

Abstract
In cardiac muscle the process of excitation-contraction coupling (ECC) describes the chain of events that links action potential induced myocyte membrane depolarization, surface membrane ion channel activation, triggering of Ca2+ induced Ca2+ release from the sarcoplasmic reticulum (SR) Ca2+ store to activation of the contractile machinery that is ultimately responsible for the pump function of the heart. Here we review similarities and differences of structural and functional attributes of ECC between atrial and ventricular tissue. We explore a novel ‘fire-diffuse-uptake-fire’ paradigm of atrial ECC and Ca2+ release that assigns a novel role to the SR SERCA pump and involves a concerted ‘tandem’ activation of the ryanodine receptor Ca2+ release channel by cytosolic and luminal Ca2+. We discuss the contribution of the inositol 1,4,5-trisphosphate (IP3) receptor Ca2+ release channel as an auxiliary pathway to Ca2+ signaling, and we review IP3 receptor induced Ca2+ release involvement in beat-to-beat ECC, nuclear Ca2+ signaling and arrhythmogenesis. Finally, we explore the topic of electromechanical and Ca2+ alternans and its ramifications for atrial arrhythmia.
Keywords: excitation-contraction coupling, atrium, Ca2+ release, IP3 receptor Ca2+ signaling, alternans, arrhythmia
Introduction
Beat-to-beat Ca2+ signaling in atrial and ventricular muscle shows similarities, but also significant structural and functional differences. Here we review the mechanisms of atrial and ventricular excitation-contraction coupling (ECC) and Ca2+ release, the role of a secondary pathway of Ca2+ release via IP3 receptor Ca2+ release channels for atrial ECC and atrial function, and finally the manifestations and functional implications of atrial alternans for atrial arrhythmogenesis.
Atrial excitation-contraction coupling
In the heart, ECC refers to the process that links electrical activation to cardiac contraction. The sequence of events that constitutes ECC initiates with membrane depolarization by an action potential (AP), followed by opening of voltage-gated Ca2+ channels in the surface membrane and Ca2+ entry, which in turn triggers Ca2+ release from the sarcoplasmic reticulum (SR) Ca2+ store through ryanodine receptor (RyR) Ca2+ release channels by a process known as Ca2+-induced Ca2+ release (CICR). The elevation of cytosolic [Ca2+] ([Ca2+]i) subsequently activates the contractile elements that results in cardiac muscle contraction. Most of the mechanistic details of ECC known to date were determined in ventricular muscle. While atrial and ventricular ECC clearly have important similarities, there are critical differences. Atrial and ventricular cells have unique sets of ion channels [78,107] leading to distinctive AP morphologies that in turn affect Ca2+ transient (CaT) triggering efficiency and SR Ca2+ store loading [48]. In addition, atrial myocytes exhibit lower expression of phospholamban that results in higher activity of the sarco/endoplasmic reticulum ATPase (SERCA) [6]. It is well known that changes in phopholamban expression levels have profound effects on CaTs and ECC. As we have shown previously, phospholamban ablation caused accelerated decay of CaTs and Ca2+ sparks in mouse ventricular myocytes, increased SR Ca2+ load and frequently led to Ca2+ waves that were spatially narrower and often aborted after propagating over only a short distance [40]. However, aside from differences in phospholamban levels, one of the most important difference between atrial and ventricular myocytes is the system of transverse- (t) tubules. These surface membrane invaginations extend in a sarcomeric pattern throughout the entire ventricular myocyte, but not in atrial cells (Fig. 1A). The t-tubule system is an integral part of the surface membrane (or the sarcolemma), that allows the placement of voltage-gated L-type Ca2+ channels (LCCs) in close vicinity to RyR clusters. RyR clusters are considered SR Ca2+ release units (CRUs; [28]) which give rise to elementary intracellular Ca2+ release events, also known as Ca2+ sparks. AP induced whole cell CaTs are the spatial and temporal summation of Ca2+ sparks from thousands of CRUs. In the presence of a t-tubular system the vast majority of CRUs has its own source of activator Ca2+ in form of a small number of adjacent LCCs. As a consequence of these structural arrangements, Ca2+ release during ventricular ECC is spatially homogeneous throughout the cell. Atrial myocytes, however, have only a rather sparse and irregular t-tubule system or are even entirely lacking any t-tubules [98,41,63,7] with important consequences for atrial Ca2+ dynamics during ECC. Because of these structural features AP induced Ca2+ release in atrial cells is characterized by pronounced spatial inhomogeneities [41,8,5,94,4]. Elevation of [Ca2+]i starts in the cell periphery where the opening of LCCs provides the required Ca2+ to induce CICR from the most peripheral SR Ca2+ release sites [55,94]. This generates Ca2+ gradients that are large enough to overcome endogenous cytosolic Ca2+ buffering [92] and allow for centripetal Ca2+ diffusion and activation of CICR from SR release sites in progressively more central regions of the cell. Thus, during atrial ECC [Ca2+]i rises by propagating CICR from the periphery to the center of the cell in a Ca2+ wave-like fashion by a diffusion-reaction process or a ‘fire-diffuse-fire’ mechanism [94,51]. The propagating nature of atrial Ca2+ release results in complex [Ca2+]i inhomogeneities and subcellular [Ca2+]i gradients.
Fig. 1. Ca2+ signaling during ECC in atrial myocytes.
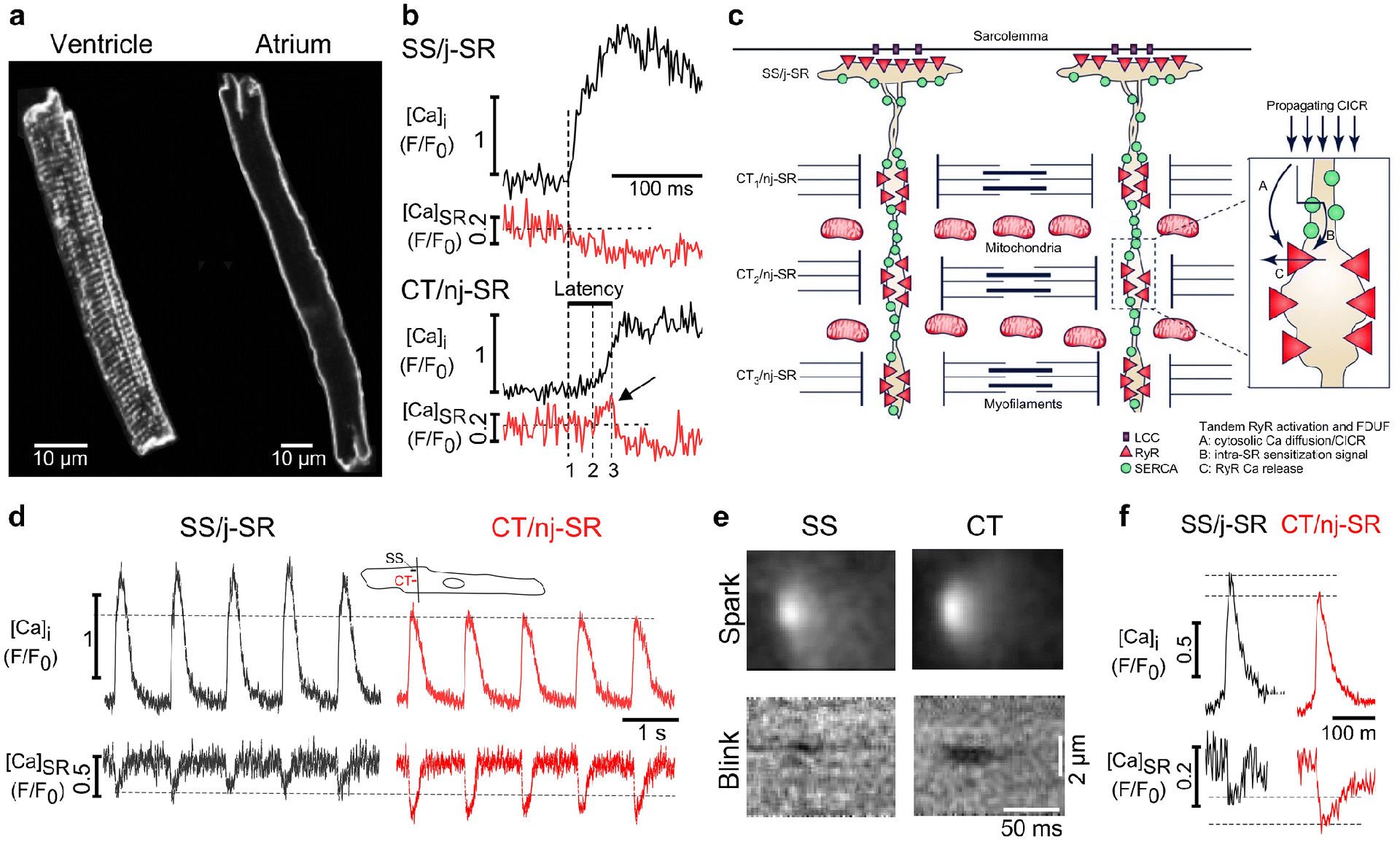
A, Membrane staining with the fluorescent probe Di-8-ANEPPS reveals the regular structure of the t-tubule system in ventricular myocytes (left) and the absence of t-tubules in atrial cells (right). B, Atrial ECC. Subsarcolemmal SS/j-SR (top) and central CT/nj-SR (bottom) [Ca2+]i and [Ca2+]SR transients from individual CRUs. Intra-SR Ca2+ sensitization signal (arrow) and latency of CT/nj-SR Ca2+ release. 1, begin SS CaT; 2, begin Ca2+ sensitization signal (arrow) and latency of CT/nj-SR Ca2+ release are shown. 1, begin SS Ca2+ release; 2, begin nj-SR Ca2+ sensitization signal; 3, start of nj-SR [Ca2+]SR decline. C, Current paradigm of atrial ECC. AP-induced Ca2+ release from SS/j-SR by LCC activation starts in the cell periphery followed by centripetally propagating CICR from CT/nj-SR CRUs (CT1→CT2→CT3→….). Inset: FDUF mechanism; Tandem RyR activation by cytosolic CICR (A) and luminal RyR sensitization (B), resulting in Ca2+ release (C). D, Subcellular SS/j-SR and CT/nj-SR AP-induced [Ca2+]i and [Ca2+]SR transients. Cytosolic CaT amplitude in central regions are smaller, however SR depletion levels are lower in the nj-SR. Inset: line scan position and SS and CT regions of interest (1 μm wide). E, Averaged confocal line scan images (F/F0) of Ca2+ sparks and corresponding Ca2+ blinks originating from SS/j-SR and CT/nj-SR. F, Averaged Ca2+ spark and Ca2+ blink profiles from j-SR and nj-SR CRUs from images in panel E. Reproduced and modified from [67].
The extent of t-tubule endowment of atrial myocytes shows considerable species differences (for reference see [85,104]). A putative role of an intracellular axial membrane structure recently described in certain species further enhances the complexity of atrial Ca2+ signals during ECC [9]. However, in cat and rabbit atrial cells for example, the t-tubular system is entirely absent [41,67]. The absence or paucity of t-tubules divides the atrial SR Ca2+ store into two types of SR based on the proximity to the sarcolemma: junctional SR (j-SR) is found in close association with the sarcolemmal membrane, whereas the much more abundant non-junctional SR (nj-SR) is found distant from the sarcolemma in more central regions of the cell. Importantly, RyR Ca2+ release channels are abundant in the membranes of both j-SR and nj-SR and participate in physiological ECC [110,11,63,98,7,55]. RyRs are organized in a 3-dimensional array of channel clusters or CRUs [41,55,94,92]. The j-SR forms close physical associations with the sarcolemma known as peripheral couplings. Here, the sarcolemma hosts voltage-gated LCCs that are facing clusters of RyRs in the SR membrane across a narrow inter-membrane cleft, similar to dyads in ventricular myocytes [55,68]. Thus, the CRUs of the j-SR are functionally organized like a ‘classical couplon’ [100,90]. Ca2+ entry through LCCs in response to an AP raises [Ca2+]i in the cleft fast and high enough to activate CICR from j-SR RyRs. In contrast, the fact that the quantitatively much more abundant nj-SR in central regions does not associate with the sarcolemma raises the conceptual question of how RyRs of the nj-SR are activated in the first place. This conundrum is anchored in the fact that the Ca2+-sensitivity [70,82,116] of the cardiac-specific isoform of the RyR (RyR type-2, or RyR2) is low, and compared to the j-SR the activating Ca2+ signal for the nj-SR CRUs is slower, diffuser and lower in amplitude. Given the facts that bulk cytosolic CaT amplitude barely exceeds 1 μM and RyR Ca2+ sensitivity is low, in principle would preclude activation of nj-SR Ca2+ release. Nonetheless, during atrial ECC robust nj-SR Ca2+ release indeed occurs and actually provides the bulk Ca2+ supply for atrial contraction.
A similar situation is found for Ca2+ waves observed in atrial and ventricular cells under pathological conditions, especially during SR Ca2+ overload. Waves propagate through the cytosol by CICR in the absence of LCC activation and depend primarily on RyR properties [65], thus raising the same question how, without the LCC Ca2+ influx, CICR can be activated efficiently. We observed that in ventricular myocytes cell-wide propagation of spontaneous Ca2+ waves depends on an intra-SR Ca2+ ‘sensitization’ signal [66]. During wave propagation, the elevation of [Ca2+]i at the wave front leads to local Ca2+ uptake by SERCA which results in a local increase of [Ca2+]SR that sensitizes the RyR to cytosolic CICR via its luminal Ca2+-dependence [32]. By this mechanism the cytosolic Ca2+ sensitivity of the RyR is shifted to lower levels and brings the threshold for CICR into the range of the amplitude of a propagating cytosolic Ca2+ wave. A mechanism of wave propagation involving regulation of cytosolic Ca2+ sensitivity of the RyR by luminal Ca2+ during wave propagation has been proposed based on indirect experimental conclusions [52] and theoretical considerations [84], and was confirmed empirically with direct simultaneous measurements of [Ca2+]i and [Ca2+]SR [66].
These observations from ventricular myocytes fertilized a novel paradigm of atrial ECC. We extended the concept of an intra-SR Ca2+ sensitization signal to atrial myocytes [67] with the goal to unravel the aforementioned baffling conundrum of atrial ECC. By measuring simultaneously [Ca2+]i and [Ca2+]SR with high-resolution confocal fluorescence imaging we determined cytosolic CaTs and [Ca2+]SR depletion signals in atrial myocytes during AP induced Ca2+ release (Fig. 1B). The CaT initiated in the cell periphery through release of Ca2+ from j-SR is characterized by coinciding increase of [Ca2+]i and decline of [Ca2+]SR, reminiscent of ventricular cells where the rise of [Ca2+]i and decline of [Ca2+]SR also occur simultaneously and are highly synchronized throughout the entire myocyte. In stark contrast to ventricular cells, atrial Ca2+ release from central regions (nj-SR) lags behind peripheral release due to the time required for the activation to propagate to the center of the cell. The rise of central [Ca2+]i is slower and peaks at a lower level at a point in time when peripheral [Ca2+]i is already declining. Furthermore, the cell center revealed a temporal dispersion between onset of the cytosolic CaT and the decline of [Ca2+]SR. The time interval between rise of peripheral subsarcolemmal (SS) [Ca2+]i and begin of decline of central (CT) [Ca2+]SR was defined as latency (Δt between dashed vertical lines 1 and 3 in Fig. 1B). Along the transverse axis the latency steadily increased with increasing distance from the cell membrane, and the [Ca2+]SR signal revealed a unique and surprising feature. Instead of an immediate decline, a rise of [Ca2+]SR occurred before [Ca2+]SR began to decrease. This transient increase of [Ca2+]SR was highly reproducible in amplitude and kinetics, and could be observed reliably from beat to beat at the same CRU. Ca2+ uptake by SERCA at the propagation front was responsible for this rise of [Ca2+]SR during the latency period and generated - analogous to the previous observation for Ca2+ waves - an intra-SR Ca2+ sensitization signal that via luminal action lowers the activation threshold of the RyR to cytosolic CICR from nj-SR. The higher luminal [Ca2+]SR also lengthens RyR open time [12] and increases RyR unitary Ca2+ flux. Together, these luminal Ca2+ actions promote RyR activation and inter-RyR CICR and sustain robust propagating CICR through a mechanism termed ‘tandem activation’ of the nj-SR RyRs by cytosolic and luminal Ca2+. Additional experiments further confirmed the central role of SERCA in this process. β-adrenergic stimulation with isoproterenol to increase SERCA activity increased amplitude, duration and latency of the Ca2+ sensitization signal. In contrast, SERCA inhibition with cyclopiazonic acid abolished the Ca2+ sensitization signal [67].
Based on these experimental findings we proposed a novel paradigm of atrial ECC termed ‘fire-diffuse-uptake-fire’ or FDUF mechanism (Fig. 1C). In summary, atrial ECC consists of the following sequence of key events: atrial ECC is initiated by AP dependent membrane depolarization leading to LCC activation, Ca2+ influx and subsequent CICR from subsarcolemmal j-SR CRUs (or peripheral couplings). The rise of subsarcolemmal [Ca2+]i establishes a robust Ca2+ gradient that drives centripetal Ca2+ movement that subsequently triggers CICR from the first array of nj-SR CRUs which further initiates CICR from progressively more centrally located nj-SR CRUs. The process of propagation of CICR through the nj-SR network is sustained by the FDUF mechanism and the aforementioned tandem activation of nj-SR CRUs. Propagating CICR ultimately results in a cell-wide elevation of [Ca2+]i that initiates and sustains contraction.
There are additional features unique to atrial ECC and Ca2+ release. Comparison of AP-induced cytosolic CaTs and corresponding SR Ca2+ depletion signals revealed important differences between j-SR and nj-SR (Fig. 1D). Closer inspection of [Ca2+]i and [Ca2+]SR signals at individual CRUs revealed the largest cytosolic CaT amplitude in the cell periphery reflecting the initial AP-induced release of Ca2+ from the j-SR. Once activation reached the first nj-SR CRU (CT1) the cytosolic CaT amplitude decreased significantly, with further small progressive decline along the centripetal direction of propagation. In contrast and contrary to expectation, the depletion signal was smallest in the cell periphery (j-SR) and became significantly larger in the nj-SR despite a smaller amplitude of the cytosolic signal. The same pattern was found to apply to spontaneous elementary cytosolic Ca2+ release (Ca2+ sparks) and corresponding Ca2+ depletion events (Ca2+ blinks; [10]) measured simultaneously from individual CRUs (Figs. 1E, 1F). Spontaneous Ca2+ sparks originating from j-SR have a larger amplitude than nj-SR sparks consistent with earlier findings [93], however nj-SR Ca2+ blinks depleted to a lower [Ca2+]SR level than j-SR blinks and the depletion amplitude was larger (Fig. 1F). Thus, spontaneous elementary Ca2+ release and depletion events from individual CRUs mirror the differential properties of AP-induced CaTs originating from j-SR and nj-SR.
The subcellular differences in cytosolic CaT and SR Ca2+ depletion amplitudes raise interesting questions. Does the lower depletion level of the nj-SR suggest more effective CICR at nj-SR CRUs? The ability of a smaller cytosolic Ca2+ signal to trigger a larger depletion is advantageous for centripetal propagation of CICR. The requirement for the magnitude of the cytosolic trigger Ca2+ signal for nj-SR CICR appears to be less stringent, and fractional Ca2+ release, i.e. the relationship between magnitude of trigger and amount of released Ca2+, is larger for the nj-SR. Several potential mechanisms can be envisioned for the more pronounced depletion of nj-SR release sites. One possibility is that the pool size of releasable Ca2+ of an individual CRU is different in j-SR and nj-SR. The difference in [Ca2+]SR depletion levels in j-SR and nj-SR might also be related to intra-SR Ca2+ buffering. Intra-SR Ca2+ buffering is provided by the endogenous Ca2+ buffer calsequestrin (CASQ). CASQ buffers SR Ca2+ in a [Ca2+]SR dependent fashion and thereby determines Ca2+ storage capacity of the SR and the functional size of the Ca2+ store [103]. Furthermore, CASQ is also involved in luminal regulation of RyR gating [33,53,34]. High resolution studies revealed subcellular differences in CASQ endowment in atrial cells. RyR and CASQ co-localize to a lower degree and less CASQ staining is detected in the nj-SR [89], suggesting less CASQ mediated RyR inhibition and higher RyR excitability in the interior of atrial cells that facilitates the spread of excitation from the periphery to the center. Furthermore, lower CASQ levels and less Ca2+ buffering allow for depletion to lower [Ca2+]SR levels, consistent with our observations.
Furthermore, geometrical and structural factors contribute to the larger cytosolic Ca2+ signal in the cell periphery. In the cell periphery Ca2+ is released into the narrow cleft of the j-SR peripheral couplings. The narrow cleft geometry assures that [Ca2+]i in this confined space reaches high levels rapidly, while the same amount of Ca2+ release from a source that lacks surrounding membranes will dissipate rapidly and fail to reach comparable peak levels. This is indeed reflected in Ca2+ spark properties. Ca2+ sparks are the quintessential measure of Ca2+ release of an individual CRU. In the periphery of atrial cells Ca2+ sparks are spatially asymmetrical and show an elongation in longitudinal direction by ~1.7 compared to the transverse dimension [94]. In contrast, Ca2+ sparks from nj-SR are symmetrical. After surface membrane permeabilization the asymmetry and the amplitude of j-SR sparks is reduced (presumably by disrupting the physical integrity of the narrow cleft of the peripheral couplings), but the spatial dimensions and amplitude [93] of nj-SR sparks are unaffected. These data clearly show that the geometry of the narrow cleft of the j-SR peripheral couplings shapes the local Ca2+ signal. Furthermore, Ca2+ entry through LCCs makes a sizable contribution to cleft [Ca2+] [55]. Obviously, this Ca2+ source is absent in the center of a cell lacking t-tubules. Finally, the aforementioned lower phospholamban expression level in atrial cells is favorable for the buildup of the intra-SR Ca2+ sensitization signal, and differences in Ca2+ removal pathways contribute to peripheral and central CaTs [36] since only the j-SR is in the vicinity of plasmalemmal Na+/Ca2+ exchange (NCX), the main Ca2+ removal system in cardiac cells. Restoration of diastolic [Ca2+]i in central cell regions, however depends predominantly on SR Ca2+ reuptake, Ca2+ diffusion and buffering rather than Ca2+ extrusion. Further contributions to cardiac ECC come from mitochondria. Mitochondria contribute to ECC by cycling and buffering cytosolic Ca2+ which in turn shapes the cytosolic CaT. Mitochondria provide ATP for the contractile apparatus, ion pumps and alter the activity of Ca2+ handling proteins. Mitochondria are also a source of reactive oxygen species (ROS) which determine the redox environment of ECC Ca2+ handling proteins [17]. Mitochondria occupy approximately a third of the volume of a cardiac myocytes and have a large capacity to take up and sequester Ca2+. However, it has remained a matter of debate whether and how mitochondria might play a [Ca2+]i regulatory role on a beat-to-beat basis. An open question is whether the repetitive cytosolic CaTs are mirrored in robust oscillatory changes of mitochondrial Ca2+ ([Ca2+]mito), or whether the magnitude of mitochondrial Ca uptake on a beat-to-beat basis is small and changes of [Ca2+]mito reflect an integrative signal of cytosolic Ca2+ spiking (for discussion see [77]). Mitochondrial Ca2+ buffering also profoundly modulates ECC and CaTs in atrial myocytes. Inhibition of mitochondrial Ca2+ uptake or collapsing the negative mitochondrial membrane potential significantly accelerates the centripetal SR Ca2+ release propagation from the peripheral j-SR through the network of the nj-SR and increases the CaT amplitude of nj-SR release [37]. Furthermore, atrial cells from failing hearts (left-ventricular heart failure) have a reduced mitochondrial density and a decreased capacity to buffer Ca2+, resulting in CaTs of increased amplitude [38]. For efficient atrial ECC the propagation of CICR from the j-SR to the first array of nj-SR release sites is critical and the mechanism is controversial. In rat atrial myocytes mitochondria located between j-SR and nj-SR have been suggested to curtail Ca2+ passage by sequestering Ca2+ and acting as ‘mitochondrial firewall’ [8], however in rabbit atrial myocyte we found that this subcellular region is actually largely devoid of mitochondria and Ca2+ movement through this region occurs nearly twice as fast as through the central regions occupied by nj-SR [37].
IP3 receptor-induced Ca2+ release
Atrial myocytes are endowed with a second, less abundant Ca2+ release channel, the inositol-1,4,5-trisphosphate receptor (IP3R). Three isoforms of the IP3R are known, with the IP3R type 2 (IP3R2) being the predominant isoform in heart muscle. IP3Rs are outnumbered by RyRs ~1:50 at the protein level, however IP3R expression level in atrial cells is higher than in ventricular cells [57,19]. IP3R activation depends on G protein coupled receptor-mediated activation of phospholipase C (PLC). The subsequent hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) leads to formation of IP3 (Fig. 2A) and diacylglycerol (DAG), an activator of protein kinase C (PKC). In the atria increased [IP3] has been reported after vasoactive agonist stimulation (Fig. 2B), stretch, ischemia/reperfusion and in dilated cardiomyopathy. Several vasoactive agonists (angiotensin II, endothelin-1, phenylephrine) are capable of IP3R induced Ca2+ release (IICR) activation, indicative of the fact that atrial Ca2+ signaling during ECC is subject to humoral regulation. The role of IICR in heart has long been debated (reviewed in [57]), and involves participation in ECC but also non-ECC functions (e.g. regulation of nuclear Ca2+ [37,113] and transcription factors [86,111]). There is strong evidence that the nucleus and especially the nuclear envelope Ca2+ store is a distinct IICR signaling domain. Experiments performed in intact and membrane permeabilized myocytes as well as in isolated nuclei revealed that stimulation of IP3Rs elicited larger nuclear Ca2+ signals than RyR activation, and resting nuclear [Ca2+] increased more and with a different time course than cytosolic [Ca2+]. Also, agonist-dependent IICR stimulation increased AP-induced CaTs throughout the entire cell but had the most pronounced effect in the nuclear region, and the highest frequency of Ca2+ puffs, the elementary IICR events from IP3R clusters (Fig. 2C), was found in the nucleus [37,113]. Compared to RyR mediated Ca2+ sparks, Ca2+ puffs have an approximately 5-fold smaller amplitude, 3-fold longer duration and a 2- to 3-fold slower rise time [114,38]. In atrial myocytes IICR participation in ECC results in positive inotropy by increasing the amplitude of the AP-induced CaT (Figs. 2B, 2D). Enhancement of CaTs occurs through a cytosolic Ca2+-dependent sensitization of the RyR, whereas Ca2+ release through IP3Rs may also add directly to the CaT. But IICR also has proarrhythmic actions [114,61,62,19,50,64]. It increases diastolic [Ca2+]i, enhances the propensity of spontaneous Ca2+ release, results in spontaneous APs and arrhythmogenic Ca2+ waves (Fig. 2E), and leads to Ca2+ alternans (Figs. 2F, 2G). IP3R is upregulated in cardiac disease [38] and IICR acquires a more prominent role in atrial ECC by enhancing SR Ca2+ release from the nj-SR. Enhanced IICR under pathological conditions supports the important hemodynamic duties of the atria. Atrial contraction contributes to ventricular filling, referred to as ‘atrial kick’ or atrial booster function [69,39,104]. The contribution to ventricular filling amounts to 20–40% of end-diastolic filling and is subject to atrial remodeling in disease. We have shown previously that in earlier stages of ventricular failure when ventricular CaTs are already deteriorating, atrial CaTs are enhanced [38,36] and improve atrial contractile function, ventricular filling and thus ejection fraction and cardiac output. Upregulation of IICR and recruitment of IICR predominantly affects the nj-SR which is hemodynamically favorable since nj-SR Ca2+ release is the main Ca2+ supplier for atrial contraction.
Fig. 2. IP3 receptor induced Ca2+ release.
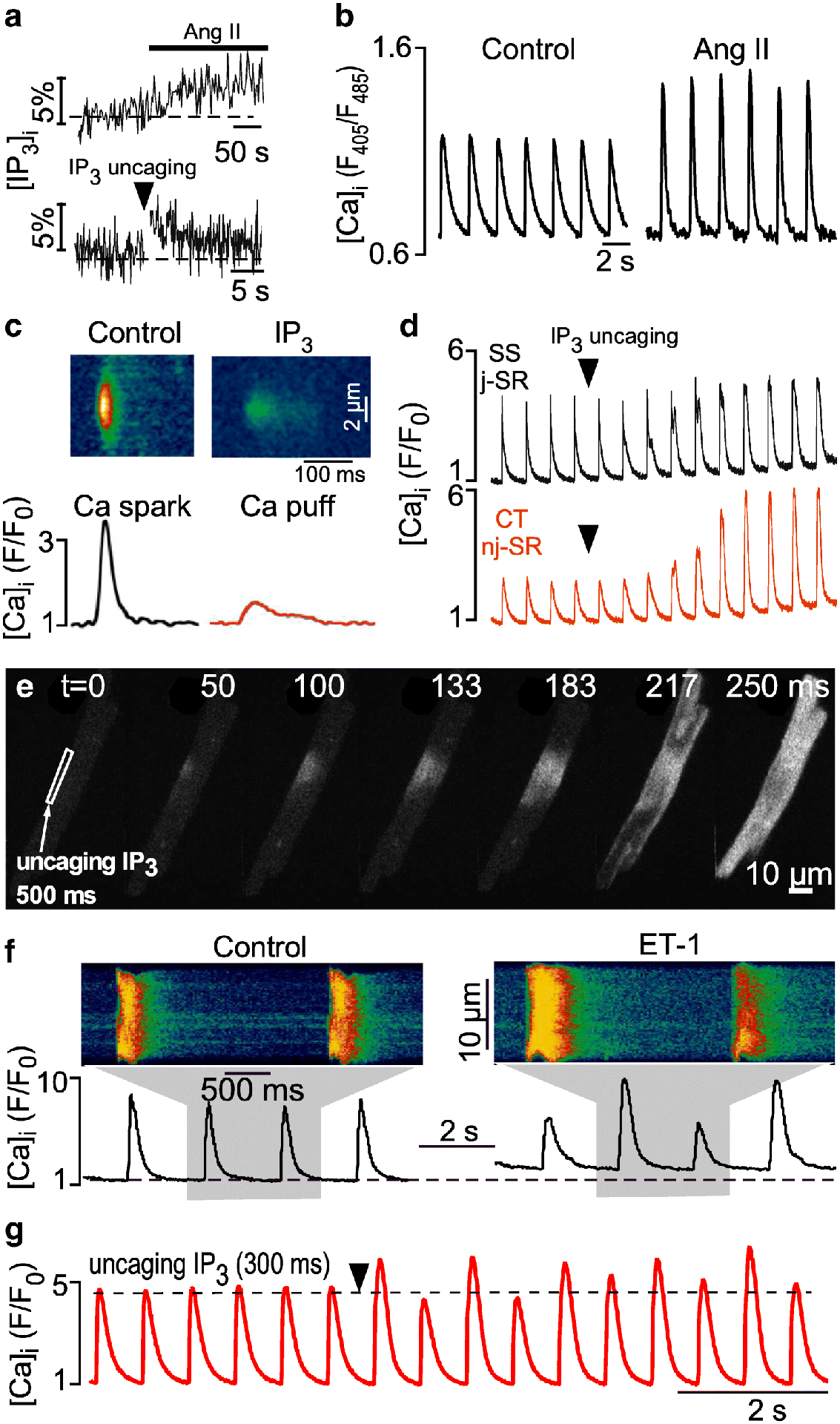
A, [IP3]i measurements with the FRET(CFP/YFP)-based IP3 sensor FIRE in response to Angiotensin II (Ang II) and photorelease of caged IP3 (cag-IP3 PM). [IP3]i expressed as % change of F530/F488. B, Ang II increased the amplitude of electrically evoked CaTs. C, average line scan images (top) and [Ca2+]i profiles (bottom) of Ca2+ sparks (black) and IP3- (red) mediated Ca2+ release events (Ca2+ puffs) recorded from membrane permeabilized atrial myocytes. Ca2+ puffs were elicited with IP3 in the presence of tetracaine to eliminate RyR mediated Ca2+ sparks. D, SS/j-SR and CT/nj-SR CaTs after IP3 uncaging. CaTs originating from nj-SR regions are enhanced to a larger degree by IICR than peripheral SS/j-SR CaTs. E, IP3 uncaging in a narrow subsarcolemmal region triggers a propagating Ca2+ wave. F, CaTs and CaT alternans recorded in control and in the presence of endothelin-1 (ET-1). G, IP3 uncaging elicits CaT alternans. Arrow head: exposure to 405 nm laser light for photolysis of caged IP3. Panels B and D reproduced and modified from [38]. Panels C and F reproduced and modified from [114]. Panels E and G reproduced and modified from [95].
Atrial electromechanical and Ca2+ alternans
As mentioned above one of the manifestations of Ca signaling disturbances in response to IICR is Ca2+ alternans (Figs. 2F, 2G). Alternans is a recognized risk factor for cardiac arrhythmia - including atrial fibrillation (AF) [14,27,74,1,105] - and sudden cardiac death [106,102,80]. Aside from IICR, a plethora of experimental and pathological conditions can cause cardiac alternans, suggesting a multifactorial process (for reviews see [108,109,13,60,22,87,23,5,71,20]). At the cellular level cardiac alternans is defined as cyclic, beat-to-beat alternation in contraction amplitude (mechanical alternans), AP duration (APD or electrical alternans) and Ca2+ transient amplitude (Ca2+ alternans) at constant stimulation frequency (Fig. 3A). APD and CaT are closely linked. This is due to the fact that the regulation of [Ca2+]i and Vm is bi-directionally coupled ([Ca2+]i↔Vm) and governed by complex feedback pathways. Vm→[Ca2+]i coupling refers to the notion that Vm contributes to [Ca2+]i regulation and Ca2+ signaling through AP attributes and APD restitution properties. The AP and the AP-dependent changes of Vm determine voltage-dependent Ca2+ fluxes. APD restitution is a time-dependent process and becomes especially critical at short cycle lengths when beat-to-beat differences in APD restitution and thus AP morphology profoundly affect Ca2+ release. [Ca2+]i→Vm coupling is determined by the effect of [Ca2+]i dynamics, Ca2+ fluxes, Ca2+ carrying membrane currents and Ca2+-dependent ion currents and transporters on Vm and APD. It is generally agreed that this bi-directional coupling represents a key causative factor for alternans. It is however a matter of ongoing debate whether Vm→[Ca2+]i or [Ca2+]i→Vm coupling is the predominant mechanism. While there are arguments and experimental data supporting both directions as the primary cause, recent results (including our own [45,96]) and computational findings tend to favor the prospect that Ca2+ signaling disturbances are the primary cause of alternans (Figs. 3B, 3C), although the debate is far from settled (for reviews and pertinent experimental work see [13,60,22,87,23,5,30]). Nonetheless, there is also experimental evidence that membrane conductances drive Ca2+ alternans, for example via LCCs [48], Ca2+-activated Cl− channels [46,47] and several K+ channels [49]. AP duration is an important determinant of susceptibility to alternans. AP prolongation is a risk factor for alternans and AP shortening has been proposed as a therapeutic strategy for alternans protection at the cellular and organ level [49], a strategy that could be especially effective in conditions of long QT syndrome [91].
Fig. 3. Electromechanical and Ca2+ alternans.
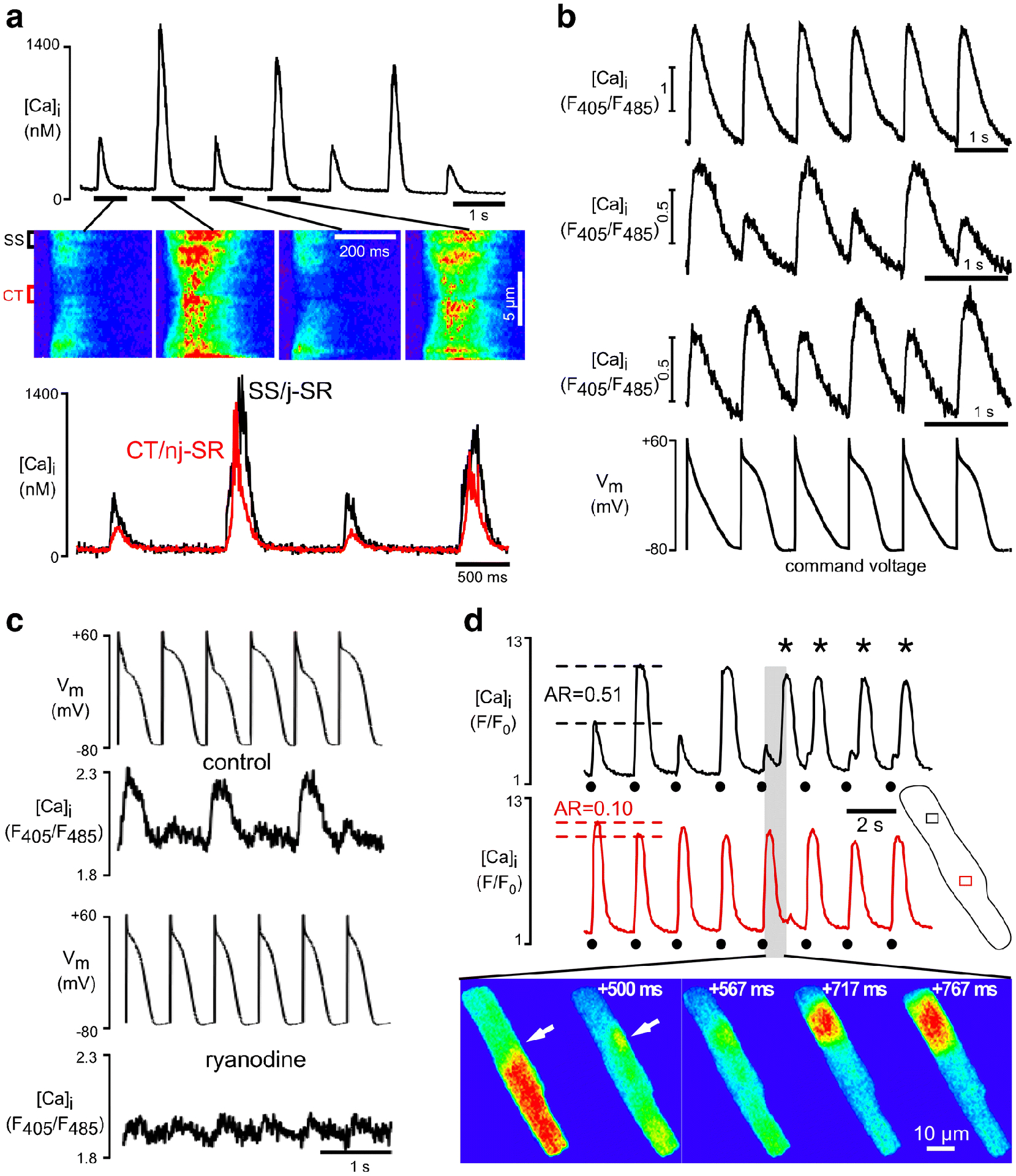
A, Confocal transverse line scan recordings of CaT alternans. Whole cell CaTs (top), selected line scan images (middle) and local (SS, subsarcolemmal, release from j-SR; CT, central, release from nj-SR) subcellular [Ca2+]i profiles (bottom) during alternans. B, CaTs recorded from atrial cells under voltage clamp conditions with an AP alternans command voltage protocol. AP alternans voltage clamp elicits no CaT alternans (top), CaT alternans where the large amplitude CaT coincides with the short AP (middle), and CaT alternans where the small amplitude CaT coincides with the short AP (bottom). The data indicate that CaT alternans can develop irrespective of Vm morphology or presence of AP alternans, indicative of a primary disturbance of Ca2+ signaling ([Ca2+]i→Vm coupling). C, Simultaneous recording of [Ca2+]i and Vm in current clamped atrial myocytes. Pacing induced AP and Ca2+ alternans in control (top). Bottom: SR Ca2+ release inhibition with ryanodine abolishes AP alternans, indicating that CaT alternans drives electrical alternans. D, Subcellular out-of-phase Ca2+ alternans with longitudinal alternans ratio (AR) gradient. The degree of alternans is quantified by the AR. AR=1-CaTSmall/CaTLarge, where CaTSmall and CaTLarge are the amplitudes of the small and large CaTs of a pair of alternating CaTs. Top portion of the cell reveals an approximately 5-fold higher AR than lower half of the cell. Repetitive propagating Ca2+ waves are initiated at the junction of out-of-phase alternating subcellular regions (white arrows). Circles: triggered CaTs. Asterisks: Ca2+ waves. Panel A reproduced and modified from [42]. Panels B and C reproduced and modified from [45]. Panel D reproduced and modified from [54].
Two parameters (for review and references see [108,109]) have emerged as being critically relevant to [Ca2+]i→Vm coupling and the origin of alternans: 1) SR Ca2+ load/fractional Ca2+ release, and 2) the efficiency of cytosolic Ca2+ sequestration. The non-linear relationship between Ca2+ sequestration and load/fractional release determines the vulnerability to alternans [108]. Here, Ca2+ sequestration refers to the net efficiency of clearing the cytosolic compartment of Ca2+. It includes Ca2+ reuptake into the SR via SERCA, extrusion via NCX and plasmalemmal Ca2+-ATPase, cytosolic buffering and mitochondrial uptake, but it also accounts for diastolic SR Ca2+ leak (via RyRs, IP3Rs and other pathways; see [115]) which counteracts any Ca2+ removal pathways. The relationship predicts that factors increasing Ca2+ load and fractional release promote, whereas factors increasing Ca2+ sequestration protect against alternans. A prominent role in alternans etiology is played by mitochondria [3,97,31]. Inhibition of various mitochondrial functions and signaling pathways (dissipation of mitochondrial membrane potential, inhibition of mitochondrial F1/F0-ATP synthase, electron transport chain (ETC), Ca2+-dependent dehydrogenases and mitochondrial Ca2+ uptake and extrusion) all enhanced CaT alternans [25,26], whereas stimulation of Ca2+ uptake via the mitochondrial Ca2+ uniporter complex improved CaT alternans [79]. The beat-to-beat dynamics of both Ca2+ sequestration and load/fractional release are critically dependent on restitution properties and refractory kinetics of the SR Ca2+ release mechanism. The amount of Ca2+ released during a given heartbeat is determined by the recovery of the trigger for CICR, SR Ca2+ load, and the release mechanism itself. APD restitution (including recovery of LCCs) has been recognized as a causative and/or contributing factor to Ca2+ alternans and may play a role particularly at high heart rates (reviewed in [108,109]). The role of SR Ca2+ reuptake and re-establishment of Ca2+ load have been the subject of numerous investigations [44,112,16,81] together with the controversial question whether cardiac alternans requires beat-to-beat alternations in SR Ca2+ content and end-diastolic SR filling [18] or not [42]. While it has been suggested that instability in the beat-to-beat feedback control of SR content leads to Ca2+ alternans [21], we revealed with direct dynamic measurements of [Ca2+]SR that alternans can occur with and without significant end-diastolic [Ca2+]SR fluctuations [81,96,20]. Refractoriness or recovery from inactivation of the Ca2+ release machinery also plays a key role for alternans. Recovery, when examined at the level of whole-cell CaTs as well as Ca2+ sparks [10,99,101,58], occurs on a time scale that overlaps with the stimulation frequencies where Ca2+ alternans typically occurs [2,58]. Ion channels participating in ECC, including the RyR, have time-dependent characteristics of recovery from inactivation, typically referred to as restitution properties. Restitution refers to the time interval required for the SR Ca2+ release to overcome refractoriness and to become fully available again after a previous release event. In atrial myocytes restitution of Ca2+ release from nj-SR is slower than from j-SR, reflecting another important difference in Ca2+ handling between j-SR and nj-SR [96], and SR release restitution properties have been demonstrated to play a key role in alternans.
Recently, an overarching conceptual model for cardiac alternans has been forwarded, termed ‘3R theory’ [88,76,83]. The 3R theory links Ca2+ spark properties (i.e. the properties of Ca2+ release from individual CRUs) to whole-cell Ca2+ alternans. The 3R theory states Ca2+ alternans occurs due to instabilities in the relationship of 3 critical spark attributes: Randomness, Recruitment, and Refractoriness. The theory predicts (based on numerical computations) that alternans occurs when the probability of a spontaneous primary spark is intermediate (intermediate randomness) but coupling between CRUs is strong (high degree of recruitment), and the degree of refractoriness is high. This unifying theoretical framework predicts how ECC Ca2+ handling proteins (LCC, RyR, SERCA, NCX, Ca2+ buffers) affect the 3 R’s and SR Ca2+ load, and thus predict Ca2+ alternans probability.
Atrial myocytes are particularly susceptible to Ca2+ alternans [5,42,54,56,45]. In contrast to ventricular myocytes, atrial Ca2+ alternans is subcellularly inhomogeneous (Fig. 3A) with transverse and longitudinal subcellular gradients in the degree of Ca2+ alternans, including subcellular regions alternating out-of-phase (Fig. 3D). This spatiotemporal heterogeneity of Ca2+ alternans is unique to atrial myocytes and - together with the unique atrial ECC mechanism - hints that the alternans mechanism is also distinctly different from that in ventricle. Complex subcellular [Ca2+]i inhomogeneities of atrial alternans generate a substrate for spontaneous (i.e. not electrically triggered) proarrhythmic Ca2+ release pointing towards a mechanistic link to atrial arrhythmia at the cellular level [54].
The beat-to-beat alternations in the time course of ventricular AP repolarization are reflected in the ECG as T-wave alternans (TWA). Even subtle TWA in the microvolt range (referred as microvolt TWA) has emerged as a valuable prognostic tool for ventricular arrhythmia risk stratification [72]. The clinical use of atrial AP repolarization alternans as a diagnostic tool, however, is hindered by the fact that in the conventional ECG recordings of the atrial repolarization signal is masked by the ventricular QRS complex. Nevertheless, several experimental [43,105] and clinical studies [35,73,74,59] using monophasic AP electrodes to monitor atrial repolarization alternans in-vivo have provided convincing evidence that AP alternans in atria may lead directly to atrial fibrillation or its transition from atrial flutter. Taken together, there is strong evidence that AP alternans precedes development of AF and has prognostic value for arrhythmia prediction.
Conclusions
Here we discussed unique structural and functional features of atrial ECC and Ca2+ signaling at the cellular level. While there are commonalities between atrial and ventricular ECC, structural differences with respect to arrangement of surface (t-tubules) and internal (SR) membranes, tissue-specific endowment with ion channels and electrophysiological attributes result in the discussed differential features of atrial and ventricular ECC. We emphasized the FDUF mechanism of tandem activation of SR Ca2+ release in atrial cells and the integral role played by the IP3R and IICR. Furthermore, we reviewed atria specific manifestations of alternans, its underlying mechanisms and relationship to atrial arrhythmia, especially AF. AF is the most prevalent manifestation of atrial arrhythmia and a frequent complication of heart failure [24,29] that carries a poor prognosis. The mechanisms and conditions leading to AF are complex and far from being completely understood [75], and the clinical magnitude of the problem, the high prevalence of the disease [24], and the prospect that the burden will likely only increase as the population ages and AF prevalence rises [15] constitute a grave health problem. Accumulating clinical evidence and a growing number of experimental studies indicate that AF is often related to episodes of atrial electromechanical alternans, however the alternans-AF link is often just phenomenological, leaving a mechanistic underpinning between alternans and AF elusive. Understanding the cellular mechanisms of electromechanical alternans has the potential to open a window not only towards a better understanding of AF mechanisms, but also novel therapeutic approaches of AF treatment.
Funding
This work was supported by National Institutes of Health grants HL057832 (LAB), HL128330 (KB), HL132871 (LAB, KB) and HL134781 (LAB).
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Akar FG (2014) Emergence of atrial repolarization alternans at late stages of remodeling: the “second factor” in atrial fibrillation progression? Journal of cardiovascular electrophysiology 25:428–430. doi: 10.1111/jce.12377 [DOI] [PubMed] [Google Scholar]
- 2.Alvarez-Lacalle E, Cantalapiedra IR, Penaranda A, Cinca J, Hove-Madsen L, Echebarria B (2013) Dependency of calcium alternans on ryanodine receptor refractoriness. PloS one 8:e55042. doi: 10.1371/journal.pone.0055042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aon MA (2013) Mitochondrial dysfunction, alternans, and arrhythmias. Front Physiol 4:83. doi: 10.3389/fphys.2013.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berlin JR (1995) Spatiotemporal changes of Ca2+ during electrically evoked contractions in atrial and ventricular cells. Am J Physiol 269:H1165–1170 [DOI] [PubMed] [Google Scholar]
- 5.Blatter LA, Kockskamper J, Sheehan KA, Zima AV, Huser J, Lipsius SL (2003) Local calcium gradients during excitation-contraction coupling and alternans in atrial myocytes. J Physiol 546:19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boknik P, Unkel C, Kirchhefer U, Kleideiter U, Klein-Wiele O, Knapp J, Linck B, Luss H, Muller FU, Schmitz W, Vahlensieck U, Zimmermann N, Jones LR, Neumann J (1999) Regional expression of phospholamban in the human heart. Cardiovasc Res 43:67–76 [DOI] [PubMed] [Google Scholar]
- 7.Bootman MD, Higazi DR, Coombes S, Roderick HL (2006) Calcium signalling during excitation-contraction coupling in mammalian atrial myocytes. J Cell Sci 119:3915–3925. doi: 10.1242/jcs.03223 [DOI] [PubMed] [Google Scholar]
- 8.Bootman MD, Smyrnias I, Thul R, Coombes S, Roderick HL (2011) Atrial cardiomyocyte calcium signalling. Biochimica et biophysica acta 1813:922–934. doi: 10.1016/j.bbamcr.2011.01.030 [DOI] [PubMed] [Google Scholar]
- 9.Brandenburg S, Kohl T, Williams GS, Gusev K, Wagner E, Rog-Zielinska EA, Hebisch E, Dura M, Didie M, Gotthardt M, Nikolaev VO, Hasenfuss G, Kohl P, Ward CW, Lederer WJ, Lehnart SE (2016) Axial tubule junctions control rapid calcium signaling in atria. J Clin Invest 126:3999–4015. doi: 10.1172/JCI88241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brochet DX, Yang D, Di Maio A, Lederer WJ, Franzini-Armstrong C, Cheng H (2005) Ca2+ blinks: rapid nanoscopic store calcium signaling. Proc Natl Acad Sci U S A 102:3099–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ Jr., Vaghy PL, Meissner G, Ferguson DG (1995) Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. J Cell Biol 129:672–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen H, Valle G, Furlan S, Nani A, Gyorke S, Fill M, Volpe P (2013) Mechanism of calsequestrin regulation of single cardiac ryanodine receptor in normal and pathological conditions. The Journal of general physiology 142:127–136. doi: 10.1085/jgp.201311022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clusin WT (2008) Mechanisms of calcium transient and action potential alternans in cardiac cells and tissues. Am J Physiol Heart Circ Physiol 294:H1–H10. doi:00802.2007 [DOI] [PubMed] [Google Scholar]
- 14.Comtois P, Nattel S (2012) Atrial repolarization alternans as a path to atrial fibrillation. Journal of cardiovascular electrophysiology 23:1013–1015. doi: 10.1111/j.1540-8167.2012.02391.x [DOI] [PubMed] [Google Scholar]
- 15.Coyne KS, Paramore C, Grandy S, Mercader M, Reynolds M, Zimetbaum P (2006) Assessing the direct costs of treating nonvalvular atrial fibrillation in the United States. Value in health : the journal of the International Society for Pharmacoeconomics and Outcomes Research 9:348–356. doi: 10.1111/j.1524-4733.2006.00124.x [DOI] [PubMed] [Google Scholar]
- 16.Cutler MJ, Wan X, Laurita KR, Hajjar RJ, Rosenbaum DS (2009) Targeted SERCA2a gene expression identifies molecular mechanism and therapeutic target for arrhythmogenic cardiac alternans. Circ Arrhythm Electrophysiol 2:686–694. doi:CIRCEP.109.863118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dedkova EN, Blatter LA (2013) Calcium signaling in cardiac mitochondria. Journal of molecular and cellular cardiology 58:125–133. doi: 10.1016/j.yjmcc.2012.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diaz ME, O’Neill SC, Eisner DA (2004) Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res 94:650–656 [DOI] [PubMed] [Google Scholar]
- 19.Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA (2008) IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol 294:H596–604 [DOI] [PubMed] [Google Scholar]
- 20.Edwards JN, Blatter LA (2014) Cardiac alternans and intracellular calcium cycling. Clinical and Experimental Pharmacology and Physiology 41:524–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisner DA, Diaz ME, Li Y, O’Neill SC, Trafford AW (2005) Stability and instability of regulation of intracellular calcium. Exp Physiol 90:3–12 [DOI] [PubMed] [Google Scholar]
- 22.Eisner DA, Li Y, O’Neill SC (2006) Alternans of intracellular calcium: mechanism and significance. Heart Rhythm 3:743–745 [DOI] [PubMed] [Google Scholar]
- 23.Euler DE (1999) Cardiac alternans: mechanisms and pathophysiological significance. Cardiovasc Res 42:583–590 [DOI] [PubMed] [Google Scholar]
- 24.Feinberg WM, Blackshear JL, Laupacis A, Kronmal R, Hart RG (1995) Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Archives of internal medicine 155:469–473 [PubMed] [Google Scholar]
- 25.Florea SM, Blatter LA (2010) The role of mitochondria for the regulation of cardiac alternans. Front Physio 1:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Florea SM, Blatter LA (2012) Regulation of cardiac alternans by beta-adrenergic signaling pathways. American journal of physiology Heart and circulatory physiology 303:H1047–1056. doi: 10.1152/ajpheart.00384.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franz MR, Jamal SM, Narayan SM (2012) The role of action potential alternans in the initiation of atrial fibrillation in humans: a review and future directions. Europace 14 Suppl 5:v58–v64. doi: 10.1093/europace/eus273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franzini-Armstrong C, Jorgensen AO (1994) Structure and development of E-C coupling units in skeletal muscle. Annu Rev Physiol 56:509–534. doi: 10.1146/annurev.ph.56.030194.002453 [DOI] [PubMed] [Google Scholar]
- 29.Go AS, Hylek EM, Phillips KA, Chang Y, Henault LE, Selby JV, Singer DE (2001) Prevalence of diagnosed atrial fibrillation in adults: national implications for rhythm management and stroke prevention: the AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 285:2370–2375 [DOI] [PubMed] [Google Scholar]
- 30.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN (2005) Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res 96:459–466 [DOI] [PubMed] [Google Scholar]
- 31.Gordan R, Fefelova N, Gwathmey JK, Xie LH (2016) Involvement of mitochondrial permeability transition pore (mPTP) in cardiac arrhythmias: Evidence from cyclophilin D knockout mice. Cell Calcium 60:363–372. doi: 10.1016/j.ceca.2016.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gyorke I, Gyorke S (1998) Regulation of the cardiac ryanodine receptor channel by luminal Ca2+ involves luminal Ca2+ sensing sites. Biophysical journal 75:2801–2810. doi: 10.1016/S0006-3495(98)77723-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gyorke S, Stevens SC, Terentyev D (2009) Cardiac calsequestrin: quest inside the SR. J Physiol 587:3091–3094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gyorke S, Terentyev D (2008) Modulation of ryanodine receptor by luminal calcium and accessory proteins in health and cardiac disease. Cardiovascular research 77:245–255. doi: 10.1093/cvr/cvm038 [DOI] [PubMed] [Google Scholar]
- 35.Hiromoto K, Shimizu H, Furukawa Y, Kanemori T, Mine T, Masuyama T, Ohyanagi M (2005) Discordant repolarization alternans-induced atrial fibrillation is suppressed by verapamil. Circ J 69:1368–1373 [DOI] [PubMed] [Google Scholar]
- 36.Hohendanner F, DeSantiago J, Heinzel FR, Blatter LA (2016) Dyssynchronous calcium removal in heart failure-induced atrial remodeling. Am J Physiol Heart Circ Physiol 311:H1352–H1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hohendanner F, Maxwell JT, Blatter LA (2015) Cytosolic and nuclear calcium signaling in atrial myocytes: IP3-mediated calcium release and the role of mitochondria. Channels 9:129–138. doi: 10.1080/19336950.2015.1040966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohendanner F, Walther S, Maxwell JT, Kettlewell S, Awad S, Smith GL, Lonchyna VA, Blatter LA (2015) Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation-contraction coupling in atrial myocytes from normal and failing hearts. The Journal of Physiology 593:1459–1477. doi: 10.1113/jphysiol.2014.283226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoit BD (2014) Left atrial size and function: role in prognosis. Journal of the American College of Cardiology 63:493–505. doi: 10.1016/j.jacc.2013.10.055 [DOI] [PubMed] [Google Scholar]
- 40.Huser J, Bers DM, Blatter LA (1998) Subcellular properties of [Ca2+]i transients in phospholamban-deficient mouse ventricular cells. Am J Physiol 274:H1800–1811 [DOI] [PubMed] [Google Scholar]
- 41.Huser J, Lipsius SL, Blatter LA (1996) Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol 494 (Pt 3):641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA (2000) Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol 524 Pt 3:795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jousset F, Tenkorang J, Vesin JM, Pascale P, Ruchat P, Rollin AG, Fromer M, Narayan SM, Pruvot E (2012) Kinetics of atrial repolarization alternans in a free-behaving ovine model. Journal of Cardiovass Electrophysiol 23:1003–1012. doi: 10.1111/j.1540-8167.2012.02336.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kameyama M, Hirayama Y, Saitoh H, Maruyama M, Atarashi H, Takano T (2003) Possible contribution of the sarcoplasmic reticulum Ca2+ pump function to electrical and mechanical alternans. J Electrocardiol 36:125–135 [DOI] [PubMed] [Google Scholar]
- 45.Kanaporis G, Blatter LA (2015) The Mechanisms of Calcium Cycling and Action Potential Dynamics in Cardiac Alternans. Circ Res 116:846–856. doi: 10.1161/CIRCRESAHA.116.305404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanaporis G, Blatter LA (2016) Ca2+-activated chloride channel activity during Ca2+ alternans in ventricular myocytes. Channels (Austin) 10:507–517. doi: 10.1080/19336950.2016.1207020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanaporis G, Blatter LA (2016) Calcium-activated chloride current determines action potential morphology during calcium alternans in atrial myocytes. The Journal of Physiology 594.3:699–714. doi: 10.1113/JP271887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanaporis G, Blatter LA (2017) Membrane potential determines calcium alternans through modulation of SR Ca2+ load and L-type Ca2+ current. J Mol Cell Cardiol 105:49–58. doi: 10.1016/j.yjmcc.2017.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanaporis G, Kalik ZM, Blatter LA (2019) Action potential shortening rescues atrial calcium alternans. J Physiol 597:723–740. doi: 10.1113/JP277188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kapur N, Banach K (2007) Inositol-1,4,5-trisphosphate-mediated spontaneous activity in mouse embryonic stem cell-derived cardiomyocytes. J Physiol 581:1113–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keizer J, Smith GD, Ponce-Dawson S, Pearson JE (1998) Saltatory propagation of Ca2+ waves by Ca2+ sparks. Biophysical journal 75:595–600. doi: 10.1016/S0006-3495(98)77550-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keller M, Kao JP, Egger M, Niggli E (2007) Calcium waves driven by “sensitization” wave-fronts. Cardiovascular research 74:39–45. doi: 10.1016/j.cardiores.2007.02.006 [DOI] [PubMed] [Google Scholar]
- 53.Knollmann BC (2009) New roles of calsequestrin and triadin in cardiac muscle. J Physiol 587:3081–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kockskamper J, Blatter LA (2002) Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J Physiol 545:65–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kockskamper J, Sheehan KA, Bare DJ, Lipsius SL, Mignery GA, Blatter LA (2001) Activation and propagation of Ca2+ release during excitation-contraction coupling in atrial myocytes. Biophys J 81:2590–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kockskamper J, Zima AV, Blatter LA (2005) Modulation of sarcoplasmic reticulum Ca2+ release by glycolysis in cat atrial myocytes. J Physiol 564:697–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kockskamper J, Zima AV, Roderick HL, Pieske B, Blatter LA, Bootman MD (2008) Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol 45:128–147. doi:S0022–2828(08)00451–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kornyeyev D, Petrosky AD, Zepeda B, Ferreiro M, Knollmann B, Escobar AL (2012) Calsequestrin 2 deletion shortens the refractoriness of Ca2+ release and reduces rate-dependent Ca2+ -alternans in intact mouse hearts. Journal of molecular and cellular cardiology 52:21–31. doi: 10.1016/j.yjmcc.2011.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lalani GG, Schricker AA, Clopton P, Krummen DE, Narayan SM (2013) Frequency analysis of atrial action potential alternans: a sensitive clinical index of individual propensity to atrial fibrillation. Circulation Arrhythmia and electrophysiology 6:859–867. doi: 10.1161/CIRCEP.113.000204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laurita KR, Rosenbaum DS (2008) Cellular mechanisms of arrhythmogenic cardiac alternans. Prog Biophys Mol Biol 97:332–347. doi:S0079–6107(08)00025–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li X, Zima AV, Sheikh F, Blatter LA, Chen J (2005) Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res 96:1274–1281 [DOI] [PubMed] [Google Scholar]
- 62.Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD (2000) Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol 10:939–942 [DOI] [PubMed] [Google Scholar]
- 63.Mackenzie L, Bootman MD, Berridge MJ, Lipp P (2001) Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol 530:417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mackenzie L, Bootman MD, Laine M, Berridge MJ, Thuring J, Holmes A, Li WH, Lipp P (2002) The role of inositol 1,4,5-trisphosphate receptors in Ca2+ signalling and the generation of arrhythmias in rat atrial myocytes. J Physiol 541:395–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martinez-Hernandez E, Blatter LA (2020) Effect of carvedilol on atrial excitation-contraction coupling, Ca2+ release, and arrhythmogenicity. Am J Physiol Heart Circ Physiol 318:H1245–H1255. doi: 10.1152/ajpheart.00650.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maxwell JT, Blatter LA (2012) Facilitation of cytosolic calcium wave propagation by local calcium uptake into the sarcoplasmic reticulum in cardiac myocytes. The Journal of physiology 590:6037–6045. doi: 10.1113/jphysiol.2012.239434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maxwell JT, Blatter LA (2017) A novel mechanism of tandem activation of ryanodine receptors by cytosolic and SR luminal Ca2+ during excitation-contraction coupling in atrial myocytes. J Physiol 595:3835–3845. doi: 10.1113/JP273611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McNutt NS, Fawcett DW (1969) The ultrastructure of the cat myocardium. II. Atrial muscle. J Cell Biol 42:46–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mehrzad R, Rajab M, Spodick DH (2014) The three integrated phases of left atrial macrophysiology and their interactions. Int J Mol Sci 15:15146–15160. doi: 10.3390/ijms150915146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meissner G, Henderson JS (1987) Rapid calcium release from cardiac sarcoplasmic reticulum vesicles is dependent on Ca2+ and is modulated by Mg2+, adenine nucleotide, and calmodulin. The Journal of biological chemistry 262:3065–3073 [PubMed] [Google Scholar]
- 71.Merchant FM, Armoundas AA (2012) Role of substrate and triggers in the genesis of cardiac alternans, from the myocyte to the whole heart: implications for therapy. Circulation 125:539–549. doi: 10.1161/CIRCULATIONAHA.111.033563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Merchant FM, Sayadi O, Moazzami K, Puppala D, Armoundas AA (2013) T-wave alternans as an arrhythmic risk stratifier: state of the art. Curr Cardiol Rep 15:398. doi: 10.1007/s11886-013-0398-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Narayan SM, Bode F, Karasik PL, Franz MR (2002) Alternans of atrial action potentials during atrial flutter as a precursor to atrial fibrillation. Circulation 106:1968–1973 [DOI] [PubMed] [Google Scholar]
- 74.Narayan SM, Franz MR, Clopton P, Pruvot EJ, Krummen DE (2011) Repolarization alternans reveals vulnerability to human atrial fibrillation. Circulation 123:2922–2930. doi: 10.1161/CIRCULATIONAHA.110.977827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nattel S (2002) New ideas about atrial fibrillation 50 years on. Nature 415:219–226 [DOI] [PubMed] [Google Scholar]
- 76.Nivala M, Qu Z (2012) Calcium alternans in a couplon network model of ventricular myocytes: role of sarcoplasmic reticulum load. American journal of physiology Heart and circulatory physiology 303:H341–352. doi: 10.1152/ajpheart.00302.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.O’Rourke B, Blatter LA (2009) Mitochondrial Ca2+ uptake: tortoise or hare? J Mol Cell Cardiol 46:767–774. doi:S0022–2828(08)01443–0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ordög B, Brutyó E, Puskás LG, Papp JG, Varró A, Szabad J, Boldogkoi Z (2006) Gene expression profiling of human cardiac potassium and sodium channels. Int J Cardiol 111:386–393. doi: 10.1016/j.ijcard.2005.07.063 [DOI] [PubMed] [Google Scholar]
- 79.Oropeza-Almazan Y, Blatter LA (2020) Mitochondrial Calcium Uniporter Complex Activation Protects Against Calcium Alternans in Atrial Myocytes. Am J Physiol Heart Circ Physiol. doi: 10.1152/ajpheart.00375.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS (1999) Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 99:1385–1394 [DOI] [PubMed] [Google Scholar]
- 81.Picht E, DeSantiago J, Blatter LA, Bers DM (2006) Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res 99:740–748 [DOI] [PubMed] [Google Scholar]
- 82.Qin J, Valle G, Nani A, Chen H, Ramos-Franco J, Nori A, Volpe P, Fill M (2009) Ryanodine receptor luminal Ca2+ regulation: swapping calsequestrin and channel isoforms. Biophysical journal 97:1961–1970. doi: 10.1016/j.bpj.2009.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qu Z, Nivala M, Weiss JN (2013) Calcium alternans in cardiac myocytes: Order from disorder. Journal of molecular and cellular cardiology 58:100–109. doi: 10.1016/j.yjmcc.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ramay HR, Jafri MS, Lederer WJ, Sobie EA (2010) Predicting local SR Ca2+ dynamics during Ca2+ wave propagation in ventricular myocytes. Biophysical journal 98:2515–2523. doi: 10.1016/j.bpj.2010.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM (2011) Transverse tubules are a common feature in large mammalian atrial myocytes including human. American journal of physiology Heart and circulatory physiology 301:H1996–2005. doi: 10.1152/ajpheart.00284.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rinne A, Blatter LA (2010) Activation of NFATc1 is directly mediated by IP3 in adult cardiac myocytes. Am J Physiol 299:H1701–H1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rosenbaum DS (2001) T wave alternans: a mechanism of arrhythmogenesis comes of age after 100 years. J Cardiovasc Electrophysiol 12:207–209 [DOI] [PubMed] [Google Scholar]
- 88.Rovetti R, Cui X, Garfinkel A, Weiss JN, Qu Z (2010) Spark-Induced Sparks As a Mechanism of Intracellular Calcium Alternans in Cardiac Myocytes. Circ Res 106:1582–1591. doi:CIRCRESAHA.109.213975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Schulson MN, Scriven DR, Fletcher P, Moore ED (2011) Couplons in rat atria form distinct subgroups defined by their molecular partners. J Cell Sci 124:1167–1174. doi: 10.1242/jcs.080929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scriven DR, Asghari P, Moore ED (2013) Microarchitecture of the dyad. Cardiovascular research 98:169–176. doi: 10.1093/cvr/cvt025 [DOI] [PubMed] [Google Scholar]
- 91.Shah SR, Park K, Alweis R (2019) Long QT Syndrome: A Comprehensive Review of the Literature and Current Evidence. Current problems in cardiology 44:92–106. doi: 10.1016/j.cpcardiol.2018.04.002 [DOI] [PubMed] [Google Scholar]
- 92.Sheehan KA, Blatter LA (2003) Regulation of junctional and non-junctional sarcoplasmic reticulum calcium release in excitation-contraction coupling in cat atrial myocytes. J Physiol 546:119–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sheehan KA, Zima AV, Blatter LA (2006) Regional differences in spontaneous Ca2+ spark activity and regulation in cat atrial myocytes. J Physiol 572:799–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shkryl VM, Blatter LA (2013) Ca2+ release events in cardiac myocytes up close: insights from fast confocal imaging. PloS one 8:e61525. doi: 10.1371/journal.pone.0061525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shkryl VM, Maxwell JT, Blatter LA (2012) A novel method for spatially complex diffraction-limited photoactivation and photobleaching in living cells. J Physiol 590:1093–1100. doi:jphysiol.2011.223446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shkryl VM, Maxwell JT, Domeier TL, Blatter LA (2012) Refractoriness of sarcoplasmic reticulum Ca release determines Ca alternans in atrial myocytes. Am J Physiol Heart Circ Physiol 302:H2310–2320. doi:ajpheart.00079.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Smith RM, Visweswaran R, Talkachova I, Wothe JK, Tolkacheva EG (2013) Uncoupling the mitochondria facilitates alternans formation in the isolated rabbit heart. American journal of physiology Heart and circulatory physiology. doi: 10.1152/ajpheart.00915.2012 [DOI] [PubMed] [Google Scholar]
- 98.Smyrnias I, Mair W, Harzheim D, Walker SA, Roderick HL, Bootman MD (2010) Comparison of the T-tubule system in adult rat ventricular and atrial myocytes, and its role in excitation-contraction coupling and inotropic stimulation. Cell calcium 47:210–223. doi: 10.1016/j.ceca.2009.10.001 [DOI] [PubMed] [Google Scholar]
- 99.Sobie EA, Song LS, Lederer WJ (2005) Local recovery of Ca2+ release in rat ventricular myocytes. J Physiol 565:441–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR, Rios E (1999) Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J Gen Physiol 113:469–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Szentesi P, Pignier C, Egger M, Kranias EG, Niggli E (2004) Sarcoplasmic Reticulum Ca2+ Refilling Controls Recovery From Ca2+-Induced Ca2+ Release Refractoriness in Heart Muscle. Circ Res 95:807–813 [DOI] [PubMed] [Google Scholar]
- 102.Ter Keurs HE, Boyden PA (2007) Calcium and arrhythmogenesis. Physiol Rev 87:457–506. doi:87/2/457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S (2003) Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc Natl Acad Sci U S A 100:11759–11764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Trafford AW, Clarke JD, Richards MA, Eisner DA, Dibb KM (2013) Calcium signalling microdomains and the t-tubular system in atrial mycoytes: potential roles in cardiac disease and arrhythmias. Cardiovascular research 98:192–203. doi: 10.1093/cvr/cvt018 [DOI] [PubMed] [Google Scholar]
- 105.Verrier RL, Fuller H, Justo F, Nearing BD, Rajamani S, Belardinelli L (2016) Unmasking atrial repolarization to assess alternans, spatiotemporal heterogeneity, and susceptibility to atrial fibrillation. Heart Rhythm 13:953–961. doi: 10.1016/j.hrthm.2015.11.019 [DOI] [PubMed] [Google Scholar]
- 106.Walker ML, Rosenbaum DS (2003) Repolarization alternans: implications for the mechanism and prevention of sudden cardiac death. Cardiovasc Res 57:599–614 [DOI] [PubMed] [Google Scholar]
- 107.Wang Z, Yue L, White M, Pelletier G, Nattel S (1998) Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation 98:2422–2428. doi: 10.1161/01.cir.98.22.2422 [DOI] [PubMed] [Google Scholar]
- 108.Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z (2006) From pulsus to pulseless: the saga of cardiac alternans. Circ Res 98:1244–1253 [DOI] [PubMed] [Google Scholar]
- 109.Weiss JN, Nivala M, Garfinkel A, Qu Z (2011) Alternans and arrhythmias: from cell to heart. Circ Res 108:98–112. doi:108/1/98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Woo SH, Cleemann L, Morad M (2003) Spatiotemporal characteristics of junctional and nonjunctional focal Ca2+ release in rat atrial myocytes. Circ Res 92:e1–11 [DOI] [PubMed] [Google Scholar]
- 111.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM (2006) Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest 116:675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xie LH, Sato D, Garfinkel A, Qu Z, Weiss JN (2008) Intracellular Ca alternans: coordinated regulation by sarcoplasmic reticulum release, uptake, and leak. Biophys J 95:3100–3110. doi:S0006–3495(08)78450–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zima AV, Bare DJ, Mignery GA, Blatter LA (2007) IP3-dependent nuclear Ca2+ signalling in the mammalian heart. J Physiol 584:601–611. doi:jphysiol.2007.140731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zima AV, Blatter LA (2004) Inositol-1,4,5-trisphosphate-dependent Ca2+ signalling in cat atrial excitation-contraction coupling and arrhythmias. J Physiol 555:607–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zima AV, Bovo E, Bers DM, Blatter LA (2010) Ca2+ spark-dependent and - independent sarcoplasmic reticulum Ca2+ leak in normal and failing rabbit ventricular myocytes. J Physiol 588:4743–4757. doi:jphysiol.2010.197913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zima AV, Kockskamper J, Mejia-Alvarez R, Blatter LA (2003) Pyruvate modulates cardiac sarcoplasmic reticulum Ca2+ release in rats via mitochondria-dependent and -independent mechanisms. J Physiol 550:765–783 [DOI] [PMC free article] [PubMed] [Google Scholar]