

Abstract
Background:
ATP1A2 mutations cause hemiplegic migraine with or without epilepsy or acute reversible encephalopathy. Typical onset is in adulthood or older childhood without subsequent severe long-term developmental impairments.
Aim:
Describe the manifestations of early onset severe ATP1A2-related epileptic encephalopathy and its underlying mutations in a cohort of seven patients.
Methods:
Retrospective chart review of a cohort of seven patients. Response to open label memantine therapy, used off-label due to its NMDA receptor antagonist effects, was assessed by the Global Rating Scale of Change (GRSC) and Clinical Global Impression Scale of Improvement (CGI-I) methodologies. Molecular modeling was performed using PyMol program.
Results:
Patients (age 2.5-20 years) had symptom onset at an early age (6 days-1 year). Seizures were either focal or generalized. Drug resistance, recurrent status epilepticus, severe developmental delay with episodes of acute severe encephalopathy often with headaches, dystonias, hemiplegias, seizures, and developmental regression were common features. All had variants predicted to be disease causing (p.Ile293Met, p.Glu1000Lys, c.1017+5G>A, p.Leu809Arg, and 3 patients with p.Met813Lys). Modeling revealed that mutations interfered with ATP1A2 ion binding and translocation sites. Memantine, given to five, was tolerated in all (mean treatment: 2.3 years, range 6 weeks-4.8 years) with some improvements reported in all five.
Conclusions:
Our observations describe a distinctive clinical profile of seven unrelated probands with early onset severe ATP1A2-related epileptic encephalopathy, provide insights into structure-function relationships of ATP1A2 mutations, and support further studies of NMDAR antagonist therapy in ATP1A2-encephalopathy.
Keywords: ATP1A2, Alternating Hemiplegia of Childhood, Familial Hemiplegic Migraine, Encephalopathy, NMDA Receptor Antagonist, Memantine
1. Introduction
ATP1A2 codes for the α2-subunit of the Na+/K+-ATPase pump,[1] which is responsible for maintenance and restoration of membrane potential in astrocytes. [2] The underlying pathophysiology is presumed to be related, at least in part, to failure of the ATP1A2 Na+/K+-ATPase pump function. This results in the subsequent inability of astrocytes to uptake released glutamate from synaptic clefts. [2, 3] Variations in this gene have been associated with Familial Hemiplegic Migraine-Type 2 (FHM2), [4, 5] at times with epilepsy [6, 7] and less commonly with reversible epileptic encephalopathy. [7, 8] Previous case series analyzing the relationship between variants in the ATP1A2 gene and the development of encephalopathy describe patients with FHM who develop encephalopathy as a symptom occurring in conjunction with hemiplegia and migraine. [8–10] Additional case series describe related proband patients with FHM who present with acute reversible encephalopathy as their first presenting symptom. [11, 12] Typical onset has been in older children or in adulthood without subsequent severe long-term developmental impairments, irreversible severe regression or intractable epilepsy. [6–12] Here, we report the clinical manifestations of seven patients with early onset severe ATP1A2-related epileptic encephalopathy, their underlying mutations and their response to therapy.
2. Methods
2.1. Case Series data collection and analysis methods:
Retrospective review of data of seven consecutive patients who had both ATP1A2 mutations and epileptic encephalopathy with symptoms starting before the age of 2.5 years. Data from our center as well as from care at other centers were analyzed. Guardians of all patients provided written consent and data were entered into our Institutional Review Board approved database. These included type and age of symptom onset, type and severity of epilepsy, response to therapies and additional disease manifestations. Certain medications, including memantine (5 patients) and ketamine (one patient), were used off-label based on the understanding of the presumptive underlying pathophysiology of ATP1A2 mutations. These medications were titrated according to doses used for other indications, as well as patients’ tolerance. Data regarding side effects and response were collected and summarized. In order to develop a better appreciation of the apparent response to medications, while at the same time recognizing the open label nature of the data, we utilized two validated instruments for measuring response. The Global Rating Scale of Change (GRSC) is a validated scale designed to be completed by the patient or guardian. [13] The scale ranges from −5 to +5 with a rating of −5 indicating severe worsening, a rating of 0 indicating no change, and a rating of +5 indicating complete recovery. The guardian of the patient was asked each question and then reported their perception of a specific aspect of their child’s development after starting memantine. The Clinical Global Impression Scale of Improvement (CGI-I) is validated for completion by trained healthcare providers. [14] The scale ranges from 1 to 7 with a rating of 1 indicating a very substantial improvement, a score of 4 indicating no change, and a score of 7 indicating severe symptom exacerbation. Provider observations consisted of evaluations by members the treating team of board-certified neurologists and supporting providers.
2.2. Molecular Testing and Modeling:
Whole exome sequencing was performed on all patients. When positive findings were detected these were confirmed by Sanger sequencing. In silico variant analysis software, to predict pathogenicity and SNAP2 software were used to predict pathogenicity and likelihood of loss of protein function, respectively. Molecular visualization of ATP1A2 protein structure and analysis of location and potential impact of the mutations on protein function were performed using PyMol. [15] In addition, to further demonstrate the pathogenicity of the M813K mutation, we also generated a knock-in mouse model carrying that mutation. The methods used to generate and test the mouse and the results of that testing are included in the Supplementary data section. [16–18]
3. Results
3.1. Case Reports:
Please see Tables 1 and 2 for a summary of the GRSC and CGI-I scores in response to memantine therapy. In addition, we have included more detailed descriptions of the changes that occurred after start of this therapy in the case histories below. We would like to emphasize here again that these are descriptions of open label data, subject to confounding variables and to observers’ bias, and not the results of a controlled trial.
Table 1.
Summary of patient dose, duration, and side effects to memantine
| Patient | Final Dose | Duration | Side Effects |
|---|---|---|---|
| 1 | 10 mg/day | 3.10 years (began at age 11.7 years) | Ceased temporarily due to nausea and vomiting; restarted with no additional side effects |
| 2 | 20 mg/day | 2.3 years (began at age 8.5 years) | None reported |
| 3 | 20 mg/day | 11 months (began at age 4.5 years) | None reported |
| 4 | 5 mg/day | 1.4 years (began at age 16.11 years) | None reported |
| 5 | 40 mg/day | 4.8 years (began at age 5.3 years) | None reported |
Table 2.
Summary of guardian-perceived responses to memantine utilizing the GRSC and healthcare provider-perceived responses to memantine utilizing the CGI-I Scale.
| Patient | Question 1 With respect to your child’s motor function, how would you rate after starting memantine? |
Question 2 With respect to your child’s behavior, how would you rate it after starting memantine? |
Question 3 With respect to your child’s learning abilities how would you rate it after memantine? |
|---|---|---|---|
| Global Rating Scale of Change (GRSC): The scale ranges from −5 to +5 with a rating of −5 indicating severe worsening, a rating of 0 indicating no change, and a rating of +5 indicating complete recovery. | |||
| 1 | 3 | 2 | 5 |
| 2 | 5 | 0 | 5 |
| 3 | 3 | 0 | 0 |
| 4* | - | - | - |
| 5 | 4 | 3 | 3 |
| Clinical Global Impression of Improvement (CGI-I): The scale ranges from 1 to 7 with a rating of 1 indicating a very substantial improvement, a score of 4 indicating no change, and a score of 7 indicating severe symptom exacerbation. | |||
| 1 | 3 | 3 | 2 |
| 2 | 2 | 2 | 2 |
| 3 | 2 | 4 | 2 |
| 4 | 5 | 4 | 2 |
| 5 | 2 | 2 | 2 |
The guardians of patient 4 were unable to be contacted to complete the GRSC.
Patient 1 is a 15.5-year-old male bom to healthy parents. Development was mildly delayed in infancy and childhood. His first words were at 15-18 months of age and walking was at 16 months of age. Starting at the age of 18 months, till the age of 4 years, he had recurrent tonic-clonic seizures that occurred, approximately, once every two weeks. During that period, he was also noted to lag even more in his development being always 1-2 years behind. He also had hemiplegias that occurred either in association with the seizures or independently. EEGs revealed frequent spike and wave activity in the left occipital lobe. He received therapy with phenytoin and lamotrigine without much success, but his seizures could eventually be controlled at the age of four years on Depakote, which was tapered at the age of 9 years. At 11 years old, he started reporting headaches localized to the forehead, lasting 1-2 hours, and associated with fatigue, photophobia and phonophobia. Additionally, during that period of time, he had regression of his function. His handwriting became illegible. His academic performance declined with his reading level regressing by two levels. Teachers and parents reported worsening of memory and comprehension. He also experienced severe anxiety with accompanying trichotillomania. MRI of the brain showed symmetrical high T2 signals in the peritrigonal region, and borderline cerebellar cortical atrophy. Exome sequencing revealed a de novo heterozygous variant of the ATP1A2 gene (c.879C>G; p.Ile293Met, de novo). He was started on 2 mg/day of memantine at the age of 11.7 years. Memantine dosage was progressively increased to 4 mg/day, 8 mg/day, and finally, 10 mg/day over a 13-month period. During the first month of starting memantine, it had to be temporarily stopped due to nausea and vomiting, but it was restarted several days later and was well tolerated with no side effects. One month after restarting, parents reported that the patient’s sentences were longer, and speech was clearer. His energy level was better, and his verbal learning and memory were noted to be better in school. Three months after starting memantine, teachers, therapists, neighbors, and family members all noted continued improvement in overall functioning speech and learning.
Patient 2 is a 10.8-year-old female born to healthy parents. Features noted in infancy included hypotonia bilateral eyelid ptosis and mild developmental delay. She manifested occasional generalized tonic-clonic and myoclonic seizures usually with subsequent left sided hemiparesis as of the age of 6 months and frequent headaches from the age of 5 years. Between the ages of 6-8 months, her seizures were associated with fevers but later occurred without fevers. She initially failed levetiracetam and oxcarbazepine but was eventually treated with levetiracetam which controlled her seizures for several years and was then tapered off. However, at the age of 7.5 years she had an episode of status epilepticus associated with severe neurological regression. MRI of the brain at that time revealed non-specific hyperintense signals in the right parietal white matter. EEG at that time showed post ictal changes. Whole exome sequencing revealed a heterozygous variant of the ATP1A2 gene, likely pathogenic (c.2998G>A; p.Glu1000Lys, de novo). Subsequent to this result, she was started on memantine therapy at 5 mg/day at the age of 8.5 years. In the following month, her parents reported that she improved in her attention, focus, and then increased her counting ability from none up to 20. Prior to that she had to repeat kindergarten and was reading at the lowest first grade reading level, level D. In the few weeks following the start of memantine she achieved level E first grade reading level. The guided reading levels consist of levels A-Z with A being the easiest. On average, children require several months, rather than a few weeks, to progress reading levels. Her dosage was increased to 20 mg/day one month later. On further follow up, six months after starting memantine, improvements in handwriting, academics, and reading level from level E to G were noted. Additionally, there was an observed improvement in attention, and in performance in her occupational therapy program.
Patient 3 is a 5.4-year-old male born to a healthy mother, and a father with history of hemiplegic migraine associated with the same ATP1A2 variant in the child. Patient 3 was born at 35 weeks due to pre-eclampsia and had to undergo intubation. He manifested bilateral myoclonic and clonic seizures from age 6 days to 8 months of age with EEG showing bilateral centrotemporal spikes. He also had mild developmental delay in infancy. At the age of 1 year, he developed occasional episodes of left or right independent hemiplegias, dystonia spells, nystagmus and less frequent quadriplegias. At the the age of 1.5 years, he was suspected of having headaches due to episodes of head banging and irritability. These did not respond to cyproheptadine but did respond to topiramate, recurred when it was stopped and resolved again when it was restarted. At the the age of 1.6 years, his seizures took the form of staring with impaired awareness occurring from several times per week to once every few months. MRI at the age of 2 years was normal. EEG at the age of 3.4 years revealed bursts of generalized spike wave discharges. He has failed sequential therapies with phenobarbital, topiramate, gabapentin and cannabidiol. Whole Exome Sequencing revealed a heterozygous splicing variant of the ATP1A2 gene (c.1017+5G>A, IVS8+5G>A). He was started on 1 mg/kg/day of memantine at the age of 3.4 years, and in the following months, parents noted marked improvement as he started to regularly use two-word phrases. After being on memantine for 9 months, his occupational therapist noted continued improvement in his functional speech and ability to follow instructions. His dose was increased to 20 mg/day and over a 12-month period this was followed by him using three-word sentences, being able to recognize 5 colors and count two objects when he could not do that before.
Patient 4 is an 18.3-year-old male born to healthy parents. Pregnancy was slightly complicated by manual manipulation of breech presentation and ultimately a C-section delivery that occurred at 37 weeks gestation. At one year of age, he developed recurrent drug resistant focal motor seizures that occurred every few weeks and that usually were associated with regression of milestones over several days requiring him several weeks to recover and regain previous function. Combined or independent episodes of hemiplegia, dystonia, nystagmus and autonomic dysfunction later also occurred but were less of a problem. Initial childhood development, other than the reversible regressions noted above, was otherwise within normal, but he plateaued cognitively after the age of 6-7 years and failed to progress. In addition, after that age, the intermittent episodes of seizures with regression became more impairing. It started to take him several months to regain only some of the cognitive and motor functions that were being lost without ever regaining his prior developmental trajectory. Whereas his initial seizures were suspected to be febrile seizures in type, it was later recognized that the fever always developed afterward suggesting that the fever was part of the episodes. He failed sequential therapies with valproate, topiramate, zonisamide, lacosamide, levetiracetam and lamotrigine. It was found that the combination of the last two medications offered him the best, albeit incomplete, control. EEG at age 15.3 years revealed focal seizures arising from the right temporal region and rapidly spreading to the left, rare interictal sharp waves in the right frontal region, and evidence for a global encephalopathy with abnormal background due to diffuse slowing with maximal slowing over the left hemisphere. MRI at the age of 16-years-old showed areas of increased T2 signal in the left thalamus and cerebral cortex and an area of SWI hypointensity in the right cerebral hemisphere cortex. Exome sequencing revealed a variant in the ATP1A2 gene (c.2426T>G; p.Leu809Arg, de novo status unknown due to absence of parental testing). He was started on 5 mg/day of memantine at age 16.11 years. The parents reported improvement in energy level, social skills and in his performance in school, but he continued to have difficulties with fine motor skill tasks.
Patient 5 is a 10.9-year-old male born to healthy parents. At 15 months of age, he started to manifest episodes of daily drug resistant focal seizures followed by postictal Todd’s paralysis. Concurrent, and at times independent, episodes of hemiplegia also occurred. These would also often precede the onset of the seizures. Initial development was within normal with walking and first word occurring at the age 15 months. However, at the age of 4 years, he regressed significantly losing all gross motor, fine motor, and language skills when he developed an episode of refractory status epilepticus requiring hospitalization. He improved slightly and then similarly regressed again with a similar episode at the age of 6.8 years. These episodes were associated with severe encephalopathy fluctuating unilateral hemiplegia on either side and less frequently dystonic posturing of the neck and body. A vagal nerve stimulation (VNS) device was implanted at 6.5 years of age. MRIs obtained at ages 6 and 6.5 years showed moderate to severe cortical and white matter atrophy and increased signal in the periventricular white matter bilaterally. Whole exome sequencing revealed a de novo variant for the ATP1A2 gene (c.2438T>A; p.Met813Lys, de novo). After the hospitalization at the age of 6.8 years which lasted 2 months, he was discharged on memantine and ketogenic diet. This combination was followed by marked improvements in motor control, alertness, and speech. Upon re-evaluation at the age of 7.3 years, improvements in motor control, awareness, and speech continued and no seizures were observed at that time. Specifically, his head control had improved, he was able to sit independently. He was able to transition to a seated position from a laying position also independently. He could stand from a seated position with minimal help, used his right arm/hand more than before, and walked with support, better and for longer distances than before. He also experienced steady improvement in his communication, such as using yes/no cards and short responses to questions. After his dose of memantine was increased to 40 mg/day, parents reported further increases in his functionality. He initially became sleepy during the titration but with gradual escalation was able to reach the 40 mg/day dose. Specifically, he was able to use an eye-gaze device to answer questions, turn lights on/off, and drink minimal liquids. More importantly, he also displayed further improved motor control after having no progress for a few months before that. Namely, he could reach out push and manipulate objects and use pincer grasp with either hand, with his left hand displaying a better grasp than his right. Also, he started using a gait trainer at home and was able take a few steps with it. At 8.3-years-old, his parents noted that a dose of ketamine given prior to sedation for a dental procedure was followed by a marked improvement in alertness and general functioning for a couple weeks. Thus, intravenous ketamine infusions were initiated with a dosage of 0.5mg/kg over a 40-minute period once per week for the first month, and then to once per month after that. The dosing schedule for ketamine infusions was based on the 2018 article by Hofler and Trinka, and the 2003 article by Mewasingh et al. for other indications. [19, 20] The use of memantine and ketamine was based on the known underlying pathophysiology of ATP1A2 dysfunction and their known effects. [21–25] While on the above ketamine regimen he was able to sustain two episodes of pneumonia and a urinary tract infection without hemiplegia, without seizures or regression, and without hospital admission for over 1.5 years. Previously, such infections would consistently precipitate severe spells of encephalopathy, seizures and regression. Additionally, he improved in weight-bearing, in reaching grasping and manipulating objects and started peddling his tricycle. He also became more communicative with his assistive device and mastered identification of the alphabet, numbers 1-10, and sorting objects by colors and type. He has now transitioned to IM Ketamine injections at home at the same dose. Over the past 2 year although he has experienced 4 clinical seizure events and was admitted for three of them because they progressed to status epilepticus with hemiplegia, his development has not regressed after these episodes.
In addition to the above five patients who received memantine therapy, we describe below two other patients with the M813K mutation who manifested a similar phenotype but have not received memantine.
Patient 6 is a 2-year 3-month old male who started to have 10-45-minute hemiplegic episodes at the age of 1 year. At times, these would progress to total paralysis and would occur once every 1-2 months. He was also noted to have delay in expressive speech after the onset of those spells. At the age of 2 years and 2 months, he presented with altered mental status, fever, vomiting, and left body focal motor secondary generalized seizures that necessitated intubation for several hours in addition to intravenous antiseizure medications. Cerebrospinal fluid was negative and EEG monitoring during the subsequent six hours revealed bilateral slowing higher amplitude on the right, subclinical seizures, one per hour, from the right posterior quadrant and interictal spikes and sharp wave in the left frontal and central regions. MRI showed right sided cerebral edema. His seizures subsequently subsided, however, one month later he had a similar episode of status epilepticus with right hemisphere onset clinical and subclinical seizures that was similarly managed.
Patient 7 is a 20-year-old male born to healthy patients. His first left-sided tonic-clonic seizure occurred at 2 weeks of age. His development was normal until the age of 14 months when he experienced prominent seizures and two episodes of status epilepticus with regression of developmental milestones. He continued to experience seizures and right-sided hemiplegia with right-sided visual field deficit. Antiepileptic medications proved ineffective and VNS had a transient partial benefit. From the age 11-12 years, his seizures worsened in frequency from 1 cluster every 2-3 weeks to 2-3 clusters per week with each cluster lasting 5-10 minutes and characterized by limb jerking and stiffening and head drops. Seizure frequency decreased at age 15 following initiation of ketogenic diet. From the age of 16-17 years, he continued to have seizures that included 0-2 tonic seizures per night or quick facial jerking. From ages 18-20 years, his seizure frequency fluctuated while on VNS ketogenic diet, felbamate and brivaracetam. He currently uses an eye gaze communication device to answer yes/no questions. He can stand, take 10-15 steps with assistance, but uses driving wheelchair for daily ambulation.
3.2. ATP1A2 Variants Structural Analysis and their Potential Pathophysiological Implications:
The four missense variants identified in our patients are in regions of the protein that are critical for proper interaction with sodium ions. Figures 1 and 2A show the relationship of the mutations to the Na+/K+ binding sites and their legends discuss the potential implications on ion transport. Figures 2B and C show the relative position of Ile293 in the α-subunit of the sodium potassium ATPase; Ile293 lies in TM3 and with the side chain pointing toward the interior of the ion pathway. Ile293 lies about 8Å from the nearest bound sodium ion and is 4.2Å from Tyr851. Tyr851 is 4.2Å from Asn780, which forms a contact with abound sodium ion. Glu1000 lies in TM10 and is located in a hydrophilic cavity facing the intracellular side on the membrane (Figures 2C–E). This region has been shown to be involved in ion binding and transport. [26] Glu1000 makes polar contacts with Arg1002, Lys1003, and Arg1007. Mutations in Asp999 and Arg1002 have been identified as disease forming in ATP1A2. [27, 28] Glu1000 is about 3.5Å from Phe996 which forms a π-π interaction with Phe942 and which bridges TM8 with TM9. Figures 2B and C also show the relative positions of Leu809 and Met813. These residues are located in TM6 and are just one turn from each other in the α-helix. TM6 forms a central component of the ion translocation pathway and abuts TM4 and TM8 which also are central to the ion translocation pathway. Asp 807 and 811 are adjacent to both Leu809 and Met813 and are involved in binding sodium ion. The c,1017+5G>A mutation is in intron 8, between exons 8 and 9. The mutation is in the donor site and replaces a G for an A. While G is the preferred base at +5, A is also allowed. [28] This replacement would likely reduce, but would not eliminate, the level of correct splicing. Thus, the level of protein is likely reduced below 100%, but is likely higher than 50%, in a heterozygote as compared to the normal. This variant is expected to cause a haploinsufficiency disease phenotype that is less than what would be expected from a complete loss of function from a nonsense or deletion mutation. In the event that the mutation causes aberrant splicing, there are a number of possibilities including skipping of exon 8, skipping intron 8, or uncovering a cryptic donor site. [28, 29]
Figure 1:
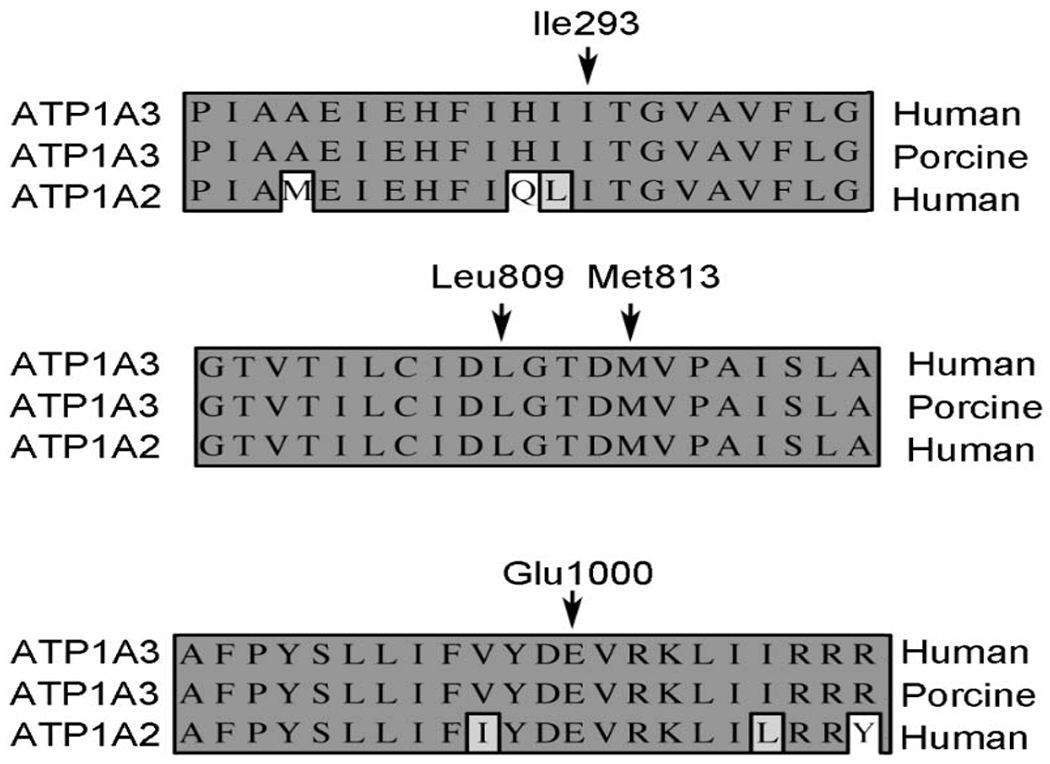
Homology of α-subunit isoforms. The primary sequence comparison is shown for human α2 isoform relative to the α3 human and porcine isoforms. The numbering is that of the human α2 isoform.
Figure 2:
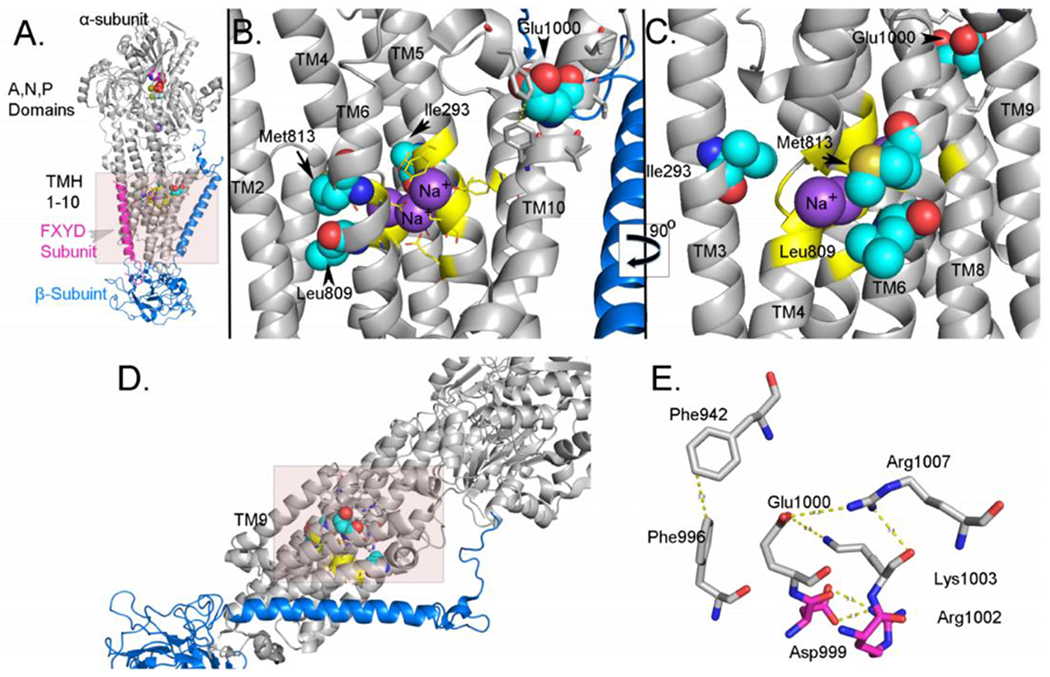
A. Cartoon representation of the structure of the sodium potassium ATPase. The α-subunit is colored in grey, the β-subunit is colored in blue, and the FXYD subunit is colored in magenta. The atoms of nucleotide bound to the N-site are shown as spheres. The carbon atoms of residues Ile293, Leu809, Met813 and Glu1000 are spheres and colored light blue. The sodium ions are spheres and colored purple. B. Cartoon representation of the position of Ile293, Leu809, Met813 and Glu1000. The perspective is that of the shaded area in A but TM8 and TM9 are not shown. The color coding is the same as in A. C. Cartoon representation of the position of Ile293, Leu809, Met813 and Glu1000. The perspective is that shown in B after rotation along the y-axis by 90°. In this image, TM8 and TM9 are shown while TM1 and TM2 are not shown. D. Cartoon representation of the structure of the sodium potassium ATPase. The shaded area represents the perspective in E. E. Structural interactions at the C-terminal region involving Glu1000. The polar interactions related to Glu1000 are shown. The carbon atoms of residues that have been identified as residues that have been identified as mutated in diseases are colored in magenta. These images were generated from the pdb: 3WUG, [26] which is the structure of the porcine enzyme with the α3-isoform. The porcine enzyme is highly homologous to the human enzyme and the α2-isoform is highly homologous to the α3-isoform, especially in TM1-10. The images were generated using Pymol. [15] Please see section 3.2 for further details.
The American College of Medical Genetics and Genomics (ACMG) criteria and Combined Annotation Dependent Deletion (CADD) were applied for each variant found in each patient (Table 3). That table also shows the amino acid changes resulting from the mutations. The pathogenicity of the Met813Lys variant is strengthened by our findings in the knock-in mouse model (Supplementary Material and Supplementary Figure 1), and by the fact that we have three cases fitting the phenotypic profile of our series and with the same Met813Lys variant. This indicates that the Met813 mutation is a recurring mutation manifesting the described phenotype.
Table 3.
Summary of variants of each patient and the corresponding amino acid change, American College of Medical Genetics and Genomics (ACMG) criteria met and Combined Annotation Dependent Deletion (CADD) for each variant.
| Patient/s | Variant | Amino Acid Change | ACMG classification | ACMG criteria met | *CADD Score |
|---|---|---|---|---|---|
| 1 | de novo c.879C>G; p.I293M | Isoleucine is a nonpolar, non-charged, branched chain amino acid, while methionine is a nonpolar, non-charged, sulfur-containing amino acid with no branched chain characteristics. This substitution is a non-conservative change in a well-conserved residue. In silico analysis, including SNAP2, predicts that this variant likely impacts the secondary structure of its respective protein. | Pathogenic | PM2, PP2, PP5, PS2 | 25.4 |
| 2 | de novo c.2998G>A; p.Glu1000Lys | Glutamate is a negatively charged amino acid with high tendency for interactions, while lysine is a positively charged and basic amino acid. This substitution is a non-conservative change in a well-conserved residue. In silico analysis predicts that tliis variant likely impacts the secondary structure of its respective protein. | Likely pathogenic | PM2, PP3, PP2, PS2 | 32 |
| 3 | c.1017+5G>A paternally inherited, father has hemiplegia migraine | Splicing mutation in the ATP1A2 gene. Contributing to the pathogenicity of the c. 1017+5G>A splicing variant is the fact that the patient’s father has the same variant and has hemiplegic migraine. When examining the splicing change of the intron. the effect of +5G>A is expected to impact the splice efficiency and thus reduce the cellular level of the correctly spliced mRNA. | VUS | PM2, PP5, PP3 | 22.5 |
| 4 | de novo status unknown c.2426T>G; p.Leu809Arg | Leucine is a nonpolar, non-charged, branched chain amino acid, while arginine is a positively charged and basic amino acid. This substitution is a non-conservative change in a well-conserved residue. In silico analysis predicts that this variant likely impacts the secondary structure of its respective protein. | VUS | PM2, PP3, PP2 | 25.3 |
| 5, 6, 7 | de novo c.2438T>A; Met813Lys | Methionine is a nonpolar, non-charged, sulfur containing amino acid, while lysine is a positively charged and basic amino acid. This substitution is a non-conservative change in a well-conserved residue. In silico analysis predicts that this variant likely impacts the secondary structure of its respective protein. | Pathogenic | PM2, PP5, PP3, PP2, PS2 | 27 |
CADD score >20 indicates that these variants are among the 1% most delirious substitutions in humans
4. Discussion
4.1. Expanding the spectrum of ATP1A2-related encephalopathy:
The seven patients in our cohort demonstrated a clinical course characterized by initial normal or near normal early development in infancy, with development of hemiplegias and seizures during the first 2.5 years of life associated with subsequent significant developmental slowing and recurrent episodes of acute encephalopathy with regression in milestones (without regaining the original level of function), and at times headaches. These episodes were associated with seizures, hemiplegias, status epilepticus, and at times, dystonias. Seizures were either generalized or focal in nature. Previous case series and case reports of the phenotypes associated with ATP1A2 mutations have reported the occurrence of hemiplegic migraine with acute reversible encephalopathy. In those patients, unlike our patients, the usual age of onset of symptoms was in adulthood or later childhood and severe long-term developmental impairments or intractable epilepsy were not characteristic features. [6–12] Thus, whereas our patients show some similarities with prior cases, the differences they demonstrate point to a distinctive, though related, phenotype notably characterized by early onset and more severe phenotype.
4.2. Potential therapy with NMDAR antagonists
Patients in our cohort had reported improvement in at least one of the following categories when memantine treatment was initiated: motor, behavior, or learning abilities. Memantine is a noncompetitive NMDAR antagonist that is used in Alzheimer’s disease. [21] Ketamine, an anesthetic, blocks NMDARs, has neuroprotective anti-glutamatergic effects, and has been used as a therapeutic agent to treat depression. [22] Previous studies in animal models show that ATPase is involved in protecting neurons from glutamate and NMDAR-related excitotoxicity, which supports the notion that neuronal dysfunction found in patients with ATP1A2-related encephalopathy is secondary to glutamate toxicity. [3, 23–25] The side effects of memantine were minimal and were limited to one patient needing to temporarily cease treatment shortly upon initiation due to nausea and vomiting; however, he restarted memantine shortly afterwards with no further side effects. While the patient utilizing ketamine injections did not report any side effects, some potential side effects of ketamine include short term amnesia, injection site irritation, dizziness and other potential complications related to sedation. [30]
There are three single case reports we are aware of that reported the use of anti-NMDAR therapy in ATP1A2-encephalopathy. One reported an improvement in handwriting, increase in focus, and academic performance in a patient that fits the above profile. [31] Additionally, one study using a knock-in ATP1A2 mouse model resulting in FMH2 reported improved behavioral functioning with the use of an NMDAR antagonist. [23] The third case report discussed a patient with familial hemiplegic migraine type due to an ATP1A2 mutation. Utilization of memantine resulted in improvement of the patient’s migraine and hemiparalysis, and reduction of edema on magnetic resonance imaging. [32] Given these previous studies coupled with our current study, we believe that controlled prospective studies of NMDAR antagonist therapies in the treatment of ATP1A2-related encephalopathy are warranted.
4.3. Structure-function relationships
Variants in the ATP1A2 gene interfere with proper functioning of the alpha-2 subunit of the Na+/K+ catalytic subunit of ATPase. [1, 3] We found that four of the five mutations were missense variants that are within the transmembrane helix and about the Na+/K+ binding site, while the fifth (patient 3) had a splicing mutation with likely complex effects on protein structure and function. The four patients with missense variants are located in the region of the protein that is critical for proper interaction with sodium ions. Therapies aimed at correcting abnormalities in this interaction may prove useful in development of future therapies for ATP1A2-related encephalopathy. Such approaches targeting the ion transport functions rather than the downstream effects of glutamatergic toxicity may offer an opportunity for more specific precision therapy. [33, 34]
4.4. Limitations
The present study is retrospective in design out of necessity due to the rarity of ATP1A2-related encephalopathy. Another limitation is the open label nature of the therapeutic trials. The observed improvements could have been due to natural developmental progression that occurs with age or to other ongoing therapies such as the ketogenic diet in patient 5.
5. Conclusions
Our observations demonstrate that the current spectrum of ATP1A2-related encephalopathy should be expanded to include patients with early onset severe epileptic encephalopathy. Our examination of the potential structural and pathophysiological implications of the mutations support pathogenicity of the observed variants and could potentially help development of future therapies for ATP1A2-related epileptic encephalopathy. Finally, our data also suggest that NMDAR antagonist therapy may be beneficial in ATP1A2-encephalopathy, and support the performance of prospective controlled studies of the use of NMDAR antagonist therapy in this disorder.
Supplementary Material
The ATP1A2 spectrum should include early onset severe epileptic encephalopathy.
The phenotype consists of hemiplegia and seizures starting by 2.5 years of age
It also includes developmental delay and recurrent encephalopathy with regression
NMDAR antagonist therapy was associated with apparent improvement in all 5 patients
Acknowledgements:
This study was supported by Duke Fund numbers 4410161 and 3912247, and by a donation by the Cure AHC Foundation for ATP1A2 related research (MAM) and by grant NIH 1R35GM131731 to D.M.M. We also acknowledge Dwight Koeberl MD PhD for his assistance with genetic evaluation of patients and the Duke Alternating Hemiplegia of Childhood and Related Disorders Multidisciplinary Team for the excellent care of patients.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of Interest: Mohamad Mikati MD and Arsen Hunanyan PhD have a pending patent application for gene therapy of ATPase related diseases.
References
- [1].Vanmolkot KRJ, Kors EE, Hottenga J-J, Terwindt GM, Haan J, Hoefnagels WAJ, Black DF, Sandkuijl LA, Frants RR, Ferrari MD, Van Den Maagdenberg AMJM. Novel mutations in the Na+, K+-ATPase pump gene ATP 1A2 associated with familial hemiplegic migraine and benign familial infantile convulsions. Ann Neurol 2003;54: 360–366. doi: 10.1002/ana.10674. [DOI] [PubMed] [Google Scholar]
- [2].de Lores Arnaiz GR, Ordieres MGL. Brain Na(+), K(+)-ATPase Activity In Aging and Disease. Int J Biomed Sci 2014;10: 85–102. [PMC free article] [PubMed] [Google Scholar]
- [3].Illarionava NB, Brismar H, Aperia A, Gunnarson E. Role of Na,K-ATPase α1 and α2 Isoforms in the Support of Astrocyte Glutamate Uptake. PLoS ONE 2014;9: 1–10. doi: 10.1371/journal.pone.0098469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gritz SM, Radcliffe RA. Genetic effects of ATP1A2 in familial hemiplegic migraine type II and animal models. Human Genomics 2013;7: 8. doi: 10.1186/1479-7364-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Vanmolkot KRJ, Kors EE, Turk U, Turkdogan D, Keyser A, Broos LAM, Kia SK, van den Heuvel JJMW, Black DF, Haan J, Frants RR, Barone V, Ferrari MD, Casari G, Koenderink JB, van den Maagdenberg AMJM. Two de novo mutations in the Na,K-ATPase gene ATP1A2 associated with pure familial hemiplegic migraine. Eur J Hum Genet 2006;14: 555–560. doi: 10.1038/sj.ejhg.5201607. [DOI] [PubMed] [Google Scholar]
- [6].Gallanti A, Tonelli A, Cardin V, Bussone G, Bresolin N, Bassi MT. A novel de novo nonsense mutation in ATP1A2 associated with sporadic hemiplegic migraine and epileptic seizures. J Neurol Sci 2008;273: 123–126. doi: 10.1016/j.jns.2008.06.006. [DOI] [PubMed] [Google Scholar]
- [7].Deprez L, Weckhuysen S, Peeters K, Deconinck T, Claeys KG, Claes LRF, Suls A, Van Dyck T, Palmini A, Matthijs G, Van Paesschen W, De Jonghe P. Epilepsy as part of the phenotype associated with ATP1A2 mutations. Epilepsia 2008;49: 500–508. doi: 10.1111/j.1528-1167.2007.01415.x. [DOI] [PubMed] [Google Scholar]
- [8].Merwick A, Fernandez D, McNamara B, Harrington H. Acute encephalopathy in familial hemiplegic migraine with ATP1A2 mutation. BMJ Case Reports 2013;2013: 10.1136/bcr-2013-009750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hart AR, Trinick R, Connolly DJ, Mordekar SR. Profound encephalopathy with complete recovery in three children with familial hemiplegic migraine. J Paediatr Child Health 2009;45: 154–157. doi: 10.1111/j.1440-1754.2009.01465.x. [DOI] [PubMed] [Google Scholar]
- [10].Murphy OC, Merwick A, O’Mahony O, Ryan AM, McNamara B. Familial Hemiplegic Migraine With Asymmetric Encephalopathy Secondary to ATP1A2 Mutation: A Case Series. J Clin Neurophysiol 2018;35: e3–e7. doi: 10.1097/wnp.0000000000000387. [DOI] [PubMed] [Google Scholar]
- [11].Spacey SD, Vanmolkot KRJ, Murphy C, Van Den Maagdenberg AMJM, Hsiung RGY. Familial Hemiplegic Migraine Presenting as Recurrent Encephalopathy in a Native Indian Family. Headache: J Head Face Pain 2005;45: 1244–1249. doi: 10.1111/j.1526-4610.2005.00249.x. [DOI] [PubMed] [Google Scholar]
- [12].Ohmura K, Suzuki Y, Saito Y, Wada T, Goto M, Seto S. Sporadic hemiplegic migraine presenting as acute encephalopathy. Brain Dev 2012;34: 691–695. doi: 10.1016/j.braindev.2011.11.002. [DOI] [PubMed] [Google Scholar]
- [13].Kamper SJ, Maher CG, Mackay G. Global rating of change scales: a review of strengths and weaknesses and considerations for design. J Man Manip Ther 2009;17: 163–170. doi: 10.1179/jmt.2009.17.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Busner J, Targum SD. The clinical global impressions scale: applying a research tool in clinical practice. Psychiatry 2007;4: 28–37. [PMC free article] [PubMed] [Google Scholar]
- [15].The PyMOL Molecular Graphics System. In: Delano Scientific, San Carlos; 2002. [Google Scholar]
- [16].Raveux A, Vandormael-Poumin S, Cohen-Tannoudji M. Optimization of the production of knock-in alleles by CRISPR/Cas9 microinjection into the mouse zygote. Scientific Reports 2017;7: 42661. doi: 10.1038/srep42661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hunanyan AS, Fainberg NA, Linabarger M, Arehart E, Leonard AS, Adil SM, Helseth AR, Swearingen AK, Forbes SL, Rodriguiz RM, Rhodes T, Yao X, Kibbi N, Hochman DW, Wetsel WC, Hochgeschwender U, Mikati MA. Knock-in mouse model of alternating hemiplegia of childhood: Behavioral and electrophysiologic characterization. Epilepsia 2015;56: 82–93. doi: 10.1111/epi.12878. [DOI] [PubMed] [Google Scholar]
- [18].Helseth AR, Hunanyan AS, Adil S, Linabarger M, Sachdev M, Abdelnour E, Arehart E, Szabo M, Richardson J, Wetsel WC, Hochgeschwender U, Mikati MA. Novel E815K knock-in mouse model of alternating hemiplegia of childhood. Neurobiol Dis 2018; 119: 100–112. doi: 10.1016/j.nbd.2018.07.028. [DOI] [PubMed] [Google Scholar]
- [19].Höfler J, Trinka E. Intravenous ketamine in status epilepticus. Epilepsia 2018;59: 198–206. doi: 10.1111/epi.14480. [DOI] [PubMed] [Google Scholar]
- [20].Mewasingh LD, Sékhara T, Aeby A, Christiaens FJ, Dan B. Oral ketamine in paediatric non-convulsive status epilepticus. Seizure 2003;12: 483–9. doi: 10.1016/s1059-1311(03)00028-1. [DOI] [PubMed] [Google Scholar]
- [21].Hosenbocus S, Chahal R. Memantine: a review of possible uses in child and adolescent psychiatry. J Can Acad Child and Adolesc Psychiatry 2013;22: 166–171. [PMC free article] [PubMed] [Google Scholar]
- [22].Peltoniemi MA, Hagelberg NM, Olkkola KT, Saari TI. Ketamine: A Review of Clinical Pharmacokinetics and Pharmacodynamics in Anesthesia and Pain Therapy. Clin Pharmacokinet 2016;55: 1059–1077. doi: 10.1007/s40262-016-0383-6. [DOI] [PubMed] [Google Scholar]
- [23].Bøttger P, Glerup S, Gesslein B, Illarionova NB, Isaksen TJ, Heuck A, Clausen BH, Füchtbauer E-M, Gramsbergen JB, Gunnarson E, Aperia A, Lauritzen M, Lambertsen KL, Nissen P, Lykke-Hartmann K. Glutamate-system defects behind psychiatric manifestations in a familial hemiplegic migraine type 2 disease-mutation mouse model. Scientific Reports 2016;6: 22047. doi: 10.1038/srep22047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ikeda K, Onaka T, Yamakado M, Nakai J, Ishikawa T-o, Taketo MM, Kawakami K. Degeneration of the Amygdala/Piriform Cortex and Enhanced Fear/Anxiety Behaviors in Sodium Pump α2 Subunit ATP1A2-Deficient Mice. J Neurosci 2003;23: 4667–4676. doi: 10.1523/jneurosci.23-11-04667.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kinoshita PF, Leite JA, Orellana AM, Vasconcelos AR, Quintas LE, Kawamoto EM, Scavone C. The Influence of Na(+), K(+)-ATPase on Glutamate Signaling in Neurodegenerative Diseases and Senescence. Front Physiol 2016;7: 195. doi: 10.3389/fphys.2016.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kanai R, Ogawa H, Vilsen B, Cornelius F, Toyoshima C. Crystal structure of a Na+-bound Na+,K+-ATPase preceding the E1P state. Nature 2013;502: 201–206. doi: 10.1038/naturel2578. [DOI] [PubMed] [Google Scholar]
- [27].Poulsen H, Khandelia H, Morth JP, Bublitz M, Mouritsen OG, Egebjerg J, Nissen P. Neurological disease mutations compromise a C-terminal ion pathway in the Na+/K+-ATPase. Nature 2010;467: 99–102. doi: 10.1038/nature09309. [DOI] [PubMed] [Google Scholar]
- [28].Sheth N, Roca X, Hastings ML, Roeder T, Krainer AR, Sachidanandam R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Research 2006;34: 3955–3967. doi: 10.1093/nar/gkl556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Needham PG, Guerriero CJ, Brodsky JL. Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb Perspect Biol 2019;11. doi: 10.1101/cshperspect.a033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sinner B, Graf BM. Ketamine. In: Schüttler J, Schwilden H, editors. Modem Anesthetics. Berlin, Heidelberg: Springer Berlin Heidelberg; 2008, p. 313–333. [Google Scholar]
- [31].Ueda K, Serajee F, Huq AM. Clinical Benefit of NMD A Receptor Antagonists in a Patient With ATP1A2 Gene Mutation. Pediatrics 2018;141: S390. doi: 10.1542/peds.2017-0852. [DOI] [PubMed] [Google Scholar]
- [32].Ying Du CL, Feng-ju Duan, Chao Zhao, Wei Zhan. Early Treatment in Acute Severe Encephalopathy Caused by ATP1A2 Mutation of Familial Hemiplegic Migraine Type 2: Case Report and Literature Review. Neuropediatrics 2020;87. [DOI] [PubMed] [Google Scholar]
- [33].Mesraoua B, Deleu D, Kullmann DM, Shetty AK, Boon P, Perucca E, Mikati MA, Asadi-Pooya AA. Novel therapies for epilepsy in the pipeline. Epilepsy Behav 2019;97: 282–290. doi: 10.1016/j.yebeh.2019.04.042. [DOI] [PubMed] [Google Scholar]
- [34].Hunanyan AS, Helseth AR, Abdelnour E, Kherallah B, Sachdev M, Chung L, Masoud M, Richardson J, Li Q, Nadler JV, Moore SD, Mikati MA. Mechanisms of increased hippocampal excitability in the Mashl+/− mouse model of Na+/K+-ATPase dysfunction. Epilepsia 2018;59: 1455–1468. doi: 10.1111/epi.14441 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.