

ABSTRACT
The primary cilium and the immunological synapse are both specialized functional plasma membrane domains that share several similarities. Signalling output of membrane domains is regulated, spatially and temporally, by segregating and focusing lipids and proteins. ARL3, a small GTPase, plays a major role in concentrating lipid-modified proteins in both the immunological synapse and the primary cilia. Here in this review we will introduce the role of ARL3 in health and disease and its role in polarizing signalling at the primary cilia and immunological synapses.
KEYWORDS: Primary cilia, immune synapse, ARL3, signalling, cargo
Introduction
The role of primary cilia in health and disease continues to rapidly expand. The finding of new players in ciliogenesis, cilia structure and cilia function has led to an increased understanding of the function of this organelle in development, tissue maintenance and disease. Despite being seen as a mere protrusion from the plasma membrane, the ciliary composition, including both the ciliary membrane and its inside, is distinct from the rest of the cell. Integral membrane, membrane-associated and soluble proteins are among the molecules that can be selectively enriched in the cilia. For integral membrane proteins a combination of diffusion barriers and other mechanisms including selective targeting, exclusion and retention have been proposed (reviewed in [1–3]). For soluble proteins a size-dependent diffusion barrier in the cilium has been proposed which shows parallels to the nuclear pore complex (NPC) or the base of dendritic spines [4].
Prenylated and myristoylated proteins can, in most cases, switch from membrane-bound to soluble forms, either by binding to chaperones, which sequester their lipid groups, or via conformation changes which result in the retraction of lipid groups. Recently, it has been proposed that the small GTPases ARL3 and ARL13B play a role in modulating the solubility of prenylated and myristoylated proteins and thus their access to cilia [5–7]. The importance of the ARL3 in cilia is underscored by the ciliary phenotypes associated with ARL3 mutations, including non-syndromic dominant retinitis pigmentosa [8,9] and Joubert syndrome (JBTS) a retinal-cerebello-renal multisystem ciliopathy syndrome [10].
Lymphocytes are among the few cell types that do not possess primary cilia; nevertheless they form structures called immune synapses, which share similarities with primary cilia. Both are specialized functional membrane domains that maintain protein and lipid segregation (reviewed in [11,12]). Recently, it was shown that ARL3 plays a critical role in the concentration of immune signalling molecules at this location [13].
Primary cilia
Primary cilia are solitary, highly conserved organelles and can be found on almost all mammalian cell types [14]. These 1–10 μm antenna-like structures project from the basal body of the cell where their role is to detect extracellular signals such as mechanical flow and chemical stimulation, and to mediate signalling of growth factors and morphogens; which they then transduce into the cell [15,16]. Examples of the signalling pathways mediated by the cilium are platelet derived growth factor (PDGF), Hedgehog (Hh), Wnt and planar cell polarity (PCP) [17–19]; which are essential during development, establishing the left-right axis, and maintaining cellular homoeostasis [7,20–22] (Figure 1). Tucker et al. showed that primary cilia were assembled in vitro when cells were serum starved, as this caused the cells to exit the cell cycle at the G0 phase [23]. The cilium is also disassembled and resorbed in vitro when cells are stimulated with serum. The deciliation of cells corresponds to the G0/G1 transition, suggesting that primary cilia cross-talk with the cell cycle.
Figure 1.
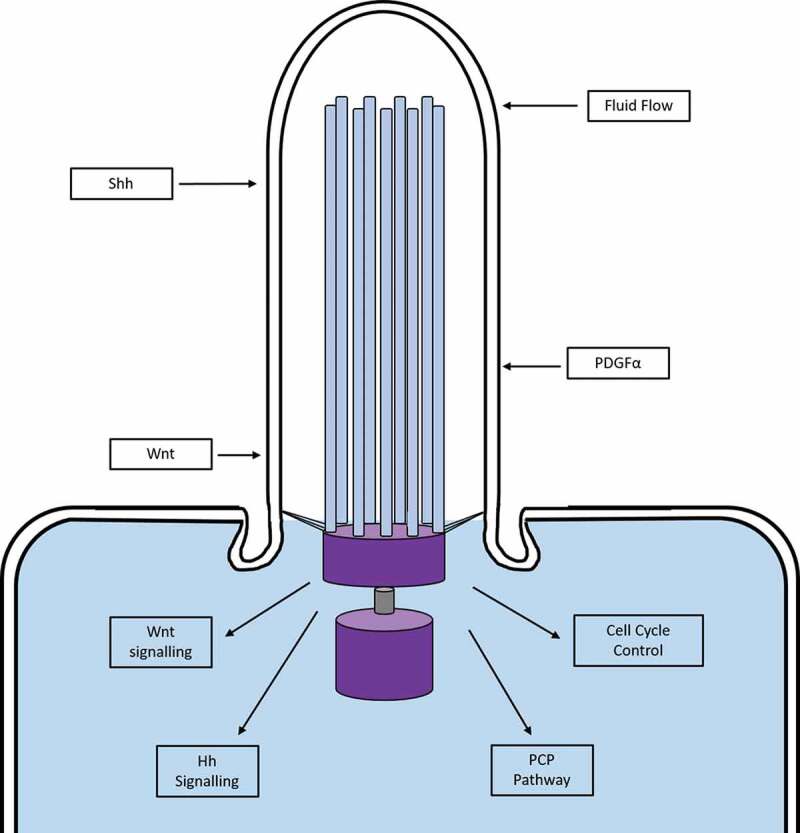
Signalling pathways in the primary cilium. Schematic diagram demonstrating some of the signalling pathways transduced by the primary cilium. Mechanical and chemical extracellular signals are sensed by proteins in the membrane of the cilium, which are then transduced into the cell to activate a variety of intracellular pathways. Figure adapted from [32]. Hh, Hedgehog; PCP, planar cell polarity; PDGFα, Platelet Derived Growth Factor Subunit alpha; Shh, Sonic Hedgehog
Primary cilia have been adapted by cells in the retina; the outer segment of retinal photoreceptors are specialized sensory primary cilia for light detection, and are the largest of mammalian cilia [24–27]. In these cilia, the elongated transition zone is known as the connecting cilium through which phototransduction proteins (rhodopsin and iodopsin) are transported to the outer segment [26].
Primary cilia also act as mechanosensors in many tissues throughout the body. In the kidney, primary cilia are present on the apical surface of epithelial cells in the tubule which extend into the lumen [28]. Regulated fluid flow through the tubules and collecting ducts is sensed by kidney epithelial cell primary cilia [29,30]. This is achieved by bending of the cilium due to increased fluid flow which is postulated to increase intracellular Ca2+, mediated by the polycystin-1/polycystin-2 (PC1/2) ion channel complex [30]. PC1 is a membrane protein in the cilium that is a regulator of the PC2 channel; disruption of this interaction, or correct cilia formation, causes autosomal dominant polycystic kidney disease [31].
Ciliopathies
Diseases and syndromes that are caused by dysfunction of cilia are termed ciliopathies. There are sets of diseases secondary to dysfunction of motile cilia (known confusingly as primary ciliary dyskinesias) as well as disorders of the primary cilia which include tissue specific disorders such as retinitis pigmentosa and multisystem disorders such as ciliary chondrodysplasias (e.g. short rib-polydactyly syndromes), neurodevelopmental disorders (e.g. Meckel Syndrome (MKS) and Joubert syndrome (JBTS)), obesity syndromes (e.g. Bardet-Biedl syndrome (BBS)) and renal cystic disorders (e.g. nephronophthisis (NPHP)) [33]. Mutations in genes that are involved in primary ciliary formation can have different effects on cilium length and associated dysfunction. For instance, gene mutations encoding IFT-B proteins cause a loss of cilia formation due to defective trafficking of key cilia-building components into the cilium, resulting in short, non-functioning primary cilia [34]. Whereas mutations in genes encoding IFT-A proteins cause accumulation of distal proteins at the ciliary tip, forming bulbous primary cilia [35,36].
As primary cilia are involved in many fundamental signalling pathways, disturbances to the function of the cilium leads to ciliopathies which can affect multiple organ systems in varying degrees of severity, causing phenotypic overlap between diseases and syndromes [37]. Primary ciliopathies are often characterized by overlapping phenotypes involving the brain, eye and kidney. Renal and hepatic disease has been documented in Senior-Loken syndrome (SLS), NPHP, MKS, BBS and JBTS; retinopathies are seen with SLS, NPHP, MKS, BBS and JBTS, and cerebellar malformations are associated with JBTS. Other phenotypes such as polydactyly, obesity and mental retardation can also be associated with some of these ciliopathies [38]. As a result of these closely related and overlapping phenotypes, diagnosing a ciliopathy is complex and can lead to misdiagnosis. However, the overlap of phenotypes also suggests that there are closely related mechanisms of pathogenicity, that may be caused by differences in gene and genetic background. Therefore, investigations into mechanistic action of each of the known ciliopathy-causing genes are essential to fully understand how each syndrome is caused.
Joubert syndrome (JBTS)
JBTS is an archetypal primary ciliopathy syndrome. There are at least 35 genes (https://www.omim.org/phenotypicSeries/PS213300) that cause JBTS and new genetic causes of JBTS continue to be described [39]. JBTS occurs in 1 in every 100,000 live births, however this could be an underestimation due to the heterogeneity of the disease and the overlap of phenotypes with other ciliopathies including NPHP and SLS [40,41]. Marie Joubert first described the typical phenotype of patients with JBTS in 1968 [42]. The hallmark of a JBTS patient is a molar tooth sign on brain MRI scanning, which can be evident from 14–16 weeks gestation. This is caused by a set of mid and hindbrain malformations including cerebellar vermis hypoplasia, abnormal superior cerebellar peduncles, and an unusually deep interpeduncular fossa [43]. Cerebellar vermis hypoplasia in JBTS is thought to be a result of disturbed proliferation and migration of germinal zone granular cell precursors [41]. The symptoms associated with the syndrome include dysmorphic features (prominent forehead, upturned nose, and open mouth) and neurological symptoms (hypotonia, ataxia, developmental delay, intellectual disability, and abnormal ocular movements). During the neonatal period, patients can have an altered respiratory pattern with episodes of apnoea and hyperpnoea. The features described above are typical of ‘pure’ JBTS however, patients that have pathognomonic and neuroradiological features similar to JBTS but also various organ dysfunction (e.g. brain, eye, kidney) are described as having a Joubert syndrome related disorder (JSRD) [44]. Renal disorders affect ~25% of patients with JSRD. This often presents as tubulointerstitial disease with irregular, thickened tubular basement membranes, progressive interstitial fibrosis, and small cysts at the corticomedullary junction. The renal component of JSRD manifests as a juvenile-onset cystic kidney disease, nephronophthisis, progressing to end-stage kidney disease [45]. What is fascinating is that the growing number of genetic causes of JBTS are revealing new mechanistic insights into how ciliary dysfunction causes human disease. As well as the genetic heterogenity, there is a huge range of clinical phenotypes which emphasizes the developmental and tissue maintenance roles of primary cilia.
Ciliary ARL3
Arf like (ARL) proteins constitute a large subfamily of small GTPases, over 20 members, nevertheless little is known about their regulation and functions. Mutations in ARL3, ARL6, ARL13B and ARL2BP have been reported in primary ciliopathies including JBTS [10,46] and BBS [47] indicating that the involvement of ARF-like proteins in cilia can be a general feature of several ARLs. An open question is what makes ARF-like proteins ciliary related. One possible answer might lie in the N-terminus end of ciliary ARLs. All Arf proteins possess a myristoyl group in addition to an amphipathic helix to associate with membranes in a GTP-dependent manner. ARL2, ARL3 and ARL6 are not myristoylated whereas ARL2 and ARL3 retain the amphipathic helix, ARL6 seems to have a hydrophobic helix [48]. ARL13B has a palmitoylation motif instead which plays a critical role in its localization to the cilia. These differences in the N-terminus might dictate how these ARLs gain access to the cilia [49].
ARL3 is highly conserved in ciliated organisms, and has been shown to localize throughout the cell and is enriched in the primary cilium [50–52]. ARL3 interacts and regulates a family of proteins called GDI-like solubilizing factors (GSFs). Structural analysis showed that GSFs, such as PDE6D, UNC119a and its paralog UNC119b, possess a hydrophobic pocket that can accommodate prenyl or myristol groups respectively [6,53,54]. Indeed there are several reports showing the involvement of GSFs in transport of lipid modified proteins [55]. It was shown in cells that UNC119b is required for NPHP3 targeting to the cilia [5]. Deletion of UNC119a resulted in mislocalisation of transducin alpha subunit in C. elegans [53]. PDE6D plays a role in targeting GRK1 and PDE6 to the outer segments of photoreceptors in mice [56].
Based on biochemical and structural data, ARL3 has been shown to allosterically regulate the interaction of GSFs with lipid-modified proteins [5,6,57]. A model has been put forward where ARL3 releases lipid modified proteins into the cilia (Figure 2). Recently, the Wittinghofer group have shown that ARL13b functions, in vitro, as a GEF for ARL3 which would create an active node of ARL3GTP within cilia [7]. This mechanism shares similarity to the small GTPase RAN (RAs-related Nuclear protein) regulated nucleo-cytoplasmic transport. The nuclear RAN GTP destabilizes the interaction of import receptors to their cargo and stabilizes the interaction of export receptors to cargo [58]. The RAN GEF RCC1 is localized in the nucleus whereas the RAN GAP localizes in the cytoplasm which ensure the directionality of the transport [59].
Figure 2.
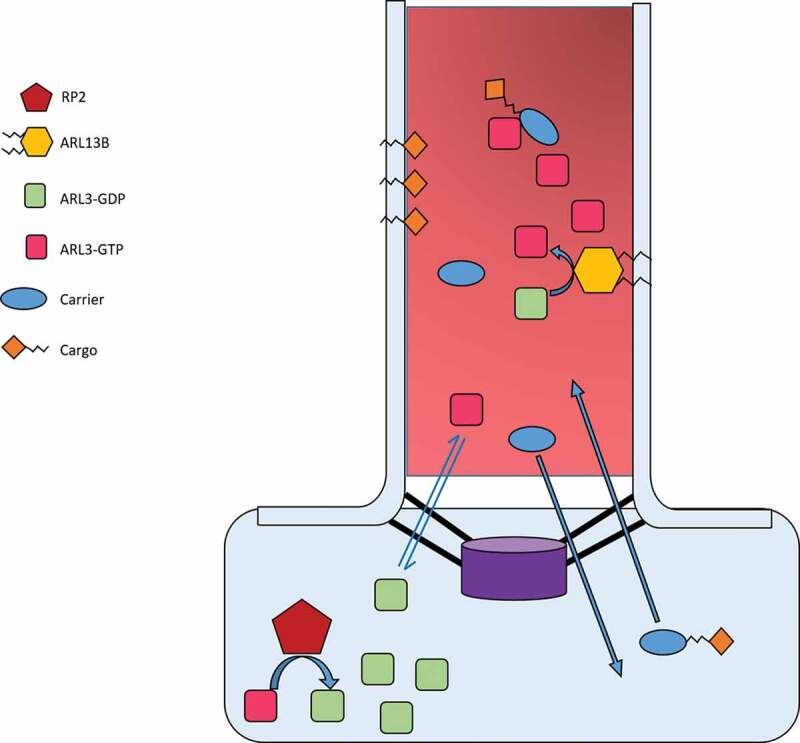
Proposed interaction and function of ARL3 in the cilium
ARL3-GDP has a high concentration in the cell whereas ARL3-GTP has a high concentration in the cilium; this is thought to be achieved by the localization of ARL13B in the cilium, and RP2 outside the cilium. This would create a concentration gradient for movement of ARL3-GDP/GTP in and out of the cilium to maintain homoeostasis. In the cilium, ARL13B hydrolyses GDP-bound ARL3 into active ARL3-GTP. Trafficking of cargo and its carrier into the cilium allows interaction with ARL3-GTP which causes release of the cargo from the carrier. The carrier then passes back into the cell to bind other cargo, along its concentration gradient. ARL3-GTP also exits the cilium down its concentration gradient into the cell where it interacts with RP2 and is converted into ARL3-GDP.
The ARL3 GAP, XRP2 (also known as RP2) has been reported to localize in the preciliary region [60], which supports the proposed model, nevertheless other studies have shown XRP2 to localize inside the cilia [61,62]. It would be interesting to study the interplay between the XRP2 localization and the ciliary lipid modified cargo localization.
ARL3 dysfunction
In murine models, global knockout of Arl3 is either embryonically lethal or early postnatally lethal, with pups only surviving ~21 days. Arl3 −/- mice developed multi-organ phenotypes including cystic kidney disease and photoreceptor degeneration [63].
To further understand the role of ARL3 in photoreceptors, rod-specific and retina-specific Arl3−/- mouse models were used [64]. The rod-specific knockout mice had Cre expression post-ciliogenesis which allows the outer segment of the photoreceptor to form; whereas the retina-specific knockout mice had Cre expression during embryogenesis, preventing ciliogenesis. The results showed that trafficking of lipidated proteins in the rod-specific knockout mice was impaired in the retina. Specifically, prenylated proteins mistrafficked to the inner segment, outer nuclear layer and synaptic terminals; causing an accumulation of proteins in these areas, outer segment shortening, and retinal degeneration. In the retina-specific knockout mice, photoreceptors failed to form connecting cilia or an outer segment membrane which caused protein accumulation in the inner segment, and rapid retinal degeneration. Hanke-Gogokhia et al were able to rescue the retinal phenotype by harvesting degenerated retinas and injecting ARL3-EGFP AAV particles into the sub-retinal space [64]. Patients with non-syndromic autosomal dominant retinitis pigmentosa have recently been found to have a heterozygous missense mutation p.(Tyr90Cys) in ARL3 [9] confirming earlier reports of this missense mutation causing retinal disease [8]. The missense change is predicted, using in silico models to disrupt protein folding and function.
For detailed studies of ciliary protein transport, hTERT-RPE cells were used to investigate the role of ARL3 and XRP2 in the trafficking of ciliary tip kinesin KIF17. By performing knockdowns of either XRP2 or ARL3, they found significantly reduced levels of KIF7 and KIF17 at ciliary tips, suggesting that correct ARL3 transport is essential for trafficking of KIF7 and KIF17 [65]. These findings are important as KIF7 is known to regulate the dynamics of microtubule plus ends and thereby control the structure and organization of primary cilia tips [66]. Protein interaction assays showed that KIF17 formed a complex with both ARL3-GDP and XRP2 but not a heterocomplex with either protein, suggesting that KIF17 and KIF7 cilia trafficking could be mediated by interaction with XRP2 and ARL3 complexes [65]. The knockdown of KIF7 in hTERT-RPE GFP-KIF17 expressing cells showed a decrease in the level of KIF17 at the cilia tip [65]. Together these findings suggest that the ARL3/XRP2 trafficking system is employed in the regulation of ciliary tip kinesins KIF7 and KIF17 [65].
Beyond the retina, genome wide association studies have linked the loci of ARL3 (10q24.32) to diseases such as autism, schizophrenia, scoliosis and high blood pressure [67–69], highlighting the potential breadth of physiological functions ARL3 may be involved in. These studies suggest a role for ARL3 in gene regulation. Indeed, ARL3 was identified as a STAT3–binding partner in vivo. Activated STAT3 translocates to the nucleus and binds to specific promoter sequences to regulate transcription of various genes that are essential for cell proliferation, survival and migration [70–73]. STAT3 has also been reported to be essential in embryonic development, and in maintaining stem cell pluripotency [74–76]. Togi et al found that ARL3 binds to the C-terminal region of STAT3, yet specific knock-down of ARL3 via siRNA had no effect on STAT3 protein levels. However, they reported that STAT3-mediated transactivation, after stimulation with IL-6, was reduced; suggesting that ARL3 regulates transactivation of STAT3. To further validate this, ARL3 knockdown was also performed in HeLa cells. Knockdown cells were found to have a reduced capacity for cell migration (a process STAT3 has previously been shown to be essential for) suggesting that ARL3 mediates cell migration through regulation of STAT3 transactivation [77].
The most compelling data for a role of ARL3 in multisystem disease was recently reported where autozygosity mapping and whole-exome sequencing identified ARL3 missense mutations in two JBTS-affected families [10]. The first family consisted of Saudi Arabian first-cousin parents with 6 children, 1 of which was affected (II-1). II-1 was a 5 year old male with developmental delay, multicystic dysplastic left kidney, night blindness and mild dysmorphic features. MRI of the brain showed severe vermis hypoplasia and abnormally thick cerebellar peduncles, causing the classical molar tooth sign. The second family was consanguineous and originated from Pakistan. Of the 6 children, 3 were affected with JBTS. The eldest (II-1) had hypotonia and psychomotor delay, as well as night blindness and bilateral vision loss by 4 years of age. MRI revealed the molar tooth sign and retinal dystrophy. The other 2 affected siblings (II-4 and II-5) had similar presentations of predominately brain and retinal features. II-1 and II-5 also had renal presentations of recurrent urinary tract infections, bilateral renal scarring and unequal kidneys. Remarkably, the mutation of both families occurred at the same residue (Arg149) yet code for different missense mutations. The first family had a c.445C>T, p.(Arg149Cys) homozygous missense mutation and the second family had a c.446G>A (p.Arg149His) homozygous missense mutation. The Arg149 residue is highly conserved in ARL3 throughout evolution and both variants are located in a loop between the α4 and β6 domains. Alkanderi et al predicted that the mutation disrupts the interaction of Arl3 with Arl13b at the conserved Glu88 residue (where Arl3 ionically bonds) (Figure 3). This hypothesis was tested by GEF fluorescence-based polarization experiments where Arl13b failed to accelerate the nucleotide exchange of mutant p.(Arg149His) murine Arl3 bound to fluorescently labelled GDP in the presence of excess GTP. The morphology of the primary cilia from the second JBTS family with ARL3 mutation p.(Arg149His) was assessed by harvesting fibroblasts from the 3 affected members, as well as controls from both parents and an unaffected sibling. The primary cilia were of normal length in the affected patients, and there was no difference in ciliation rates from the controls. However, cilia in ARL3 patient cells had significantly less prenylated INPP5E and myristolyated NPHP3 suggesting an impairment in the release of this cargo by ARL3. These results were the first to implicate ARL3 mutations in patients with JBTS and show how the mutation affects the functionality of ARL3 [10]. Breslow et al has recently reported that Arl3 was among the top 40 positive regulators of Hh signalling in a CRISPR-based Hh screen and siRNA knockdown of Arl3 in cells caused a decrease in Hh signalling [78]. Nethertheless in ARL3 JBTS paitents, GLI3 was transported normally, indicating that Hh signalling was unaffected. Further investigations to uncover the requirement of ARL3 in Hh signalling within different tissues and timepoints are required. In ARL3 patient cells compensatory mechanisms are likely to be in place which may lead to different results when compared to the relatively acute knocking down or editing out of Arl3. Furthermore, the ARL3 patients studied had a missense mutation which might have been less severe than a complete null. Rescue experiments of Arl3 deleted cells using a Arl3 missense mutant gene would be very interesting to determine if Hh signalling is restored.
Figure 3.
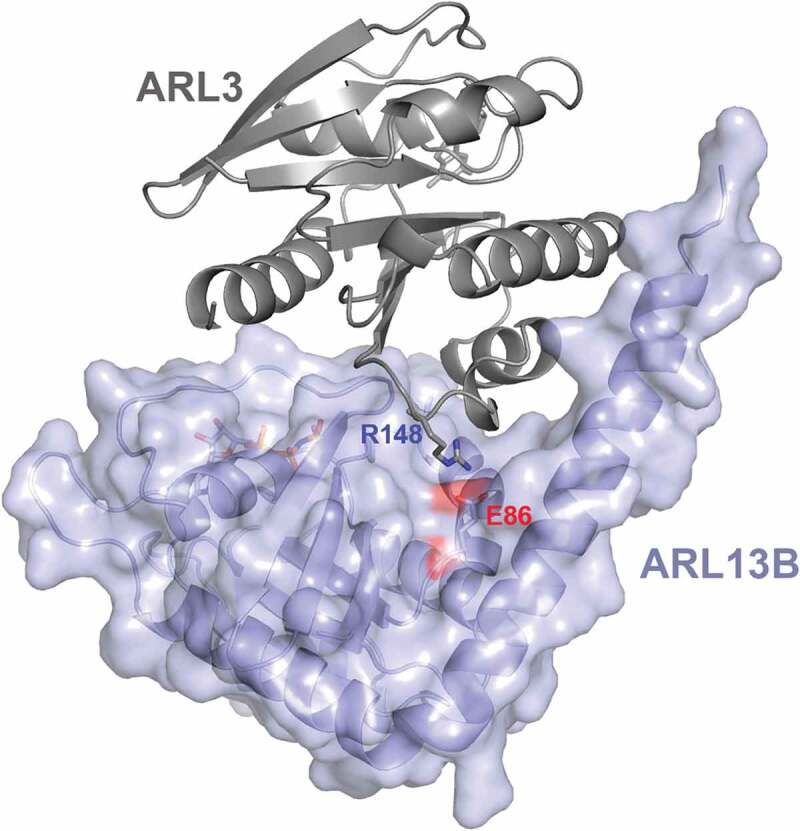
ARL3 missense mutations at a conserved site affects the interaction of ARL3 with ARL13B
Ribbon and surface representation of ARL3 protein (in grey) interacting with ARL13B protein in violet. ARL3 Arg148 and ARL13B Glu86 side chains are shown as sticks.
A role for ARL3 in the immune synapse
The interface between T cells and antigen presenting cells or target cells is called the immunological synapse. A mature canonical immune synapse is formed of three concentric rings forming a bull's eye structure. The central supramolecular activation centre (cSMAC) where the T cell receptors (TCRs) are clustered surrounded by the peripheral SMAC where the adhesion molecules lymphocyte function-associated antigen (LFA) is concentrated and finally the distal SMAC where actin is concentrated [79]. The immunological synapse is a region of active endocytosis and exocytosis where cells communicate via direct interactions and secreted particles [80,81]. The endocytosis and exocytosis has been shown to be directed by the plasma membrane docked centrosome, which shows resemblance to ciliogenesis [80]. Futhermore, components of the IFT were found to be expressed in these cells. This is intriguing as IFT proteins were thought to be exclusively required for cilia. It is now known that IFT proteins in T cells play a role in recycling TCRs to the immune synapse (reviewed in [11]). Indeed, the membrane composition of cilia and immune synapse also share striking similarities (Figure 4). Recently, the common phosphoinositides signature and how the interplay between phosphoinositide composition and actin recruitment can be a common mechanism to regulate signalling at both immune synapse and cilia has been highlighted [12,82]. This resemblance may suggest localization of similar proteins in immune synapse and cilia [80]. More research is needed here, especially in membrane associated proteins that can sense lipid composition.
Figure 4.
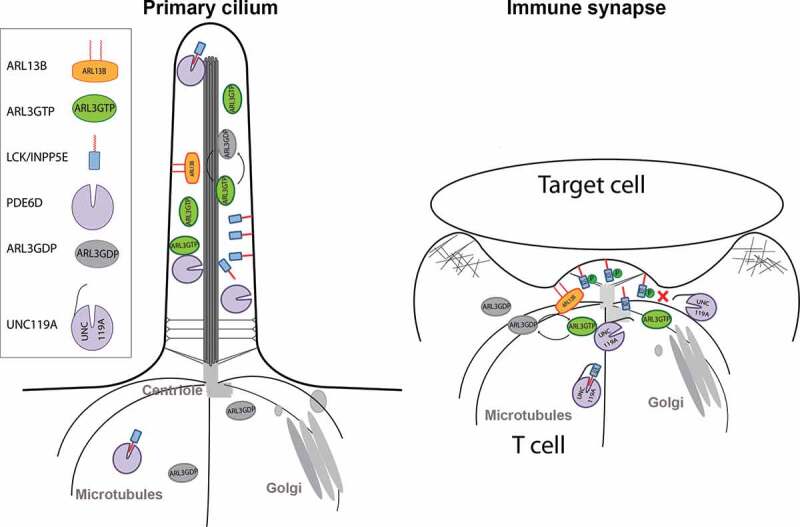
A common role for ARL3, ARL13B and UNC119A in the primary cilia and the immunological synapse
A model of transporting prenylated and myristoylated proteins to the primary cilium (on the left) and a canonical immune synapse on the right, where docking of the centriole at the plasma membrane and polarization of the Golgi are common features. ARL13B, the ARL3 GEF, is enriched in both the cilium and immune synapse, which results in a high concentration of active GTP loaded ARL3. UNC119A, UNC119B or PDE6D solubilize the lipid modified cargo by forming a complex with the cargo and sequestering the hydrophobic lipid group. The complex is then disrupted by ARL3GTP resulting in the exposure of the lipid group which then associates with the ciliary or immune synapse membranes. Phosphorylation of the LCK at tyrosine 394 interferes with the interaction with UNC119A.
In brief, both cilia and immune synapses are specialized functional membrane domains that can serve as platforms for signalling. Indeed it was shown that Hh components are polarized to the immune synapse [83] and deletion of Smoothened impaired T lymphocyte cytotoxicity [84]. This again highlights common features and functions between the cilia and the immune synapse. A precise link between ciliopathies such as JBTS, which lead to structural and developmental disorders of the brain and degenerative disorders of other tissues including the retina and kidney and the processes underlying the immune response, in particular the T cell synapse is currently not evident. Patients with ARL3 mutations identified to date do not have obvious immune deficiency phenotypes. To answer this question in more detail, ex vivo T cell studies from patients with ARL3 mutations and other related conditions will be informative. These studies may identify new mechanisms and allow sub-clinical immunity phenotypes to be revealed.
The lipid-modified, myristoylated, lymphocyte specific SRC kinase LCK plays a critical role in initiating TCR signalling. In unstimulated T cells LCK is localized on the plasma membrane and a minor fraction on the RAB11 positive endosomes [85,86]. Nevertheless, upon immune synapse formation LCK is seen enriched and focused at the immunological synapse. Recently, using structural and biochemical approaches, it was shown that the ciliary machinery, composed of UNC119, ARL3 and ARL13B, play a role in the enrichment of LCK at the immune synapse [13]. Furthermore, the authors show that UNC119 contacts the LCK kinase domain, and that this interaction is inhibited by activation phosphorylation of Tyr394. Vesicle trafficking is known to target LCK to the immune synapse; this ciliary transport mechanism might help vesicle trafficking to direct LCK to the immune synapse. Another possibility is that the UNC119 mediated transport facilitates the exit of the LCK from vesicles to the immune synapse. Temporal and functional studies will precisely define the significance of this mechanism in cells.
UNC119A, ARL13B, and ARL3 are all involved in ciliopathies. Interestingly a mutation in UNC119A has been reported in a patient with CD4 lymphopenia, who showed disruption of LCK localization [87]. Disruption of LCK localisation might be responsible for impairment of T cell activation and proliferation. Other cargoes might also be affected by UNC119 and more studies should be performed in the light of the new role of UNC119A in transporting myristoylated proteins to the immune synapse. It will be very interesting to study the ARL13B and ARL3 ciliopathy-associated mutations' effect on cellular immune response. Localization of LCK and other myristoylated immune synapse proteins might be impaired in ciliopathy patient mutations and thus may influence the function of T lymphocytes. Defining the exact mechanistic roles and molecular basis of these proteins is essential to investigate these mutations in the context of immunity and to ask the right biological questions. For example, not all ARL13B mutations impair GEF catalytic activity. These mutations might have cilia specific implications. Nevertheless, mutations affecting catalytic activity of the protein are more likely to have implications in both cilia and immunity. Another layer of complexity is the interplay between cilia function and inflammation. Indeed, it was reported that a module of ciliary proteins, which include NPHP1, regulate the expression of the chemokine CCL2 [88]. Based on this study, it was proposed that inflammation and CCL2 mediated macrophage recruitment may contribute to disease progression in ciliopathy syndromes.
Conclusions
The small GTPase ARL3 has an important role in primary cilia and ARL3mutations lead to a classical ciliopathy phenotype. A role for ARL3 in the immune synapse has also recently been established, suggesting repurposing of ARL3 for this role. It remains to be established whether ciliopathies such as JBTS will have specific immunodeficiency phenotypes. A link between immunological disorders and ciliopathies needs to be investigated further. Specific drugs targeting ARL3 and its role in the cilium and immune synapse now need to be explored as potential therapies.
Funding Statement
This work was funded by Kidney Research UK (J.A.S); Northern Counties Kidney Research Fund (J.A.S. and L.P) and Cancer Research UK [core funding award A19257 to S.I.].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Nachury MV, Seeley ES, Jin H.. Trafficking to the ciliary membrane: how to get across the periciliary diffusion barrier? Annu Rev Cell Dev Biol. 2010;26:59–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Garcia-Gonzalo FR, Reiter JF.. Scoring a backstage pass: mechanisms of ciliogenesis and ciliary access. J Cell Biol. 2012;197(6):697–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nachury MV, Mick DU. Establishing and regulating the composition of cilia for signal transduction. Nat Rev Mol Cell Biol. 2019;20(7):389–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Lin YC, Niewiadomski P, Lin B, et al. Chemically inducible diffusion trap at cilia reveals molecular sieve-like barrier. Nat Chem Biol. 2013;9(7):437–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wright KJ, Baye LM, Olivier-Mason A, et al. An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 2011;25(22):2347–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ismail SA, Chen YX, Miertzschke M, et al. Structural basis for Arl3-specific release of myristoylated ciliary cargo from UNC119. Embo J. 2012;31(20):4085–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gotthardt K, Lokaj M, Koerner C, et al. A G-protein activation cascade from Arl13B to Arl3 and implications for ciliary targeting of lipidated proteins. eLife. 2015;4:e11859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Strom SP, Clark MJ, Martinez A, et al. De novo occurrence of a variant in ARL3 and apparent autosomal dominant transmission of retinitis pigmentosa. PLoS One. 2016;11(3):e0150944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Holtan JP, Teigen K, Aukrust I, et al. Dominant ARL3-related retinitis pigmentosa. Ophthalmic Genet. 2019;40(2):124–128. [DOI] [PubMed] [Google Scholar]
- [10].Alkanderi S, Molinari E, Shaheen R, et al. ARL3 mutations cause Joubert syndrome by disrupting ciliary protein composition. Am J Hum Genet. 2018;103(4):612–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Cassioli C, Baldari CT. A ciliary view of the immunological synapse. Cells. 2019;8(8):789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gawden-Bone CM, Griffiths GM. Phospholipids: pulling back the actin curtain for granule delivery to the immune synapse. Front Immunol. 2019;10:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stephen LA, ElMaghloob Y, McIlwraith MJ, et al. The ciliary machinery is repurposed for T cell immune synapse trafficking of LCK. Dev Cell. 2018;47(1):122–32.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Berbari NF, O’Connor AK, Haycraft CJ, et al. The primary cilium as a complex signaling center. Curr Biol. 2009;19(13):R526–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Malicki JJ, Johnson CA. The cilium: cellular antenna and central processing unit. Trends Cell Biol. 2017;27(2):126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Christensen ST, Pedersen LB, Schneider L, et al. Sensory cilia and integration of signal transduction in human health and disease. Traffic (Copenhagen, Denmark). 2007;8(2):97–109. [DOI] [PubMed] [Google Scholar]
- [18].Hildebrandt F, Otto E. Cilia and centrosomes: a unifying pathogenic concept for cystic kidney disease? Nat Rev Genet. 2005;6(12):928–940. [DOI] [PubMed] [Google Scholar]
- [19].Wheway G, Parry DA, Johnson CA. The role of primary cilia in the development and disease of the retina. Organogenesis. 2014;10(1):69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Gillingham AK, Munro S. The small G proteins of the Arf family and their regulators. Annu Rev Cell Dev Biol. 2007;23:579–611. [DOI] [PubMed] [Google Scholar]
- [21].Bologna G, Yvon C, Duvaud S, et al. N-terminal myristoylation predictions by ensembles of neural networks. Proteomics. 2004;4(6):1626–1632. [DOI] [PubMed] [Google Scholar]
- [22].Pasqualato S, Renault L, Cherfils J. Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for ‘front-back’ communication. EMBO Rep. 2002;3(11):1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tucker RW, Pardee AB, Fujiwara K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell. 1979;17(3):527–535. [DOI] [PubMed] [Google Scholar]
- [24].De Robertis E. Electron microscope observations on the submicroscopic organization of the retinal rods. J Biophys Biochem Cytol. 1956;2(3):319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Allen RA. Isolated cilia in inner retinal neurons and in retinal pigment epithelium. J Ultrastruct Res. 1965;12(5):730–747. [DOI] [PubMed] [Google Scholar]
- [26].Yang J, Gao J, Adamian M, et al. The ciliary rootlet maintains long-term stability of sensory cilia. Mol Cell Biol. 2005;25(10):4129–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pan J, Wang Q, Snell WJ. Cilium-generated signaling and cilia-related disorders. Lab Invest. 2005;85(4):452–463. [DOI] [PubMed] [Google Scholar]
- [28].Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18(5):1381–1388. [DOI] [PubMed] [Google Scholar]
- [29].Leyssac PP. Changes in single nephron renin release are mediated by tubular fluid flow rate. Kidney Int. 1986;30(3):332–339. [DOI] [PubMed] [Google Scholar]
- [30].Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol. 2003;191(1):69–76. [DOI] [PubMed] [Google Scholar]
- [31].Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13(10):2508–2516. [DOI] [PubMed] [Google Scholar]
- [32].D’Angelo A, Franco B. The dynamic cilium in human diseases. PathoGenetics. 2009;2(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].May-Simera HL, Kelley MW. Cilia, Wnt signaling, and the cytoskeleton. Cilia. 2012;1(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xin D, Christopher KJ, Zeng L, et al. IFT56 regulates vertebrate developmental patterning by maintaining IFTB complex integrity and ciliary microtubule architecture. Development. 2017;144(8):1544–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liem KF Jr., Ashe A, He M, et al. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol. 2012;197(6):789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Qin J, Lin Y, Norman RX, et al. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci U S A. 2011;108(4):1456–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137(1):32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol (Berlin, Germany). 2011;26(7):1039–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Latour BL, Van De Weghe JC, Rusterholz TDS, et al. ARMC9 and TOGARAM1 define a Joubert syndrome-associated protein module that regulates axonemal post-translational modifications and cilium stability. bioRxiv. 2019;817213. [Google Scholar]
- [40].Malicdan MC, Vilboux T, Stephen J, et al. Mutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes. J Med Genet. 2015;52(12):830–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Travaglini L, Brancati F, Silhavy J, et al. Phenotypic spectrum and prevalence of INPP5E mutations in Joubert syndrome and related disorders. Eur J Human Genet. 2013;21(10):1074–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology. 1968;18(3):302–303. [PubMed] [Google Scholar]
- [43].Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009;16(3):143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet. 2008;51(1):1–23. [DOI] [PubMed] [Google Scholar]
- [45].Srivastava S, Ramsbottom SA, Molinari E, et al. A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies. Hum Mol Genet. 2017;26(23):4657–4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Humbert MC, Weihbrecht K, Searby CC, et al. ARL13B, PDE6D, and CEP164 form a functional network for INPP5E ciliary targeting. Proc Natl Acad Sci U S A. 2012;109(48):19691–19696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fan Y, Esmail MA, Ansley SJ, et al. Mutations in a member of the Ras superfamily of small GTP-binding proteins causes Bardet-Biedl syndrome. Nat Genet. 2004;36(9):989–993. [DOI] [PubMed] [Google Scholar]
- [48].Wiens CJ, Tong Y, Esmail MA, et al. Bardet-Biedl syndrome-associated small GTPase ARL6 (BBS3) functions at or near the ciliary gate and modulates Wnt signaling. J Biol Chem. 2010;285(21):16218–16230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Roy K, Jerman S, Jozsef L, et al. Palmitoylation of the ciliary GTPase ARL13b is necessary for its stability and its role in cilia formation. J Biol Chem. 2017;292(43):17703–17717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Avidor-Reiss T, Maer AM, Koundakjian E, et al. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117(4):527–539. [DOI] [PubMed] [Google Scholar]
- [51].Blacque OE, Perens EA, Boroevich KA, et al. Functional genomics of the cilium, a sensory organelle. Curr Biol. 2005;15(10):935–941. [DOI] [PubMed] [Google Scholar]
- [52].Zhou C, Cunningham L, Marcus AI, et al. Arl2 and Arl3 regulate different microtubule-dependent processes. Mol Biol Cell. 2006;17(5):2476–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhang H, Constantine R, Vorobiev S, et al. UNC119 is required for G protein trafficking in sensory neurons. Nat Neurosci. 2011;14(7):874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hanzal-Bayer M, Renault L, Roversi P, et al. The complex of Arl2-GTP and PDE delta: from structure to function. Embo J. 2002;21(9):2095–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stephen LA, Ismail S. Shuttling and sorting lipid-modified cargo into the cilia. Biochem Soc Trans. 2016;44(5):1273–1280. [DOI] [PubMed] [Google Scholar]
- [56].Zhang H, Hanke-Gogokhia C, Jiang L, et al. Mistrafficking of prenylated proteins causes retinitis pigmentosa 2. Faseb J. 2015;29(3):932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ismail SA, Chen YX, Rusinova A, et al. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat Chem Biol. 2011;7(12):942–949. [DOI] [PubMed] [Google Scholar]
- [58].Cavazza T, Vernos I. The RanGTP pathway: from nucleo-cytoplasmic transport to spindle assembly and beyond. Front Cell Dev Biol. 2015;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Yamada M, Mattaj IW, Yoneda Y. An ATP-dependent activity that releases RanGDP from NTF2. J Biol Chem. 2004;279(35):36228–36234. [DOI] [PubMed] [Google Scholar]
- [60].Evans RJ, Schwarz N, Nagel-Wolfrum K, et al. The retinitis pigmentosa protein RP2 links pericentriolar vesicle transport between the Golgi and the primary cilium. Hum Mol Genet. 2010;19(7):1358–1367. [DOI] [PubMed] [Google Scholar]
- [61].Stephan A, Vaughan S, Shaw MK, et al. An essential quality control mechanism at the eukaryotic basal body prior to intraflagellar transport. Traffic (Copenhagen, Denmark). 2007;8(10):1323–1330. [DOI] [PubMed] [Google Scholar]
- [62].Hurd TW, Fan S, Margolis BL. Localization of retinitis pigmentosa 2 to cilia is regulated by Importin beta2. J Cell Sci. 2011;124(Pt 5):718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Schrick JJ, Vogel P, Abuin A, et al. ADP-ribosylation factor-like 3 is involved in kidney and photoreceptor development. Am J Pathol. 2006;168(4):1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hanke-Gogokhia C, Wu Z, Gerstner CD, et al. Arf-like protein 3 (ARL3) regulates protein trafficking and ciliogenesis in mouse photoreceptors. J Biol Chem. 2016;291(13):7142–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Schwarz N, Lane A, Jovanovic K, et al. Arl3 and RP2 regulate the trafficking of ciliary tip kinesins. Hum Mol Genet. 2017;26(13):2480–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].He M, Subramanian R, Bangs F, et al. The kinesin-4 protein KIF7 regulates mammalian Hedgehog signalling by organizing the cilium tip compartment. Nat Cell Biol. 2014;16(7):663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhu Z, Tang NL, Xu L, et al. Genome-wide association study identifies new susceptibility loci for adolescent idiopathic scoliosis in Chinese girls. Nat Commun. 2015;6:8355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Li C, Kim YK, Dorajoo R, et al. Genome-wide association study meta-analysis of long-term average blood pressure in East Asians. Circ Cardiovasc Genet. 2017;10(2):e001527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yu H, Yan H, Li J, et al. Common variants on 2p16.1, 6p22.1 and 10q24.32 are associated with schizophrenia in Han Chinese population. Mol Psychiatry. 2017;22(7):954–960. [DOI] [PubMed] [Google Scholar]
- [70].Darnell JE Jr. STATs and gene regulation. Science (New York, NY). 1997;277(5332):1630–1635. [DOI] [PubMed] [Google Scholar]
- [71].Jove R. Preface: STAT signaling. Oncogene. 2000;19(21):2466–2467. [DOI] [PubMed] [Google Scholar]
- [72].Levy DE, Darnell JE Jr.. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3(9):651–662. [DOI] [PubMed] [Google Scholar]
- [73].Bromberg J, Darnell JE Jr.. The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19(21):2468–2473. [DOI] [PubMed] [Google Scholar]
- [74].Takeda K, Noguchi K, Shi W, et al. Targeted disruption of the mouse STAT3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94(8):3801–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19(21):2607–2611. [DOI] [PubMed] [Google Scholar]
- [76].Okita K, Yamanaka S. Intracellular signaling pathways regulating pluripotency of embryonic stem cells. Curr Stem Cell Res Ther. 2006;1(1):103–111. [DOI] [PubMed] [Google Scholar]
- [77].Togi S, Muromoto R, Hirashima K, et al. A new STAT3-binding partner, ARL3, enhances the phosphorylation and nuclear accumulation of STAT3. J Biol Chem. 2016;291(21):11161–11171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Breslow DK, Hoogendoorn S, Kopp AR, et al. A CRISPR-based screen for Hedgehog signaling provides insights into ciliary function and ciliopathies. Nat Genet. 2018;50(3):460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Dustin ML. The immunological synapse. Cancer Immunol Res. 2014;2(11):1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Griffiths GM, Tsun A, Stinchcombe JC. The immunological synapse: a focal point for endocytosis and exocytosis. J Cell Biol. 2010;189(3):399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Stinchcombe JC, Griffiths GM. Communication, the centrosome and the immunological synapse. Philos Trans R Soc London, Ser B. 2014;369(1650):20130463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Gawden-Bone CM, Frazer GL, Richard AC, et al. PIP5 kinases regulate membrane phosphoinositide and actin composition for targeted granule secretion by cytotoxic lymphocytes. Immunity. 2018;49(3):427–37.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Dustin ML. T-cells play the classics with a different spin. Mol Biol Cell. 2014;25(11):1699–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].de la Roche M, Ritter AT, Angus KL, et al. Hedgehog signaling controls T cell killing at the immunological synapse. Science (New York, NY). 2013;342(6163):1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Gorska MM, Liang Q, Karim Z, et al. Uncoordinated 119 protein controls trafficking of LCK via the RAB11 endosome and is critical for immunological synapse formation. J Iimmunol (Baltimore, Md: 1950). 2009;183(3):1675–1684. [DOI] [PubMed] [Google Scholar]
- [86].Zlatkine P, Mehul B, Magee AI. Retargeting of cytosolic proteins to the plasma membrane by the LCK protein tyrosine kinase dual acylation motif. J Cell Sci. 1997;110(Pt 5):673–679. [DOI] [PubMed] [Google Scholar]
- [87].Gorska MM, Alam R. A mutation in the human Uncoordinated 119 gene impairs TCR signaling and is associated with CD4 lymphopenia. Blood. 2012;119(6):1399–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Viau A, Bienaime F, Lukas K, et al. Cilia-localized LKB1 regulates chemokine signaling, macrophage recruitment, and tissue homeostasis in the kidney. Embo J. 2018;37(15):e98615. [DOI] [PMC free article] [PubMed] [Google Scholar]