

Abstract
Embryonic stem cells can propagate indefinitely in a pluripotent state, able to differentiate into all types of specialized cells when restored to the embryo. What sustains their pluripotency during propagation remain unclear. Here, we show that core pluripotency factors Oct4 and Sox2 suppress chaperone-mediated autophagy (CMA), a selective form of autophagy, until the initiation of differentiation. Low CMA activity promotes embryonic stem cell self-renewal, while its upregulation enhances differentiation. CMA degrades isocitrate dehydrogenases IDH1 and IDH2 and reduces levels of intracellular α-ketoglutarate, an obligatory co-factor for various histone and DNA demethylases involved in pluripotency. These findings suggest that CMA mediates the effect of core pluripotency factors on metabolism, thereby shaping the epigenetic landscape of stem cells and governing the balance between self-renewal and differentiation.
One Sentence Summary:
CMA mediates the effect of core pluripotency factors on metabolism and epigenome and influences stem cell fate decisions
Embryonic stem (ES) cells can relentlessly self-renew while retaining the ability to differentiate into any cell type of the developing embryo (1, 2). This property is governed not only by a small set of core transcription factors (1, 2), but also by metabolism (3–5). However, it remains unclear how transcriptional circuitry is linked with metabolism to regulate self-renewal and differentiation of ES cells. Mammalian cells depend on chaperone-mediated autophagy (CMA) (6). Unlike macroautophagy, which delivers proteins and organelles in bulk to the lysosome for degradation (7), CMA targets a subset of cytoplasmic proteins individually. CMA substrates contain a KFERQ-like pentapeptide motif, which is recognized by the cytosolic chaperone Hsc70 (8, 9). The substrate-chaperone complex is recruited to the external surface of the lysosome through an interaction with lysosome-associated membrane protein type 2A (LAMP2A), which mediates subsequent translocation of the substrate into the lumen for degradation (10). This selectivity permits CMA to regulate intracellular processes. Nevertheless, both the regulation and physiological functions of CMA remain unclear. While macroautophagy is involved in the maintenance of the pluripotency of stem cells (11, 12), the role of CMA in stem cells is undefined.
CMA is upregulated during ES cell differentiation
To examine CMA activity in mouse ES cells and their differentiating derivatives, we cultured cells from the ES D3 cell line in medium deprived of leukemia inhibitory factor (LIF). As expected, D3 cells progressively downregulated expression of the pluripotency transcription factors Oct4, Sox2, and Nanog and reactivity of alkaline phosphatase (AP) typical of undifferentiated ES cells (Fig. 1, A to C, and fig. S1, A to C), and lost the compact stem cell colony morphology (Fig. 1B). LAMP2A is the rate-limiting component of CMA (10). Levels of LAMP2A protein, which were low in undifferentiated D3 cells, increased over time and eventually became very high (Fig. 1, A and B, and fig. S1, A and C). Concurrently, LAMP2A mRNA levels rose significantly (Fig. 1C). We also cultured D3 cells in medium supplemented with retinoic acid (RA) at a low concentration that did not inhibit CMA but sufficed to effectuate differentiation (13) (fig. S1, D and E). Again, D3 cells upregulated LAMP2A protein and mRNA levels while reducing the expression of pluripotency markers (Fig. 1, A to C, and fig. S1, A to C). To corroborate these results, we tested cells from ES E14TG2a (E14) line. As E14 cells gradually lost their ES cell identity in LIF-deprived or RA-supplemented medium, they concurrently increased expression of LAMP2A protein and mRNA (fig. S1, F to K). We analyzed gene expression profiles of three other genetically distinct mouse ES cell lines (R1, J1, and V6.5) (14), and found that all of them contained LAMP2A at a higher abundance upon differentiation (fig. S1, L to N).
Fig. 1. Activation of CMA during ES cell differentiation.
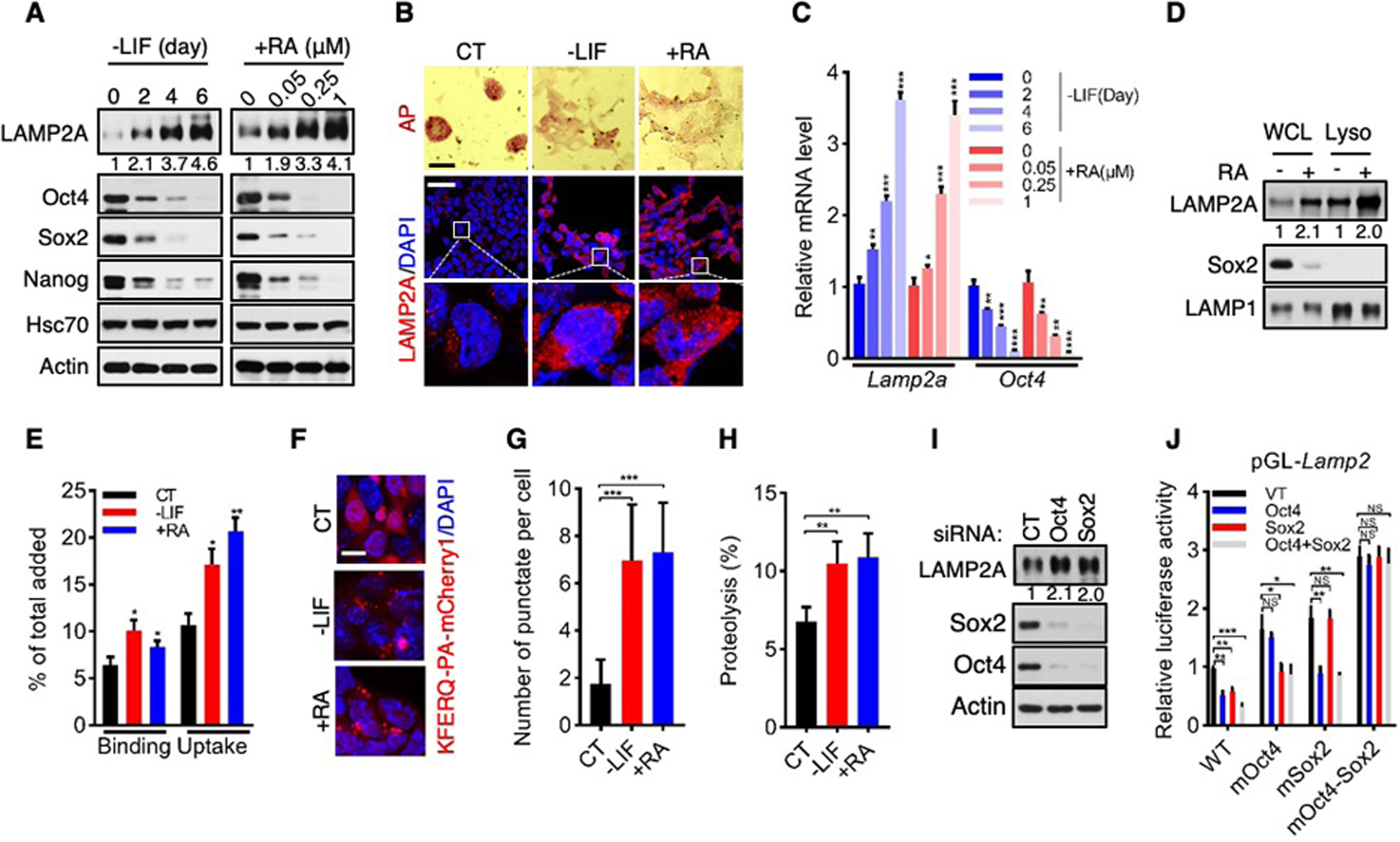
(A and C) Levels of proteins (A, with relative LAMP2A/actin ratios indicated) and mRNAs (C) in D3 cells cultured in medium deprived of LIF (-LIF) for the indicated time, or supplemented with RA (+RA) at the indicated concentrations for 2 days.
(B) Representative images of AP staining (top; scale bar, 100 μm) and LAMP2A immunofluorescence (middle and bottom; scale bar, 25 μm) of D3 cells cultured in -LIF or +RA medium. CT, undifferentiated control ES cells.
(D) LAMP2A levels in whole cell lysates (WCL) and lysosomes (Lyso) of D3 cells treated with or without RA. Relative LAMP2A/LAMP1 ratios are indicated
(E) Relative binding and uptake of GAPDH by lysosomes isolated from control (CT) and differentiating D3 cells.
(F and G) Representative cell images (F; scale bar, 10 μm) and quantification of CMA flux (G; n = 20 randomly chosen cells) of D3 cells expressing KFERQ-PA-mCherry1.
(H) CMA-mediated proteolysis in control and differentiating D3 cells.
(I) Suppression of LAMP2A protein level by Sox2 and Oct4 in D3 cells.
(J) Oct4/Sox2-mediated suppression of luciferase reporter gene driven by wild-type, but not mutant, Oct4/Sox2-binding motif.
Data are mean ± SD (n = 3 biological replicates unless otherwise indicated). *P < 0.05, **P < 0.01, ***P < 0.001, NS, no significant, unpaired Student’s t-test.
The amount of LAMP2A on lysosomes determines CMA activity (15). In differentiating D3 and E14 cells, LAMP2A accumulated on lysosomes despite a steady lysosomal content (Fig. 1D and fig. S2). Lysosomes isolated from these cells displayed ~50–100% stronger binding with, and uptake of, the model CMA substrate glyceraldehyde-3-phosphate (GAPDH) relative to lysosomes from undifferentiated ES cells (Fig. 1E and fig. S3, A to D). To assess CMA flux in live cells, we used a photo-activatable (PA) reporter, KFERQ-PA-mCherry1 (16). Differentiating D3 and E14 cells displayed more than 2-fold increase in CMA flux compared to their undifferentiated counterparts (Fig. 1, F and G, and fig. S3, E and F). We also evaluated CMA-dependent global protein degradation (17), and observed that differentiating D3 and E14 cells exhibited ~70–100% higher rate of CMA-dependent proteolysis (Fig. 1H and fig. S3G). These results demonstrate that mouse ES cells normally maintain CMA at low levels, but up-regulate CMA activity during differentiation by increasing LAMP2A levels.
Oct4, Sox2, and Nanog govern the pluripotent state of ES cells in part by restricting the expression of genes involved in differentiation (1, 2). The inverse correlation between these core pluripotency factors and LAMP2A prompted us to test whether they suppress expression of the Lamp2a gene. We examined the Lamp2 gene sequence and identified one putative binding motif for the Oct4-Sox2 heterodimer in the distal promoter region (fig. S4A), but none for Nanog. An analysis of public chromatin immunoprecipitation-sequencing (ChIP-Seq) datasets (18, 19) revealed that Oct4 and Sox2 co-occupy the Lamp2 genomic region encompassing this motif in mouse ES cells (fig. S4B). Upon differentiation, association of Oct4 and Sox2 with Lamp2 declined (fig. S4, C and D). When Oct4 or Sox2 was knocked down via small interfering (si) RNA, levels of LAMP2A protein and mRNA increased (Fig. 1I, and fig. S4, E to I). In a reporter gene assay, Oct4 and Sox2 inhibited expression of a luciferase gene driven by the wild-type Oct4-Sox2-binding motif, but not mutant motifs in which their corresponding binding sites were altered (Fig. 1J, and fig. S4, J and K). These results indicate that the expression of Lamp2a is suppressed in ES cells by Oct4 and Sox2 until differentiation is initiated.
CMA suppresses the pluripotency of ES Cells
To evaluate the functional role of CMA in pluripotency, we overexpressed LAMP2A in D3 cells to accentuate CMA flux (Fig. 2A and fig. S5, A to F). This led to a reduction in Oct4, Sox2, and Nanog levels and AP reactivity (~50%; Fig. 2, A to C, and fig. S5A), and to the loss of ES cell morphology (Fig. 2D and fig. S5G). Concurrently, expression of genes associated with differentiation was enhanced, including the endodermal markers Gata4, the mesodermal marker Brachyury, and the ectodermal marker Fgf5 (Fig. 2B). Likewise, forced LAMP2A expression increased CMA flux and inhibited pluripotency of E14 cells (fig. S5, H to R). D3 and E14 ES cells treated with the small molecule Qx77 to activate CMA (20) down-regulated pluripotency factors and AP reactivity and lost the characteristic ES cell morphology (Fig. 2, E and F, and fig. S6). Therefore, a higher CMA activity impedes self-renewal and promotes differentiation of ES cells.
Fig. 2. CMA suppresses the pluripotency of ES Cells.
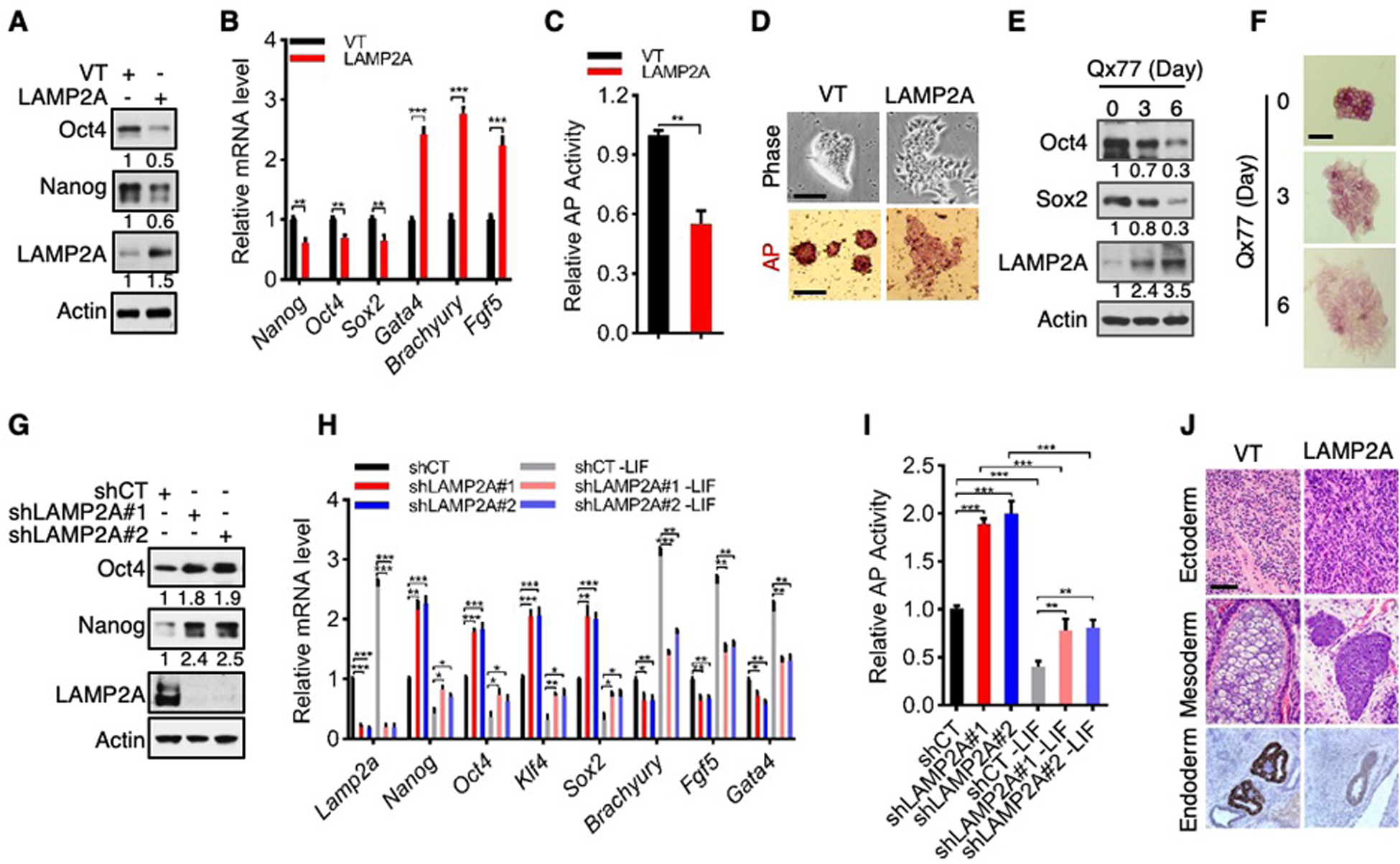
(A to D) Protein (A) and mRNA (B) levels, AP activity (C), and representative phase-contrast (D, top) and AP staining (D, bottom) images of D3 cells stably expressing vector control (VT) or LAMP2A. Scale bars: 100 μm.
(E and F) Protein levels (E) and representative images of AP staining (F) of D3 cells treated with 10 μM Qx77. Scale bars: 100 μm.
(G to I) Protein (G) and mRNA (H) expression, and AP activity (I), in control and LAMP2A-knockown D3 cells cultured in normal (G to I) or LIF-deprived medium.
(J) H&E (top and middle) and Gata4 IHC (bottom) staining of teratomas formed by control and LAMP2A-overexpressing D3 cells. Scale bar: 200 μm.
Data are mean ± SD (n = 3 biological replicates). *P < 0.05, **P < 0.01, ***P < 0.001, unpaired Student’s t-test.
Conversely, we stably knocked down LAMP2A using independent short hairpin (sh) RNAs to abate CMA activity (Fig. 2G and fig. S7, A to E). This augmented the expression of pluripotency factors (~80–140%) (Fig. 2G and fig. S7, A to C) and AP reactivity (~60–80%) (fig. S7, F and G). It also delayed differentiation induced by LIF withdrawal, as shown by the retention of the characteristic stem cell morphology (fig. S7, H to K), slower downregulation in pluripotency factors and AP reactivity (Fig. 2, H and I, and fig. S7, L and M), and subdued upregulation of differentiation markers (Fig. 2H and fig. S7L). Additionally, we knocked out Lamp2a in D3 and E14 cells using CRISPR-mediated gene editing. Compared to wild-type (Lamp2a+/+) cells, Lamp2a-knockout (Lamp2a−/−) cells displayed higher levels of stem cell markers and AP reactivity and lower expression of differentiation markers (fig. S8), and a delay in differentiation (fig. S9). Therefore, suppression of CMA reinforces the self-renewal of ES cells.
To extend these observations, we used an ES cell line expressing enhanced green fluorescent protein (GFP) under the control of the endogenous Oct4 promoter (Oct4-GFP) (21). Forced expression of LAMP2A reduced, while knockdown of LAMP2A increased, GFP fluorescence intensity and protein levels in Oct4-GFP cells (fig. S10, A to F), again indicating an inhibitory effect of CMA on the pluripotent state. To evaluate the effect of LAMP2A on pluripotency in vivo, we performed a teratoma formation assay. Control D3 cells formed teratomas that contained tissues derived from all three embryonic germ layers (endoderm, mesoderm, and ectoderm) (Fig. 2J). LAMP2A-knockdown ES cells also produced tissues derived from these germ layers albeit with reduced expression of differentiation markers (fig. S10, G to I), suggesting that their differentiation potential is impaired. In contrast, LAMP2A-overexpressing D3 cells formed teratomas of poorly differentiated tissues, containing mainly primitive cells in immature mesenchyme and lacking structures of an endodermal origin (Fig. 2J).
CMA regulates intracellular αKG levels
Next, we investigated the mechanism by which CMA regulates pluripotency. CMA-deficient cells can upregulate other forms of autophagy (22). However, in cells devoid of a key macroautophagy regulator (Atg7), or an ATPase important for microautophagy (Vps4), LAMP2A knockdown was still able to elevate stemness markers (fig. S11, A and B). We also considered the possibility that CMA might target the core pluripotency factors for lysosomal degradation. However, Oct4, Sox2, and Nanog were not found on lysosomes in ES cells or their differentiating derivatives (fig. S11, C and D), and their levels were increased by the proteasome inhibitor MG132, but not lysosome inhibitors LN (fig. S11, E and F).
Although potential substrates for CMA are numerous (6), metabolic enzymes constitute a substantial fraction of known CMA substrates (23). Specific metabolic states are important for the pluripotency of mouse ES cells (24–27). We therefore performed liquid chromatography-mass spectrometry (LC-MS)-based metabolomics on control and LAMP2A-knockdown D3 cells. Many metabolites were present at higher abundance in LAMP2A-knockdown cells compared to control cells (Fig. 3A and fig. S12A). Among them were most amino acids and nucleotides—suggesting that a reduction in CMA flux augments the supply of anabolic precursors for stem cell proliferation, and glycolytic metabolites—consistently with the observation that ES cells prefer a high rate of glycolysis (28, 29). Several glycolytic enzymes are CMA substrates in non-stem cells (23); however, expression of these enzymes, as well as other glycolytic enzymes, remained unchanged in D3 cells upon LAMP2A knockout (fig. S12, B and C).
Fig. 3. CMA regulates intracellular αKG levels in ES cells.
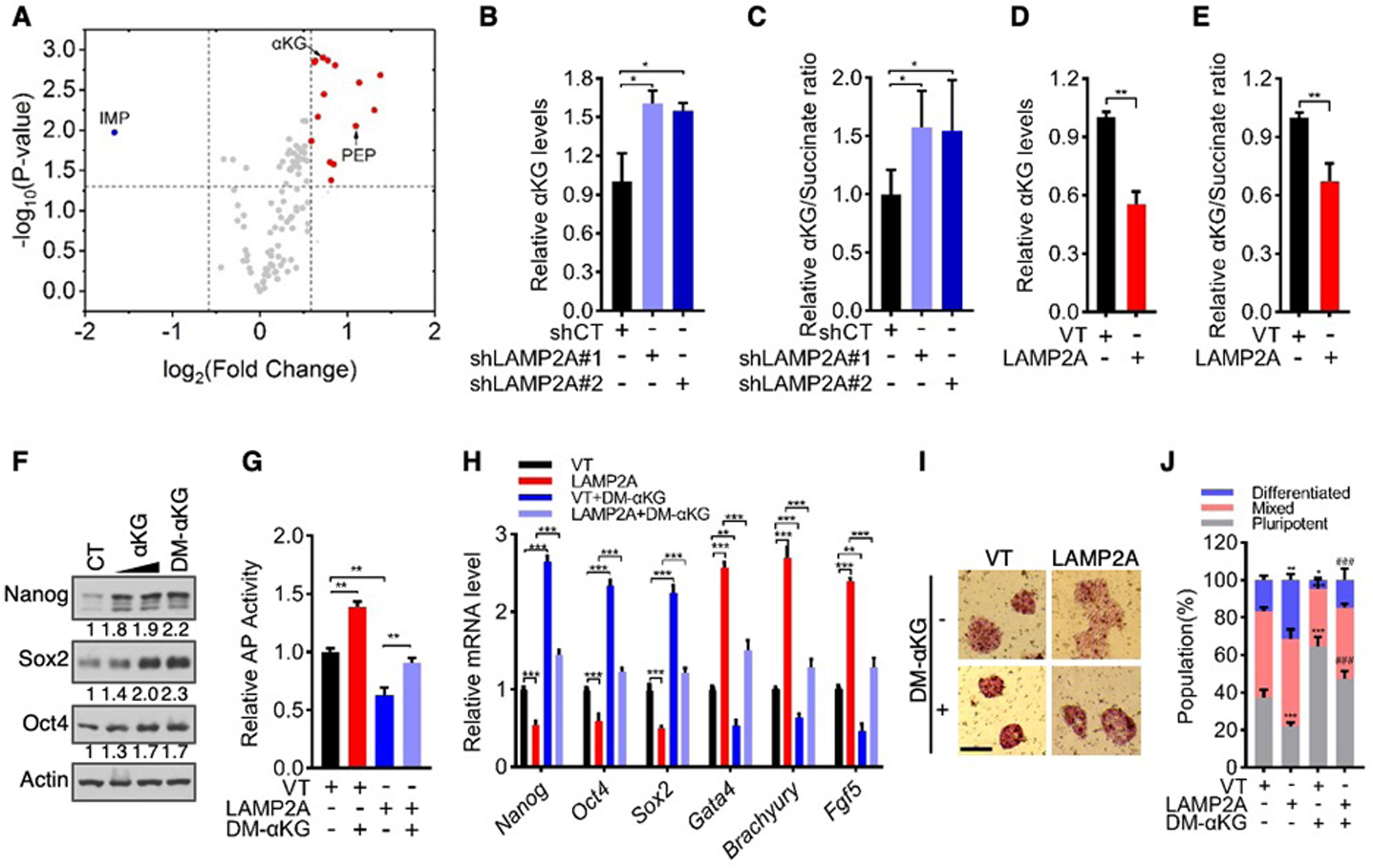
(A) Volcano plot of metabolites that were altered by LAMP2A knockdown. Blue dots represent metabolites with change < 1.5-fold and P-value < 0.05, and red dots represent metabolites with change > 1.5 fold and P-value < 0.05. IMP, inosine monophosphate. PEP, phosphoenolpyruvate.
(B to E) Relative αKG levels (B and D) and αKG/succinate ratios (C and E) in LAMP2A-knockdown (B and C), LAMP2A-overexpressing (D and E), and the corresponding control D3 cells.
(F) Western blot analysis of D3 cells treated with vehicle (CT), αKG (2 and 4 mM), or DM-αKG (4 mM) for 72 h.
(G to J) Control and LAMP2A-overexpressing D3 cells were cultured in medium with or without 4 mM DM-αKG for 3 days. Shown are relative AP activity (G), mRNA levels of pluripotency and differentiation genes (H), representative images of AP staining (I; scale bars, 100 μm), and quantification of differentiated, mixed, and pluripotent colonies (J).
Data shown are mean ± SD (n = 3 biological replicates). *P < 0.05, **P < 0.01, ***P < 0.001. ###P < 0.001 vs. LAMP2A in (J). Two-way ANOVA for (J), and unpaired Student’s t-test for the rest.
Intracellular levels of the tricarboxylic acid cycle (TCA) intermediate α-ketoglutarate (αKG) and its ratio to succinate regulate pluripotency of mouse ES cells (26, 27). αKG was among the most highly elevated metabolites in LAMP2A-depleted D3 cells (Fig. 3A). The αKG/succinate ratio also increased in these cells, albeit to a lesser extent (fig. S12, D to G). These metabolic changes were similarly observed in LAMP2A-knockdown E14 cells using an LC-MS-based assay of central carbon metabolites (fig. S12H), and further verified in both LAMP2A-knockdown and -knockout E3 and E14 cells using an enzyme-based assay (Fig. 3, B and C, and fig. S12, I to L). On the contrary, αKG levels and αKG/succinate ratio were reduced in LAMP2A-overexpressing D3 and E14 cells (Fig. 3, D and E, and fig. S12, M and N). These results show that CMA reduces intracellular αKG levels.
Consistent with the previous findings (26, 27), treatment with the cell membrane-permeable dimethyl-αKG (DM-αKG) enhanced the pluripotency of stem cells (Fig. 3, F to J, and fig. S13, A to G). Knocking down αKG dehydrogenase (also known as 2-oxoglutarate dehydrogenase or OGDH), a key component of the oxoglutarate dehydrogenase complex that consumes αKG, phenocopied the effect of DM-αKG and bolstered stemness (fig. S13, H to M). The positive impact of DM-αKG on stemness was neutralized by the inhibition of succinate dehydrogenase (SDH) through a competitive inhibitor dimethyl-malonate (DMM) (fig. S14) or siRNA-mediated knockdown (fig. S15), both of which elevated intracellular succinate levels and reduce the αKG/succinate ratio.
If CMA inhibits pluripotency of ES cells by lowering intracellular αKG levels, forced increase in αKG is expected to counteract the effect of CMA. Indeed, supplementation of DM-αKG restored stemness markers and ES cell morphology and decreased differentiation genes in LAMP2A-overexpressing cells, all to levels seen in control ES cells (Fig. 3, G to J, and fig. S13, A to G). DM-αKG also returned GFP expression in LAMP2A-overexpressing Oct4-GFP cells to levels seen in control Oct4-GFP cells (fig. S16A). Similarly, addition of exogenous αKG, which can be imported into cells (30), neutralized the effect of LAMP2A overexpression and enforced pluripotency (Fig. 3F, Fig. 3,G to J, and fig. S13, A to G, and fig. S16, B to I). These results suggest that CMA inhibits the pluripotency of ES cells by reducing intracellular αKG levels and αKG/succinate ratio.
CMA targets IDH1/2 for lysosomal degradation
αKG can be produced through various enzymes in the TCA cycle, the serine biosynthesis pathway, and amino acid metabolism (fig. S17A). To identify which enzyme(s) is subject to CMA-mediated proteolysis, we analyzed the sequences of αKG-generating enzymes for a putative KFERQ-like motif(s) and, for those with such a motif(s) (Table S1), we compared their levels in control and LAMP2A-knockout cells. Levels of IDH1 and IDH2, but not any other enzymes, were augmented in LAMP2A-knockout D3 cells (Fig. 4A and fig. S17, B to D). Similarly, IDH1/2 protein levels were increased in LAMP2A-knockout E14 cells (fig. S17, E and F), as well as in LAMP2A-knockdown D3 and E14 cells (fig. S17, G and H). Concurrently, total IDH1 and IDH2 activity was elevated (fig. S17, I to L). On the contrary, IDH1/2 protein levels and total activity declined in LAMP2A-overexpression D3 and E14 cells (Fig. 4B and fig. S18, A to E). An isotope tracing experiment with [1, 2-13C]glucose showed that LAMP2A knockout increased, while LAMP2A overexpression decreased, the enrichment of 13C in αKG, but not (iso)citrate (Fig. 4, C and D, and fig. S18, F and G), suggesting that CMA attenuates the metabolic flux through IDHs. Therefore, CMA diminishes cellular levels of IDH1 and IDH2 and their enzymatic function.
Fig. 4. CMA targets IDH1/2 for lysosomal degradation.
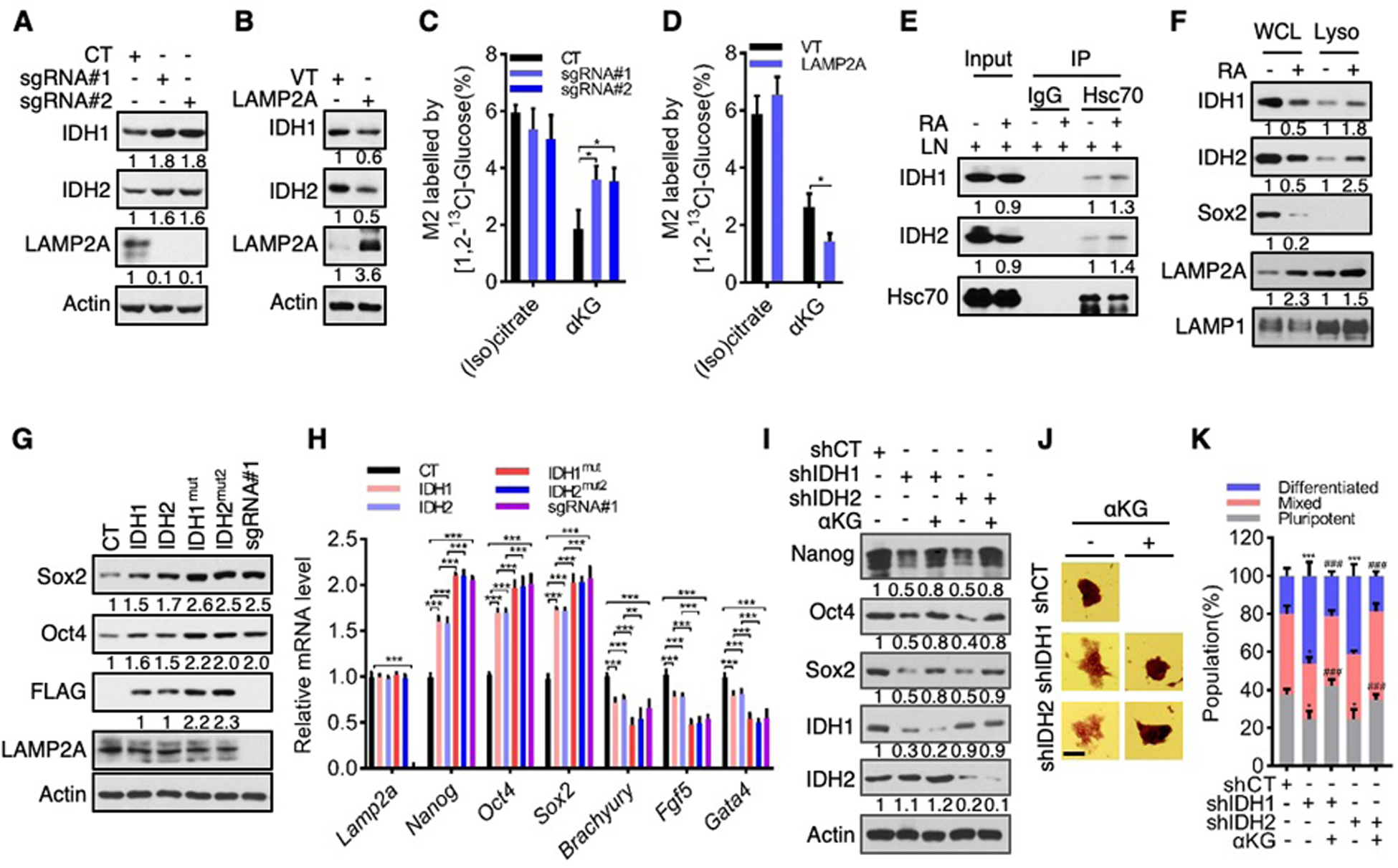
(A to D) Effect of LAMP2A knockout (A and C) and overexpression (B and D) on IDH1/2 protein levels (A and B), or M2 enrichment of (iso)citrate and αKG upon culturing in [1, 2-13C]glucose-containing medium (C and D).
(E) Interaction between endogenous IDH1/2 and Hsc70 in D3 cells treated with or without RA.
(F) IDH1/2 protein levels in whole cell lysate (WCL) and the lysosomal fraction (Lyso) of D3 cells treated with or without RA.
(G and H) Protein (G) and mRNA (H) levels in D3 cells overexpressing wild-type or mutant IDH1 or IDH2, or depleted of LAMP2A (sgRNA#1).
(I to K) Protein expression (I), representative images of AP staining (J; scale bars: 100 μm), and quantification of pluripotent, differentiated, and mixed colonies (K) of control or IDH1/2-knockdown D3 cells treated with or without αKG.
Data shown are mean ± SD (n = 3 biological replicates). *P < 0.05, **P < 0.01, ***P < 0.001. ###P < 0.001 vs. shIDH1/2. Two-way ANOVA for (K), unpaired Student’s t-test for the other panels.
Consistent with CMA-mediated degradation of IDH1/2, treatment with LN, but not MG132, enhanced levels of IDH1 and IDH2 (fig. S19, A to D). LN also increased the association of IDH1/2 with lysosomes in a LAMP2A-dependent manner (fig. S19, E to J), presumably due to stalled degradation of IDH1/2. As CMA substrates are recruited to the lysosome by Hsc70 (8), we tested a potential Hsc70-IDH1/2 interaction. Endogenous Hsc70 interacted with Flag-tagged IDH1/2 in HEK293T cells (fig. S20, A and B) and with endogenous IDH1/2 in D3 and E14 cells (Fig. 4E and fig. S20, B to D). IDH1 contains one KFERQ-like motif, while IDH2 contains three such motifs (Table S1). Mutations in the only motif in IDH1 (IDH1mut) and in the second motif in IDH2 (IDH2mut2) abolished the interaction of these enzymes with Hsc70 (fig. S20, E to H). When D3 and E14 cells underwent differentiation in the presence of RA or Qx77, IDH1/2 expression declined, along with αKG levels (~50%; Fig. 4F, and fig. S21). This was accompanied by increased association of IDH1/2 with Hsc70 (Fig. 4E and fig. S20, B to D) and lysosomes (Fig. 4F and fig. S21A). CMA-mediated regulation of IDH2 is consistent with previous findings that nuclear-encoded mitochondrial proteins can be degraded by CMA, presumably prior to their translocation into mitochondria (23). These results indicate that IDH1 and IDH2 are selected by Hsc70 for lysosomal degradation during ES cell differentiation.
To evaluate the functional role of IDH1/2, we ectopically expressed them and CMA-resistant mutants individually in D3 and E14 cells (Fig. 4G and fig. S22, A to C). IDH1/2 augmented the expression of pluripotency genes and suppressed the expression of differentiation genes (Fig. 4G and fig. S22, D and E). IDH1mut and IDH2mut2 were expressed at higher levels than their wild-type counterparts (Fig. 4G and fig. S22, A to C). These mutants also displayed a more robust impact on stemness, which was comparable to that of LAMP2A knockout (Fig. 4H and fig. S22, A to C, and F to J). Moreover, treatment of LN elevated levels of IDH1 and IDH2, but not IDH1mut and IDH2mut2, thereby equalizing them (fig. S23, A to D). Under this condition, ES cells expressing IDH1 and IDH1mut or IDH2 and IDH2mut2 contained comparable levels of αKG and stemness/differentiation markers (fig. S23). Conversely, we knocked down IDH1 and IDH2 in D3 and E14 cells. This led to a decline in pluripotency factors, loss of ES cell morphology, and an increase in differentiation markers (Fig. 4, I to K, and fig. S24). Therefore, IDH1/2 enhances the pluripotent state of ES cells.
In the ES cells devoid of IDH1 or IDH2, silencing LAMP2A did not affect IDH1/2 activity and αKG levels, or promote stemness (Fig. 5, A and B, and fig. S25, A to L). Likewise, in Oct4-GFP ES cells devoid of IDH1 or IDH2, silencing LAMP2A was unable to increase Oct4 promoter activity (fig. S25M). In contrast, downregulation of LAMP2A in cells where two other αKG-generating enzymes, PAST1 or GLUD1, were depleted augmented intracellular αKG levels and bolstered stemness (fig. S26). Therefore, CMA and IDH1/2 likely act in the same pathway to regulate the pluripotent state of ES cells, with IDH1/2 being the downstream effectors.
Fig. 5. CMA regulates pluripotency and epigenetic and transcriptional states of ES cells by reducing IDH1/2 and αKG levels.
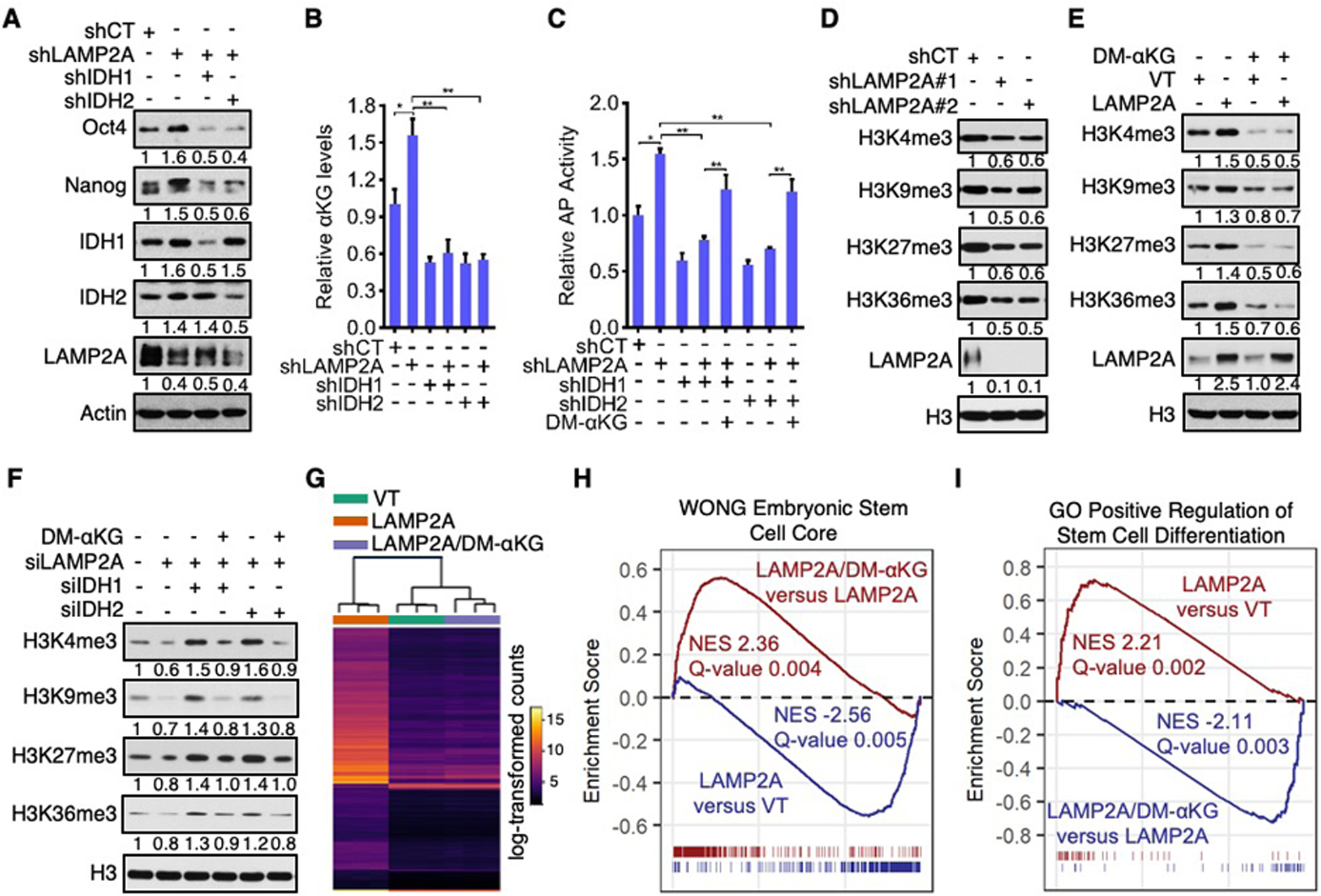
(A to C) Protein expression (A), αKG levels (B), and AP activity (C) of D3 cells depleted of LAMP2A and/or IDH1/2 and cultured in the absence (A to C) or presence (C) of 4 mM DM-αKG.
(D and E) Histone methylation in control, LAMP2A-knockdown (D), and LAMP2A-overexpressing (E) D3 cells treated with or without 4 mM DM-αKG.
(F) Histone trimethylation in D3 cells transfected with the indicated siRNAs and treated with or without 4 mM DM-αKG for 72 h.
(G) The hierarchical clustering of gene expression in control (VT), LAMP2A, and LAMP2A/DM-αKG D3 cells.
(H and I) Leading edge plots for gene sets of Wong embryonic stem cell core (H) and GO positive regulation of stem cell differentiation (I). Gene sets enriched by up-regulated genes are shown in dark red and by down-regulated genes are shown in blue. Genes that exist in the corresponding pathways are shown by ticks. NES, normalized enrichment score.
Data are mean ± SD (n = 3 biological replicates). *P < 0.05, **P < 0.01, unpaired Student’s t-test.
Supplementation with αKG was able to restore pluripotency of IDH1/2-knockdown cells, increasing levels of stem cell markers and pluripotent cell populations and reducing the expression of differentiation markers (Fig. 4, I to K, and fig. S24, E to M). Addition of DM-αKG to D3 and E14 cells with LAMP2A/IDH1 or LAMP2A/IDH2 double knockdown also effectively restored the expression of pluripotency factors and AP reactivity (Fig. 5C, and fig. S25, G to L). Moreover, DM-αKG rescued Oct4 promoter activity in LAMP2A/IDH1 or LAMP2A/IDH2 double knockdown Oct4-GFP cells (fig. S25M). Collectively, these data show that CMA suppresses pluripotency by reducing IDH1/2-mediated production of αKG.
CMA regulates epigenetic and transcriptional states of ES cells
αKG is an essential cofactor and succinate is a competitive inhibitor for a large family of dioxygenases including Jumonji C (JmjC)-domain-containing histone demethylases and the ten-eleven translocation (TET) family of DNA demethylases (31). Congruent with their effects on αKG, LAMP2A knockdown decreased, while LAMP2A overexpression increased, global histones trimethylation including histone H3 lysine 4 trimethylation (H3K4me3), H3K9me3, H3K27me3, and H3K36me3 (Fig. 5, D and E, and fig. S27, A to F). Supplementation with DM-αKG reversed the epigenetic changes in LAMP2A-overexpressing cells (Fig. 5E and fig. S27, D to F), and this effect of DM-αKG was in turn abrogated by DMM (fig. S27, G to J). In addition, the increase in histone trimethylation in LAMP2A/IDH1 and LAMP2A/IDH2 double knockdown ES cells was negated by DM-αKG supplementation (Fig. 5F and fig. S27K). Therefore, CMA regulates the epigenome of ES cells by reducing αKG levels.
αKG-dependent histone and DNA demethylases influence the expression of pluripotency-associated genes (32, 33). To characterize changes in global gene expression engendered by a higher CMA flux and their relevance to intracellular αKG levels, we performed an RNA-sequencing (RNA-Seq) analysis of three populations of ES cells: control D3 cells, LAMP2A-overexpressing D3 cells (LAMP2A), and LAMP2A-overexpressing D3 cells treated with DM-αKG (LAMP2A/DM-αKG). LAMP2A cells displayed a gene expression profile distinct from, while LAMP2A/DM-αKG cells display a gene expression profile similar to, that of control D3 cells (Fig. 5G). Gene set enrichment analysis (GSEA) confirmed that upon LAMP2A overexpression, genes involved in stem cell maintenance were down-regulated, while genes involved in stem cell differentiation were up-regulated. However, these changes engendered by LAMP2A were effectively revered by DM-αKG (Fig. 5, H and I, and fig. S27L). Therefore, CMA regulates epigenetic and transcriptional states of ES cells by reducing αKG levels.
Outlook
The continuous proliferation and unrestricted developmental potential of ES cells are not only maintained by a core set of transcription factors (1, 2), but also enabled by metabolism (3–5). Our results reveal a mechanism by which CMA links these processes. We find that CMA activity is kept at the minimum due to Oct4/Sox2-mediated suppression of LAMP2A. This maintains IDH1/2 levels and promotes αKG production, permitting demethylation of repressive chromatin markers and reinforcing pluripotency. With differentiation, LAMP2A is upregulated due to the decline in Oct4 and Sox2 expression. Given that differentiation is often accompanied by higher reactive oxygen species (ROS) levels (4, 34) and that CMA can be activated by oxidative stresses (6), CMA may be further stimulated by ROS in differentiating ES cells. A higher CMA activity suppresses αKG levels through the degradation of IDHs, increasing histone trimethylation and favoring differentiation. Thus, the suppression of CMA enables core pluripotency factors to coordinately regulate metabolism and epigenome, thereby impacting the self-renewal and differentiation of stem cells.
CMA and αKG in the context of cellular pluripotency represent targets for genetic or pharmacological manipulation that may lead to improved protocols for ES cell maintenance and differentiation. Distinct from the other forms of autophagy, which are conserved in eukaryotes, CMA is demonstrated only in mammals (6). CMA might have evolved later during evolution to regulate increasingly elaborate processes, including the long-term maintenance and controlled differentiation of stem cells, that characterize mammals.
Supplementary Material
Acknowledgments
We thank H. Yan for providing plasmids and R. Wang, I. Asangani, and Q. Deng for technical assistance.
Funding: Supported by NIH grants R01CA182675, R01CA184867, R01CA235760, P30ES013508, and DOD grant W81XWH-15-1-0678.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: The RNA-seq data generated from this study are available in the Gene Expression Omnibus (GEO) repository under the accession number GSE144093. All data are available in the main paper, the supplementary materials, or at GEO. Requests for materials should be addressed to X.Y.
Supplementary Materials
Materials and Methods
Figures S1-S27
Tables S1
References (35–53)
References and Notes:
- 1.Martello G, Smith A, The nature of embryonic stem cells. Annu Rev Cell Dev Biol 30, 647–675 (2014). [DOI] [PubMed] [Google Scholar]
- 2.De Los Angeles A et al. , Hallmarks of pluripotency. Nature 525, 469–478 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Shyh-Chang N, Daley GQ, Cantley LC, Stem cell metabolism in tissue development and aging. Development 140, 2535–2547 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chandel NS, Jasper H, Ho TT, Passegue E, Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol 18, 823–832 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Zhang J et al. , Metabolism in Pluripotent Stem Cells and Early Mammalian Development. Cell Metab 27, 332–338 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Kaushik S, Cuervo AM, The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19, 365–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He C, Klionsky DJ, Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43, 67–93 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang HL, Terlecky SR, Plant CP, Dice JF, A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246, 382–385 (1989). [DOI] [PubMed] [Google Scholar]
- 9.Dice JF, Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci 15, 305–309 (1990). [DOI] [PubMed] [Google Scholar]
- 10.Cuervo AM, Dice JF, A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273, 501–503 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Prat L et al. , Autophagy maintains stemness by preventing senescence. Nature 529, 37–42 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Ho TT et al. , Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anguiano J et al. , Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat Chem Biol 9, 374–382 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hailesellasse Sene K et al. , Gene function in early mouse embryonic stem cell differentiation. BMC Genomics 8, 85 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuervo AM, Dice JF, Regulation of lamp2a levels in the lysosomal membrane. Traffic 1, 570–583 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Koga H, Martinez-Vicente M, Macian F, Verkhusha VV, Cuervo AM, A photoconvertible fluorescent reporter to track chaperone-mediated autophagy. Nature communications 2, 386 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juste YR, Cuervo AM, Analysis of Chaperone-Mediated Autophagy. Methods Mol Biol 1880, 703–727 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whyte WA et al. , Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu ZY, Kraus WL, Catalytic-Independent Functions of PARP-1 Determine Sox2 Pioneer Activity at Intractable Genomic Loci. Molecular Cell 65, 589-+ (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J et al. , Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J Biol Chem, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loh YH et al. , The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38, 431–440 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Massey AC, Kaushik S, Sovak G, Kiffin R, Cuervo AM, Consequences of the selective blockage of chaperone-mediated autophagy. Proc Natl Acad Sci U S A 103, 5805–5810 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneider JL, Suh Y, Cuervo AM, Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20, 417–432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J et al. , Dependence of mouse embryonic stem cells on threonine catabolism. Science 325, 435–439 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shyh-Chang N et al. , Influence of threonine metabolism on S-adenosylmethionine and histone methylation. Science 339, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB, Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518, 413–416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang IY et al. , Psat1-Dependent Fluctuations in alpha-Ketoglutarate Affect the Timing of ESC Differentiation. Cell Metab 24, 494–501 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Varum S et al. , Energy metabolism in human pluripotent stem cells and their differentiated counterparts. PLoS One 6, e20914 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J et al. , UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J 30, 4860–4873 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.TeSlaa T et al. , alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell Metabolism 24, 485–493 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaelin WG Jr., McKnight SL, Influence of metabolism on epigenetics and disease. Cell 153, 56–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tee WW, Reinberg D, Chromatin features and the epigenetic regulation of pluripotency states in ESCs. Development 141, 2376–2390 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beerman I, Rossi DJ, Epigenetic Control of Stem Cell Potential during Homeostasis, Aging, and Disease. Cell Stem Cell 16, 613–625 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bigarella CL, Liang R, Ghaffari S, Stem cells and the impact of ROS signaling. Development 141, 4206–4218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.