

Summary
Cells have evolved an elaborate DNA repair network to ensure complete and accurate DNA replication. Defects in these repair machineries can fuel genome instability and drive carcinogenesis while creating vulnerabilities that may be exploited in therapy. Here, we use nascent chromatin capture (NCC) proteomics to characterize the repair of replication-associated DNA double-strand breaks (DSBs) triggered by topoisomerase 1 (TOP1) inhibitors. We reveal profound changes in the fork proteome, including the chromatin environment and nuclear membrane interactions, and identify three classes of repair factors according to their enrichment at broken and/or stalled forks. ATM inhibition dramatically rewired the broken fork proteome, revealing that ataxia telangiectasia mutated (ATM) signalling stimulates DNA end resection, recruits PLK1, and concomitantly suppresses the canonical DSB ubiquitination response by preventing accumulation of RNF168 and BRCA1-A. This work and collection of replication fork proteomes provide a new framework to understand how cells orchestrate homologous recombination repair of replication-associated DSBs.
Keywords: replication stress, ATM, BRCA1-A, PLK1, NDRG3, UBAP2, homologous recombination, NHEJ, Camptothecin, nascent chromatin capture
Graphical abstract
Highlights
-
•
Comprehensive proteomics of replication forks damaged by TOP1 inhibition
-
•
Broken and stalled forks show distinct repairomes and chromatin environments
-
•
Rewiring of the broken fork proteome by ATM inhibition toward DSB ubiquitination
-
•
PLK1, NDRG3, and UBAP2 are promoting repair of broken forks by HR
By systematic proteomics profiling of replication forks challenged by the topoisomerase I inhibitor camptothecin, Nakamura et al. identify dedicated repair factors for broken replication forks, characterize their chromatin environment, and reveal that ATM and PLK1 promote homologous recombination by suppressing the canonical DNA double-strand break ubiquitination response at broken forks.
Introduction
Maintenance and inheritance of genomic and epigenomic information is fundamental for genome stability and function in eukaryotes (Alabert and Groth, 2012). Replication stress threatens genome stability and drives cancer development and progression (Macheret and Halazonetis, 2015), and cells have evolved an elaborate network of DNA repair mechanisms to counter this threat. Endogenous sources of replication stress include oncogenic signaling, reactive oxygen species, replication-transcription collisions, DNA secondary structures such as G-quadruplexes, DNA and protein crosslinks, and deoxyribonucleotide triphosphate (dNTP) exhaustion (Zeman and Cimprich, 2014). Replication stress can also be induced by a wide range of exogenous genotoxic agents (Zeman and Cimprich, 2014), many of which are potent chemotherapeutic agents (Lecona and Fernández-Capetillo, 2014). Replication stress responses are therefore attractive targets for cancer therapy, with the potential to develop more effective and targeted treatment as well as overcome chemotherapy resistance.
Replication stress refers broadly to replication fork challenges, but it is becoming increasingly clear that fork repair mechanisms are highly tailored toward specific types of damage and replication intermediates (Cortez, 2019). The highly specialized fork repair mechanisms act in concert with shared global protective responses that include dormant origin firing and cell cycle checkpoints. Key replication fork-associated repair mechanisms include fork reversal and nascent strand protection, translesion synthesis, mismatch repair, lesion skipping/bypass and repriming, break-induced replication, and homologous recombination (HR) (Cortez, 2019), which have differing prevalence toward distinct fork damage. The interplay of these repair processes and how they are regulated in response to different types of replication stress remains to be fully understood. Proteomics analysis of replication forks by isolation of proteins on nascent DNA (iPOND) has uncovered a large number of repair and checkpoint signaling factors recruited in response to hydroxyurea (HU)-induced nucleotide depletion that stalls fork progression (Dungrawala et al., 2015). The repair proteome of DNA interstrand crosslink (ICL) repair has also been characterized using cell-free DNA replication in Xenopus egg extracts (Räschle et al., 2015). However, protein dynamics at replication forks challenged by topoisomerase 1 (TOP1) inhibitors has not been systematically characterized despite their widespread clinical use to treat colorectal, lung, ovarian, cervical, and pancreatic cancers (Thomas and Pommier, 2019). TOP1 relaxes positive supercoiling ahead of DNA and RNA polymerases and enables DNA translocation during replication and transcription. TOP1 generates a single-stranded DNA (ssDNA) nick by formation of a transient TOP1-DNA cleavage complex (TOP1cc) that self-resolves to religate the DNA strand. TOP1 inhibitors, such as camptothecin (CPT), bind the interface of TOP1cc and prevent reversal, resulting in a unique type of ssDNA break associated with DNA-protein crosslinks (DPCs) that block DNA metabolic processes. Upon replisome encounter, such lesions generate highly toxic replication-associated single-ended double-strand breaks (seDSBs), referred to as replication fork breakage (Ray Chaudhuri et al., 2012; Thomas and Pommier, 2019). seDSBs are repaired primarily by HR (Arnaudeau et al., 2001), and a hallmark of HR-deficient tumors is exquisite sensitivity to TOP1 inhibitors, probably because of toxic non-homologous end joining (NHEJ) (Adachi et al., 2004; Balmus et al., 2019; Thomas and Pommier, 2019). How the balance of HR over NHEJ is achieved at seDSBs remains unclear, although ataxia telangiectasia mutated (ATM) signaling and the BRCA1-A complex have recently been found to be involved in pathway choice (Balmus et al., 2019).
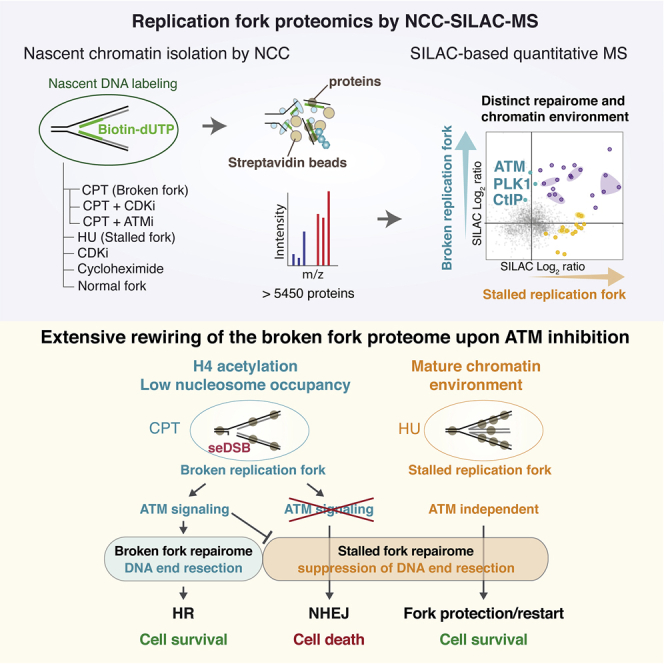
We performed a comprehensive investigation of protein dynamics at replication forks challenged by CPT to determine the broken fork proteome and its unique regulation of repair pathway choice. As a discovery approach, we used nascent chromatin capture (NCC) for comprehensive isolation of proteins enriched at replication forks and nascent chromatin (Alabert et al., 2014; Cortez, 2017). We combined NCC with stable isotope labeling by amino acids in cell culture (SILAC) (Ong et al., 2002) to quantitatively identify proteome dynamics in response to replication-associated seDSBs by mass spectrometry (NCC-SILAC-MS). Comparing CPT and HU replication fork proteomes, we identified three classes of fork repair factors according to their recruitment dynamics in response to replication fork breakage upon CPT treatment and fork stalling in response to HU. ATM was specifically recruited to broken forks, consistent with the presence of seDSBs. NCC-SILAC-MS showed extensive rewiring of the broken fork repairome upon ATM inhibition, revealing that this master kinase promotes recruitment of HR factors while suppressing the canonical DSB ubiquitination responses and NHEJ. We also demonstrated the value of our large datasets as a resource for discovery of novel DNA repair factors by identifying NDRG3 and UBAP2 as novel HR factors required for CPT resistance. This provides a new framework to understand seDSB repair as well as cancer vulnerabilities and resistance mechanisms relevant for clinical use of TOP1 inhibitors.
Results
Protein composition of broken replication forks
To characterize the broken fork proteome, we used CPT, a clinically relevant and well-described inducer of seDSBs (Thomas and Pommier, 2019). We treated cells with CPT and purified replication forks by NCC. To maximize the number of replication forks encountering a lesion and minimize secondary effects, S phase-synchronized cells were briefly exposed to a high dose of CPT, conditions known to induce replication-dependent DSBs (Hsiang et al., 1989; Neelsen and Lopes, 2015; Ray Chaudhuri et al., 2012; Thomas and Pommier, 2019). This treatment induced a large number of lesions while only moderately reducing DNA synthesis (Figure S1A). To achieve comparable biotin-dUTP (b-dUTP) incorporation in control and CPT-treated cells, we slightly extended the labeling time for CPT samples (Figure 1A; Figure S1B). Analysis of fork composition by immunoblotting showed CPT-specific phosphorylation of histone H2AX at Ser139 (γH2AX) and RAD51 recruitment, validating our strategy for isolation of CPT-damaged replication forks (Figure 1B; Figure S1C). This setup was combined with SILAC (Ong et al., 2002) labeling, and we performed NCC-SILAC-MS in biological triplicates to quantitatively compare the protein composition of normal and damaged forks (Table S1). We identified more than 4,000 proteins across three independent replicates (Figures S7A–S7C).
Figure 1.

Protein composition of broken replication forks
(A) SILAC-NCC-MS strategy for proteomics analysis of broken replication forks. Cells were released from a single thymidine block into mid-S phase and labeled with biotin-dUTP (b-dUTP) in the absence (untreated [Unt]) or presence of CPT (1 μM) prior to NCC purification.
(B) Left: NCC pull-downs analyzed by western blotting. Unt samples were harvested immediately after b-dUTP labeling (0 h) or 2 h. Right: b-dUTP-labeled S-phase cells show CPT-induced DNA damage (γH2AX and pRPAS33). An arrowhead indicates b-dUTP-negative cells (outside of S phase). Scale bar, 15 μm.
(C) GO analysis of proteins recruited (top 10% based on H/L ratio) or depleted (bottom 10% based on H/L ratio) at CPT-damaged forks. All identified proteins were used as background. p vvalue is shown with fold enrichment.
(D) Illustration of dormant origin firing in response to CPT and suppression of new origin firing by roscovitine (rosco).
(E) Experimental design for NCC-SILAC-MS using roscovitine to block CPT-induced dormant origin firing.
(F) GO analysis as in (C).
(G) Heatmap of replication and DDR factor enrichment, indicating the mean of three independent experiments. Not detected is indicated by gray coloring.
See also Figure S1.
For an unbiased assessment of CPT-induced changes in the replication fork proteome, we performed a Gene Ontology (GO) analysis of the 10% most enriched or depleted proteins (Figure 1C). Proteins involved in the DNA damage response (DDR) such as HR and DNA damage signaling were significantly enriched in CPT, verifying successful isolation of proteins surrounding CPT-induced seDSBs. In addition, we found enrichment of DNA/RNA helicases, likely reflecting induction of replication-transcription collision and R-loop formation caused by CPT-dependent inhibition of transcription (Tuduri et al., 2009). The most depleted categories included NHEJ, the repressive PRC1 complex, and nuclear inner membrane proteins. While inspecting the data for replication factors, we noticed a strong CPT-induced skew between leading- and lagging-strand proteins. Leading-strand proteins were moderately depleted, whereas lagging-strand proteins were enriched in CPT (Figure S1D). Because this might be a result of dormant origin firing to compensate for CPT-induced fork slow-down (Haahr et al., 2016; Ray Chaudhuri et al., 2012; Wang et al., 2014; Woodward et al., 2006), we carried out a new round of NCC-SILAC-MS in the presence of the CDK2 inhibitor roscovitine to suppress new origin firing (Petermann et al., 2010a) and normalize fork numbers (Figures 1D and 1E). Roscovitine did not affect the replication elongation rate or DNA damage induction by CPT (Figures S1E and S1F), and a separate NCC-SILAC-MS analysis showed that roscovitine treatment alone did not substantially change fork composition (Figures S1G and S7A–S7C). The NCC-SILAC-MS analysis of cells treated with CPT and roscovitine identified more than 4,000 proteins across three highly concordant replicates (Figure 1E; Figure S7A–S7C). A GO analysis also revealed CPT-induced enrichment for HR repair and DNA damage signaling and depletion of interactions with nuclear inner membrane proteins in the presence of roscovitine (Figure 1F). However, replication factors and associated DNA mismatch repair (MMR) proteins were depleted when CPT treatment was combined with roscovitine (Figure 1F), reflecting moderate depletion of leading- and lagging-strand proteins in this setting (Figure 1G). The observed CPT-induced enrichment of lagging-strand factors is thus roscovitine sensitive (Figure 1G; Figure S1D), consistent with trapping of newly fired dormant origins where primer elongation by polymerase delta (without CMG interaction) establishes leading-strand replication (Aria and Yeeles, 2018; Yeeles et al., 2017). When dormant origin firing is blocked and similar numbers of replication intermediates are compared, leading- and lagging-strand replication proteins are overall depleted upon CPT treatment. This argues that the canonical replication machinery is disassembled at a fraction of CPT-damaged forks.
Distinct protein dynamics at broken and stalled replication forks
Detailed inspection of the broken fork proteome revealed enrichment of a large panel of DNA repair, fork-processing and checkpoint-signaling factors (Figures S2A and S2B). This is not surprising because CPT induces a variety of replication problems, such as fork reversal and transcription-replication collision, in addition to fork breakage (Neelsen and Lopes, 2015). Thus, to pinpoint specific factors responding to fork breakage, we carried out NCC-SILAC-MS of stalled replication forks using a short high HU dose to fully block DNA synthesis and promote fork reversal without fork breakage (Petermann et al., 2010b; Figures S1E, S2C, S2D, and S7A–S7C). Under these conditions, HU treatment immediately blocked DNA synthesis (Figure S2E), consistent with previous work showing that dormant origins do not fire upon short exposure to high doses of HU (Dungrawala et al., 2015). A GO analysis showed high enrichment of DDR processes (Figure S2F) and a high overlap with previous analysis of HU arrested forks by iPOND (Dungrawala et al., 2015; Figure S2G; Table S2). With the exception of PCNA unloading (Sirbu et al., 2011), we saw no evidence of replisome disassembly in HU (Figure S2H), suggesting that this is a CPT-specific response.
We identified three distinct classes of fork repair factors by comparing the replication fork proteomes of HU- and CPT-treated cells (Figures 2A and 2B). Class I factors recruited only in CPT, class II factors recruited only in HU, and class III factors showed enrichment in CPT and HU (Figure 2A). The latter highlights the commonality between CPT- and HU-induced responses, consistent with observations that ATR signaling and replication fork reversal occur in response to both types of replication stresses (Ray Chaudhuri et al., 2012). Class I and II factors are the most intriguing, revealing unique responses to CPT and HU at a proportion of replication forks. Indeed, identification of classic DSB signaling (ATM) and HR factors (CtIP) in class I (Bensimon et al., 2011; Sartori et al., 2007) confirms induction of replication-associated seDSBs upon CPT treatment (Chanut et al., 2016; Huertas and Jackson, 2009). The role of PLK1 in this response is unexpected and deserves further exploration. NHEJ factors, including Ku70/80 and DNA-PKcs were not enriched upon CPT or HU treatment, but RIF1 was lost from broken forks. A substantial number of repair factors travel with the replication fork (Alabert et al., 2014) and are present at stressed replication forks without necessarily being enriched further (Figure 2B, red gradient fill color). In CPT, a set of those factors (Rad18-SLF1-SLF2) involved in post-replication repair is selectively depleted, suggesting that their function is adversary in the face of a broken fork. Class II proteins include a group of well-known proteins with functions in ubiquitin signaling at DSBs (RNF168 and RNF169), including the BRCA1-A complex, which antagonizes HR by suppressing DNA end resection (Coleman and Greenberg, 2011; Hustedt and Durocher, 2016). These factors are not recruited to CPT-damaged forks in the absence or presence of roscovitine (Figure S1H). These results indicate that repair of broken replication forks is facilitated by recruitment of ATM, PLK1, and CtIP (Chanut et al., 2016), whereas DSB-associated ubiquitin signaling and the BRCA1-A deubiquitinase complex are more prevalent at stalled forks.
Figure 2.

Protein dynamics in response to fork breakage and fork stalling
(A) Left: heatmap of replication and DDR factor enrichment at broken (CPT+Rosco/Rosco) and stalled (HU/Unt) replication forks. To normalize for fork numbers, CPT+Rosco/Rosco data were used. The mean of three independent experiments is shown. Right: enrichment of DDR proteins across CPT and HU NCC-SILAC-MS datasets. Broken forks, CPT+Rosco/Rosco; stalled fork, HU/Unt.
(B) Overview of DDR factor recruitment to broken forks (blue bars, CPT+Rosco/Rosco) and stalled forks (yellow bars, HU/Unt). The box gradient fill color indicates enrichment at undamaged forks (red) over mature chromatin (blue), as illustrated on the right (Alabert et al., 2014). Well-established protein complexes (red lines), functional groups (dotted line), and interactions (gray lines) are shown. Not detected (ND) is indicated by gray coloring.
See also Figure S2.
TOP1 inhibition challenges chromatin assembly and nuclear attachment
Consistent with the GO analysis (Figures 1C and 1F), a focused view of more than 40 inner nuclear membrane (INM) and nuclear pore complexes (NPCs) showed clear CPT-specific dissociation of nascent chromatin from the INM compartment, in particular lamins and NPCs (Figure 3A). These changes are likely to reflect specific changes in engagement or proximity of replication forks to the INM because the cytoplasmic components of the NPC did not show a similar trend. The PRC1 complex was also lost from CPT-treated forks, according to the GO analysis (Figure 1C), and similar behavior was observed for histone H1, the PRC2 complex, and the histone H2A.Z replacement variant (Figure 3B). In contrast, these chromatin regulators were enriched at HU stalled forks (Figure 3B). Under unperturbed conditions, these proteins are recruited during chromatin restoration (Alabert et al., 2014) in the hours after fork passage. Chromatin restoration and the associated histone H4 deacetylation proceed in the presence of HU (Annunziato, 1989; Sirbu et al., 2011; Figure S2C), but NCC-pulse SILAC-MS showed that new histone H4 remained hyperacetylated at CPT-damaged forks (Figure 1B; Figure S3A). We also analyzed the methylation status of histone H4 lysine 20 (H4K20) because it is critical for the balance between HR and NHEJ at canonical DSBs (Nakamura et al., 2019; Saredi et al., 2016). However, H4K20 methylation was not altered by CPT treatment; old histones were methylated uniformly at H4K20 (mainly di-methylated [me2]], >80%), whereas new histones were unmethylated at H4K20 (H4K20me0) (Figure S3A). In accordance, CPT treatment does not alter global H4K20 methylation levels in chromatin (Saredi et al., 2016). These data show that the chromatin environments of stalled and broken forks are highly distinct; chromatin maturation continues in the face of fork stalling, whereas fork breakage somehow delays this process.
Figure 3.

Distinct chromatin environment at broken and stalled replication forks
(A and B) Heatmap showing enrichment of inner nuclear membrane (INM) proteins (A), nuclear pore complex (NPC; A), and chromatin regulators (B) at broken (CPT+Rosco/Rosco) and stalled (HU/Unt) replication forks. The mean of three independent experiments is shown.
(C) Enrichment of canonical histones across all NCC-SILAC-MS data (see Figures S7A and S7B for details). Mean is shown with SEM; n = 3. CHX, cycloheximide.
(D) Left: the ratio of newly synthesized (new) and old recycled (old) histone H4 at replication forks isolated from CPT- and CHX-treated cells. New and old histones were analyzed by pulse-SILAC labeling followed by NCC-MS (Figure S3A). The CHX dataset is from Alabert et al. (2015). Mean is shown with SEM; n = 3 (CHX), 2 (CPT). Right: illustration of new and old histones in nascent chromatin.
See also Figure S3.
To understand why chromatin maturation was delayed at broken forks, we analyzed the overall abundance of histones in nascent chromatin. For comparison, we also performed NCC-SILAC-MS of cells treated short term with cycloheximide (CHX) to block new histone biosynthesis (Mejlvang et al., 2014; Figures S7A–S7C). In response to CPT, nucleosome occupancy was reduced at replication forks almost to the same extent as preventing new histone deposition with CHX (Figure 3C). However, although histone occupancy was reduced in response to CPT and CHX, the relative ratio of new and old histones differed. In response to CHX, parental histones were strongly enriched as expected, but CPT did not dramatically alter the new/old ratio (Figure 3D). Therefore, reduced nucleosome occupancy at CPT-damaged forks reflects a defect in deposition of new and old histones behind the fork. Indeed, we found that CPT treatment resulted in gradual enrichment of histone H3.3 in global chromatin (Figure S3B; Table S3), probably reflecting gap filling during chromatin restoration (Ray-Gallet et al., 2011). Collectively, these data argue that nucleosome occupancy is reduced at CPT-damaged replication forks and that this, in turn, may delay association of chromatin regulators involved in repression (PRC1, PRC2, and histone H1).
Dissecting ATM function at broken replication forks
ATM, a master regulator of the DSB response, was recruited to broken replication forks but not detected at HU stalled forks (Figures 2A and 2B). Consistent with this, CPT activates ATM signaling (Shiloh, 2014), and ATM deficient cells are hypersensitive to TOP1 inhibitors (Smith et al., 1989). To dissect how ATM promotes repair and cell survival, we directly compared the broken fork proteome in the absence and presence of ATM signaling using a highly specific inhibitor (ATMi; AZD0156) currently in clinical trials (NCT02588105). AZD0156 abrogated ATM autophosphorylation in response to CPT more efficiently than the commonly used ATMi KU55933 and reduced γH2AX, phospho-RPA, and RPA accumulation in response to CPT but not HU (Figures S4A–S4F). NCC-SILAC-MS identified more than 4,000 proteins with high reproducibility across six biological replicates (Figure 4A; Figures S7A–S7C) and revealed major rewiring of the broken fork proteome in the absence of ATM signaling (Figure 4B).
Figure 4.

ATM inhibition rewires the broken replication fork proteome toward a stalled fork response
(A) Experimental design of NCC-SILAC-MS analysis of ATM function at CPT-damaged forks (CPT+ATMi/CPT). The ATMi AZD0156 (250 nM) was added 5 min before CPT (1 μM) treatment.
(B) Enrichment of DDR (left) and chromatin (right) proteins, shown as log2 SILAC ratios of CPT+ATMi (heavy) over CPT (light) (CPT+ATMi/CPT). The mean of six independent experiments is shown. NE, nuclear envelope.
(C) Enrichments of DDR proteins from (B), grouped according to function. Symbol size indicates the number of replicates in which a protein was identified. ∗, RPA binding proteins.
(D and E) High-content microscopy of U-2-OS cells exposed to CPT or HU for 1 h with or without ATMi added 5 min before. Pre-extracted cells were stained for γH2AX and BRCA1 (D) or RPA (E).
(D) BRCA1 foci in γH2AX positive cells are shown relative to cells treated with CPT alone. Error bars indicate SEM; n = 3. Individual measurements are indicated by dots and correspond to the mean of more than 1,092 cells. ∗p = 0.0303; NS, not significant by ratio-paired two-sided t test. See the gating strategy for γH2AX positive cells in Figure S4I.
(E) RPA intensity shown as mean (+), with whiskers indicating 10th–90th percentiles; from left, n = 3,858, 3,402, 3,398, 3,249, 8,946, 9,509, 8,525, and 8,068 cells. See the gating strategy for RPA-positive cells in Figure S4J.
(F) Correlation plot showing DDR proteins identified in NCC-SILAC-MS as indicated. Pearson correlations (r) are shown.
See also Figure S4.
INM proteins showed limited changes upon ATM inhibition, whereas a panel of chromatin regulators, including PRC1, H3.3, DNMT3a, and G9a/GLP-ZNF644-Wiz were suppressed by ATM signaling. Most prominently, ATM inhibition caused massive deregulation of factors involved in protein ubiquitination during DSB repair, promoting recruitment of the positive H2A ubiquitination (H2A-Ub) regulators MDC1, RNF8, RNF168, RNF169, and L3MBTL2 and expelling the negative regulator POH1 (Figure 4C; Figure S4G; Butler et al., 2012; Kakarougkas et al., 2013; Nowsheen et al., 2018; Uckelmann and Sixma, 2017). Upon ATM inhibition, we also found recruitment of UBQLN4, a negative regulator of MRE11 (Jachimowicz et al., 2019), and the E3/E4 ubiquitin ligases UBE4A, which contribute to sustain K48- and K63-linked ubiquitin chains at DSBs (Baranes-Bachar et al., 2018). In addition, several factors otherwise preferentially enriched at stalled forks, such as FANCD2-I, BLM, and members of the BRCA1-A complex (BRCA1, BRCC45, ABRAXAS, RAP80, and MERIT40) were recruited to CPT-damaged forks upon ATM inhibition (Figure 4C). The BRCA1-A complex limits DNA end resection, and high-content microscopy confirmed that BRCA1 recruitment to CPT-damaged forks is suppressed by ATM signaling (Figure 4D; Figures S4H and S4I), consistent with recent reports (Balmus et al., 2019). RAP80, a reader of K63-ubiquitin chains, was among the most affected proteins in the BRCA1-A complex (Figure 4C), as expected if enhanced recruitment of BRCA1-A is a response to deregulation of ubiquitin signaling. The core NHEJ factors Ku70/80 and DNA-PKcs were not recruited, but the two anti-HR factors RIF1 and REV7 accumulated at broken forks upon ATM inhibition (Figure 4C; Setiaputra and Durocher, 2019). This argues that ATM activity promotes HR repair of broken replication forks in part by restricting protein ubiquitination and, thereby, recruitment of anti-HR factors. Importantly, this also explains the recent finding that deletion of BRCA1-A complex components (BRCC45, ABRAXAS, RAP80, MERIT40, and BRCC36) rescues sensitivity to TOP1 inhibitors in ATM-deficient cells (Balmus et al., 2019).
Consistent with rewiring away from HR, RPA accumulation at broken forks was suppressed by ATM inhibition, and, concomitantly, the RPA binding proteins ETAA1, SMARCAL1, ATR, and TONSL-MMS22L were lost (Figure 4C). High-content microscopy confirmed that ATM signaling and CtIP-dependent end resection were required for RPA accumulation at CPT-treated forks (Figure 4E; Figure S4J), as also reported by others (Balmus et al., 2019; Chanut et al., 2016). A correlation analysis across DNA repair proteins in our NCC-SILAC-MS data showed that the overall repair proteome shifted away from seDSB repair toward a stalled fork composition when ATM kinase activity was inhibited (Figure 4F; Figure S4K). We thus hypothesize that HR repair is suppressed at stalled forks through recruitment of the H2A ubiquitination machinery and the BRCA1-A complex, among others.
ATM inhibition significantly reduced restart of CPT-damaged replication forks but was dispensable for restart of HU-stalled forks (Figure 5A; Figure S5A). Upon ATM inhibition, RNF168 and RAP80 accumulated at CPT-induced repair foci (Figures 5B and 5C; Figures S5B and S5C), consistent with our NCC-SILAC-MS analysis (Figure 4C). RAP80 accumulation was dependent on RNF168 (Figure 5D), arguing that RNF168-dependent H2A ubiquitination facilitates BRCA1-A recruitment via RAP80 binding to K63-ubiquitin chains. Because our NCC-SILAC-MS data indicated that PLK1 is recruited to CPT-induced seDSBs in an ATM-dependent manner (Figures 2A and 4C), we also included the PLK1 inhibitor volasertib in these experiments. Volasertib is a potent inhibitor of PLK1 but can also inhibit PLK2 and PLK3 at higher doses. Inhibition of PLK kinase activity largely mimicked the effect of ATM inhibition on RNF168 and RAP80 recruitment. Collectively, this argues that ATM and PLK1 counteract toxic NHEJ by suppressing RNF168-dependent H2A ubiquitination and BRCA1-A recruitment (Figure 5E).
Figure 5.

ATM and PLK1 suppress RNF168 recruitment and downstream accumulation of RAP80
(A) DNA fiber analysis of replication fork restart. Top: experimental design. Fork restart, fork arrest, and new origin firing were scored as shown in Figure S5A. Bottom: percentage of labeled DNA tracks is shown as the mean, with error bars indicating SEM. Individual measurements are indicated by dots (CPT, n = 3; HU, n = 2). From the left, ∗∗p = 0.0046, 0.0028, and 0.2658; NS, not significant by ratio-paired two-sided t test.
(B–D) High-content microscopy of RNF168 and RAP80 recruitment to CPT-induced DNA repair foci. U-2-OS cells were exposed to CPT for 1 h in the presence or absence of ATMi (AZD0156) and PLK1 inhibitor (volasertib). In (D), cells were treated with control or RNF168 siRNAs for 48 h prior to drug treatment. Pre-extracted cells were stained for γH2AX and RNF168 (B) or RAP80 (C and D). RNF168 or RAP80 foci in γH2AX-positive cells are shown relative to cells treated with CPT alone. Error bars indicate SEM; n = 4 (B), n = 5 (C), n = 3 (D). Individual measurements are indicated by dots and correspond to the mean of more than 894 cells. A representative experiment is shown in Figures S5B and S5C. ∗p < 0.05, ∗∗p < 0.01 by ratio-paired two-sided t test.
(E) Model illustrating suppression of H2A ubiquitination by ATM and PLK1 kinase activity at broken forks.
See also Figure S5.
Novel HR factors required for repair of CPT damaged replication forks
Analysis of protein enrichment across all NCC-SILAC-MS datasets identified a number of novel putative fork repair proteins (Figure 6A; Table S4). To validate this resource, we focused on two factors, NDRG3 (N-myc downstream-regulated gene 3) and UBAP2 (ubiquitin-binding associated protein 2), identified previously as putative phosphorylation targets of ATM/ATR (Balmus et al., 2019; Boeing et al., 2016; Matsuoka et al., 2007; Stokes et al., 2007). NDRG3 was predominantly cytosolic and showed increased abundance in the nuclear fraction following CPT treatment, whereas UBAP2 was present in the nuclear and cytosolic fraction regardless of CPT treatment (Figure S6A). Depletion of NDRG3 or UBAP2 by small interfering RNA (siRNA) or CRISPRi reduced cell viability in response to CPT and led to enhanced accumulation of γH2AX (Figures 6B and 6C; Figures S6B–S6F). Consistent with this, NDRG3 and UBAP2 depletion impaired HR repair of canonical DSBs (Figure 6D) in the well-established DR-GFP HR reporter assay (Pierce et al., 1999). These data identify NDRG3 and UBAP2 as novel HR regulators required for CPT resistance, validating our collection of NCC-SILAC-MS data as a resource for pinpointing novel DDR candidates.
Figure 6.

NDRG3 and UBAP2 are new HR factors required for CPT resistance
(A) Overview of putative novel DDR factors identified by NCC-SILAC-MS. Recruitment to broken forks (blue, CPT+Rosco/Rosco) and stalled forks (yellow, HU/Unt) is indicated as in Figure 2B. A red dotted line encircles ATM/ATR substrates (Boeing et al., 2016; Matsuoka et al., 2007; Stokes et al., 2007). Factors were selected based on the enrichment of a higher than 0.3 log2 SILAC ratio and being putative ATM/ATR targets (Table S4).
(B) Colony formation analysis. Left: siRNA depletion of NDRG3 in U-2-OS cells treated with CPT (50 nM) for 24 h. Mean with SEM, n = 3. Right: CRISPRi depletion of UBAP2 in U-2-OS cells treated with CPT (50 nM) for 24 h. Mean with SEM, n = 3. ∗p < 0.05, ∗∗p < 0.01 by ratio-paired two-sided t test.
(C) High-content microscopy of γH2AX in siRNA-transfected U-2-OS cells exposed to CPT for 1 h. Mean intensity relative to control siRNA (siControl) is shown for γH2AX-positive cells, gated as shown in Figure S6F. Error bars indicate SEM, n = 4. Individual data points are indicated by dots and correspond to the means of more than 1,871 cells.
(D) HR efficiency measured in DR-GFP U-2-OS reporter cells. The HR efficiency is shown as percent of control siRNA. Mean is shown with SEM, n = 3. ∗p < 0.05, ∗∗p < 0.01 by ratio-paired two-sided t test.
(E) High-content microscopy of RPA in siRNA-transfected U-2-OS cells exposed to CPT for 1 h. Mean intensity relative to control siRNA (siControl) is shown for γH2AX-positive cells, gated as shown in Figure S6C. Error bars indicate SEM, n = 3. Individual data points are indicated by dots and correspond to the means of more than 1,871 cells.
(F) High-content microscopy of RAD51 foci. Left: representative images of RAD51 and γH2AX. Scale bar, 10 μm. Right: bar diagram showing RAD51 foci in γH2AX-positive cells relative to control siRNA treatment. Mean with error bars showing SEM, n = 3. Data points are indicated by dots and correspond to the mean of more 449 cells. A representative experiment is shown in Figure S6H. siRNA-transfected U-2-OS cells were exposed to CPT for 3 h.
See also Figure S6.
To address NDRG3 function in further detail, we asked whether this novel HR factor works up- or downstream of DNA end resection and Rad51 loading. CPT-induced phosphorylation of RPA and RPA accumulation on chromatin were not altered by absence of NDRG3 (Figure 6E; Figure S6G), suggesting that DNA end resection was normal. However, RAD51 loading at CPT-damaged forks was decreased substantially in NDRG3-depleted cells (Figure 6F; Figure S6H), and this was not due to changes in the cell cycle or RAD51 protein expression (Figures S6B and S6F). Because NDRG3 was also recruited to HU-arrested replication forks (Figure 6A), we monitored RAD51 accumulation in response to HU. In this setting, NDRG3 was also required for efficient RAD51 loading (Figure S6I). This indicates that NDRG3 is a novel HR factor facilitating RAD51 loading at different replication damage intermediates.
Discussion
In this work, we provide a comprehensive proteomics characterization of replication fork responses to TOP1 inhibitors, widely used in the clinic to treat colorectal, lung, ovarian, cervical, and pancreatic cancers (Thomas and Pommier, 2019). This coordinated replication fork proteome analysis, including six novel large datasets, provides unprecedented insights into the composition of replication forks challenged by seDSBs and topological stress. This is a valuable resource to track the behavior of known replication, repair, and chromatin proteins and, importantly, allows identification of novel repair factors, as we demonstrate for NDRG3 and UBAP2. Our NCC-SILAC-MS approach identifies a high number of chromatin and nuclear architectural proteins, revealing an unexpected difference in the environment of CPT- and HU-damaged forks, spanning from nucleosome occupancy to nuclear membrane interactions. Comparing the “repairome” of CPT- and HU-challenged forks, we identify three classes of fork repair factors, with classes I and II being specific to broken and stalled forks, respectively. Class I included ATM, the master regulator of DSB responses required for genome repair and cell survival in response to CPT. Upon ATM inhibition, NCC-SILAC-MS revealed that the broken fork proteome was rewired toward that of stalled forks and anti-HR activities. These pervasive changes argue that ATM promotes HR-dependent fork restart by concomitantly promoting DNA end resection and suppressing canonical DSB-associated ubiquitin signaling.
This comprehensive study of replication fork responses to TOP1 inhibitors highlights the strength of NCC-SILAC-MS for defining replication fork repairomes and revealing the complex changes in chromatin environment resulting from fork damage. In previous work, iPOND-SILAC-MS technology, involving 5-ethynyl-2’-deoxyuridine (EdU)-labeling and click-it chemistry (in place of b-dUTP labeling used in NCC), has been instrumental in characterizing the response to fork stalling (Dungrawala et al., 2015; Mukherjee et al., 2019). One study analyzed CPT-treated cells by iPOND and non-quantitative proteomics (Ribeyre et al., 2016) but identified less than 30 proteins that show little overlap with the large number of DDR factors enriched at CPT-challenged forks in our dataset. This probably reflects the importance of taking a quantitative proteomics approach, as highlighted by a comparison across published iPOND and NCC data (Cortez, 2017). NCC-SILAC-MS was developed specifically to comprehensively define the proteome of replication forks and newly assembled chromatin relative to post-replication mature chromatin (Alabert et al., 2014, 2015). To robustly trap chromatin components, NCC includes stronger protein-DNA crosslinking as compared to iPOND, explaining why more proteins are identified by NCC-SILAC-MS. NCC and iPOND thus are complementary technologies, and intersection of NCC-SILAC-MS (Alabert et al., 2014) and iPOND-SILAC-MS (Dungrawala et al., 2015) datasets previously allowed identification of a high-confidence mammalian replication fork proteome (Cortez, 2017). Similarly, overlapping the NCC-SILAC-MS (this work) and iPOND-SILAC-MS (Dungrawala et al., 2015) analysis of forks stalled by short HU treatment identifies a high-confidence stalled replication fork proteome, including 52 proteins, 26 of which are not known DNA repair factors (Figure S2G; Table S2).
The chromatin and nuclear environments of replication forks challenged by HU and CPT are dramatically different. As chromatin restoration proceeds, we envision that the environment of stalled forks is rather heterogeneous and largely determined by the local chromatin landscape (e.g., repressive or active chromatin). In contrast, active and CPT-damaged forks are embedded in highly acetylated nascent chromatin. In response to CPT, nucleosome occupancy was reduced in a manner affecting new and old histones similarly and maintaining the balance of H4K20me0 and H4K20me2 specific to replicated chromatin. This could be a consequence of seDSB processing or topological stress interfering with nucleosome assembly (Hammond et al., 2017; Hauer et al., 2017). The levels of the variant histone H3.3 increased during recovery from CPT, consistent with gap filling by HIRA (Ray-Gallet et al., 2011). CPT treatment also strongly reduced the interactions of replicating chromatin with the INM and nuclear pores, suggesting that seDSBs or topological stress triggers dissociation of forks from the nuclear envelope. This was largely independent of ATM signaling, suggesting that it originates from topological stress, low chromatin compaction, and/or interference with processes such as gene gating at nuclear pores (Bermejo et al., 2011). Mutation in nuclear envelope proteins in the premature aging diseases Hutchinson-Gilford progeria syndrome and restrictive dermopathy causes DNA damage and genome instability (Carrero et al., 2016). Comprehensive characterization of CPT-induced changes in the fork proteome provides an entry point to mechanistically address how interactions with the nuclear periphery are involved in fork progression and repair.
There was a substantial overlap in the repairome of CPT- and HU-challenged forks (class III), many of which are involved in ATR-dependent checkpoint signaling and RPA binding, arguing for their presence at different stressed fork intermediates. Consistent with this, these replication checkpoint factors are also recruited to replicating chromatin upon induction of DNA ICLs (Räschle et al., 2015). The CPT-specific factors included only ATM, CtIP, and PLK1. The requirement of ATM and CtIP in seDSBs repair is well established (Balmus et al., 2019; Chanut et al., 2016; Sartori et al., 2007; Smith et al., 1989), but the specific ATM-dependent recruitment of PLK1 to this type of lesion was unexpected. PLK1 can restrict NHEJ in mitosis by phosphorylation of 53BP1 (Benada et al., 2015) and XRCC4 (Terasawa et al., 2014). Local PLK1 activity at broken forks may thus prevent deleterious NHEJ. Consistent with this idea, PLK1 inhibition sensitizes cells to CPT analogs (Zuco et al., 2015), and PLK1 is also recruited to ICLs in replicating chromatin, where NHEJ is unwarranted (Räschle et al., 2015). We find that PLK1 operates downstream of ATM to suppress the H2A ubiquitination response and BRCA1-A recruitment. ATM inhibition diminished RPA accumulation at broken forks, consistent with previous work showing ATM-dependent DNA end resection at seDSBs (Balmus et al., 2019; Chanut et al., 2016). Strikingly, ATM not only promoted HR via positive regulation, but it actively restricted accumulation of a large group of proteins involved in NHEJ and protein ubiquitination, including REV7, SMCHD1, BRCA1-A, UBQLN4, RNF168, RNF169, RNF8, and Rad18. Many of these factors were enriched at HU-stalled forks (RNF168, RNF169, BRCA1-A, and Rad18), suggesting that ATM signaling extensively rewires the broken fork proteome to reduce activities that may act to limit HR at stalled forks (Figures 4F and 7). Many of these factors were also recruited during ICL repair in replicating Xenopus chromatin, including RNF168, Rad18, and BRCA1-A (Räschle et al., 2015), whereas the MRE11-RAD50-NBS1 (MRN) complex and CtIP were not enriched at ICL repair sites. RNF8 and RNF168 regulate ubiquitin signaling at canonical DSBs, where they contribute to recruitment of NHEJ and HR factors in a complex fashion (Uckelmann and Sixma, 2017). At canonical DSBs, ATM-dependent phosphorylation of MDC1 and γH2AX is upstream of the ubiquitination response (Kolas et al., 2007). In contrast, ATM and PLK1 signaling restrict this response at fork-associated seDSBs. We hypothesize that initiation of DNA end resection at seDSBs normally “deactivates” the DSB ubiquitination response but that ATM inactivation disables this feedback loop and leads to hyperubiquitin signaling despite reduced γH2AX (Figure 7). Our results argue that the BRCA1-A complex is recruited erroneously to seDSBs via RAP80 binding to excess ubiquitin chains that accumulate in the absence of ATM signaling (Kim et al., 2007; Sobhian et al., 2007; Wang et al., 2007). This, in turn, explains why genetic deletion of BRCA1-A rescues DNA end resection and CPT sensitivity in ATM-null cells (Balmus et al., 2019).
Figure 7.

Model of proteome dynamics at broken replication forks
Detailed model illustrating ATM function in protein recruitment at broken forks (this work) integrated with repair functions established in the literature (see text for references). ATM is recruited to broken forks, where it recruits PLK1 and promotes CtIP-dependent DNA end resection to facilitate HR repair. In parallel, ATM and PLK1 suppress H2A monoubiquitination and K63-linked poly-ubiquitination and, thereby, recruitment of BRCA1-A. ATM also promotes accumulation of POH1, a K63-specific deubiquitinase, and restricts accumulation of NHEJ factors such as SMCHD1, RIF1, and REV7.
The NCC-SILAC-MS resource also identifies novel fork repair factors, advancing our understanding of replication stress responses and providing new potential targets for drug discovery. We identify a panel of uncharacterized factors enriched at CPT- and HU-challenged forks, a high proportion of which are putative ATM/ATR targets, consistent with a DDR function. Functional analysis revealed NDRG3 and UBAP2 as novel HR-promoting factors. NDRG3 is required for efficient Rad51 loading and cell survival in response to CPT. NDRG3 is expressed ubiquitously, and in testes, NDRG3 is localized to the outer layers of seminiferous epithelium (Zhao et al., 2001), suggesting that it could be involved in HR at damaged forks and during meiotic recombination in spermatogenesis. NDRG3 upregulation correlates with poor prognosis in prostate and liver cancer (Lee et al., 2016) and has been linked to drug resistance (Du et al., 2017; Yu et al., 2019), which could be linked to an HR function. Our results also suggest that mutations in the H2A ubiquitination machinery may promote resistance to ATM inhibitors and provide important insights relevant to ongoing clinical investigations of PLK1 inhibitors in cancer therapy (Liu et al., 2017). We thus suggest that integration of replication fork proteomics with cancer genomics can be explored as an entry point to identify resistance mechanisms and cancer vulnerabilities that can be exploited in treatment.
Limitations of study
In this study, we perform comprehensive proteomics profiling of damaged replication forks using NCC-SILAC-MS. The repairome of CPT-induced broken replication forks is highly distinct from proteomics analyses of replication forks challenged by nucleotide depletion or DNA ICLs (Dungrawala et al., 2015; Mukherjee et al., 2019; Räschle et al., 2015). The response to CPT-induced fork breakage is also distinct from fork collapse triggered by ATR inhibition in combination with HU, which is permissive for NHEJ (Dungrawala et al., 2015), as indicated by recruitment of DNA-PKcs and KU70-KU80. Fork breakage induced by other genotoxic agents (e.g., PARP inhibitors) will likely share some but not all features with CPT-induced fork breakage, because most agents challenge replisome stability though a number of mechanisms. In addition to fork breakage, TOP1 inhibitors introduce topological stress at the replication fork and enhance the likelihood of replication-transcription collision. The response to these stresses is also captured in our study of the CPT-specific fork repairome, which is critical to integrate these molecular data with the clinical response. We focus our analysis on replication forks exposed for a short time to CPT to capture the initial response to seDSBs. Building on these findings, we expect that a time-resolved analysis of the CPT response and fork recovery could inform further on repair dynamics in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BRCA1 | Santa Cruz Biotechnology | sc-6954; RRID: AB_626761 |
| Phospho-H2A.X (S139) | Cell Signaling Technology | 2577; RRID: AB_2118010 |
| Phospho-H2A.X (S139) | Millipore | 05-636; RRID: AB_309864 |
| RAD51 | Bio Academia | 70-001; RRID: AB_2177110 |
| RAD51 | Santa Cruz Biotechnology | sc-8349; RRID: AB_2253533 |
| Phospho-RPA (S33) | Bethyl | A300-246A; RRID: AB_2180847 |
| Phospho-RPA (S4/S8) | Bethyl | A300-245A; RRID: AB_210547 |
| Phospho-ATM (S1981) | Millipore | MAB3806; RRID: AB_569379 |
| PCNA | Santa Cruz Biotechnology | sc-56; RRID: AB_628110 |
| Histone H3 | abcam | ab1791; RRID: AB_302613 |
| Histone H4K5ac | abcam | ab51997; RRID: AB_2264109 |
| Streptavidin, Alexa Fluor 488 Conjugate | Thermo Scientific | S32354; RRID: AB_2315383 |
| RPA | Thermo Scientific | MA1-26418; RRID: AB_795362 |
| BrdU (CldU) | Eurobio | ABC117-7513; RRID: AB_2314033 |
| BrdU (IdU) | BD Bioscience | 347580; RRID: AB_10015219 |
| ssDNA | Millipore | MAB3034; RRID: AB_94645 |
| NDRG3 | Santa Cruz Biotechnology | sc-514561 |
| RAP80 | Novus | NBP1-87156; RRID: AB_10999813 |
| RNF168 | R&D BIosystems | AF7217; RRID: AB_10971653 |
| Chemicals, peptides, and recombinant proteins | ||
| EdU | Invitrogen | Cat No. A10044 |
| Biotin-16-dUTP | IBA oligonucleotides | N/A |
| MyC1 Streptavidin beads | Invitrogen | Cat No. 65001 |
| RPMI 1640 Medium | Thermo Scientific | Cat No. 89984 |
| Dialyzed FBS | Invitrogen | Cat No. 26400-036 |
| MEM non-essential amino acid mix | Invitrogen | Cat No. 11140 |
| Glutamax | Invitrogen | Cat No. 35050-038 |
| Lysine | Invitrogen | Cat No. L8662 |
| Arginine | Invitrogen | Cat No. A8094 |
| Heavy Lysine | Cambridge isotope laboratories | Cat No. CNLM-291 |
| Heavy Arginine | Cambridge isotope laboratories | Cat No. CLM-2265 |
| CldU | MP Biomedicals | Cat No. 0 2105478 |
| IdU | Sigma | Cat No. I7125 |
| Thymidine | Sigma | Cat No. T1895 |
| Deoxycytidine | Sigma | Cat No. D0776 |
| Roscovitine | Sigma | Cat No. R7772 |
| Camptothecin | Sigma | Cat No. C9911 |
| Hydroxyurea | Sigma | Cat No. H8627 |
| Cycloheximide | Sigma | Cat No. C7698 |
| AZD0156 | Active Biochem | Cat No. A-6008 |
| Volasertib (BI 6727) | Selleckchem | Cat No. S2235 |
| AZ20 | MedChemExpres | Cat No. HY-15557 |
| Critical commercial assays | ||
| Click-IT Alexa Fluor 488 imaging kit | Thermo Fisher Scientific | Cat No. C10337 |
| Deposited data | ||
| Histone PTM analysis | This paper | PRIDE: PXD018220 |
| NCC-SILAC-MS | This paper | PRIDE: PXD018092 |
| Raw image files | Mendeley Data | https://dx.doi.org/10.17632/4ynntcp4xj.1 |
| Experimental models: cell lines | ||
| HeLa S3 | ATCC | Cat. No. CCL-2-2 |
| U-2-OS | Laboratory of Jiri Bartek | N/A |
| U-2-OS dCAS9-mCherry | This paper | N/A |
| Oligonucleotides | ||
| siNDRG3#1, 5′- CCAAUGUUGUGGACAUUAUtt −3′ | This paper | N/A |
| siNDRG3#2, 5′- GCCUUGUGCUCAUUAAUGUtt −3′ | This paper | N/A |
| siUBAP2#1, 5′- CCUAGCUAAUAAUCCAUAUtt −3′ | This paper | N/A |
| siUBAP2#2, 5′- GCAUUGAUCUGGUAGCCUUtt −3′ | This paper | N/A |
| negative control siRNA (NC1), 5′- UAACGACGCGAC GACGUAAtt −3′ |
Thermo Fisher Scientific | Cat. No. 4390844 |
| siNBS1, 5′- GUACGUUGUUGGAAGGAAAtt −3′ | Nakamura et al., 2011 | N/A |
| siCtIP, 5′- GCUAAAACAGGAACGAAUCtt −3′ | Sartori et al., 2007 | N/A |
| siRNF168, 5′- GGCGAAGAGCGAUGGAAGAtt −3′ | Thorslund et al., 2015 | N/A |
| sgRNAs | ||
| UBAP2, 5′- GAAGCTCGGCCCGGTCCTCAC −3′ | This paper | N/A |
| negative control,5′ - GCGCCAAACGTGCCCTGACGG −3′ | Gilbert et al., 2014 | N/A |
| Recombinant DNA | ||
| pHR-SFFV-KRAB-dCas9-2A- CHERRY | Gilbert et al., 2014 | Addgene (cat.no 60954) |
| pU6-sgRNA- EF1α-puro-T2A-BFP | Gilbert et al., 2014 | Addgene (cat.no 60955) |
| Software and algorithms | ||
| Prism8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R (3.4.1) | R Project | https://www.r-project.org |
| DAVID Bioinformatics Resources 6.8 | Huang et al., 2009b, 2009a | https://david.ncifcrf.gov |
| MaxQuant (1.5.8.3) | Cox and Mann, 2008 | https://www.maxquant.org |
| Volocity image analysis software | PerkinElmer | N/A |
| softWoRx (6.5.2) | Applied Precision | N/A |
| ScanR analysis software (2.6.1.0) | Olympus | N/A |
| Cytoscape | (Shannon et al., 2003) | N/A |
| Perseus 1.6.0.7 | Tyanova et al., 2016 | https://maxquant.net/perseus/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anja Groth (anja.groth@cpr.ku.dk)
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019). The accession number for the histone PTM analysis reported in this paper is PRIDE: PXD018220 and NCC-SILAC-MS is PRIDE: PXD018092.
Original image data have been deposited to Mendeley Data:
Experimental model and subject details
Cell culture
U-2-OS and HeLa S3 described previously (Alabert et al., 2014) were grown in Dulbecco’s modified Eagle’s medium (GIBCO) containing 10% fetal bovine serum (HyClone), 1% penicillin/streptomycin and drugs for selection. All cell lines used in this study tested negative for Mycoplasma contamination. SILAC-labeling of cells was performed as described previously (Alabert et al., 2014). Briefly, HeLa S3 cells in spinners were grown in SILAC medium depleted for arginine and lysine (Thermo Scientific RPMI 1640 Medium) and supplemented with dialyzed FBS (#26400-036 Invitrogen), MEM non-essential amino acid mix (#11140 Invitrogen), Glutamax (#35050-038 Invitrogen), 66 mg l−1 of arginine and 100 mg l−1 of lysine (SIGMA). Heavy medium was complemented with heavy lysine and arginine (Cambridge Laboratory number CNLM-291-0.25, number CLM-2265-0). Cells were maintained for eight to nine divisions in heavy or light medium before NCC analysis. Catalytically dead Cas9 (dCAS9) expressing U-2-OS cells were sorted based on mCherry expression level (Figure S6D) and used for CRISPRi.
Method details
Synchronization and drug treatment
HeLa S3 cells were synchronized at the G1/S border by single thymidine block (2 mM, 17 h) and released into fresh media containing deoxycytidine (24 μM) for 3 h as described (Alabert et al., 2014). The following drugs were used: 1 μM CPT (Sigma), 3 mM HU (Sigma), 100 μg/ml CHX (Sigma), ATMi; 250 nM AZD0156 (Active Biochem), ATRi; 300 nM AZ20 (MedChemExpress (MCE)), CDKi; 25 μM roscovitine (Sigma), PLK1i; 1 μM Volasertib (Selleckchem).
Nascent chromatin capture (NCC)
NCC was performed as described previously (Alabert et al., 2014) with the following modifications. For biotin–dUTP labeling, cells were incubated for 5 min in a hypotonic buffer (50 mM KCl and 10 mM HEPES) containing biotin–dUTP and resuspended in fresh cell culture medium containing biotin–dUTP. Labeling and drug treatment is detailed in Figure S6A. Cells were fixed 15 min in 2% formaldehyde and crosslinking was stopped by addition of glycine to a final concentration of 1% and incubation for 5 min at room temperature. Nuclei were mechanically isolated in a sucrose buffer (0.3 M sucrose, 10 mM HEPES–NaOH at pH 7.9, 1% Triton X-100 and 2 mM MgOAc), and chromatin was solubilized by sonication in a Diagenode Bioruptor at 4◦C in sonication buffer (10 mM HEPES–NaOH at pH 7.9, 100 mM NaCl, 2 mM EDTA at pH 8, 1 mM EGTA at pH 8, 0.2% SDS, 0.1% sodium sarkosyl and 1 mM phenylmethylsulphonyl fluoride; Bioruptor setting: High, 28 cycles of 30 s sonication and 90 s pause). Biotinylated chromatin fragments were purified on streptavidin-coated magnetic beads (MyC1 Streptavidin beads) by overnight end-over-end rotation at 4◦C and five stringent washes (10 mM HEPES–NaOH at pH 7.9, 100 mM NaCl, 2 mM EDTA at pH 8, 1 mM EGTA at pH 8, 0.1% SDS, and 1 mM phenylmethylsulphonyl fluoride). For SILAC analysis, nascent and mature samples were mixed in the last wash. To release chromatin and reverse the crosslink, beads were boiled in LSB for 40 min at 100◦C, including a brief vortex and short spin every 10 min to prevent drying.
Mass spectrometry
NCC samples were in-gel digested with trypsin as previously described (Alabert et al., 2014). Tryptic peptides were acidified with orthophosphoric acid and subjected to strong cation exchange (SCX) fractionation, using a polySULFOETHYL A column on an Ultimate 3000 Dionex LC system (Thermo Fisher Scientific). Mobile phase A consisted of 5mM KH2PO4 in 10% acetonitrile at pH 3; mobile phase B was 5 mM KH2PO4, 1 M KCl, and 10% acetonitrile, pH 3. The flow rate was constant at 200 μl/min. Fractions were collected at 1 min intervals. SCX fractions were diluted with an equal volume of 0.1% TFA and spun onto StageTips as described (Rappsilber et al., 2003). Peptides were eluted in 20 μl 80% acetonitrile in 0.1% TFA and concentrated down to less than 4 μl by vacuum centrifugation to evaporate acetonitrile. Mass spectrometry analyses were performed on a Q Exactive Mass spectrometer (Thermo Fisher Scientific), coupled with Ultimate 3000 RSLC nano Systems (Thermo Fisher Scientific). Peptides were loaded directly onto a spray emitter analytical column (75 μm inner diameter, 8 μm opening, 250 mm length; New Objectives) packed with C18 material (ReproSil-Pur C18-AQ 3 μm; Dr Maisch GmbH) using an air pressure pump (Proxeon Biosystems) (Ishihama et al., 2006). Mobile phase A consisted of water and 0.1% formic acid, and mobile phase B consisted of 80% acetonitrile and 0.1% formic acid. The gradient used was 180 min. The peptides were loaded onto the column at a flow rate of 0.5 μl/min and eluted at a flow rate of 0.2 μl/min according to the following gradient: 2%–40% buffer B in 180 min, then to 95% in 16 min. MS1 spectra were recorded at 70,000 resolution with scan range 350–1400 m/z. Precursor ions with top 10 intensity and charge state ≥ 2 were isolated and fragmented using high energy collision dissociation (HCD) of 25% normalized collision energy. The fragmentation spectra were recorded with a resolution of 17,500, isolation window of 2.0 m/z, maximum injection time of 60 ms and AGC target of 5e4. Dynamic exclusion was enabled with single repeat count and 60 s exclusion duration.
NCC raw file processing
NCC-related mass spectrometry raw files were processed using MaxQuant 1.5.8.3. The following default MaxQuant search parameters were used: MS1 tolerance for the first Andromeda search: 20 ppm, MS1 tolerance for the main Andromeda search: 4.5 ppm, FTMS MS2 match tolerance: 20 ppm, ITMS MS2 match tolerance: 0.5 Da, Variable modifications: acetylation of protein N-termini, oxidation of methionine, Fixed modifications: carbamidomethylation of cysteine, Decoy mode set to reverse, Minimum peptide length: 7 and Max missed cleavages set to 2. The following non-default settings were used: In group-specific parameters, match type was set to “No matching” and SILAC labels were set to Arg6 and Lys8. The “Re-quantify” option was enabled. Under global parameters, the minimum ratio count was set to 1 and “Discard unmodified counterpart peptide” was disabled. Also in global parameters, writing of large tables was disabled. Canonical and isoform protein sequences were downloaded from UniProt (Bateman, 2019) on 29th March 2017, considering only reviewed SwissProt entries that were part of the human proteome. To simplify quantitation of core histones, 29 redundant core histone sequences were removed from the database, as described before (Alabert et al., 2014).
NCC data analysis
Data analysis was performed in R 3.4.1. The scripts used to process these data are available in GitHub (https://github.com/Rappsilber-Laboratory/NCC). Protein fold-changes were extracted from the MaxQuant proteinGroups file. Proteins labeled as contaminants and reverse hits and those only identified by a modification site were discarded. Normalized SILAC ratios were considered for downstream analysis, log2 transformed and median-normalized. For each protein, the mean SILAC ratio and the standard error of the mean were calculated across the three replicates (six replicates in the case of NCC-ATMi), considering only SILAC ratios that had been detected with at least two ratio counts. The complete NCC-SILAC proteomics dataset is presented in Table S1. Interphase chromatin probabilities (Kustatscher et al., 2014) are included in the table to facilitate identification of new candidate DDR and replication factors as described (Alabert et al., 2014).
Gene ontology (GO) analysis
Gene ontology enrichment analysis for NCC-SILAC-MS assays was performed using DAVID 6.8 (Huang et al., 2009b, 2009a). Only proteins detected in two or more experiments were considered. Enrichment of terms from the GO aspects biological process, cellular component and molecular function, and enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and protein domains (InterPro) were calculated, using all identified proteins as background. Terms enriched with a p value lower than 0.05 were considered statistically significant. Ribosomal proteins are very abundant and can be found as contaminants in NCC samples regardless of whether replication forks are stalled or stressed. We therefore excluded GO terms related to ribosomes.
Histone PTM analysis
NCC isolated histones were separated in a 16% gel and processed with either propionic anhydride (Figure S3B) or deuterated-6 acetic anhydride (Figure 3D; Figure S3A) as described before with minor modifications (Völker-Albert et al., 2018a). For LC-MS/MS purposes, desalted peptides were injected in an Ultimate 3000 RSLCnano system (Thermo), separated in a 15-cm analytical column (75 μm ID home-packed with ReproSil-Pur C18-AQ 2.4 μm from Dr. Maisch) with a 50-min gradient from 5 to 60% acetonitrile in 0.1% formic acid. The effluent from the HPLC was directly electrosprayed into a LTQ-Orbitrap mass spectrometer XL (Figure S3B) or a Q Exactive HF (Figure 3D; Figure S3A) (Thermo). For LTQ-Orbitrap measurements, the instrument was operated in data dependent mode to automatically switch between full scan MS and MS/MS acquisition. Survey full scan MS spectra (from m/z 300 – 2000) were acquired in the Orbitrap with resolution R = 60,000 at m/z 400 (after accumulation to a ‘target value’ of 500,000 in the linear ion trap). The six most intense peptide ions with charge states between 2 and 4 were sequentially isolated to a target value of 10,000 and fragmented in the linear ion trap by collision induced dissociation (CID). All fragment ion spectra were recorded in the LTQ part of the instrument. For all measurements with the Orbitrap detector, 3 lock-mass ions from ambient air were used for internal calibration as described before (Olsen et al., 2005). Typical MS conditions were: spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 200°C; normalized CID energy 35%; activation q = 0.25; activation time = 30 ms.
For Q Exactive HF measurements, the mass spectrometer was operated in the PRM mode to identify and quantify the light and heavy (K8R10) versions of the H4 4-17 peptide carrying one, two or three naturel acetyl groups6. In this mode, the mass spectrometer automatically switched between one survey scan and 6 MS/MS acquisitions of the m/z values described in the inclusion list containing the precursor ions, modifications, and fragmentation conditions. Survey full scan MS spectra (from m/z 260–760) were acquired with resolution R = 30,000 at m/z 400 (AGC target of 3x106). The ions from the inclusion list were sequentially isolated to a target value of 2x105 (isolation window 1.3 m/z), and fragmented at 27% normalized collision energy. Typical mass spectrometric conditions were: spray voltage, 1.5 kV; no sheath and auxiliary gas flow; heated capillary temperature, 250°C; ion selection threshold, 33.000 counts. Raw data were analyzed using Skyline or QuanBrowser as described previously (Feller et al., 2015; Völker-Albert et al., 2018b). The peak areas corresponding to the peptides and modifications of interest were exported to csv and the statistical analysis were done with GraphPad Prism 8.
siRNA transfection
siRNA transfection was performed with RNAiMax reagent (Invitrogen) according to the manufacturer’s protocol. siRNA sequences (Ambion, Thermo Fisher scientific) were: siNDRG3#1, 5′-CCAAUGUUGUGGACAUUAUtt-3′; and siNDRG3#2, 5′-GCCUUGUGCUCAUUAAUGUtt-3′; negative control siRNA (NC1), 5′-UAACGACGCGACGACGUAAtt-3′; siNBS1, 5′-GUACGUUGUUGGAAGGAAAtt-3′; siCtIP, 5′-GCUAAAACAGGAACGAAUCtt-3′; siUBAP2#1, 5′-CCUAGCUAAUAAUCCAUAUtt-3′; siUBAP2#2, 5′-GCAUUGAUCUGGUAGCCUUtt-3′.
HR assay
DR-GFP-U-2-OS cells were described previously (Nakamura et al., 2019). Briefly, 3.75 × 105 cells/well were seeded on 6-well plates. After 24 h, the cells were transfected with siRNAs and incubate another 24 h before transfection with I-SceI (pCBASce) and RFP expression vectors. 24 h after transfection, cells were washed and replated on 100-mm dishes. Flow cytometric analysis to quantify the presence of GFP-positive cells was performed 2 days later on a FACSCalibur using Cellquest Pro software (BD Biosciences). For each sample, 30,000 cells were analyzed and the percentage of GFP-positive cells out of total transfected RFP positive cells was calculated as HR repaired cells.
Colony formation
U-2-OS cells were transfected with siRNA and 24 h later cells were trypsinised and seeded in technical triplicates of 300 to 10,000 cells. After 24 h, CPT was added for 24 h at the indicated concentrations. Cells were then cultured in fresh medium for 7–15 days before fixation and staining with MeOH/Crystal Violet. Colony formation efficiency was determined by manual colony counting and normalized to non-drug treated control. Each data point (biological replicate) represents the mean of technical triplicates.
DNA fiber assay
DNA fiber assay was performed as described previously (Mejlvang et al., 2014) with the following modifications. U-2-OS or HeLa-S3 cells were labeled with 150 μM IdU (Sigma-Aldrich) and 150 μM CldU (MP Biomedicals) as shown in the figures. 2 μL of cells resuspended in ice-cold PBS was deposited on a microscope slide and incubated with 7 μL of spreading buffer (200 mM Tris-HCl, pH 7.5, 0.5% SDS, and 50 mM EDTA) for 3 min. The slides were tilted 15° to stretch the DNA fibers. After fixation with methanol/acetic acid (3:1), DNA was denatured with 2.5 M HCl and blocked (PBS with 1% BSA and 0.1% Triton X-100) before staining with primary antibodies, anti-CldU (eurobio), anti-IdU (BD) and corresponding secondary antibodies conjugated with Alexa Fluor 488 (Chicken anti-Rat IgG) or 568 (Goat anti-Mouse IgG). To enhance the IdU-Alexa Fluor 568 signal, cells were additionally incubated Alexa Fluor 568 (Donkey anti-Goat IgG). ssDNA was detected by anti-ssDNA (MAB3034, Millipore) and corresponding secondary antibodies conjugated with Alexa 647 (Invitrogen). Images were captured on a DeltaVision system (Applied Precision) and analyzed with softWoRx 6.5.2 software (Applied Precision).
Immunofluorescence and high-content microscopy
Immunofluorescence was performed as described previously (Nakamura et al., 2019). Briefly, U-2-OS cells were grown on glass coverslips or 96-well plates and either directly fixed in 4% paraformaldehyde for 10 min or washed in cytoskeleton buffer (CSK; 10 mM PIPES pH 7, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2), pre-extracted for 5 min with CSK/0.5% Triton X-100 (on ice) and rinsed with CSK and phosphate buffered saline before fixation in 4% paraformaldehyde for 10 min. For B-dUTP incorporation, images were acquired using a DeltaVision system (Applied Precision) and analyzed by Volocity image analysis software (PerkinElmer). For high-content quantitative analysis, fluorescence images were acquired using an Olympus ScanR high-content microscope and processed on the ScanR analysis software (Olympus).
sgRNA cloning
sgRNA cloning was performed as described in Gilbert et al. (2014). The oligos were annealed in annealing buffer (200 mM potassium acetate, 60 mM HEPES-KOH pH 7.4, 4 mM magnesium acetate) and ligated into BstXI + BlpI digested pU6-sgRNA-EF1α-puro-T2A-BFP.
Lentiviral transduction
HEK293FT cells were co-transfected with pU6-sgRNA- EF1α-puro-T2A-BFP or pHR-SFFV-KRAB-dCas9-P2A-mCHERRY and pAX8 and pCMV-VSV using a standard calcium phosphate protocol. The viral supernatant was collected 72 h after HEK293FT transfection and used for transduction. Transduction was performed in presence of polybrene at 8 μg/ml. 24 h after transduction with sgRNA- encoding lentiviral plasmids, puromycin was added at 1 μg/ml for U-2-OS cells.
Quantification and statistical analysis
Statistics were analyzed with GraphPad Prism 8. All statistics were evaluated by either independent or paired two-tailed Student’s t test and two-tailed Mann–Whitney U-test. Pearson’s correlation and Student’s t test were performed under the assumption of normality. The exact sample sizes (n) used to calculate statistics are provided in the figure captions. p values are provided in the figure legends and captions. Whiskers indicate 10th–90th percentiles, and outliers are plotted as individual points in box-and-whisker plots. All experiments were reproduced with similar results a minimum two times.
Acknowledgments
We thank S. Graziano, and K.R. Stewart-Morgan for critical reading of this manuscript; members of the Groth laboratory, D. Walter, and C.S. Sørensen for fruitful discussions; J. Zou and C. Spanos for assistance with mass spectrometry; Y. Antoku for assistance with microscopy; and S. Miyagi for assistance with CRISPRi. Research in the Groth lab was supported by the Danish Cancer Society, the European Research Council (ERC StG no. 281765 and ERC CoG no. 724436), Independent Research Fund Denmark (7016-00042B and 4092-00404B), the Novo Nordisk Foundation (NNF14OC0012839), the NEYE Foundation, and the Lundbeck Foundation (R198-2015-269). Research in the Rappsilber group was supported by the Wellcome Trust through a senior research fellowship to J.R. (grant 103139). The Wellcome Centre for Cell Biology is supported by core funding from the Wellcome Trust (grant 798 203149). Research in the Choudhary lab was supported by the Danish Cancer Society and Independent Research Fund Denmark (8020-00221B). The Novo Nordisk Foundation Center for Protein Research is financially supported by the Novo Nordisk Foundation (NNF14CC0001).
Author contributions
A.G. and K.N. conceived the project and designed the study. K.N., C.A., and M.H. performed NCC experiments and analyzed data. G.K., S.S., C.C., and J.R. performed mass spectrometry, and G.K. analyzed NCC-SILAC-MS data. I.F., M.V.-A., and A.I. performed histone PTM mass spectrometry and analyzed data. T.E.B. performed cell fractionation. N.M. contributed to HU NCC-SILAC-MS design. The manuscript was written by K.N. and A.G. and commented on by all authors.
Declaration of interests
A.G. is cofounder and CSO of Ankrin Therapeutics and inventor on a filed patent application covering the therapeutic targeting of TONSL for cancer therapy. A.I. and M.VA are cofounders of EpiQMAx.
Published: January 14, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.molcel.2020.12.025.
Contributor Information
Juri Rappsilber, Email: juri.rappsilber@tu-berlin.de.
Anja Groth, Email: anja.groth@cpr.ku.dk.
Supporting Citations
The following references appear in the supplemental information: Fang et al., 2014, Jasencakova et al., 2010, Ju et al., 2010, Sabatel et al., 2012.
Supplemental information
References
- Adachi N., So S., Koyama H. Loss of nonhomologous end joining confers camptothecin resistance in DT40 cells. Implications for the repair of topoisomerase I-mediated DNA damage. J. Biol. Chem. 2004;279:37343–37348. doi: 10.1074/jbc.M313910200. [DOI] [PubMed] [Google Scholar]
- Alabert C., Groth A. Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol. 2012;13:153–167. doi: 10.1038/nrm3288. [DOI] [PubMed] [Google Scholar]
- Alabert C., Bukowski-Wills J.-C., Lee S.-B., Kustatscher G., Nakamura K., de Lima Alves F., Menard P., Mejlvang J., Rappsilber J., Groth A. Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat. Cell Biol. 2014;16:281–293. doi: 10.1038/ncb2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alabert C., Barth T.K., Reverón-Gómez N., Sidoli S., Schmidt A., Jensen O.N., Imhof A., Groth A. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev. 2015;29:585–590. doi: 10.1101/gad.256354.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato A.T. Inhibitors of topoisomerases I and II arrest DNA replication, but do not prevent nucleosome assembly in vivo. J. Cell Sci. 1989;93:593–603. doi: 10.1242/jcs.93.4.593. [DOI] [PubMed] [Google Scholar]
- Aria V., Yeeles J.T.P. Mechanism of Bidirectional Leading-Strand Synthesis Establishment at Eukaryotic DNA Replication Origins. Mol. Cell. 2018;73:199–211. doi: 10.1016/j.molcel.2018.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaudeau C., Lundin C., Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol. 2001;307:1235–1245. doi: 10.1006/jmbi.2001.4564. [DOI] [PubMed] [Google Scholar]
- Balmus G., Pilger D., Coates J., Demir M., Sczaniecka-Clift M., Barros A.C., Woods M., Fu B., Yang F., Chen E. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019;10:87. doi: 10.1038/s41467-018-07729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranes-Bachar K., Levy-Barda A., Oehler J., Reid D.A., Soria-Bretones I., Voss T.C., Chung D., Park Y., Liu C., Yoon J.-B. The Ubiquitin E3/E4 Ligase UBE4A Adjusts Protein Ubiquitylation and Accumulation at Sites of DNA Damage, Facilitating Double-Strand Break Repair. Mol. Cell. 2018;69:866–878.e7. doi: 10.1016/j.molcel.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A., UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi: 10.1093/nar/gky1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benada J., Burdová K., Lidak T., von Morgen P., Macurek L. Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle. 2015;14:219–231. doi: 10.4161/15384101.2014.977067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A., Aebersold R., Shiloh Y. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 2011;585:1625–1639. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Bermejo R., Capra T., Jossen R., Colosio A., Frattini C., Carotenuto W., Cocito A., Doksani Y., Klein H., Gómez-González B. The replication checkpoint protects fork stability by releasing transcribed genes from nuclear pores. Cell. 2011;146:233–246. doi: 10.1016/j.cell.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeing S., Williamson L., Encheva V., Gori I., Saunders R.E., Instrell R., Aygün O., Rodriguez-Martinez M., Weems J.C., Kelly G.P. Multiomic Analysis of the UV-Induced DNA Damage Response. Cell Rep. 2016;15:1597–1610. doi: 10.1016/j.celrep.2016.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler L.R., Densham R.M., Jia J., Garvin A.J., Stone H.R., Shah V., Weekes D., Festy F., Beesley J., Morris J.R. The proteasomal de-ubiquitinating enzyme POH1 promotes the double-strand DNA break response. EMBO J. 2012;31:3918–3934. doi: 10.1038/emboj.2012.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrero D., Soria-Valles C., López-Otín C. Hallmarks of progeroid syndromes: lessons from mice and reprogrammed cells. Dis. Model. Mech. 2016;9:719–735. doi: 10.1242/dmm.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanut P., Britton S., Coates J., Jackson S.P., Calsou P. Coordinated nuclease activities counteract Ku at single-ended DNA double-strand breaks. Nat. Commun. 2016;7:12889. doi: 10.1038/ncomms12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman K.A., Greenberg R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D. Proteomic Analyses of the Eukaryotic Replication Machinery. In: Eichman Brandt F., editor. Methods in Enzymology. Academic Press; 2017. pp. 33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D. Replication-Coupled DNA Repair. Mol. Cell. 2019;74:866–876. doi: 10.1016/j.molcel.2019.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Du Z., Niu S., Xu X., Xu Q. MicroRNA31-NDRG3 regulation axes are essential for hepatocellular carcinoma survival and drug resistance. Cancer Biomark. 2017;19:221–230. doi: 10.3233/CBM-170568. [DOI] [PubMed] [Google Scholar]
- Dungrawala H., Rose K.L., Bhat K.P., Mohni K.N., Glick G.G., Couch F.B., Cortez D. The Replication Checkpoint Prevents Two Types of Fork Collapse without Regulating Replisome Stability. Mol. Cell. 2015;59:998–1010. doi: 10.1016/j.molcel.2015.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L., Choudhary S., Zhao Y., Edeh C.B., Yang C., Boldogh I., Brasier A.R. ATM regulates NF-κB-dependent immediate-early genes via RelA Ser 276 phosphorylation coupled to CDK9 promoter recruitment. Nucleic Acids Res. 2014;42:8416–8432. doi: 10.1093/nar/gku529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller C., Forné I., Imhof A., Becker P.B. Global and specific responses of the histone acetylome to systematic perturbation. Mol. Cell. 2015;57:559–571. doi: 10.1016/j.molcel.2014.12.008. [DOI] [PubMed] [Google Scholar]
- Gilbert L.A., Horlbeck M.A., Adamson B., Villalta J.E., Chen Y., Whitehead E.H., Guimaraes C., Panning B., Ploegh H.L., Bassik M.C. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haahr P., Hoffmann S., Tollenaere M.A., Ho T., Toledo L.I., Mann M., Bekker-Jensen S., Räschle M., Mailand N. Activation of the ATR kinase by the RPA-binding protein ETAA1. Nat. Cell Biol. 2016;18:1196–1207. doi: 10.1038/ncb3422. [DOI] [PubMed] [Google Scholar]
- Hammond C.M., Strømme C.B., Huang H., Patel D.J., Groth A. Histone chaperone networks shaping chromatin function. Nat. Rev. Mol. Cell Biol. 2017;18:141–158. doi: 10.1038/nrm.2016.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauer M.H., Seeber A., Singh V., Thierry R., Sack R., Amitai A., Kryzhanovska M., Eglinger J., Holcman D., Owen-Hughes T., Gasser S.M. Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat. Struct. Mol. Biol. 2017;24:99–107. doi: 10.1038/nsmb.3347. [DOI] [PubMed] [Google Scholar]
- Hsiang Y.H., Lihou M.G., Liu L.F. Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P., Jackson S.P. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009;284:9558–9565. doi: 10.1074/jbc.M808906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N., Durocher D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2016;19:1–9. doi: 10.1038/ncb3452. [DOI] [PubMed] [Google Scholar]
- Ishihama Y., Rappsilber J., Mann M. Modular stop and go extraction tips with stacked disks for parallel and multidimensional Peptide fractionation in proteomics. J. Proteome Res. 2006;5:988–994. doi: 10.1021/pr050385q. [DOI] [PubMed] [Google Scholar]
- Jachimowicz R.D., Beleggia F., Isensee J., Velpula B.B., Goergens J., Bustos M.A., Doll M.A., Shenoy A., Checa-Rodriguez C., Wiederstein J.L. UBQLN4 Represses Homologous Recombination and Is Overexpressed in Aggressive Tumors. Cell. 2019;176:505–519.e22. doi: 10.1016/j.cell.2018.11.024. [DOI] [PubMed] [Google Scholar]
- Jasencakova Z., Scharf A.N.D., Ask K., Corpet A., Imhof A., Almouzni G., Groth A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell. 2010;37:736–743. doi: 10.1016/j.molcel.2010.01.033. [DOI] [PubMed] [Google Scholar]
- Ju J., Naura A.S., Errami Y., Zerfaoui M., Kim H., Kim J.G., Abd Elmageed Z.Y., Abdel-Mageed A.B., Giardina C., Beg A.A. Phosphorylation of p50 NF-kappaB at a single serine residue by DNA-dependent protein kinase is critical for VCAM-1 expression upon TNF treatment. J. Biol. Chem. 2010;285:41152–41160. doi: 10.1074/jbc.M110.158352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarougkas A., Ismail A., Katsuki Y., Freire R., Shibata A., Jeggo P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013;41:10298–10311. doi: 10.1093/nar/gkt802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Chen J., Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Kolas N.K., Chapman J.R., Nakada S., Ylanko J., Chahwan R., Sweeney F.D., Panier S., Mendez M., Wildenhain J., Thomson T.M. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637–1640. doi: 10.1126/science.1150034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustatscher G., Hégarat N., Wills K.L.H., Furlan C., Bukowski-Wills J.-C., Hochegger H., Rappsilber J. Proteomics of a fuzzy organelle: interphase chromatin. EMBO J. 2014;33:648–664. doi: 10.1002/embj.201387614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecona E., Fernández-Capetillo O. Replication stress and cancer: it takes two to tango. Exp. Cell Res. 2014;329:26–34. doi: 10.1016/j.yexcr.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.Y., Chun Y.S., Shin H.W., Park J.W. Potential role of the N-MYC downstream-regulated gene family in reprogramming cancer metabolism under hypoxia. Oncotarget. 2016;7:57442–57451. doi: 10.18632/oncotarget.10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Sun Q., Wang X. PLK1, A potential target for cancer therapy. Transl. Oncol. 2017;10:22–32. doi: 10.1016/j.tranon.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheret M., Halazonetis T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R., 3rd, Hurov K.E., Luo J., Bakalarski C.E., Zhao Z., Solimini N., Lerenthal Y. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- Mejlvang J., Feng Y., Alabert C., Neelsen K.J., Jasencakova Z., Zhao X., Lees M., Sandelin A., Pasero P., Lopes M., Groth A. New histone supply regulates replication fork speed and PCNA unloading. J. Cell Biol. 2014;204:29–43. doi: 10.1083/jcb.201305017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee C., Tripathi V., Manolika E.M., Heijink A.M., Ricci G., Merzouk S., de Boer H.R., Demmers J., van Vugt M.A.T.M., Ray Chaudhuri A. RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat. Commun. 2019;10:3287. doi: 10.1038/s41467-019-11246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K., Kato A., Kobayashi J., Yanagihara H., Sakamoto S., Oliveira D.V.N.P., Shimada M., Tauchi H., Suzuki H., Tashiro S. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell. 2011;41:515–528. doi: 10.1016/j.molcel.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Nakamura K., Saredi G., Becker J.R., Foster B.M., Nguyen N.V., Beyer T.E., Cesa L.C., Faull P.A., Lukauskas S., Frimurer T. H4K20me0 recognition by BRCA1-BARD1 directs homologous recombination to sister chromatids. Nat. Cell Biol. 2019;21:311–318. doi: 10.1038/s41556-019-0282-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen K.J., Lopes M. Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat. Rev. Mol. Cell Biol. 2015;16:207–220. doi: 10.1038/nrm3935. [DOI] [PubMed] [Google Scholar]
- Nowsheen S., Aziz K., Aziz A., Deng M., Qin B., Luo K., Jeganathan K.B., Zhang H., Liu T., Yu J. L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nat. Cell Biol. 2018;20:455–464. doi: 10.1038/s41556-018-0071-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J.V., de Godoy L.M.F., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- Ong S.-E., Blagoev B., Kratchmarova I., Kristensen D.B., Steen H., Pandey A., Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Woodcock M., Helleday T. Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA. 2010;107:16090–16095. doi: 10.1073/pnas.1005031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A.J., Johnson R.D., Thompson L.H., Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J., Ishihama Y., Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003;75:663–670. doi: 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Räschle M., Smeenk G., Hansen R.K., Temu T., Oka Y., Hein M.Y., Nagaraj N., Long D.T., Walter J.C., Hofmann K. DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science. 2015;348:1253671. doi: 10.1126/science.1253671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K.J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., Lopes M. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 2012;19:417–423. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- Ray-Gallet D., Woolfe A., Vassias I., Pellentz C., Lacoste N., Puri A., Schultz D.C., Pchelintsev N.A., Adams P.D., Jansen L.E.T., Almouzni G. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell. 2011;44:928–941. doi: 10.1016/j.molcel.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Ribeyre C., Zellweger R., Chauvin M., Bec N., Larroque C., Lopes M., Constantinou A. Nascent DNA Proteomics Reveals a Chromatin Remodeler Required for Topoisomerase I Loading at Replication Forks. Cell Rep. 2016;15:300–309. doi: 10.1016/j.celrep.2016.03.027. [DOI] [PubMed] [Google Scholar]
- Sabatel H., Di Valentin E., Gloire G., Dequiedt F., Piette J., Habraken Y. Phosphorylation of p65(RelA) on Ser(547) by ATM represses NF-κB-dependent transcription of specific genes after genotoxic stress. PLoS ONE. 2012;7:e38246. doi: 10.1371/journal.pone.0038246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saredi G., Huang H., Hammond C.M., Alabert C., Bekker-Jensen S., Forne I., Reverón-Gómez N., Foster B.M., Mlejnkova L., Bartke T. H4K20me0 marks post-replicative chromatin and recruits the TONSL–MMS22L DNA repair complex. Nature. 2016;534:714–718. doi: 10.1038/nature18312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori A.A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., Jackson S.P. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiaputra D., Durocher D. Shieldin - the protector of DNA ends. EMBO Rep. 2019;20:e47560. doi: 10.15252/embr.201847560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y. ATM: expanding roles as a chief guardian of genome stability. Exp. Cell Res. 2014;329:154–161. doi: 10.1016/j.yexcr.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Sirbu B.M., Couch F.B., Feigerle J.T., Bhaskara S., Hiebert S.W., Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev. 2011;25:1320–1327. doi: 10.1101/gad.2053211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.J., Makinson T.A., Watson J.V. Enhanced sensitivity to camptothecin in ataxia-telangiectasia cells and its relationship with the expression of DNA topoisomerase I. Int. J. Radiat. Biol. 1989;55:217–231. doi: 10.1080/09553008914550271. [DOI] [PubMed] [Google Scholar]
- Sobhian B., Shao G., Lilli D.R., Culhane A.C., Moreau L.A., Xia B., Livingston D.M., Greenberg R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes M.P., Rush J., Macneill J., Ren J.M., Sprott K., Nardone J., Yang V., Beausoleil S.A., Gygi S.P., Livingstone M. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA. 2007;104:19855–19860. doi: 10.1073/pnas.0707579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa M., Shinohara A., Shinohara M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 2014;10:e1004563. doi: 10.1371/journal.pgen.1004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A., Pommier Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019;25:6581–6589. doi: 10.1158/1078-0432.CCR-19-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorslund T., Ripplinger A., Hoffmann S., Wild T., Uckelmann M., Villumsen B., Narita T., Sixma T.K., Choudhary C., Bekker-Jensen S., Mailand N. Histone H1 couples initiation and amplification of ubiquitin signalling after DNA damage. Nature. 2015;527:389–393. doi: 10.1038/nature15401. [DOI] [PubMed] [Google Scholar]
- Tuduri S., Crabbé L., Conti C., Tourrière H., Holtgreve-Grez H., Jauch A., Pantesco V., De Vos J., Thomas A., Theillet C. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009;11:1315–1324. doi: 10.1038/ncb1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Uckelmann M., Sixma T.K. Histone ubiquitination in the DNA damage response. DNA Repair (Amst.) 2017;56:92–101. doi: 10.1016/j.dnarep.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Völker-Albert M.C., Schmidt A., Forne I., Imhof A. Analysis of Histone Modifications by Mass Spectrometry. Curr. Protoc. Protein Sci. 2018;92:e54. doi: 10.1002/cpps.54. [DOI] [PubMed] [Google Scholar]
- Völker-Albert M.C., Schmidt A., Barth T.K., Forne I., Imhof A. Detection of Histone Modification Dynamics during the Cell Cycle by MS-Based Proteomics. In: Guillermo A.Orsi, Geneviève Almouzni., editors. Methods in Molecular Biology. Humana Press; 2018. pp. 61–74. [DOI] [PubMed] [Google Scholar]
- Wang B., Matsuoka S., Ballif B.A., Zhang D., Smogorzewska A., Gygi S.P., Elledge S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.H., Lahusen T.J., Chen Q., Xu X., Jenkins L.M., Leo E., Fu H., Aladjem M., Pommier Y., Appella E., Deng C.X. SIRT1 deacetylates TopBP1 and modulates intra-S-phase checkpoint and DNA replication origin firing. Int. J. Biol. Sci. 2014;10:1193–1202. doi: 10.7150/ijbs.11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward A.M., Göhler T., Luciani M.G., Oehlmann M., Ge X., Gartner A., Jackson D.A., Blow J.J. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J. Cell Biol. 2006;173:673–683. doi: 10.1083/jcb.200602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeeles J.T.P., Janska A., Early A., Diffley J.F.X. How the Eukaryotic Replisome Achieves Rapid and Efficient DNA Replication. Mol. Cell. 2017;65:105–116. doi: 10.1016/j.molcel.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J., Shen J., Qiao X., Cao L., Yang Z., Ye H., Xi C., Zhou Q., Wang P., Gong Z. SNHG20/miR-140-5p/NDRG3 axis contributes to 5-fluorouracil resistance in gastric cancer. Oncol. Lett. 2019;18:1337–1343. doi: 10.3892/ol.2019.10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeman M.K., Cimprich K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Tang R., Huang Y., Wang W., Zhou Z., Gu S., Dai J., Ying K., Xie Y., Mao Y. Cloning and expression pattern of the human NDRG3 gene. Biochim. Biophys. Acta. 2001;1519:134–138. doi: 10.1016/s0167-4781(01)00210-x. [DOI] [PubMed] [Google Scholar]
- Zuco V., De Cesare M., Zaffaroni N., Lanzi C., Cassinelli G. PLK1 is a critical determinant of tumor cell sensitivity to CPT11 and its inhibition enhances the drug antitumor efficacy in squamous cell carcinoma models sensitive and resistant to camptothecins. Oncotarget. 2015;6:8736–8749. doi: 10.18632/oncotarget.3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019). The accession number for the histone PTM analysis reported in this paper is PRIDE: PXD018220 and NCC-SILAC-MS is PRIDE: PXD018092.
Original image data have been deposited to Mendeley Data: