n/m = 11/6 animals; F(1,46) = 8.6; p < 0.01, two-way ANOVA; p < 0.01, saline nontransduced vs cocaine nontransduced; p = 0.73, cocaine transduced vs saline transduced; p < 0.01, cocaine transduced vs cocaine nontransduced; Bonferroni post-test). These results indicate an essential role of GluN2B NMDARs in the generation of silent synapses after cocaine.
Role of AMPARs in silent synapse generation
Generation of silent synapses in the NAcSh requires multiple days of exposure to cocaine (Huang et al., 2009). In our current procedure, animals were trained daily for 5 d with cocaine self-administration over a 2 h session, followed by a 22 h withdrawal period (Figs. 1A, 2A), leading to a possibility that the cocaine versus withdrawal experience on each training day plays differential roles in the generation of silent synapses. To test this possibility, we tailored the procedure such that the rats were sampled right after (within 1 h; operationally defined as withdrawal day 0) the last self-administration session, thus, without the 22 h withdrawal on training day 5 (Fig. 2A,R,S). At this time point, NMDAR EPSCs in NAcSh MSNs exhibited prolonged decay kinetics in cocaine-trained rats compared with saline-trained rats, suggesting that GluN2B-enriched synapses had been generated (Fig. 2B,C; saline: 36.2 ± 2.4 ms, n/m = 19/4; cocaine: 45.3 ± 1.6, n/m = 17/3; t(34) = 3.1, p < 0.01, two-tailed t test). However, the percentage of silent synapses in NAcSh MSNs was comparable between cocaine-trained and saline-trained rats (Fig. 2D–F; saline: 14.4 ± 3.9%, n/m = 9/4; cocaine: 14.6 ± 4.6%, n/m = 8/3; t(15) = 0.05, p = 0.96, two-tailed t test). This lack of increase in silent synapses was not likely because of a lack of cocaine-generated new synapses, because, at this time point, the total density of dendritic spines was higher than the basal level in age-matched rats without cocaine experience (Fig. 2G,H; control: 1.6 ± 0.1, n/m = 54/15; cocaine: 2.0 ± 0.1, n/m = 34/8; t(86) = 5.4, p < 0.01). These results led to a working hypothesis that new, GluN2B-enriched synapses were generated and accumulated at this time point, but they also contained AMPARs as well and were thus not detected as silent synapses; however, AMPARs at these synapses were not stable, and internalized during the subsequent withdrawal period, resulting in a high percentage of silent synapses on withdrawal day 1 (Fig. 2I).
Figure 2.
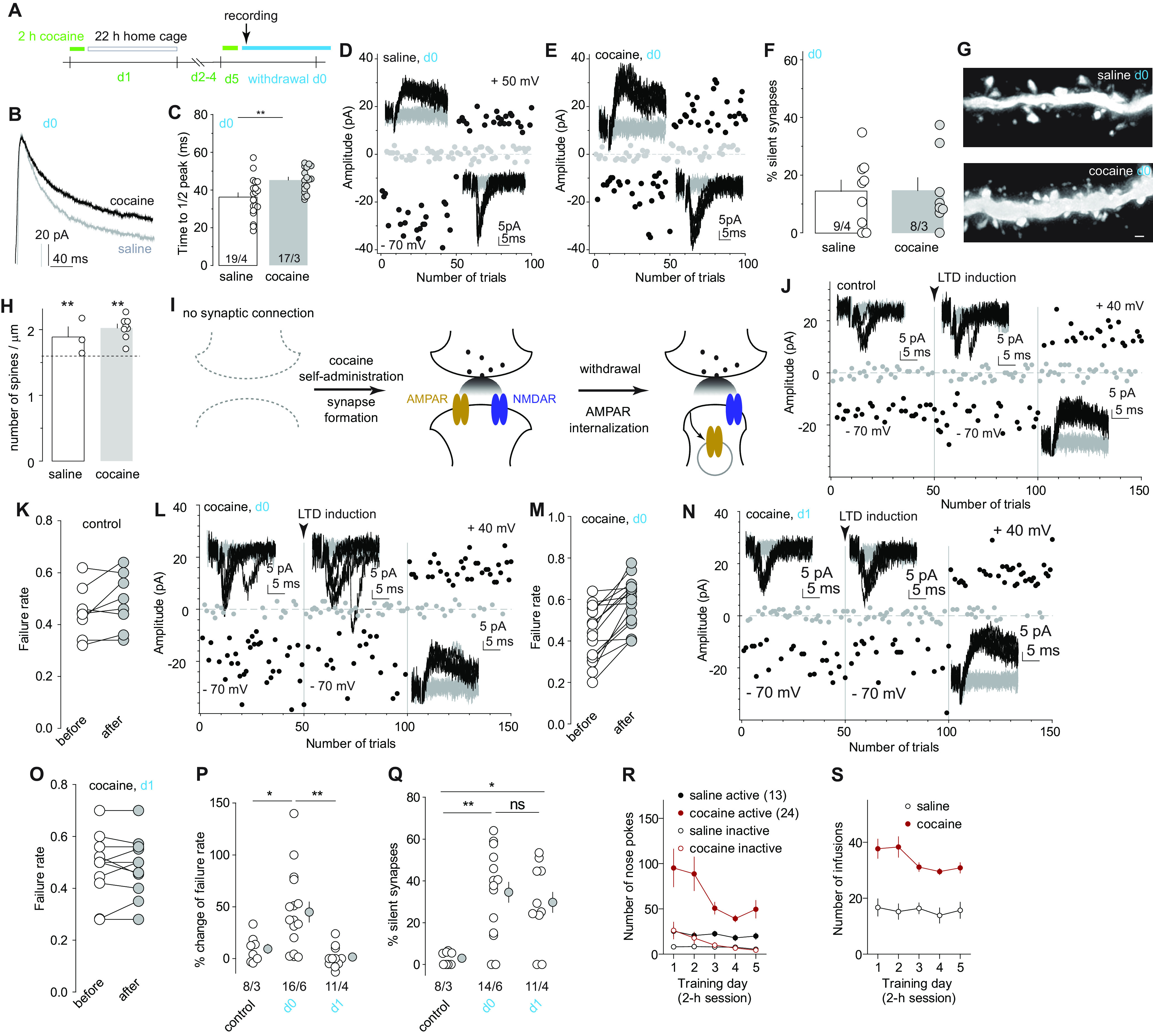
LTD unmasks cocaine-generated silent synapses. A, Diagram showing experimental timeline, in which the silent synapses assessment was ∼1 h after the last cocaine session on training day 5 (operationally defined as withdrawal day 0). B, C, Example traces (B) and summary (C) showing that on withdrawal day 0, NAcSh MSNs from cocaine-trained rats exhibited prolonged decay kinetics of NMDAR EPSCs compared with saline-trained rats. D, E, Example EPSCs evoked at −70 or +50 mV (insets) over 100 consecutive trials in the minimum stimulation assay from recordings on withdrawal day 0 after saline (D) or cocaine (E) self-administration. F, Summary showing a comparably low percentage of silent synapses in NAcSh MSNs between saline- and cocaine-trained rats on withdrawal day 0. G, Example NAcSh dendrites from saline-trained rats (top) and cocaine-trained rats (bottom) on withdrawal day 0. H, Summary showing that on withdrawal day 0, spine densities of NAcSh MSNs in saline- and cocaine-trained rats were both higher than age-matched naive control rats (indicated by the dashed line; control: 1.6 ± 0.1, n/m = 54/15; cocaine: 2.0 ± 0.1, n/m = 34/8; t(86) = 5.4, p < 0.01). I, Schematic illustration depicting the hypothesized two-step process of cocaine-induced generation of silent synapses, including the first step of generation of synapses containing both AMPARs and NMDARs during cocaine experience, and the second step of internalization of AMPARs from newly generated synapses after short-term withdrawal. J–O, Example EPSCs and trials in the minimum stimulation assay and summary of failure rates after a mild LTD induction in NAcSh MSNs from naive rats (J, K), cocaine-trained rats on withdrawal day 0 (L, M), or cocaine-trained rats on withdrawal day 1 (N, O). P, Summary showing that a mild LTD induction selectively increased the failure rate of AMPAR EPSCs in NAcSh MSNs from cocaine-trained rats on withdrawal day 0, but not on withdrawal day 1 or in drug-naive rats. Q, Summary showing in cocaine-trained rats that the low percentage of NAcSh silent synapses on withdrawal day 0 was increased by LTD induction to a level comparable to that on withdrawal day 1. R, S, Self-administration results of rats whose electrophysiological and morphologic results are presented in B–Q. *p < 0.05; **p < 0.01; ns, no significance.
To test this hypothesis, we optimized a moderate LTD induction protocol (1 Hz, 20 times), which, in the minimal stimulation assay (see Materials and Methods), did not affect the failure rate of AMPAR-mediated EPSCs or the percentage of silent synapses in NAcSh MSNs in age-compatible drug-naive rats, verifying that this LTD protocol did not affect the basal synapses unassociated with cocaine (Fig. 2J,K,P,Q; statistical summary for Fig. 2P: control, 9.3 ± 4.5%, n/m = 8/3; cocaine-day 0, 44.9 ± 9.7%, n/m = 16/6; cocaine-day 1, 1.5 ± 3.2%, n/m = 11/4, F(2,32) = 9.2, p < 0.01, one-way ANOVA; p = 0.02, control vs day 0; p > 0.99, control vs day 1; p = 0.01, day 0 vs day 1, Bonferroni post-tests). However, this protocol increased the failure rate of AMPAR EPSCs in rats on withdrawal day 0 after cocaine self-administration and increased the percentage of silent synapses to the level comparable to withdrawal day 1 (Fig. 2L,M,P,Q; statistical summary for Fig. 2Q: control, 2.9 ± 1.1%, n/m = 8/3; cocaine-day 0, 34.6 ± 5.7%, n/m = 14/6; cocaine-day 1, 29.6 ± 6.1%, n/m = 10/3; F(2,29) = 8.5, p < 0.01, one-way ANOVA; p < 0.01, control vs day 0; p = 0.01, control vs day 1; p > 0.99, day 0 vs day 1, Bonferroni post-tests). On withdrawal day 1 when the percentage of silent synapses was high, this LTD protocol did not affect the failure rate of AMPAR EPSCs and did not further increase the percentage of silent synapses (Fig. 2N–Q). These results suggest the following two-step process in the generation of silent synapse: (1) insertion of both AMPARs and NMDARs to new synaptic locations during cocaine experience; followed by (2) internalization of AMPARs through an LTD-like process from these new synapses during short-term (i.e., 22 h) withdrawal from cocaine (Fig. 2I).
Role of GluA2 AMPARs in silent synapse generation
GluA2-containing AMPAR subunits drive spinogenesis (Passafaro et al., 2003) and are enriched in the NAcSh in rats with or without cocaine experience (Boudreau et al., 2007), while GluA2-lacking AMPARs accumulate in NAcSh MSNs only after more prolonged withdrawal from cocaine (Conrad et al., 2008; Wolf, 2016). Based on these temporal patterns, we speculate that GluA2-containing AMPARs are the key AMPAR subtypes that trafficked during the two-step silent synapse generation process after cocaine. To test this hypothesis, we selectively interfered with the GluA2 AMPAR internalization during step 1 versus step 2 by intravenous injection of Tat-GluA23Y peptide. This synthetic peptide translocates into neurons and blocks experience-dependent endocytosis of GluA2-containing AMPARs with minimal effects on basal synaptic transmission (Ahmadian et al., 2004). These functional specificities of GluA23Y, as well as the lack of function of its scrambled control peptide, have been verified in NAc MSNs (Brebner et al., 2005; Graziane et al., 2016; Wright et al., 2020). After intravenous injection at 1.5 nmol/kg, GluA23Y peptide reaches peak concentrations in rat brain by 1.5 h and declines to one-half of the peak concentration in another 2–3 h (Brebner et al., 2005).
Taking advantage of these concentration dynamics, we administered GluA23Y 30 min before each cocaine self-administration session, which ensured GluA23Y to be present in the brain at high concentrations during cocaine exposure but to decline to low levels with diminished efficacy during the subsequent 22 h withdrawal (Fig. 3A,S–U). On withdrawal day 1, we observed an increased percentage of NAcSh silent synapses in cocaine-trained rats with the administration of either scrambled control peptide or GluA23Y compared with saline-trained rats (Fig. 3B–F; saline-scrambled: 7.8 ± 3.7%, n/m = 7/5; cocaine-scrambled: 34.6 ± 7.0%, n/m = 10/5; saline-GluA23Y: 10.5 ± 3.1%, n/m = 10/6; cocaine-GluA23Y: 30.1 ± 5.0%, n/m = 15/7; cocaine effect: F(1,38) = 18.0, p < 0.01; peptide effect: F(1,38) = 0.03, p = 0.86, two-way ANOVA; p < 0.01, saline-scrambled vs cocaine scrambled; p = 0.02, saline-GluA23Y vs cocaine-GluA23Y, Bonferroni post-tests). Thus, preventing GluA2 AMPAR internalization during cocaine experience did not prevent cocaine-induced generation of silent synapses. This result echoes our previous results showing that GluA23Y-mediated inhibition of GluA2-containing AMPAR internalization did not affect silent synapse generation induced by noncontingent cocaine procedures (Graziane et al., 2016). Consistently, densities of NAcSh dendritic spines were also increased in cocaine-trained rats with administration of either GluA23Y or scrambled peptide compared with saline-trained rats (Fig. 3G,H; number per micrometer: saline-scrambled: 1.6 ± 0.1, n/m = 35/8; cocaine-scrambled: 2.3 ± 0.1, n/m = 20/3; saline-GluA23Y: 1.6 ± 0.1, n/m = 21/4; cocaine-GluA23Y: 2.3 ± 0.1, n/m = 12/3; cocaine effect: F(1,84) = 96.3, p < 0.01; peptide effect: F(1,84) = 0.27, p = 0.60, two-way ANOVA; p < 0.01, saline-scrambled vs cocaine-scrambled; p < 0.01, saline-GluA23Y vs cocaine-GluA23Y, Bonferroni post-tests). Thus, preventing GluA2-containing AMPAR internalization during cocaine experience did not prevent the generation of silent synapses.
Figure 3.
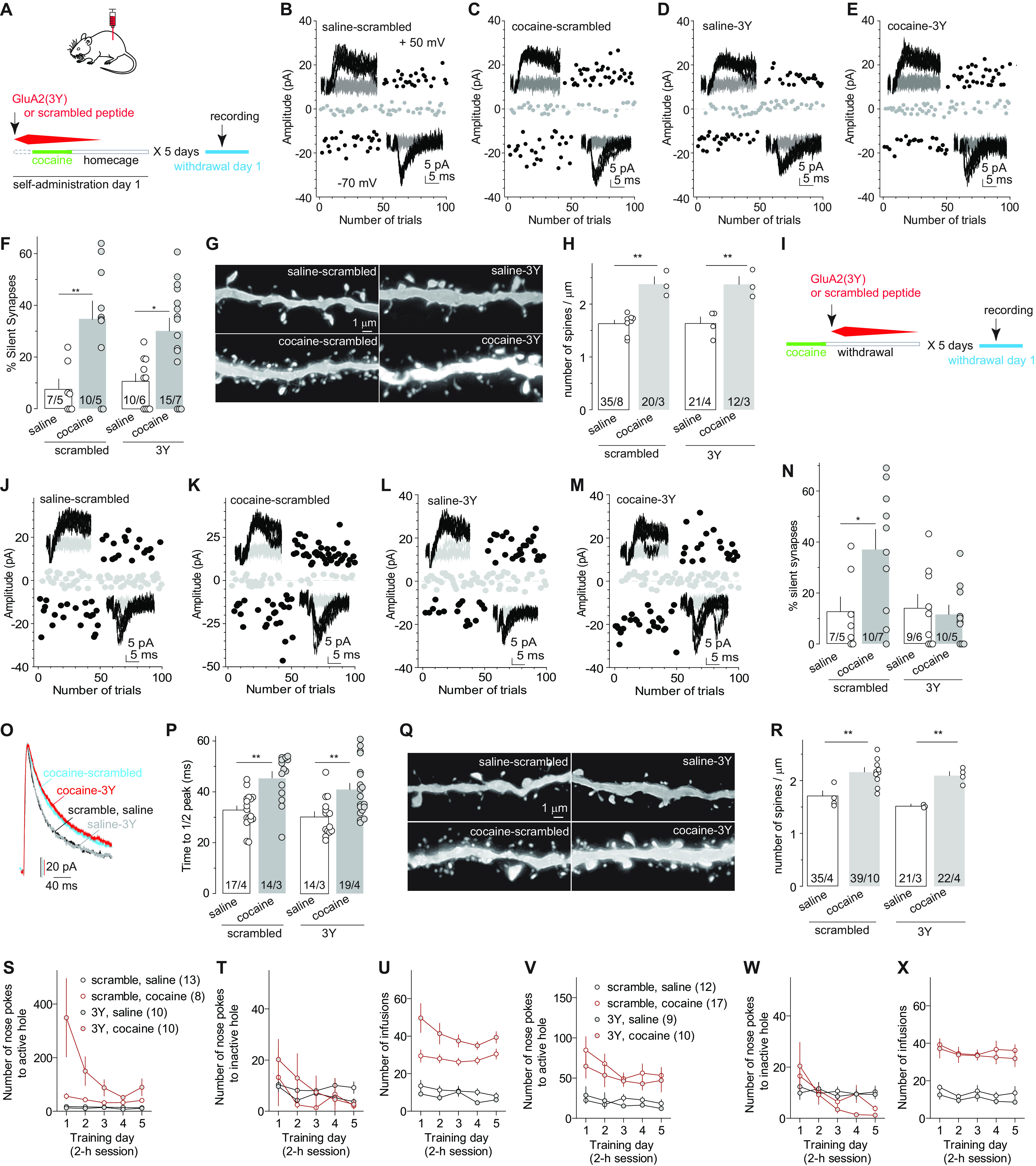
AMPAR internalization silences cocaine-generated synapses. A, Diagram showing the experimental timeline, in which GluA23Y or scrambled control peptide was injected 30 min before each daily self-administration session. B–E, Example EPSCs in NAcSh MSNs evoked at −70 or +50 mV (insets) over 100 consecutive trials in the minimum stimulation assay from rats after the procedure described in A. F, Summary showing that disrupting AMPAR internalization during cocaine self-administration training did not prevent cocaine-induced generation of silent synapses tested on withdrawal day 1. G, Example NAcSh dendrites from saline- and cocaine-trained rats with the administration of scramble or GluA23Y peptide 30 min before each training session. H, Summary showing that compromising AMPAR internalization during training did not prevent cocaine-induced increases in the density of total spines tested on withdrawal day 1. I, Diagram showing experimental timeline, in which the GluA23Y or scrambled peptide was administered 30 min after the daily self-administration session. J–M, Example EPSCs in the minimum stimulation assay of withdrawal day 1 saline- or cocaine-trained rats with the administration of scrambled or GluA23Y peptide 30 min after daily self-administration session. N, Summary showing that compromising AMPAR internalization during the short-term withdrawal period following a daily self-administration session resulted in a low percentage of silent synapses tested on withdrawal day 1. O, P, Example traces (O) and summary (P) showing that compromising AMPAR internalization either before or after a daily self-administration session did not prevent cocaine-induced elongation of the decay kinetics of NMDAR EPSCs in NAcSh MSNs. Q, Example NAcSh dendrites from saline- and cocaine-trained rats with the administration of scramble or GluA23Y peptide 30 min after daily self-administration session. R, Summary showing that compromising AMPAR internalization during daily short-term withdrawal did not prevent the cocaine-induced increase in densities of total spines tested on withdrawal day 1. S–U, Self-administration results of rats whose electrophysiological and morphologic results are presented in B–H. V–X Self-administration results of rats whose electrophysiological and morphologic results are presented in J–R. *p < 0.05; **p < 0.01.
Next, instead of the cocaine exposure periods, we selectively interfered with GluA2-containing AMPAR internalization during the daily 22 h withdrawal periods by administering GluA23Y 30 min after the daily 2 h self-administration session (Fig. 3I,V–X). On withdrawal day 1, while cocaine-trained rats with scrambled peptide exhibited an increased percentage of NAcSh silent synapses compared with saline-trained rats, as expected, the percentage of NAcSh-silent synapses in cocaine-trained rats with GluA23Y was low, comparable to that in saline-trained rats (Fig. 3J–N; saline-scrambled: 12.7 ± 5.8%, n/m = 7/5; cocaine-scrambled: 37.2 ± 7.8%, n/m = 10/7; saline-GluA23Y: 14.3 ± 5.2%, n/m = 9/6; cocaine-GluA23Y: 11.7 ± 3.7%, n/m = 10/5; F(1,32) = 5.2, p = 0.03, two-way ANOVA; p = 0.02, saline-scrambled vs cocaine-scrambled; p > 0.99, saline-GluA23Y vs cocaine-GluA23Y, Bonferroni post-tests). Despite the low percentage, cocaine-trained rats with GluA23Y exhibited prolonged decay kinetics of NMDAR EPSCs in NAcSh MSNs compared with saline-trained rats, similar to cocaine-trained rats with scrambled peptide (Fig. 3O,P; saline-scrambled: 32.9 ± 1.6 ms, n/m = 17/4; cocaine-scrambled: 45.5 ± 2.5 ms, n/m = 14/3; saline-GluA23Y: 30.2 ± 2.0 ms, n/m = 14/3; cocaine-GluA23Y: 40.9 ± 2.4 ms, n/m = 19/4; cocaine effect: F(1,60) = 28.1, p < 0.01; peptide effect: F(1,60) = 2.7, p = 0.11, two-way ANOVA; p < 0.01, saline-scrambled vs cocaine-scrambled; p < 0.01, saline-GluA23Y vs cocaine-GluA23Y, Bonferroni post-tests). These results support the two-step model—that is, new synapses that contained both AMPARs and NMDARs were generated by cocaine experience, but preventing AMPAR internalization during the daily short-term withdrawal maintained AMPARs at these synapses, resulting in a low percentage of silent synapses. This notion was further supported by the observation that cocaine-trained rats with either scrambled peptide or GluA23Y exhibited similarly higher densities of dendritic spines compared with saline-trained rats (Fig. 3Q,R; number per micrometer: saline-scrambled: 1.6 ± 0.1, n = 35/4; cocaine-scrambled: 2.2 ± 0.1, n/m = 39/10; saline-GluA23Y: 1.6 ± 0.1, n/m =21/3; cocaine-GluA23Y: 2.1 ± 0.1, n/m = 22/4; cocaine effect: F(1,113) = 131.7, p < 0.01; peptide effect: F(1,113) = 1.39, p = 0.24, two-way ANOVA; p < 0.01, saline-scrambled vs cocaine scrambled; p < 0.01, saline-GluA23Y vs cocaine-GluA23Y, Bonferroni post-tests).
Removal of GluN2B NMDARs is required for silent synapse maturation
Cocaine-generated silent synapses mature 7–10 d after withdrawal from cocaine by recruiting AMPARs (Lee et al., 2013). This maturation process strengthens newly generated spines and synapses, and results in a return of silent synapses to low levels (Graziane et al., 2016; Wright et al., 2020). On withdrawal day 7 after cocaine self-administration, overall densities of NAcSh dendritic spines remained higher in cocaine-trained rats compared with saline-trained rats (Fig. 4A,B,C,I,J; number per micrometer: saline: 1.6 ± 0.1, n/m = 22/6; cocaine: 2.2, n/m = 26/9; t(46) = 5.5, p < 0.01, two-tailed t test). In addition, the decay kinetics of NMDAR EPSCs in NAcSh MSNs in cocaine-trained rats returned to the level in saline-trained rats, suggesting that previously increased GluN2B NMDARs at synapses were replaced by non-GluN2B NMDARs (Fig. 4D,E; saline: 49.3 ± 2.6 ms, n/m = 30/9; cocaine: 47.6 ± 1.5 ms, n/m = 41/10; t(69) = 0.59, p = 0.56, two-tailed t test). This switch of NMDAR subtypes was also observed in previous studies after withdrawal from cocaine (Wright et al., 2020). In parallel, the percentage of silent synapses in cocaine-trained rats also returned to basal levels as in saline-trained rats (Fig. 4F–H; saline: 12.0 ± 3.3%, n/m = 18/11; cocaine: 10.9 ± 3.0%, n/m = 17/12; t(33) = 0.23, p = 0.82, two-tailed t test). These results suggested that, at this withdrawal time point, a portion of cocaine-generated silent synapses recruited AMPARs and matured, a process correlated with a switch of NMDAR subtypes. In the developing brain, although the maturation of silent synapses relies on the activation of NMDARs and NMDAR-coupled CaMKII signaling (Liao et al., 1995; Durand et al., 1996; Wu et al., 1996), basal activities of GluN2B NMDARs inhibit AMPAR insertion (Adesnik et al., 2008; Gray et al., 2011). Our following experiments therefore tested whether the removal of GluN2B NMDARs was essential for the maturation of cocaine-generated silent synapses.
Figure 4.

Maturation of cocaine-generated silent synapses coincides with removal of GluN2B NMDARs. A, Diagram showing experimental timeline, in which silent synapse assessment was performed on withdrawal day 7. B, Example of NAcSh dendrites from saline- and cocaine-trained rats tested on withdrawal day 7. C, Summary showing that the density of total spines remained higher on withdrawal day 7 in cocaine-trained rats compared with saline-trained rats. D, E, Example traces (D) and summary (E) showing that on withdrawal day 7, the decay kinetics of NMDAR EPSCs in NAcSh MSNs from cocaine-trained rats were comparable to saline-trained rats. F, G, Example EPSCs and trials in the minimum stimulation assay from withdrawal day 7 saline-trained (F) and cocaine-trained (G) rats. H, Summary showing that the percentage of silent synapses in cocaine-trained rats returned to low levels comparable to saline controls on withdrawal day 7. I, J, Self-administration results of rats whose electrophysiological and morphologic results are presented in B–H. **p < 0.01.
If removal of GluN2B NMDARs is a prerequisite of silent synapse maturation after withdrawal from cocaine, experimentally locking GluN2B NMDARs at cocaine-generated silent synapses should maintain the silent state of these synapses after prolonged drug withdrawal. To test this possibility, we adopted an siP-based strategy. Synaptic removal of GluN2B NMDARs is primed by a Cdk5 (cyclin-dependent kinase 5)-mediated phosphorylation, which relies on the interaction between Cdk5 and the C-terminal motif 1111RRPPRSPDHKRYFRDKE1127 of GluN2B (Plattner et al., 2014). An siP, RRPPRSPDHKRYFRDKE, compromises Cdk5–GluN2B interactions and selectively attenuates synaptic removal of GluN2B without affecting Cdk5-mediated phosphorylation of other substrates (Plattner et al., 2014).
We packaged this siP, fused to eYFP, into an AAV, and bilaterally injected it into rat NAcSh. After the self-administration procedure, we recorded transduced NAcSh MSNs, in which synaptic removal of GluN2B NMDARs was presumably impaired by siP. Nontransduced MSNs were recorded as controls. On withdrawal day 1, both nontransduced and transduced NAcSh MSNs exhibited prolonged decay kinetics of NMDAR EPSCs in cocaine-trained rats compared with saline-trained rats, indicating that the presence of siP did not prevent cocaine-induced synaptic insertion of GluN2B NMDAR (Fig. 5A,B,Q,R; saline-nontransduced: 44.8 ± 1.1 ms, n/m = 20/5; cocaine-nontransduced: 54.9 ± 3.9 ms, n/m = 16/5; saline-transduced: 41.9 ± 2.6 ms, n/m = 15/4; cocaine-transduced: 55.9 ± 3.3 ms, n/m = 12/6; cocaine effect: F(1,59) = 18.7, p < 0.01, two-way ANOVA; p = 0.01, saline-nontransduced vs cocaine-nontransduced; p < 0.01, saline-transduced vs cocaine-transduced, Bonferroni post-tests). On withdrawal day 7, the decay kinetics of NMDAR EPSCs in nontransduced MSNs from cocaine-trained rats returned to basal, saline control levels, as expected (Fig. 5C,D; saline-nontransduced: 43.8 ± 1.4 ms, n/m = 32/8; cocaine-nontransduced: 42.9 ± 1.6 ms, n/m = 20/7; saline-transduced: 37.7 ± 2.2 ms, n/m = 15/6; cocaine-transduced: 51.5 ± 2.8 ms, n/m = 22/8; F(1,85)= 12.4, p < 0.01, two-way ANOVA; p > 0.99, saline-nontransduced vs cocaine-nontransduced; p < 0.01, saline-transduced vs cocaine-transduced, Bonferroni post-tests). However, NMDAR EPSCs in transduced MSNs continued to exhibit prolonged kinetics in cocaine-trained rats (Fig. 5C,D). Thus, the expression of siP effectively locked cocaine-induced GluN2B NMDARs at synapses after drug withdrawal.
Figure 5.
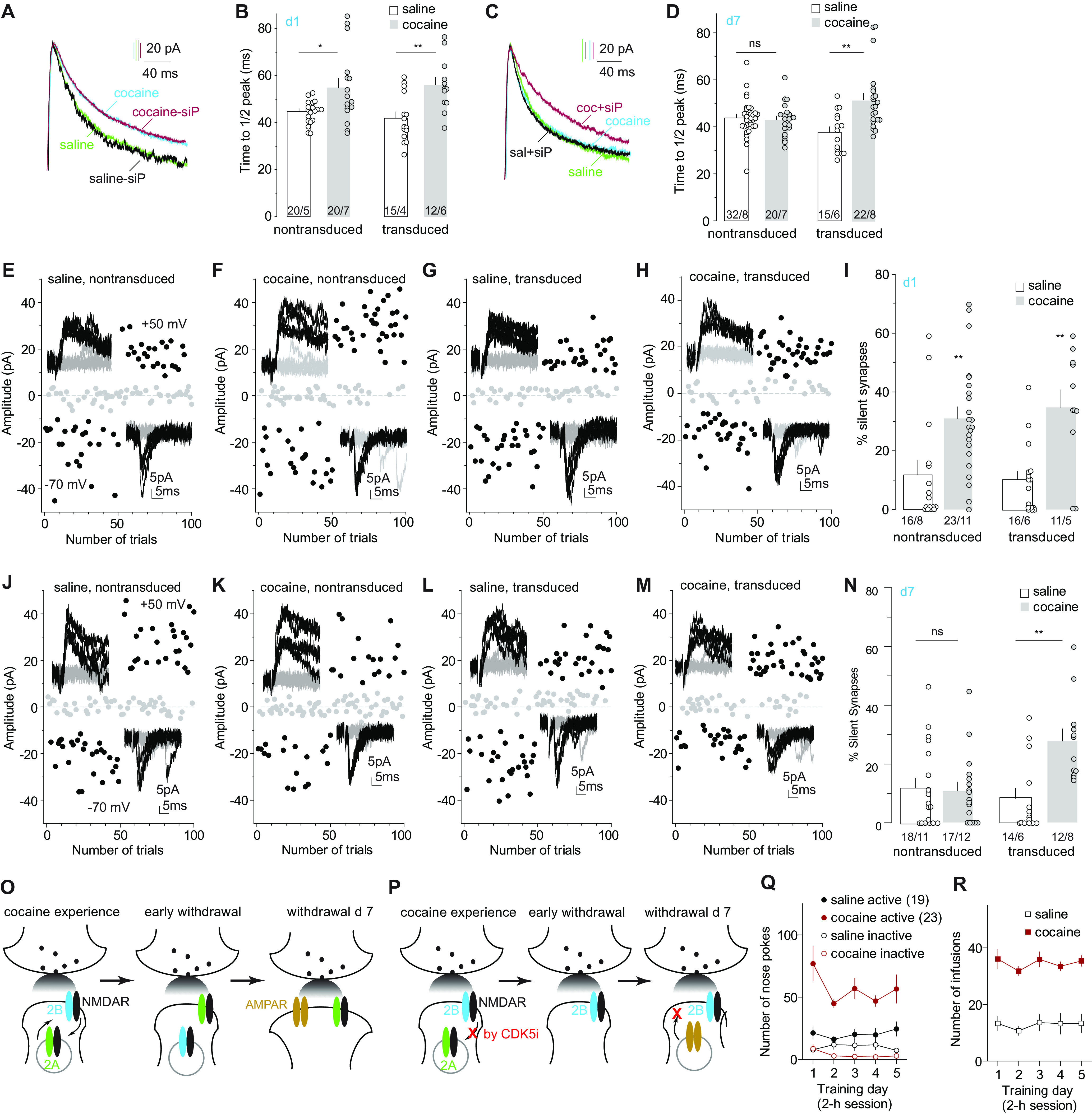
Locking GluN2B NMDARs prevents maturation of cocaine-generated silent synapses. A, B, Example traces (A) and summary (B) showing that on withdrawal day 1, transduced Cdk5 siP-expressing NAcSh MSNs and nontransduced MSNs exhibited similarly prolonged kinetics of NMDAR EPSCs in cocaine-trained rats compared with saline-trained rats. C, D, Example traces (C) and summary (D) showing that on withdrawal day 7, the decay kinetics of NMDAR EPSCs in nontransduced NAcSh MSNs in cocaine-trained rats returned to levels in saline control rats, whereas the prolonged kinetics of NMDAR EPSCs persisted in transduced Cdk5 siP-expressing MSNs. E–H, Example EPSCs and trials in the minimum stimulation assay of transduced and nontransduced NAcSh MSNs in saline- and cocaine-trained rats on withdrawal day 1. I, Summary showing that on withdrawal day 1, the percentage of silent synapses was increased in both nontransduced and transduced MSNs in cocaine-trained rats compared with saline controls. J–M, Example EPSCs and trials in the minimum stimulation assay of transduced and nontransduced NAcSh MSNs in saline- and cocaine-trained rats on withdrawal day 7. N, Summary showing that on withdrawal day 7, the percentage of silent synapses in nontransduced MSNs in cocaine-trained rats decreased to low levels comparable to saline controls, but this percentage remained high in transduced MSNs. O, P, Illustration of the hypothesized, two-step maturation of cocaine-generated silent synapses, with the first step of switching GluN2B NMDARs to non-GluN2B, potentially GluN2A, NMDARs (O), and the second step of recruiting AMPARs, while preventing this NMDAR switch prevents AMPAR insertion and maturation of silent synapses after cocaine withdrawal (P). Q, R, Self-administration results of rats whose electrophysiological results are presented in A–N. *p < 0.05; **p < 0.01; ns, no significance.
We next examined whether locking GluN2B NMDARs prevented maturation of cocaine-generated silent synapses. On withdrawal day 1, both transduced siP-expressing MSNs and nontransduced MSNs exhibited similarly increased percentage of silent synapses in cocaine-trained rats compared with saline controls, indicating that siP did not affect the generation of silent synapses (Fig. 5E–I; saline-nontransduced: 11.8 ± 4.7%, n/m = 16/8; cocaine-nontransduced: 31.0 ± 3.9%, n/m = 23/11; saline-transduced: 10.0 ± 3.0%, n/m = 16/6; cocaine-transduced: 34.7 ± 6.0%, n/m = 11/5; cocaine effect: F(1,62) = 24.0, p < 0.01, two-way ANOVA; p < 0.01, saline-nontransduced vs cocaine-nontransduced; p < 0.01, saline-transduced vs cocaine-transduced, Bonferroni post-tests). On withdrawal day 7, the percentage of silent synapses in nontransduced MSNs returned to basal levels in cocaine-trained rats, as expected (Fig. 5J–N; saline-nontransduced: 12.0 ± 3.3%, n/m = 18/11; cocaine-nontransduced: 10.9 ± 3.0%, n/m = 17/12; saline-transduced: 8.4 ± 3.4%, n/m = /14/6; cocaine-transduced: 27.9 ± 4.1%, n/m = 12/8; F(1,57) = 8.8, p < 0.01, two-way ANOVA; p > 0.99 saline-nontransduced vs cocaine-nontransduced; p < 0.01, saline-transduced vs cocaine-transduced, Bonferroni post-tests). However, the percentage remained at high levels in transduced MSNs in cocaine-trained rats on withdrawal day 7 (Fig. 5J–N). These results support our hypothesis that GluN2B NMDARs function to maintain cocaine-generated synapses in the AMPAR-silent state, while the switch of GluN2B NMDARs to presumably GluN2A NMDARs allows AMPAR insertion and maturation of cocaine-generated silent synapses (Fig. 5O,P). Thus, the switch of NMDAR subtypes and the insertion of AMPARs comprise a two-step model for the maturation of cocaine-generated silent synapses.
Role of CaMKII signaling in GluN2B NMDAR removal
In many brain regions, the GluN2B-to-GluN2A switch of synaptic NMDARs marks the maturation of glutamatergic synapses during postnatal development (Carroll and Zukin, 2002; Cull-Candy and Leszkiewicz, 2004; Vieira et al., 2020). Among several potential mechanisms underlying this switch, we focused on CaMKII signaling. GluN2B differs from GluN2A in its enhanced capacity of binding and recruiting CaMKII, and maintaining CaMKII activation (Strack and Colbran, 1998; Mayadevi et al., 2002; Halt et al., 2012). CaMKII can phosphorylate GluN2B, an effect that operates with other signaling pathways to promote the removal of GluN2B NMDARs (Omkumar et al., 1996; O'Leary et al., 2011; Sanz-Clemente et al., 2013). To test the role of CaMKII signaling, we used a KI mouse line, in which the endogenous GluN2B was replaced by a mutant with reduced ability to activate CaMKII signaling (Halt et al., 2012; Wang et al., 2018). We continued to use the 5 d cocaine procedure but extended the withdrawal time to 21 d, to explore the potentially persistent effects of GluN2B on silent synapses.
We trained the KI and wild-type control mice with the same self-administration procedure as in the rat experiments above. KI and wild-type mice exhibited comparable acquisition of operant responding and cocaine consumption. In cocaine-trained wild-type mice, NAcSh MSNs exhibited prolonged decay kinetics of NMDAR EPSCs on withdrawal day 1, and this change diminished on withdrawal day 21 (Fig. 6A,B,M,N; control: 36.4 ± 1.3 ms, n/m = 25/8; day 1: 48.9 ± 2.2 ms, n/m = 27/8; day 21, 41.8 ± 1.2 ms, n/m = 32/6; F(2,81)= 14.5, p < 0.01, one-way ANOVA; p < 0.01, control vs day 1; p < 0.01 day 1 vs day 21, Bonferroni post-tests). Meanwhile, the percentage of silent synapses in NAcSh MSNs was increased in cocaine-trained wild-type mice on withdrawal day 1 and returned to saline control levels on withdrawal day 21 (Fig. 6C–F; control: 13.4 ± 4.3%, n/m = 17/12; day 1, 41.5 ± 5.6%, n/m = 14/7; day 21, 7.5 ± 2.3%, n/m = 7/5; F(2,35) = 13.0, p < 0.01, one-way ANOVA; p < 0.01, control vs day 1; p < 0.01, day 1 vs day 21, Bonferroni post-tests). Thus, GluN2B-mediated generation and subsequent maturation of silent synapses were confirmed in mice.
Figure 6.
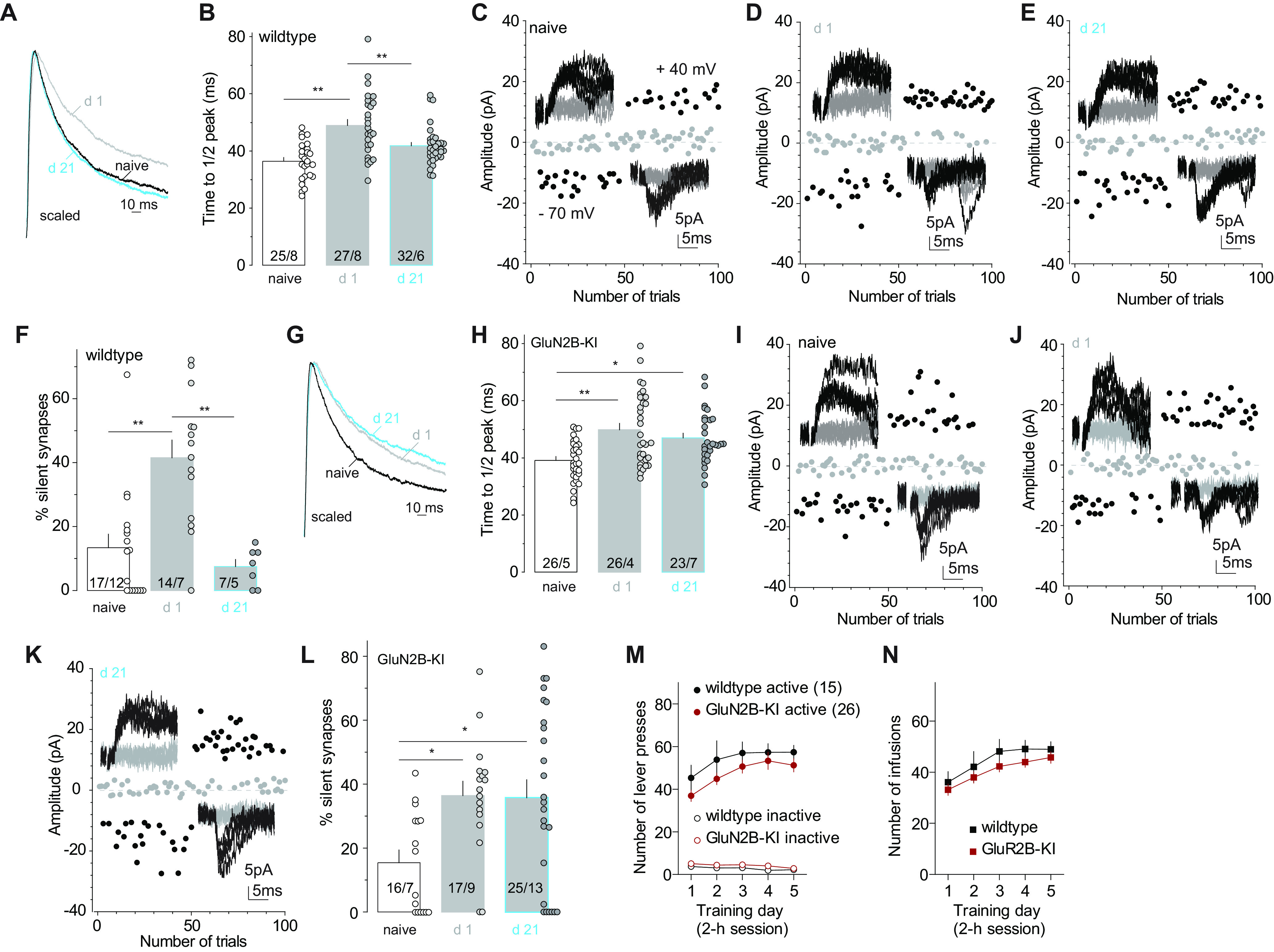
Disrupting GluN2B–CaMKII interaction prevents the removal of GluN2B NMDARs and maturation of silent synapses after cocaine withdrawal. A, B, Example traces (A) and summary (B) showing that in wild-type mice the decay kinetics of NMDAR EPSCs in NAcSh MSNs was prolonged after 1 d withdrawal from cocaine self-administration, and returned to basal levels after 21 d withdrawal. C–E, Example EPSCs and trials in the minimum stimulation assay of control and cocaine-trained mice after 1 or 21 d withdrawal. F, Summary showing in wild-type mice that the percentage of silent synapses in NAcSh MSNs was increased on withdrawal day 1 and declined to control levels on withdrawal day 21. G, H, Example traces (G) and summary (H) showing that in GluN2B-KI mice, the decay kinetics of NMDAR EPSCs in NAcSh MSNs was prolonged after 1 d withdrawal from cocaine self-administration and remained slow on withdrawal day 21. I–K, Example EPSCs and trials in the minimum stimulation assay of control and cocaine-trained mice after 1 or 21 d withdrawal. L, Summary showing that in GluN2B-KI mice, the percentage of silent synapses in NAcSh MSNs was increased on withdrawal day 1 and remained at high levels on withdrawal day 21. M, N, Self-administration results of mice whose electrophysiological results are presented in A–L. *p < 0.05; **p < 0.01.
In KI mice, NAcSh MSNs exhibited prolonged decay kinetics of NMDAR EPSCs on both withdrawal days 1 and 21 after cocaine self-administration compared with saline controls (Fig. 6G,H; control: 40.2 ± 1.3 ms, n/m = 26/5; day 1: 49.1 ± 2.4 ms, n/m = 26/4; day 21: 47.6 ± 1.9 ms, n/m = 23/7; F(2,72) = 6.3, p < 0.01, one-way ANOVA; p < 0.01, control vs day 1; p = 0.03, control vs day 21, Bonferroni post-test). These results indicate that, with compromised coupling to CaMKII, mutant GluN2B NMDARs were still inserted into synapses by cocaine experience, but they could not be removed during withdrawal periods. On the other hand, on both withdrawal days 1 and 21, the percentage of silent synapses in cocaine-trained KI mice was higher than in saline-trained mice (Fig. 6I–L; control: 15.5 ± 4.0%, n/m = 16/7; day 1, 36.4 ± 4.5%, n/m = 17/9; day 21, 35.8 ± 5.6%, n/m = 25/13; F(2,55) = 4.8, p = 0.01, one-way ANOVA; p = 0.03, control vs day 1; p = 0.02, control vs d 21; Bonferroni post-tests). These results suggest that an interaction with CaMKII is required for GluN2B NMDAR removal after withdrawal from cocaine, and, again, GluN2B NMDARs maintain cocaine-generated synapses in the AMPAR-silent state. A caveat using this global KI mice is that, beyond NAcSh MSNs, CaMKII-dependent processes in other brain regions might also contribute to the GluN2B NMDAR removal and silent synapse maturation through potential network effects.
Discussion
Generated by cocaine experience, silent synapses in NAcSh MSNs may stand as a distinct population of synapses encoding cocaine-specific memory traces. They are not essential for establishing operant responses to cocaine (Wang et al., 2020), but are critically important for cue-conditioned cocaine seeking that may be mediated by cue–cocaine associative memories (Lee et al., 2013; Ma et al., 2016; Wright et al., 2020). Our current study characterized the basic cellular processes underlying the generation and maturation of these synapses. These results establish a conceptual foundation for understanding the cellular and circuit mechanisms underlying cocaine memories and a knowledge basis to manipulate cocaine memories for clinical benefit.
Cocaine-induced versus developmental synaptogenesis
Increasing evidence suggests that cocaine-induced generation of silent synapses in NAcSh is a synaptogenesis process using similar cellular mechanisms that mediate the formation of new synapses throughout forebrain during development (Dong and Nestler, 2014). For example, cocaine-induced generation of silent synapses requires increased activities of CREB (Brown et al., 2011), a transcription factor critically implicated in synaptogenesis during development (Lonze and Ginty, 2002). Cocaine-generated silent synapses are enriched in GluN2B NMDARs (Huang et al., 2009; Brown et al., 2011), a signature feature distinguishing developmental from mature glutamatergic synapses (Cull-Candy and Leszkiewicz, 2004). Moreover, after cocaine self-administration, an astrocyte-mediated synaptogenic signaling, namely, thrombospondin-α2δ−1 signaling, is engaged in the adult NAcSh for silent synapse generation (Wang et al., 2020). This astrocytic signaling actively mediates synaptogenesis during development but declines to low levels in adulthood (Baldwin and Eroglu, 2017). As such, cocaine experience may reopen some of the developmental mechanisms to redevelop synaptic connections and network connectivity, embedding persistent drug-associated memory traces, as elaborated in the “rejuvenation hypothesis” (Dong and Nestler, 2014; Huang et al., 2015).
Despite the general difference of cellular environments between developing and adult brains, key synaptic components in the adult NAcSh may also undergo certain degrees of the rejuvenation process in response to cocaine. For example, SAP102 and PSD-95 are two signaling scaffold proteins that regulate the stability and number of AMPARs at glutamatergic synapses (Elias et al., 2006; Bonnet et al., 2013), and, especially, PSD-95 promotes the maturation of silent synapses (Shukla et al., 2017; Favaro et al., 2018). SAP102 predominates in the developing brain and might thus preferentially regulate synaptic AMPARs in nascent synapses (Elias et al., 2008; Favaro et al., 2018). In the adult NAcSh, although PSD-95 dominates in naive animals (Shukla et al., 2017), it is decreased after repeated exposure to cocaine (Yao et al., 2004). Although the effects of cocaine on NAcSh SAP102 has not been examined directly, evidence exists showing that reduced functionality of PSD-95 is accompanied by increased protein levels of SAP102 in the striatum (Vickers et al., 2006). Such a potential switch of the SAP102/PSD-95 balance may be part of the rejuvenation signaling cascades that regulate postsynaptic arrangements as well as trafficking of AMPARs and NMDARs at cocaine-generated silent synapses.
Role of AMPARs in generation and maturation of NAcSh-silent synapses
During development, AMPARs are in proximity with NMDARs for the initial postsynaptic landscaping when new glutamatergic synapses are formed. For example, in cultures of hippocampal neurons, transport packets of AMPARs and NMDARs are both detected to move toward postsynaptic membranes during the formation of nascent synapses (Washbourne et al., 2002). Although packets containing AMPARs are less mobile than those of NMDARs (Washbourne et al., 2002), both AMPARs and NMDARs are detected at postsynaptic membranes with kinetics similar to that of PSD-95-mediated clustering (Friedman et al., 2000). Functionally, both AMPAR- and NMDAR-mediated EPSCs are detected in the hippocampus from late embryonic periods to early postnatal periods, suggesting the existence of functional AMPARs at newborn synapses (Diabira et al., 1999; Verhage et al., 2000; Groc et al., 2002). However, these observations appear to contradict the fact that the developing brain is highly enriched in AMPAR-silent synapses during synaptogenesis periods (Durand et al., 1996; Wu et al., 1996; Kerchner and Nicoll, 2008; Hanse et al., 2013). This discrepancy is reconciled by a seminal study demonstrating a quick internalization of AMPARs at presumably newborn synapses after a moderate train of activity (Xiao et al., 2004). These and other results lead to a hypothesis that, rather than being “absent,” AMPARs at nascent synapses are instead highly labile, and are internalized or functionally muted on moderate synaptic activities but stabilized or functionally sound on strong synaptic activities (e.g., LTP induction; Groc et al., 2006; Hanse et al., 2009, 2013).
Our current results suggest striking similarities of AMPAR properties between cocaine-generated synapses in the adult NAcSh and newborn synapses in the developing forebrain. Silent synapses are generated gradually and accumulatively over repeated exposure to cocaine (Huang et al., 2009). However, the high percentage of silent synapses can only be detected after a short-term withdrawal period (e.g., withdrawal days 1 or 2), but not right after (e.g., withdrawal day 0) cocaine experience (Figs. 1, 2). Despite a low percentage of silent synapses, the increased weight of synaptic GluN2B NMDARs and elevated spine densities remained on withdrawal day 0 (Fig. 2). In light of the AMPAR trafficking at developing synapses, we propose that AMPARs are present initially at new synapses generated by cocaine experience, but they are prone to internalization on basal synaptic activities during early withdrawal periods. This notion is substantiated by our subsequent results that preventing AMPAR internalization during withdrawal from cocaine, but not during exposure to cocaine, led to a low electrophysiological readout (percentage) of silent synapses without affecting the high densities of dendritic spines in cocaine-trained rats (Fig. 3).
After generation, a portion of silent synapses mature by recruiting GluA2-lacking AMPARs after a relatively long period of withdrawal (Lee et al., 2013; Ma et al., 2014; Shukla et al., 2017; Wright et al., 2020). Our current results suggest that AMPARs operating in the initial synapse formation are GluA2-containing, rather than GluA2-lacking, AMPARs. Specifically, the 3Y peptide, which contains a portion of GluA2 C terminus and selectively prevents GluA2-containing AMPAR internalization, prevented the silencing process of cocaine-generated synapses during short-term withdrawal from cocaine (Fig. 3). This role of GluA2-containing AMPARs is consistent with the general synaptogenic role of GluA2, as demonstrated in cultures of developing hippocampal neurons, in which GluA2 promotes large-scale spinogenesis (Passafaro et al., 2003). As such, two distinct sets of AMPARs, and likely two distinct sets of postsynaptic signaling cascades, are differentially involved in the generation and maturation of NAcSh-silent synapses after cocaine experience.
Further analysis of our current results suggests that AMPAR trafficking at cocaine-generated synapses can be highly dynamic. Specifically, a low percentage of silent synapses was observed right after exposure on the fifth day (withdrawal day 0; Fig. 2). However, silent synapses are generated gradually and cumulatively, and are expected to reach a relatively high level after the first 4 d of exposure to cocaine (Huang et al., 2009). The silent synapses generated during the first 4 d exposure were not likely to be eliminated on withdrawal day 0, given the elevated level of spine density at this time point (Fig. 2). Instead, they may become temporarily unsilenced on re-exposure to cocaine. We speculate that the exposure to cocaine on the fifth day initiates another round of GluA2-mediated synaptogenesis, which may not only drive GluA2-containing AMPARs to newly generated synapses but also to pregenerated silent synapses, which represent in essence “open slots” prone for AMPAR recruitment (Malenka and Nicoll, 1997; Kerchner and Nicoll, 2008). As discussed below, the presence of GluN2B NMDARs prevents a stabilization of inserted AMPARs, leading to the resilencing of these synapses subsequently.
It is worth noting that our current (Figs. 4, 5) and previous (Lee et al., 2013) results suggest that after 7–10 d of withdrawal from cocaine self-administration, NAcSh-silent synapses mature by recruiting CP-AMPARs potentially. If this is the case, this insertion of CP-AMPARs occurs before, and therefore is different from, the massive upregulation of synaptic CP-AMPARs observed after long-term (e.g., >33 d) withdrawal from cocaine self-administration (Wolf, 2016; Nestler and Lüscher, 2019). The delayed CP-AMPAR upregulation likely occurs at NAc excitatory synapses in an across-the-board manner, mediated in part through altered mGluR1-signaling (Conrad et al., 2008; Loweth et al., 2014; Wolf, 2016; Nestler and Lüscher, 2019).
Role of NMDARs in generation and maturation of NAcSh-silent synapses
In the developing hippocampus, overexpression of GluN2B-containing NMDARs or GluN2B C termini increases the density of dendritic spines and the percentage of AMPAR-silent synapses (Barria and Malinow, 2002, 2005; Gambrill and Barria, 2011). On the other hand, the deletion of GluN2B decreases spine densities in the developing hippocampus (Gray et al., 2011). These synaptogenic features of GluN2B NMDARs are also detected in the adult NAcSh after cocaine experience. In adult rats, cocaine experience delivers GluN2B-containing NMDARs to NAcSh synapses, and the selective inhibition of GluN2B NMDARs decreases the percentage of silent synapses to levels similar to those in saline-treated rats (Huang et al., 2009; Brown et al., 2011). In our current experiments, knockdown of GluN2B prevented cocaine-induced generation of silent synapses (Fig. 1). Thus, GluN2B NMDARs are an essential part of the cellular mechanisms underlying both developmental and cocaine-induced synaptogenesis.
A prominent developmental change of synaptic NMDARs is the switch from GluN2B- to GluN2A-enriched subtypes (Cull-Candy and Leszkiewicz, 2004). In the developing brain, although experience-dependent NMDAR signaling generally promotes synaptic recruitment of AMPARs and the maturation of nascent synapses (Liao et al., 1995; Durand et al., 1996), this process is staggered at nascent synapses that contain GluN2B NMDARs, which, indeed, function to restrict synaptic AMPAR contents (Adesnik et al., 2008; Gray et al., 2011). In contrast, experimentally upregulating GluN2A-coupled signaling terminates the ongoing increase in spine number and size during development (Gambrill and Barria, 2011). It is thus tempting to speculate that the GluN2B-to-GluN2A switch gates the developmental maturation of nascent glutamatergic synapses. Our previous and current results support this speculated scenario for cocaine-generated silent synapses in NAcSh. First, cocaine-generated silent synapses in this region are enriched in GluN2B NMDARs during early withdrawal periods (Huang et al., 2009; Brown et al., 2011). Second, cocaine-generated synapses mature after longer withdrawal periods, accompanied by a return of synaptic GluN2B to basal levels (Fig. 4; Graziane et al., 2016; Wright et al., 2020). Third, locking GluN2B NMDARs at newly generated synapses prevents maturation of these synapses (Figs. 5, 6). Thus, although the recruitment of AMPARs to nascent synapses can also be mediated by NMDAR-independent mechanisms during development (Friedman et al., 2000; Ultanir et al., 2007), our results pinpoint NMDARs, particularly the GluN2B-to-GluN2A switch of NMDARs, that gate the maturation process of cocaine-generated synapses.
It should be noted that GluN2B NMDARs are commonly detected at mature synapses, including NAcSh synapses in both saline- and cocaine-treated animals after long-term withdrawal, albeit at low levels (Huang et al., 2009; Graziane et al., 2016; Wright et al., 2020). As such, the “switch” discussed above may reflect a sharp change in the relative weights of GluN2A versus GluN2B rather than a complete replacement. Furthermore, although a large portion of cocaine-generated silent synapses mature by recruiting GluA2-lacking AMPARs (Lee et al., 2013; Shukla et al., 2017; Wright et al., 2020), little evidence exists showing a preferential link of GluN2A signaling to this type of AMPARs. Instead, GluN2A may simply remove the inhibition of AMPAR insertion exerted by GluN2B and its associated synaptic scaffolds without involvement in receptor subtype selection.
Upregulation of GluN2B is also observed at excitatory synapses on midbrain dopamine neurons after cocaine exposure, but instead of GluN1 these subunits are assembled with GluN3A subunits, forming noncanonical NMDARs that promote the synaptic recruitment of CP-AMPARs (Yuan et al., 2013). Similar GluN2B/GluN3A NMDARs are expressed in the NAc core after extended access cocaine self-administration (Dong et al., 2017). It is important for future studies to determine whether these GluN2B/GluN3A NMDARs are also expressed during early phases of cocaine exposure (e.g., 5 d), whether they are present at cocaine-generated silent synapses, and whether they contribute to synaptic recruitment of AMPARs, as demonstrated in midbrain dopamine neurons (Yuan et al., 2013).
After maturation
By making new connections, cocaine-generated synapses are hypothesized to create new connectivity patterns of NAc circuits that promote drug seeking (Dong and Nestler, 2014; Wright and Dong, 2020). After long-term withdrawal, these synapses mature and are expected to be both biochemically and biophysically similar to pre-existing glutamatergic synapses that have undergone cross-the-board upregulation of CP-AMPARs. They may similarly respond to the dysregulated homeostasis of ambient glutamate (Kalivas, 2009) and altered dopamine tone (Volkow et al., 2017), and operate in concert with pre-existing synapses in the NAc to promote cocaine seeking and relapse (Wolf, 2016). However, on re-exposure to cocaine-associated cues, while randomly sampled NAc glutamatergic synapses (the majority of which are presumably non-cocaine-generated synapses) are transiently strengthened (Gipson et al., 2013), matured cocaine-generated synapses are weakened and become AMPAR silent again (Wright et al., 2020), indicating differential behavioral implications (Wright and Dong, 2020). Furthermore, dopamine D1 versus D2 receptor-expressing neurons represent the two major subtypes of NAc MSNs, which undergo differential adaptive changes in response to cocaine and differentially contribute to cocaine-induced behaviors (Lobo and Nestler, 2011; Gangarossa et al., 2013; Calipari et al., 2016). AMPAR-mediated transmissions from several key limbic regions to NAc D1 MSNs are preferentially strengthened after cocaine withdrawal (Britt et al., 2012; Pascoli et al., 2014), promoting cocaine seeking and locomotor responses. This projection- and cell type-specific synaptic strengthening as well as their resulting behavioral consequences are partly mediated by the maturation of silent synapses within the amygdala, prefrontal cortex, and thalamus (Lee et al., 2013; Ma et al., 2014; Neumann et al., 2016).
Concluding remarks
Cocaine-generated silent synapses potentially serve as initial connections in forming new network patterns encoding addition-related memories (Wang et al., 2020; Wright et al., 2020). Our current study characterizes the key trafficking properties of AMPARs and NMDARs during the generation and maturation of nascent synapses after cocaine self-administration. These results not only depict a detailed cellular cascade underlying cocaine-induced synaptogenesis, but also pinpoint key molecular substrates to manipulate drug memories for therapeutic benefit.
Footnotes
The study was supported by National Institutes of Health (NIH)/National Institute on Drug Abuse (NIDA) Grants DA-014133 (to E.J.N.), DA-040620 (to E.J.N. and Y.D.), DA-043826 (to Y.H.H.), DA-046491 (to Y.H.H.), DA-047861 (to Y.D.), DA-023206 (to Y.D.), and NS-107604 (to O.M.S). Cocaine was provided by the NIH NIDA drug supply program. We thank Jaryd Ross for excellent technical support, and Dr. Johannes Hell for providing the GluN2B KI mouse line.
The authors declare no competing financial interests.
References
- Adesnik H, Li G, During MJ, Pleasure SJ, Nicoll RA (2008) NMDA receptors inhibit synapse unsilencing during brain development. Proc Natl Acad Sci U S A 105:5597–5602. 10.1073/pnas.0800946105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, Taghibiglou C, Wang Y, Lu J, Wong TP, Sheng M, Wang YT (2004) Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J 23:1040–1050. 10.1038/sj.emboj.7600126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin KT, Eroglu C (2017) Molecular mechanisms of astrocyte-induced synaptogenesis. Curr Opin Neurobiol 45:113–120. 10.1016/j.conb.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35:345–353. 10.1016/s0896-6273(02)00776-6 [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R (2005) NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 48:289–301. 10.1016/j.neuron.2005.08.034 [DOI] [PubMed] [Google Scholar]
- Bonnet SAD, Akad DS, Samaddar T, Liu Y, Huang X, Dong Y, Schlüter OM (2013) Synaptic state-dependent functional interplay between postsynaptic density-95 and synapse-associated protein 102. J Neurosci 33:13398–13409. 10.1523/JNEUROSCI.6255-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau AC, Reimers JM, Milovanovic M, Wolf ME (2007) Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci 27:10621–10635. 10.1523/JNEUROSCI.2163-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, Phelps L, Phillips AG, Wang YT (2005) Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 310:1340–1343. 10.1126/science.1116894 [DOI] [PubMed] [Google Scholar]
- Britt JP, Benaliouad F, McDevitt RA, Stuber GD, Wise RA, Bonci A (2012) Synaptic and behavioral profile of multiple glutamatergic inputs to the nucleus accumbens. Neuron 76:790–803. 10.1016/j.neuron.2012.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TE, Lee BR, Mu P, Ferguson D, Dietz D, Ohnishi YN, Lin Y, Suska A, Ishikawa M, Huang YH, Shen H, Kalivas PW, Sorg BA, Zukin RS, Nestler EJ, Dong Y, Schlüter OM (2011) A silent synapse-based mechanism for cocaine-induced locomotor sensitization. J Neurosci 31:8163–8174. 10.1523/JNEUROSCI.0016-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill ME, Browne CJ, Wang J, Hamilton PJ, Dong Y, Nestler EJ (2018) Withdrawal from repeated morphine administration augments expression of the RhoA network in the nucleus accumbens to control synaptic structure. J Neurochem 147:84–98. 10.1111/jnc.14563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calipari ES, Bagot RC, Purushothaman I, Davidson TJ, Yorgason JT, Peña CJ, Walker DM, Pirpinias ST, Guise KG, Ramakrishnan C, Deisseroth K, Nestler EJ (2016) In vivo imaging identifies temporal signature of D1 and D2 medium spiny neurons in cocaine reward. Proc Natl Acad Sci U S A 113:2726–2731. 10.1073/pnas.1521238113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Zukin RS (2002) NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci 25:571–577. 10.1016/s0166-2236(02)02272-5 [DOI] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME (2008) Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature 454:118–121. 10.1038/nature06995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cull-Candy SG, Leszkiewicz DN (2004) Role of distinct NMDA receptor subtypes at central synapses. Sci STKE 2004:re16. 10.1126/stke.2552004re16 [DOI] [PubMed] [Google Scholar]
- Diabira D, Hennou S, Chevassus-Au-Louis N, Ben-Ari Y, Gozlan H (1999) Late embryonic expression of AMPA receptor function in the CA1 region of the intact hippocampus in vitro. Eur J Neurosci 11:4015–4023. 10.1046/j.1460-9568.1999.00831.x [DOI] [PubMed] [Google Scholar]
- Dong Y (2016) Silent synapse-based circuitry remodeling in drug addiction. Int J Neuropsychopharmacol 19:pyv136. 10.1093/ijnp/pyv136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nestler EJ (2014) The neural rejuvenation hypothesis of cocaine addiction. Trends Pharmacol Sci 35:374–383. 10.1016/j.tips.2014.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Taylor JR, Wolf ME, Shaham Y (2017) Circuit and synaptic plasticity mechanisms of drug relapse. J Neurosci 37:10867–10876. 10.1523/JNEUROSCI.1821-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand GM, Kovalchuk Y, Konnerth A (1996) Long-term potentiation and functional synapse induction in developing hippocampus. Nature 381:71–75. 10.1038/381071a0 [DOI] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA (2006) Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron 52:307–320. 10.1016/j.neuron.2006.09.012 [DOI] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA (2008) Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci U S A 105:20953–20958. 10.1073/pnas.0811025106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favaro PD, Huang X, Hosang L, Stodieck S, Cui L, Liu YZ, Engelhardt KA, Schmitz F, Dong Y, Löwel S, Schlüter OM (2018) An opposing function of paralogs in balancing developmental synapse maturation. PLoS Biol 16:e2006838. 10.1371/journal.pbio.2006838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, Sinha N, Zuber J (2013) An optimized microRNA backbone for effective single-copy RNAi. Cell Rep 5:1704–1713. 10.1016/j.celrep.2013.11.020 [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE (2000) Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron 27:57–69. 10.1016/s0896-6273(00)00009-x [DOI] [PubMed] [Google Scholar]
- Gambrill AC, Barria A (2011) NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A 108:5855–5860. 10.1073/pnas.1012676108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangarossa G, Espallergues J, de Kerchove d'Exaerde A, El Mestikawy S, Gerfen CR, Hervé D, Girault J-A, Valjent E (2013) Distribution and compartmental organization of GABAergic medium-sized spiny neurons in the mouse nucleus accumbens. Front Neural Circuits 7:22. 10.3389/fncir.2013.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, Kalivas PW (2013) Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron 77:867–872. 10.1016/j.neuron.2013.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Shi Y, Usui H, During MJ, Sakimura K, Nicoll RA (2011) Distinct modes of AMPA receptor suppression at developing synapses by GluN2A and GluN2B: single-cell NMDA receptor subunit deletion in vivo. Neuron 71:1085–1101. 10.1016/j.neuron.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziane NM, Sun S, Wright WJ, Jang D, Liu Z, Huang YH, Nestler EJ, Wang YT, Schlüter OM, Dong Y (2016) Opposing mechanisms mediate morphine- and cocaine-induced generation of silent synapses. Nat Neurosci 19:915–925. 10.1038/nn.4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Gustafsson B, Hanse E (2002) Spontaneous unitary synaptic activity in CA1 pyramidal neurons during early postnatal development: constant contribution of AMPA and NMDA receptors. J Neurosci 22:5552–5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Gustafsson B, Hanse E (2006) AMPA signalling in nascent glutamatergic synapses: there and not there! Trends Neurosci 29:132–139. 10.1016/j.tins.2006.01.005 [DOI] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW (2012) CaMKII binding to GluN2B is critical during memory consolidation. EMBO J 31:1203–1216. 10.1038/emboj.2011.482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanse E, Taira T, Lauri S, Groc L (2009) Glutamate synapse in developing brain: an integrative perspective beyond the silent state. Trends Neurosci 32:532–537. 10.1016/j.tins.2009.07.003 [DOI] [PubMed] [Google Scholar]
- Hanse E, Seth H, Riebe I (2013) AMPA-silent synapses in brain development and pathology. Nat Rev Neurosci 14:839–850. 10.1038/nrn3642 [DOI] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schlüter OM, Zukin RS, Dong Y (2009) In vivo cocaine experience generates silent synapses. Neuron 63:40–47. 10.1016/j.neuron.2009.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Ishikawa M, Lee BR, Nakanishi N, Schlüter OM, Dong Y (2011) Searching for presynaptic NMDA receptors in the nucleus accumbens. J Neurosci 31:18453–18463. 10.1523/JNEUROSCI.3824-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Schlüter OM, Dong Y (2015) Silent synapses speak up: updates of the neural rejuvenation hypothesis of drug addiction. Neuroscientist 21:451–459. 10.1177/1073858415579405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC (1995) Evidence for silent synapses: implications for the expression of LTP. Neuron 15:427–434. 10.1016/0896-6273(95)90046-2 [DOI] [PubMed] [Google Scholar]
- Kalivas PW (2009) The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci 10:561–572. 10.1038/nrn2515 [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA (2008) Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci 9:813–825. 10.1038/nrn2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger JM, Favaro PD, Liu M, Kitlinska A, Huang X, Raabe M, Akad DS, Liu Y, Urlaub H, Dong Y, Xu W, Schlüter OM (2013) Differential roles of postsynaptic density-93 isoforms in regulating synaptic transmission. J Neurosci 33:15504–15517. 10.1523/JNEUROSCI.0019-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BR, Ma Y-Y, Huang YH, Wang X, Otaka M, Ishikawa M, Neumann PA, Graziane NM, Brown TE, Suska A, Guo C, Lobo MK, Sesack SR, Wolf ME, Nestler EJ, Shaham Y, Schlüter OM, Dong Y (2013) Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat Neurosci 16:1644–1651. 10.1038/nn.3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R (1995) Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature 375:400–404. 10.1038/375400a0 [DOI] [PubMed] [Google Scholar]
- Lobo MK, Nestler EJ (2011) The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat 5:41. 10.3389/fnana.2011.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonze BE, Ginty DD (2002) Function and regulation of CREB family transcription factors in the nervous system. Neuron 35:605–623. 10.1016/s0896-6273(02)00828-0 [DOI] [PubMed] [Google Scholar]
- Löw K, Aebischer P, Schneider BL (2013) Direct and retrograde transduction of nigral neurons with AAV6, 8, and 9 and intraneuronal persistence of viral particles. Hum Gene Ther 24:613–629. 10.1089/hum.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loweth JA, Scheyer AF, Milovanovic M, LaCrosse AL, Flores-Barrera E, Werner CT, Li X, Ford KA, Le T, Olive MF, Szumlinski KK, Tseng KY, Wolf ME (2014) Synaptic depression via mGluR1 positive allosteric modulation suppresses cue-induced cocaine craving. Nat Neurosci 17:73–80. 10.1038/nn.3590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y-Y, Lee BR, Wang X, Guo C, Liu L, Cui R, Lan Y, Balcita-Pedicino JJ, Wolf ME, Sesack SR, Shaham Y, Schlüter OM, Huang YH, Dong Y (2014) Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron 83:1453–1467. 10.1016/j.neuron.2014.08.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y-Y, Wang X, Huang Y, Marie H, Nestler EJ, Schluter OM, Dong Y (2016) Re-silencing of silent synapses unmasks anti-relapse effects of environmental enrichment. Proc Natl Acad Sci U S A 113:5089–5094. 10.1073/pnas.1524739113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1997) Silent synapses speak up. Neuron 19:473–476. 10.1016/S0896-6273(00)80362-1 [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC (2005) Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron 45:741–752. 10.1016/j.neuron.2005.01.039 [DOI] [PubMed] [Google Scholar]
- Mayadevi M, Praseeda M, Kumar KS, Omkumar RV (2002) Sequence determinants on the NR2A and NR2B subunits of NMDA receptor responsible for specificity of phosphorylation by CaMKII. Biochim Biophys Acta 1598:40–45. 10.1016/s0167-4838(02)00315-1 [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Lüscher C (2019) The molecular basis of drug addiction: linking epigenetic to synaptic and circuit mechanisms. Neuron 102:48–59. 10.1016/j.neuron.2019.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann PA, Wang Y, Yan Y, Wang Y, Ishikawa M, Cui R, Huang YH, Sesack SR, Schlüter OM, Dong Y (2016) Cocaine-induced synaptic alterations in thalamus to nucleus accumbens projection. Neuropsychopharmacology 41:2399–2410. 10.1038/npp.2016.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary H, Liu WH, Rorabaugh JM, Coultrap SJ, Bayer KU (2011) Nucleotides and phosphorylation bi-directionally modulate Ca2+/calmodulin-dependent protein kinase II (CaMKII) binding to the N-methyl-D-aspartate (NMDA) receptor subunit GluN2B. J Biol Chem 286:31272–31281. 10.1074/jbc.M111.233668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omkumar RV, Kiely MJ, Rosenstein AJ, Min KT, Kennedy MB (1996) Identification of a phosphorylation site for calcium/calmodulindependent protein kinase II in the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem 271:31670–31678. 10.1074/jbc.271.49.31670 [DOI] [PubMed] [Google Scholar]
- Paoletti P, Bellone C, Zhou Q (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14:383–400. 10.1038/nrn3504 [DOI] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O'Connor EC, Lüscher C (2014) Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature 509:459–464. 10.1038/nature13257 [DOI] [PubMed] [Google Scholar]
- Passafaro M, Nakagawa T, Sala C, Sheng M (2003) Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature 424:677–681. 10.1038/nature01781 [DOI] [PubMed] [Google Scholar]
- Plattner F, Hernández A, Kistler TM, Pozo K, Zhong P, Yuen EY, Tan C, Hawasli AH, Cooke SF, Nishi A, Guo A, Wiederhold T, Yan Z, Bibb JA (2014) Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron 81:1070–1083. 10.1016/j.neuron.2014.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA, Roche KW (2013) Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep 3:607–614. 10.1016/j.celrep.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R (2001) Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell 105:331–343. 10.1016/s0092-8674(01)00321-x [DOI] [PubMed] [Google Scholar]
- Shukla A, Beroun A, Panopoulou M, Neumann PA, Grant SG, Olive MF, Dong Y, Schlüter OM (2017) Calcium-permeable AMPA receptors and silent synapses in cocaine-conditioned place preference. EMBO J 36:458–474. 10.15252/embj.201695465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonntag F, Köther K, Schmidt K, Weghofer M, Raupp C, Nieto K, Kuck A, Gerlach B, Böttcher B, Müller OJ, Lux K, Hörer M, Kleinschmidt JA (2011) The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol 85:12686–12697. 10.1128/JVI.05359-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Colbran RJ (1998) Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl- D-aspartate receptor. J Biol Chem 273:20689–20692. 10.1074/jbc.273.33.20689 [DOI] [PubMed] [Google Scholar]
- Suska A, Lee BR, Huang YH, Dong Y, Schlüter OM (2013) Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proc Natl Acad Sci U S A 110:713–718. 10.1073/pnas.1206287110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ultanir SK, Kim JE, Hall BJ, Deerinck T, Ellisman M, Ghosh A (2007) Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc Natl Acad Sci U S A 104:19553–19558. 10.1073/pnas.0704031104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287:864–869. 10.1126/science.287.5454.864 [DOI] [PubMed] [Google Scholar]
- Vickers CA, Stephens B, Bowen J, Arbuthnott GW, Grant SG, Ingham CA (2006) Neurone specific regulation of dendritic spines in vivo by post synaptic density 95 protein (PSD-95). Brain Res 1090:89–98. 10.1016/j.brainres.2006.03.075 [DOI] [PubMed] [Google Scholar]
- Vieira M, Yong XLH, Roche KW, Anggono V (2020) Regulation of NMDA glutamate receptor functions by the GluN2 subunits. J Neurochem 154:121–143. 10.1111/jnc.14970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wise RA, Baler R (2017) The dopamine motive system: implications for drug and food addiction. Nat Rev Neurosci 18:741–752. 10.1038/nrn.2017.130 [DOI] [PubMed] [Google Scholar]
- Wang J, Ishikawa M, Yang Y, Otaka M, Kim JY, Gardner GR, Stefanik MT, Milovanovic M, Huang YH, Hell JW, Wolf ME, Schluter OM, Dong Y (2018) Cascades of homeostatic dysregulation promote incubation of cocaine craving. J Neurosci 38:4316–4328. 10.1523/JNEUROSCI.3291-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Li KL, Shukla A, Beroun A, Ishikawa M, Huang X, Wang Y, Wang YQ, Yang Y, Bastola ND, Huang HH, Kramer LE, Chao T, Huang YH, Sesack SR, Nestler EJ, Schluter OM, Dong Y (2020) Cocaine triggers astrocyte-mediated synaptogenesis. Biol Psychiatry 89:386–397. 10.1016/j.biopsych.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washbourne P, Bennett JE, McAllister AK (2002) Rapid recruitment of NMDA receptor transport packets to nascent synapses. Nat Neurosci 5:751–759. 10.1038/nn883 [DOI] [PubMed] [Google Scholar]
- Wolf ME (2016) Synaptic mechanisms underlying persistent cocaine craving. Nat Rev Neurosci 17:351–365. 10.1038/nrn.2016.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WJ, Dong Y (2020) Psychostimulant-induced adaptations in nucleus accumbens glutamatergic transmission. Cold Spring Harb Perspect Med 10:a039255. 10.1101/cshperspect.a039255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WJ, Graziane NM, Neumann PA, Hamilton PJ, Cates HM, Fuerst L, Spenceley A, MacKinnon-Booth N, Iyer K, Huang YH, Shaham Y, Schlüter OM, Nestler EJ, Dong Y (2020) Silent synapses dictate cocaine memory destabilization and reconsolidation. Nat Neurosci 23:32–46. 10.1038/s41593-019-0537-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT (1996) Maturation of a central glutamatergic synapse. Science 274:972–976. 10.1126/science.274.5289.972 [DOI] [PubMed] [Google Scholar]
- Xia SH, Yu J, Huang X, Sesack SR, Huang YH, Schlüter OM, Cao JL, Dong Y (2020) Cortical and thalamic interaction with amygdala-to-accumbens synapses. J Neurosci 40:7119–7132. 10.1523/JNEUROSCI.1121-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MY, Wasling P, Hanse E, Gustafsson B (2004) Creation of AMPA-silent synapses in the neonatal hippocampus. Nat Neurosci 7:236–243. 10.1038/nn1196 [DOI] [PubMed] [Google Scholar]
- Yao WD, Gainetdinov RR, Arbuckle MI, Sotnikova TD, Cyr M, Beaulieu JM, Torres GE, Grant SG, Caron MG (2004) Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity. Neuron 41:625–638. 10.1016/s0896-6273(04)00048-0 [DOI] [PubMed] [Google Scholar]
- Yuan T, Mameli M, O'Connor EC, O' Connor EC, Dey PN, Verpelli C, Sala C, Perez-Otano I, Lüscher C, Bellone C (2013) Expression of cocaine-evoked synaptic plasticity by GluN3A-containing NMDA receptors. Neuron 80:1025–1038. 10.1016/j.neuron.2013.07.050 [DOI] [PubMed] [Google Scholar]