

Supplemental Digital Content is available in the text.
Keywords: Air pollution, Temperature, Myocardial damage, Cardiac troponin
Abstract
The mechanisms whereby ambient air pollution and temperature changes promote cardiac events remain incompletely described. Seventy-three nonsmoking healthy adults (mean age 23.3, SD 5.4 years) were followed with up to four repeated visits across 15 months in Beijing in 2014–2016. Biomarkers relevant to myocardial damage (high-sensitivity cardiac troponin I [hs-cTnI]), inflammation (growth differentiation factor-15 [GDF-15]), and oxidative stress (8-hydroxy-2′-deoxyguanosine [8-OHdG]) were measured at each visit, while ambient air pollution and temperature were monitored throughout the study. Linear mixed-effects models coupled with distributed lag nonlinear models were used to assess the impacts of each exposure measure on study outcomes. During follow-up, average daily concentrations of fine particulate matter and outdoor temperature were 62.9 µg/m3 (8.1–331.0 µg/m3) and 10.1 °C (−6.5°C to 29.5°C). Serum hs-cTnI levels were detectable in 18.2% of blood samples, with 27.4% of individuals having ≥1 detectable values. Higher levels of ambient particulates and gaseous pollutants (per interquartile range) up to 14 days before clinical visits were associated with significant alterations in hs-cTnI levels of 22.9% (95% CI, 6.4, 39.4) to 154.7% (95% CI, 94.4, 215.1). These changes were accompanied by elevations of circulating GDF-15 and urinary 8-OHdG levels. Both low (5th percentile, −2.5 °C) and high (95th percentile, 24.8°C) outdoor temperatures, with breakpoint at ~13.0°C as the reference level, were also associated with elevations of hs-cTnI levels. Short-term exposure to ambient air pollution and temperature was associated with cardiac troponin, a biomarker of myocardial damage, along with increased inflammation and oxidative stress responses. These findings extend our understanding of the biological mechanisms linking pervasive environmental exposure to adverse cardiac events.
Introduction
Global environmental and climate changes have heightened concerns about the adverse health effects of anthropogenic air pollution and extreme temperature.1 A large body of studies have reported that ambient air pollution and outdoor temperature are both associated with increases in risks of hospital admissions and mortality due to cardiovascular events (e.g., myocardial infarctions, strokes, and heart failure).2,3 While the entire adult population is at risk through a variety of biological mechanisms in response to ambient air pollution and temperature exposures, patients with preexisting cardiac diseases are known to be particularly susceptible.1,4 Further, epidemiological and experimental studies have shown that adverse cardiac remodeling such as ventricular dilatation and hypertrophy are also associated with various environmental stimuli (e.g., particulate matter [PM], outdoor temperature).5–7 These findings support that intermittent or chronic myocardial damage may be a mechanism linking air pollution and temperature to the onset of overt cardiovascular events.
What this study adds?
Cardiac troponin measured with high-sensitivity cardiac troponin I assay (hs-cTnI) is highly specific for myocardial damage and has become the preferred diagnostic biomarker. We show here that short-term exposure to ambient air pollution and outdoor temperature was associated with significant increases in serum hs-cTnI levels along with elevations of circulating GDF-15 and urinary 8-OHdG levels in healthy adults. Our findings support that pervasive environmental exposures may promote myocardial damage and systemic inflammation and oxidative stress responses, which thereby heighten the risk for cardiovascular events in general population.
Prior studies have demonstrated linkages between ambient air pollutants and ECG evidence of myocardial ischemia; however, the modest degree of ST-segment depression observed tended to be of uncertain clinical relevance.8,9 On the other hand, increases in circulating cardiac troponin measured with high-sensitivity assays [high-sensitivity cardiac troponin (hs-cTn)] are highly specific for myocardial damage and have thus become the preferred diagnostic biomarker.10–12 As a unique structural protein of cardiac myocytes, cTn can be released from functionally free cytosolic pool of the heart due to sublethal myocardial injury and irreversible cell death.13 It is worth noting that at least half of the individuals without active myocardial ischemia can still have detectable circulating levels of hs-cTn.14,15 Mounting studies further have shown that previously unmeasurable concentrations of troponin, even at low levels well-below diagnostic thresholds for an acute myocardial infarction, are independent predictors of future cardiovascular events in both healthy and ill populations.14–17 These existing evidence suggests that other pathophysiological stimuli might be responsible for the release of cardiac troponin in people free of heart diseases.
Mechanistically, oxidative stress caused by production of reactive oxygen species (ROS) is capable of triggering cardiac myocyte membrane permeability dysfunction and ongoing DNA damage, thereby contributing to the development of cellular death and subsequent appearance of cardiac cell proteins in circulation system.18–20 Indeed, in patients with cardiomyopathies or heart failure, 8-OHdG (a marker of oxidative DNA damage) has been shown in elevation with damaged cardiac myocytes.21,22 Moreover, emerging evidence indicates that stress-responsive cytokines (e.g., growth differentiation factor-15 [GDF-15]) can exert prominent cardio-protective effects against ischemia/reperfusion injuries, including anti-inflammatory and anti-apoptotic actions.23 Activation of cardiac myocytes has been considered as the major sources of GDF-15 expression, and persisting increases in GDF levels have shown in association with circulating troponin as evidence of myocardial damage.23,24 It is therefore plausible that air pollution and temperature, established instigators of oxidative-inflammatory damage,2 may be partially responsible for myocardial damage.
In prior analyses, collectively we have observed increases in numerous cardiovascular responses in association with increased exposure to air pollution in healthy adults,25–29 including changes in biomarkers of lipid metabolism, atherosclerotic plaque stability, blood thrombogenicity, and electrophysiological abnormalities, as well as bunted blood pressure responses. In the present analysis of Beijing AIRCHD Study (Air Pollution and Cardiovascular Dysfunction in Healthy Adults in Beijing), we hypothesized that short-term exposure to ambient air pollution and outdoor temperature could alter biomarkers of myocardial damage, inflammation, and oxidative stress.
Methods
Participants and study design
The protocol of the Beijing AIRCHD Study has been described in details previously.26–29 In brief, 73 nonsmoking healthy adults (mean age 23.3, SD 5.4 years) including 48 females were recruited for 4 repeated visits in 2014–2016 (visit 1: November 2014 to January 2015; visit 2: April 2015 to June 2015; visit 3: September 2015 to November 2015; and visit 4: December 2015 to January 2016). At each clinical visit, each participant completed a standardize questionnaire survey on individual demographic information and received clinical examinations including vascular function assessment, blood withdrawal, and urine collection. All clinic visits were conducted at the Clinic of Peking University Health Science Center (PUHSC) in the morning (between 8 and 10 am) after overnight fasting. Approximately 93% of participants lived within 1 kilometer from the fixed air pollution monitoring station locating on PUHSC campus. This study was approved by the Institutional Review Board of PUHSC, and a written informed consent was obtained from each participant.
Biomarker measurements
Health outcomes
Blood samples were collected using BD Vacutainer SST tubes for serum separation, and serum concentrations of high-sensitive cardiac troponin I (hs-cTnI) were analyzed by the ARCHITECHSTAT i2000SR platform using the Abbott ARCHITECT STAT High Sensitive Troponin I assay (Abbott Laboratories, Abbott Park, IL), with a limit of detection (LoD) of 1.20 ng/L (assay range 0–50,000 ng/L).14,15 Serum GDF-15 levels were determined by an enzyme-linked immunosorbent assay (ELISA) approach (R&D Systems Inc., Minneapolis, MN). Spot urine samples were analyzed for 8-hydroxy-2′-deoxyguanosine (8-OHdG) using an ELISA approach (BioTSZ, San Francisco, CA). For each outcome measure, samples obtained from each participant were analyzed simultaneously in the same assay to reduce the potential impact of inter-assay variability.
Covariates
Blood samples were collected using lithium-heparin tubes (PST, Vacutainer PST, BD Diagnostics, Franklin Lakes, NJ) for biochemical analyses including creatinine, glucose, high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C), and triglycerides, and these biomarkers were analyzed within 4 hours of collection by an automatic analyzer (Beckman AU5800, Beckman Coulter, Fullerton, CA) at Peking University First Hospital. The estimated glomerular filtration rate (eGFR) was calculated by creatinine levels with the Modified Diet in Renal Disease Equation.30 Urinary cotinine (a proxy of environmental tobacco smoke exposure) and cortisol (a biomarker of the stress response of human body) were measured using a commercially available ELISA kit (Pomona, CA and LIUHEBIO, Wuhan, China, respectively).
The measurement of urinary biomarkers may be potentially affected by urinary dilution. In this study, urinary creatinine, which is the most commonly used correcting measure of urinary dilution in spot samples,31 was applied to normalize urinary concentrations of 8-OHdG, cotinine, and cortisol. The results of all urinary biomarkers were reported as nanograms/milligram creatinine.
Air pollution and meteorologic parameter measurements
Hourly concentrations of ambient PM2.5 were continuously measured using a beta attenuation mass monitor (BAM-1020; Met One Instruments Inc., Washington). The Fast Mobility Particle Size Spectrometer (FMPS Model 3091, TSI Inc., Shoreview, MN) was used to measure 5-minute particulate number concentrations (PNC) in size range of 5.6–560 nm, and PNC in sizes of 5–50 nm (PNC5–50), 50–100 nm (PNC50–100), and 100–560 nm (PNC100–560) were calculated. Minute-to-minute black carbon (BC) concentrations were measured using a dual wavelength aethalometer (AE-33, Magee Scientific, Berkeley, CA), and concentrations of sulfur dioxide (SO2), nitrogen oxides (NOx), carbon monoxide (CO), and ozone (O3) were measured by EC9800 series ambient gas analyzers (EcoTech Pty. Ltd., VIC, Melbourne, Australia). Hourly ambient temperature (Temp) and relative humidity (RH) were measured by a Met One unit (Met One Instruments Inc., Washington) at the same station. The reliability of pollutants measured at the fixed monitoring station as an air pollution exposure proxy for individuals has been validated.26 Further, minute-to-minute measurements of ambient dew point temperature, wind speed, and barometric pressure were obtained from Tsinghua University meteorologic monitoring station, ~3 kilometers in distance to the PUHSC Clinic. Apparent temperature (AT), a metric of the ambient heat perceived by humans, was calculated using Temp and dew point temperature.32 Daily averages of all environmental measurements were calculated from 9 am to 9 am the next day when hourly measurements with at least 18 hours for a given day were available. Due to routine instrument maintenance, limited missing hourly exposure data were reported and not imputed in the analysis.
Statistical analysis
Descriptive statistics were calculated for outcomes and environmental measurements. Spearman correlations were performed across measurements of air pollutants and meteorologic parameters. All health outcome variables were normalized through log transformation because of their skewed distributions (eFigure 1A–C; http://links.lww.com/EE/A68). In the present analysis, concentrations of hs-cTnI between 0.10 and 1.20 ng/L were also included, because studies suggested that cTn values below the LoD were indicative of cardiovascular physiology.33–35 Linear mixed-effects (LME) models with a continuous first-order autoregressive covariance structure were used to assess the lagged associations of each outcome with averaged concentrations of environmental factors including the maximum lag up to 14 days for ambient air pollutants and up to 7 days for temperatures
We first built the basic LME model without including environmental factors for each health outcome. To explain the potential impacts of meteorologic variables, all individual time lags of Temp, RH, wind speed, and barometric pressure were considered; but only Temp and RH levels averaged over the last 24 hours before clinical visits were chosen by minimizing Akaike Information Criterion (AIC). All LME models were further controlled for seasonality using a harmonic function (sine and cosine terms of calendar dates of blood withdrawal) based on the criteria of lower AIC. In addition, as certain covariates might have unequal influences on each health outcome, we thus identify the key covariates for individual outcome separately. Tested covariates were selected according to literature and biological plausibility,14,17 including age, sex, body mass index, day of week of clinical visits, blood pressure, heart rate, HDL-C, LDL-C, triglycerides, eGFR, creatinine-adjusted urine cotinine, and cortisol. These covariates were only included in final LME models if they yielded lower AIC
Single-pollutant LME models were then developed by including environmental factors to assess the lagged associations of measured outcomes with air pollutants and temperatures. However, considering potentially lagged and non-linear effects of environmental exposures, distributed lag nonlinear models (DLNM) were constructed, which allowed a flexible modeling strategy to identify potentially delayed effects of environmental factor.36 For this purpose, two-dimensional matrices for the space of environmental factors and the space of effect time lags were established using cross-basis functions, with linear functions for air pollution and penalized regression splines for temperature. In our initial analysis, the shapes of associations between air pollutants and all outcome measures were approximately linear, whereas “U-shaped” relationships with temperatures were observed for hs-cTnI, and inverted “U-shaped” associations were observed for 8-OHdG. We did not find consistent relationships between temperatures and GDF-15 over multiple-day lags of exposure. To quantify the effects of temperature on health outcomes, temperature breakpoints with minimum and maximum effects on hs-cTnI and 8-OHdG were set as the reference levels for temperature exposure metrics, respectively.36,37 For GDF-15, the median values of temperatures during the study period were defined as the reference levels.38 Finally, cross-basis DLNM functions in the framework of LME models were constructed to estimate the lagged associations of environmental factors. For temperature exposures, we also included corresponding exposure periods of PM2.5 and O3 in final LME models because studies have showed that temperature effects on cardiovascular events could be significantly modified by ambient particulates and oxidant air pollutants (e.g., O3).39,40 Because humidity is part of the calculation of AT, we did not control the RH while assessing AT effects.
To check the robustness of the observed associations obtained in main models, we performed several sensitivity analyses. Given the distributions of air pollutants showing skewed distributions (eFigure 1D–L; http://links.lww.com/EE/A68), we repeated regression analyses using log-transformed air pollutant concentrations as exposure metrics instead of the raw pollution data. We ran additional LME models by: (1) restricting participants living within 1 kilometer from the fixed monitoring station to reduce potential exposure misclassification; (2) excluding participants with urinary cotinine levels above the 75th percentile (4.51 ng/mg creatinine) to evaluate whether the observed associations could be partially affected by potential passive smoking.
All estimates of air pollution exposures are presented as percent changes with 95% CIs associated with interquartile range (IQR) increases in air pollutant concentrations. The results of temperatures are reported as the relative risks (a 1°C change) of a given temperature value relative to the referent level. Statistical significance was determined at P-value <0.05. All statistical analyses were performed using R, version 3.4.3 (R Project for Statistical Computing).
Results
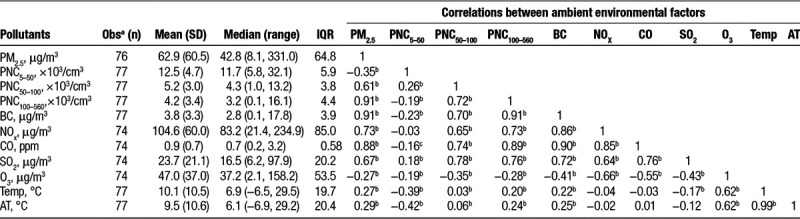
Of 73 participants, 56 completed all four visits, three completed three visits, seven participants completed two visits, and seven participants only had one visit. The characteristics of measured biomarkers across all clinical visits are shown in Table 1. In this healthy and young study population, the circulating levels of hs-cTnI were detectable in 18.2% of blood samples (above LoD), with 27.4% of individuals having ≥1 detectable values. Further, when participants were grouped based on the LoD of hs-cTnI, individuals with detectable levels exhibited significantly higher levels of systolic blood pressure and triglycerides, and lower levels of heart rate (eTable 1; http://links.lww.com/EE/A68). Table 2 summarizes descriptive statistics and correlation matrixes of averaged environmental measurements during the last 24 hours before each participant’s clinic visit. Average daily concentrations of PM2.5 and PNC <100 nm before clinical visits were 62.9 µg/m3 and 17.7 × 103/cm3, respectively. Further, large variabilities in ambient air pollutants and meteorological factors (e.g., PM2.5, NOx, and Temp) were observed, as reflected in larger IQR and significant seasonal change for each environmental measurement (Table 2 and eFigure 2; http://links.lww.com/EE/A68). Among the pollutants, correlations were higher between ambient PM2.5 and PNC100-560 and BC with correlation coefficients of 0.91 and between CO and PNC100-560 and BC (coefficients of 0.89 and 0.90, respectively), whereas O3 was negatively correlated with other pollutants.
Table 1.
Participant characteristics (N = 73, 48 females).
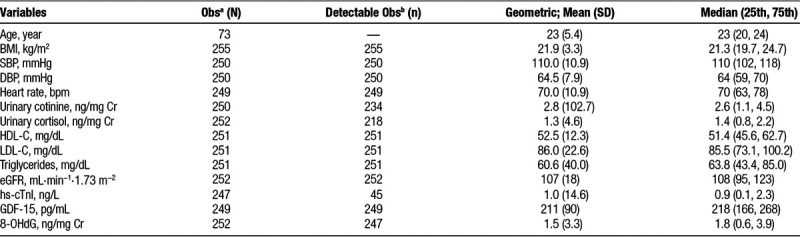
Table 2.
Characteristics of ambient 24-hour mean concentrations of air pollutants and temperatures before clinical visits.
Air pollution associations
As shown in eFigure 3; http://links.lww.com/EE/A68, the shapes of the relationships between measured outcomes and ambient air pollutants appeared to be approximately linear over multiple-day lags of exposure. In regression analyses, the adjusted percent changes in the circulating levels of hs-cTnI were significantly associated with IQR increases in exposure to multiple air pollutants (Figure 1). In associations with IQR increases in different size-fractioned particulates including PM2.5 and PNC in sizes of 5 to 560 nm over cumulative lags of 0 to 13 days, we observed significant increases in hs-cTnI levels of 25.1% (95% CI, 0.4, 49.7) to 154.7% (95% CI, 94.4, 215.1). For traffic-related air pollutants BC, CO, and NOx, significant increases in hs-cTnI levels were observed at various exposure periods, with the largest estimate effects ranging from 105.6% (95% CI, 53.9, 157.3) to 149.8% (95% CI, 50.8, 248.8). In addition, a positive association between hs-cTnI levels and combustion pollutant SO2 was observed at lag days 0–13, whereas a significant inverse association was found with the secondary pollutant O3 at the same exposure time lag.
Figure 1.
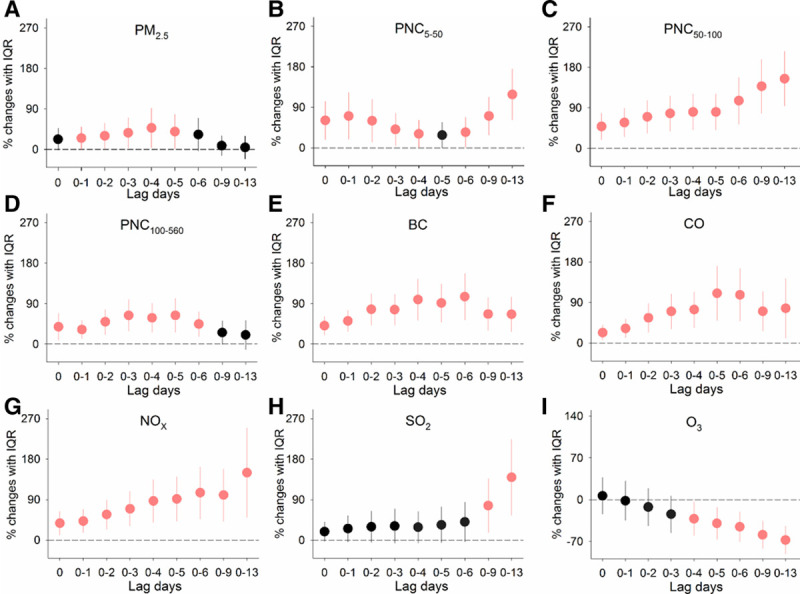
Percent changes in circulating hs-cTnI levels associated with IQR increases in exposure to ambient air pollutants over cumulative lags of 0–13 days. Error bars indicate 95% confidence intervals. Significant associations (P-value < 0.05) are shown in red. The lagged association estimates were derived from linear mixed-effects models coupled with distributed lag non-linear models, with adjustments for body mass index, urinary cortisol, low-density lipoprotein cholesterol, season, ambient temperature, and RH. lag 0, averaged pollutant concentrations over the last 24 hours before each participant’s clinic visit; lag 0–1, 1 to 2 days; lag 0–2, 1 to 3 days and so on up to lag 0–13; PM2.5, fine particulate matter; PNCx, particulate number concentrations in given size ranges (nm).
For biomarkers relevant to inflammation and oxidative stress, as shown in Figure 2, we observed significant increases in GDF-15 of 5.4% (95% CI, 0.8, 9.9) to 14.6% (95% CI, 7.2, 22.1) associated with IQR increases in PNC in sizes of 5 to 560 nm over cumulative lags of 1–13 days, but no significant effects were observed for PM2.5 at all of the time periods examined. Further, circulating GDF-15 levels were significantly increased by 4.4% (95% CI, 0.8, 8.0) to 16.9% (95% CI, 6.8, 27.0) in association with IQR increases in exposure to BC and SO2 over cumulative lags of 0–13 days. As hypothesized, significant elevations in urinary 8-OHdG levels of 43.2% (95% CI, 5.8, 80.6) to 95.3% (95% CI, 28.6, 162.1) were observed in association with IQR increases in exposure to PNC in sizes of 50–560 nm, BC, and CO over cumulative lags of 3–9 days (Figure 3). Greater magnitude of elevated 8-OHdG was observed for SO2 at various lag day periods of exposure, with significant association estimates ranging from 71.7% (95% CI, 2.3, 141.1) to 157.3% (95% CI, 39.5, 275.1). Additionally, significant inverse associations of 8-OHdG with O3 were observed at lag days 0–9 and 0–13 of exposure.
Figure 2.
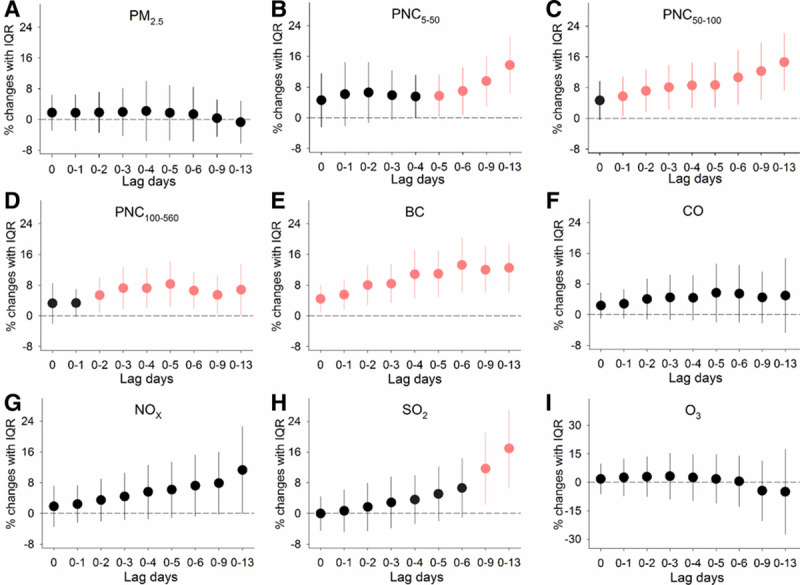
Percent changes in circulating GDF-15 levels associated with IQR increases in exposure to ambient air pollutants over cumulative lags of 0–13 days. Error bars indicate 95% confidence intervals. Significant associations (P-value < 0.05) are shown in red. The lagged association estimates were derived from linear mixed-effects models coupled with distributed lag non-linear models, with adjustments for body mass index, urinary cotinine, season, ambient temperature, and RH. lag 0, averaged pollutant concentrations over the last 24 hours before each participant’s clinic visit; lag 0–1, 1 to 2 days; lag 0–2, 1 to 3 days and so on up to lag 0–13; PM2.5, fine particulate matter; PNCx, particulate number concentrations in given size ranges (nm).
Figure 3.
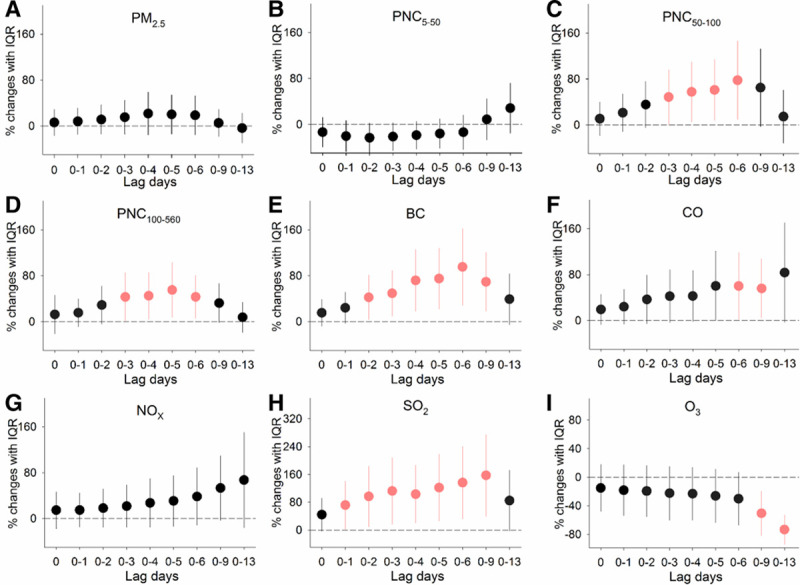
Percent changes in urinary 8-OHdG levels associated with IQR increases in exposure to ambient air pollutants over cumulative lags of 0–13 days. Error bars indicate 95% confidence intervals. Significant associations (P-value < 0.05) are shown in red. The lagged association estimates were derived from linear mixed-effects models coupled with distributed lag non-linear models, with adjustments for sex, high-density lipoprotein cholesterol, day of week of clinical visits, season, ambient temperature, and RH. 8-OHdG, 8-hydroxy-2′-deoxyguanosine; lag 0, averaged pollutant concentrations over the last 24 hours before each participant’s clinic visit; lag 0–1, 1 to 2 days; lag 0–2, 1 to 3 days and so on up to lag 0–13; PM2.5, fine particulate matter; PNCx, particulate number concentrations in given size ranges (nm).
Temperature associations
For Temp exposures, non-linear “U shaped” relationships were observed for alterations in hs-cTnI levels at different lag periods (Figure 4). Overall, both lower and higher Temp were positively associated with hs-cTnI at the short time lag, but the associations only appeared to be persistent significances with exposure to lower Temp over longer lags. For example, at lag day 0 of Temp exposures, comparing the 5th percentile (−2.5 °C) or 95th percentile (24.8 °C) level to the reference level (13.0 °C), the RR of elevated hs-cTnI was 2.29 (95% CI, 1.45, 3.14) and 2.01 (95% CI, 1.08, 2.94), respectively. For 8-OHdG, we observed inverted “U-shaped” relationships with Temp. In specific, exposure to higher Temp was significantly and negatively associated with urinary 8-OHdG levels, and the RRs of 8-OHdG at 95th percentile (24.8 °C) relative to the reference level (3.0 °C) over cumulative lags of 1–6 days ranged from 0.03 (95% CI, 0.005, 0.06) to 0.48 (95% CI, 0.16, 0.81; Figure 5). However, relationships between Temp and GDF-15 at different lag day periods were not consistent (eFigure 4; http://links.lww.com/EE/A68). For AT, the cumulative exposure–response relationship curves and lagged association patterns were similar to Temp exposures (eFigures 5–7; http://links.lww.com/EE/A68 ).
Figure 4.
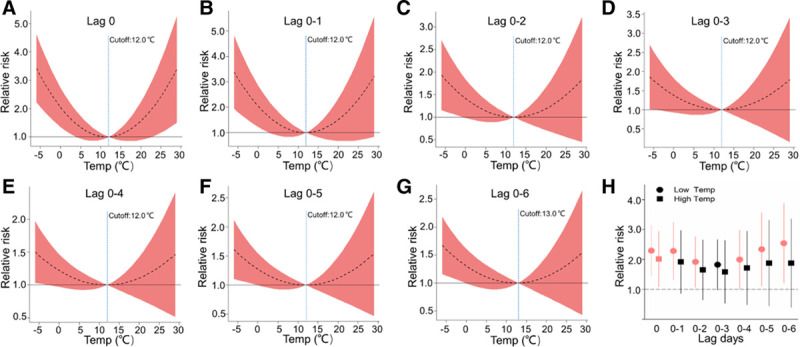
Exposure–response relationship curves (A–G) and the lag structures in the relative risks of hs-cTnI in low (5th percentile) and high (95th percentile) Temp exposures (H) over cumulative lags of 0–6 days. The black lines indicate mean effect estimates, and red regions are 95% confidence intervals. Blue reference line indicates the Temp breakpoints. The lagged association estimates of Temp are presented as relative risks (a 1°C change) of 5th percentile (−2.5°C) or 95th percentile (24.8°C) relative to the referent level (Temp breakpoint at lag 0–6, 13.0°C). Error bars indicate 95% confidence intervals. Significant associations (P-value < 0.05) are shown in red. All models were ran controlling the same covariates as the pollutant models but additional adjustments for corresponding exposure periods of fine particulate matter and O3. lag 0, averaged levels of Temp over the last 24 hours before each participant’s clinic visit; lag 0–1, 1 to 2 days; lag 0–2, 1 to 3 days and so on up to lag 0–6.
Figure 5.
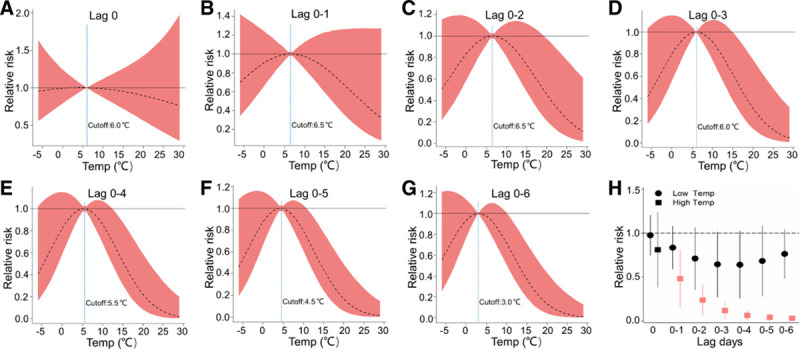
Exposure–response relationship curves (A–G) and the lag structures in the relative risks of 8-OHdG in low (5th percentile) and high (95th percentile) Temp exposures (H) over cumulative lags of 0–6 days. The black lines indicate mean effect estimates, and red regions are 95% confidence intervals. Blue reference line indicates the Temp breakpoints. The lagged association estimates of Temp are presented as relative risks (a 1°C change) of 5th percentile (−2.5°C) or 95th percentile (24.8°C) relative to the referent level (Temp breakpoint at lag 0–6, 3.0°C). Error bars indicate 95% confidence intervals. Significant associations (P-value < 0.05) are shown in red. All models were ran controlling the same covariates as the pollutant models but additional adjustments for corresponding exposure periods of fine particulate matter and O3. 8-OHdG, 8-hydroxy-2′-deoxyguanosine; lag 0, averaged levels of Temp over the last 24 hours before each participant’s clinic visit; lag 0–1, 1 to 2 days; lag 0–2, 1 to 3 days and so on up to lag 0–6.
Sensitivity analyses
As shown in eTables 2–4; http://links.lww.com/EE/A68, the overall biomarker-pollutant associations remained robust when repeated analyses were conducted using log-transformed air pollutant concentrations as exposure metrics, excluding participants with urinary cotinine levels >75th percentile, or restricting the analyses to those who lived within 1 kilometer from air monitoring station.
Discussion
In this study, we show here that short-term exposure to higher levels of ambient particulates and gaseous pollutants was associated with significant increases in the circulating hs-cTnI levels, along with heightened GDF-15 and 8-OHdG levels. Extremes in outdoor temperatures, both higher and lower temperatures, were also positively associated with hs-cTnI levels, whereas only higher temperature exposures were observed for significant reductions in 8-OHdG levels. These findings provide important evidence that air pollution and outdoor temperature might instigate myocardial damage and thereby heighten the risk for cardiovascular events.
There is some, albeit inconsistent, evidence that laboratory generated particulates are related to alterations in circulating troponin levels.41–43 In an experimental study, elevations in cTnI levels were observed in male Sprague–Dawley rats following 1 day of diesel exhaust particles exposures.41 However, no significant changes in serum cTnI levels were observed in spontaneously hypertensive rats, followed by inhalation of synthetic particulates for 4 hours. Further, no consistent changes in cTnI levels were reported in 26 participants with heart failure exposed to extremely high levels of diesel exhaust particles for 15 minutes.42 We speculated that the discrepancy in findings is likely attributed to the limited exposure durations that was not adequate to trigger additional myocardial damage. In this study of healthy adults residing in a highly polluted area, we showed significant elevations of hs-cTnI levels associated with short-term exposures (days) to numerous ambient pollutants, including particulates (PM2.5, particles in size fractions of 5–560 nm, and BC) and gaseous pollutants (CO, NOX, and SO2). Interestingly, O3 exposures were inversely and significantly associated with hs-cTnI levels. These discrepancies might be due to negative correlations between O3 and NOX and other air pollutants.25 We recognized that the generalizability of our findings to a broader clinical setting can be limited by the low detection rate of hs-cTnI occurred in this healthy population. Given that cTn levels are usually below the LoD under normal physiological conditions in the majority of individuals, emerging evidence suggests that cTn levels below the LoD could provide potentially important data on cardiovascular physiology.33–35 A recent population study has shown that cTn levels between the limit of blank (namely highest apparent cTn levels in the analyte-free sample) and LoD are associated with increased cardiovascular risk burdens.35 Nevertheless, our results provide preliminary but important evidence with regard to the ambient air pollution effects on myocardial damage based on the measure of cardiac troponin, and further studies are warranted to confirm our observational findings in general populations.
Apart from air pollution, people are generally co-exposed to other environmental factors, such as outdoor temperatures. A meta-analysis of epidemiological studies has shown that both extremes in temperatures (higher and lower levels) are associated with increases in cardiovascular events.44 Our findings match this well-known “U-shaped” relationship by demonstrating a nadir of risk for serum hs-cTnI elevations at a middle temperature of ~12°C for Temp and 11°C –13°C for AT exposures, which suggests that either temperature extreme can promote myocardial damage. These observations are novel as few prior studies have been published.45,46 For example, elevations in cTnI levels were observed during a heat wave in an elderly hyperthermic patient population.45 Increased levels of hs-cTnI have been observed among healthy fire fighters after a training exercise in a simulated scenario of fire suppression.46 Furthermore, significant elevations of serum cTnI were observed following cold air exposure in cardiovascular disease patients.47 However, these findings in response to extreme heat and cold exposures are not generalizable to the general population experiencing more typical Temp variations in outdoor environment. In addition, aging, disease states, medications, and stress response can also affect the release of cardiac troponin.18,48 In line with our findings, accumulating epidemiological evidence indicates that higher temperature-associated cardiovascular mortality was more often observed over shorter term lag days of exposure, and effects of lower temperature could be persisted over longer cumulative lag days.38,49,50 Herein, concentrations of cardiac troponin above the LoD in this healthy individuals were mainly observed during the first clinical visit, which was conducted in the cold and more polluted season of the year. Though underlying mechanisms remained unclear, we speculated that co-exposure to lower temperature and higher levels of air pollution might be partially responsible for the observed elevations of circulating hs-cTnI. Collectively, we show here that outdoor temperature levels in a real-world setting were associated with hs-cTnI elevations, even in a healthy population at low cardiovascular risk; however, further studies with larger sample size and long-term follow-up are warranted.
The underlying biological mechanisms by which environmental exposure alter circulating levels of cardiac troponin remain poorly understood. Existing evidence suggests that heightened inflammation and oxidative stress may play a role via triggering of myocardial necrosis and transient alterations of the membrane permeability of cardiomyocytes.18 Studies in a rat model of myocardial infarction have reported that oxidative stress-induced DNA damage (e.g., 8-OHdG) is implicated the pathogenesis of cardiomyocyte death.51 Persistent elevations in GDF-15, which has anti-inflammatory and anti-apoptotic actions on cardiomyocytes,23 are related to increased cTnI levels.24 Recent experiments reported that the cardiomyocyte membrane damage following particulate air pollutants exposures is likely due to ROS production.52,53 In this study, elevations in hs-cTnI, 8-OHdG, and GDF-15 levels over days of exposure suggested that air pollution associated myocardial damage involving inflammation and oxidative stress may require a longer period of time to elicit.54 Additionally, in line with our findings, a previously study reported that heat exposures were associated with significant reductions in urinary 8-OHdG levels among heat-acclimated participants.55 An animal model has shown that expression of 8-OHdG in ischemia-reperfusion liver can be swiftly suppressed after exposure to heat shock precondition.56 Considering all of these evidence, our results support that inflammation and oxidative stress provoked by environmental exposures,2 may be partially responsible for elevations of cardiac troponin attributable to ambient air pollutants and outdoor temperature exposures.
Our study has several strengths. This is the first study to explore the effects of a broad array of real-world environmental exposures on a state-of-the-art (fifth generation) hs-cTnI assay. The design of repeated measures across cold and warm months as well as detailed exposure characterizations provided a unique opportunity to assess the critical impacts of co-exposure to high levels of air pollution and outdoor temperature. Despite study strengths, several limitations should also be noted interpretation of our findings. The sample size of 73 participants was relatively small, and air pollutants were measured by a fixed monitoring station which may introduce exposure misclassification; however, these limitations tend to bias the effect estimates toward null. Further, it should be noted that the generalizability of our findings to a broader clinical setting (e.g., elderly, those with cardiovascular disease) is limited due to the low detectable rate of cardiac troponin in our study population. However, it is worth noting that the inclusion of only nonsmoking healthy adults provided a powerful approach to investigate the impacts of exposures on myocardial damage by removing the effects of other factors including preexisting disease and age-related susceptibility. Finally, it is possible that some unmeasured time-varying factors (e.g., exercise, diet) could pose potential confounding impacts; however, we believe it is unlikely that unmeasured temporally varying factors would be co-linear with either (or both) ambient air pollutants and temperatures in a manner that fully explains our reported associations (e.g., act as true confounders). Future studies should enroll more diverse populations and measure temporally variable factors such as activity levels to assure that they are not in fact responsible.
Conclusions
Pervasive environmental exposures were associated with significant increases in hs-cTnI levels that were accompanied by increased systematic inflammation and oxidative status. These results provide important evidence that both air pollution and temperature exposures can promote myocardial damage and thereby heighten the risk for cardiovascular events.
Conflicts of interest statement
The author/authors declares/declare that they have no conflicts of interest with regard to the content of this report.
The results reported herein correspond to specific aims of grant 81773381 to investigator Wei Huang from National Natural Science Foundation of China. This work was (also) supported by grants from National Natural Science Foundation of China (81470025) and Peking University Health Science Center-University of Michigan Health System Joint Institute for Clinical and Translational Research, and Peking University Infrastructure Fund for Interdisciplinary Studies (2013-3-02).
Acknowledgements
We would like to thank all study participants.
Supplementary Material
Footnotes
Published online 2 December 2019
The data that support the findings of this study are available from the corresponding author on reasonable request.
Supplemental digital content is available through direct URL citations in the HTML and PDF versions of this article (www.environepidem.com).
References
- 1.Claeys MJ, Rajagopalan S, Nawrot TS, Brook RD. Climate and environmental triggers of acute myocardial infarction. Eur Heart J 201738955–960 [DOI] [PubMed] [Google Scholar]
- 2.Rajagopalan S, Al-Kindi SG, Brook RD. Air pollution and cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol 2018722054–2070 [DOI] [PubMed] [Google Scholar]
- 3.Chen R, Yin P, Wang L, et al. Association between ambient temperature and mortality risk and burden: time series study in 272 main Chinese cities. BMJ 2018363k4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen AJ, Brauer M, Burnett R, et al. Estimates and 25-year trends of the global burden of disease attributable to ambient air pollution: an analysis of data from the Global Burden of Diseases Study 2015. Lancet 20173891907–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aung N, Sanghvi MM, Zemrak F, et al. Association between ambient air pollution and cardiac morpho-functional phenotypes. Circulation 20181–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wold LE, Ying Z, Hutchinson KR, et al. Cardiovascular remodeling in response to long-term exposure to fine particulate matter air pollution. Circ Heart Fail 20125452–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Li L, Hua Y, et al. Cardiac-specific knockout of ET(A) receptor mitigates low ambient temperature-induced cardiac hypertrophy and contractile dysfunction. J Mol Cell Biol 2012497–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chuang KJ, Coull BA, Zanobetti A, et al. Particulate air pollution as a risk factor for ST-segment depression in patients with coronary artery disease. Circulation 20081181314–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delfino RJ, Gillen DL, Tjoa T, et al. Electrocardiographic ST-segment depression and exposure to traffic-related aerosols in elderly subjects with coronary artery disease. Environ Health Perspect 2011119196–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Twerenbold R, Boeddinghaus J, Nestelberger T, et al. Clinical use of high-sensitivity cardiac troponin in patients with suspected myocardial infarction. J Am Coll Cardiol 201770996–1012 [DOI] [PubMed] [Google Scholar]
- 11.Thygesen K, Alpert JS, Jaffe AS, et al. ; Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction Fourth universal definition of myocardial infarction (2018). Circulation 2018138e618–e651 [DOI] [PubMed] [Google Scholar]
- 12.Holzmann MJ. Clinical implications of high-sensitivity cardiac troponins. J Intern Med 201828450–60 [DOI] [PubMed] [Google Scholar]
- 13.White HD. Pathobiology of troponin elevations: do elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol 2011572406–2408 [DOI] [PubMed] [Google Scholar]
- 14.Zhu K, Knuiman M, Divitini M, et al. High-sensitivity cardiac troponin I and risk of cardiovascular disease in an Australianpopulation-based cohort. Heart 2018104895–903 [DOI] [PubMed] [Google Scholar]
- 15.Adamson PD, Anderson JA, Brook RD, et al. Cardiac troponin I and cardiovascular risk in patients with chronic obstructive pulmonary disease. J Am Coll Cardiol 2018721126–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeller T, Tunstall-Pedoe H, Saarela O, et al. ; MORGAM Investigators High population prevalence of cardiac troponin I measured by a high-sensitivity assay and cardiovascular risk estimation: the MORGAM biomarker project Scottish cohort. Eur Heart J 201435271–281 [DOI] [PubMed] [Google Scholar]
- 17.Eggers KM, Venge P, Lindahl B, Lind L. Cardiac troponin I levels measured with a high-sensitive assay increase over time and are strong predictors of mortality in an elderly population. J Am Coll Cardiol 2013611906–1913 [DOI] [PubMed] [Google Scholar]
- 18.Giannitsis E, Katus HA. Cardiac troponin level elevations not related to acute coronary syndromes. Nat Rev Cardiol 201310623–634 [DOI] [PubMed] [Google Scholar]
- 19.Nie J, Close G, George KP, Tong TK, Shi Q. Temporal association of elevations in serum cardiac troponin T and myocardial oxidative stress after prolonged exercise in rats. Eur J Appl Physiol 20101101299–1303 [DOI] [PubMed] [Google Scholar]
- 20.L’Ecuyer T, Sanjeev S, Thomas R, et al. DNA damage is an early event in doxorubicin-induced cardiac myocyte death. Am J Physiol Heart Circ Physiol 2006291H1273–H1280 [DOI] [PubMed] [Google Scholar]
- 21.Nimata M, Kishimoto C, Shioji K, et al. Upregulation of redox-regulating protein, thioredoxin, in endomyocardial biopsy samples of patients with myocarditis and cardiomyopathies. Mol Cell Biochem 2003248193–196 [DOI] [PubMed] [Google Scholar]
- 22.Kono Y, Nakamura K, Kimura H, et al. Elevated levels of oxidative DNA damage in serum and myocardium of patients with heart failure. Circ J 2006701001–1005 [DOI] [PubMed] [Google Scholar]
- 23.Kempf T, Eden M, Strelau J, et al. The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res 200698351–360 [DOI] [PubMed] [Google Scholar]
- 24.Kahli A, Guenancia C, Zeller M, et al. Growth differentiation factor-15 (GDF-15) levels are associated with cardiac and renal injury in patients undergoing coronary artery bypass grafting with cardiopulmonary bypass. PLoS One 20149e105759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rich DQ, Kipen HM, Huang W, et al. Association between changes in air pollution levels during the Beijing Olympics and biomarkers of inflammation and thrombosis in healthy young adults. JAMA 20123072068–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu H, Wang T, Liu S, et al. Extreme levels of air pollution associated with changes in biomarkers of atherosclerotic plaque vulnerability and thrombogenicity in healthy adults. Circ Res 2019124e30–e43 [DOI] [PubMed] [Google Scholar]
- 27.Li J, Zhou C, Xu H, et al. Ambient air pollution is associated with HDL (High-Density Lipoprotein) dysfunction in healthy adults. Arterioscler Thromb Vasc Biol 201939513–522 [DOI] [PubMed] [Google Scholar]
- 28.Xu H, Chen J, Zhao Q, et al. Ambient air pollution is associated with cardiac repolarization abnormalities in healthy adults. Environ Res 2019171239–246 [DOI] [PubMed] [Google Scholar]
- 29.Huang W, Wang L, Li J, et al. Short-term blood pressure responses to ambient fine particulate matter exposures at the extremes of global air pollution concentrations. Am J Hypertens 201831590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levey AS, Stevens LA, Schmid CH, et al. ; CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) A new equation to estimate glomerular filtration rate. Ann Intern Med 2009150604–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muscat JE, Liu A, Richie JP, Jr.. A comparison of creatinine vs. specific gravity to correct for urinary dilution of cotinine. Biomarkers 201116206–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilker EH, Yeh G, Wellenius GA, Davis RB, Phillips RS, Mittleman MA. Ambient temperature and biomarkers of heart failure: a repeated measures analysis. Environ Health Perspect 20121201083–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ungerer JP, Pretorius CJ. A fit-for-purpose approach to analytical sensitivity applied to a cardiac troponin assay: time to escape the ‘highly-sensitive’ trap. Clin Chem Lab Med 201452553–556 [DOI] [PubMed] [Google Scholar]
- 34.Boeckel JN, Palapies L, Zeller T, et al. Estimation of values below the limit of detection of a contemporary sensitive troponin I assay improves diagnosis of acute myocardial infarction. Clin Chem 2015611197–1206 [DOI] [PubMed] [Google Scholar]
- 35.Parikh RH, Seliger SL, de Lemos J, et al. Prognostic significance of high-sensitivity cardiac troponin T concentrations between the limit of blank and limit of detection in community-swelling adults: a metaanalysis. Clin Chem 2015611524–1531 [DOI] [PubMed] [Google Scholar]
- 36.Li H, Bai H, Yang C, et al. Acute effects of ambient temperature and particulate air pollution on fractional exhaled nitric oxide: a panel study among diabetic patients in Shanghai, China. J Epidemiol 201727584–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hampel R, Schneider A, Brüske I, et al. Altered cardiac repolarization in association with air pollution and air temperature among myocardial infarction survivors. Environ Health Perspect 20101181755–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dang TN, Seposo XT, Duc NH, et al. Characterizing the relationship between temperature and mortality in tropical and subtropical cities: a distributed lag non-linear model analysis in Hue, Viet Nam, 2009-2013. Glob Health Action 2016928738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Analitis A, De’ Donato F, Scortichini M, et al. Synergistic effects of ambient temperature and air pollution on health in Europe: results from the PHASE project. Int J Environ Res Public Health 201815E1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen K, Wolf K, Breitner S, et al. ; UF&HEALTH Study Group Two-way effect modifications of air pollution and air temperature on total natural and cardiovascular mortality in eight European urban areas. Environ Int 2018116186–196 [DOI] [PubMed] [Google Scholar]
- 41.Huang CH, Lin LY, Tsai MS, et al. Acute cardiac dysfunction after short-term diesel exhaust particles exposure. Toxicol Lett 2010192349–355 [DOI] [PubMed] [Google Scholar]
- 42.Vieira JL, Guimaraes GV, de Andre PA, Cruz FD, Saldiva PH, Bocchi EA. Respiratory filter reduces the cardiovascular effects associated with diesel exhaust exposure: a Randomized, Prospective, Double-Blind, Controlled Study of Heart Failure: the FILTER-HF Trial. JACC Heart Fail 2016455–64 [DOI] [PubMed] [Google Scholar]
- 43.Farraj AK, Hazari MS, Haykal-Coates N, et al. ST depression, arrhythmia, vagal dominance, and reduced cardiac micro-RNA in particulate-exposed rats. Am J Respir Cell Mol Biol 201144185–196 [DOI] [PubMed] [Google Scholar]
- 44.Ye X, Wolff R, Yu W, Vaneckova P, Pan X, Tong S. Ambient temperature and morbidity: a review of epidemiological evidence. Environ Health Perspect 201212019–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hausfater P, Doumenc B, Chopin S, et al. Elevation of cardiac troponin I during non-exertional heat-related illnesses in the context of a heatwave. Crit Care 201014R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunter AL, Shah AS, Langrish JP, et al. Fire simulation and cardiovascular health in firefighters. Circulation 20171351284–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang X, Zhang S, Wang C, Wang B, Guo P. Effects of moderate strength cold air exposure on blood pressure and biochemical indicators among cardiovascular and cerebrovascular patients. Int J Environ Res Public Health 2014112472–2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lazzarino AI, Hamer M, Gaze D, Collinson P, Steptoe A. The association between cortisol response to mental stress and high-sensitivity cardiac troponin T plasma concentration in healthy adults. J Am Coll Cardiol 2013621694–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu W, Xiao Y, Li G, et al. Temperature-mortality relationship in four subtropical Chinese cities: a time-series study using a distributed lag non-linear model. Sci Total Environ 2013449355–362 [DOI] [PubMed] [Google Scholar]
- 50.Hajat S, Kovats RS, Lachowycz K. Heat-related and cold-related deaths in England and Wales: who is at risk? Occup Environ Med 20076493–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Suematsu N, Tsutsui H, Wen J, et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 20031071418–1423 [DOI] [PubMed] [Google Scholar]
- 52.Holland NA, Fraiser CR, Sloan RC, 3rd, Devlin RB, Brown DA, Wingard CJ. Ultrafine particulate matter increases cardiac ischemia/reperfusion injury via mitochondrial permeability transition pore. Cardiovasc Toxicol 201717441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tong F, Zhang H. Pulmonary exposure to particulate matter (PM2.5) affects the sensitivity to myocardial ischemia/reperfusion injury through farnesoid-X-Receptor-Induced autophagy. Cell Physiol Biochem 2018461493–1507 [DOI] [PubMed] [Google Scholar]
- 54.Routledge HC, Ayres JG. Air pollution and the heart. Occup Med (Lond) 200555439–447 [DOI] [PubMed] [Google Scholar]
- 55.Huang YK, Lin CW, Chang CC, et al. Heat acclimation decreased oxidative DNA damage resulting from exposure to high heat in an occupational setting. Eur J Appl Physiol 20121124119–4126 [DOI] [PubMed] [Google Scholar]
- 56.Yamagami K, Yamamoto Y, Toyokuni S, Hata K, Yamaoka Y. Heat shock preconditioning reduces the formation of 8-hydroxy-2’-deoxyguanosine and 4-hydroxy-2-nonenal modified proteins in ischemia-reperfused liver of rats. Free Radic Res 200236169–176 [DOI] [PubMed] [Google Scholar]