

Abstract
Arabinogalactan proteins (AGPs) are plant proteoglycans with functions in growth and development. However, these functions are largely unexplored, mainly because of the complexity of the sugar moieties. These carbohydrate sequences are generally analyzed with the aid of glycoside hydrolases. The exo-β-1,3-galactanase is a glycoside hydrolase from the basidiomycete Phanerochaete chrysosporium (Pc1,3Gal43A), which specifically cleaves AGPs. However, its structure is not known in relation to its mechanism bypassing side chains. In this study, we solved the apo and liganded structures of Pc1,3Gal43A, which reveal a glycoside hydrolase family 43 subfamily 24 (GH43_sub24) catalytic domain together with a carbohydrate-binding module family 35 (CBM35) binding domain. GH43_sub24 is known to lack the catalytic base Asp conserved among other GH43 subfamilies. Our structure in combination with kinetic analyses reveals that the tautomerized imidic acid group of Gln263 serves as the catalytic base residue instead. Pc1,3Gal43A has three subsites that continue from the bottom of the catalytic pocket to the solvent. Subsite −1 contains a space that can accommodate the C-6 methylol of Gal, enabling the enzyme to bypass the β-1,6–linked galactan side chains of AGPs. Furthermore, the galactan-binding domain in CBM35 has a different ligand interaction mechanism from other sugar-binding CBM35s, including those that bind galactomannan. Specifically, we noted a Gly → Trp substitution, which affects pyranose stacking, and an Asp → Asn substitution in the binding pocket, which recognizes β-linked rather than α-linked Gal residues. These findings should facilitate further structural analysis of AGPs and may also be helpful in engineering designer enzymes for efficient biomass utilization.
Keywords: glycoside hydrolase family 43; carbohydrate-binding module family 35 exo-β-13-galactanase; arabinogalactan-protein; Phanerochaete chrysosporium; enzyme structure; carbohydrate metabolism; carbohydrate-binding protein; plant cell wall; biodegradation; carbohydrate-binding module family 35; exo-β-1,3-galactanase
Arabinogalactan proteins (AGPs) are proteoglycans characteristically localized in the plasma membrane, cell wall, and intercellular layer of higher land plants (1), in which they play functional roles in growth and development (2). The carbohydrate moiety of AGPs is composed of a β-1,3-d-galactan main chain and β-1,6-d-galactan side chain, decorated with arabinose, fucose, and glucuronic acid residues (1, 2). The chain lengths and frequencies of side chains are different among plant species, organs, and stages of development (3), and the overall structures of the carbohydrate moieties of AGPs are not yet fully understood. Degradation of polysaccharides using specific enzymes is one approach to investigate their structures and roles. In this context, exo-β-1,3-galactanase (EC 3.2.1.145) specifically cleaves the nonreducing end β-1,3–linked galactosyl linkage of β-1,3-galactans to release d-galactose (Gal). In particular, it releases β-1,6-galactooligosaccharides together with Gal from AGPs (4, 5) and is therefore useful for structural analysis of AGPs.
The basidiomycete Phanerochaete chrysosporium produces an exo-β-1,3-galactanase (Pc1,3Gal43A; GenBankTM accession no. BAD98241) that degrades the carbohydrates of AGPs when grown with β-1,3-galactan as a carbon source (6). Pc1,3Gal43A consists of a glycoside hydrolase (GH) family 43 subfamily 24 (GH43_sub24) catalytic domain and a carbohydrate-binding module (CBM) belonging to family 35 (designated as PcCBM6 in (6)) based on the amino acid sequences in the Carbohydrate-Active enZymes (CAZy) database (RRID:SCR012935) (6, 7, 8). The properties of the enzyme have been analyzed using recombinant Pc1,3Gal43A expressed in the methylotrophic yeast Pichia pastoris (6). The CBM35 of Pc1,3Gal43A was characterized as the first β-1,3-galactan–binding module, and Pc1,3Gal43A showed typical GH43_sub24 activity. The enzyme cleaves only β-1,3 linkages of oligosaccharides and polysaccharides but produces β-1,6-galactooligosaccharides together with Gal. Thus, Pc1,3Gal43A specifically recognizes β-1,3–linked Gal but can accommodate β-1,6–bound side chains (6).
Glycoside hydrolases are classified into families based on sequence similarity, whereas they are also divided into two major groups according to their catalytic mechanisms (i.e. inverting enzymes and retaining enzymes) (9, 10). Inverting enzymes typically utilize two acidic residues that act as an acid and a base, respectively, and a hydroxyl group connected to anomeric carbon inverts from the glycosidic linkage after the reaction. GH43 enzymes are members of the inverting group and share conserved Glu and Asp as the catalytic acid and base, respectively (8), but GH43_sub24 enzymes lack the catalytic base Asp (8, 11, 12). In Ct1,3Gal43A (from Clostridium thermocellum), Glu112 was thought to be the catalytic base (13), but in BT3683 (from Bacteroides thetaiotamicron), Glu367 (corresponding to Glu112 of Ct1,3Gal43A) was found not to act as a base but to be involved in recognition of the C-4 hydroxyl group of the nonreducing terminal Gal, and instead, Gln577 is predicted to be the catalytic base in the form of an unusual tautomerized imidic acid (12). An example of GH lacking a catalytic base, endoglucanase V from P. chrysosporium (PcCel45A), is already known, and based on the mechanism proposed for this enzyme, it is possible that tautomerized Gln functions as a base in GH43_sub24 or that this Gln stabilizes nucleophilic water. PcCel45A lacks the catalytic base Asp that is conserved in other GH45 subfamilies (14), but it uses the tautomerized imidic acid of Asn as the base, as indicated by neutron crystallography (15). However, it is difficult to understand the situation in GH43_sub24, because no holo structure with a ligand at the catalytic center has yet been solved in this family. Moreover, no structure of eukaryotic GH43_sub24 has yet been reported.
The CBM35 module is composed of ∼140 amino acids. This family includes modules with various binding characteristics and decorated with xylans, mannans, β-1,3-galactans, and glucans (16, 17, 18, 19, 20, 21). The family members are divided into four clusters based on their sequences and binding specificities (17). The structures of CBM35s binding with xylan, mannan, and glucan have already been solved (16, 17, 18, 19, 20, 21), but no structure of β-1,3-galactan–binding CBM35 has yet been reported.
In the present work, we solved the apo and liganded structures of Pc1,3Gal43A. Based on the results, we discuss the catalytic mechanism and the mode of ligand binding to CBM35 in the two-domain structure.
Results
Overall structure of Pc1,3Gal43A
The crystal structure of the SeMet derivative of Pc1,3Gal43A was first determined by means of the multiwavelength anomalous dispersion method, and this was followed by structure determination of the ligand-free WT, the WT bound with Gal (WT_Gal), the E208Q mutant co-crystallized with β-1,3-galactotriose (Gal3; E208Q_Gal3), and the E208A mutant co-crystallized with Gal3 (E208A_Gal3). Data collection statistics and structural refinement statistics are summarized in Table 1, Table 2, respectively.
Table 1.
Data collection statistics
Values in parentheses are for the highest-resolution shell.
| Data | WT | SeMet |
WT Gal3 soaking | E208Q Gal3 co-crystal | E208A Gal3 co-crystal | |||
|---|---|---|---|---|---|---|---|---|
| Peak | Edge | Low remote | High remote | |||||
| Space group | P1 | P21 | P21 | P21 | P21 | P212121 | P21 | P3221 |
| Unit-cell parameters | ||||||||
| a, b, c (Å) | 40.5, 66.3, 74.0 | 66.4, 50.5, 75.8 | 50.8, 66.6, 106.4 | 66.1, 50.4, 75.7 | 156.7, 156.7, 147.7 | |||
| α, β, γ (degrees) | 72.0, 84.7, 82.1 | 90.0, 111.9, 90.0 | 90.0, 90.0, 90.0 | 90.0, 111.3, 90.0 | 90.0, 120.0, 90.0 | |||
| Beam line | PF BL-5 | PF BL-6A | PF BL-6A | PF BL-6A | PF BL-6A | PF-AR NW12 | PF-AR NE3 | PF-AR NE3 |
| Detector | ADSC Q315 | ADSC Q4R | ADSC Q210 | ADSC Q270 | ADSC Q270 | |||
| Wavelength (Å) | 0.90646 | 0.97882 | 0.97950 | 0.98300 | 0.96400 | 1.0000 | 1.0000 | 1.0000 |
| Resolution (Å) | 50–1.40(1.45–1.40) | 50.0–1.80 (1.86–1.80) | 50.0–2.00 (2.07–2.00) | 50.0–2.00 (2.07–2.00) | 50.0–2.00 (2.07–2.00) | 100.0–1.50 (1.55–1.50) | 50.0–2.50 (2.54–2.50) | 100.0–2.30 (2.38–2.30) |
| Rsym | 0.054 (0.370) | 0.079 (0.672) | 0.061 (0.307) | 0.060 (0.303) | 0.062 (0.307) | 0.046 (0.109) | 0.143 (0.399) | 0.167 (0.627) |
| Completeness (%) | 95.6 (89.0) | 100.0 (99.9) | 100.0 (100.0) | 100.0 (100.0) | 100.0 (100.0) | 97.5 (94.9) | 96.2 (83.0) | 99.1 (92.0) |
| Multiplicity | 3.8 (3.1) | 14.0 (12.6) | 7.2 (6.9) | 7.2 (6.9) | 7.2 (7.0) | 9.2 (8.9) | 4.4 (3.0) | 9.7 (5.1) |
| Average I/σ(I) | 24.4 (2.8) | 36.6 (4.7) | 30.9 (8.3) | 30.8 (8.2) | 31.3 (8.2) | 48.9 (21.0) | 13.5 (2.7) | 17.9 (2.7) |
| Unique reflections | 136,692 (12,747) | 43,643 (4,353) | 31,744 (3,139) | 31,760 (3,144) | 31,780 (3,146) | 57,278 (5,493) | 16,007 (702) | 92,497 (8,510) |
| Observed reflections | 520,085 | 613,162 | 227,158 | 228,381 | 228,595 | 524,957 | 69,939 | 900,469 |
| Z | 2 | 1 | 1 | 1 | 4 | |||
Table 2.
Refinement statistics
Values in parentheses are for the highest-resolution shell.
| Data | WT | WT_Gal | E208Q_Gal3 | E208A_Gal3 |
|---|---|---|---|---|
| Resolution range | 7.997–1.398 (1.448–1.398) | 41.56–1.500 (1.554–1.500) | 29.79–2.499 (2.588–2.499) | 30.66–2.300 (2.382–2.300) |
| Completeness (%) | 95.46 (87.82) | 97.51 (94.80) | 96.41 (85.67) | 98.78 (92.17) |
| Wilson B-factor | 12.76 | 10.11 | 29.91 | 30.40 |
| Reflections used in refinement | 136,655 (12,497) | 57,105 (5,474) | 15,762 (1,381) | 92,011 (8,507) |
| Reflections used for R-free | 6,862 (630) | 2,884 (272) | 799 (64) | 4,568 (441) |
| R-work (%) | 15.47 (22.50) | 13.43 (12.71) | 16.62 (25.54) | 16.10 (22.39) |
| R-free (%) | 18.56 (26.28) | 16.00 (17.93) | 24.39 (42.53) | 21.43 (28.28) |
| No. of nonhydrogen atoms | 7,966 | 3,923 | 3,576 | 14,570 |
| Macromolecules | 6,615 | 3,290 | 3,235 | 12,886 |
| Ligands | 109 | 121 | 114 | 678 |
| Solvent | 1,242 | 512 | 227 | 1,006 |
| Protein residues | 2,106 | 427 | 428 | 1,708 |
| r.m.s. (bonds) | 0.008 | 0.006 | 0.008 | 0.011 |
| r.m.s. (angles) | 1.22 | 0.87 | 0.94 | 1.05 |
| Ramachandran favored (%) | 97.29 | 97.41 | 94.13 | 95.76 |
| Ramachandran allowed (%) | 2.71 | 2.59 | 5.87 | 4.24 |
| Ramachandran outliers (%) | 0 | 0 | 0 | 0 |
| Rotamer outliers (%) | 0.81 | 0.55 | 0.29 | 0.36 |
| Clash score | 2.06 | 1.95 | 6.94 | 3.50 |
| Average B-factor (Å2) | 17.21 | 12.45 | 30.48 | 32.98 |
| Macromolecules | 14.97 | 10.57 | 29.77 | 31.60 |
| Ligands | 29.38 | 23.33 | 52.26 | 56.11 |
| Solvent | 28.09 | 22.02 | 29.74 | 35.03 |
| PDB code | 7BYS | 7BYT | 7BYV | 7BYX |
The recombinant Pc1,3Gal43A molecule is composed of a single polypeptide chain of 428 amino acids (Gln21–Tyr448) with two extra amino acids, Glu19 and Phe20, derived from the restriction enzyme cleavage site, which are disordered and thus were not observed. The protein is decorated with N-glycans because it was expressed in Pichia yeast. Up to three sugar chains are attached at Asn79, Asn194, and Asn389; the attached chains vary in position and structure, and most contain one or two GlcNAc moieties.
Pc1,3Gal43A is composed of two domains, and ligands introduced by soaking or co-crystallization are located in a subsite of the catalytic domain or the binding site of CBM35 (Fig. 1). The N-terminal catalytic domain consists of a five-bladed β-propeller (Gln21–Gly325), as in other GH clan-F enzymes, and the C-terminal domain (PcCBM35) takes a β-jellyroll fold (Thr326–Tyr448) structure, as in previously reported CBM35s (16, 17, 18, 19, 20, 21, 22, 23, 24, 25). PcCBM35 contains one calcium ion near the end of the first β-strand on a different domain surface from the plane to which the ligand binds (Fig. 1). The structure of PcCBM35 is similar to those of other known CBM35s. The interface area is 686 Å2 and includes many water molecules. The PDBePISA server (RRID:SCR015749) indicates that the enzyme forms a complex in the crystal, but this is an effect of crystallization, and the enzyme exists as a monomer in solution (data not shown).
Figure 1.

Overall structure of Pc1,3Gal43A. In the three-dimensional structure of Pc1,3Gal43A, the five blades of the catalytic domain are shown in blue (Gln21–Leu87), cyan (Ser88–Asp155), green (Ser156–Gly204), yellow (Ala205–Ser247), and orange (Ala248–Asp297) with successive roman numerals. The CBM (The326–Val448) is shown in orange. The linker connecting the two domains (Phe298–Gly325) is shown in gray.
Sugar-binding structure of the Pc1,3Gal43A catalytic domain
The five-bladed β-propeller exhibits an almost spherical structure, and two central cavities are located at the ends of the pseudo-5-fold axis (Fig. 1). One of them contains the catalytic site and it is common in almost all GH43 enzymes. The catalytic site is located in the center of the five-bladed β-propeller, whose blades are formed by Gln21 or Asn22–Leu87 (I in Fig. 1), Ser88–Asp155 (II in Fig. 1), Ser156–Gly204 (III in Fig. 1), Ala205–Ser247 (IV in Fig. 1), and Ala248–Asp297 (V in Fig. 1).
As shown in Fig. 2, the Gal3 molecule co-crystallized with the E208Q mutant occupies subsites −1, +1, and +2 of the catalytic site, from the nonreducing end to the reducing end. Gal−1 is located at the bottom of the catalytic cavity, and Gal+1 and Gal+2 extend linearly outwards. Gal+1 is half-buried in the cavity, whereas Gal+2 is exposed at the surface (Fig. 2A).
Figure 2.

Gal3-binding mode at the catalytic site.A, surface structure of the catalytic center. Gal3 is represented as green (carbon) and red (oxygen) sticks. B, schematic diagram showing the interaction mode at the catalytic center. Black, red, and blue, carbon, oxygen, and nitrogen, respectively. Red lines indicate the hydrophobically interacting residues. This diagram was drawn with LigPlot+ (version 1.4.5). C, the 2Fo − Fc omit map is drawn as a blue mesh (0.8σ). Residues are shown in white (carbon), red (oxygen), and blue (nitrogen). Gal3 is shown in green (carbon) and red (oxygen). Yellow dots, hydrogen bonds and/or hydrophobic interaction; red spheres, water molecules interacting with ligands or residues.
Gal−1 adopts a 1S3 skew boat conformation and interacts with many residues via hydrogen bonds and hydrophobic interactions. As shown in Fig. 2 (B and C), the C-2 hydroxyl group of Gal−1 forms hydrogen bonds with NH2 of Arg103 and with OE1 of Gln263 via water. In addition, this water molecule is bound with O of Gly228. The C-3 hydroxyl group of Gal−1 also forms a hydrogen bond with OE2 of Glu57 via water. Glu102, Tyr126, Asp158, Gln208, Thr226, Trp229, and Gln263 interact with Gal3 through hydrophobic interactions. Notably, Trp229 supports the flat C3-C4-C5-C6 structure of Gal−1, and Tyr126 recognizes the C-6 methylol and C-4 hydroxyl groups, whereas Glu102 recognizes the C-3 hydroxyl and C-4 hydroxyl groups. In Gal+1 (as shown in Fig. 2, B and C), the C-2 hydroxyl group forms a hydrogen bond with NE2 of Gln208 and N of Gly228, whereas O5 forms a hydrogen bond with ND2 of Asn180, and C-6 hydroxyl group forms a hydrogen bond with OD1 of Asn179 via water. Tyr126, Arg157, Asn180, and Gln208 interact hydrophobically with Gal. In Gal+2 (Fig. 2, B and C), the C-2 and C-4 hydroxyl groups form hydrogen bonds with OG1 of Thr226 and ND2 of Asn180, respectively. In addition, Thr226 interacts with Gal+2 through hydrophobic interaction. Furthermore, the glycosidic oxygen between Gal+1 and Gal+2 interacts with ND2 of Asn180 through a hydrogen bond.
In the structure of WT_Gal, one Gal was found at subsite −1, taking a 4C1 chair conformation with α-anomeric conformation of the C-1 hydroxyl group (data not shown). The binding mode of Gal−1 is almost the same as that in E208Q_Gal3, but the C-1 hydroxyl group in the axial position forms hydrogen bonds with Gly228 and Gln263. No Gal3 molecule was observed at the catalytic domain in the structure of the Gal3 co-crystallized E208A mutant.
To identify the catalytic residues, we examined the relative activity of WT and the six mutants toward β-1,3-galactobiose (Gal2) and Gal3. WT showed 5.58 ± 0.35 and 11.15 ± 0.39 units of activity (μmol of Gal/min/nmol of enzyme) toward Gal2 and Gal3, respectively, whereas the six mutants showed no detectable activity (Fig. S1), suggesting that these residues are all essential for the catalysis.
Sugar-binding structure of CBM35 in Pc1,3Gal43A
Pc1,3Gal43A has one CBM35 domain at the C terminus. We previously reported that this enzyme has a CBM6-like domain (6), but it has been reclassified into the CBM35 family (7). The β-jellyroll fold domain is accompanied by a single calcium ion–binding site on a domain surface different from the surface to which the ligand at the end of the first β chain binds, and this corresponds to a conserved calcium ion–binding site in CBM35s. Some CBM35 modules bind another calcium ion at a site at the top of domain (16), but PcCBM35 lacks this second calcium ion–binding site (Fig. 1).
In E208A_Gal3, electron density of Gal3 was observed in the ligand-binding site of PcCBM35. As illustrated in Fig. 3A and Fig. S2, 2Fo − Fc omit maps showed that the binding mode of PcCBM35 with ligands is “exo-type,” corresponding to type-C CBM (26). The asymmetric unit of E208A_Gal3 contained four Pc1,3Gal43A molecules, and each molecule binds to the nonreducing end of Gal3 (called Gal_site 1), as in other CBM35 modules. However, the middle Gal (Gal_site 2) and the reducing end Gal (Gal_site 3) are found in two main locations (Fig. 3), although residues involved in the interactions with the ligand in each molecule were mostly shared. The Gal_site 1 forms hydrogen bonds with Tyr355 and Arg388 and interacts hydrophobically with Leu342, Gly354, Tyr438, and Asp441. The Gal_site 2 interacts hydrophobically with Gly383 and Asp384. The main ligand interaction in the Gal_site 3 involves Gly409 and Gly410, but in addition to these residues, Asn411 is also involved in ligand recognition in chain C (Fig. 4).
Figure 3.

Surface structures of the CBM.A, substrate-binding mode at CBM35. Green, cyan, magenta, and yellow, carbons of chains A, B, C, and D of E208A_Gal3, respectively; red, oxygen. Left, nonreducing end of Gal3; right, reducing end. B, calcium ion–binding mode at CBM35. Calcium ion is represented as green spheres, and interacting residues are shown as stick models. Yellow dots, interaction.
Figure 4.

Ligand interaction mode at CBM35.A and E, B and F, C and G, and D and H, chains A, B, C, and D of E208A, respectively. A–D, interaction modes between ligand and CBM35 residues. Atoms are indicated in the same colors as in Fig. 2. E–G, schematic diagram showing the interaction mode at CBM35. Atoms are indicated in the same colors as in Fig. 2. Sugar-binding sites are named Gal_site 1, Gal_site 2, and Gal_site 3 from the nonreducing end of the sugar, and in this figure, they are labeled 1, 2, and 3, respectively.
Ensemble refinement
To understand the fluctuation of ligands, ensemble refinements were performed with the refined models. This method produces ensemble models by employing a combination of X-ray structure refinement and molecular dynamics. These models can simultaneously account for anisotropic and anharmonic distributions (27). Four different pTLS values of 0.6, 0.8, 0.9, and 1.0% were set for each model. Table 3 shows the statistical scores of the refinement with the most appropriate pTLS value for each model. Focused views of the catalytic site in the catalytic domain and the ligand-binding site of the CBM are shown in Figure 5, Figure 6, respectively. Note that structures containing multiple molecules in the asymmetric unit (WT and E208A_Gal3) are found for all molecules in this paper.
Table 3.
Refinement statistics of ensemble refinement
Values in parentheses are for the highest-resolution shell.
| Data | WT | WT_Gal | E208Q_Gal3 | E208A_Gal3 |
|---|---|---|---|---|
| Resolution range | 7.997–1.398 (1.448-1.398) | 41.56–1.500 (1.554–1.500) | 29.79–2.499 (2.588–2.499) | 30.66–2.300 (2.382– 2.300) |
| Completeness (%) | 95.97 (82) | 97.52 (95) | 96.47 (88) | 98.93 (87) |
| pTLS (%) | 0.9 | 0.9 | 0.9 | 1.0 |
| Tx | 1.0 | 0.9 | 0.3 | 0.4 |
| Wilson B-factor | 12.8 | 10.1 | 29.9 | 30.4 |
| Reflections used in refinement | 136,649 | 57,112 | 15,759 | 91,994 |
| Reflections used for R-free | 6,862 | 2,885 | 799 | 4,569 |
| R-work | 13.81 (24.36) | 12.08 (10.68) | 17.82 (24.73) | 15.92 (22.56) |
| R-free | 17.08 (26.30) | 15.29 (17.10) | 23.33 (32.75) | 20.71 (28.84) |
| r.m.s. (bonds) | 0.008 | 0.010 | 0.007 | 0.008 |
| r.m.s. (angles) | 1.171 | 1.312 | 1.078 | 1.090 |
| Ramachandran favored (%) | 94.06 | 95.39 | 88.98 | 92.62 |
| Ramachandran allowed (%) | 5.08 | 4.03 | 9.19 | 7.24 |
| Ramachandran outliers (%) | 0.86 | 0.58 | 1.83 | 0.74 |
| Rotamer outliers (%) | 7.45 | 7.00 | 11.05 | 7.85 |
| Clash score | 0 | 0 | 0 | 0 |
| Average B-factor (Å2) | 13.65 | 9.55 | 28.32 | 32.83 |
| Macromolecules | 13.63 | 9.54 | 28.30 | 32.66 |
| Ligands | 14.97 | 9.82 | 28.98 | 36.05 |
| Molprobity score | 1.56 | 1.45 | 1.87 | 1.64 |
| Model number | 100 | 103 | 20 | 34 |
Figure 5.

Results of ensemble refinement at the catalytic site. Each model is divided into three parts for clarity. A (E and I), B (F and J), C (G and K), and D (H and L) show WT, WT_Gal, E208Q_Gal3, and E208A_Gal3, respectively. Although WT and E208A_Gal3 contained multiple molecules in an asymmetric unit, the results obtained with multiple molecules were considered as an ensemble of one molecule in the present study. Letters indicate the chain names. Atoms are indicated in the same colors as in Fig. 2. Gal3 of the structure of E208Q_Gal3 obtained by X-ray crystallography is arranged in each figure to maximize ease of comparison.
Figure 6.

Results of ensemble refinement at the CBM ligand-binding site.A, B, C, and D, residues related to ligand interaction. In this figure, Gal3 of chain A of refined E208A_Gal3 is drawn for comparison. E, F, G, and H, the ligands of each chain. Green, cyan, and yellow are used in order from the nonreducing terminal Gal. A and E, B and F, C and G, and D and H represent chains A, B, C, and D of E208A_Gal3, respectively. Atoms are indicated in the same colors as in Fig. 2.
In the catalytic site, the vibration levels of some residues were significantly different between the apo and holo forms. As shown in Fig. 5, Tyr126, Arg157, Asp158, Asn179, Asn180, Gln181, Trp229, and Gln263 in the liganded structures (Fig. 5, B, C, F, G, J, and K) showed smaller vibrations than in the apo structures (Fig. 5, A, D, E, H, I, and L). These results indicate that side-chain fluctuations converge upon ligand binding. Comparison of the Gal-bond structure (i.e. WT_Gal; Fig. 5, B, F, and J) with the Gal3-bond structure (i.e. E208Q_Gal3; Fig. 5, C, G, and K) showed that the fluctuations of Glu(Gln)208, Asn179, and Thr226 of E208Q_Gal3 were smaller than these in WT_Gal. Therefore, it can be inferred that these residues recognize the ligands at the plus subsites. The catalytic acid, Glu208, has two major conformations in WT and WT_Gal. These two conformations were also reported in the BT3683 structure (12). Thus, the movement of this residue appears to be important for catalysis. Gln263 shows one conformation (Fig. 5, A–D) that is identical to the result of the ensemble refinement of Asn92, known as imidic acid in PcCel45A (Fig. S3). Glu102 may distinguish nonreducing terminal Gal, because it interacts with the axial C-4 hydroxyl group of Gal−1 (12). The vibration degree of Glu102 was different between WTs and mutants, so its conformation does not depend on the ligand localization, but reflects interaction with Glu208, which serves as a general acid. Asp158 of WT and E208A_Gal3 show greater vibration than WT_Gal and E208Q_Gal3. The role of Asp158 is thought to be a pKa modulator; therefore, its function and conformational stability might be related. Focusing on Fig. 5 (I–L), there were large differences in the fluctuation level of Trp229. E208Q_Gal3 (Fig. 5K) showed small movements of Trp229, but other structures showed much larger fluctuations (Fig. 5, I, J, and L). These results suggest that this Trp is normally flipped and forms a π-π interaction to anchor the ligand in the proper position upon arrival. A histogram of the dihedral angle is shown in Fig. S4.
As regards the ligand-binding site of the CBM, a comparison of each chain of the E208A_Gal3 asymmetric unit showed no significant difference in the vibration levels of each residue involved in ligand binding (Fig. 6). However, ensemble refinement revealed that Gal_site 1 and Gal_site 2 do not show huge fluctuations, whereas Gal_site 3 has many conformations. They include the same conformation of each Gal chain in X-ray crystallography. Interestingly, a spatial difference in fluctuations was observed between ligands bound to the catalytic site and to the ligand-binding site of CBM35 (Fig. 7). At the catalytic site, Gal−1 is anchored in the appropriate position, and Gal+2 appears to fluctuate in a planar fashion as it interacts with the surrounding residues. In the CBM, it was inferred that Gal_site 1 is fixed and Gal_site 3 is adsorbed at the appropriate location at the binding site while fluctuating in three dimensions.
Figure 7.

Ligand conformation of ensemble refinement at a glance.A, ligand conformation of E208Q_Gal3 ensemble model. B, ligand conformation of E208A_Gal3 ensemble models with four chains aligned. Green, cyan, and yellow are used in order from the nonreducing terminal Gal.
Discussion
Most exo-β-1,3-galactanases belonging to GH43_sub24 possess CBMs that can be classified into CBM35 or CBM13 (8). In this study, we elucidated the structure of a β-1,3-galactan–binding module for the first time by solving the structure of a GH43_sub24 containing CBM35 and obtained the ligand-bound structures of both the catalytic and sugar-binding domains of Pc1,3Gal43A. This is also the first study to reveal the structure of a eukaryotic exo-β-1,3-galactanase. This information will be useful to understand how the CBM35 module interacts with β-1,3-galactan in combination with the GH43_sub24 catalytic module.
How does Pc1,3Gal43A hydrolyze β-1,3-galactan?
Although catalytic residues such as Glu and Asp are conserved in GH43 as a catalytic acid and base, respectively, GH43_sub24 lacks such a base residue. Cartmell et al. (12) suggested that GH43_sub24 may use Gln in the base role via conversion to imidic acid or use an exogenous base or utilize the Grotthuss mechanism of catalysis (8, 12). In this study, we measured the enzyme activity of six variants of the three residues speculated to be involved in the catalytic reaction. As shown in Fig. S1, production of Gal by the mutants was not detected by means of HPLC analysis, suggesting that all three residues are essential for the catalytic activity of Pc1,3Gal43A. Glu102, Glu208, and Gln263 are speculated to serve in C-4 hydroxyl group recognition, as a catalytic acid, and as a catalytic base, respectively. These residues are well-conserved in GH43_sub24, as shown in Fig. S5.
In GH43_sub24, only bacterial enzyme structures have been solved so far (http://www.cazy.org/GH43_24.html). To understand the catalytic mechanism of Pc1,3Gal43A, we compared its structure with those of BT3683 and Ct1,3Gal43A (Fig. 8). Most of the residues that interact with ligands are conserved in these three enzymes. In subsite −1, all residues, Glu57, Glu102, Arg103, Tyr126, Asp158, Glu208, Trp229, and Gln263, of Pc1,3Gal43A are conserved, indicating that the binding mode at subsite −1 is fully conserved in GH43_sub24. Based on the results of ensemble refinement, Trp229 showed huge fluctuation, especially in the apo structure (Fig. 5, I–L). Trp541 of BT3683, which corresponds to Trp229 of Pc1,3Gal43A, has a polar interaction with Gal (12). Trp229 fluctuates in solution and plays a role in holding the substrate at the catalytic site through polar interactions. On the other hand, Asn179 and Thr226 of Pc1,3Gal43A are replaced by Asp490 and Cys538 in BT3683 and by Glu199 and Cys247 in Ct1,3Gal43A. Because all of these enzymes can accommodate a β-1,6–branched side chain (6, 12, 28), we considered that these residues are not related to the mechanism of side-chain accommodation.
Figure 8.

Catalytic domain structure comparison.A, visualization of the degree of preservation of GH43_sub24. The degree of conservation of amino acid residues in the catalytic domain of GH43_sub24 was visualized using the ConSurf server (RRID:SCR002320), the query for BLAST was set to Pc1,3Gal43A, and the conservation degree was analyzed based on 150 amino acid sequences in the ConSurf server (47,48, 49, 50,51). The conservation degree is shown in graded color. Preservation degrees are shown in a gradient with cyan for the lowest degree of preservation and blue for the highest. B, catalytic domain comparison of Pc1,3Gal43A and two GH43_sub24 galactanases. Shown are the catalytic centers of E208Q_Gal3 of Pc1,3Gal43A (white, PDB code 7BYV), BT3683 (cyan, PDB code 6EUI), and Ct1,3Gal43A (pink, PDB code 3VSF). Red, blue, and yellow, oxygen atoms, nitrogen atoms, and sulfur atoms, respectively. Residue names are shown for Pc1,3Gal43A/BT3683/Ct1,3Gal43A.
The bypass mechanism of Pc1,3Gal43A, which enables accommodation of the β-1,6-galactan side chain so that the β-1,3-galactan main chain can be cleaved, appears to depend on the orientation of the C-6 methylol group of Gal3 at each subsite. The C-6 methylol group of Gal−1 is exposed to the solvent, so that the side chain can be accommodated externally. The C-6 methylol groups of Gal+1 and Gal+2 are also exposed to the solvent, so that the enzyme should be able to cleave the β-1,3 linkage of continuously β-1,6–substituted galactan, and a similar situation has been reported for BT3683 (12). Moreover, there are spaces near the nonreducing terminal Gal in these enzymes (12). This enables the enzymes to degrade the main chain, even if the side chain contains multiple carbohydrates. Similarly, β-1,3-glucanases belonging to GH55 also bypass the β-1,6-glucan side chain and degrade β-1,3-glucan from the nonreducing end (29, 30). Comparing the surface structure of the catalytic site of Pc1,3Gal43A with that of these GH55 exo-β-1,3-glucanases from P. chrysosporium (PcLam55A), we see that Pc1,3Gal43A has a small pocket-like space capable of accepting the C-6 side chain of Gal at subsite −1 (Fig. 9, A and B). In addition, the C-6 methylol group of Gals, located at the positive subsites of Pc1,3Gal43A, are exposed to solvent in a similar manner to that reported for SacteLam55A, GH55 exo-β-1,3-glucanase from Streptomyces sp. SirexAA-E (Fig. 9, A and C). Structures capable of accepting nonreducing terminal Gal with β-1,6–linked Gal are conserved among GH43_sub24 of known structure (Fig. 8 and Fig. S5). In the nonbypassing GH3 Hypocrea jecorina β-glucosidase (HjCel3A), the C-6 hydroxyl group of nonreducing glucose is oriented toward the enzyme, introducing steric hindrance (Fig. 9D) (31). In other words, enzymes bypassing side chains have a space adjacent to C-6 of the nonreducing terminal sugar, and the positive subsites are particularly wide, allowing side chains of the substrate to be accommodated. In contrast, enzymes unable to bypass the side chain have no space next to the −1 subsite and have a narrow entrance to the catalytic site, so that they are unable to accommodate side chains (Fig. 9D).
Figure 9.

Structure comparison of the catalytic sites of Pc1,3Gal43A (A), GH55 exo-β-1,3-glucanase from P. chrysosporium (B; PcLam55A; PDB code3EQO), GH55 exo-β-1,3-glucanase from Streptomyces sp. SirexAA-E (C; SacteLam55A; PDB code4PF0), and GH3 β-glucosidase from H. jecorina (D, HjCel3A; PDB code3ZYZ). A, B, and C hydrolyze the main chain of β-1,3-galactan or β-1,3-glucan, bypassing β-1,6–branched side chains (6, 29, 30). D hydrolyzes four types of β-bonds, and it does not bypass side chains (31, 52). The top panel shows the overall surface structure, and the bottom panel shows an enlarged view of the catalytic region. Orange dashed circles, space near the C-6 position of Gal or glucose at the nonreducing end.
Although the electron density of Gal3 was observed in the present study, Pc1,3Gal43A is proposed to have four subsites ranging from −1 to +3, based on biochemical experiments (6). As mentioned above, although Pc1,3Gal43A has a structure capable of accepting the C-6 side chain, its degradation activity toward β-1,3/1,6-galactan is only approximately one-fifth that of the linear β-1,3-galactan (6). This difference in reactivity may be due to the structure of the sugar. The β-1,3-galactan in solution has a right-handed triple helical structure with 6–8 Gal residues per turn (32, 33), with the C-6 methylol group pointing outward to avoid collisions between the β-1,6–bonded Gal side chains (32). However, as shown in Fig. S6, Gal3 bound to the catalytic site of Pc1,3Gal43A is anchored to the enzyme, so that the helix of the glycans differs from the usual state in solution. Therefore, the reason why the hydrolytic activity of Pc1,3Gal43A toward β-1,3/1,6-galactan is lower than that toward β-1,3-galactan may be interference between the β-1,6-Gal side chains as a result of changes in the helical state of the main chain.
How does PcCBM35 recognize β-1,3-galactan?
Although the amino acid sequence similarity of CBM35s is not so high, important residues involved in ligand binding are well-conserved (17). The modules belonging to CBM35 can be divided into four clades according to the mode of ligand binding, and the diversity in ligand binding and in the calcium ion–coordinating residue account for the various ligand-binding specificities (17) (Fig. 10A). Moreover, the residues involved in ligand binding of PcCBM35 differ from those of CBM35, which binds to α-Gal of galactomannan. This CBM is one part of a protein predicted to be the β-xylosidase of C. thermocellum cellulosomal protein (Cte_2137; Fig. 10), which belongs to the same cluster as PcCBM35 (17). There are some differences between the residues interacting with α-Gal of Cte_2137 and those interacting with β-Gal of PcCBM35. For instance, the regions of Ala352–Tyr355 and Tyr438–Asp441 of PcCBM35 correspond to Val39–Gly42 and Ser136–Asn140 of Cte_2137, which are related to ligand specificity (Fig. 10). Especially, Asn140 of Cte_2137 is not conserved but replaced by Asp441 in PcCBM35 and is located at the bottom of the ligand-binding site. Furthermore, Trp108 of Cte_2137 plays a key role in sacking the pyranose ring (17), whereas in CBM35 of Pc1,3Gal43A, this Trp residue is replaced with Gly (Fig. 10B). In other words, although PcCBM35 and Cte_2137 are in the same cluster, the residues involved in ligand recognition are different, and this difference affects the discrimination between β-Gal and α-Gal and between galactan and galactomannan. It is still unclear how CBM35s acquire such variation of binding specificity within a similar binding architecture. However, a detailed understanding of the molecular mechanisms of polysaccharide recognition by CBM35 will be essential for efficient utilization of various types of biomass.
Figure 10.
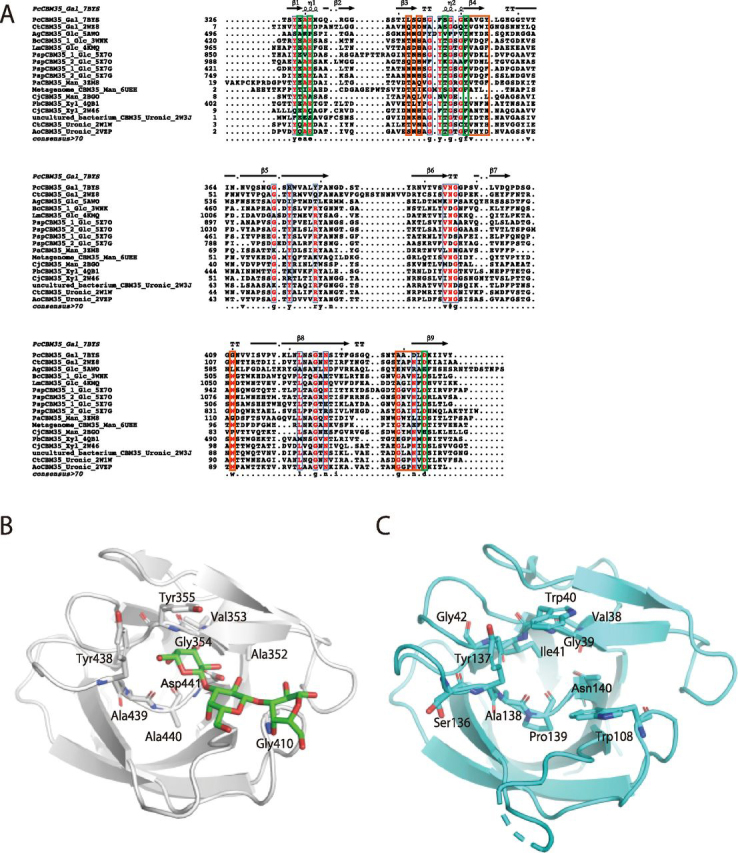
Sequence alignment of known CBM35s (A) and structure comparison between CBM35s of Pc1,3Gal43A (B) and Cte_2137 (C).A, taxon names are shown as scientific names, ligand specificity, and PDB code only for brevity. When the same enzyme contains two CBM35 domains, the taxon name is indicated with 1 on the N terminus and 2 on the C terminus. Gal, Glc, Man, Xyl, and Uronic, ligand specificities for Gal, glucose, mannose, xylose, and glucronic acid and/or galacturonic acid, respectively. Among these, 3ZM8, 6UEH, and 2BGO, which bind to Man, are type B CBMs, which show endo-type binding, whereas the other 14 are all type C CBMs, which show exo-type binding. The alignment was built by using MUSCLE on MEGAX: Molecular Evolutionary Genetics Analysis (53, 54), and the figure was generated with ESPrint 3.0 (RRID:SCR006587) (55). Orange and green boxes represent ligand-binding and calcium ion–binding residues, respectively. B and C, ligand-binding residues of Pc1,3Gal43A (chain A of E208A_Gal3) and Cte_2137 (PDB code 2WZ8). Red and blue, oxygen and nitrogen, respectively. The green stick model represents Gal3.
In conclusion, we have determined the crystal structure of the catalytic and binding domains of Pc1,3Gal43A with the aim of reaching a detailed understanding of the mechanism of substrate accommodation by side chain–bypassing galactanase. Pc1,3Gal43A uses Glu as the catalytic acid and Gln as the catalytic base and has a structure in which the side chain of the substrate does not interfere with the catalytic reaction, thus making it possible to degrade the β-1,3-galactan main chain of AGPs despite the presence of the β-1,6-galactan side chain. Thus, although polysaccharides have a variety of molecular decorations, it appears that the structures of the degrading enzymes enable them to recognize specific features of the substrate while accommodating the variations. The introduction of mutations in substrate recognition residues to create enzymes with altered substrate recognition properties is expected to be helpful in the structural analysis of AGP glycans and also for the preparation of useful oligosaccharides.
Experimental procedures
Expression of Pc1,3Gal43A and its mutants
The E208Q, E208A, E102Q, E102A, Q263E, and Q263A mutants were constructed by inverse PCR using PrimeSTAR MAX (Takara, Tokyo, Japan). For crystallization, Pc1,3Gal43A WT, E208Q, and E208A from P. chrysosporium were expressed in P. pastoris and purified as reported previously (7). For a reactivity assay, WT and mutants were purified by using SkillPak TOYOPEARL Phenyl-650M (c.v. = 5 ml, Tosoh, Tokyo, Japan) equilibrated with 20 mm sodium acetate buffer, pH 4.0, containing 1 m ammonium sulfate, and the enzymes were eluted with 20 mm sodium acetate buffer, pH 4.0, containing 0.7 m ammonium sulfate. SeMet-labeled Pc1,3Gal43A was expressed as reported previously (7).
Preparation for β-1,3-galactooligosaccharides and crystallization of Pc1,3Gal43A
Gal2 and Gal3 were prepared as reported previously (6). The protein solution was concentrated to 4.9–6.9 mg/ml and used for the crystallization setup. The WT plate crystal used for data collection was obtained from a reservoir of 2.1 m ammonium sulfate, 0.1 m citrate buffer, pH 5.5. Other WT crystals were obtained from solutions in 16% (w/v) PEG 10000, 0.1 m ammonium sulfate, 0.1 m bis-tris, pH 5.5, and 5.0% (v/v) glycerol. SeMet crystals were obtained from 16% (w/v) PEG 10000, 95 mm ammonium sulfate, 95 mm bis-tris, pH 5.5, and 4.8% (v/v) glycerol. Two types of crystals, thin plate crystals (space group P21) and rod crystals (P212121), appeared under the same condition. Cocrystallization of the E208Q mutant with 10 mm Gal3 in 16% (w/v) PEG 10000, 95 mm ammonium sulfate, 95 mm bis-tris, pH 5.5, and 4.8% (v/v) glycerol afforded thin plate crystals. The E208A mutant was cocrystallized with 10 mm Gal3 in 0.2 m potassium nitrate, 15% (w/v) PEG 6000, 20 mm sodium citrate, pH 4.5, and 5% glycerol to afford bipyramidal crystals.
Data collection and structure determination
Diffraction experiments for Pc1,3Gal43A crystals were conducted at the beamlines of the Photon Factory (PF) or Photon Factory Advanced Ring (PF-AR), High Energy Accelerator Research Organization, Tsukuba, Japan (Table 1). Diffraction data were collected using CCD detectors (Area Detector Systems Corp., Poway, CA, USA). Crystals were cryocooled in a nitrogen gas stream to 95 K. For data collection of the WT enzyme complexed with Gal3, Pc1,3Gal43A crystals were soaked in a drop containing 1% (w/v) Gal3 for 10 min before the diffraction experiment. The data were integrated and scaled using the programs DENZO and SCALEPACK in the HKL2000 program suite (34).
Crystal structure was determined by means of the multiwavelength anomalous dispersion method using a SeMet-labeled crystal (7). Initial phases were calculated using the SOLVE/RESOLVE program (35) from five selenium atom positions. The resultant coordinates were subjected to the automodeling ARP/wARP program (36) in the CCP4 program suite (37), and manual model building and molecular refinement were performed using Coot (version 0.8.9, University of Oxford, Oxfordshire, UK) (38), REFMAC5 (version 7.0.063, Science and Technology Facilities Council, Swindon, UK) (39), phenix.refine (40), and phenix.ensemble_refinement (27, 41, 42) in the Phenix suite of programs (version 1.13-2998-000, Lawrence Berkeley National Laboratory, Berkeley, CA, USA) (43). The refinement statistics are summarized in Table 2.
For the analyses of WT and ligand-bound structures, structural determination was conducted by the molecular replacement method with the MOLREP program (44) in the CCP4 program suite using the SeMet or ligand-free structure as the starting model. Bound sugars, water molecules, and crystallization agents were modeled into the observed electron density difference maps. Calcium ion was modeled based on the electron density map and the coordination distances. Three N-glycans were observed, and the identified sugars were modeled. The stereochemistry of the models was analyzed with LigPlot + (version 1.4.5) (45, 46), and structural drawings were prepared using PyMOL (version 2.2.3, Schrödinger, LLC, New York).
Enzymatic activity assay of Pc1,3Gal43A and its mutants
To evaluate the reactivity toward Gal2 and Gal3 of WT and each mutant, 20 nm enzyme was incubated with 0.263 or 0.266 mm galactooligosaccharides in 20 mm sodium acetate, pH 5.0, for 30 min at 30 °C, respectively. The reaction was stopped by heating at 95 °C for 5 min. The supernatant was separated with 75% (v/v) acetonitrile on a Shodex Asahipak NH2P-50 4E column (Showa Denko, Tokyo, Japan), and the amount of released Gal was determined by HPLC (LC-2000 series; Jasco, Tokyo, Japan) with a Corona charged aerosol detector (ESA Biosciences, now Thermo Fisher Scientific). One unit of enzyme activity was defined as the amount of enzyme that releases 1 μmol of Gal/1 min/1 nmol of enzyme under our experimental conditions.
Data availability
The structures presented in this paper have all been deposited in the Protein Data Bank (PDB) with the following codes: 7BYS, 7BYT, 7BYV, and 7BYX. All remaining data are contained within the article.
Acknowledgments
We thank Dr. Takuya Ishida (Japan Aerospace Exploration Agency) for helping with the crystallization and structure refinement. We also thank the staff of Photon Factory for X-ray data collection.
MEXT | Japan Society for the Promotion of Science (JSPS) (19H03013) to Kiyohiko Igarashi
Ministry of Education, Culture, Sports, Science and Technology (MEXT) (18H05494) to Kiyohiko Igarashi
Business Finland (BioAD) to Kiyohiko Igarashi
Edited by Joseph M. Jez
Footnotes
This article contains supporting information.
Author contributions—K. M., K. I., and S. K. conceptualization; K. M., N. K., Z. F., N. S., and K. I. data curation; K. M., N. K., Z. F., and K. I. formal analysis; M. S., K. I., and S. K. supervision; K. I. funding acquisition; K. M., N. K., Z. F., and K. I. validation; K. M., K. I., and S. K. writing-original draft; K. M., N. K., and K. I. project administration; K. M., K. I., and S. K. writing-reviewand editing; N. S. methodology; T. K. and Y.T. resources.
Funding and additional information—This work was supported in part by Grant-in-Aid for Scientific Research (B) 19H03013 (to K. I.) from the Japan Society for the Promotion of Science (JSPS) and a Grant-in-Aid for Innovative Areas 18H05494 from the Japanese Ministry of Education, Culture, Sports, and Technology (MEXT) (to K. I.). In addition, K.I. was supported by Business Finland (BF, formerly the Finnish Funding Agency for Innovation (TEKES)) via the Finland Distinguished Professor (FiDiPro) Program “Advanced approaches for enzymatic biomass utilization and modification (BioAD).”
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- AGP
- arabinogalactan protein
- Gal
- d-galactose
- Pc1,3Gal43A
- exo-β-1,3-galactanase from Phanerochaete chrysosporium
- GH
- glycoside hydrolase
- GH43_sub24
- GH family 43 subfamily 24
- CBM
- carbohydrate-binding module
- Ct1,3Gal43A
- exo-β-1,3-galactanase from Clostridium thermocellum
- BT3683
- β-1,3-galactosidase from Bacteroides thetaiotaomicron VPI-5482
- PcCel45A
- endoglucanase V from P. chrysosporium
- SeMet
- selenomethionine
- Gal3
- β-1,3-galactotriose
- WT_Gal
- WT bound with Gal
- E208Q_Gal3
- E208Q bound with Gal3
- E208A_Gal3
- E208A bound with Gal3
- PcCBM35
- CBM35 domain of Pc1,3Gal43A
- Gal−1
- Gal residue–occupied subsite −1
- Gal+1
- Gal residue–occupied subsite +1
- Gal+2
- Gal residue–occupied subsite +2
- Gal2
- β-1,3-galactobiose
- Gal_site 1
- the nonreducing terminal Gal residue of Gal3 bound to PcCBM35
- Gal_site 2
- middle Gal residue of Gal3 bound to PcCBM35
- Gal_site 3
- reducing terminal Gal residue of Gal3 bound to PcCBM35
- PcLam55A
- exo-β-1,3-glucanase from P. chrysosporium
- SacteLam55A
- GH55 exo-β-1,3-glucanase from Streptomycs sp.
- Cte_2137
- CBM35 of C. thermocellum cellulosomal protein
- r.m.s.
- root mean square
- bis-tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PF
- Photon Factory
- PF-AR
- Photon Factory Advanced Ring
- PDB
- Protein Data Bank.
Supplementary Material
References
- 1.Tsumuraya Y., Hashimoto Y., Yamamoto S., Shibuya N. Structure of l-arabino-d-galactan-containing glycoproteins from radish leaves. Carbohydr. Res. 1984;134:215–228. doi: 10.1016/0008-6215(84)85039-9. [DOI] [Google Scholar]
- 2.Majewska-Sawka A., Nothnagel E.A. The multiple roles of arabinogalactan proteins in plant development. Plant Physiol. 2000;122:3–9. doi: 10.1104/pp.122.1.3. 10631243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ellis M., Egelund J., Schultz C.J., Bacic A. Arabinogalactan-proteins: key regulators at the cell surface? Plant Physiol. 2010;153:403–419. doi: 10.1104/pp.110.156000. 20388666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsumuraya Y., Mochizuki N., Hashimoto Y., Kovác P. Purification of an Exo-β-(1-3)-d-galactanase of Irpex lacteus (Polyporus tulipiferae) and its action on arabinogalactan-proteins. J. Biol. Chem. 1990;265:7207–7215. 2158993. [PubMed] [Google Scholar]
- 5.Pellerin P., Brillouet J.M. Purification and properties of an exo-(1→3)-β-d-galactanase from Aspergillus niger. Carbohydr. Res. 1994;264:281–291. doi: 10.1016/S0008-6215(05)80012-6. 7805066. [DOI] [PubMed] [Google Scholar]
- 6.Ichinose H., Yoshida M., Kotake T., Kuno A., Igarashi K., Tsumuraya Y., Samejima M., Hirabayashi J., Kobayashi H., Kaneko S. An exo-β-1,3-galactanase having a novel β-1,3-galactan-binding module from Phanerochaete chrysosporium. J. Biol. Chem. 2005;280:25820–25829. doi: 10.1074/jbc.M501024200. 15866877. [DOI] [PubMed] [Google Scholar]
- 7.Ishida T., Fujimoto Z., Ichinose H., Igarashi K., Kaneko S., Samejima M. Crystallization of selenomethionyl exo-β-1,3-galactanase from the basidiomycete Phanerochaete chrysosporium. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009;65:1274–1276. doi: 10.1107/S1744309109043395. 20054127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mewis K., Lenfant N., Lombard V., Henrissat B. Dividing the large glycoside hydrolase family 43 into subfamilies: a motivation for detailed enzyme characterization. Appl. Environ. Microbiol. 2016;82:1686–1692. doi: 10.1128/AEM.03453-15. 26729713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davies G., Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3:853–859. doi: 10.1016/S0969-2126(01)00220-9. 8535779. [DOI] [PubMed] [Google Scholar]
- 10.Rye C.S., Withers S.G. Glycosidase mechanisms. Curr. Opin. Chem. Biol. 2000;4:573–580. doi: 10.1016/S1367-5931(00)00135-6. 11006547. [DOI] [PubMed] [Google Scholar]
- 11.Cartmell A., McKee L.S., Peña M.J., Larsbrink J., Brumer H., Kaneko S., Ichinose H., Lewis R.J., Viksø-Nielsen A., Gilbert H.J., Marles-Wright J. The structure and function of an arabinan-specific α-1,2-arabinofuranosidase identified from screening the activities of bacterial GH43 glycoside hydrolases. J. Biol. Chem. 2011;286:15483–15495. doi: 10.1074/jbc.M110.215962. 21339299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cartmell A., Muñoz-Muñoz J., Briggs J.A., Ndeh D.A., Lowe E.C., Baslé A., Terrapon N., Stott K., Heunis T., Gray J., Yu L., Dupree P., Fernandes P.Z., Shah S., Williams S.J. A surface endogalactanase in Bacteroides thetaiotaomicron confers keystone status for arabinogalactan degradation. Nat. Microbiol. 2018;3:1314–1326. doi: 10.1038/s41564-018-0258-8. 30349080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang D., Fan J., Wang X., Zhao Y., Huang B., Liu J., Zhang X.C. Crystal structure of 1,3Gal43A, an exo-β-1,3-galactanase from Clostridium thermocellum. J. Struct. Biol. 2012;180:447–457. doi: 10.1016/j.jsb.2012.08.005. 22960181. [DOI] [PubMed] [Google Scholar]
- 14.Igarashi K., Ishida T., Hori C., Samejima M. Characterization of an endoglucanase belonging to a new subfamily of glycoside hydrolase family 45 of the basidiomycete Phanerochaete chrysosporium. Appl. Environ. Microbiol. 2008;74:5628–5634. doi: 10.1128/AEM.00812-08. 18676702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakamura A., Ishida T., Kusaka K., Yamada T., Fushinobu S., Tanaka I., Kaneko S., Ohta K., Tanaka H., Inaka K., Higuchi Y., Niimura N., Samejima M., Igarashi K. “Newton's cradle” proton relay with amide–imidic acid tautomerization in inverting cellulase visualized by neutron crystallography. Sci. Adv. 2015;1 doi: 10.1126/sciadv.1500263. 26601228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Montanier C., Van Bueren A.L., Dumon C., Flint J.E., Correia M.A., Prates J.A., Firbank S.J., Lewis R.J., Grondin G.G., Ghinet M.G., Gloster T.M., Herve C., Knox J.P., Talbot B.G., Turkenburg J.P. Evidence that family 35 carbohydrate binding modules display conserved specificity but divergent function. Proc. Natl. Acad. Sci. U. S. A. 2009;106:3065–3070. doi: 10.1073/pnas.0808972106. 19218457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Correia M.A.S., Abbott D.W., Gloster T.M., Fernandes V.O., Prates J.A.M., Montanier C., Dumon C., Williamson M.P., Tunnicliffe R.B., Liu Z., Flint J.E., Davies G.J., Henrissat B., Coutinho P.M., Fontes C.M.G.A. Signature active site architectures illuminate the molecular basis for ligand specificity in family 35 carbohydrate binding module. Biochemistry. 2010;49:6193–6205. doi: 10.1021/bi1006139. 20496884. [DOI] [PubMed] [Google Scholar]
- 18.Couturier M., Roussel A., Rosengren A., Leone P., Stålbrand H., Berrin J.G. Structural and biochemical analyses of glycoside hydrolase families 5 and 26 β-(1,4)-mannanases from Podospora anserina reveal differences upon manno-oligosaccharide catalysis. J. Biol. Chem. 2013;288:14624–14635. doi: 10.1074/jbc.M113.459438. 23558681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okazawa Y., Miyazaki T., Yokoi G., Ishizaki Y., Nishikawa A., Tonozuka T. Crystal structure and mutational analysis of isomalto-dextranase, a member of glycoside hydrolase family 27. J. Biol. Chem. 2015;290:26339–26349. doi: 10.1074/jbc.M115.680942. 26330557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujimoto Z., Kishine N., Suzuki N., Suzuki R., Mizushima D., Momma M., Kimura K., Funane K. Isomaltooligosaccharide-binding structure of Paenibacillus sp. 598K cycloisomaltooligosaccharide glucanotransferase. Biosci. Rep. 2017;37 doi: 10.1042/bsr20170253. 28385816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki N., Fujimoto Z., Kim Y.M., Momma M., Kishine N., Suzuki R., Suzuki S., Kitamura S., Kobayashi M., Kimura A., Funane K. Structural elucidation of the cyclization mechanism of α-1,6-glucan by Bacillus circulans T-3040 cycloisomaltooligosaccharide glucanotransferase. J. Biol. Chem. 2014;289:12040–12051. doi: 10.1074/jbc.M114.547992. 24616103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Light S.H., Cahoon L.A., Halavaty A.S., Freitag N.E., Anderson W.F. Structure to function of an α-glucan metabolic pathway that promotes Listeria monocytogenes pathogenesis. Nat. Microbiol. 2016;2 doi: 10.1038/nmicrobiol.2016.202. 27819654. [DOI] [PubMed] [Google Scholar]
- 23.Fujimoto Z., Suzuki N., Kishine N., Ichinose H., Momma M., Kimura A., Funane K. Carbohydrate-binding architecture of the multi-modular α-1,6-glucosyltransferase from Paenibacillus sp. 598K, which produces α-1,6-glucosyl-α-glucosaccharides from starch. Biochem. J. 2017;474:2763–2778. doi: 10.1042/BCJ20170152. 28698247. [DOI] [PubMed] [Google Scholar]
- 24.Ji S., Dix S.R., Aziz A.A., Sedelnikova S.E., Baker P.J., Rafferty J.B., Bullough P.A., Tzokov S.B., Agirre J., Li F.L., Rice D.W. The molecular basis of endolytic activity of a multidomain alginate lyase from Defluviitalea phaphyphila, a representative of a new lyase family, PL39. J. Biol. Chem. 2019;294:18077–18091. doi: 10.1074/jbc.RA119.010716. 31624143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mandelli F., De Morais M.A.B., De Lima E.A., Oliveira L., Persinoti G.F., Murakami M.T. Spatially remote motifs cooperatively affect substrate preference of a ruminal GH26-type endo-β-1,4-mannanase. J. Biol. Chem. 2020;295:5012–5021. doi: 10.1074/jbc.RA120.012583. 32139511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guillén D., Sánchez S., Rodríguez-Sanoja R. Carbohydrate-binding domains: multiplicity of biological roles. Appl. Microbiol. Biotechnol. 2010;85:1241–1249. doi: 10.1007/s00253-009-2331-y. 19908036. [DOI] [PubMed] [Google Scholar]
- 27.Burnley B.T., Afonine P.V., Adams P.D., Gros P. Modelling dynamics in protein crystal structures by ensemble refinement. Elife. 2012;2012 doi: 10.7554/elife.00311. 23251785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ichinose H., Kuno A., Kotake T., Yoshida M., Sakka K., Hirabayashi J., Tsumuraya Y., Kaneko S. Characterization of an exo-β-1,3-galactanase from Clostridium thermocellum. Appl. Environ. Microbiol. 2006;72:3515–3523. doi: 10.1128/AEM.72.5.3515-3523.2006. 16672498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bianchetti C.M., Takasuka T.E., Deutsch S., Udell H.S., Yik E.J., Bergeman L.F., Fox B.G. Active site and laminarin binding in glycoside hydrolase family 55. J. Biol. Chem. 2015;290:11819–11832. doi: 10.1074/jbc.M114.623579. 25752603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishida T., Fushinobu S., Kawai R., Kitaoka M., Igarashi K., Samejima M. Crystal structure of glycoside hydrolase family 55 β-1,3-glucanase from the basidiomycete. J. Biol. Chem. 2009;284:10100–10109. doi: 10.1074/jbc.M808122200. 19193645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karkehabadi S., Helmich K.E., Kaper T., Hansson H., Mikkelsen N.E., Gudmundsson M., Piens K., Fujdala M., Banerjee G., Scott-Craig J.S., Walton J.D., Phillips G.N., Sandgren M. Biochemical characterization and crystal structures of a fungal family 3 β-glucosidase, Cel3A from Hypocrea jecorina. J. Biol. Chem. 2014;289:31624–31637. doi: 10.1074/jbc.M114.587766. 25164811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chandrasekaran R., Janaswamy S. Morphology of Western larch arabinogalactan. Carbohydr. Res. 2002;337:2211–2222. doi: 10.1016/S0008-6215(02)00223-9. 12433485. [DOI] [PubMed] [Google Scholar]
- 33.Kitazawa K., Tryfona T., Yoshimi Y., Hayashi Y., Kawauchi S., Antonov L., Tanaka H., Takahashi T., Kaneko S., Dupree P., Tsumuraya Y., Kotake T. β-Galactosyl Yariv reagent binds to the β-1,3-galactan of arabinogalactan proteins. Plant Physiol. 2013;161:1117–1126. doi: 10.1104/pp.112.211722. 23296690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otwinowski Z., Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/s0076-6879(97)76066-x. 27799103. [DOI] [PubMed] [Google Scholar]
- 35.Terwilliger T.C., Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999;55:849–861. doi: 10.1107/s0907444999000839. 10089316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perrakis A., Morris R., Lamzin V.S. Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 1999;6:458–463. doi: 10.1038/8263. 10331874. [DOI] [PubMed] [Google Scholar]
- 37.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., McNicholas S.J., Murshudov G.N., Pannu N.S., Potterton E.A., Powell H.R. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. 21460441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. 20383002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. 15299926. [DOI] [PubMed] [Google Scholar]
- 40.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix. refine research papers. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. 22505256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burnley B.T., Gros P. phenix.ensemble_refinement: a test study of apo and holo BACE1. Comput. Crystallogr. Newsl. 2013;4:51–58. [Google Scholar]
- 42.Forneris F., Burnley B.T., Gros P. Ensemble refinement shows conformational flexibility in crystal structures of human complement factor D. Acta Crystallogr. D Biol. Crystallogr. 2014;70:733–743. doi: 10.1107/S1399004713032549. 24598742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebschner D., Afonine P.V., Baker M.L., Bunkóczi G., Chen V.B., Croll T.I., Hintze B., Hung L.W., Jain S., McCoy A.J., Moriarty N.W., Oeffner R.D., Poon B.K., Prisant M.G., Read R.J. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 2019;75:861–877. doi: 10.1107/S2059798319011471. 31588918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vagin A., Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010;66:22–25. doi: 10.1107/S0907444909042589. 20057045. [DOI] [PubMed] [Google Scholar]
- 45.Wallace A.C., Laskowski R.A., Thornton J.M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. 7630882. [DOI] [PubMed] [Google Scholar]
- 46.Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. 21919503. [DOI] [PubMed] [Google Scholar]
- 47.Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38:529–533. doi: 10.1093/nar/gkq399. 20478830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landau M., Mayrose I., Rosenberg Y., Glaser F., Martz E., Pupko T., Ben-Tal N. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. 15980475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Celniker G., Nimrod G., Ashkenazy H., Glaser F., Martz E., Mayrose I., Pupko T., Ben-Tal N. ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013;53:199–206. doi: 10.1002/ijch.201200096. [DOI] [Google Scholar]
- 50.Ashkenazy H., Abadi S., Martz E., Chay O., Mayrose I., Pupko T., Ben-Tal N. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–W350. doi: 10.1093/nar/gkw408. 27166375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glaser F., Pupko T., Paz I., Bell R.E., Bechor-Shental D., Martz E., Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. 12499312. [DOI] [PubMed] [Google Scholar]
- 52.Korotkova O.G., Semenova M.V., Morozova V.V., Zorov I.N., Sokolova L.M., Bubnova T.M., Okunev O.N., Sinitsyn A.P. Isolation and properties of fungal β-glucosidases. Biochemistry. 2009;74:569–577. doi: 10.1134/s0006297909050137. 19538132. [DOI] [PubMed] [Google Scholar]
- 53.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. E. 2018;35:1547–1549. doi: 10.1093/molbev/msy096. 29722887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stecher G., Tamura K., Kumar S. Molecular evolutionary genetics analysis (MEGA) for macOS. Mol. Biol. Evol. 2020;37:1237–1239. doi: 10.1093/molbev/msz312. 31904846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. 24753421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Uncited reference
- 56.Bohne A., Lang E., von der Lieth C.W. W3-SWEET: carbohydrate modeling by internet. J. Mol. Model. 1998;4:33–43. doi: 10.1007/s008940050068. [DOI] [Google Scholar]
- 57.Bohne A., Lang E., von der Lieth C.W. SWEET—WWW-based rapid 3D construction of oligo- and polysaccharides. Bioinformatics. 1999;15:767–768. doi: 10.1093/bioinformatics/15.9.767. 10498779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structures presented in this paper have all been deposited in the Protein Data Bank (PDB) with the following codes: 7BYS, 7BYT, 7BYV, and 7BYX. All remaining data are contained within the article.