

Abstract
Sudden cardiac death due to malignant ventricular arrhythmias remains the major cause of mortality in the postindustrial world. Defective intracellular Ca2+ homeostasis has been well established as a key contributing factor to the enhanced propensity for arrhythmia in acquired cardiac disease, such as heart failure or diabetic cardiomyopathy. More recent advances provide a strong basis to the emerging view that hereditary cardiac arrhythmia syndromes are accompanied by maladaptive remodeling of Ca2+ homeostasis which substantially increases arrhythmic risk. This brief review will focus on functional changes in elements of Ca2+ handling machinery in cardiomyocytes that occur secondary to genetic mutations associated with catecholaminergic polymorphic ventricular tachycardia, and long QT syndrome.
Keywords: Calcium-dependent arrhythmia, Calcium homeostasis remodeling, Heart failure, Catecholaminergic polymorphic ventricular tachycardia, Long QT syndrome
Introduction
Cardiac contractility relies on the coordinated actions of intracellular Ca2+ cycling machinery in cardiomyocytes, including the sarcoplasmic reticulum (SR) Ca2+ release channel ryanodine receptor (RyR2), SR Ca2+ ATPase (SERCa), the electrogenic plasmalemmal Na+/Ca2+ exchanger (NCX1), and the L-Type Ca2+ channel (LTCC) [6, 28]. The tight control of coupling between excitation and Ca2+-dependent contraction of the heart is essential for meeting variable metabolic demands of the body. Inherited mutations in ion channels, auxiliary or structural proteins that alter cardiac cell electrophysiology or cardiac conduction, manifesting as arrhythmia syndromes, usually do not dramatically change basal cardiac contractile function [28]. This strongly suggests that adaptive remodeling of intracellular Ca2+ transport machinery occurs to ensure long-term survival. However, under certain conditions such as stress, functional changes in Ca2+ handling proteins become problematic, exacerbating arrhythmia burden.
Bidirectional control of SR Ca2+ release and sarcolemmal ion fluxes
During early stages of the action potential (AP), a small amount of Ca2+ enters the myocyte via LTCCs and NCX1 in reverse mode. This small amount of Ca2+ is sufficient to activate RyR2s, resulting in a massive Ca2+ release from the main intracellular storage organelle, the SR. Released Ca2+ instantaneously feeds back on sarcolemmal ionic conductance, playing important roles in shaping AP [6]. The large increase in subsarcolemmal [Ca2+] during the Ca2+ transient, which can reach 20–40 μM at its peak [71], effectively inactivates LTCCs, reducing the depolarizing force of Ca2+ current (ICa). At the same time, activation of electrogenic NCX1 in forward mode, which injects 3 Na+ for each 1 Ca2+ removed from the cell, contributes to depolarization and prolongs AP duration (APD). In addition, [Ca2+]i can shape AP via enhancement of Ca2+-dependent K+ and Cl− channels promoting repolarization and shortening APD [39, 40]. Therefore, depending on the specific composition of ionic fluxes, SR Ca2+ release can either prolong or shorten APD. This is especially well illustrated during APD alternans; beat to beat alterations in APD [27]. Concordant alternans exhibit long APD when SR Ca2+ release is large and short when Ca2+ release in small. During discordant alternans, this relationship is reversed. Increase in depolarizing ICa and INCX and decrease in repolarizing currents along with untimely RyR2-mediated Ca2+ release cause arrhythmogenic disturbances in membrane potential called delayed or early afterdepolarizations (DADs and EADs, respectively) that underlie triggered activity at the whole heart level [64].
Pharmacological interventions to rapidly change activity of sarcolemmal ion channels and transporters are known to elicit profound effects on SR Ca2+ release [6]. Pharmacological inhibition of repolarizing K+ channels to prolong APD permits larger [Ca2+] influx via LTCCs, resulting in much larger Ca2+ release. Enhancement of Na+ conductance increases depolarization and induces rapid accumulation of Na+ in the cell, and consequently drives Ca2+ “overload” via inhibition of forward mode NCX1. This can result in increased amplitude of Ca2+ release during systole and generation of spontaneous Ca2+ waves during diastole. However, genetic mutations modifying the same ion fluxes often produce minimal changes in net intracellular Ca2+ cycling under basal conditions both in human patients and animal models. Furthermore, even mutations in components of SR Ca2+ release machinery are relatively well tolerated and manifestation in the form of deadly arrhythmias is a rare, primarily occurring under stress [15, 41, 69]. Therefore, constant change in electrical or mechanical properties, either acquired or inherited, or even change in activity of a single member of Ca2+ handling machinery must cause secondary adaptive changes that allow preservation of a primary heart function, i.e., contractility for as long as possible.
Balance of cellular Ca2+ fluxes
At steady state, the amount of Ca2+ entering the cell via LTCCs during each beat must be equal to Ca2+ extruded by NCX1 [28]. Similarly, the amount of Ca2+ released from the SR by RyR2s must be matched by SERCa-mediated sequestration. Given the key function of rhythmic Ca2+ cycling in cardiomyocytes, there are several self-regulation mechanisms to maintain steady state. The most powerful mechanism is based on the ability of RyR2 to sense Ca2+ not only on the cytosolic side but also in the SR lumen as well. A decrease in luminal [Ca2+] during the Ca2+ transient directly or indirectly forces the cessation of RyR2 cluster activity, eliciting the termination of SR Ca2+ release [30, 73, 75]. Increased RyR2s activity leads to diminished SR Ca2+ content given the loss of Ca2+ during diastole, named SR Ca2+ leak [28]. However, this has a limited impact on the amplitude of systolic Ca2+ release because more active RyR2s remain open at substantially lower intra-SR [Ca2+]. As a result, RyR2-mediated SR Ca2+ leak must be sufficiently large to reduce Ca2+ transient amplitude. Notably, enhancement of RyR2 activity is the most common finding throughout the whole spectrum of acquired cardiac diseases including heart failure (HF), myocardial infarct (MI), diabetic cardiomyopathy, and age-related cardiac dysfunction [31, 32, 60, 84].
Another important self-limiting mechanism is Ca2+-dependent inactivation of LTCCs [6]. Increased ICa significantly increases myocyte loading, with Ca2+ consequently increasing systolic SR Ca2+ release and thereby accelerating LTCC inactivation. Pharmacologically-mediated reduction in NCX1 activity leads to similar effects on SR Ca2+ release and LTCC inactivation [36], which might explain why NCX1 inhibitors do not produce massive Ca2+ overload when used to attenuate EADs and DADs that underlie triggered activity [64]. Importantly, when the metabolic demand of the body increases, such as during stress, self-regulatory mechanisms are overridden to increase cardiac contractility [6]. During stress, the catecholamine-induced increase in LTCC-mediated Ca2+ influx and SERCa-mediated SR Ca2+ sequestration outpaces NCX1-mediated Ca2+ removal and RyR2-mediated diastolic Ca2+ leak, reaching a new steady state with increased systolic SR Ca2+ transient amplitude [28]. Failure to match the fluxes and deficiencies of self-regulatory mechanisms leads to impaired cardiac contractility and an enhanced propensity to Ca2+-dependent arrhythmia.
Regulatory mechanisms of modulation of intracellular Ca2+ homeostasis
As HF is accompanied by profound changes in ionic currents and increased arrhythmogenesis, it is likely there is a substantial overlap of mechanisms underlying the remodeling of Ca2+ homeostasis in hereditary arrhythmia syndromes. Years of research studying remodeling of Ca2+ handling in HF and other models of acquired cardiac disease have revealed a number of fundamental mechanisms affecting function of Ca2+-handling complexes. Increased NCX1 activity in HF has been attributed to increased expression levels and an indirect effect of cytosolic Na+/Ca2+ overload given increased late Na+ current (INaL) [22, 23, 65]. The expression levels of the α1c pore forming subunit of LTCC are decreased in human HF [16, 80]. However, baseline ICa amplitude is not affected because PKA-dependent phosphorylation of the channel, which enhances channel activity, is increased. This results in reduced responsiveness of LTCC to β-adrenergic stimulation in HF. Depressed SERCa activity in HF has been ascribed to decreased expression levels and reduced phosphorylation of auxiliary negative SERCa regulator, phospholamban (PLB) [14, 35, 79]. Increased localized activity of Serine/Threonine phosphatase PP1 underlies hypo-phosphorylation of PLB in HF [14]. This interferes with the relief of PLB’s inhibitory action on SERCa under basal conditions and during catecholaminergic stimulation. Likewise, changes in intracellular signaling cascades are involved in modulation of RyR2 activity in HF [60, 84]. Enhanced PKA- and CaMKII-dependent phosphorylation increase RyR2 activity [1, 52]. Increased RyR2 phosphorylation has been attributed to the increased activity of kinases and the dissociation of opposing phosphatases PP1 and PP2a from the complex [1, 5, 47, 54]. In addition, changes in redox state, metabolism, mitochondrial function, and subcellular structural remodeling are thought to affect Ca2+ homeostasis as well [60, 84]. Recent advances provide growing evidence that many of these mechanisms are similarly involved in Ca2+ handling remodeling in hereditary ventricular arrhythmia syndromes.
Inherited cardiac arrhythmia syndromes and Ca2+ homeostasis remodeling
Catecholaminergic polymorphic ventricular tachycardia
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a highly malignant arrhythmogenic disorder, manifesting as polymorphic or bidirectional VT after emotional stress or exercise in patients with structurally normal hearts [15, 82]. Mutations linked to CPVT are typically associated with gain of function of RyR2 SR Ca2+ release complex that promotes arrhythmogenic spontaneous SR Ca2+ release (Fig. 1). CPVT type 1 is primarily caused by gain of function mutations in RyR2. CPVT types 2–6 have been attributed to loss-of function mutations in auxiliary proteins regulating RyR2 activity. CPVT types 2 and 5 are caused by mutations in SR luminal proteins calsequestrin (CASQ2) and triadin (TRDN) respectively [15], and characterized by loss of control of RyR2 complex activity by luminal Ca2+. Mutations in calmodulin (Calm) 1 and 3 (underlying CPVT types 4 and 6, respectively) and more recently Calm2, which tether to RyR2 at the cytosolic side, interfere with the complex responsiveness to activation by cytosolic Ca2+ [82]. CPVT type 3 has been linked to mutations in trans-2,3-enoyl-CoA reductase-like (TECRL) [24, 59], an enzyme residing primarily in the SR, but the mechanism of action is yet to be defined.
Fig. 1.
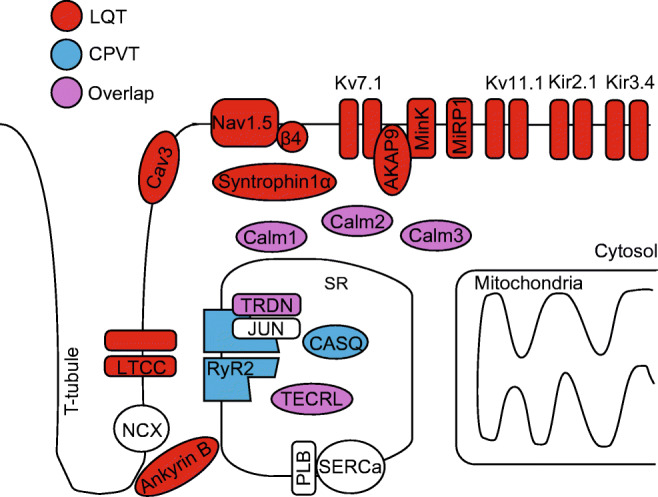
Proteins of cardiac excitation-contraction coupling associated with long QT syndrome or catecholaminergic polymorphic ventricular tachycardia, caused by pathogenic mutation. Proteins with mutations associated with long QT syndrome are colored red; proteins with mutations associated with CPVT are colored blue; proteins with mutations that can cause long QT syndrome or CPVT are colored purple. Kv7.1; KCNQ1 gene, α-subunit of IKs channel, mutations underlie LQT1. Kv11.1; KCNH2 gene, α-subunit of IKr channel, mutation underlies LQT2. Nav1.5; SCN5a gene, α-subunit of INa channel, mutations underlie LQT3. Ankyrin B; ANK2 gene, functions as an adaptor protein, mutations underlie LQT4. minK; KCNE1 gene, β-subunit of IKs channel, mutations underlie LQT5. MiRP1; KCNE2 gene, β-subunit of IKr channel, mutations underlie LQT6. Kir2.1; KCNJ2 gene, α-subunit of IK1 channel, mutations underlie LQT7. LTCC; CACNA1C gene, mutations in α-subunit of ICa,L channel underlie LQT8 (Timothy syndrome). Cav3; CAV3 gene, caveolin-3 protein is a component of caveolae that co-localizes with Nav1.5, mutations underlie LQT9. β4; SCN4B gene, β-subunit of INa channel, mutation underlies LQT10. AKAP9; AKAP9 gene, protein mediates Kv7.1 phosphorylation, mutations underlie LQT11. Syntrophin1α; SNTA1 gene, protein regulates INa function, mutations underlie LQT12. Kir3.4; KCNJ5 gene, subunit of KACh channel, mutations underlie LQT13. Calm1; CALM1 gene, calmodulin serves as a Ca2+-binding messenger protein, mutations underlie LQT14 and CPVT4. Calm2; CALM2 gene, mutations underlie LQT15 and phenotype overlaps with CPVT. Calm3; CALM3 gene, mutations underlie LQT16 and CPVT6. TRDN; TRDN gene, triadin is an accessory protein of RyR2, mutations underlie LQT17, and phenotype overlaps with CPVT5. TECRL; TECRL gene, trans-2,3-enoyl-CoA reductase like protein belongs to the steroid 5-alpha reductase family, mutations underlie CPVT3 and LQT18. RyR2; RYR2 gene, ryanodine receptor is the major sarcoplasmic reticulum Ca2+ release channel, mutations underlie CPVT1. CASQ; CASQ2 gene, calsequestrin2 is an accessory protein of RyR2, mutations underlie CPVT2. JUN; ASPH gene, junctin is an accessory protein of RyR2, no CPVT or LQT-associated mutations reported. SERCa; ATP2A2 gene, protein functions as the sarcoplasmic reticulum Ca2+-ATPase, no CPVT or LQT-associated mutations reported. PLB; PLN gene, phospholamban functions as an inhibitory protein of SERCa, no CPVT or LQT-associated mutations reported
Ca2+ homeostasis and post-translational remodeling
Data accumulated over almost 20 years suggest that CPVT mutations causative of RyR2-mediated SR Ca2+ leak have minimal impact on Ca2+ transient amplitude under basal conditions. Major changes become obvious under β-adrenergic stimulation, including diminished Ca2+ transient amplitude and, importantly, the incidence of spontaneous diastolic Ca2+ waves that drive EADs and DADs [15, 41]. More direct treatment strategies targeting the RyR2 macromolecular complex that have been successfully tested using animal models include (1) pharmacological inhibition of RyR2 (dantrolene [44], flecainide [81], JTV-519 [48, 83]); (2) overexpression of WT form of accessory protein (i.e., CASQ) to reduce impact of a recessive CPVT mutation [52]; (3) gene editing-mediated disruption or siRNA-mediated suppression of a dominant CPVT mutation disease-causing allele [8, 62]; and (4) expression of exogenous plant form of regulatory protein CALM with enhanced ability to stabilize RyR2 [50].
Importantly, several indirect approaches to reduce arrhythmogenicity and improve Ca2+ homeostasis in CPVT models were also proven to be successful. For example, the Radwanski group reported that inhibition of late Na+ current is sufficient to alleviate catecholamine-induced arrhythmia in CASQ2-R33Q knock in (KI) CPVT mice [67]. Liu et al. [49] demonstrated that CaMKII inhibition with pharmacological inhibitor KN93 or inhibitory peptide AIP reduces spontaneous SR Ca2+ release and thereby triggered activity in the form of DADs in cardiomyocytes from RyR2-R4496C(+/−) KI CPVT mice. CaMKII inhibition with KN93 completely alleviated catecholamine-induced sustained ventricular tachyarrhythmia in this model. The efficacy of CaMKII-suppression-based therapy was further validated in experiments using CPVT patient-derived iPSCs and CPVT mice with AAV-mediated overexpression of AIP [7, 25]. Moreover, experiments using CRISPR/CAS9 technology recently showed that phosphorylation at RyR2 CaMKII-specific site Serine-2814 is necessary to reveal CPVT phenotype [63]. Experiments using isolated channels from a CPVT RyR2-V2475F(+/−) KI mouse model showed that phosphorylation at PKA RyR2 phosphorylation site Serine-2030 is increased in response to PKA application, while phosphorylation of CaMKII site Serine-2814 was not changed under similar conditions [51]. Taken together, these findings raise the possibility that in CPVT RyR2 complex loses association with resident phosphatases PP1 and PP2A that counter local activities of PKA and CaMKII, the phenomenon described in HF [1, 5].
Mitochondrial dysfunction
The information regarding CPVT-related changes in mitochondria SR-communication remains limited. Electron microscopy studies have revealed subcellular structural changes in the RyR2-A4860G(+/−) mouse model of CPVT, suggesting altered tunneling and thereby communication patterns between jSR and mitochondria [45]. To our knowledge, there are no reports yet as to whether there are differences in expression levels of mitofusins, the scaffolding proteins that tether SR and mitochondria [68]; and mitochondrial Ca2+ handling proteins including mitochondrial Na+/Ca2+ exchanger (NCLX), and partners of mitochondrial Ca2+ uniporter (MCU), including Micu1, Micu2, and EMRE [29]. Our recent study [34] showed unchanged MCU expression and increased expression of MCU inhibitory paralog MCUb in CASQ2 KO CPVT mouse hearts. We demonstrated that disturbances in the RyR2 SR Ca2+ release complex profoundly affect mitochondrial function, causing excessive production of mitochondrial reactive oxygen species (ROS) such as superoxide and hydrogen peroxide [10, 34]. The role of less reactive of the two, hydrogen peroxide, as a second messenger is well established [72]. Given it can diffuse several microns in the cell milieu from the source [56], mitochondria-derived H2O2 can reach RyR2 clusters which are situated at a distance as close as 20 nm in ventricular myocytes [19]. Increased mito-ROS emission results in oxidation of RyR2, further increasing its activity. Importantly, mito-ROS scavenging with the mitochondrial-targeted antioxidant mito-TEMPO reduced RyR2 oxidation, restored SR Ca2+ content, and reduced incidence of pro-arrhythmic spontaneous Ca2+ waves in β-adrenergic agonist-treated cardiomyocytes from the CASQ2 knock out (KO) CPVT mouse model [34]. Earlier studies using a canine model of tachypacing-induced HF demonstrated increased RyR2 oxidation in ventricular cardiomyocytes [76]. Furthermore, mito-ROS scavenging using mito-TEMPO attenuated RyR2 oxidation and arrhythmogenic spontaneous Ca2+ release in a rabbit model of aging [18]. Together, these studies establish a direct link between RyR2 complex hyperactivity, RyR2 oxidation, and excessive mitochondrial-mediated ROS production, a common phenomenon in both hereditary CPVT and acquired cardiac diseases.
Of note, there is ongoing debate whether mitochondria can shape intracellular Ca2+ cycling serving as a Ca2+ buffer, in addition to being source of ROS [61]. Interestingly, pharmacological enhancement of inner mitochondrial membrane-residing Ca2+ uniporter (MCU) complex, or outer mitochondrial membrane residing channel VDAC, reduced spontaneous Ca2+ release incidence in myocytes from CPVT mice [70]. However, this beneficial effect conflicts with recent data where pharmacological facilitation of mitochondria Ca2+ accumulation was shown to produce mito-ROS surge, exacerbating RyR2 hyperactivity and thereby spontaneous Ca2+ release [33]. Furthermore, analysis of temporal parameters of spontaneous Ca2+ waves in this work showed that both inhibition and facilitation of mitochondrial Ca2+ uptake have no discernible effects on wave propagation velocity, suggesting a minimal role of mitochondria as Ca2+ buffer in adult ventricular myocytes [33]. Changes in Ca2+ wave incidence and frequencies caused by facilitation and inhibition of mito-Ca2+ uptake reported in this manuscript were attributable to the changes in RyR2 oxidation levels by mito-ROS. These results are in line with the view accepted by several leading groups that mitochondria Ca2+ buffering ability in terminally differentiated VMs is very low in comparison to contractile apparatus or SERCa [4, 9, 28, 53].
Subcellular structural remodeling
Typically CPVT mutations do not cause structural remodeling of the heart [15]. However, there is a growing evidence of CPVT-associated changes in ventricular myocyte subcellular organization. The first indications of such phenomena have been obtained using CASQ2 KO CPVT mouse model where Knollmann et al. [43] documented dramatic increase in SR volume, potentially a compensatory change to preserve SR Ca2+ buffering capacity in the absence of CASQ2, a major luminal Ca2+ buffer. An elegant study from this group which followed demonstrated that KO of luminal accessory protein TRDN (to mimic CPVT-linked loss-of TRDN-function mutations) causes profound changes in RyR2 complexes and subcellular structural organization, leading to almost 50% loss of contacts between T-tubules and junctional SR [17]. Loss of contacts between T-tubules and jSR is a recurrent finding in HF [12, 16, 74]. Importantly, prevention of proteasomal degradation of misfolded proteins by an inhibitor of mannosidase-I kifunensine successfully reduced CPVT occurrence in TRDN-KO mice [13]. The loss of jSR-T-tubular contacts in TRDN KO cardiomyocytes results in reduced Ca2+-dependent inactivation of LTCCs, enhancing Ca2+ influx through the plasmalemma. Interestingly, later studies revealed that CPVT linked to TRDN mutations exhibit features consistent with long QT syndrome (LQTS) as well [2], which is not surprising given LTCC inactivation impairment. The overlap with LQTS was also noticed for CPVT TECRL loss-of function mutations manifested by QTc prolongation in patients under catecholaminergic surge [24, 59].
Taken together, these works provide strong support for the concept that initial insult by CPVT mutations causes profound secondary changes in the following: (a) posttranslational control of RyR2 activity; (b) mitochondrial function; and (c) intracellular structural organization. Evidently, these secondary changes are key to revealing the arrhythmogenic phenotype in CPVT.
LQT syndrome and Ca2+ release
Long QT syndrome (LQTS) is a malignant arrhythmogenic disorder, characterized by QT prolongation accompanied with ventricular tachyarrhythmias typically in the form of torsade de pointes (TdP) and polymorphic VT [3, 15, 41, 69]. Arrhythmic events in LQTS usually occur in patients during emotional stress or exercise and less frequently during sleep. Mutations in three genes are responsible for the vast majority of LQTS cases in humans, namely KCNQ1 encoding Kv7.1 channel α-subunit (LQT1, 35% of cases), KCNH2 encoding Kv11.1 channel α-subunit (LQT2, 30% of cases), and SCN5A encoding Nav1.5 Na+ channel α-subunit (LQT3, 10% of cases). Loss-of-function K+ channel mutations reduce repolarizing K+ currents IKs (LQT1) and IKr (LQT2) leading to AP prolongation, similarly to gain-of-function LQT3 mutations in Na+ channel which promote depolarization. As mentioned above, acute pharmacologically induced AP prolongation in ventricular myocytes leads to severe intracellular Ca2+ overload, enhancing both systolic and arrhythmogenic spontaneous SR Ca2+ release. Increased Ca2+ transient amplitude increases cardiac contraction. However, a robust increase in cardiac function is not a common observation in inherited LQTS. Available literature documents mechanical changes in human LQTS patients and large animal models consistent with diastolic dysfunction [41, 69], which implies adaptive remodeling of Ca2+ homeostasis occurs. Given HF is accompanied by a loss of repolarizing currents and increase in INaL, the mechanisms underlying changes in Ca2+ handling may have substantial overlap with those in LQTS.
LQT2
Notably, IKs and IKr have minimal roles in repolarization in rodents [6]. Therefore, the studies using large animal models of LQT1 and LQT2 provide vital opportunities to delineate arrhythmia mechanisms and potential role of changes in Ca2+ homeostasis secondary to mutation-induced AP prolongation [3]. Transgenic rabbits overexpressing LQT2-linked mutant KCNH2 (HERG-G628S) in the heart exhibited significant AP prolongation and high incidence of SCD (> 50% at 1 year) due to polymorphic VT, recapitulating human LQTS [11]. Experiments using isolated ventricular myocytes from LQT2 hearts revealed decrease in SR Ca2+ content and Ca2+ transient amplitude, particularly noticeable under β-adrenergic stimulation [77]. Further analysis showed unchanged ICa and NCX1 function, while SR SERCa-mediated Ca2+ uptake and RyR2-mediated SR Ca2+ leak were accelerated in LQT2 ventricular myocytes. Increased SERCa activity in LQT2 has been attributed to an increase in PKA PLB phosphorylation under baseline conditions [77]. Typically SERCa activity is reduced in HF; however, increased PLB phosphorylation was previously reported in a rabbit pressure-overload-induced model of HF [20]. Enhanced RyR2 activity in LQTS has been ascribed to an increase in PKA and CaMKII phosphorylation of the channel due to the loss of phosphatases PP1 and PP2a from the complex. Identical results were reported earlier in rabbit and canine HF models [1, 5].
Enhanced RyR2-mediated loss of SR Ca2+ during diastole facilitates NCX1-mediated Ca2+ removal to balance increased LTCC-mediated influx during longer AP [6]. More active SERCa plays a primary role in shortening the Ca2+ transient during AP plateau when membrane potentials are close to NCX1 reversal potential. Together, these events prevent a substantial increase in Ca2+ transient amplitude in LQT2 ventricular myocytes under basal conditions, in contrast to pharmacological IKr block. However, in the presence of β-adrenergic agonist isoproterenol, enhanced RyR2 activity becomes the major contributor to triggered activity in the form of arrhythmogenic EADs [67]. Stabilization of RyR2 function by pharmacological inhibition of CaMKII is sufficient to completely alleviate Ca2+-dependent afterdepolarizations in LQT2 ventricular myocytes [67]. Partial inhibition of NCX1 activity either directly by using pharmacological NCX inhibitor SEA400 [55] or indirectly by blocking Late INa with GS967 [37] also effectively eliminates EADs in this model. However, chronic use of NCX1 or INaL inhibitors for arrhythmia prevention in LQT2 requires extensive testing to ensure no adverse effects of such treatments.
The data whether or LQT1 or 2 induces subcellular structural remodeling is lacking. However, proteomics analysis demonstrated significant changes in expression levels and activities of enzymes involved in ATP generation via glucose utilization and fatty acids β-oxidation pathways [38], suggesting increased energy demand and increased supply in LQT1 and LQT2 transgenic rabbit hearts.
LQT3
LQT3 is associated with SCN5A gain-of-function mutations that impede inactivation of the channel, leading to increased INaL [3, 11, 41]. Unlike most LQTS, arrhythmia episodes in LQT3 occur during sleep or rest in the absence of increased catecholaminergic tone. A decrease in heart rhythm provokes profound lengthening of AP and increases incidence of tdP and polymorphic VTs in human patients. Pharmacological INaL enhancement to model LQT3 in rabbit ventricular myocytes induces intracellular Na+/Ca2+ overload, which accelerates mitochondrial ROS production [78]. Increased ROS leads to oxidation and thereby activation of CaMKII. Activated CaMKII phosphorylates RyR2 increasing its activity, which underlies an increase in pro-arrhythmic spontaneous Ca2+ release. In this work, both antioxidants and CaMKII inhibition restored diminished Ca2+ transients and reduced diastolic [Ca2+] and spontaneous Ca2+ waves, similar to the effects in mouse ventricular myocytes with pressure-overload induced HF [78]. Experiments using LQT3 mutation mouse models suggest that increased INaL increases SR Ca2+ load and this increase promotes arrhythmogenic spontaneous waves [46]. Interestingly, in mice with LQT3 evoked by deletion residues 1510–1512 (ΔQKP) in the Scn5a gene, SERCa activity was depressed due to increased PLB expression and its reduced phosphorylation [58]. Furthermore, NCX1 expression and activity were unaltered. This is an interesting finding because Na+ overload is expected to impede forward mode NCX1. The simplest explanation of these phenomena is that INaL enhancement is insufficient to significantly alter intracellular [Na+] despite the profound effect on APD. If this is the case, prolonged LTCC-mediated Ca2+ influx during the long AP is sufficient to increase SR Ca2+ content in LQT3. Indeed, mouse models of LQT8 (Timothy Syndrome) linked to LTCC gain-of-function mutations in CACNA1C also show increase in SR Ca2+ content and increased frequency of spontaneous Ca2+ waves in ventricular myocytes [26]. Remarkably, in the Scn5a ΔQKP LQT3 model, RyR2 phosphorylation remained unchanged and no evidence of enhanced activity of the channel was presented despite an increase in spontaneous Ca2+ waves [58], which is not the case in HF. However, LQT3 Ca2+ transients exhibited longer time-to-peak, suggesting subcellular dyadic structural remodeling: a hallmark of HF.
Notably, to our knowledge, a large animal model of hereditary LQT3 is yet to be created. Given substantial differences in Ca2+ cycling patterns between mice and larger animals, mechanisms of secondary remodeling uncovered in mice may differ greatly than those in humans. In small rodents, an increase in stimulation frequency decreases Ca2+ transient amplitude, e.g., a negative staircase. In rabbits or humans, increased stimulation frequency increases Ca2+ transients [6]. Accordingly, the SR loses Ca2+ at slower rates due to higher NCX1 activity and lower SERCa activity in large animals and humans vs mice. Therefore, given that arrhythmia episodes in human LQT3 patients are prevalent during slower heart rates and assuming that spontaneous Ca2+ release is a key element of trigger [58], there is a good chance that SERCa activity is increased, in stark contrast to mice. Indeed, Xiao Yan Qi et al. [66] showed enhanced SERCa activity and PLB phosphorylation due to enhanced activity of CaMKII in rabbit hearts with slowed heart rate induced by AV block 2 weeks after the procedure. At the cellular level, bradycardia was accompanied by AP prolongation resulting in enhanced LTCC-mediated Ca2+ influx, increased SR Ca2+ load, increased Ca2+ transient amplitude, increased contraction, and importantly, arrhythmogenic EADs at very slow pacing rates in the absence of β-adrenergic stimulation. At higher stimulation rates, presence of β-agonist was necessary for EADs induction.
Other inherited arrhythmia syndromes
The list of genes associated with LQTS is rapidly expanding. Although most of these genes encode proteins that regulate K+ and Na+ conductance, the list of mutations in genes directly involved in Ca2+ handling that manifest as LQTS continues to grow. Calm1, Calm2, Calm3, TRDN, and TECRL (LQT14–18) are recent additions to the CACNA1C gain-of-function mutations associated with LQT8 [2, 24, 26, 41, 58] (Fig. 1). Although much remains to be done to delineate specific mechanism underlying electrical defects triggered by these mutations, it is unequivocally obvious how tightly changes in electrical activity are coupled with changes in Ca2+ handling. Furthermore, the key roles of secondary to initial insult Ca2+ remodeling become widely recognized in other forms of hereditary arrhythmias including Arrhythmogenic Right Ventricular Hypertrophy [21] and Brugada Syndrome [57].
Perspective
Sudden cardiac death remains a major health problem in the postindustrial world. Over the last quarter of century, significant progress has been made in identification of genetic components of malignant cardiac arrhythmia and improved diagnostics. This lead to rapid development of effective therapies; however, further advancement in this area requires a significantly new level of mechanistic understanding. The body of evidence accumulated over the last decade provided strong foundation for a new paradigm-shifting concept when it became obvious that the impact of a single point mutation goes far beyond elementary modification of a certain enzyme or ion channel function. Instead, mutation can induce systemic changes affecting numerous cellular signaling cascades, energy production, protein expression and degradation, and Ca2+ homeostasis. This remodeling, in an attempt to provide long-term preservation of basic contractile cardiac function, ultimately exacerbates arrhythmic potential under certain conditions such as stress. Notably, since the main goal is the same, i.e., preservation of contractile function, remodeling pathways evoked by arrhythmogenic mutations in genes encoding proteins involved in Ca2+ transport, cell electrical activity, or structural elements that underlie cardiac conduction often converge, bearing resemblance of those in structural heart disease and each other (see Fig. 2).
Fig. 2.

Comparison of proarrhythmic changes in action potentials and Ca2+ homeostasis in HF, CPVT, and LQTS 2 and 3 ventricular myocytes. a Schematic of action potentials, Ca2+ transients, and changes in intra-SR Ca2+ in a healthy human ventricular myocyte under β-adrenergic stimulation. Grey dashed lines indicate minimum and maximum Ca2+ levels reached in healthy myocytes. b In HF, APD is prolonged due to decrease in K+ currents and increase in late INa. Ca2+-dependent EADs/DADs underlie arrhythmogenesis under β-adrenergic stimulation. Enhanced sensitivity of RyR2 to intra-SR [Ca2+] due to increased phosphorylation and oxidation of the channel leads to termination of systolic Ca2+ release at reduced intra-SR [Ca2+]. Faster RyR2-mediated SR Ca2+ leak and reduced refractoriness of RyR2 also contributes to the enhanced propensity for proarrhythmic spontaneous Ca2+ release. Enhanced NCX1 activity, depressed SERCa activity and SR Ca2+ leak underlie reduced intra-SR [Ca2+] and diminished Ca2+ transient amplitude. Loss of dyadic contacts between T-tubular LTCCs and jSR RyR2s impedes Ca2+ transient rise. c Under β-adrenergic stimulation, CPVT myocytes exhibit spontaneous Ca2+ release via defective RyR2 complexes, leading to reduced Ca2+ transient amplitude and reduced intra-SR [Ca2+]. Posttranslational remodeling, mitochondrial dysfunction, and subcellular structural remodeling contribute to the hyperactivity of RyR2 caused by CPVT-associated mutations. Proarrhythmic activity of RyR2 drives NCX1 activity, causing a depolarizing inward current and DADs. Uncoupling of LTCCs and RyR2s due to dyad remodeling may increase Ca2+ transient rise time and reduce LTCC Ca2+-dependent inactivation which can result in longer APD. d In LQT2, loss-of-function mutation in KCNH2 reduces outward IKr and prolongs APD during β-adrenergic stimulation. SR Ca2+ leak is accelerated due to hyperphosphorylation of RyR2. SERCa-mediated SR Ca2+ uptake is accelerated at baseline due to PLB phosphorylation. Enhanced activity of hyperphosphorylated RyR2s contributes to a reduction of SR [Ca2+], Ca2+ transients amplitude, and arrhythmogenic EADs under β-adrenergic stimulation. e In LQT3, gain-of-function mutation in SCN5A increases inward late INa and prolongs APD. Arrhythmogenic activity occurs at rest, in the absence of β-adrenergic stimulation. Longer APD increases LTCC-mediated Ca2+ influx. Na+/Ca2+ overload and increased activity of SERCa due to PLB phosphorylation underlies increase in SR Ca2+ content, Ca2+ transient amplitude, and spontaneous RyR2-mediated Ca2+ release thereby EADs at slow rates
Given that arrhythmia syndromes are accompanied by cell systems remodeling, there is great promise in expansion of classical reductionist approaches with rapidly developing new techniques including proteomics, transcriptomics, metabolomics, and big data analytical tools to identify druggable nodes. This is expected to facilitate development of brand new classes of antiarrhythmic agents with improved efficacy and reduced adverse effects. The understanding that remodeling secondary to initial insult caused by a specific mutation has an enormous impact on arrhythmogenesis points to a necessity for expanded genetic screening panels of patients with idiopathic arrhythmias to a new level far beyond classical suspects. Also, although valuable information is being obtained generated using patient-induced pluripotent stem cell (iPSC)–derived cardiomyocytes, this experimental platform needs further development to ensure the highest maturation degree of subcellular structure, metabolic, and signaling cascades, given their key roles in revealing arrhythmogenic phenotype [42]. Finally, the value in future development of engineered tissues and large animal models of hereditary arrhythmias to study mechanisms and test antiarrhythmic therapies cannot be overstated.
Funding
This work was supported by the Ohio State University President’s Postdoctoral Scholars Award (SH) and NIH NHLBI R01HL142588 (DT).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
This article is part of the special issue on Calcium Signal Dynamics in Cardiac Myocytes and Fibroblasts: Mechanisms in Pflügers Archiv—European Journal of Physiology
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ai X, Curran JW, Shannon TR, Bers DM, Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97(12):1314–1322. doi: 10.1161/01.RES.0000194329.41863.89. [DOI] [PubMed] [Google Scholar]
- 2.Altmann HM, Tester DJ, Will ML, Middha S, Evans JM, Eckloff BW, Ackerman MJ. Homozygous/compound heterozygous triadin mutations associated with autosomal-recessive long-QT syndrome and pediatric sudden cardiac arrest: elucidation of the triadin knockout syndrome. Circulation. 2015;131(23):2051–2060. doi: 10.1161/CIRCULATIONAHA.115.015397. [DOI] [PubMed] [Google Scholar]
- 3.Baczkó I, Hornyik T, Brunner M, Koren G, Odening KE. Transgenic rabbit models in proarrhythmia research. Front Pharmacol. 2020;11:853. doi: 10.3389/fphar.2020.00853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bassani JW, Bassani RA, Bers DM. Relaxation in rabbit and rat cardiac cells: species-dependent differences in cellular mechanisms. J Physiol. 1994;476(2):279–293. doi: 10.1113/jphysiol.1994.sp020130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Györke S, Terentyev D. MicroRNA-1 and -133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RyR2 complex. PLoS One. 2011;6(12):e28324. doi: 10.1371/journal.pone.0028324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bers DM. Excitation-contraction coupling and cardiac contractile force. 2001, Springer, 2nd Edition, Volume 237 ISBN : 978–0–7923-7158-8
- 7.Bezzerides VJ, Caballero A, Wang S, Ai Y, Hylind RJ, Lu F, Heims-Waldron DA, Chambers KD, Zhang D, Abrams DJ, Pu WT. Gene therapy for catecholaminergic polymorphic ventricular tachycardia by inhibition of Ca2+/calmodulin-dependent kinase II. Circulation. 2019;140(5):405–419. doi: 10.1161/CIRCULATIONAHA.118.038514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bongianino R, Denegri M, Mazzanti A, Lodola F, Vollero A, Boncompagni S, Fasciano S, Rizzo G, Mangione D, Barbaro S, Di Fonso A, Napolitano C, Auricchio A, Protasi F, Priori SG. Allele-specific silencing of mutant mRNA rescues ultrastructural and arrhythmic phenotype in mice carriers of the R4496C mutation in the ryanodine receptor gene (RYR2) Circ Res. 2017;121(5):525–536. doi: 10.1161/CIRCRESAHA.117.310882. [DOI] [PubMed] [Google Scholar]
- 9.Boyman L, Chikando AC, Williams GSB, Khairallah RJ, Kettlewell S, Ward CW, Smith GL, Kao JPY, Lederer WJ. Calcium movement in cardiac mitochondria. Biophys J. 2014;107(6):1289–1301. doi: 10.1016/j.bpj.2014.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016;100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 11.Brunner M, Peng X, Liu GX, Ren XQ, Ziv O, Choi BR, Mathur R, Hajjiri M, Odening KE, Steinberg E, Folco EJ, Pringa E, Centracchio J, Macharzina RR, Donahay T, Schofield L, Rana N, Kirk M, Mitchell GF, Poppas A, Zehender M, Koren G. Mechanisms of cardiac arrhythmias and sudden death in transgenic rabbits with long QT syndrome. J Clin Invest. 2008;118(6):2246–2259. doi: 10.1172/JCI33578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryant SM, Kong CHT, Watson J, Cannell MB, James AF, Orchard CH. Altered distribution of ICa impairs Ca release at the t-tubules of ventricular myocytes from failing hearts. J Mol Cell Cardiol. 2015;86:23–31. doi: 10.1016/j.yjmcc.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cacheux M, Fauconnier J, Thireau J, Osseni A, Brocard J, Roux-Buisson N, Brocard J, Fauré J, Lacampagne A, Marty I. Interplay between Triadin and Calsequestrin in the pathogenesis of CPVT in the mouse. Mol Ther. 2020;28(1):171–179. doi: 10.1016/j.ymthe.2019.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, Breeden K, Jing SL, Allen PB, Greengard P, Yatani A, Hoit BD, Grupp IL, Hajjar RJ, DePaoli-Roach AA, Kranias EG. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002;22(12):4124–4135. doi: 10.1128/mcb.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cerrone M, Napolitano C, Priori SG. Catecholaminergic polymorphic ventricular tachycardia: a paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm. 2009;6(11):1652–1659. doi: 10.1016/j.hrthm.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 16.Chen X, Piacentino V, 3rd, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91(6):517–524. doi: 10.1161/01.res.0000033988.13062.7c. [DOI] [PubMed] [Google Scholar]
- 17.Chopra N, Yang T, Asghari P, Moore ED, Huke S, Akin B, Cattolica RA, Perez CF, Hlaing T, Knollmann-Ritschel BE, Jones LR, Pessah IN, Allen PD, Franzini-Armstrong C, Knollmann BC. Ablation of triadin causes loss of cardiac Ca2+ release units, impaired excitation-contraction coupling, and cardiac arrhythmias. Proc Natl Acad Sci U S A. 2009;106(18):7636–7641. doi: 10.1073/pnas.0902919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper LL, Li W, Lu Y, Centracchio J, Terentyeva R, Koren G, Terentyev D. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J Physiol. 2013;591(23):5895–5911. doi: 10.1113/jphysiol.2013.260521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Csordás G, Thomas AP, Hajnóczky G. Calcium signal transmission between ryanodine receptors and mitochondria in cardiac muscle. Trends Cardiovasc Med. 2001;11(7):269–275. doi: 10.1016/s1050-1738(01)00123-2. [DOI] [PubMed] [Google Scholar]
- 20.Currie S, Smith GL. Enhanced phosphorylation of phospholamban and downregulation of sarco/endoplasmic reticulum Ca2+ ATPase type 2 (SERCA 2) in cardiac sarcoplasmic reticulum from rabbits with heart failure. Cardiovasc Res. 1999;41(1):135–146. doi: 10.1016/s0008-6363(98)00241-7. [DOI] [PubMed] [Google Scholar]
- 21.Delmar M, McKenna WJ. The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ Res. 2010;107(6):700–714. doi: 10.1161/CIRCRESAHA.110.223412. [DOI] [PubMed] [Google Scholar]
- 22.Despa S, Bers DM. Na+ transport in the normal and failing heart - remember the balance. J Mol Cell Cardiol. 2013;61:2–10. doi: 10.1016/j.yjmcc.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105(21):2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 24.Devalla HD, Gélinas R, Aburawi EH, Beqqali A, Goyette P, Freund C, Chaix MA, Tadros R, Jiang H, Le Béchec A, Monshouwer-Kloots JJ, Zwetsloot T, Kosmidis G, Latour F, Alikashani A, Hoekstra M, Schlaepfer J, Mummery CL, Stevenson B, Kutalik Z, de Vries AA, Rivard L, Wilde AA, Talajic M, Verkerk AO, Al-Gazali L, Rioux JD, Bhuiyan ZA, Passier R., TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med. 2016 Dec 1;8(12):1390-1408. doi: 10.15252/emmm.201505719 [DOI] [PMC free article] [PubMed]
- 25.Di Pasquale E, Lodola F, Miragoli M, Denegri M, Avelino-Cruz JE, Buonocore M, Nakahama H, Portararo P, Bloise R, Napolitano C, Condorelli G, Priori SG. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013;4(10):e843. doi: 10.1038/cddis.2013.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drum BM, Dixon RE, Yuan C, Cheng EP, Santana LF. Cellular mechanisms of ventricular arrhythmias in a mouse model of Timothy syndrome (long QT syndrome 8) J Mol Cell Cardiol. 2014;66:63–71. doi: 10.1016/j.yjmcc.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edwards JN, Blatter LA. Cardiac alternans and intracellular calcium cycling. Clin Exp Pharmacol Physiol. 2014;41(7):524–532. doi: 10.1111/1440-1681.12231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. 2017;121(2):181–195. doi: 10.1161/CIRCRESAHA.117.310230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feno S, Rizzuto R, Raffaello A, Vecellio RD. The molecular complexity of the mitochondrial calcium uniporter. Cell Calcium. 2020;93:102322. doi: 10.1016/j.ceca.2020.102322. [DOI] [PubMed] [Google Scholar]
- 30.Guo T, Gillespie D, Fill M. Ryanodine receptor current amplitude controls Ca2+ sparks in cardiac muscle. Circ Res. 2012;111(1):28–36. doi: 10.1161/CIRCRESAHA.112.265652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton S, Terentyev D. Proarrhythmic remodeling of calcium homeostasis in cardiac disease; Implications for diabetes and obesity Front Physiol 2018 Oct 30;9:1517. doi: 10.3389/fphys.2018.01517. eCollection 2018 [DOI] [PMC free article] [PubMed]
- 32.Hamilton S, Terentyev D. Altered intracellular calcium homeostasis and arrhythmogenesis in the aged heart. Int J Mol Sci. 2019;20(10):2386. doi: 10.3390/ijms20102386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamilton S, Terentyeva R, Kim TY, Bronk P, Clements RT, O-Uchi J, Csordas G, Choi BR, Terentyev D. Pharmacological modulation of mitochondrial Ca2+ content regulates sarcoplasmic reticulum Ca2+ release via oxidation of the ryanodine receptor by mitochondria-derived reactive oxygen species. Front Physiol. 2018;9:1831. doi: 10.3389/fphys.2018.01831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, Bonilla IM, Knollmann BC, Radwański PB, Györke S, Belevych AE, Terentyev D. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. 2020;115(4):38. doi: 10.1007/s00395-020-0797-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasenfuss G, Reinecke H, Studer R, Meyer M, Pieske B, Holtz J, Holubarsch C, Posival H, Just H, Drexler H. Relation between myocardial function and expression of sarcoplasmic reticulum Ca2+-ATPase in failing and nonfailing human myocardium. Circ Res. 1994;75:434–442. doi: 10.1161/01.RES.75.3.434. [DOI] [PubMed] [Google Scholar]
- 36.Hobai IA, Maack C, O’Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95(3):292–299. doi: 10.1161/01.RES.0000136817.28691.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang J, Kim TY, Terentyev D, Zhong M, Kabakov AY, Bronk P, Arunachalam K, Belardinelli L, Rajamani S, Kunitomo Y, Pfeiffer Z, Lu Y, Peng X, Odening KE, Qu Z, Karma A, Koren G, Choi BR. Late I(Na) blocker GS967 Supresses polymorphic ventricular tachycardia in a transgenic rabbit model of long QT type 2. Circ Arrhythm Electrophysiol. 2020;13(8):e006875. doi: 10.1161/CIRCEP.118.006875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jindal HK, Merchant E, Balschi JA, Zhangand Y, Koren G. Proteomic analyses of transgenic LQT1 and LQT2 rabbit hearts elucidate an increase in expression and activity of energy producing enzymes. J Proteome. 2012;75(17):5254–5265. doi: 10.1016/j.jprot.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanaporis G, Blatter LA. Calcium-activated chloride current determines action potential morphology during calcium alternans in atrial myocytes. J Physiol. 2016;594(3):699–714. doi: 10.1113/JP271887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kennedy M, Bers DM, Chiamvimonvat N, Sato D. Dynamical effects of calcium-sensitive potassium currents on voltage and calcium alternans. J Physiol. 2017;595(7):2285–2297. doi: 10.1113/JP273626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kistamás K, Veress R, Horváth B, Bányász T, Nánási PP, Eisner DA. Calcium handling defects and cardiac arrhythmia syndromes. Front Pharmacol. 2020;11:72. doi: 10.3389/fphar.2020.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knollmann BC. Induced pluripotent stem cell-derived cardiomyocytes: boutique science or valuable arrhythmia model? Circ Res. 2013;112(6):969–976. doi: 10.1161/CIRCRESAHA.112.300567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BE, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C, Pfeifer K. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest. 2006;116(9):2510–2520. doi: 10.1172/JCI29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi S, Yano M, Uchinoumi H, Suetomi T, Susa T, Ono M, Xu X, Tateishi H, Oda T, Okuda S, Doi M, Yamamoto T, Matsuzaki M. Dantrolene, a therapeutic agent for malignant hyperthermia, inhibits catecholaminergic polymorphic ventricular tachycardia in a RyR2(R2474S/+) knock-in mouse model. Circ J. 2010;74(12):2579–2584. doi: 10.1253/circj.cj-10-0680. [DOI] [PubMed] [Google Scholar]
- 45.Lavorato M, Iyer VR, Dewight W, Cupo RR, Debattisti V, Gomez L, De la Fuente S, Zhao YT, Valdivia HH, Hajnóczky G, Franzini-Armstrong C. Increased mitochondrial nanotunneling activity, induced by calcium imbalance, affects intermitochondrial matrix exchanges. Proc Natl Acad Sci U S A. 2017;114(5):E849–E858. doi: 10.1073/pnas.1617788113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lemoine MD, Duverger JE, Naud P, Chartier D, Qi XY, Comtois P, Fabritz L, Kirchhof P, Nattel S. Arrhythmogenic left atrial cellular electrophysiology in a murine genetic long QT syndrome model. Cardiovasc Res. 2011;92(1):67–74. doi: 10.1093/cvr/cvr166. [DOI] [PubMed] [Google Scholar]
- 47.Little SC, Curran J, Makara MA, Kline CF, Ho HT, Xu Z, Wu X, Polina I, Musa H, Meadows AM, Carnes CA, Biesiadecki BJ, Davis JP, Weisleder N, Györke S, Wehrens XH, Hund TJ, Mohler PJ. Protein phosphatase 2A regulatory subunit B56α limits phosphatase activity in the heart. Sci Signal. 2015 Jul 21;8(386):ra72. doi: 10.1126/scisignal.aaa5876 [DOI] [PMC free article] [PubMed]
- 48.Liu N, Colombi C, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99(3):292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 49.Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, Napolitano C, Coetzee WA, Priori SG. Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/−) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011;50(1):214–222. doi: 10.1016/j.yjmcc.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Liu B, Walton SD, Ho HT, Belevych AE, Tikunova SB, Bonilla I, Shettigar V, Knollmann BC, Priori SG, Volpe P, Radwański PB, Davis JP, Györke S. Gene transfer of engineered calmodulin alleviates ventricular arrhythmias in a calsequestrin-associated mouse model of catecholaminergic polymorphic ventricular tachycardia. J Am Heart Assoc. 2018;7(10):e008155. doi: 10.1161/JAHA.117.008155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loaiza R, Benkusky NA, Powers PP, Hacker T, Noujaim S, Ackerman MJ, Jalife J, Valdivia HH. Heterogeneity of ryanodine receptor dysfunction in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2013;112(2):298–308. doi: 10.1161/CIRCRESAHA.112.274803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lodola F, Morone D, Denegri M, Bongianino R, Nakahama H, Rutigliano L, Gosetti R, Rizzo G, Vollero A, Buonocore M, Napolitano C, Condorelli G, Priori SG, Di Pasquale E. Adeno-associated virus-mediated CASQ2 delivery rescues phenotypic alterations in a patient-specific model of recessive catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2016;7(10):e2393. doi: 10.1038/cddis.2016.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ Res. 2013;112(3):424–431. doi: 10.1161/CIRCRESAHA.111.300501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 55.Milberg P, Pott C, Fink M, Frommeyer G, Matsuda T, Baba A, Osada N, Breithardt G, Noble D, Eckardt L. Inhibition of the Na+/Ca2+ exchanger suppresses torsades de pointes in an intact heart model of long QT syndrome-2 and long QT syndrome-3. Heart Rhythm. 2008;5(10):1444–1452. doi: 10.1016/j.hrthm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 56.Mishina NM, Tyurin-Kuzmin PA, Markvicheva KN, Vorotnikov AV, Tkachuk VA, Laketa V, Schultz C, Lukyanov S, Belousov VV. Does cellular hydrogen peroxide diffuse or act locally? Antioxid Redox Signal. 2011;14(1):1–7. doi: 10.1089/ars.2010.3539. [DOI] [PubMed] [Google Scholar]
- 57.Monasky MM, Pappone C, Piccoli M, Ghiroldi A, Micaglio E, Anastasia L. Calcium in Brugada syndrome: questions for future research. Front Physiol. 2018;9:1088. doi: 10.3389/fphys.2018.01088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Montnach J, Chizelle FF, Belbachir N, Castro C, Li L, Loussouarn G, Toumaniantz G, Carcouët A, Meinzinger AJ, Shmerling D, Benitah JP, Gómez AM, Charpentier F, Baró I. Arrhythmias precede cardiomyopathy and remodeling of Ca2+ handling proteins in a novel model of long QT syndrome. J Mol Cell Cardiol. 2018;123:13–25. doi: 10.1016/j.yjmcc.2018.08.019. [DOI] [PubMed] [Google Scholar]
- 59.Moscu-Gregor A, Marschall C, Müntjes C, Schönecker A, Schuessler-Hahn F, Hohendanner F, Parwani AS, Boldt LH, Ott CE, Bennewiz A, Paul T, Krause U, Rost I. Novel variants in TECRL cause recessive inherited CPVT type 3 with severe and variable clinical symptoms. J Cardiovasc Electrophysiol. 2020;31(6):1527–1535. doi: 10.1111/jce.14446. [DOI] [PubMed] [Google Scholar]
- 60.Niggli E, Ullrich ND, Gutierrez D, Kyrychenko S, Polakova E, Shirokova N. Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC-coupling. Biochim Biophys Acta. 2013;1833(4):866–875. doi: 10.1016/j.bbamcr.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.O’Rourke B, Blatter LA. Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol. 2009;46(6):767–774. doi: 10.1016/j.yjmcc.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pan X, Philippen L, Lahiri SK, Lee C, Park SH, Word TA, Li N, Jarrett KE, Gupta R, Reynolds JO, Lin J, Bao G, Lagor WR, Wehrens XHT. In vivo Ryr2 editing corrects catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2018;123(8):953–963. doi: 10.1161/CIRCRESAHA.118.313369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park SJ, Zhang D, Qi Y, Li Y, Lee KY, Bezzerides VJ, Yang P, Xia S, Kim SL, Liu X, Lu F, Pasqualini FS, Campbell PH, Geva J, Roberts AE, Kleber AG, Abrams DJ, Pu WT, Parker KK. Insights into the pathogenesis of catecholaminergic polymorphic ventricular tachycardia from engineered human heart tissue. Circulation. 2019;140(5):390–404. doi: 10.1161/CIRCULATIONAHA.119.039711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med. 2004;14(2):61–66. doi: 10.1016/j.tcm.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 65.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85(11):1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 66.Qi X, Yeh YH, Chartier D, Xiao L, Tsuji Y, Brundel BJ, Kodama I, Nattel S. The calcium/calmodulin/kinase system and arrhythmogenic afterdepolarizations in bradycardia-related acquired long-QT syndrome. Circ Arrhythm Electrophysiol. 2009;2(3):295–304. doi: 10.1161/CIRCEP.108.815654. [DOI] [PubMed] [Google Scholar]
- 67.Radwański PB, Brunello L, Veeraraghavan R, Ho HT, Lou Q, Makara MA, Belevych AE, Anghelescu M, Priori SG, Volpe P, Hund TJ, Janssen PM, Mohler PJ, Bridge JH, Poelzing S, Györke S. Neuronal Na+ channel blockade suppresses arrhythmogenic diastolic Ca2+ release. Cardiovasc Res. 2015;106(1):143–152. doi: 10.1093/cvr/cvu262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell. 2016;61(5):683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 69.Schwarz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5(4):868–877. doi: 10.1161/CIRCEP.111.962019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schweitzer MK, Wilting F, Sedej S, Dreizehnter L, Dupper NJ, Tian Q, Moretti A, My I, Kwon O, Priori SG, Laugwitz KL, Storch U, Lipp P, Breit A, Schnitzler MMY, Gudermann T, Schredelseker J. Suppression of arrhythmia by enhancing mitochondrial Ca 2+ uptake in catecholaminergic ventricular tachycardia models. JACC Basic Transl Sci. 2017;2(6):737–747. doi: 10.1016/j.jacbts.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sies H, Berndt C, Jones DP. Oxidative stress. Annu Rev Biochem. 2017;86:715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 73.Sobie EA, Dilly KW, dos Santos CJ, Lederer WJ, Jafri MS. Termination of cardiac Ca(2+) sparks: an investigative mathematical model of calcium-induced calcium release. Biophys J. 2002;83(1):59–78. doi: 10.1016/s0006-3495(02)75149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci U S A. 2006;103(11):4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Terentyev D, Viatchenko-Karpinski S, Valdivia HH, Escobar AL, Gyorke S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ Res. 2002;91(5):414–420. doi: 10.1161/01.res.0000032490.04207.bd. [DOI] [PubMed] [Google Scholar]
- 76.Terentyev D, Györke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Györke S. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res. 2008;103(12):1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Terentyev D, Rees CM, Li W, Cooper LL, Jindal HK, Peng X, Lu Y, Terentyeva R, Odening KE, Daley J, Bist K, Choi BR, Karma A, Koren G. Hyperphosphorylation of RyRs underlies triggered activity in transgenic rabbit model of LQT2 syndrome. Circ Res. 2014;115(11):919–928. doi: 10.1161/CIRCRESAHA.115.305146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Viatchenko-Karpinski S, Kornyeyev D, El-Bizri N, Budas G, Fan P, Jiang Z, Yang J, Anderson ME, Shryock JC, Chang CP, Belardinelli L, Yao L. Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J Mol Cell Cardiol. 2014;76:247–256. doi: 10.1016/j.yjmcc.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Waggoner JR, Kranias EG. Role of phospholamban in the pathogenesis of heart failure. Heart Fail Clin. 2005;1(2):207–218. doi: 10.1016/j.hfc.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 80.Wang W, Zhang H, Gao H, Kubo H, Berretta RM, Chen X, Houser SR. {beta}1-Adrenergic receptor activation induces mouse cardiac myocyte death through both L-type calcium channel-dependent and -independent pathways. Am J Physiol Heart Circ Physiol. 2010;299(2):H322–H331. doi: 10.1152/ajpheart.00392.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Watanabe H, Chopra N, Laver D, Hwang HS, Davies SS, Roach DE, Duff HJ, Roden DM, Wilde AA, Knollmann BC [DOI] [PMC free article] [PubMed]
- 82.Wleklinski MJ, Kannankeril PJ, Knollmann BC. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J Physiol. 2020;598(14):2817–2834. doi: 10.1113/JP276757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang XH, Wei H, Xia Y, Morad M. Calcium signaling consequences of RyR2 mutations associated with CPVT1 introduced via CRISPR/Cas9 gene editing in human-induced pluripotent stem cell-derived cardiomyocytes: Comparison of RyR2-R420Q, F2483I, and Q4201R. Heart Rhythm. 2020 Sep 12;S1547–5271(20)30889–4. doi: 10.1016/j.hrthm.2020.09.007 [DOI] [PMC free article] [PubMed]
- 84.Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch. 2014;466(6):1129–1137. doi: 10.1007/s00424-014-1469-3. [DOI] [PMC free article] [PubMed] [Google Scholar]