

Abstract
Pompe disease is a lysosomal and neuromuscular disorder caused by deficiency of acid alpha-glucosidase (GAA), and causes classic infantile, childhood onset, or adulthood onset phenotypes. The biochemical diagnosis is based on GAA activity assays in dried blood spots, leukocytes, or fibroblasts. Diagnosis can be complicated by the existence of pseudodeficiencies, i.e., GAA variants that lower GAA activity but do not cause Pompe disease. A large-scale comparison between these assays for patient samples, including exceptions and borderline cases, along with clinical diagnoses has not been reported so far. Here we analyzed GAA activity in a total of 1709 diagnostic cases over the past 28 years using a total of 2591 analyses and we confirmed the clinical diagnosis in 174 patients. We compared the following assays: leukocytes using glycogen or 4MUG as substrate, fibroblasts using 4MUG as substrate, and dried blood spots using 4MUG as substrate. In 794 individuals, two or more assays were performed. We found that phenotypes could only be distinguished using fibroblasts with 4MUG as substrate. Pseudodeficiencies caused by the GAA2 allele could be ruled out using 4MUG rather than glycogen as substrate in leukocytes or fibroblasts. The Asian pseudodeficiency could only be ruled out in fibroblasts using 4MUG as substrate. We conclude that fibroblasts using 4MUG as substrate provides the most reliable assay for biochemical diagnosis and can serve to validate results from leukocytes or dried blood spots.
Subject terms: Diagnosis, Disease genetics
Introduction
Pompe disease is an autosomal recessive metabolic disorder caused by acid α-glucosidase (GAA) deficiency, which leads to intralysosomal accumulation of glycogen causing progressive damage especially to cardiac and skeletal muscles [1–3]. Pompe disease presents as a spectrum of phenotypes. The classic infantile form is associated with rapidly progressive general muscle weakness and hypertrophic cardiomyopathy, culminating in death within the first year of life if left untreated. Less severe and less progressive forms can manifest at any age from infancy to late adulthood and present with proximal muscle weakness and/or respiratory problems with minimal or no cardiac involvement [4–7]. These patients may eventually lose ambulation and/or become ventilator dependent. Patients with less severe forms and with the same GAA genotype can have broad phenotypic variation, with symptom onset ranging from early infantile to late adult [8]. This indicates the presence of environmental, epigenetic, or genetic modifying factors. Recently we identified the silent, cis-acting c.510C>T GAA variant as a genetic modifier of symptom onset and splicing in Pompe disease [9]. Symptoms in other tissues and organs also occur including in smooth muscle, visceral organs, and, in the classic infantile form, the central nervous system. Enzyme replacement therapy with recombinant human GAA is available and is often applied in combination with immunomodulation in classic infantile cases to reduce the chance of antibody formation [10–13]. Early diagnosis including phenotype prediction is required to optimize counseling and to ensure a timely start of treatment.
Current diagnostic guidelines recommend the establishment of GAA enzyme deficiency with additional confirmation of two disease-associated GAA variants [14, 15]. Disease-associated variants, recently linked to clinical phenotypes, are listed in the open access Pompe disease GAA variant database [www.pompevariantdatabase.nl] [16, 17]. Different enzymatic diagnostic assays are available in which the biological material and the choice of substrate are variables.
Biological materials can be leukocytes, dried blood spots (DBSs), fibroblasts derived from skin biopsies, and muscle tissue. Leukocytes and DBSs are easily obtained, which is important for timely screening, diagnosis, and treatment of classic infantile patients. Newborn screening (NBS) programs are based on DBS assays and positive cases require a second-tier assay for confirmation [18]. Muscle biopsies can be used for diagnosis however, these are not always available and involve a rather invasive and sometimes painful procedure. In addition, in the case of fat replacement of muscle, which can occur in Pompe disease, it can be difficult to obtain muscle cells from a muscle biopsy. Substrates include the natural substrate glycogen, and the artificial substrates 4-methylumbelliferyl-α-D-glucopyranoside (4MUG) [19] and (GAA-S), a substrate used in tandem mass spectrometry for NBS [20]. In certain assays such as using leukocytes or DBSs, the neutral hydrolase glucoamylase activity needs to be inhibited using acarbose as it interferes with the measurement of GAA activity [21–23].
Additional evidence for the diagnosis of Pompe disease can be obtained by measuring urine tetrasaccharide Glc4 (TGLC), which can be measured using HPLC with UV detection or using mass spectrometry. This biomarker has been found to be sensitive but it is not specific to Pompe disease, as it is also elevated in liver abnormalities or in response to food intake. In borderline cases, it might help to establish the diagnosis [24–26].
For the interpretation of diagnostic outcome, it is important to consider the following information on GAA DNA variants. (1) At least two (combinations of) GAA variants can lead to pseudodeficiency. In the Caucasian population, the GAA2 (c.271G>A) pseudodeficiency variant lowers the activity of GAA for the natural substrate glycogen, but not for the artificial substrate 4MUG [27, 28]. In the Asian population, the common GAA pseudodeficiency variants c.[1726G>A; 2065G>A] can lower GAA activity to levels that come close to the disease threshold, and their presence can lead to a false positive diagnosis in certain assays [23, 29, 30]. (2) Standard DNA diagnostic analysis may fail to identify GAA variants, in these cases extended analysis is required as described [31–33].
Over the past 28 years, our laboratory has processed 1709 diagnostic cases and has diagnosed over 250 patients with Pompe disease using various assays separately and in parallel. In this article we are presenting and reviewing all our test results of blood-based and fibroblast-based assays from 1990 until 2018 allowing to compare the various methods for enzymatic diagnosis and their pitfalls.
Materials and methods
Nomenclature and Pompe variant database
The variant nomenclature is according to HGVS standards [34]. The reference sequences used for the annotation of GAA cDNA variants were NM_000152.5 and LRG_673t1.1. Position c.1 represents the first nucleotide of the translation start codon ATG located in exon 2. Exon numbering was according to Niño et al., 2019 [17]. NP_000143.2 was used for annotation of GAA protein variants. Variant information was retrieved from the Pompe disease GAA variant database at www.pompevariantdatabase.nl and the Leiden open variation database (http://lovd.nl/gaa).
Diagnostic materials and assay procedures
GAA activity assays were performed when there was a suspicion of Pompe disease based on clinical symptoms (described below). Results were obtained over the period 1990–2018. Reference ranges reflect those of the Diagnostic Department of the Erasmus MC and were established for each assay based on at least 20 individuals per phenotype (classic infantile, childhood, adulthood, healthy controls) with a confirmed clinical diagnosis. Values in between these ranges are specified as ‘gray zone’, which is defined as GAA enzyme activities above the patient range but below the control range. For fibroblasts, the reference ranges were derived from data collected over a 40 years period. For the other assays, including GAA activity in leukocytes and bloodspots, and tetrasaccharide 6-α-D-glucopyranosyl-maltotriose (Glc4) concentration in urine by mass spectrometry, data collection of at least 8-years was used [24]. All reference ranges for patients, healthy controls, and the gray zones are indicated in the legends of the figures. The study was conducted according to the Declaration of Helsinki. The Medical Ethical Committee at Erasmus University MC approved the study protocol, and all patients, or their parents or legal guardians, provided written informed consent.
Activity assays were performed as previously described [22, 24, 35–37]. A final concentration of 3 µmol/L acarbose (in the reaction mixture) was used for the glycogen assay and 8 µmol/L acarbose for the 4MUG assay to inhibit glucoamylase activity present in mixed leukocytes and which interferes with GAA activity measurements. Performance of the assays was determined for each individual assay by including an internal standard sample. According to standard diagnostic procedures, a maximal deviation of less than 10% of the internal standard was required to use values as diagnostic values. For all assays, no false positive or false negative values of the internal standard were obtained. Typical standard deviations were: leukocytes/4MU: 5.1% (n = 29); leukocytes/glycogen: 6.2% (n = 72); fibroblasts/4MUG: 7.1% (n = 78); DBS (in our Diagnostic Center not used as diagnostic assay): 11.3% (n = 7). The GAA activity in DBS was determined in the presence of 8 µmol/L acarbose for glucoamylase inhibition and after precipitation of hemoglobin as previously described [35, 36].
Clinical diagnosis
We classified patients with classic-infantile Pompe disease when they presented symptoms at or under 12 months of age. Symptoms in these patients consist of muscle weakness combined with a hypertrophic cardiomyopathy. The phenotype of childhood and adulthood onset Pompe disease is dominated by a progressive limb-girdle myopathy that leads to severe functional limitations, while respiratory muscle dysfunction limits patients’ average life span. Childhood onset is here defined as symptom onset before the age of 18 years, adult onset at 18 years or older. The diagnosis was established based on combined evidence consisting of these clinical symptoms, GAA enzyme deficiency, and DNA analysis.
Results
Over the past 28 years, we analyzed a total of 1709 individuals using a total of 2591 assays (Fig. 1 and Table S1). In the majority of individuals, leukocytes were used with both glycogen and 4MUG as substrate (n = 637). In 539 individuals, fibroblasts using 4MUG as substrate were used as single assay, while in 375 individuals, leukocytes with glycogen were used as single assay. Only one individual was analyzed with leukocytes using 4MUG as substrate as single assay. Eighty-eight individuals were analyzed with all three assays, while 69 individuals were analyzed with both leukocytes/glycogen and fibroblasts/4MUG. As part of this study we traced back the clinical data of all patients with a confirmed enzymatic diagnosis. The following categories for the diagnoses were defined: ‘Classic Infantile’, ‘Childhood onset’ (symptom onset before the age of 18 years), ‘Adult onset’ (symptom onset at 18 years and older), ‘no Pompe disease’, and ‘Unknown’. For enzymatic assays, the following additional categories were defined: ‘Unknown/Deficient’ (i.e., the value of the assay was deficient but it was unknown whether the individual had Pompe disease), ‘No Pompe/Deficient’ (i.e., the value of the assay was deficient but Pompe disease was excluded based on other assays), ‘Asymptomatic/Deficient’ (i.e., the value of the assay was deficient but the individual was asymptomatic), and ‘Gray Zone’ (the values above patient ranges but below normal ranges).
Fig. 1. Venn diagram showing how many diagnostic cases were processed with each of the three different enzymatic methods.
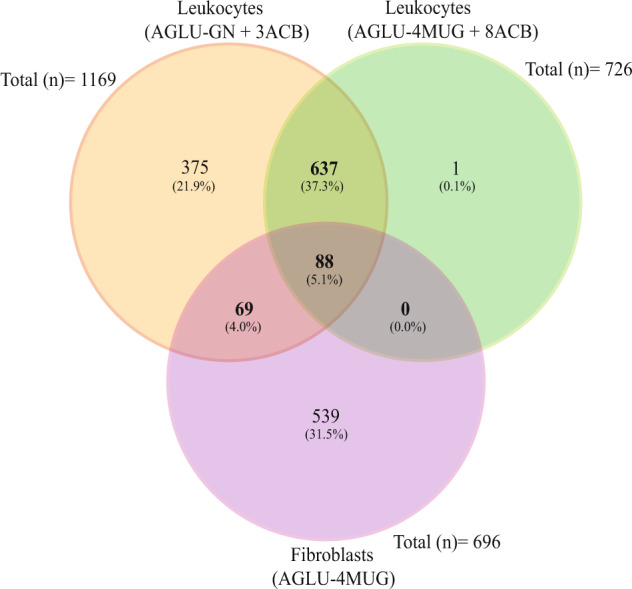
The figure shows the similarities and differences of individuals whereby the GAA activity was measured in leukocytes with glycogen as well as with 4MUG as substrate, and in fibroblast with 4MUG. Of note 11 assay results (five from leukocytes with glycogen four from leukocytes with 4MUG, and two from fibroblast with 4MUG) were not included in this comparison since they are biological replicates. DBS assays (20 assays from 18 individuals) are not shown.
GAA activity assay in leukocytes using glycogen as substrate
Figure 2A shows GAA activities using glycogen as substrate that were measured in 1169 individuals. Within the range of 0–3.5 nmol/mg/h for classic infantile patients, there were 20 classic infantile patients, 15 with childhood onset, and 111 with adult onset Pompe disease. Two individuals were ‘unknown deficient’. Within the range of 3.5–10 nmol/mg/h, which is the higher activity range for childhood/adult onset patients (range 0–10 nmol/h/mg), there were 1 classic infantile patient, 6 with childhood onset, 20 with adult onset, 7 ‘no Pompe/deficient’, and 3 ‘unknown/deficient’. This indicated that the range of 0–3.5 nmol/mg/h contained 95% of the classic infantile, 71% of the childhood onset, and 84% of the adulthood onset patients. The 3.5–10 nmol/mg/h range contained 5% of the classic infantile, 29% of the childhood onset, and 16% of the adulthood onset patients. In both ranges, putative false positives were found to a total of 12 individuals. A subset of these false positives was followed up using leukocytes/4MUG and fibroblasts/4MUG assays, and these showed mainly gray zone (seven assays) and normal (nine assays) values, while one assay in leukocytes/4MUG showed an unknown/deficient value (see Table S1 for raw data). Within the range of 10–40 nmol/mg/h, 52 individuals were classified with gray zone (above the patient range but below the control range) values, while 1 classic infantile patient was present. This patient showed values in the patient range in leukocytes/4MUG (3.46 nmol/mg/h) and in the classic infantile range in fibroblasts/4MUG (0.2 nmol/mg/h). In the normal ranges above 40 nmol/mg/h, a total of 921 healthy individuals were present. In conclusion, the leukocytes/glycogen assay has limited ability to discriminate between patients with classic infantile and late onset phenotypes, has a considerable risk of false positive, and a low risk of false negative outcomes.
Fig. 2. Distribution of GAA activity in leukocytes using.

A Glycogen as substrate in the presence of 3 µmol/L acarbose (AGLU-GN+ 3ACB assay). A total of 1169 individuals suspected of Pompe disease were included. B 4MUG as substrate in the presence of 8 µmol/L acarbose (AGLU-4MUG+ 8ACB assay). A total of 726 individuals suspected of Pompe disease were included in this series.
GAA activity assay in leukocytes using 4MUG as substrate
In Fig. 2B, the activities in leukocyte assays using 4MUG as substrate, measured in 726 individuals, are shown. In the patient range of 0.0–4.9 nmol/h/mg, there were 19 classic infantile patients, 10 with childhood and 66 with adulthood onset, 2 were no Pompe/deficient, and 17 were unknown/deficient. The two individuals that were no Pompe/deficient and the 17 individuals that were unknown/deficient showed either gray zone and/or normal values when tested in leukocytes/4MUG and/or fibroblasts/4MUG. This showed that the leukocyte/4MUG assay did not distinguish classic infantile from late onset phenotypes. The range 4.9–6.7 showed 1 childhood onset patient, 1 adult onset patient, and 57 gray zone individuals. The childhood onset patient had borderline activity just above the threshold (5.14 nmol/mg/h) and tested in the patient range (0.365 nmol/mg/h) in the leukocyte/glycogen assay. The healthy range > 6.7 nmol/mg/h included 548 healthy individuals and 5 individuals with somewhat increased enzyme activities. This indicated that the leukocyte/4MUG assay could faithfully discriminate between healthy and diseased individuals but could also result in false positive outcomes.
GAA activity assay in fibroblasts with 4MUG as substrate
In Fig. 3, the activities in fibroblasts using 4MUG as substrate are shown. In the range 0–3 nmol/h/mg for classic infantile patients, there were 77 classic infantile patients, 8 patients with childhood and 3 with adulthood onset. One patient with childhood onset had an activity in between 3 and 4.2. In the range 4.2–20 nmol/mg/h for childhood/adult onset patients, there were 1 classic infantile patient, 18 with childhood onset and 108 with adulthood onset. There were six asymptomatic/deficient individuals. These individuals may develop symptoms later in life, as symptom onset in Pompe disease can be highly variable. Two of these individuals were also tested using leukocytes/glycogen and 4MUG, and in both individuals, these two assays yielded gray zone values. This indicated that the fibroblasts/4MUG assay could distinguish classic infantile from childhood/adulthood onset patients to a large extent, and between childhood and adulthood patients to a lesser extent. There were 61 individuals with enzyme activities in the gray zone range of >20–45 nmol/h/mg. All individuals with activities in the healthy range >45 nmol/h/mg were healthy. This showed that the fibroblast/4MUG assay can distinguish both healthy from diseased individuals and classic infantile from childhood/adulthood onset phenotypes.
Fig. 3. Distribution of GAA activity in fibroblast using the artificial substrate 4-methylumbelliferyl-α-D-glucoside (4MUG) (AGLU-4MUG assay).

A total of 695 individuals suspected of Pompe disease were included in this series.
GAA activity assay in DBS
Table 1 shows the results of DBS assays in 18 individuals with Pompe disease. In 15 cases (12 classic infantile, 2 childhood, 1 with an unknown phenotype), GAA activity was in the patient range of 11–56 pmol/17 h/punch, while three individuals (patients 30–32, who had classic infantile Pompe disease) were in the gray zone of 56–94 pmol/17 h/punch. However, all individuals including individuals 30–32 tested in the patient range in leukocytes with glycogen as substrate. One sample, from patient 32 was measured three times, of which one was in the patient range and 2 in the gray zone. Fibroblasts from this patient showed activity in the patient range and DNA analysis of this patient and patients 30 and 31 confirmed the presence of two severe GAA disease-associated variants (Table 2 and Fig S1). These results indicated that in this cohort the DBS assay resulted in relatively frequent false negative outcomes (16.7% in this analysis) although the number of DBS samples analyzed here (n = 18) was low.
Table 1.
GAA activity in Pompe patients measured in DBS and leukocytes.
| Patient | Dried blood spot (DBS) | LEUKOCYTES | Clinical diagnosis | ||
|---|---|---|---|---|---|
| AGLU-4MUG + 8ACB (pmol/17 h/punch) | BGAL (pmol/17 h/punch) | AGLU-GN+3ACB (nmol/h/mg) | BGAL (nmol/h/mg) | ||
| 15 | 30.8 | 4730 | 1.6 | 152 | Classic infantile Pompe disease |
| 16 | 48.8 | 2480 | 1.0 | 131 | Childhood onset Pompe disease |
| 17 | 13.8 | 2860 | –0.1 | 162 | Classic infantile Pompe disease |
| 18 | 25.2 | 2520 | 0.8 | 129 | Classic infantile Pompe disease |
| 19 | 17.4 | 1660 | 5.0 | 147 | Classic infantile Pompe disease |
| 20 | 11.4 | 1380 | 1.2 | 192 | Classic infantile Pompe disease |
| 21 | 10.6 | 1780 | –1.1 | 174 | Classic infantile Pompe disease |
| 22 | 27.8 | 3100 | –0.2 | 192 | Classic infantile Pompe disease |
| 23 | 14.6 | 2040 | –1.6 | 167 | Classic infantile Pompe disease |
| 24 | 12.9 | 2950 | 0.3 | 207 | Classic infantile Pompe disease |
| 25 | 25.4 | 3920 | 0.1 | 172 | Classic infantile Pompe disease |
| 26 | 28.6 | 3710 | 2.8 | 104 | Childhood onset Pompe disease |
| 27 | 25 | 4990 | 0.9 | 222 | Unknown |
| 28 | 8.51 | 3060 | 1.2 | 161 | Classic infantile Pompe disease |
| 29 | 41.5 | 4700 | –0.4 | 181 | Classic infantile Pompe disease |
| 30 | 59.4 | 3620 | –2.0 | 225 | Classic infantile Pompe disease |
| 31 | 58.6 | 6050 | –1.1 | 169 | Classic infantile Pompe disease |
| 32* | 85.3 | 4710 | –0.7 | 139 | Classic infantile Pompe disease |
| 68 | 5230 | Classic infantile Pompe disease | |||
| 48.6 | 3250 | Classic infantile Pompe disease | |||
| Normal range | 94–448 | 476–4680 | >40–250 | 50–326 | |
| Patient range | 11–56 | 88 | Classic infantile: 0–3.5 | 0.6–6.3 | |
| Childhood/Adult: 0–10 | |||||
Cases 30–32 were borderline inconclusive using the DBS, but were positively confirmed with leukocytes as sample source. *Patient 32 showed variable measurements of GAA activity and BGAL in DBS (technical replicates were performed). Patient 16 and 26 were diagnosed with childhood Pompe disease, and individual 27 was classified as Unknown/Deficient due lack of clinical information.
Italic: values within the patient range.
Table 2.
Follow up of three cases with inconclusive DBS data.
| Patient | Genotype DNA (protein) | Predicted severity (Pompe disease GAA variant database) | ACMG variant classification | Fibroblast (GAA activity) | Clinical diagnosis |
|---|---|---|---|---|---|
| AGLU-4MUG (nmol/h/mg) | |||||
| 30 | c.1460T>C p.(Phe487Ser); | Potentially less severe | Likely pathogenic | 0.432 | Classic infantile |
| c.1460T>C p.(Phe487Ser) | Potentially less severe | Likely pathogenic | Pompe disease | ||
| 31 | c.379_380del p.(Cys127Leufs*18); | Very severe | Pathogenic | 0.35 | Classic infantile |
| c.525del p.(Glu176Argfs*45) | Very severe | Pathogenic | Pompe disease | ||
| 32 | c.2481+102_2646+31del p.(Gly828_Asn882del); | Very severe | Pathogenic | 0.415 | Classic infantile |
| c.525del p.(Glu176Argfs*45) | Very severe | Pathogenic | Pompe disease | ||
| Ranges (GAA activity) | Normal range | >45–180 | |||
| Classic infantile | 0–3 | ||||
| Childhood/Adult | 4.2–20 | ||||
DNA analysis and GAA activity in cultured fibroblasts left no doubt that all three patients had classic infantile Pompe disease.
GAA2 pseudodeficiency
Individuals with pseudodeficiencies can give false positive outcomes in GAA enzyme assays. To illustrate this, we compared individuals with confirmed presence of the GAA2 (c.271G>A) and Asian c.[1726G>A; 2065G>A] pseudodeficiencies using different GAA activity assays.
In four individuals with the GAA2 variant at heterozygous state without other known GAA disease-associated variants, activities were in the gray zone (two cases) or in the normal range (two cases) in leukocytes/glycogen (Table 3). In leukocytes/4MUG, one case had activity in the gray zone, and three in the normal range. In fibroblasts/4MUG, three patients were tested, two of whom were in the normal range and one (individual 4) in the patient range. This indicated that the GAA2 allele at heterozygous state can already lower GAA activity in leukocyte-based assays to values in the gray zone. Individual 4 had a medical record of hypertrophic cardiomyopathy. This individual underwent metabolic screening as no cause for his hypertrophic cardiomyopathy could initially be found. Genetic testing using WES led to the identification of a disease-associated variant in exon 19 (c.1831G>A, p.Gly611Ser) of the MYBPC1 gene (Myosin Binding Protein C, slow type), which plays an important role in muscle contraction and cardiac conduction. No muscle biopsy or MRI was performed, CK was normal (91 U/l). He had an incomplete traumatic cervical spinal cord lesion; he can walk but suffers from spasms due to this condition. General and neurological examination showed no other abnormalities, and there were no signs or symptoms of Pompe disease.
Table 3.
GAA enzyme activity in leukocytes and fibroblast from individuals carrying the GAA2 pseudodeficiency allele (c.271G>A p.(Asp91Asn).
| Individual/patient (gender) | Genotype DNA (protein) | Enzymatic assay | Clinical diagnosis | ||
|---|---|---|---|---|---|
| Leukocytes | Fibroblast | ||||
| AGLU-GN + 3ACB | AGLU-4MUG+ 8ACB | AGLU-4MUG | |||
| (nmol/h/mg) | (nmol/h/mg) | (nmol/h/mg) | |||
| 1 (M) | c.271G>A p.(Asp91Asn) | 36.2 | 7.9 | 61.7 | No Pompe disease |
| 2 (M) | c.271G>A p.(Asp91Asn) | 46.6 | 8.5 | 50.4 | No Pompe disease |
| 3 (M) | c.271G>A p.(Asp91Asn) | 77.6 | 12.2 | N.D. | No Pompe disease |
| 4 (M)a | c.271G>A p.(Asp91Asn) | 24.0 | 5.7 | 19.5 | No Pompe disease |
| 5 (M) | c.271G>A p.(Asp91Asn); c.271G>A p.(Asp91Asn) | 7.6 | 10.9 | 49.8 | No Pompe disease |
| 6 (M)b | c.271G>A p.(Asp91Asn); c.271G>A p.(Asp91Asn) | 4.4/7.7 | 11.1 | 86.9 | No Pompe disease |
| 7 (F) | c.271G>A p.(Asp91Asn); c.-32-13T>G p.(=), p.(0) | 2.2 | 6.1 | 25.9 | No Pompe disease |
| 8 (M) | c.271G>A p.(Asp91Asn); c.-32-13T>G p.(=), p.(0) | 4.1 | N.D. | 51.0 | No Pompe disease |
| 9 (F) | c.271G>A p.(Asp91Asn); c.-32-13T>G p.(=), p.(0) | 4.7 | 6.5 | 53.5 | No Pompe disease |
| 10 (F) | c.271G>A p.(Asp91Asn); c.-32-13T>G p.(=), p.(0) | 6.6 | N.D. | 53.1 | No Pompe disease |
| 11 (F)c | c.271G>A p.(Asp91Asn); c.-32-13T>G p.(=), p.(0); c.1076-22T>G p.? | 5.7 | N.D. | 33.3 | No Pompe disease |
| 12 (F) | c.271G>A p.(Asp91Asn); c.-32-13T>G p.(=), p.(0); c.1447G>A p.(Gly483Arg) | 1.5 | 1.9 | 7.6 | Adult onset Pompe disease |
| Ranges | Normal range | >40–250 | >6.7–27 | >45–180 | |
| Patient range | Classic infantile: 0–3.5 | 1.1–4.9 | Classic infantile: 0–3 | ||
| Childhood/Adult: 0–10 | Childhood/Adult: 4.2–20 | ||||
Italic: values within the patient range.
aUnaffected by Pompe disease, but a disease-associated variant was found in the MYBPC1 gene, which is associated with cardiac hypertrophy.
bThe biological replicate was 7.7 nmol/h/mg in leukocytes using glycogen as substrate.
cUnaffected by Pompe disease.
In individuals that were homozygous for GAA2 (number 5 and 6), GAA activities were in the patient range in leukocytes/glycogen, but in the normal range in leukocytes/4MUG and in fibroblasts/4MUG (Table 3). In individuals that were compound heterozygous for GAA2 in combination with a second disease-associated GAA variant (numbers 7–11), GAA activities were in the patient range in leukocytes/glycogen in all cases (individual 11 contained an additional GAA variant classified as childhood when combined with a null allele according to www.pompevariantdatabase.nl) (Table 3). In leukocytes/4MUG and fibroblasts/4MUG, values were either in the gray zone or in the normal range. This highlights that the GAA2 pseudodeficiency at homozygous or compound heterozygous state can give false positive outcomes when using the leukocyte/glycogen assay, while correct outcomes can be obtained by using either the leukocytes/4MUG or fibroblast/4MUG assays. Patient 12 contained 2 GAA disease-associated variants in addition to the GAA2 allele at heterozygous state, and showed GAA values in the patient range in all three assays consistent with the diagnosis of Pompe disease (adulthood onset).
Asian c.[1726G>A; 2065G>A] pseudodeficiency
Individual 13 was compound heterozygous and individual 14 homozygous for the Asian c.[1726G>A; 2065G>A] pseudodeficiency (Table 4). In both cases, assays using leukocytes, either with glycogen or with 4MUG as substrate, resulted in activities in the patient range, while activities in fibroblasts/4MUG were in the gray zone, just above the patient range. Additional diagnostic analyses showed that individual 13 had normal CK, ASAT, and ALAT levels, but slightly elevated TGLC levels. Individual 14 did have elevated CK, ASAT, and ALT levels but normal TGLC levels. Both individuals were diagnosed not to have Pompe disease based on the diagnostic information and the lack of clinical signs associated with Pompe disease. This indicated that the presence of the Asian pseudodeficiency can seriously affect the diagnostic outcome of enzymatic assays that are based on leukocytes, independent of the substrate used.
Table 4.
Lack of diagnosis in cases with the Asian pseudodeficiency (c.1726G>A and c.2065G>A).
| Individual (gender, age in years) | Genotype DNA (protein) | Enzymatic assay | Ancillary studies | Clinical diagnosis | |||||
|---|---|---|---|---|---|---|---|---|---|
| Leukocytes | Fibroblast | Plasma | Urine | ||||||
| AGLU-GN+ 3ACB | AGLU-4MUG+8ACB | AGLU-4MUG | CK | ASAT | ALAT | TGLC | |||
| (nmol/h/mg) | (nmol/h/mg) | (nmol/h/mg) | (U/l) | (U/l) | (U/l) | (mmol/mol creatinine) | |||
| 13 (F, 74 years) |
c.1726G>A p.(Gly576Ser); c.2065G>A p.(Glu689Lys); c.-32-13T>G p.(=), p.(0) |
9.6 | 3 | 22 | 58–104 | 27–34 | 19–32 | 2.3a | No Pompe disease |
| 14 (M, 53 years) |
c.1726G>A p.(Gly576Ser); c.2065G>A p.(Glu689Lys); c.1726G>A p.(Gly576Ser); c.2065G>A p.(Glu689Lys) |
18.7 | 3.9 | 20.75 | 992 | 50 | 57 | 0.8 | No Pompe disease |
| Ranges | Normal range | >40–250 | >6.7–27 | >45–180 | W > 17 years: <170 | W > 17 years: <31 | W > 17 years: <31 | >20 years: 0–2.2 | |
| M > 17 years: <200 | M > 17 years: <37 | M > 17 years: <41 | |||||||
| Patient range | Classic infantile: 0–3.5 | 1.1–4.9 | Classic infantile: 0–3 | >20 years: 2.3–130 | |||||
| Childhood/Adult: 0–10 | Childhood/Adult: 4.2–20 | ||||||||
GAA activities were measured both in leukocytes using the natural and artificial substrates as well as in cultured fibroblasts.
Italic: values within the patient range.
a2th measurement of TGLC was 5.1 and a 3rd measurement was 11.6 mmol/mol creatinine.
Discussion
We compared the outcome of different assay procedures for measuring the GAA activity in various sample types using different substrates measured at the Erasmus MC in the last 28 years. The pro’s and con’s of the different diagnostic procedures are discussed below.
Blood-based assays
Leukocytes
We used leukocyte pellets for diagnostic purposes and applied both glycogen as well as 4MUG as substrate [36]. The results presented in Fig. 2A, B illustrate that a diagnosis can be established in most cases and with both substrates if proper cutoff values are chosen. As previously suggested by van Diggelen et al. [36], our long-term data also demonstrate that the dynamic range, i.e., the difference in values between patients and healthy individuals, is broader with glycogen than with 4MUG. None of the two assays fully discriminate between classic and milder (childhood, adult) phenotypes, but by using both assays in parallel, pseudodeficiencies caused by the Caucasian GAA2 allele can be excluded (Fig. 4B) [27, 28]. Another pseudodeficiency that complicates the assay is the Asian c.[1726G>A; 2065G>A] pseudodeficiency [23, 29, 30]. This problem applies to all enzymatic procedures used.
Fig. 4. Flow chart for diagnosis of Pompe disease.

A General flow chart. B Flow chart for cases with pseudodeficiency.
Bloodspots
In our small patient cohort, 3 (16.7%) of 18 patients (assays 30–32 in Fig. S1) came out as false negative in the DBS assay but were correctly diagnosed with other diagnostic methods. Analysis of the activity of a second lysosomal enzyme (for instance BGAL) as reference enzyme may help to judge the outcome of the assay. For example, a normal GAA activity should not be trusted if the reference enzyme shows activity outside of the normal ranges. In such an event additional assays are required. In our diagnostic center we do not use the DBS assay routinely as we have fast and standard procedures for leukocyte-based assays. DBS-based assays prove to be very valuable tools for NBS. In both individual cases and in NBS, a second assay is required to confirm the diagnosis.
Despite the ten times higher activity of GAA for glycogen compared to 4MUG, the small sample size of the bloodspot assay precludes the use of glycogen for DBS testing due to the lower sensitivity of the colorimetric assay used with glycogen compared to the fluorimetric detection using 4MUG. The tandem mass spectrometry methodology, employing GAA-S has proven to be a valuable alternative [20]. A recent refinement of that method was reported to separate 96% of the Taiwanese newborns with GAA pseudodeficiency and all Pompe disease carriers from patients with Pompe disease [38]. A recent study from Japan supports this claim [39].
Skin fibroblast assays
Figures 2A, B and 3 clearly demonstrate that the combination of fibroblasts as diagnostic material and the fluorimetric assay using 4MUG as artificial substrate provide the most robust and reliable enzymatic assay for diagnosing Pompe disease. In the majority of cases it distinguishes classic infantile Pompe disease from childhood/adulthood onset phenotypes on the basis of residual activity (although exceptions exist), while none of the other methods do. Assays with intermediate GAA activity were likely derived from Pompe disease carriers. DNA analysis usually provides a clear answer in these cases, but some remained unsolved. For instance, in individual 13, that carried the Asian pseudodeficiency c.[1726G>A; 2065G>A] in combination with the IVS1 variant, biochemical results suggested Pompe disease while clinical signs did not, but it leaves doubt whether adult/late onset Pompe disease can be fully ruled out in this patient.
Conclusions and recommendations
We conclude from our data that cultured skin fibroblasts provide the only sample source that can distinguish classic infantile from childhood/adulthood phenotypes, although exceptions can occur, as noted previously [40]. The laborious procedure of biopsy and culturing makes this assay not suitable for a fast diagnosis. Leukocytes isolated from peripheral blood offer a fast diagnostic sample source, provided that the reaction mixture contains acarbose to inhibit glucoamylase. The use of glycogen as natural substrate enhances the resolution between affected and unaffected, but GAA2 pseudodeficiency occurs in the Caucasian population and has to be excluded by also using 4MUG as substrate in case of exceptionally low GAA activity (Fig. 4B). The implementation of DBSs has enabled (newborn) screening, and several analytical methods have proven their value as first-tier test. There is general agreement that additional tests remain necessary to finally establish the diagnosis. For reasons mentioned in the text, we advise to conduct genetic testing in addition to any type of enzymatic testing, as recommended recently by the EPOC consortium and others [18, 41].
The recommended enzymatic and molecular diagnostic flow at our Center is as follows: (1) start with leukocytes using 4MUG and glycogen as substrate; (2) confirm with DNA analysis, in which two disease-associated variants should be identified, while a pseudodeficiency does not qualify as disease-associated variant; (3) if there is doubt, for example when values are close to the gray zone or when a DNA variant is unidentified or novel with unknown significance unknown, use fibroblasts as confirmation (Fig. 4A, B).
Supplementary information
Acknowledgements
We thank Dr. Arnold Reuser for invaluable discussions.
Funding
This work was supported by Departamento Administrativo de Ciencia, Tecnología e Innovación (Colciencias), Colombia (Administrative Department of Science, Technology and Innovation, Colciencias, Colombia), and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brasil), Zeldzame Ziekten Fonds/WE Foundation, the Sophia Foundation for Medical Research (SSWO; project number s17-32), Metakids (project number 2016–063 and 2018–082), and Stofwisselkracht.
Compliance with ethical standards
Conflict of interest
ATvdP has provided consulting services for various industries in the field of Pompe disease under an agreement between these industries and Erasmus MC, Rotterdam, The Netherlands. All other authors do not declare any conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Mark Wijgerde, Douglas Oliveira Soares de Faria
Supplementary information
The online version of this article (10.1038/s41431-020-00752-2) contains supplementary material, which is available to authorized users.
References
- 1.Dasouki M, Jawdat O, Almadhoun O, Pasnoor M, McVey AL, Abuzinadah A, et al. Pompe disease: literature review and case series. Neurol Clin. 2014;32:751–76. doi: 10.1016/j.ncl.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hers HG. alpha-Glucosidase deficiency in generalized glycogenstorage disease (Pompe’s disease) Biochem J. 1963;86:11–6. doi: 10.1042/bj0860011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reuser A, Hirschhorn R, Kroos M. Pompe disease: glycogen storage disease type II, acid α-glucosidase (acid maltase) deficiency. NY: McGraw-Hill Medical; 2018.
- 4.Herzog A, Hartung R, Reuser AJ, Hermanns P, Runz H, Karabul N, et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J Rare Dis. 2012;7:35. doi: 10.1186/1750-1172-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laforet P, Laloui K, Granger B, Hamroun D, Taouagh N, Hogrel JY, et al. The French Pompe registry. Baseline characteristics of a cohort of 126 patients with adult Pompe disease. Rev Neurol. 2013;169:595–602. doi: 10.1016/j.neurol.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 6.van Capelle CI, van der Meijden JC, van den Hout JM, Jaeken J, Baethmann M, Voit T, et al. Childhood Pompe disease: clinical spectrum and genotype in 31 patients. Orphanet J Rare Dis. 2016;11:65. doi: 10.1186/s13023-016-0442-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Ploeg AT, Reuser AJ. Pompe’s disease. Lancet. 2008;372:1342–53. doi: 10.1016/S0140-6736(08)61555-X. [DOI] [PubMed] [Google Scholar]
- 8.Wens SC, van Gelder CM, Kruijshaar ME, de Vries JM, van der Beek NA, Reuser AJ, et al. Phenotypical variation within 22 families with Pompe disease. Orphanet J Rare Dis. 2013;8:182. doi: 10.1186/1750-1172-8-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergsma AJ, In ‘t Groen SLM, van den Dorpel JJA, van den Hout H, van der Beek N, Schoser B, et al. A genetic modifier of symptom onset in Pompe disease. EBioMedicine. 2019;43:553–61. doi: 10.1016/j.ebiom.2019.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elder ME, Nayak S, Collins SW, Lawson LA, Kelley JS, Herzog RW, et al. B-Cell depletion and immunomodulation before initiation of enzyme replacement therapy blocks the immune response to acid alpha-glucosidase in infantile-onset Pompe disease. J Pediatr. 2013;163:847–54. e841. doi: 10.1016/j.jpeds.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poelman E, Hoogeveen-Westerveld M, Kroos-de Haan MA, van den Hout JMP, Bronsema KJ, van de Merbel NC, et al. High sustained antibody titers in patients with classic infantile Pompe disease following immunomodulation at start of enzyme replacement therapy. J Pediatr. 2018;195:236–43. e233. doi: 10.1016/j.jpeds.2017.11.046. [DOI] [PubMed] [Google Scholar]
- 12.Poelman E, Hoogeveen-Westerveld M, van den Hout JMP, Bredius RGM, Lankester AC, Driessen GJA, et al. Effects of immunomodulation in classic infantile Pompe patients with high antibody titers. Orphanet J Rare Dis. 2019;14:71. doi: 10.1186/s13023-019-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stenger EO, Kazi Z, Lisi E, Gambello MJ, Kishnani P. Immune tolerance strategies in siblings with infantile Pompe disease-advantages for a preemptive approach to high-sustained antibody titers. Mol Genet Metab Rep. 2015;4:30–4. doi: 10.1016/j.ymgmr.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Musumeci O, Toscano A. Diagnostic tools in late onset Pompe disease (LOPD) Ann Transl Med. 2019;7:286. doi: 10.21037/atm.2019.06.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van der Ploeg AT, Kruijshaar ME, Toscano A, Laforet P, Angelini C, Lachmann RH, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017;24:768–e31.. doi: 10.1111/ene.13285. [DOI] [PubMed] [Google Scholar]
- 16.Kroos M, Hoogeveen-Westerveld M, Michelakakis H, Pomponio R, Van der Ploeg A, Halley D, et al. Update of the pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants. Hum Mutat. 2012;33:1161–5. doi: 10.1002/humu.22108. [DOI] [PubMed] [Google Scholar]
- 17.Nino MY, In ‘t Groen SLM, Bergsma AJ, van der Beek N, Kroos M, Hoogeveen-Westerveld M, et al. Extension of the Pompe mutation database by linking disease-associated variants to clinical severity. Hum Mutat. 2019;40:1954–67. doi: 10.1002/humu.23854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bodamer OA, Scott CR, Giugliani R. Pompe disease newborn screening working G. Newborn screening for Pompe disease. Pediatrics. 2017;140:S4–13. doi: 10.1542/peds.2016-0280C. [DOI] [PubMed] [Google Scholar]
- 19.Robinson D, Price RG, Dance N. Rat-urine glycosidases and kidney damage. Biochem J. 1967;102:533–8. doi: 10.1042/bj1020533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y, Scott CR, Chamoles NA, Ghavami A, Pinto BM, Turecek F, et al. Direct multiplex assay of lysosomal enzymes in dried blood spots for newborn screening. Clin Chem. 2004;50:1785–96. doi: 10.1373/clinchem.2004.035907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gelb MH, Turecek F, Scott CR, Chamoles NA. Direct multiplex assay of enzymes in dried blood spots by tandem mass spectrometry for the newborn screening of lysosomal storage disorders. J Inherit Metab Dis. 2006;29:397–404. doi: 10.1007/s10545-006-0265-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okumiya T, Keulemans JL, Kroos MA, Van der Beek NM, Boer MA, Takeuchi H, et al. A new diagnostic assay for glycogen storage disease type II in mixed leukocytes. Mol Genet Metab. 2006;88:22–8. doi: 10.1016/j.ymgme.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 23.Shigeto S, Katafuchi T, Okada Y, Nakamura K, Endo F, Okuyama T, et al. Improved assay for differential diagnosis between Pompe disease and acid alpha-glucosidase pseudodeficiency on dried blood spots. Mol Genet Metab. 2011;103:12–7. doi: 10.1016/j.ymgme.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Sluiter W, van den Bosch JC, Goudriaan DA, van Gelder CM, de Vries JM, Huijmans JG, et al. Rapid ultraperformance liquid chromatography-tandem mass spectrometry assay for a characteristic glycogen-derived tetrasaccharide in Pompe disease and other glycogen storage diseases. Clin Chem. 2012;58:1139–47. doi: 10.1373/clinchem.2011.178319. [DOI] [PubMed] [Google Scholar]
- 25.Piraud M, Pettazzoni M, de Antonio M, Vianey-Saban C, Froissart R, Chabrol B, et al. Urine glucose tetrasaccharide: a good biomarker for glycogenoses type II and III? A study of the French cohort. Mol Genet Metab Rep. 2020;23:100583. doi: 10.1016/j.ymgmr.2020.100583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Young SP, Zhang H, Corzo D, Thurberg BL, Bali D, Kishnani PS, et al. Long-term monitoring of patients with infantile-onset Pompe disease on enzyme replacement therapy using a urinary glucose tetrasaccharide biomarker. Genet Med. 2009;11:536–41. doi: 10.1097/GIM.0b013e3181a87867. [DOI] [PubMed] [Google Scholar]
- 27.Martiniuk F, Bodkin M, Tzall S, Hirschhorn R. Identification of the base-pair substitution responsible for a human acid alpha glucosidase allele with lower “affinity” for glycogen (GAA 2) and transient gene expression in deficient cells. Am J Hum Genet. 1990;47:440–5. [PMC free article] [PubMed] [Google Scholar]
- 28.Swallow DM, Kroos M, Van der Ploeg AT, Griffiths B, Islam I, Marenah CB, et al. An investigation of the properties and possible clinical significance of the lysosomal alpha-glucosidase GAA*2 allele. Ann Hum Genet. 1989;53:177–84. doi: 10.1111/j.1469-1809.1989.tb01782.x. [DOI] [PubMed] [Google Scholar]
- 29.Kumamoto S, Katafuchi T, Nakamura K, Endo F, Oda E, Okuyama T, et al. High frequency of acid alpha-glucosidase pseudodeficiency complicates newborn screening for glycogen storage disease type II in the Japanese population. Mol Genet Metab. 2009;97:190–5. doi: 10.1016/j.ymgme.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 30.Tajima Y, Matsuzawa F, Aikawa SI, Okumiya T, Yoshimizu M, Tsukimura T, et al. Structural and biochemical studies on Pompe disease and a “pseudodeficiency of acid alpha-glucosidase”. J Hum Genet. 2007;52:898–906. doi: 10.1007/s10038-007-0191-9. [DOI] [PubMed] [Google Scholar]
- 31.Bergsma AJ, Kroos M, Hoogeveen-Westerveld M, Halley D, van der Ploeg AT, Pijnappel WW. Identification and characterization of aberrant splicing in Pompe disease using a generic approach. Hum Mutat. 2015;36:57–68. doi: 10.1002/humu.22705. [DOI] [PubMed] [Google Scholar]
- 32.Hauser F, Gokce S, Werner G, Danckwardt S, Sollfrank S, Neukirch C, et al. A non-invasive diagnostic assay for rapid detection and characterization of aberrant mRNA-splicing by nonsense mediated decay inhibition. Mol Genet Metab. 2020;130:27–35. doi: 10.1016/j.ymgme.2020.03.002. [DOI] [PubMed] [Google Scholar]
- 33.In ‘t Groen SLM, de Faria DOS, Iuliano A, van den Hout JMP, Douben H, Dijkhuizen T, et al. Novel GAA variants and mosaicism in Pompe disease identified by extended analyses of patients with an incomplete DNA diagnosis. Mol Ther Methods Clin Dev. 2020;17:337–48. doi: 10.1016/j.omtm.2019.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37:564–9. doi: 10.1002/humu.22981. [DOI] [PubMed] [Google Scholar]
- 35.Oemardien LF, Boer AM, Ruijter GJ, van der Ploeg AT, de Klerk JB, Reuser AJ, et al. Hemoglobin precipitation greatly improves 4-methylumbelliferone-based diagnostic assays for lysosomal storage diseases in dried blood spots. Mol Genet Metab. 2011;102:44–8. doi: 10.1016/j.ymgme.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 36.van Diggelen OP, Oemardien LF, van der Beek NA, Kroos MA, Wind HK, Voznyi YV, et al. Enzyme analysis for Pompe disease in leukocytes; superior results with natural substrate compared with artificial substrates. J Inherit Metab Dis. 2009;32:416–23. doi: 10.1007/s10545-009-1082-3. [DOI] [PubMed] [Google Scholar]
- 37.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 38.Liao HC, Chan MJ, Yang CF, Chiang CC, Niu DM, Huang CK, et al. Mass spectrometry but not fluorimetry distinguishes affected and pseudodeficiency patients in newborn screening for Pompe disease. Clin Chem. 2017;63:1271–7. doi: 10.1373/clinchem.2016.269027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mashima R, Okuyama T. Enzyme activities of alpha-glucosidase in Japanese neonates with pseudodeficiency alleles. Mol Genet Metab Rep. 2017;12:110–4. doi: 10.1016/j.ymgmr.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wens SC, Kroos MA, de Vries JM, Hoogeveen-Westerveld M, Wijgerde MG, van Doorn PA, et al. Remarkably low fibroblast acid alpha-glucosidase activity in three adults with Pompe disease. Mol Genet Metab. 2012;107:485–9. doi: 10.1016/j.ymgme.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 41.van der Ploeg AT, Kruijshaar ME, Toscano A, Laforêt P, Angelini C, Lachmann RH, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017;24:768–e-31. doi: 10.1111/ene.13285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.