

Abstract
Schizophrenia (SZ) is a debilitating disease that impacts 1% of the population worldwide. Association studies have shown that inherited genetic mutations account for a portion of disease risk. However, environmental factors play an important role in the pathophysiology of the disease by altering cellular epigenetic marks at the level of chromatin. Postmortem brain studies of SZ subjects suggest that the dynamic equilibrium between DNA methylation and demethylation network components is disrupted at the level of individual SZ target genes. Herein, we review the role of DNA methylation and demethylation in the context of what is currently known regarding SZ. Furthermore, we describe the deficits that accompany two mouse models of SZ. The chronic methionine mouse model of SZ is predicated on the administration of methionine to SZ patients and controls in the context of clinical studies that were carried out during the 1960s and 1970s. The prenatal restraint stress model of SZ is based on a prolonged stress paradigm administered to pregnant dams during gestation days 7–21. The adult offspring of these dams show various behavioral and biochemical deficits in adulthood. Both models are epigenetic in origin and mimic the positive and negative symptoms, as well as the cognitive endophenotypes commonly observed in SZ patients. We also discuss the utility of typical and atypical antipsychotic drugs in alleviating these symptoms in each model.
1. THE EPIGENETIC HYPOTHESIS OF PSYCHIATRIC DISEASE
Current thinking suggests that both genetic predisposition and environmental factors have interacting roles in the pathophysiology of the more common psychiatric disorders, such as schizophrenia (SZ), bipolar illness (BP), and major depression. Familial studies support a strong hereditary component, whereas twin studies implicate the role of additional environmental variables, such as prenatal and early life stressors. There is evidence that environmental stressors impact the development of key brain structures and circuitries, and can lead to the development of psychiatric disease. Genetic studies have implicated some 108 distinct genomic loci associated with the development of SZ.1 However, association is not the same as causation and each genome variant is likely present in only a small number of individuals (1–2%) that are diagnosed with the disorder. He gene mutations that have been identified are intimately involved in the biology and pathogenesis of the disorder but are not, by themselves, sufficient to cause the disease. As described in the sections that follow, environmental factors interact with the expression of multiple genes that show overlap with these 108 loci impacting the levels of expression of these genes in the brain. This suggests that fundamental epigenetic mechanisms are operative in regulating finite gene networks that, when dysregulated, lead to the development of psychiatric disorders. The epigenetic hypothesis of SZ postulates that interactions between environmental factors and the genetic landscape of affected individuals are ultimately responsible for the pathogenesis of the disorder.2 The aforementioned concepts will be elaborated upon in subsequent sections.
1.1. Neurodevelopmental Models—Multiple Hit Hypothesis
Most psychiatric disorders exhibit complex patterns of inheritance. That is, their inheritance is non-Mendelian indicating that it likely involves the interaction of one or more genes with environmental factors. For purposes of this article, we will focus primarily on the effects of epigenetics in the context of SZ, which is a devastating and chronic neuropsychiatric disorder. Moreover, it is triggered by an array of interacting genetic, developmental, and environmental factors that interfere with normal brain development. SZ has a worldwide prevalence of 1% and is a major socioeconomic burden. With some exceptions, it is usually diagnosed in young adults following the first episode of psychosis. Symptoms of SZ are classified as positive, negative, or cognitive. Positive symptoms include hallucinations that can be visual and/or auditory. Delusions are beliefs that may arise from the hallucinations that are also part of the spectrum of positive symptoms. People with SZ have difficulty in concentrating and often have trouble keeping track of their thoughts. Negative symptoms of SZ often appear temporally before onset of the disorder (during the so-called prodromal period). People experiencing negative symptoms may lose interest and motivation in life and activities, including relationships and sex. They become asocial and often feel uncomfortable with people, avoiding conversations, and so on. Patients with SZ over longer periods of time experience declines in cognitive capacity and executive function. Because of the complexities of neuropsychiatric disorders like SZ, modeling these diseases in animals is not an exact science. Nevertheless, there are behaviors that can be used to assess various aspects of social activity in rodents that mimic behaviors reminiscent of those observed in patients diagnosed with SZ. For example, deficits in social interaction can be assessed by examining the amount of active interaction time a mouse spends with a novel or nonfamiliar mouse. These will be discussed in more detail as the various animal models are described.
In the multihit hypothesis of SZ, the initial hit is presumed to be genetic. Environmental factors (stressors) that are triggered by detrimental events act progressively and the effects of these accumulate during the pre- and postadolescent lifespan of the individual (Fig. 1).3 How these stressors impact brain development is currently under intense scrutiny in a large number of laboratories worldwide. Both the intensity of the stressor and the developmental timing of when the stress occurs are important factors that determine the ultimate consequences to the mature brain. Prenatal stresses often include maternal stress during pregnancy (physical or psychological abuse, trauma), maternal infections during pregnancy, obstetrical complications both before and during delivery, exposure to drugs of abuse in utero, maternal and paternal age at conception, and malnutrition and exposure to environmental pollutants in utero. Early postnatal stresses, which occur following birth, directly affect the developing brain and some of the major factors include exposure to physical and psychological abuse and trauma, malnutrition, and poverty. In addition to physical and psychological abuse, poverty, urban city living, later postnatal environmental influences also include abnormal hormonal influences, exposure to drugs of abuse such as cannabis or psychostimulants and bullying.4 It is widely thought that any one environmental factor is not a necessary and sufficient cause of any mental disorder. Instead, it seems that cumulative exposure to multiple environmental influences at different stages in the lifespan is more likely to be predictive of psychiatric disorder risk. However, it should be pointed out that research into how environmental stressors impact the risk of psychiatric disease is at an early stage. Moreover, the list of impactful environmental factors is likely to grow considerably before we clearly understand how relevant these factors may be.4 It seems clear that one of the mechanisms by which stressors impact individual cells during development is by altering the methylation status of vulnerable genes in the genome.
Fig. 1.
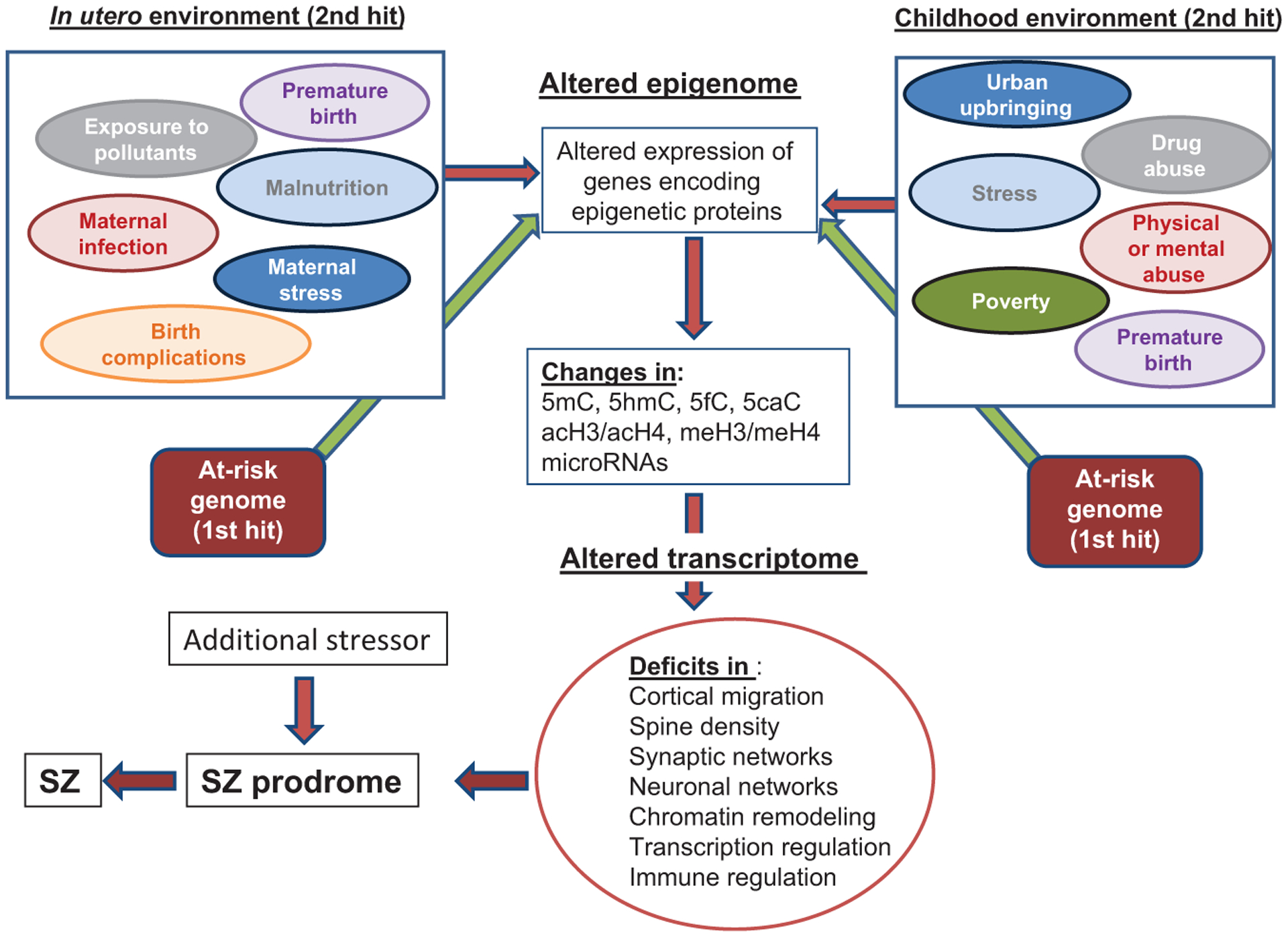
Epigenetic impact of the pre- and postnatal environment on brain development. In the two-hit hypothesis of SZ, a predisposing genome would be considered a first hit. Environmentally induced epigenetic mechanisms acting on the genome during prenatal and postnatal development facilitate genome-wide changes in the epigenome that alter cell type-specific transcriptional profiles at distinct developmental time points. These environmental insults that then act on the predisposing genome at distinct development times represent a second hit. The subsequent mRNA alterations, in turn, produce altered protein profiles that cause deficits in multiple processes including cortical migration, chromatin remodeling, transcriptional regulation, immune-response regulation, and the formation of synaptic connections. Moreover, changes at the cellular level can translate into altered neuronal network connections that ultimately impact the phenotype of the individual. Genome-wide association and whole exome sequencing studies have identified a large number of genes associated with SZ that encode proteins that function in synapse formation, transcriptional regulation, and chromatin-remodeling pathways.
1.2. DNA Methylation/Demethylation Pathway in the Brain
1.2.1. DNA Methyltransferases
DNA methylation is a postreplicative epigenetic modification of DNA that occurs at the 5 position of cytosine and is used by cells as one mechanism to control patterns of gene expression.2,5,6 The DNA methylation/demethylation pathway is schematically shown in Fig. 2. In general, DNA methylation negatively impacts transcription through an interaction with various methyl CpG-binding proteins and other repressor proteins that tend to regionally condense chromatin in the vicinity of transcriptional start sites of targeted genes. DNA methylation is carried out enzymatically by members of the DNA methyltransferase (DNMT) enzyme family which consists of DNMT1, DNMT3a, DNMT3b, and DNMT3l.6,7 The DNMTs catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to the 5 position of cytosine forming 5-methylcytosine (5mC) and S-adenosylhomocysteine (SAH). For many years, it was thought that only the cytosine in the dinucleotide pair CpG is methylated. However, we now know that non-CpG methylation is also abundant in the brain and other tissues and is present throughout the genome.8
Fig. 2.
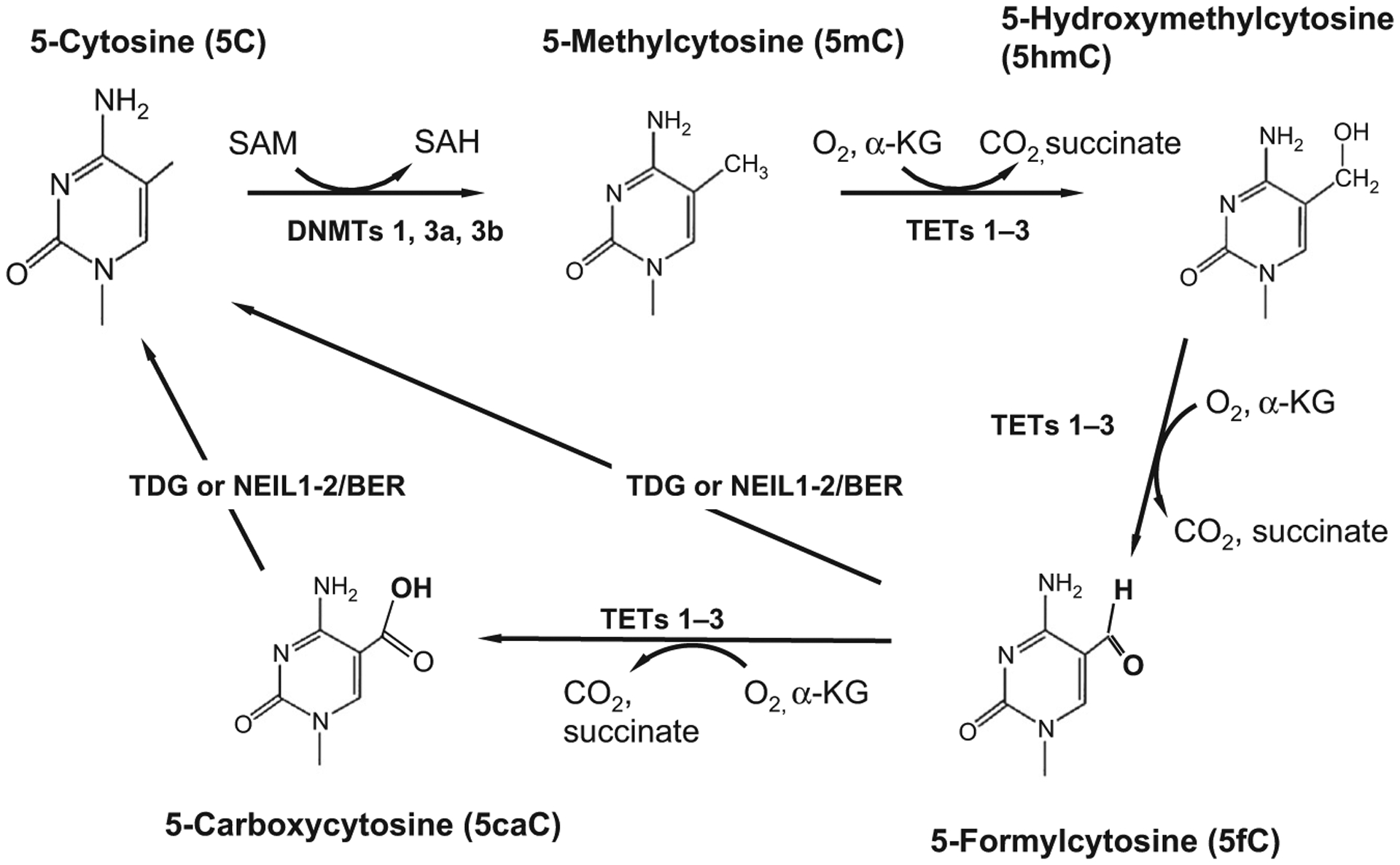
The DNA demethylation pathway. The DNA methyltransferase enzymes (DNMTs 1, 3a, and 3b) that catalyze DNA methylation utilize the universal methyl donor SAM (S-adenosylmethionine) to transfer a methyl group to the 5 position of the heterocyclic aromatic ring of cytosine (5C). Methylcytosine (5mC) is a repressive chromatin mark that negatively correlates with gene expression. Cytosine demethylation, which often reactivates transcription, occurs through several steps, the first of which involves hydroxylation of the methylated cytosine. DNA hydroxymethylation is catalyzed by members of the TET (1–3) family of heme-containing Fe(II)/α-ketoglutarate-dependent dioxygenases. TET proteins further oxidize the hydroxymethyl group to form 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC). Both 5fC and 5caC can be excised by thymine deglycosylase generating an abasic site which is subsequently replaced by base excision repair (BER) enzymes which regenerate the unmodified cytosine base. Unmodified cytosines (5C) are recognized and bound by proteins such as DNMT1 and TET1 that contain a CXXC motif in their zinc finger DNA-binding domain.9 Methyl-binding domain proteins, like MeCP2 and MBD2, bind to 5mC with high affinity.
The DNMT proteins have a similar domain structure, although there are considerable differences between DNMT1, the maintenance methyl transferase, and the de novo methyltransferases (DNMTs 3a and 3b).10 DNMT1 differs significantly at the amino terminus where it has numerous protein domains that allow it to interact with multiple additional proteins that are necessary for it to target replication foci while carrying out its function during DNA replication. That is, DNMT1 serves as a maintenance methyltransferase that methylates hemimethylated DNA by adding a methyl group to the complementary strand of DNA. In this way, methylation patterns are faithfully maintained following DNA replication. It is present at high levels in mature, postmitotic GABAergic neurons of the motor and piriform cortices, striatum, CA1 region of the hippocampus, dentate gyrus, and basolateral amygdala of mouse brains.11 The DNMT3 proteins share sequence and structural similarity and are responsible for adding new (de novo) methylation marks to previously nonmethylated DNA. DNMT3a is predominantly present in postmitotic neurons and oligodendrocytes, whereas DNMT3b is expressed during early stages of neurogenesis.12 That is, both DNMT1 and DNMT3a are present in mature differentiated neurons, whereas DNMT3b expression is either low or is not detectable in these cells. Mice with conditional knockouts of both DNMT1 and DNMT3a in excitatory forebrain neurons show abnormal long-term potentiation, as well as learning and memory deficits.13 While no neuronal loss was detected in double DNMT1/3a knockouts, these neurons showed substantially deregulated patterns of gene expression.13 DNMT3l is most similar to the DNMT3 proteins although it is catalytically inactive.5,6,14 There is evidence showing that DNMT3l binds to the tails of histone H3 and recruits DNMT3a and 3b to methylate DNA.15 However, while DNMT3l is expressed in the developing brain, it is downregulated during neuronal differentiation and is not abundantly expressed postnatally.14,16,17
It was initially reported that DNMT1 mRNA is more highly expressed in GABAergic interneurons of the prefrontal cortex of postmortem SZ cortex (BA 10) than in nonpsychiatric subjects.17–21 This finding was accompanied by the observation that Reelin (RELN) and glutamic acid decarboxylase 1 (GAD1) mRNAs are downregulated in these same postmortem brain samples. In situ hybridization studies show that these changes occur in the same cortical GABAergic neurons, suggesting a causal link between DNA methylation and the decreased expression of RELN and GAD1. Additional evidence for this regulation was provided by a study using primary neuronal cultures in vitro,22 in which a DNMT1 antisense oligonucleotide was used to show that the downregulation of DNMT1 protein is coincident with the increased expression of RELN and GAD1 mRNAs. In addition to the increased expression of DNMT1 in the brain, it was also observed in lymphocytes of patients diagnosed with SZ as compared with nonpsychiatric control lymphocytes.23 Furthermore, using laser-capture microdissection, it was shown that DNMT1 is overexpressed in layer 1 GABAergic neurons of the PFC of SZ subjects.21 Collectively, these data form the basis for subsequent studies that demonstrate that DNA methylation is increased at selected promoters including RELN in SZ PFC.24,25
DNMT1 is the maintenance methyltransferase and it adds a methyl group to hemimethylated DNA to make a double-stranded copy of the methylated residues in the daughter strands. While DNMT1 has been shown to exhibit limited de novo activity, it favors hemimethylated DNA as a substrate over nonmethylated DNA. That is, the intrinsic preference for hemimethylated DNA as a substrate is estimated at 30- to 40-fold.26 However, siRNA knockdown of DNMT1 do not alter maintenance methylation patterns.27,28 Moreover, DNMT1 possesses a cysteine-rich CXXC protein domain which allows it to bind to unmethylated C residues with higher affinity than either hemimethylated or methylated DNA.29,30 The CXXC domain is also required for DNMT enzymatic activity.29 It seems likely that DNMT1 acts both to methylate DNA and as a general repressor protein repressing transcription in the region to where it is bound. While DNMT1 is usually downregulated in nonproliferating cells, significant expression occurs in the brain where the majority of cells are postmitotic.31 In the brain, DNMT1 is predominantly expressed in neurons11 and immunohistochemistry shows it to localize to the cytoplasmic compartment.31 This observation is particularly curious, as most of the known functions of DNMT1 reside in the nucleus. A clue to this immunohistochemical staining pattern was recently revealed when it was discovered that DNMT1 is encoded by multiple isoforms. That is, upstream and in-frame with the highly conserved amino acid sequence of DNMT1, there is the equivalent of 101 codons downstream of two upstream ATG start codons.32 These additional start codons are functional producing two isoforms in addition to the highly conserved ATG translational start codon specifying isoform 1.33 More recently, it was discovered that isoform 3 of DNMT1 is targeted to the mitochondria where it modulates mitochondrial function by methylating the mitochondrial genome.34 This alternatively spliced isoform lacks the typical nuclear localization signal normally present on mature DNMT1 protein isoforms, which likely accounts, in part, for much of the cytoplasmic DNMT1 antibody staining. In addition to the nuclear localization signal, there are numerous additional protein domains that allow it to interact with histone tails and other proteins in the formation of repressor complexes.35,36
1.2.2. DNA Methylation in Schizophrenia
DNA methylation plays an important role during development by epigenetically altering cell-specific patterns of gene expression across distinct cells of the brain. Epigenetic regulation is particularly critical during development because gene expression is dynamic during fetal and infant life and becomes more stable during later periods of the lifespan. It has long been thought that the dysregulation of epigenetic mechanisms plays a vital role in the pathogenesis of psychiatric disorders such as SZ.6 It is known that external or environmental factors facilitate changes in DNA methylation at both specific regulatory loci and also globally.37 In addition, a large body of research supports the concept that the development of SZ is dependent on environmental variables especially during fetal and perinatal development (Fig. 1).
Studies of DNA methylation in the brain show that the dorsolateral prefrontal cortex exhibits dramatic changes in DNA methylation during fetal compared with postnatal development.38,39 These methylation changes are likely due to alterations in neuronal composition that occurs rapidly during early fetal development.38 That is, the large gene expression changes that accompany early fetal development are also characterized by a decrease in progenitor-like cells and an increase in differentiated adult neurons and non-neuronal cells. These cellular composition profiles explain some of the variability observed in methylation at individual CpGs especially during the transition from prenatal to postnatal life.37,38 In SZ patients, methylated CpGs associated with this disorder strongly correlate with changes related to the prenatal to postnatal transition.37 Using the Infinium HumanMethylation450 array, the methylation status of a large number of postmortem SZ subjects and nonpsychiatric controls was evaluated.40 They identified 920 CpGs that were found to be differentially methylated at a false discovery rate of <0.2, 625 of which changed in the same direction.40 These findings are complicated by the observation that the majority of SZ subjects were on antipsychotic medications at their time of death. Antipsychotics, such as clozapine, may alter the methylation profile of brain CpGs.41 Interestingly, nearly a quarter of the significantly differentially methylated CpGs observed in SZ subjects included loci identified from genome-wide association studies.40 That is, the developmentally associated changes in DNA methylation were significantly enriched in genomic regions that confer inherited risk for SZ.40 These data, which were derived from 335 SZ patients and 108 normal subjects, support an important role for DNA methylation in the developmental origins of SZ. In addition to genome-wide studies of DNA methylation, there are numerous candidate gene reports showing DNA hyper- and hypomethylation at various selected candidate genes (recently reviewed).42
A key question in contemporary psychiatry concerns the extent to which methylation signatures observed in accessible tissues, such as blood, are also present in brain. This question is driven due to the lack of brain tissue availability from recently diagnosed patients. One study that was done to compare the methylation profiles between brain and blood showed that a large proportion of tissue-specific, differentially methylated regions were located near genes involved in functional pathways related to neurodevelopment and neuronal differentiation.43 They also showed high correlations (~70%) between blood and brain tissues for sites that demonstrate significance between individual variations in DNA methylation.43 Another study utilized methyl-binding domain enrichment sequencing to compare the profiles of haloperidol treated and untreated mice.44 They reported that haloperidol treatment leaves biomarker signatures in blood and that the methylation status of many sites in the blood mirrors those in brain.44 These authors conclude that findings in the blood have value as a proxy for brain tissue. In contrast, another group evaluated DNA methylation from the blood and brain of the same individuals suffering from chronic bouts of epilepsy.45 These investigators observed that only 7.9% of CpG sites showed a statistically significant, large correlation between blood and brain.45 A methylation-wide association study (MWAS) of over 700 SZ subjects and 700 nonpsychiatric subjects identified the top MWAS finding to be located near FAM63B, a participant in networks regulated by microRNAs linked to neuronal differentiation and dopaminergic gene expression.46 The authors report that their association studies suggest testable hypotheses relevant to disease mechanisms and yield biomarkers that could prove useful to improve disease management.46 This report was followed by a second MWAS assessing the methylation status of all common CpG SNPs using brain and blood by methyl domain-binding protein sequencing.47 Their results indicate that >90% of CpG SNPs that are methylated in brain are also methylated in blood.47 Collectively, it seems clear that the amount and distribution of 5mC reflect the effects of environmental stressors on the brain. Moreover, this is also reflected in the blood of affected individuals.
1.2.3. Ten-Eleven Translocation Methyldioxygenases
In addition to DNA methylation, DNA hydroxymethylation is a common DNA modification in mammalian cells.48 In fact, 5hmC represents between 0.3% and 0.7% of total nucleotides with highest levels present in the central nervous system.49,50 DNA hydroxymethylation is catalyzed by one of three members of the Ten-Eleven Translocation (TET) family of methyl dioxygenases. These enzymes (TETs 1, 2, and 3) are Fe(II)-dependent and use molecular oxygen and α-ketoglutarate to transfer a hydroxyl group to 5mC producing 5-hydroxymethylcytosine (5hmC) (Fig. 2). The expression of TET proteins is cell-type specific, with TET1 being abundant in embryonic stem cells and TETs 2 and 3 showing comparable expression profiles in other tissues.51,52 Both TET1 and TET3 have cysteine-rich CXXC domains in their structure which are associated with DNA binding and targeting.30,53
For many years, 5mC was thought to be thermodynamically stable and the demethylation reaction was thought to be an unfavorable reaction. However, we now know that DNA demethylation is viable and that hydroxymethylation of 5mC is the first step in the pathway. In addition to 5hmC, TET proteins can further oxidize 5hmC forming 5-formylcytosine (5fC) and also 5-carboxylcytosine (5caC).54 As shown in Fig. 2, these groups are further processed and removed by thymidine DNA deglycosylase (TDG). Depletion of TDG in murine embryonic stem cells leads to the accumulation of detectable levels of 5caC.55 5hmC, 5fC, and 5caC are each stable epigenetic marks that accumulate to different extents in the brain. 5hmC is particularly prominent in Purkinje cells of the cerebellum.29,49 At the same time, 5hmC, 5fC, and 5caC show different abundances and tissue specificities. 5hmC is from 10- to 100-fold more abundant than 5fC/5caC and is enriched in neurons.54 This is likely due to the preferences of each of the TET proteins for particular substrates and their differential distribution in distinct cell types. Both TET1 and TET2 show a preference for 5mC as substrate as compared with either 5fC or 5caC.56 Similar to DNMT1, TET3 has a cysteine-rich CXXC domain and it has a high affinity for 5caC to which it binds during 5caC excision by base repair enzymes.54,57 The observation that these additional epigenetic marks are stable and accumulate to differing extents supports the view that 5hmC, 5fC, and 5caC each play distinct roles in the epigenome.
Thymine deglycosylase is a DNA repair enzyme that removes 5fC and 5caC through the base excision repair (BER) pathway. The iterative oxidation of 5mC forming 5hmC, 5fC, and 5caC is the primary means by which active DNA demethylation of 5mC begins in mammalian cells (Fig. 2). The terminal base is removed by either TDG or members of the prototypical endonuclease VIII (Nei) or Nei-like (NEIL-1–3) proteins. NEIL1 and NEIL2 promote TDG-mediated excision of 5fC and 5caC.58 While it has been suggested that 5hmC is instead deaminated by activation-induced deaminase (AID) forming 5hmU which is subsequently removed by BER enzymes,59 no detectable deamination of 5hmC has been found even for cells overexpressing AID/APOBEC.60 A role for AID/APOBEC enzymes in demethylating 5mC or its oxidized substrates is still unclear. It has recently been shown that while APOBEC3A readily deaminates 5mC, it still has a higher catalytic rate for unmodified cytosine in the identical sequence context. It also has a vastly reduced affinity for 5hmC, 5fC, or 5caC.61 This group developed sensitive assays for detecting deamination and they showed that APOBEC3A deaminates 5hmC at a 5600-fold lower rate than 5mC.61 They note that 5fC was modified at an even lower rate while there was no detectable activity for 5caC.61 Overall, their data suggest that deamination of oxidized 5hmC and other oxidation products is highly unfavorable. It seems instead that the main pathway for DNA demethylation is the iterative oxidation of 5mC to 5fC and 5caC which is then excised by TDG or related enzymes (e.g., NEIL1–3) and replaced by BER enzymes.54
1.2.4. Distribution of 5hmC in the Genome
In the previous section, we noted that 5mC is further oxidized by members of the TETenzyme family. 5hmC is a stable epigenetic mark and accumulates to measurable levels in the genome. In the adult human brain, 13.4% of all CpGs are highly hydroxymethylated, which strongly implicates a regulatory role in the modification.39 While 5mC is symmetrically situated on both strands of DNA in CpG pairs, 5hmC is usually present on only one of two strands. That is, while one strand is hydroxymethylated, the opposing CpG site usually remains methylated indicating that 5hmC/5mCpG is one of the major states for 5hmC in the mammalian genome.39,62 Interestingly, while 5hmC is generally associated with gene transcription, it is found at high levels in the gene bodies of active genes. However, similar to what has been observed with DNA methylation and transcriptional repression, the relationship between hydroxymethylation and transcription is complex. As one example, genes that are actively transcribed have low levels of 5hmC, whereas genes that are expressed at low levels have high amounts of 5hmC at their promoters. At the same time, the distribution of 5hmC is very different between neural stem cells and embryonic stem cells. It is interesting to note that enrichment of 5hmC at promoters and proximal to transcription start sites most often associates with TET1 and 5hmC located in gene bodies and at exon boundaries correlates with TET2 activity.63 It appears that many of the studies examining 5hmC abundance and gene expression are not consistent.54 Moreover, it seems like the effect of 5hmC on gene transcription is both cell-type dependent and context dependent. It has also been shown that there is a transcription correlated bias of 5hmC on the sense strand of the DNA while there is a 5mC bias on the antisense strand.39
Whole genome mapping of 5hmC has shown that 5hmC is present in promoters, enhancers, gene bodies, repetitive DNA elements, and intergenically.39 5hmC levels are 10-fold higher in the adult brain as compared with the fetal brain. Analysis of the genomic distribution of 5hmC during developmental periods indicates that there are still a large number of CpGs that have higher 5hmC levels in the fetal brain as compared with the same sites in the adult brain. The distribution of these fetal 5hmC sites shows that they are highly enriched in enhancers and not at other genomic features consistent with a role for 5hmC at regulatory domains in the fetal brain.39 Using TET-assisted bisulfite sequencing, the distribution of 5hmC in the genome was mapped. They showed that there were some 28.4 million 5hmCs in the adult human prefrontal cortex.39 In general, poised enhancers, which are marked by H3K4me1, have the highest levels of 5hmC, followed by active enhancers (marked by H3K4me1 and H3K27ac), introns, exons, and intergenic regions.39 Prominent 5hmC peaks are associated with exon-intron boundaries where they mark 5’ splice sites.39,64 Collectively, the results imply novel roles for 5hmC in regulating mRNA splicing and gene regulation. Highly hydroxymethylated CpGs are strongly enriched at multiple genomic features where they comprise ~13% of all CpGs in the genome.
While a considerable amount of information is known regarding hydroxymethylation in the brain, considerably less information is available regarding 5hmC in the context of psychiatric disorders. There is considerable interest in the role of 5hmC in SZ, especially since the report that showed elevated levels of TET1 in the inferior parietal lobule of a group of 9 BP with psychosis and 10 SZ subjects. At the same time, a group of 11 nonpsychiatric controls and 12 major depressed subjects failed to share this increase in TET1 mRNA levels.65 Interestingly, the CXXC domain containing protein DNMT1 shows increased binding to CpG rich GABAergic promoters in the prefrontal cortex of a cohort of SZ and BP patients obtained from the Harvard Brain Tissue Resource Center.66 Studies carried out thus far on 5hmC and SZ have been primarily focused on candidate genes associated with the disease. For example, significant increases in 5hmC levels were reported in this same postmortem SZ brain cohort at the GAD1 promoter. In another study, investigators observed increased amounts of both 5mC and 5hmC at BDNF promoter IXabcd in parietal cortical samples of psychotic patients from the Stanley Foundation Neuropathology Consortium.67 These increases were accompanied by decreased expression of the BDNF IXabcd mRNA levels. While studies thus far implicate a role for aberrant hydroxymethylation in the context of SZ, this is an understudied area of research.
2. METHIONINE MOUSE MODEL OF PSYCHOSIS
2.1. Role of Methionine in Schizophrenia
The idea for the methionine (MET) mouse model of SZ originated with clinical studies that were carried out in the 1960s and 1970s in which SZ subjects were administered large amounts of the amino acid METalong with a monoamine oxidase inhibitor. MET is an essential amino acid and is a methyl group donor which, when administered exogenously, increases the levels of SAM in the brain. The transmethylation hypothesis of SZ was first proposed in 1952,68 in which it was hypothesized that increases in the abnormal metabolism of dopamine, noradrenaline, and serotonin might be responsible for the psychotic symptoms of the disease.68 In other words, the disease might be the consequence of the production of psychotomimetic methylated derivatives of catecholamines or indolealkylamines in the brain.69 Excess MET was expected to increase the activity of enzymes that transmethylate biogenic amine precursors through O-methylation.70 The intention of the 10 clinical trials that were carried out was to alleviate the positive symptoms of patients by decreasing the levels of psychomimetic biogenic amines. However, what happened, instead, was that 64% of participating SZ subjects exhibited a worsening of their symptoms.70 MET had a profound pharmacologic effect on mood, perception, and function in patients diagnosed with SZ. The response of patients varied in severity and included an increase in psychosis, auditory and visual hallucinations, confusion and disorientation, autonomic responses, and psychomotor retardation. The recrudescence of psychotic symptoms varied in severity and in the numbers of patients responding with adverse symptoms between clinical trials. Of the SZ patients administered MET, two groups were identified, reactors and nonresponders. Reactors responded with an exacerbation of their symptoms, whereas nonresponders were relatively unaffected by the treatments. Interestingly, there were no diagnostic differences between these groups that might help to explain the differential responses.71
To study the effects of exogenous METon epigenetic marks in neurons in vitro, we administered a large dose of MET (2 mM) to primary cultures of cortical neurons prepared from embryonic day 16 rat pups.22 These cultures are mixed, with primarily GABAergic neurons based on the extent of GAD1 immunoreactivity and some glial cells. The addition of MET caused a downregulation of GAD1 and RELN mRNAs with no change in the amount of neuron-specific enolase mRNA.22 This result was accompanied by an increase in the levels of DNA methylation at the RELN promoter suggesting a mechanism for the reduced expression of the corresponding mRNA. That is, it seemed likely that the downregulation of the RELN mRNA was caused by increased promoter methylation mediated by the action of one of the DNMTs. To test this hypothesis directly, we used an antisense knockdown strategy. To determine whether the increased expression of DNMT1 was responsible for the reduced expression of RELN mRNA, we designed an antisense oligo that was previously used to knockdown DNMT1 mRNA in various cancers in mice ex vivo.72 The antisense was introduced into primary cortical neurons and the primary cultures were treated with MET. The downregulation of DNMT1 mRNA and protein was confirmed by qRT-PCR and western blotting, respectively. The reduced expression of DNMT1 was accompanied by an increase in both RELN and GAD1 mRNAs in the MET-treated cultures. These data were used to provide a mechanistic hypothesis regarding the recrudescence of psychotic symptoms in the MET-treated SZ subjects. We proposed the hypothesis that MET treatment of SZ subjects facilitates changes in the genome-wide methylation profiles of genes actively participating in GABAergic and glutamatergic neurons of the brain such that the expression of genes involved in neuronal function is downregulated in response to the amino acid.73
2.2. Methionine Mouse Model of Schizophrenia
It has been known for some time that both dietary and subcutaneously administered MET is converted to SAM by the enzyme MET adenosyltrans-ferase 2A (MAT2A). SAM is the universal methyl donor in virtually all living cells.73 Our group had also recently shown that SAM levels in the prefrontal cortex (BA9) of patients with SZ and BP are increased nearly twofold when compared with the levels in nonpsychiatric controls.74 The subcutaneous injection of a large dose of MET (5.2 mmol/kg, twice daily for 5 days) in mice facilitates a more than twofold increase in both SAM and SAH levels.75 Moreover, the increased levels of SAM correlate with an increase in DNMT1 mRNA levels in GABAergic neurons of the frontal cortex.75 Preliminary experiments were used to establish the MET dose needed to yield the maximal response in terms of elevated levels of SAM and SAH for the model which was 6.6 mmol/kg (twice daily) of MET. The brain SAM and SAH levels increased with increasing MET dose and time after subcutaneous injection.75
2.2.1. Behavioral Phenotype
We showed that prepulse inhibition (PPI) of startle, which represents a measure of sensory gating in both humans and rodents, decreased at a faster rate as the prepulse/startle interval increased in MET-treated mice compared with saline-treated mice. In a follow-up to the aforementioned study, the behaviors associated with chronic MET treatment in mice were expanded to include measures of social interaction. The chronic administration of MET to mice not only impairs sensory gating (PPI), but also induces a deficit in social interaction time in treated animals.76 The deficit in social interaction is reminiscent of the negative symptoms of SZ.77 In addition, chronic injection of MET into mice prevents the development of aggressive behavior toward intruder mice following a 2–3-week period of social isolation.76 In this latter study, VPA when simultaneously administered with MET was shown to prevent both the PPI and social interaction deficits.
More recently, the MET-induced mouse model of SZ was revisited by investigators that showed several additional behaviors associated with the model.77 First, they not only replicated the deficit in sensory gating (PPI) and the social interaction deficit observed by others, but also showed that these mice do not exhibit any pathological effects due to the treatment. They also showed that chronically treated mice show greater levels of stereotyped behaviors. In the forced swim test, MET-treated mice display more immobility time than saline-treated animals indicating a lower level of motivated affection. Using the novel object recognition test, MET-treated mice exhibit impaired recognition. Finally, in the inhibitory avoidance assay, while there were no differences in latency to enter the dark chamber, following 48 h, MET-treated mice displayed an increase in the latency time to enter the dark chamber and a decreased number of animals avoided entering the dark chamber.77 The latter two tests are indicative of impaired cognition. The chronic MET treatment had no effect on body weight and did not alter their nociceptive response as measured by the tail flick assay.77 Comparison of both behavioral and biochemical deficits in the MET mouse model of SZ with SZ-like behaviors are listed in Table 1.
Table 1.
Comparison of Molecular and Behavioral Abnormalities in SZ and BP Disorder Patients With the Mouse MET and PRS Models of Psychiatric Disease.
| SZ + BP Patients | MET Mice | PRS Mice | |
|---|---|---|---|
| Molecular changes | |||
| DNMT1, DNMT3a, TET expression | ↑ | ↑ | ↑ |
| DNMT1 or MECP2 binding to GAD1, RELN, BDNF promoters | ↑ | ↑ | ↑ |
| 5mC, 5hmC enrichment at RELN or GAD1 | ↑ | ↑ | ↑ |
| Candidate gene (GAD1, RELN, BDNF) expression | ↓ | ↓ | ↓ |
| Behavioral changes | |||
| Positive symptoms (stereotyped behavior, sensitivity to NMDA receptor antagonist) | ↑ | ↑ | ↑ |
| Negative symptoms (social interaction deficits) | ↑ | ↑ | ↑ |
| Cognitive, information processing deficit (PPI, fear conditioning) | ↑ | ↑ | ↑ |
2.2.2. Biochemical Phenotype
A number of different assays were used to examine aspects of chromatin architecture in the vicinity of candidate genes relevant to SZ pathophysiology. One of the first experiments performed was to determine whether RELN and GAD1 mRNAs were reduced in MET-treated mice. Both RELN and GAD1 are reduced in this model. Another study showed that the downregulation of RELN and GAD1 was accompanied by a loss of layer III pyramidal cell dendritic spin density in the frontal cortex of treated animals.78 Using bisulfite genomic sequencing, it was next shown that the region upstream of the mouse RELN promoter was more heavily methylated, by nearly twofold, in MET-treated mice as compared with saline-treated mice.75 No change was observed in RELN promoter methylation status following a comparable injection of the amino acid glycine. Finally, coinjection of the mood stabilizer and histone deacetylase (HDAC) inhibitor, VPA, reverted the MET-induced downregulation of RELN and GAD1 through an epigenetic action involving HDACs.75 The same dose of VPA increased H3 histone acetylation in mouse brain nearly fourfold.75 The latter experiment suggested the possibility that the inhibition of HDACs might represent a suitable strategy for alleviating the epigenetic risk of individuals genetically susceptible to SZ.
Some of the aforementioned data were replicated in a separate study that showed that in addition to hypermethylation of the RELN and GAD1 promoters, MET treatment facilitates a recruitment of MeCP2 to the corresponding promoter regions in the frontal cortex of treated mice.79 No change in promoter 5mC levels at the GAD2 promoter was detected indicating a degree of gene specificity to the response. The MET-induced RELN and GAD1 mRNA downregulation is reversed after 7 days of withdrawal. Moreover, the recruitment of MeCP2 to the RELN and GAD1 promoter is time dependent and is reversed nearly completely after 6–9 days following MET withdrawal.80 Similar to previous studies, VPA and the chemically unrelated benzamide HDAC inhibitor, MS275,81 reverses the RELN and GAD1 mRNA downregulation and promoter hypermethylation.80 VPA and MS275 have overlapping but distinct inhibitory profiles and different IC50s at individual Class I HDACs. Finally, the DNMT1 inhibitor, procainamide,82 when administered simultaneously with MET, blocks the accumulation of MeCP2 present at the RELN and GAD1 promoters. These data expand the existing knowledge of MET-induced epigenetic changes at the RELN and GAD1 promoters by providing information regarding the reversibility of these effects. In addition, the studies with inhibitors now include the HDAC inhibitor MS275 and the DNMT1 inhibitor procainamide.
2.2.3. Responsiveness of the Methionine Model to Medications
To test whether VPA used in combination with typical or atypical antipsychotics might reverse the RELN and GAD1 mRNA decrease and promoter hypermethylation, clozapine (CLZ), sulpiride (SULP), haloperidol (HAL), and olanzapine (OLZ) were tested in MET-treated mice.83 CLZ, SULP, HAL, and OLZ were administered to mice following 7 days of MET treatment. Results showed that doses of CLZ and SULP, but not HAL or OLZ, facilitated a demethylation of the MET-mediated promoter hypermethylation at the RELN and GAD1 promoters in the frontal cortex and striatum.83 Interestingly, the action of these atypical antipsychotics was potentiated by the coadministration of a relevant dose of VPA.83 Neither HAL nor OLZ alone or in combination with VPA were able to elicit demethylation at these two promoters or to alter the behavioral deficits. This contrasts with another study that showed that a single injection of HAL and CLZ reverses the locomotor hyperactivity and stereotyped behavior induced by chronic MET administration.77 While these differences could be due to the doses used in the respective studies, only the more recent study examined sterotyped behaviors. Nevertheless, the chronic MET mouse model of SZ has face and predictive validity and may be useful in the preclinical search for new neuroleptic drugs.
3. PRENATAL RESTRAINT STRESS MODEL OF SCHIZOPHRENIA
As indicated earlier, SZ is characterized by an abnormal genetic load and an environmental insult that is necessary for onset of the disease. Stressors associated with gestation include malnutrition, premature birth, complications during delivery, and so on (Fig. 1). Animal studies confirm the sensitivity of the developing brain to environmental disturbances. Exposure of rats and mice to stresses, immune challenges, infection, and malnutrition during pregnancy leads to a disruption of various behavioral and neurochemical parameters in adult offspring.84 The effects of chronic hyperactivation of the hypothalamus–pituitary–adrenal axis in fetal rodents have been examined for a number of years now.85 Prenatal restraint stress in mice has been shown to produce delays in the tangential migration of inhibitory neuron progenitor cells of the developing neocortex.86 In addition, prenatal stress has been shown to impair the emotional responses to subsequent stress implicating a reduced ability to adapt to new stresses.87 This impaired adaptive response is linked to a disruption of the development of serotonergic neurons in the embryonic hindbrain.87 Moreover, chronic prenatal stress also affects lymphoid cells and alters neurotrophin and cytokine expression in lymphocytes.88 Collectively, prenatal stress affects multiple systems in the developing animal including long-lasting neurobiological and behavioral changes that are linked to neuroplasticity. Because different strains of mice respond to prenatal stress differently, it is important to test behavior in each strain at the start of experimentation.
Chronic overactivity of the HPA axis produces effects that include the repression of reproduction, growth, as well as thyroid and immune functions, which have been shown to be associated with pathological states.85,89 In addition, exposure of the brain to prolonged periods of stress in utero likely results in the hyperactivity of the stress system, defective negative feedback by glucocorticoids, alterations in cognition, novelty seeking, increased vulnerability to addictive behaviors, and mood disorders.85 Moreover, mice that have been subjected to chronic prenatal stress show evidence of behaviors reminiscent of the positive and negative symptoms of SZ.90
3.1. Behavioral Phenotype of PRS Mice
The adult offspring of dams exposed to repeated episodes of restraint stress during gestation (termed PRS mice) develop a SZ-like endophenotype. While at birth the PRS mice exhibit 10% reduced body weight and are born from 12 to 18 h earlier than nonstressed (NS) mice, there are no differences in body weight, auditory sensitivity, pain sensitivity, or motor coordination at adulthood.84 Swiss-albino ND4 mice, prenatally stressed from gestation days 7 to 21, show a robust and persistent hyperlocomotor activity as measured by horizontal activity.84 The increase observed in locomotor activity was recently replicated in another study.91,92 In addition, PRS mice exhibited a decrease in social interactions with an intruder mouse.84 More recently, the social interaction deficit was also replicated in a similar study.91,92 Social withdrawal is a negative symptom of SZ. PRS mice also show deficits in PPI, which reflects sensory information processing or sensory gating.84 PRS mice demonstrate a significant deficit in fear conditioning response in contextual fear conditioning tests.84 Moreover, PRS mice exhibit increased stereotyped behaviors which is sensitive to the administration of the NMDA subtype of glutamate receptor.84 Collectively, the behavioral deficits in PRS mice are representative of behaviors observed in patients diagnosed with SZ and BP (Table 1).
3.2. Biochemical Phenotype of PRS Mice
To better understand the underlying mechanism associated with the altered epigenetic framework in adult PRS mice, experiments were performed early in development to examine the expression of DNMT1 and DNMT3a during development following prenatal stress.84 Examination of the postnatal mRNA profile of DNMT1 and 3a in the frontal cortex shows that both mRNAs are highest at postnatal day 1 (PD1). Both mRNAs show a robust decrease by PD7 which continues until PD14. There is a gradual decline thereafter until adulthood (PD60) at which point, expression of both mRNAs is lowest.84 NS mice show a similar temporal decline in mRNAs but at each time point, mRNA levels in NS mice are about 50% of that in the PRS mice. The levels of both mRNAs were also about half of that observed in the PRS mouse hippocampus.84 These results suggest that both DNMTs are overexpressed in the frontal cortex and hippocampus, and this suggests that the levels of DNA methylation are temporally increased in PRS mice as well.
Epigenetic studies of adult PRS mice at the biochemical level show that subtypes of metabotropic glutamate receptors (mGluR) are downregulated in PRS adult offspring. In one study, it was shown that the downregulation of mGluR2 and mGluR3 in the frontal cortex of PRS mice is linked with an increased expression of DNMT1.93 There was increased DNMT1 mRNA and protein in the frontal cortex and increased DNMT1 binding to the mGluR2 and mGluR3 promoters in the frontal cortex of PRS mice.93 In addition, MeCP2 binding to these promoters increased in PRS mice suggesting that the increased DNMT1 binding was accompanied by increased levels of DNA methylation.93 It was further established that the GABAergic neuron-specific GAD1 and the neurotrophin BDNF-IX mRNAs were downregulated.93 It was also shown that there was increased binding of MeCP2 to the corresponding GAD1 and BDNF-IX promoter regions.93
In a follow-up study, these investigators extended their observations to include RELN and GAD1 expression.84 Similar to what was observed with the mGluR mRNAs, RELN and GAD1 mRNAs were downregulated and there was increased binding of DNMT1 and MeCP2 to the corresponding promoter regions. The levels of 5mC and 5hmC at these two promoters were elevated in the PRS frontal cortex relative to NS mice.84 In addition to elevated DNMT1 mRNA, a time-dependent increase in the levels of TET1 mRNA in the frontal cortex and hippocampus of PRS mice was noted.91 This was accompanied by decreased amounts of BDNF I–IV, VI, and IX in the frontal cortex and BDNF III, IV, VI, and IX mRNA transcripts in the hippocampus of PRS mice.91 The levels of 5mC and 5hmC were similarly increased at each of the BDNF promoter regions. In addition, the decrease in corresponding BDNF transcript levels was accompanied by an enrichment of 5mC and 5hmC at the respective promoters.91 Interestingly, the expression of BDNF transcripts IV and IX positively correlates with the social interaction deficits observed.91 Lastly, they observed that there was increased binding of DNMT1 to BDNF promoters III, IV, VI, and IX. These studies provide a compelling set of data for comparison with the epigenetic profile derived from postmortem human SZ brain (Table 1).
3.3. Responsiveness of the PRS Mouse to Medications
Initial studies of the PRS model were designed to test the efficacy of the preclinical compound LY379268, which was structurally related to the compound Eli Lilly was testing in phase III clinical trials for treatment of SZ, LY2140023. This potent and selective mGlu2/3 receptor antagonist, LY379268, was originally synthesized by Jim Monn and Darryl Schoep at Eli Lilly and Company.94 Systemic administration of LY379268 corrected all of the behavioral and biochemical deficits exhibited by PRS mice. LY2140023 was recently tested in a three-armed clinical trial against placebo and Zyprexa. A large body of preclinical data support a role for mGluR agonists as new antipsychotic drugs for the symptomatic relief from SZ.95 Unfortunately, the mGluR agonist failed to outperform the placebo, which actually outperformed Zyprexa, Lilly’s currently marketed SZ drug.
In addition to mGluR agonists, various epigenetic drugs have been used to evaluate their utility in treating the PRS mouse. For example, VPA was shown to correct the stereotyped behavior in PRS mice.84 This result was anticipated as VPA has the same effect in the MET-treated mouse model of SZ. While CLZ corrects the behavioral phenotype of PRS mice, HAL fails to do so. Moreover, CLZ normalizes the biochemical phenotype in PRS mice, including the BDNF IV, VI and IX, RELN, and GAD1 mRNA downregulation, the elevated 5mC and 5hmC levels, % MeCP2 binding, and DNMT1 binding at corresponding promoters.92 In addition, CLZ also normalizes the elevated DNMT1 and TET1 mRNA levels in PRS mice, whereas HAL fails to do the same. Collectively, CLZ, the gold standard in antipsychotic therapy,96 corrects both the behavioral and epigenetic alterations associated with PRS mice. CLZ is the only antipsychotic medication capable of attenuating the epigenetic components of psychiatric disorders.
The PRS model in rats, and presumably also in mice, shows sexual dimorphism in terms of the outcome of the offspring. Most studies that investigate PRS mice generally use male mice in their studies. Males exhibit a pronounced response, whereas females often show opposing responses.97 Where male rats show increased anxiety, decreased neurogenesis in the hippocampal dentate gyrus, reduced activity of mGlu1/5 activity, and increased BDNF in the hippocampus, females display reduced anxiety, improved learning in the Morris water maze, increased activity of mGlu1/5 receptor activity in the hippocampus, and no changes in hippocampal neurogenesis or BDNF levels.97 These data strongly suggest that epigenetic changes in the hippocampus of PRS rats induced by early life environmental perturbations are dependent on sex and that the behavioral effects diverge between males and females. Moreover, the effects of gender in epigenetic models of psychiatric disorders need to be examined in more detail in future studies.
4. CONCLUSIONS
SZ is a debilitating disorder that has both genetic and epigenetic components that have a role in its pathophysiology. Both the MET mouse and PRS mouse mimic the positive, negative, and cognitive aspects of SZ. The data showing that CLZ is able to reverse both the behavioral and biochemical deficits associated with these models indicate that this drug may serve as a model compound in the development of new drugs that can normalize the epigenetic aspects of these disorders. Having said this, we recognize that the mechanistic details of CLZ action in reversing these endophenotypes is still incompletely understood.
ACKNOWLEDGMENT
This work is supported in part by grant 5P50 AA022538 to DRG and AG.
REFERENCES
- 1.Schizophrenia Working Group of the Psychiatric Genomics Consortium.Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grayson DR. Schizophrenia and the epigenetic hypothesis. Epigenomics. 2010;2: 341–344. [DOI] [PubMed] [Google Scholar]
- 3.Millan MJ. An epigenetic framework for neurodevelopmental disorders: from pathogenesis to potential therapy. Neuropharmacology. 2013;68:2–82. [DOI] [PubMed] [Google Scholar]
- 4.Uher R, Zwicker A. Etiology in psychiatry: embracing the reality of poly-gene-environmental causation of mental illness. World Psychiatry. 2017;16:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurkowska RZ, Jurkowski TP, Jeltsch A. Structure and function of mammalian DNA methyltransferases. Chembiochem. 2011;12:206–222. [DOI] [PubMed] [Google Scholar]
- 6.Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38:138–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayraktar G, Kreutz MR. Neuronal DNA methyltransferases: epigenetic mediators between synaptic activity and gene expression? Neuroscientist. 2017. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jang HS, Shin WJ, Lee JE, Do JT. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes (Basel). 2017;8(6):148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long HK, Blackledge NP, Klose RJ. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem Soc Trans. 2013;41:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edwards JR, Yarychkivska O, Boulard M, Bestor TH. DNA methylation and DNA methyltransferases. Epigenetics Chromatin. 2017;10:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadriu B, Guidotti A, Chen Y, Grayson DR. DNA methyltransferases1 (DNMT1) and 3a (DNMT3a) colocalize with GAD67-positive neurons in the GAD67-GFP mouse brain. JCompNeurol. 2012;520:1951–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe D, Uchiyama K, Hanaoka K. Transition of mouse de novo methyltransferases expression from Dnmt3b to Dnmt3a during neural progenitor cell development. Neuroscience. 2006;142:727–737. [DOI] [PubMed] [Google Scholar]
- 13.Feng J, Zhou Y, Campbell SL, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moore LD, Thuc L, Fan G. DNA methylation and its basic function. Neuropsychopharmacology. 2013;38:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ooi SK1. Qiu C, Bernstein E, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee MS, Jun DH, Hwang CI, et al. Selection of neural differentiation-specific genes by comparing profiles of random differentiation. Stem Cells. 2006;24:1946–1955. [DOI] [PubMed] [Google Scholar]
- 17.Kovacheva VP, Mellott TJ, Davison JM, et al. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by upregulation of Dnmt1 expression. J Biol Chem. 2007;282:31777–31788. [DOI] [PubMed] [Google Scholar]
- 18.Veldic M, Caruncho HJ, Liu WS, et al. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proc Natl AcadSci USA. 2004;101:348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veldic M, Guidotti A, Maloku E, Davis JM, Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc Natl Acad Sci USA. 2005;102:2152–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Veldic M, Kadriu B, Maloku E, et al. Epigenetic mechanisms expressed in basal ganglia GABAergic neurons differentiate schizophrenia from bipolar disorder. Schizophr Res. 2007;91:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruzicka WB, Zhubi A, Veldic M, Grayson DR, Costa E, Guidotti A. Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol Psychiatry. 2007;12:385–397. [DOI] [PubMed] [Google Scholar]
- 22.Noh JS, Sharma RP, Veldic M, et al. DNA methyltransferase 1 regulates reelin mRNA expression in mouse primary cortical cultures. Proc Natl Acad Sci USA. 2005;102:1749–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhubi A, Veldic M, Puri NV, et al. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr Res. 2009;111:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. ProcNatl AcadSci USA. 2005;102:9341–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abdolmaleky HM, Cheng KH, usso R. et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B:60–66. [DOI] [PubMed] [Google Scholar]
- 26.Jeltsch A On the enzymatic properties of Dnmt1: specificity, processivity, mechanism of linear diffusion and allosteric regulation of the enzyme. Epigenetics. 2006;1:63–66. [DOI] [PubMed] [Google Scholar]
- 27.Ting AH, Jair KW, Schuebel KE, Baylin SB. Differential requirement for DNA methyltransferase 1 in maintaining human cancer cell gene promoter hypermethylation. Cancer Res. 2006;66:729–735. [DOI] [PubMed] [Google Scholar]
- 28.Ting AH, Jair KW, Suzuki H, Yen RW, Baylin SB, Schuebel KE. CpG island hypermethylation is maintained in human colorectal cancer cells after RNAimediated depletion of DNMT1. Nat Genet. 2004;36:582–584. [DOI] [PubMed] [Google Scholar]
- 29.Pradhan M, Estève PO, Chin HG, Samaranayke M, Kim GD, Pradhan S. CXXC domain of human DNMT1 is essential for enzymatic activity. Biochemistry. 2008;47: 10000–10009. [DOI] [PubMed] [Google Scholar]
- 30.Frauer C, Rottach A, Meilinger D, et al. Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One. 2011;6:e16627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci USA. 2011;108:3630–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inano K, Suetake I, Ueda T, et al. Maintenance-type DNA methyltransferase is highly expressed in postmitotic neurons and localized in the cytoplasmic compartment. J Biochem. 2000;128:315–321. [DOI] [PubMed] [Google Scholar]
- 33.Yoder JA, Yen RW, Vertino PM, Bestor TH, Baylin SB. New 5’ regions of the murine and human genes for DNA (cytosine-5)-methyltransferase. J Biol Chem. 1996;271:31092–31097. [DOI] [PubMed] [Google Scholar]
- 34.Saini SK, Mangalhara KC, Prakasam G, Bamezai RNK. DNA Methyltransferase1 (DNMT1) Isoform3 methylates mitochondrial genome and modulates its biology. Sci Rep. 2017;7:1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tajima S, Suetake I, Takeshita K, Nakagawa A, Kimura H. Domain structure of the Dnmt1, Dnmt3a, and Dnmt3b DNA methyltransferases. Adv Exp Med Biol. 2016;945: 63–86. [DOI] [PubMed] [Google Scholar]
- 36.Kar S, Deb M, Sengupta D, et al. An insight into the various regulatory mechanisms modulating human DNA methyltransferase 1 stability and function. Epigenetics. 2012;7:994–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jaffe AE, Gao Y, Deep-Soboslay A, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jaffe AE, Shin J, Collado-Torres L, et al. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat Neurosci. 2015;18:154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wen L, Li X, Yan L, Tan Y, et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014;15:R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Montano C, Taub MA, Jaffe A, et al. Association of DNA methylation differences with schizophrenia in an epigenome-wide association study. JAMA Psychiatry. 2016;73:506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guidotti A, Dong E, Grayson DR. Epigenetic basis of clozapine action. J Drug Des Res. 2017;4:1055. [PMC free article] [PubMed] [Google Scholar]
- 42.Pries LK, Gülöksüz S, Kenis G. DNA methylation in schizophrenia. Adv Exp Med Biol. 2017;978:211–236. [DOI] [PubMed] [Google Scholar]
- 43.Davies MN, Volta M, Pidsley R, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aberg KA, Xie LY, McClay JL, et al. Testing two models describing how methylome-wide studies in blood are informative for psychiatric conditions. Epigenomics. 2013;5:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Walton E, Hass J, Liu J, et al. Correspondence of DNA methylation between blood and brain tissue and its application to schizophrenia research. Schizophr Bull. 2016;42: 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aberg KA, McClay JL, Nerella S, et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry. 2014;71:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van den Oord EJ, Clark SL, Xie LY, et al. A whole methylome CpG-SNP association study of psychosis in blood and brain tissue. Schizophr Bull. 2016;42:1018–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams K, Christensen J, Helin K. DNA methylation: TET proteins-guardians of CpG islands? EMBO Rep. 2011;13:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Globisch D, Münzel M, Müller M, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One. 2010;5:e15367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito S, Shen L, Dai Q, et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nestor CE, Ottaviano R, Reddington J, et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu Y, Xu C, Kato A, et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi DQ, Ali I, Tang J, Yang WC. New insights into 5hmC DNA modification: generation, distribution and function. Front Genet. 2017;8:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.He YF, Li BZ, Li Z, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu L, Lu J, Cheng J, et al. Structural insight into substrate preference for TET-mediated oxidation. Nature. 2015;527:118–122. [DOI] [PubMed] [Google Scholar]
- 57.Jin SG, Zhang ZM, Dunwell TL, et al. Tet3 reads 5-carboxylcytosine through its CXXC domain and is a potential guardian against neurodegeneration. Cell Rep. 2016;14:493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146–1159. [DOI] [PubMed] [Google Scholar]
- 59.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401: 301–304. [DOI] [PubMed] [Google Scholar]
- 60.Nabel CS, Jia H, Ye Y, et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. 2012;8:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schutsky EK, Nabel CS, Davis AKF, DeNizio JE, Kohli RM. APOBEC3A efficiently deaminates methylated, but not TET-oxidized, cytosine bases in DNA. Nucleic Acids Res. 2017;45:7655–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Song CX, Diao J, Brunger AT, Quake SR. Simultaneous single-molecule epigenetic imaging of DNA methylation and hydroxymethylation. Proc Natl Acad Sci USA. 2016;113:4338–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang Y, Chavez L, Chang X, et al. Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc Natl Acad SciUSA. 2014;111:1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khare T, Pai S, Koncevicius K, et al. 5hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat Struct Mol Biol. 2012;19:1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dong E, Gavin DP, Chen Y, Davis J. Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry. 2012;2:e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dong E, Ruzicka WB, Grayson DR, Guidotti A. DNA-methyltransferase1 (DNMT1) binding to CpG rich GABAergic and BDNF promoters is increased in the brain of schizophrenia and bipolar disorder patients. Schizophr Res. 2015;167:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gavin DP, Sharma RP, Chase KA, Matrisciano F, Dong E, Guidotti A. Growth arrest and DNA-damage-inducible, beta (GADD45b)-mediated DNA demethylation in major psychosis. Neuropsychopharmacology. 2012;37:531–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Osmond H, Smythies J. Schizophrenia: a new approach. J Ment Sci. 1952;98:309–315. [DOI] [PubMed] [Google Scholar]
- 69.Smythies JR. The transmethylation hypotheses of schizophrenia re-evaluated. Trends Neurosci. 1984;7:45–47. [Google Scholar]
- 70.Grayson DR, Chen Y, Dong E, Kundakovic M, Guidotti A. From transmethylation to cytosine methylation: evolution of the methylation hypothesis of schizophrenia. Epigenetics. 2009;4:144–149. [DOI] [PubMed] [Google Scholar]
- 71.Antun FJ, Burnett GB, Cooper AJ, Daly RJ, Smythies JR, Zealley AK. The effect of l-methionine (without MAOI) in schizophrenia. J Psychiatr Res. 1971;8:63–71. [DOI] [PubMed] [Google Scholar]
- 72.Szyf M Utilization of antisense oligonucleotides to study the role of 5-cytosine DNA methyltransferase in cellular transformation and oncogenesis. Methods. 2002;27: 184–191. [DOI] [PubMed] [Google Scholar]
- 73.Costa E, Chen Y, Dong E, et al. GABAergic promoter hypermethylation as a model to study the neurochemistry of schizophrenia vulnerability. Expert Rev Neurother. 2009;9:87–98. [DOI] [PubMed] [Google Scholar]
- 74.Guidotti A, Ruzicka W, Grayson DR, et al. S-adenosyl methionine and DNA methyltransferase-1 mRNA overexpression in psychosis. Neuroreport. 2007;18:57–60. [DOI] [PubMed] [Google Scholar]
- 75.Tremolizzo L, Carboni G, Ruzicka WB, et al. An epigenetic mouse model for molecular and behavioral neuropathologies related to schizophrenia vulnerability. Proc Natl Acad Sci USA. 2002;99:17095–17100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tremolizzo L, Doueiri MS, Dong E, et al. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. [DOI] [PubMed] [Google Scholar]
- 77.Wang L, Alachkar A, Sanathara N, Belluzzi JD, Wang Z, Civelli O. A methionine-induced animal model of schizophrenia: face and predictive validity. Int J Neuropsychopharmacol. 2015;18:pyv054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tueting P, Davis JM, Veldic M, et al. L-Methionine decreases dendritic spine density in mouse frontal cortex. Neuroreport. 2010;21:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dong E, Agis-Balboa RC, Simonini MV, Grayson DR, Costa E, Guidotti A. Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc Natl Acad Sci USA. 2005;102:12578–12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dong E, Guidotti A, Grayson DR, Costa E. Histone hyperacetylation induces demethylation of reelin and 67-kDa glutamic acid decarboxylase promoters. Proc Natl Acad Sci USA. 2007;104:4676–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kundakovic M, Chen Y, Guidotti A, Grayson DR. The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol Pharmacol. 2009;75:342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee BH, Yegnasubramanian S, Lin X, Nelson WG. Procainamide is a specific inhibitor of DNA methyltransferase 1. J Biol Chem. 2005;280:40749–40756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci USA. 2008;105:13614–13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matrisciano F, Tueting P, Dalal I, et al. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology. 2013;68:184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Maccari S, Morley-Fletcher S. Effects of prenatal restraint stress on the hypothalamus-pituitary-adrenal axis and related behavioural and neurobiological alterations. Psychoneuroendocrinology. 2007;32(Suppl. 1):S10–S15. [DOI] [PubMed] [Google Scholar]
- 86.Stevens HE, Su T, Yanagawa Y, Vaccarino FM. Prenatal stress delays inhibitory neuron progenitor migration in the developing neocortex. Psychoneuroendocrinology. 2013;38: 509–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miyagawa K, Tsuji M, Ishii D, Takeda K, Takeda H. Prenatal stress induces vulnerability to stress together with the disruption of central serotonin neurons in mice. Behav Brain Res. 2015;277:228–236. [DOI] [PubMed] [Google Scholar]
- 88.Pascuan CG, Di Rosso ME, Pivoz-Avedikian JE, Wald MR, Zorrilla Zubilete MA, Genaro AM. Alteration of neurotrophin and cytokine expression in lymphocytes as novel peripheral markers of spatial memory deficits induced by prenatal stress. Physiol Behav. 2017;173:144–155. [DOI] [PubMed] [Google Scholar]
- 89.Weinstock M Prenatal stressors in rodents: effects on behavior. Neurobiol Stress. 2016;6: 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guidotti A, Dong E, Tueting P, Grayson DR. Modeling the molecular epigenetic profile of psychosis in prenatally stressed mice. Prog Mol Biol Transl Sci. 2014;128:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dong E, Dzitoyeva SG, Matrisciano F, Tueting P, Grayson DR, Guidotti A. Brain-derived neurotrophic factor epigenetic modifications associated with schizophrenia-like phenotype induced by prenatal stress in mice. Biol Psychiatry. 2015;77:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dong E, Tueting P, Matrisciano F, Grayson DR, Guidotti A. Behavioral and molecular neuroepigenetic alterations in prenatally stressed mice: relevance for the study of chromatin remodeling properties of antipsychotic drugs. Transl Psychiatry. 2016;6:e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matrisciano F, Tueting P, Maccari S, Nicoletti F, Guidotti A. Pharmacological activation of group-II metabotropic glutamate receptors corrects a schizophrenia-like phenotype induced by prenatal stress in mice. Neuropsychopharmacology. 2012;37:929–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Monn JA, Valli MJ, Massey SM, et al. Synthesis, pharmacological characterization, and molecular modeling of heterobicyclic amino acids related to (+)-2-aminobicyclo[3.1.0] hexane-2,6-dicarboxylic acid (LY354740): identification of two new potent, selective, and systemically active agonists for group II metabotropic glutamate receptors. J Med Chem. 1999;42:1027–1040. [DOI] [PubMed] [Google Scholar]
- 95.Wierońska JM, Zorn SH, Doller D, Pilc A. Metabotropic glutamate receptors as targets for new antipsychotic drugs: historical perspective and critical comparative assessment. PharmacolTher. 2016;157:10–27. [DOI] [PubMed] [Google Scholar]
- 96.Meltzer HY. Update on typical and atypical antipsychotic drugs. Annu Rev Med. 2013;64:393–406. [DOI] [PubMed] [Google Scholar]
- 97.Zuena AR, Mairesse J, Casolini P, et al. Prenatal restraint stress generates two distinct behavioral and neurochemical profiles in male and female rats. PLoS One. 2008;3:e2170. [DOI] [PMC free article] [PubMed] [Google Scholar]