

ABSTRACT
We recently identified the novel function of the small GTPase RAB-35 in apoptotic cell clearance in Caenorhabditis elegans, a process in which dying cells are engulfed and degraded inside phagosomes. We have found that RAB-35 functions in two separate steps of cell corpse clearance, cell corpse recognition and the initiation of phagosome maturation. During the latter process, RAB-35 facilitates the removal of phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) from the membranes of nascent phagosomes and the simultaneous production of phosphatidylinositol-3-P (PI(3)P) on these same membranes, a process that we have coined the PI(4,5)P2 to PI(3)P shift. RAB-35 also promotes the recruitment of the small GTPase RAB-5 to the phagosomal surface. During these processes, the activity of RAB-35 is controlled by the candidate GTPase-activating protein (GAP) TBC-10 and the candidate guanine nucleotide exchange factor (GEF) FLCN-1. Overall, RAB-35 leads a third pathway during cell corpse clearance that functions in parallel to the two known pathways, one led by the phagocytic receptor CED-1 and the other led by the CED-10/Rac1 GTPase. Here, we further report that RAB-35 acts as a robustness factor that maintains the clearance activity and embryonic viability under conditions of heat stress. Moreover, we obtained additional evidence suggesting that RAB-35 acts upstream of RAB-5 and RAB-7. To establish a precise temporal pattern for its own dissociation from phagosomal surfaces, RAB-35 controls the removal of its own GAP. We propose that RAB-35 defines a largely unexplored initial phase of phagosome maturation.
KEYWORDS: Apoptosis, phagocytosis, robustness, synthetic lethality, RAB-35, RAB-5, RAB-7, TBC-10, CED-1, PI(4, 5)P2, PI(3)P
Introduction
Mammalian Rab35 and its Caenorhabditis elegans ortholog RAB-35 have been of particular interest to researchers in recent years. Rab35 has been known to function in vesicle trafficking and cytoskeleton remodelling, regulating processes such as endocytosis and endocytic recycling [1–5], exocytosis [6,7], cytoskeletal rearrangement [8–10], cytokinesis [8,11,12], autophagy [13], and cell migration [14,15]. Mammalian Rab35 has also been implicated in phagocytosis, although many of the underlying mechanisms remain unknown [16–19]. Phagocytosis, a process that includes the recognition, engulfment, and degradation of target cells by other cells known as phagocytes, is an essential process for the clearance of apoptotic cells from animal bodies (reviewed in [20]). In an adult human, an estimated one million cells die every second as part of normal homoeostasis [21], whereas 300–500 germ cells and 131 somatic cells undergo apoptosis throughout the development of C. elegans [22–24]. Apoptotic cell clearance is essential for the proper turnover of ageing, unwanted, or damaged cells and thus aids in wound healing, helps to prevent auto-immune diseases and errant inflammation caused by secondary necrosis from apoptotic cells, promotes proper neuronal and embryonic development, and maintains proper homoeostasis [20,25]. In C. elegans, defects in apoptotic cell clearance cause a persistence and accumulation of apoptotic cell corpses that is known as a Ced (Cell death abnormal) phenotype [26].
We have identified RAB-35 in a screen to identify Rab GTPases that are required for apoptotic cell clearance among all thirty predicted Rabs in the C. elegans genome [27]. RAB-35 is enriched on extending pseudopods and exhibits a burst of further enrichment shortly after the pseudopods fuse to form a phagosome [27]. Using time lapse recording and epistasis analysis, we found that the phagosomal localization of RAB-35 is primarily controlled by TBC-10, a C. elegans ortholog of the mammalian GAPs TBC1D10A/B/C, and FLCN-1, a C. elegans ortholog of the mammalian GEF folliculin [27,28]. Both TBC-10 and FLCN-1 are transiently enriched on the phagosomal surface at the start of maturation [27]. GAPs are typically known to promote the removal of their partner Rabs from the membrane [29]. However, it is unclear whether RAB-35 is involved in the rapid removal of TBC-10 and FLCN-1 from the phagosome.
We have found that RAB-35 plays multiple roles in apoptotic cell clearance, not only facilitating the recognition of apoptotic cells by engulfing cells, but also promoting the subsequent maturation of a nascent phagosome that contains the apoptotic cell [27]. Surprisingly, RAB-35 functions in parallel to both of the canonical apoptotic cell clearance pathways, the ced-1/6/7/dyn-1 pathway and the ced-2/5/10/12 pathway [27]. The former pathway is composed of the ABC transporter homolog CED-7, which facilitates the exposure of the ‘eat me’ signal phosphatidylserine (PS), the phagocytic receptor CED-1, its adaptor CED-6, and the dynamin ortholog DYN-1 [30–35]; the latter pathway is defined by the Rac1 ortholog CED-10, its bipartite GEF composed of CED-5 and CED-12, and the adaptor protein CED-2 [36–40]. Consequently, RAB-35 defines a third and comparatively unexplored genetic pathway involved in phagocytosis [27]. While our work has provided evidence that RAB-35 likely functions downstream of the integrins INA-1, PAT-2, and PAT-3, which have previously been implicated in apoptotic cell clearance, it is possible that RAB-35 enables cell corpse recognition through other means [27,41,42].
Phagosome maturation is a process where a nascent phagosome incorporates early endosomes and lysosomes, gradually becomes more acidic, and eventually becomes a mature phagolysosome that degrades the encapsulated apoptotic cell [43,44]. We have identified that RAB-35 functions in a process known as the PI(4,5)P2 to PI(3)P shift [27]. Initially, the membrane of a nascent phagosome containing an apoptotic cell in a C. elegans embryo exhibits a high level of PI(4,5)P2 inherited from the pseudopod membrane; within minutes, PI(4,5)P2 disappears from phagosomal surfaces, and PI(3)P appears on phagosomal surfaces [45–48]. MTM-1, a PI(3)P phosphatase and a PI(4,5)P2 effector, was reported to antagonize the production of PI(3)P [45,47]. The removal of PI(4,5)P2 is proposed to release MTM-1 from the surface of the nascent phagosome [45]. PI(3)P is produced on the phagosome through the sequential function of the kinases PIKI-1 and VPS-34 [45,47]. rab-35 loss of function mutants exhibit a delay in the first wave of phagosomal PI(3)P, and this delay correlates with the persistent presence of PI(4,5)P2 and MTM-1 on the surface of the nascent phagosome [27]. We have proposed that RAB-35 facilitates PI(3)P production on phagosomal surfaces by promoting the turnover of phagosomal PI(4,5)P2 [27]. Recently, C. elegans RAB-35 was also found to promote the degradation of the male linker cell, which undergoes non-apoptotic developmental cell death after its engulfment [49]. RAB-35 was also reported to drive the removal of PI(4,5)P2 from the surface of phagosomes carrying the engulfed linker cell, consistent with our report [27,49].
A major driving force of phagosome maturation is the sequential function of multiple Rabs, primarily RAB-5 and RAB-7 (reviewed in [43]). Among RAB-2, RAB-5, RAB-7, and RAB-35, four C. elegans Rab proteins that function in phagosome maturation, RAB-35 is the first to appear on phagosomal surfaces [27]. RAB-5 begins to appear on phagosomes roughly 30–60 seconds after the burst of RAB-35 localization. Furthermore, the enrichment of RAB-5 on the phagosomal surface is promoted by the additive activities of both the RAB-35 and CED-1 pathways [27,50]. On the surface of nascent phagosomes, RAB-5 facilitates the production of PI(3)P [47], promotes the incorporation of early endosomes into phagosomes (our unpublished observation), and aids the recruitment of RAB-7 to phagosomes [51]. Based on our observation that RAB-35 is required for the timely recruitment of RAB-5 on phagosomal membranes, and on the epistasis analysis results that place RAB-35 in the same pathway as RAB-5, we concluded that RAB-35 functions upstream of RAB-5 in the context of phagosome maturation. However, whether overexpression of either RAB-5 or its downstream partner RAB-7 would affect apoptotic cell clearance in rab-35(b1013) mutants has not yet been tested.
Although RAB-35 acts in both the recognition of apoptotic cells and phagosome maturation, rab-35 loss of function mutants exhibit relatively modest cell corpse clearance phenotypes, suggesting that it might make a relatively minor contribution in apoptotic cell clearance [27]. However, inactivating rab-35 greatly enhances the clearance phenotypes of null or strong loss-of-function mutants in the genes of the ced-1/6/7/dyn-1 and ced-2/5/10/12 pathways, indicating that RAB-35 function may be more important when the canonical pathways of apoptotic cell clearance are compromised [27]. rab-35(b1013) mutants, but not wild-type animals, exhibit a moderate enhancement of the Ced phenotype in response to heat stress [27]. This, in part, has led us to propose that RAB-35 functions as a robustness factor for apoptotic cell clearance, having a minor impact on its own but ensuring the proper function of cell clearance when the underlying mechanisms are compromised. In such a scenario, rab-35(b1013) loss of function would be expected to enhance the Ced defects in response to different types of stress.
Building on our previously reported observations [27], additional temperature variants were tested. Here we report that C. elegans RAB-35 increases the robustness of apoptotic cell clearance in response to heat stresses. These results strengthen the notion that RAB-35 leads a third, independent pathway to enhance the robustness of apoptotic cell clearance. Additionally, RAB-35 facilitates phagosome maturation in a manner that cannot be compensated by overexpression of either RAB-5 or RAB-7. Moreover, we have identified a novel, RAB-35-mediated mechanism that regulates TBC-10. Coupled with our recent findings [27], our work indicates that RAB-35 acts in the initial step of phagosome maturation immediately following the completion of engulfment. In addition to its role in apoptotic cell clearance, we identified a novel role of RAB-35 in supporting embryonic development under conditions of heat stress. We propose that RAB-35 acts as a robustness factor under stress conditions in multiple developmental aspects.
Materials and methods
Mutations, strains, and transgenic arrays
C. elegans strains were grown at 20°C as previously described [52]. The N2 Bristol strain was used as the reference wild-type strain. Mutations and integrated arrays were previously described [27,53]. Plasmids were injected alongside the co-injection marker punc-76(+) into unc-76(e911) mutant adult hermaphrodites as previously described, with non-Unc animals being identified as transgenic animals [27]. rab-7(ok511) m−z− embryos, which lack both the maternal (m) and zygotic (z) activities of rab-7, are progeny of rab-7(ok511) m+z− mothers. These rab-7(ok511) m+z− animals, in turn, are produced from a strain of balanced rab-7(ok511) heterozygotes [48].
Nomarski DIC microscopy
DIC microscopy was performed using an Axionplan 2 compound microscope (Carl Zeiss, Thornwood, NY) equipped with Nomarski DIC optics, a digital camera (AxioCam MRm; Carl Zeiss), and imaging software (AxioVision; Carl Zeiss) as described previously [27]. Somatic embryonic cell corpses were scored in the head region of embryos at the 1.5-fold and late 4-fold embryonic stages, which correspond to embryos ~420 min and 700–800 min after the first cell division, respectively. Germ cell corpses were scored in one of the two gonadal arms of adult hermaphrodites 48 hours after the mid-L4 stage of development. For the prolonged heat and cold treatment, plates were incubated at 25°C for 36–48 hours and at 15°C for 72 hours, respectively. Embryos at the 1.5-fold and late 4-fold stages were scored for the number of cell corpses. For the heat shock treatment, one plate containing eggs was incubated at 33°C for 2 hours followed by incubation at room temperature for 1 hour, at which point 1.5-fold stage embryos were scored for the number of cell corpses. Another plate containing eggs was incubated at 33°C for 3.5 hours followed by incubation at room temperature for 3.5 hours, at which point late 4-fold stage embryos were scored for the number of cell corpses. The protocol for the cold shock treatment is the same as described above, except that the shock temperature is 10°C instead of 33°C.
Measuring embryonic lethality
C. elegans strains were initially grown at 20°C. Hermaphrodites at the mid-L4 stage of development were singled out on agar plates seeded with E. coli; in total, 5–10 worms were isolated for each strain that was analysed. These hermaphrodites were then incubated at either 20°C or 25°C for 24 hours, at which point each hermaphrodite was transferred to a new seeded plate. This process was repeated 48 hours after the hermaphrodites were initially singled out, producing three sets of plates representing eggs laid between 0–24 hours post-isolation, 24–48 hours post-isolation, and 48–72 hours post-isolation, respectively. After the removal of the mother, each set of plates was allowed to incubate for an additional 24 hours at either 20°C or 25°C, respectively. Each plate was then scored for the number of hatched and unhatched embryos, with the latter annotated as having died during embryonic development. The data for all three plates (0–24 hours, 24–48 hours, 48–72 hours) were aggregated for each individual hermaphrodite to calculate the fraction of each brood that died during embryonic development.
Fluorescence microscopy and quantification of cell corpse clearance events
An Olympus IX70-Applied Precision DeltaVision microscope equipped with a DIC imaging apparatus and a Photometris Coolsnap 2 digital camera was used to capture fluorescence and Differential Interference Contrast (DIC) images, while Applied Precision SoftWoRxV software was utilized for image deconvolution and processing as described previously [27]. The dynamics of various GFP, mRFP, mKate2, and mCherry reporters during the engulfment and degradation of cell corpses C1, C2, and C3 were examined using an established time-lapse recording protocol [48,54]. Recordings typically lasted 60–180 minutes, with an interval of 30 secs to 2 mins. At each time point, 10–16 serial z-sections at 0.5-μm intervals were recorded. Signs such as embryo elongation and embryo turning prior to comma stage were closely monitored under DIC to ensure that the embryo being recorded was developing normally. For animals bearing the transgene co-expressing Phsp rab-5(S33N) and Pced-1 gfp::rab-35, gravid mothers were placed onto a new plate and allowed to lay eggs for 1 hour before they were removed. The plate was then incubated at 33°C for 2 hours, after which embryos were subjected to time-lapse recording.
Results
A dominant-negative form of CED-1 greatly enhances the Ced phenotype of rab-35 null mutants
Previously, we have observed that rab-35(b1013) null mutants exhibit a comparatively mild Ced phenotype and correspondingly mild defects in cell corpse recognition and phagosome maturation [27]. Despite the mild Ced phenotype of the rab-35 single mutants, the rab-35(b1013) mutation significantly enhanced the Ced phenotypes of ced-1(n1506) and ced-6(n2095) mutants, as well as that of ced-5(n1812), ced-10(n1993), and ced-12(n3261) mutants, which represent the canonical ced-1/6/7/dyn-1 and ced-2/5/10/12 pathways involved in apoptotic cell clearance, respectively [27]. These observations are the basis for the hypothesis that RAB-35 leads a third and independent cell corpse clearance pathway. To further understand the functional relationship between RAB-35 and CED-1, we analysed the transgenic lines that overexpressed a multi-copy transgene ced-1ΔC::gfp in engulfing cells under the control of the Pced-1 promoter [31].
CED-1ΔC is a truncated form of CED-1 missing the C-terminal intracellular domain. Consequently, CED-1ΔC retains the ability of CED-1 to associate with phosphatidylserine yet loses its ability to activate downstream cell corpse clearance events [31,55]. By effectively sequestering the ‘eat me’ signal phosphatidylserine, CED-1ΔC::GFP causes a weak dominant negative effect in cell corpse clearance in the wild-type background [31,55]. Our cell corpse counts performed in 1.5-fold stage embryos and adult gonads (Materials & Methods) verified the weak dominant negative effect of CED-1ΔC (Figure 1). In contrast, overexpression of CED-1C::GFP, the GFP-tagged C-terminal intracellular domain of CED-1, under the Pced-1 promoter does not result in any persistent cell corpses in a wild-type background (Figure 1) [48]. This demonstrates that unlike CED-1ΔC::GFP, CED-1C::GFP does not interfere with the function of endogenous CED-1. Overexpression of CED-1ΔC::GFP failed to further enhance the Ced phenotype in ced-1(n1506) null mutants, suggesting that CED-1ΔC specifically interferes with the function of endogenous CED-1 (Figure 1).
Figure 1.
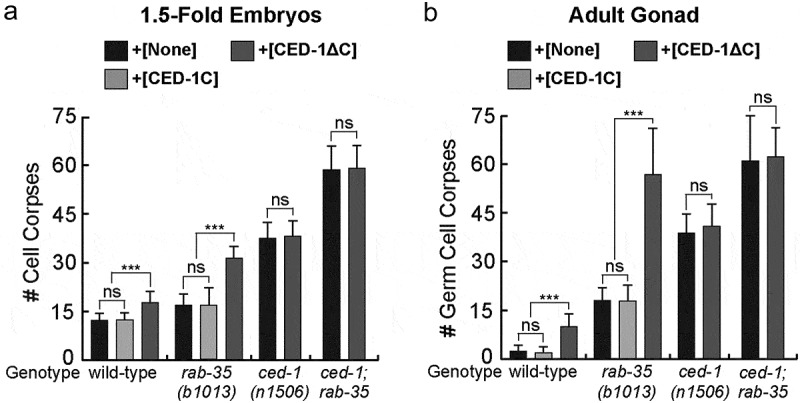
Overexpression of CED-1ΔC::GFP enhances the rab-35 phenotypes in cell corpse clearance
Animals carrying the Pced-1 ced-1ΔC::gfp or Pced-1 ced-1C::gfp transgenic arrays as well as the non-transgenic animals were scored for the number of cell corpses in 1.5-fold embryos (a) and adult gonads (b). Somatic cell corpses were scored in 1.5-fold stage embryos. Germ cell corpses were scored in one gonadal arm of each 48 hour-post L4 stage adult hermaphrodite. For each data point, at least 15 animals were scored. Bar graphs and error bars represent mean and standard deviation (sd), respectively. Brackets above the bars indicate the samples that are compared by the Student t-test. p-values are summarized as such: ***, p < 0.00001; ns, no significant difference.
Expression of CED-1ΔC::GFP in a rab-35(b1013) null mutant background greatly increases the number of cell corpses in both 1.5-fold embryos and adult gonads, whereas the expression of CED-1C::GFP does not (Figure 1). In the adult gonad, the number of persistent germ cell corpses is similar to that observed from the ced-1(n1506); rab-35(b1013) double mutants (Figure 1B), although this dominant negative effect is considerably less pronounced in embryos (Figure 1A) [27]. Moreover, as expected for its specific inhibitory effect targeting endogenous CED-1, CED-1ΔC::GFP does not further worsen the Ced phenotype in ced-1; rab-35 double mutant embryos or adults (Figure 1). The above results indicate that, in the context of apoptotic cell clearance, suppressing endogenous ced-1 function through the expression of CED-1ΔC::GFP impairs cell-corpse removal in parallel to rab-35 loss of function. These results further support the hypothesis that that rab-35 and ced-1 act in two parallel and partially redundant pathways to promote cell corpse clearance.
Rab-35 functions upstream of rab-5 and rab-7 in apoptotic cell clearance, and neither RAB-5 nor RAB-7 overexpression rescues the rab-35 loss-of-function phenotype
To further explore RAB-35’s functional relationship with RAB-5 and RAB-7, we examined the rescuing effect of gfp::rab-5 and gfp::rab-7 expressed in engulfing cells under the control of Pced-1 [48,50]. In rab-35 mutants, gfp::rab-5 or gfp::rab-7 failed to rescue cell corpse clearance defects in either embryos of different stages or in adult gonads (Figure 2A(b)). Additionally, overexpression of gfp::rab-5 or gfp::rab-7 did not change the cell corpse counts in wild-type worms (Figure 2A(a)).
Figure 2.
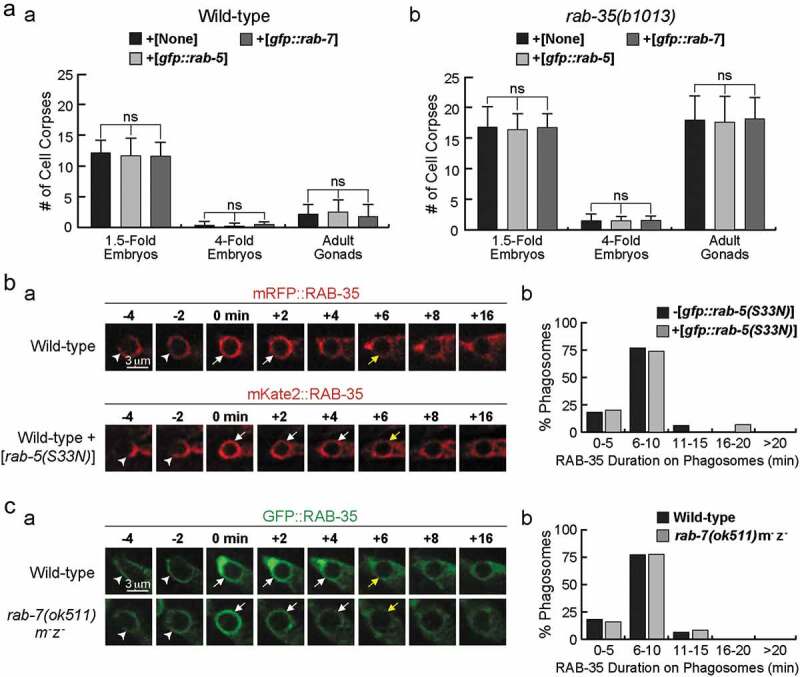
Analysing RAB-35’s relationship with RAB-5 and RAB-7 during the phagosome maturation process
(a) Overexpression of GFP::RAB-5 or GFP::RAB-7 fails to rescue the Ced phenotype of rab-35(b1013) mutants. Wild-type (a) and rab-35(b1013) mutant (b) animals expressing either Pced-1 gfp::rab-5, Pced-1 gfp::rab-7, or no transgene at all were analysed. The numbers of somatic cell corpses were scored in 1.5-fold and late 4-fold stage embryos. The numbers of germ cell corpses were scored in 48 hour-post L4 stage adult hermaphrodites. The bars and error bars represent mean and sd. For each data point, at least 15 animals were scored. Brackets above the bars cover the samples that are compared by the Student t-test. ns, no significant difference in the p-values.(B) Time-lapse recording during and after the formation of the C3 phagosome in wild-type embryos that express either Pced-1mrfp::rab-35 (top) or Pced-1mKate2::rab-35 and Pced-1rab-5(S33N) (bottom). (a) Time-lapse images. ‘0 min’ is the moment when a phagosome is just formed. Arrowheads mark extending pseudopods, white arrows mark phagosomes where either RFP::RAB-35 or mKate2::RAB-35 is visible, and yellow arrows indicate the first time point when either RFP::RAB-35 or mKate2::RAB-35 are no longer visible on phagosomes. (b) Histograms displaying the distribution of time periods during which RAB-35 is enriched on the surface of phagosomes bearing C1, C2, and C3 in wild-type embryos that express Phsp rab-5(S33N). The Pced-1 gfp::rab-35 transgene is used to monitor the dynamic localization of RAB-35 on phagosomes. The duration of RAB-35 is defined as the period between the time point when GFP::RAB-35 exhibits a burst of phagosomal enrichment (‘0 min’) and the first time point when GFP::RAB-35 signal can no longer be observed on the phagosome. At least 15 phagosomes were scored.(d) Time-lapse recording during and after the formation of the C3 phagosome in wild-type embryos that express Pced-1gfp::rab-35 in a wild-type (top) or rab-7(ok511)m−z− (bottom) background. (a) Time-lapse images. ‘0 min’ is the moment when a phagosome is just formed. Arrowheads mark extending pseudopods, white arrows mark phagosomes where GFP::RAB-35 is visible, and yellow arrows indicate the first time point when GFP::RAB-35 is no longer visible on phagosomes. (b) Histograms displaying the distribution of time periods during which RAB-35 is enriched on the surface of phagosomes bearing C1, C2, and C3 in rab-7(ok511)m−z− embryos. The Pced-1 gfp::rab-35 transgene is used to monitor the dynamic localization of RAB-35 on phagosomes. The duration of RAB-35 is defined as the period between the time point when GFP::RAB-35 exhibits a burst of phagosomal enrichment (‘0 min’) and the first time point when GFP::RAB-35 signal can no longer be observed on the phagosome. At least 15 phagosomes were scored.
Using an established time lapse recording protocol (Materials & Methods), we further monitored the dynamic phagosomal enrichment pattern of the GFP::RAB-35 reporter when endogenous rab-5 or rab-7 was inactivated [27]. Overexpression of rab-5(S33N), a dominant-negative form of rab-5, was used to inactivate endogenous rab-5 in wild-type embryos [27]. We found that this overexpression did not significantly affect the duration of GFP::RAB-35 on phagosomal surfaces (Figure 2B). Similarly, rab-7(ok511) null mutants exhibit a normal temporal pattern of the phagosomal localization of GFP::RAB-35 (Figure 2C). These results suggest that neither the overexpression nor inactivation of rab-5 or rab-7 affect the behaviour of RAB-35.
Recently, we have reported that the rab-35 null mutation significantly delays the recruitment of GFP::RAB-5 to phagosomal surfaces [27], indicating that the function of RAB-35 is required for the proper recruitment and the resulting phagosomal function of RAB-5. Epistasis assay results also indicate that rab-35 and rab-5 act in the same genetic pathway for cell corpse clearance [27]. Together, the results presented in Figure 2 and our previous observations support the notion that RAB-35 acts upstream of RAB-5 in the pathway, promoting the degradation of cell corpses.
RAB-35 regulates the dynamic phagosomal targeting of TBC-10
GAPs stimulate the GTPase activity of Rab proteins [29]. The GTP-bound form of Rabs typically localize to their target membranes, whereas their GDP-bound form is evenly distributed in the cytoplasm; this is because the GDP-bound form is a prime target for GDP dissociation inhibitors (GDIs), which remove Rabs from their target membranes [29]. We previously found that, among the three GAPs implicated to function for rab-35 (tbc-7, tbc-10, and tbc-13), only tbc-10 acts in the same genetic pathway as rab-35 in the context of cell corpse clearance [27]. In addition, in the tbc-10 loss-of-function mutant background, GFP::RAB-35 displays prolonged association with phagosomal surfaces, indicating that RAB-35 stays in its GTP-bound form longer than in the wild-type background [27]. Together, these results indicate that TBC-10 likely functions as a GAP for RAB-35.
TBC-10 is transiently enriched on the extending pseudopods and nascent phagosomes during apoptotic cell clearance, displaying a temporal pattern that is relatively coincidental to RAB-35 [27]. Here we examined whether RAB-35 played any role in the phagosomal enrichment of TBC-10. We found that in rab-35(b1013) mutant embryos, GFP::TBC-10 [27] persists on the phagosomal surface (Figure 3A,C). In wild-type embryos, TBC-10::GFP is transiently enriched on phagosomal surfaces, persisting for 10 minutes or less in all of the phagosomes observed (Figure 3C). On the contrary, in rab-35(b1013) mutant embryos, TBC-10::GFP is removed from only 18.75% of observed phagosomes within this same time frame (Figure 3C). Collectively, these results suggest that RAB-35 acts to regulate the phagosomal targeting of TBC-10. Interestingly, overexpression of the putatively GTP-bound form GFP::RAB-35(Q69L) [27] in a wild-type background does not alter either the frequency or the timing of the removal of TBC-10::RFP from the surfaces of nascent phagosomes (Figure 3B,D). Thus, the removal of TBC-10 from phagosomes requires RAB-35 activity, yet is not further affected by excessive RAB-35 activity.
Figure 3.
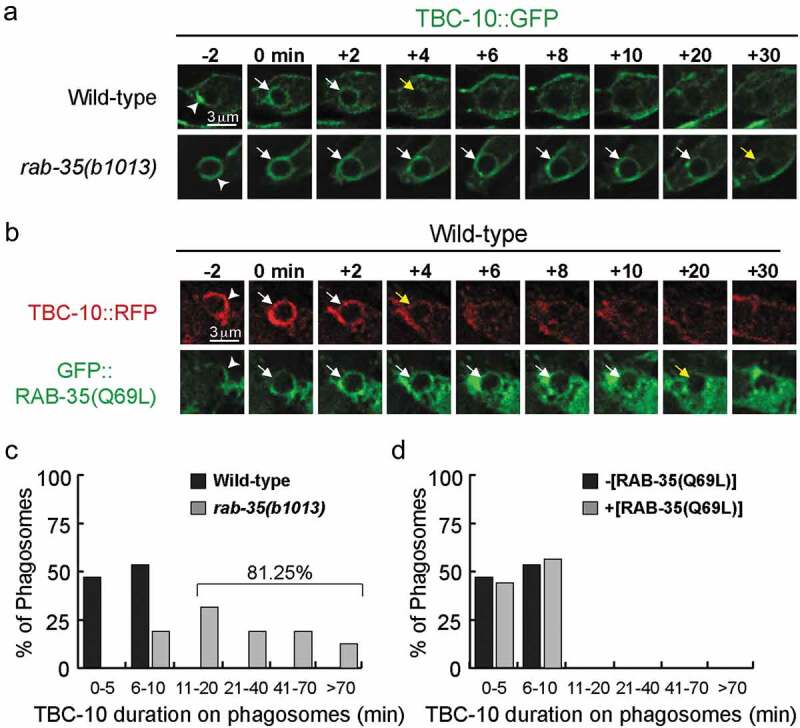
In rab-35 mutants, the disappearance of TBC-10 from phagosomal surfaces is significantly delayed
(a) Time-lapse images during and after the formation of the C3 phagosome in wild-type and rab-35(b1013) embryos expressing Pced-1 tbc-10::gfp. ‘0 min’ is the moment when a phagosome is just formed. Arrowheads mark extending pseudopods, white arrows mark phagosomes where TBC-10::GFP is visible, and yellow arrows indicate the first time point when TBC-10::GFP is no longer visible on phagosomes.(B) Time-lapse images during and after the formation of the C3 phagosome in wild-type embryos that coexpressed Pced-1 tbc-10::mrfp and Pced-1 gfp::rab-35(Q69L). ‘0 min’ is the moment when a phagosome is just formed. Arrowheads mark extending pseudopods, white arrows mark phagosomes where either TBC-10::RFP or GFP::RAB-35(Q69L) is visible, and yellow arrows indicate the first time point when either TBC-10::RFP or GFP::RAB-35(Q69L) are no longer visible on phagosomes.(c-d) Histogram displaying the range of time that TBC-10::GFP is observed on the surface of phagosomes bearing C1, C2, and C3 in wild-type and rab-35(b1013) mutant embryos (c), and in wild-type mutant embryos that either express no transgene or express Pced-1gfp::rab-35(Q69L) (d). The time interval is defined as that between when TBC-10::GFP is first observed on the phagosome (‘0 min’) and the first time point when this GFP::TBC-10 signal can no longer be observed. For each genotype, at least 15 phagosomes were scored.
In rab-35 mutants, cell corpse clearance is hypersensitive to high temperature
The ideal living temperature for C. elegans in a laboratory setting is between 15°C and 25°C, the optimal temperature being 20°C [56]. After a prolonged incubation at 25°C, neither 1.5-fold nor late 4-fold stage wild-type embryos exhibit any increase in the number of cell corpses (Figure 4A,B) (Materials & Methods). On the other hand, 1.5-fold stage rab-35(b1013) embryos exhibit a mild increase (~25%) in their average number of cell corpses (Figure 4A). In late 4-fold stage rab-35(b1013) mutant embryos, 25°C incubation induces a greater enhancement of the Ced phenotype (~62%) (Figure 4B). Similar results were found using heat shock treatment – a short incubation at 33°C – where 1.5-fold and 4-fold embryos exhibited a 13% and 57% enhancement in their average number of cell corpses, respectively (Figure 4A,B).
Figure 4.
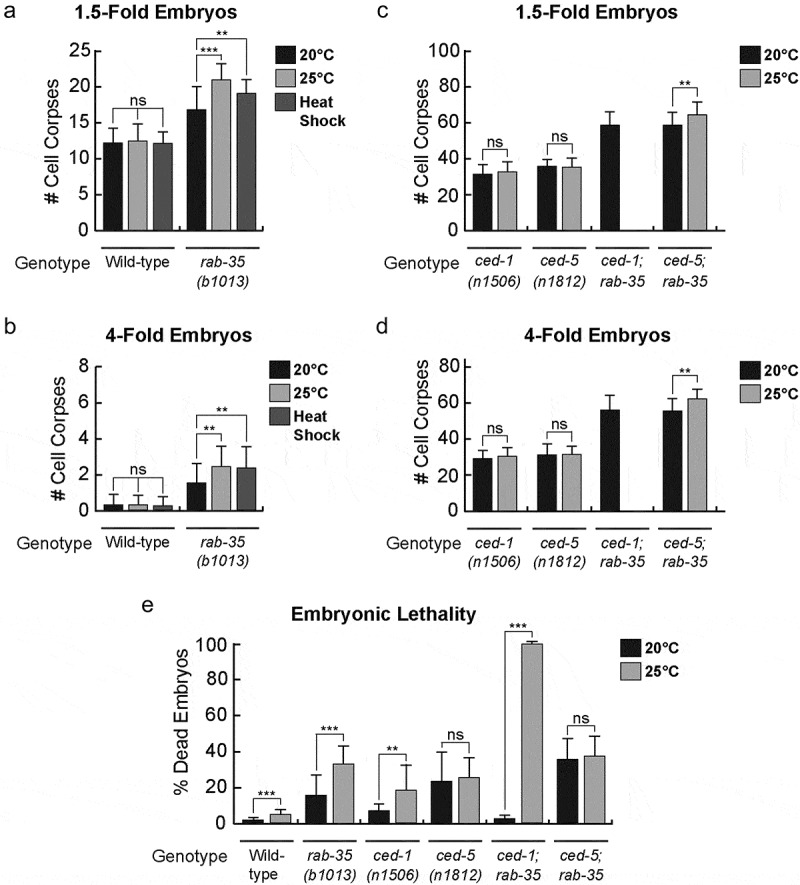
Apoptotic cell clearance in rab-35 mutants and embryonic viability are sensitive to increases in temperature
(a-d) The mean numbers of apoptotic cell corpses were scored in response to heat stress in both 1.5-fold embryos (a, b) or 4-fold embryos (c, d) of various mutant backgrounds. For each data point, at least 15 animals were scored. Error bars indicate sd. Student t-test was used for data analysis: *, 0.005 < p < 0.05; **, 0.00001 < p < 0.005; ***, p < 0.00001; ns, no significant difference. Part of (a) and (b) was reported previously [27]. 25°C data was not available for ced-1; rab-35 double mutants in (c) and (d) because of the synthetic embryonic lethality.(e) The fractions of embryos of different mutant backgrounds that died during development were scored at two different temperatures. For each data point, 5–10 broods were scored. Error bars indicate sd. Student t-test was used for data analysis: *, 0.005 < p < 0.05; **, 0.00001 < p < 0.005; ***, p < 0.00001; ns, no significant difference.
In contrast to the results observed in rab-35(b1013) mutants, prolonged incubation at 25°C failed to produce any enhancement in the number of cell corpses in either ced-1(e1735) or ced-5(n1812) mutant embryos (Figure 4C,D). The ced-5(n1812); rab-35(b1013) double mutants displayed a more severe Ced phenotype at 25°C than at 20°C (Figure 4C,D), consistent with the observation that loss of rab-35 function specifically results in an enhanced cell corpse clearance defect at 25°C. The ced-1(e1735); rab-35(b1013) double mutants exhibit nearly 100% embryonic lethality at 25°C (Figure 4E and next section), and consequently the Ced phenotype of these mutant embryos could not be scored under this condition (Figure 4E and next section). Altogether, these results suggest that compromising the rab-35 pathway, but not the ced-1 or ced-5 pathways, makes apoptotic cell clearance less robust with respect to modest heat stress.
A cold shock treatment – a short incubation at 10°C (Materials and Methods) – failed to produce a similar phenotype in either wild-type or rab-35(b1013) mutant worms (Figure 5A,B). Moreover, a prolonged incubation at 15°C failed to produce any increase in the number of cell corpses in any of the aforementioned mutant backgrounds (Figure 5C,D). These observations indicate that the loss of function of rab-35, ced-1, or ced-5 renders cell corpse clearance more sensitive to increases, but not decreases, in temperature.
Figure 5.
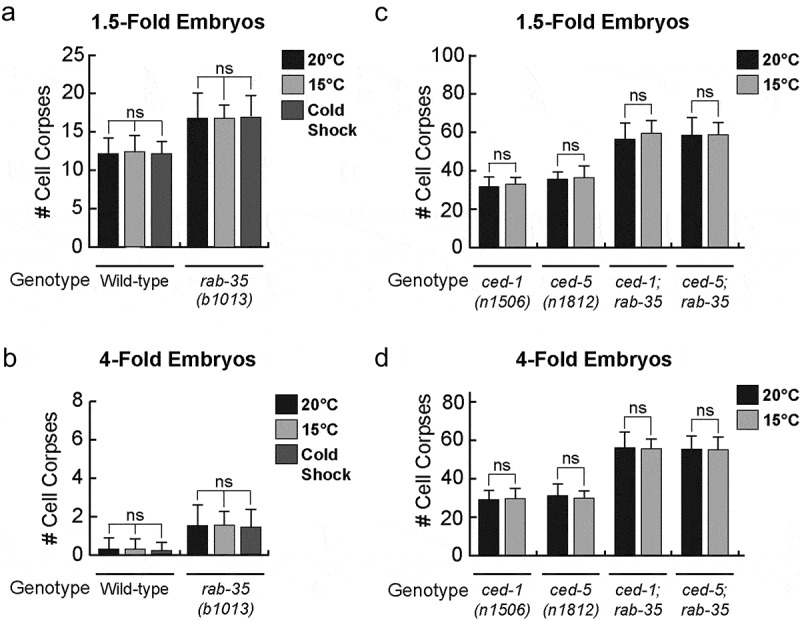
Apoptotic cell clearance in rab-35 mutants is not affected by decreases in temperature
The mean numbers of apoptotic cell corpses were scored in response to cold stress in both 1.5-fold embryos (a, b) and 4-fold embryos (c, d) of various mutant backgrounds. For each data point, at least 15 animals were scored. Error bars indicate sd. Student t-test was used for data analysis: *, 0.005 < p < 0.05; **, 0.00001 < p < 0.005; ***, p < 0.00001; ns, no significant difference.
Rab-35 and ced-1 mutants are synthetic lethal at high temperature
To investigate whether RAB-35 plays any additional roles in embryonic development besides the clearance of apoptotic cells, we quantified the embryonic lethality of rab-35 and other mutants at different temperatures (Materials and Methods). We found that at 20°C, larger fractions of embryos failed to hatch in rab-35, ced-1, and ced-5 single null mutants compared to wild-type embryos, indicating that all three genes contribute to the completion of embryonic development. The percentage of embryonic lethality increases in the order of ced-1 (6.6%), rab-35 (15.4%), and ced-5 (23.1%) mutants, indicating the quantitative difference in the contribution of each gene. Raising the temperature to 25°C resulted in 2–3 fold increases of lethality in wild-type and ced-1 and rab-35 mutants (Figure 4E), indicating that high temperature generates additional stress for embryonic development, and that both rab-35 and ced-1 play roles in maintaining normal embryonic development under conditions of stress. Remarkably, the ced-1; rab-35 double null mutants display a drastically increased embryonic lethality (99%) at 25°C in comparison to 20°C and to that of each single mutant at 25°C (Figure 4E). This synthetic lethality indicates that RAB-35 and CED-1 act synergistically to ensure normal embryonic development under heat stress.
Unlike ced-1 and rab-35 null mutants, the embryonic lethality of ced-5 null mutants at 20°C (23.1%) is not further increased at 25°C (25.3%) (Figure 4E), suggesting that ced-5 is not involved in response to heat stress. The rab-35; ced-5 double null mutants display a higher embryonic lethality than each of the single mutants at 20°C, suggesting lack of functional interaction. Consistent with the ced-5 single mutant phenotype, the rab-35; ced-5 double mutants phenotype is not further increased when temperature is increased from 20°C to 25°C (Figure 4E).
Mutations in the small GTPase ARF-6 do not impair the clearance of apoptotic cells in embryos
A recent report indicates that the small GTPase ARF-6 blocks the clearance of the male linker cell, and that RAB-35 antagonizes this inhibitory effect [49]. We have analysed both the loss-of-function (tm1447) and gain-of-function (ns388) alleles of arf-6 [49] for the presence of persistent cell corpses in both the mid-stage (1.5-fold) and late-stage (late 4-fold) embryos. The cell corpse counts in neither mutant strain is significantly different from that of the wild-type strain. Specifically, at 1.5-fold stage, wild-type, arf-6(tm1447), and arf-6(ns388) mutant embryos contain on average 12.3 ± 1.9, 12.4 ± 2.0, and 12.4 ± 1.5 cell corpses, respectively; in addition, at late 4-fold stage, wild-type, arf-6(tm1447), and arf-6(ns388) mutant embryos contain on average 0.2 ± 0.4, 0.4 ± 0.5, 0.3 ± 0.5 cell corpses, respectively. For each sample point, >15 embryos were scored. These data indicate that ARF-6 does not play an important role in the removal of embryonic cell corpses.
Discussion
Considering that the CED-1 and RAB-35 pathways both control the recognition and degradation of apoptotic cells, we characterized the genetic relationship between rab-35 and ced-1 in multiple clearance-related events [27]. The rab-35 null mutation significantly enhances the following defects characteristic of a ced-1 mutant background: (i) cell corpse recognition; (ii) the removal of PI(4,5)P2 and its effector MTM-1 from nascent phagosomes; (iii) the production of PI(3)P on phagosomal surfaces; (iv) the recruitment of RAB-5; and (v) the recruitment of early endosomes to phagosomes [27]. In this report, we found that the Ced phenotype of rab-35 null mutants, but not that of ced-1 null mutants, is significantly enhanced by the overexpression of CED-1ΔC::GFP, a truncated, dominant-negative form of CED-1. This new result further indicates that rab-35 and ced-1 function in two independent and parallel pathways, both of which regulate many of the same events essential for cell corpse clearance.
To better place RAB-35 in the pathways for cell corpse engulfment, we further investigated RAB-35’s functional relationship with RAB-5 and RAB-7, two Rab GTPases that form a Rab cascade required for the proper progression of phagosome maturation and the degradation of engulfed cell corpses (reviewed in [43]). Our previous epistasis data suggested that rab-35 and rab-5 function in the same genetic pathway to promote phagosome maturation [27]. The phagosomal enrichment of RAB-35 immediately precedes that of RAB-5, and a loss-of-function mutation of rab-35 delays the phagosomal recruitment of RAB-5, collectively indicating that RAB-35 function is required for the timely recruitment of RAB-5 [27]. Here, we further report that inactivating rab-5 or rab-7 does not affect the enrichment of RAB-35 on phagosomal surfaces, and that the overexpression of RAB-5 or RAB-7 fails to rescue the Ced phenotype of rab-35 null mutants, consistent with the conclusion that rab-35 acts upstream of the rab-5/rab-7 cascade in the regulation of phagosome maturation. All of these aforementioned observations, coupled with the early recruitment of RAB-35 to pseudopods and nascent phagosomes and the ability of RAB-35 to modulate the PI(4,5)P2 to PI(3)P shift immediately after phagosomal formation, indicate that RAB-35 is the earliest acting and most upstream Rab GTPase in cell corpse degradation. Phagosomes labelled with RAB-35 represent an important, but largely unexplored, phase of phagosome maturation that precedes later stages of maturation marked by the presence of RAB-5 and RAB-7. This early phase of phagosomal development is likely driven by RAB-35 effectors, which have yet to be identified in the context of apoptotic cell clearance.
By activating the GTPase activity of the Rab GTPases, GAPs prime the corresponding Rab proteins for removal from the membrane [29]. We found that RAB-35 must cycle between GTP- and GDP-bound states for promoting cell corpse removal [27]. As expected, the null mutation of tbc-10, which encodes the RAB-35 GAP, causes GFP::RAB-35 to persist for a period longer than normal on phagosomal surfaces [27]. Remarkably, here we observe that the dissociation of TBC-10 from phagosomal surfaces also depends on RAB-35 activity. Intriguingly, wild-type embryos that express the putative constitutively active and GTP-locked GFP::RAB-35(Q69L) mutant protein exhibit normal timing of TBC-10 removal from the phagosome, indicating that excessive RAB-35 activity does not alter the transient phagosomal enrichment pattern of TBC-10. The mutual dependence between RAB-35 and TBC-10 suggests that the association between RAB-35 and TBC-10 is required for the proper dissociation of both proteins from the phagosomal surface. Alternatively, an unknown effector of RAB-35 might act to facilitate the dissociation of TBC-10 from phagosomal surfaces. Regardless, this mechanism could act to prevent either an excessive accumulation of TBC-10 on – or an accelerated removal of RAB-35 from – phagosomal surfaces. Coupled with the fact that RAB-35 is gradually removed after TBC-10 is no longer visible on phagosomal surfaces, these observations suggest a novel mechanism that precisely controls the timing and speed of RAB-35 dissociation from phagosomal membranes.
The cell corpse clearance phenotypes of the rab-35 null mutants are relatively mild, which provokes the question regarding the physiological significance of RAB-35 in cell corpse clearance. Remarkably, loss of rab-35 function strongly enhances the Ced phenotype of single null mutants in the ced-1/-6/-7 and ced-2/-5/-10/-12 pathways as well as those of double mutants that inactivate both of these canonical pathways [27]. This suggests that the pathway led by rab-35 is essential in maintaining the cell corpse clearance activity when the function of one – or both – of the canonical pathways is compromised [27]. We thus proposed that RAB-35 functions as a ‘robustness factor’ that helps to maintain the stability and effectiveness of apoptotic cell corpse removal [27]. In this report, we provide further experimental evidence that support this ‘robustness factor’ function of RAB-35. Although prolonged incubation at 25°C does not increase the number of cell corpses observed in the wild-type, ced-1, or ced-5 null mutant animals relative to incubation at 20°C, we found that rab-35 null mutants exhibited an enhancement in their observed Ced phenotype under these same conditions. Collectively, these results suggest that the pathway defined by RAB-35 plays a unique role in ensuring that cell corpse removal occurs efficiently under physiologically stressful conditions (Figure 6). Consistent with this model, the double mutant ced-5(n1812); rab-35(b1013) displayed a more severe Ced phenotype at 25°C than at 20°C.
Figure 6.
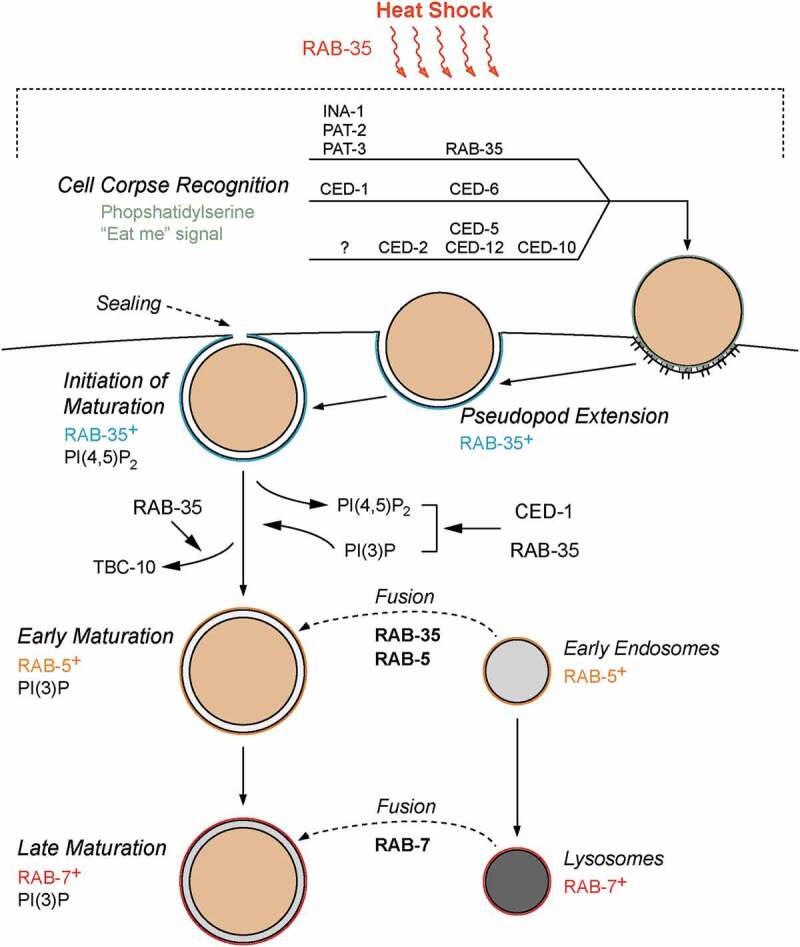
Model of RAB-35 action during apoptotic cell clearance
Diagram illustrating the various roles of RAB-35 during the recognition of apoptotic cells and the subsequent phagosome maturation and cell corpse degradation. See the ‘Discussion’ section for more details. During physiologically stressful conditions such as heat stress, RAB-35 functions to maintain the proper apoptotic cell clearance activity. RAB-35, CED-1, and CED-5 function redundantly in cell corpse recognition, while RAB-35 and CED-1 function redundantly throughout phagosome maturation. rab-35 functions in the same pathway as the integrins ina-1, pat-2, and pat-3, and in parallel to the canonical pathways ced-1/ced-6/ced-7/dyn-1 and ced-2/ced-5/ced-10/ced-12 [27]. CED-1, INA-1, PAT-2, and PAT-3 are all transmembrane receptors that recognize phosphatidylserine, an ‘eat-me’ signal exhibited by apoptotic cells that initiates engulfment. A question mark depicts that the identity of the candidate receptor activating the ced-5 pathway is still unclear. RAB-35 can be observed on the growing pseudopods alongside PI(4,5)P2, and plays a critical role in the initiation of phagosomal maturation and the removal of its GAP TBC-10. Furthermore, RAB-35 and CED-1 redundantly promote the removal of PI(4,5)P2 from, the production of PI(3)P on, and the recruitment of RAB-5 to the phagosomal membrane. RAB-35 and CED-1 were also found to independently promote the fusion of early endosomes to phagosomes, a process that is mediated by RAB-5 and PI(3)P.
Although modest increases in temperature have been found to enhance the efficiency of phagocytosis in human leukocytes [57,58], more extreme temperatures have been found to compromise this process in an irreversible and time dependent manner [59]. However, while mammalian Rab35 has been implicated in the regulation of actin remodelling [8–10,16,18], a process that is adversely impacted by incubation at 30–34°C [60], neither RAB-35 nor its mammalian ortholog have previously been implicated in heat stress resistance. Our work represents a novel role of RAB-35 and unveils a series of mechanisms that increase the robustness of apoptotic cell clearance. Essentially, while rab-35 is mostly dispensable in a wild-type background, it becomes considerably more important under conditions where apoptotic cell clearance is compromised, either due to increases in temperature or due to mutations in the pathways defined by ced-1 or ced-5.
Remarkably, at a higher temperature, ced-1 or rab-35 null mutations increase the low-level embryonic lethality observed at 20°C; furthermore, inactivation of both ced-1 and rab-35 results in a synergistically enhanced, nearly 100% embryonic lethality (99%). This is the first time such important developmental role of CED-1 is revealed. Although currently the molecular roles of RAB-35 and CED-1 in embryonic development are unknown, since neither the ced-5 single mutation nor rab-35; ced-5 double mutations result in such severe embryonic lethality under the same condition, CED-1 and RAB-35 are likely to regulate essential embryonic developmental events in addition to apoptotic cell clearance, in particular under heat stress. RAB-35 was known to function in receptor recycling [5], although whether rab-35 mutations cause any lethality was not reported previously. ced-1 mutations were not reported to affect viability. Synergistic genetic interaction is known to arise between genes that act in redundant pathways or two pathways that converge at a critical step of certain regulatory or metabolic network [61]. Further investigation is needed to reveal these novel stress-response functions of RAB-35 and CED-1.
Besides acting in a third pathway as a robustness factor for the removal of apoptotic cells ([27] and this report), RAB-35 has recently been found to promote the engulfment and degradation of the male linker cell, which undergoes a non-apoptotic developmental cell death process [49]. Unlike the clearance of apoptotic cells, the removal of the linker cell does not involve either the ced-1 or the ced-5 pathway; thus, in this particular event, RAB-35 plays a non-redundant role [49]. In addition, while Kutscher et al. [49] found that the small GTPase ARF-6 inhibits the removal of the linker cell, a process that is negatively regulated by RAB-35, our results suggest that the removal of apoptotic cells do not involve ARF-6. Together, the findings made by Kutscher et al. and by us have demonstrated that RAB-35 plays different physiological roles under different developmental contexts [27,49]. Despite its modest phenotype when knocked out under favourable conditions, our research has demonstrated that RAB-35 becomes a vital component of apoptotic cell clearance when the system is compromised either through genetic mutations or through physiological stress.
Acknowledgments
We thank Lena Kutscher and Shai Shaham, the Caenorhabditis Genetics Center (CGC), and the National BioResource Project in Japan (Shohei Mitani) for providing mutant strains.
Funding Statement
This work was supported by NIH/NIGMS grants GM067848 and GM104279 to Z.Z; National Institute of General Medical Sciences [GM104279]; National Institute of General Medical Sciences [GM067848].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Patino-Lopez G, Dong X, Ben-Aissa K, et al. Rab35 and its GAP EPI64C in T cells regulate receptor recycling and immunological synapse formation. J Biol Chem. 2008. June 27;283(26):18323–18330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Walseng E, Bakke O, Roche PA.. Major histocompatibility complex class II-peptide complexes internalize using a clathrin- and dynamin-independent endocytosis pathway. J Biol Chem. 2008. May 23;283(21):14717–14727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gao Y, Balut CM, Bailey MA, et al. Recycling of the Ca2+-activated K+ channel, KCa2.3, is dependent upon RME-1, Rab35/EPI64C, and an N-terminal domain. J Biol Chem. 2010. June 4;285(23):17938–17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sheehan P, Zhu M, Beskow A, et al. Activity-dependent degradation of synaptic vesicle proteins requires Rab35 and the ESCRT pathway. J Neurosci Off J Soc Neurosci. 2016. 17;36(33):8668–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sato M, Sato K, Liou W, et al. Regulation of endocytic recycling by C. elegans Rab35 and its regulator RME-4, a coated-pit protein. Embo J. 2008. April 23;27(8):1183–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hsu C, Morohashi Y, Yoshimura S, et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A–C. J Cell Biol. 2010. April 19;189(2):223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Biesemann A, Gorontzi A, Barr F, et al. Rab35 protein regulates evoked exocytosis of endothelial weibel-palade bodies. J Biol Chem. 2017. 14;292(28):11631–11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dambournet D, Machicoane M, Chesneau L, et al. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011. August;13(8):981. [DOI] [PubMed] [Google Scholar]
- [9].Chevallier J, Koop C, Srivastava A, et al. Rab35 regulates neurite outgrowth and cell shape. FEBS Lett. 2009;583(7):1096–1101. [DOI] [PubMed] [Google Scholar]
- [10].Zhang J, Fonovic M, Suyama K, et al. Rab35 controls actin bundling by recruiting fascin as an effector protein. Science. 2009. September 4;325(5945):1250–1254. [DOI] [PubMed] [Google Scholar]
- [11].Kouranti I, Sachse M, Arouche N, et al. Rab35 regulates an endocytic recycling pathway essential for the terminal steps of cytokinesis. Curr Biol. 2006. September 5;16(17):1719–1725. [DOI] [PubMed] [Google Scholar]
- [12].Chesneau L, Dambournet D, Machicoane M, et al. An ARF6/Rab35 GTPase cascade for endocytic recycling and successful cytokinesis. Curr Biol. 2012. January 24;22(2):147–153. [DOI] [PubMed] [Google Scholar]
- [13].Minowa-Nozawa A, Nozawa T, Okamoto-Furuta K, et al. Rab35 GTPase recruits NDP52 to autophagy targets. Embo J. 2017. 15;36(18):2790–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Allaire PD, Seyed Sadr M, Chaineau M, et al. Interplay between Rab35 and Arf6 controls cargo recycling to coordinate cell adhesion and migration. J Cell Sci. 2013. February 1;126(Pt 3):722–731. [DOI] [PubMed] [Google Scholar]
- [15].Shaughnessy R, Echard A.. Rab35 GTPase and cancer: linking membrane trafficking to tumorigenesis. Traffic Cph Den. 2018. January 4;19(4):247–252. [DOI] [PubMed] [Google Scholar]
- [16].Shim J, Lee S-M, Lee MS, et al. Rab35 mediates transport of Cdc42 and Rac1 to the plasma membrane during phagocytosis. Mol Cell Biol. 2010. March;30(6):1421–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Smith AC, Heo WD, Braun V, et al. A network of Rab GTPases controls phagosome maturation and is modulated by salmonella enterica serovar typhimurium. J Cell Biol. 2007. January 29;176(3):263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Egami Y, Fukuda M, Araki N. Rab35 regulates phagosome formation through recruitment of ACAP2 in macrophages during FcγR-mediated phagocytosis. J Cell Sci. 2011. November 1;124(21):3557–3567. [DOI] [PubMed] [Google Scholar]
- [19].Egami Y, Fujii M, Kawai K, et al. Activation-inactivation cycling of Rab35 and ARF6 is required for phagocytosis of zymosan in RAW264 macrophages. J Immunol Res. 2015; Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4502309/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010. June 28;189(7):1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011. October 28;35(4):445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sulston JE, Horvitz HR. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev Biol. 1977. March 1;56(1):110–156. [DOI] [PubMed] [Google Scholar]
- [23].Gumienny TL, Lambie E, Hartwieg E, et al. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development. 1999. March 1;126(5):1011–1022. [DOI] [PubMed] [Google Scholar]
- [24].Sulston JE, Schierenberg E, White JG, et al. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev Biol. 1983. November 1;100(1):64–119. [DOI] [PubMed] [Google Scholar]
- [25].Krysko DV, D’Herde K, Vandenabeele P. Clearance of apoptotic and necrotic cells and its immunological consequences. Apoptosis. 2006. October 1;11(10):1709–1726. [DOI] [PubMed] [Google Scholar]
- [26].Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991. September 1;129(1):79–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Haley R, Wang Y, Zhou Z. The small GTPase RAB-35 defines a third pathway that is required for the recognition and degradation of apoptotic cells. PLOS Genet. 2018. August 23;14(8):e1007558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chaineau M, Ioannou MS, McPherson PS. Rab35: GEFs, GAPs and effectors. Traffic. 2013. November 1;14(11):1109–1117. [DOI] [PubMed] [Google Scholar]
- [29].Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013. January;93(1):269–309. [DOI] [PubMed] [Google Scholar]
- [30].Yu X, Odera S, Chuang C-H, et al. C. elegans dynamin mediates the signaling of phagocytic receptor CED-1 for the engulfment and degradation of apoptotic cells. Dev Cell. 2006. June;10(6):743–757. [DOI] [PubMed] [Google Scholar]
- [31].Zhou Z, Hartwieg E. Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001. January 12;104(1):43–56. [DOI] [PubMed] [Google Scholar]
- [32].Su HP, Nakada-Tsukui K, A-C T-T, et al. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP). J Biol Chem. 2002. April 5;277(14):11772–11779. [DOI] [PubMed] [Google Scholar]
- [33].Mapes J, Chen Y-Z, Kim A, et al. CED-1, CED-7, and TTR-52 regulate surface phosphatidylserine expression on apoptotic and phagocytic cells. Curr Biol. 2012. July 24;22(14):1267–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wu YC, Horvitz HR. The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell. 1998. June 12;93(6):951–960. [DOI] [PubMed] [Google Scholar]
- [35].Venegas V, Zhou Z. Two alternative mechanisms that regulate the presentation of apoptotic cell engulfment signal in Caenorhabditis elegans. Mol Biol Cell. 2007. August;18(8):3180–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wu YC, Horvitz HR. C. elegans phagocytosis and cell-migration protein CED-5 is similar to human DOCK180. Nature. 1998. April 2;392(6675):501–504. [DOI] [PubMed] [Google Scholar]
- [37].Reddien PW, Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000. March;2(3):131–136. [DOI] [PubMed] [Google Scholar]
- [38].Gumienny TL, Brugnera E, A-C T-T, et al. CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell. 2001. October 5;107(1):27–41. [DOI] [PubMed] [Google Scholar]
- [39].Zhou Z, Caron E, Hartwieg E, et al. The C. elegans PH domain protein CED-12 regulates cytoskeletal reorganization via a Rho/Rac GTPase signaling pathway. Dev Cell. 2001. October 1;1(4):477–489. [DOI] [PubMed] [Google Scholar]
- [40].Wu YC, Tsai MC, Cheng LC, et al. C. elegans CED-12 acts in the conserved crkII/DOCK180/Rac pathway to control cell migration and cell corpse engulfment. Dev Cell. 2001. October;1(4):491–502. [DOI] [PubMed] [Google Scholar]
- [41].Hsieh -H-H, Hsu T-Y, Jiang H-S, et al. Integrin α PAT-2/CDC-42 signaling is required for muscle-mediated clearance of apoptotic cells in Caenorhabditis elegans. PLOS Genet. 2012. May 17;8(5):e1002663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hsu T-Y, Wu Y-C. Engulfment of apoptotic cells in C. elegans is mediated by integrin α/SRC signaling. Curr Biol. 2010. March 23;20(6):477–486. [DOI] [PubMed] [Google Scholar]
- [43].Lu N, Zhou Z. Membrane trafficking and phagosome maturation during the clearance of apoptotic cells. Int Rev Cell Mol Biol. 2012;293:269–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008. October;9(10):781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cheng S, Wang K, Zou W, et al. PtdIns(4,5)P2 and PtdIns3P coordinate to regulate phagosomal sealing for apoptotic cell clearance. J Cell Biol. 2015. August 3;210(3):485–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Shen Q, He B, Lu N, et al. Phagocytic receptor signaling regulates clathrin and epsin-mediated cytoskeletal remodeling during apoptotic cell engulfment in C. elegans. Development. 2013. August 1;140(15):3230–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lu N, Shen Q, Mahoney TR, et al. Two PI 3-kinases and one PI 3-phosphatase together establish the cyclic waves of phagosomal PtdIns(3)P critical for the degradation of apoptotic cells. PLoS Biol. 2012. January;10(1):e1001245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yu X, Lu N, Zhou Z. Phagocytic receptor CED-1 initiates a signaling pathway for degrading engulfed apoptotic cells. PLoS Biol. 2008. March;6(3). Available at https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2267821/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kutscher LM, Keil W, Shaham S. RAB-35 and ARF-6 GTPases mediate engulfment and clearance following linker cell-type death. Dev Cell. 2018. 22;47(2):222–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].He B, Yu X, Margolis M, et al. Live-cell imaging in Caenorhabditis elegans reveals the distinct roles of dynamin self-assembly and guanosine triphosphate hydrolysis in the removal of apoptotic cells. Mol Biol Cell. 2010. February 15;21(4):610–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kinchen JM, Ravichandran KS. Identification of two evolutionarily conserved genes regulating processing of engulfed apoptotic cells. Nature. 2010. April 1;464(7289):778–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974. May;77(1):71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Riddle DL, Blumenthal T, Meyer BJ, et al. Introduction to C. elegans. In: Riddle DL, Blumenthal T, Meyer BJ, et al., editors. C elegans II. 2nd ed. 902–1047. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press; 1997. Available at http://www.ncbi.nlm.nih.gov/books/NBK20183/ [Google Scholar]
- [54].Lu N, Yu X, He X, et al. Detecting apoptotic cells and monitoring their clearance in the nematode Caenorhabditis elegans. Methods Mol Biol. 2009;559:357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li Z, Venegas V, Nagaoka Y, et al. Necrotic cells actively attract phagocytes through the collaborative action of two distinct PS-exposure mechanisms. PLOS Genet. 2015. June 10;11(6):e1005285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wood WB, Nematode T. Caenorhabditis elegans. Cold Spring Harbor Lab. 1988;694 p. [Google Scholar]
- [57].Djaldetti M, Bessler H. High temperature affects the phagocytic activity of human peripheral blood mononuclear cells. Scand J Clin Lab Invest. 2015. August 18;75(6):482–486. [DOI] [PubMed] [Google Scholar]
- [58].Pramanik T, Thapa M, Saikia TC. Effect of temperature on phagocytic activity of neutrophils. Nepal Med Coll J NMCJ. 2004. June;6(1):39–40. [PubMed] [Google Scholar]
- [59].Utoh J, Harasaki H. Effects of temperature on phagocytosis of human and calf polymorphonuclear leukocytes. Artif Organs. 1992;16(4):377–381. [DOI] [PubMed] [Google Scholar]
- [60].Xiang W, Rensing L. Changes in cell morphology and actin organization during heat shock in Dictyostelium discoideum: does HSP70 play a role in acquired thermotolerance? FEMS Microbiol Lett. 1999. September;178(1):95–107. [DOI] [PubMed] [Google Scholar]
- [61].Pérez-Pérez JM, Candela H, Micol JL. Understanding synergy in genetic interactions. Trends Genet. 2009. August 1;25(8):368–376. [DOI] [PubMed] [Google Scholar]