

Abstract
Programmed death-ligand 1 (PD-L1) is a co-inhibitory molecule expressed on tumor cells. Immune checkpoint inhibitors focusing on the PD-L1 mechanism are now being studied for the treatment of various cancer types. However, the regulatory mechanism of PD-L1 is yet to be fully clarified, and a high-throughput system for comparing the abilities of small compounds in regulating PD-L1 has not yet been established. Therefore, we created a HiBiT-tagged lung adenocarcinoma cell line, PC9-KI, for easier and faster detection of changes in PD-L1 protein expression. Using PC9-KI cells, we screened 1,280 chemical compounds from the Library of Pharmacologically Active Compounds (LOPAC) and identified microtubule polymerization inhibitors and thapsigargin as PD-L1 upregulators and a p97 inhibitor as a PD-L1 downregulator.
Keywords: Chemical compound, endogenous protein, HiBiT, High-throughput screening, PD-L1
1. Introduction
The tumor microenvironment is composed of not only tumor cells but also of various immune cells such as infiltrated T cells, tumor-associated macrophages, and natural killer cells. In the tumor microenvironment, tumor cells express various cytokines, chemokines, and ligands, which help them survive [1]. Programmed death-ligand 1 (PD-L1) is a factor considered to be a promising target for tumor therapy. High expression of PD-L1 by tumor cells induces anergy by directly binding to PD-1 receptors, making it possible for the tumor cells to evade the immune system [2]. Immune checkpoint inhibitors such as anti-PD-1 and anti-PD-L1 antibodies are now used for many cancer types, which reflects on the clinical importance of the PD-L1 regulatory mechanisms [3]. Cytokines dependent on signaling pathways have already been reported to be important for the regulation of PD-L1 [4–9]. Currently, no techniques exist to identify chemical compounds that regulate PD-L1 protein expression by using chemical compound libraries in a cross-sectional manner. Thus, a high-throughput screening system that detects changes in PD-L1 protein expressions is of vital importance.
Herein, we present a screening system that introduces HiBiT-tagged cells as a novel strategy for identifying chemical compounds that regulate PD-L1 expression. The HiBiT tag is a peptide tag composed of only 11 amino acids that, upon insertion into the target proteins, functions as a NanoLuciferase by forming a dimer with LgBiT. By measuring the luminescence changes after inserting the peptide tag, a relatively simple procedure can enable the rapid detection of expression changes of the target proteins [10]. Using genome editing, we modified the lung adenocarcinoma cell line PC9 with HiBiT tagged at the C-terminus of PD-L1 to create PC9-KI cells. We then developed a chemical screening strategy by adding 1,280 compounds from the chemical compound library LOPAC (library of pharmacologically active compounds; Sigma Aldrich). Here, we report our success in identifying several chemical compounds that regulate the expression of PD-L1.
2. Materials and methods
2.1. Cell culture
PC9 and PC9-KI cells were maintained in Roswell Park Memorial Institute (RPMI) (Corning, NY, USA) medium supplemented with 10% fetal bovine serum (FBS) (Gibco, CA, USA), and 1% penicillin-streptomycin (Wako, Osaka, Japan) at 37°C with 5% CO2. A375 and MDAMB231 cells were maintained in DMEM (Corning) supplemented with 10%FBS (Gibco) and 1% penicillin-streptomycin (Wako) at 37°C with 5%CO2.
2.2. Creating HiBiT knocked-in cells with genome editing
HiBiT-tagged PC9 cells were established using the CRISPR-Cas9 system. ALT-R XT CRISPR RNA (crRNA) (Integrated DNA Technologies, IA, USA) was re-suspended in Nuclease-free-Duplex Buffer (Integrated DNA Technologies) to a final concentration of 100μM. Equal volumes of crRNA and trans-activating CRISPR RNA (tracrRNA) were mixed and heated for 5 minutes at 95°C. After heating, the oligo complex was gradually cooled down to room temperature. The oligo complex was then incubated at room temperature for 20 minutes with ALT-R Cas9 Nuclease V3 (61μM) (Integrated DNA Technologies) to form the Cas9 complex. The single-stranded DNA (ssDNA) oligo, including sequences of HiBiT, a complementary sequence to the C-Terminal in the CD274 genome, and the Cas9 complex were then co-transfected into the PC9 cells using the NEPA21 Super Electroporator (NEPAGENE, Chiba, Japan). After incubating the cells for a few days, single cell cloning was performed to pick up the HiBiT-tagged cells. The sequences of crRNA and ssDNA oligo are shown in Table S1.
2.3. Validation of the targeted insertion of the HiBiT tag
Insertion of the HiBiT tag sequence in the genome of the knocked-in cells was confirmed by sequencing. For further confirmation, western blotting was performed as follows. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed for approximately 50μg of protein from wild type (PC9) and HiBiT-tagged PC9 (PC9-KI) cells, which were collected using the RIPA lysis buffer (50mM Tris-HCl, 150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1%SDS). The proteins were then transferred to nitrocellulose membranes and blocked with Blocking-one (Nacalai Teque, Kyoto, Japan) for 1h. Two milliliters of LgBiT mix (PBST 2mL and LgBiT (Promega, WI, USA) 8μL) was added and reacted at 4°C for overnight. The membrane was added three times in PBST for 10 min and 1 mL of substrate mix (PBST 1mL and substrate (Promega) 3μL) was added and reacted for 10 min for detection. The chemical luminescence was captured using ImageQuant LAS4000 (GE Healthcare, MI, USA).
To compare the luminescence of wild type PC9 cells and PC9-KI cells, 4×104 of PC9 and PC9-KI cells were seeded in each well of the 96 well plates per well, and incubated for 24h. Media was discarded, then a detection mix (PBS 12.5μL, lytic buffer 12.5μL, substrate (Promega) 0.25μL, LgBiT (Promega) 0.125μL) was added and incubated for 10 min, and the luminescence was measured using ARVO X3 (PerkinElmer, MA, USA)
For further validation, siRNA knockdown was performed. Precisely 1.5×104 cells of PC9-KI cells were plated and incubated overnight. Following this, 1.5 pmol of siRNA for CD274 or the negative control (AllStars Hs Cell Death siRNA, Thermo Scientific, MA, USA) was transfected with 0.5 μg of Lipofectamine RNA iMax (Thermo Scientific, MA, USA). The siRNA sequences are shown in Table S1. After 48 h, the medium was discarded and a detection mix (PBS 25μL, lytic buffer 25μL, substrate (Promega) 0.5μL, and LgBiT (Promega) 0.25μL) was added. The plate was incubated at room temperature in the dark for 10 min, and the luminescence was measured using ARVO X3.
2.4. Pre-screening test for the HiBiT-tagged cells
To validate the effects of dimethyl sulfoxide (DMSO) on the luminescence of PC9-KI, 5×103 PC9-KI cells were seeded in 384 well plates. After 24 h, 1%, 0.5%, 0.25%, 0.125%, 0.0625%, or 0% of DMSO (final concentration) was added in RPMI medium with 10% fetal bovine serum (FBS). After 24 h, a detection mix (PBS 5μL, substrate 0.1μL, LgBiT 0.05μL) was added and incubated at room temperature in the dark for 10 min, and the luminescence was measured using ARVO X3.
To confirm the upregulation of PD-L1 by cytokines or growth factors, 4×104 of PC9-KI cells were seeded on 96 well plates. After 24 h, 1 μg/mL of epidermal growth factor (EGF) (WAKO), 100 ng/mL of tumor necrosis factor (TNF) alpha (R&D, Minnesota, USA), 20 ng/mL of interferon gamma (IFNγ) (R&D, MN, USA) or 0.1% of DMSO was added in RPMI with 10%FBS. After 24h, the medium was discarded, and a detection mix (PBS 12.5 μL, lytic buffer (Promega) 12.5 μL, substrate (Promega) 0.25 μL, LgBiT (Promega) 0.125 μL) was added and incubated at room temperature in the dark for 10min. Luminescence was measured using ARVO X3.
To further validate the dose-dependent changes in PD-L1 expression, 5×103 PC9-KI cells were seeded on a 384 well plate. Twenty-four hours later, 20 μM, 10 μM, 5 μM, 2.5 μM, 1.25 μM, 0.625 μM, or 0 μM of SB203580 (Sigma Aldrich) was added to RPMI medium with 10%FBS. After 24h, the medium was discarded, and a detection mix (PBS 5μL, substrate (Promega) 0.1μL, LgBiT (Promega) 0.05μL) was added and incubated at room temperature in the dark for 10 min. Luminescence was measured using ARVO X3.
2.5. Chemical compound screening with the LOPAC compound library
At first, 4×104 PC9-KI cells were seeded on 96 well plates. Then, after 24 h, 1μM of compounds in the LOPAC chemical compound library (Sigma Aldrich, MO, USA) were added to each well. Following 24h, the medium was discarded, and a detection mix (PBS 12.5 μL, lytic buffer 12.5 μL, substrate (Promega) 0.25 μL, and LgBiT (Promega) 0.125μL) was added to each well. The plates were incubated at room temperature for 10 min in the dark, and the luminescence was measured using ARVO X3.
2.6. Effective Concentration 50 assay with HiBiT activity measurement
First, 4×104 of PC9-KI cells were seeded on 96 well plates. After 24 h, 0.1 % of DMSO or 10−5μM, 10−4μM, 10−3μM, 10−2μM, 10−1μM, 1 μM, or 10 μM of SB203580 (Sigma Aldrich), brefeldin A (BioLegend, CA, USA), PD153035 (Selleck, TX, USA), colchicine (WAKO), vincristine (Cayman, Michigan, USA), thapsigargin (WAKO, Osaka, Japan), and ML-240 (Sigma Aldrich, MO, USA) were added. After 24 h, the medium was discarded, and detection mix (PBS 12.5 μL, lytic buffer (Promega) 12.5 μL, substrate (Promega) 0.25 μL, and LgBiT (Promega) 0.125 μL) was added to each well.
2.7. Inhibitory Concentration50 assay with Cell Titer Glo assay
Initially, 5×103 of PC9-KI, A375, and MDAMB231 cells were seeded on 96 well plates. After 24 h, 0.1 % DMSO or 10−5 μM, 10−4 μM, 10−3μM, 10−2μM, 10−1μM, 1 μM, or 10 μM of colchicine (WAKO), vincristine (Cayman), thapsigargin (WAKO), and ML-240 (Sigma Aldrich) were added. After 48h, 25 μL of Cell Titer Glo 2.0 (Promega) was added and incubated at 37°C for 30 min. Absorbance at 490 nm was measured using ARVO X3.
2.8. Treating PC9-KI cells with colchicine for validating the results of the HiBiT assay
Initially, 4×105 of PC9-KI cells were seeded on six well plates. After 24h, 100 nM of colchicine was added to the medium and incubated at 37°C overnight. Protein levels were analyzed as shown in section 2.10.
2.9. Treating cells with each compound for A375 and MDAMB231
Initially, 4×105 of A375 or MDAMB231 cells were seeded on six well plates. After 24h, 1μM of colchicine, vincristine, thapsigargin, and ML-240 were added to the medium and incubated at 37°C for 3 h. Proteins and RNA were analyzed as shown in section 2.10.
2.10. Western Blotting
Proteins were collected with the RIPA lysis buffer. Proteins in the cell lysates were separated by SDS-PAGE followed by a semi-dry transfer to a polyvinylidene fluoride (PVDF) membrane. Membranes were blocked for 1h with Blocking-One (Nacalai Tesque, Kyoto, Japan), and then reacted with PD-L1 primary antibodies (E1L3N, Cell Signaling Technology, Massachusetts, USA) or anti-beta-actin (ACTB) (AC-74, Sigma Aldrich or 010–27841, WAKO) at 4°C overnight. Subsequently, the membrane was rinsed and reacted with enhanced chemiluminescence (ECL) mouse IgG horseradish peroxidase (HRP)-conjugated whole antibody (GE Healthcare) or rabbit IgG HRP-conjugated whole antibody (GE Healthcare). The blot was developed using the ECL Select Western Blotting Detection Reagent (GE Healthcare).
2.11. RNA extraction and quantitative PCR analysis
RNA was extracted using the ReliaPrep RNA Miniprep System (Promega, Wisconsin, Japan). RNA was reverse-transcribed with Prime Script (Takara, Kusatsu, Japan), and cDNA was analyzed by quantitative PCR (qPCR). Primers are shown in Supporting Table S1.
3. Results
In order to establish a simple assay system that can rapidly evaluate the endogenous expression of PD-L1, we knocked-in the HiBiT tag at the C terminus of the CD274 gene. We used the genome editing technology-CRISPR-Cas9 and oligo ssDNA that were complementary to genome sequences in the lung adenocarcinoma cell line PC9 (Figure 1A). We selected four clones from single cell cloning, and confirmed the integration of HiBiT sequences by PCR. In our analysis, we used clone #3, which was homozygous for the targeted insertion of the HiBiT tag, in our analysis (Figure 1B). With the help of sequencing, we confirmed the targeted insertion of the HiBiT tag of the knocked-in cells (PC9-KI) (Figure 1C). We also confirmed the tagging with western blotting using LgBiT (Figure 1D). By measuring the reporter activity of the PC9-KI cells, we confirmed the highly specific luminescence activity of PC9-KI compared with that of the wild-type PC9 (Figure1E). Furthermore, by knocking-down endogenous PD-L1 using siRNA in PC9-KI cells, we confirmed a distinct decrease in luminescence, which reflected changes in the endogenous expression (Figure 1F). In this cell-based assay system, we confirmed that there were no changes in PD-L1 expression with 0%, 0.0625%, 0.125%, 0.25%, 0.5%, and 1% DMSO (Figure 1G). In addition, we found that adding EGF, TNFα, and IFNγ, which have been previously reported as PD-L1 upregulators, increased the HiBiT activities compared to when 0.1% DMSO was added (Figure 1H). Moreover, treatment with the p38 inhibitor, already reported as a downregulator of PD-L1, decreased the HiBiT activity in a dose-dependent manner (Figure 1I). Based on these results, we performed a chemical compound screening using PC9-KI cells.
Figure 1. Creating HiBiT knocked-in PC9 cells.
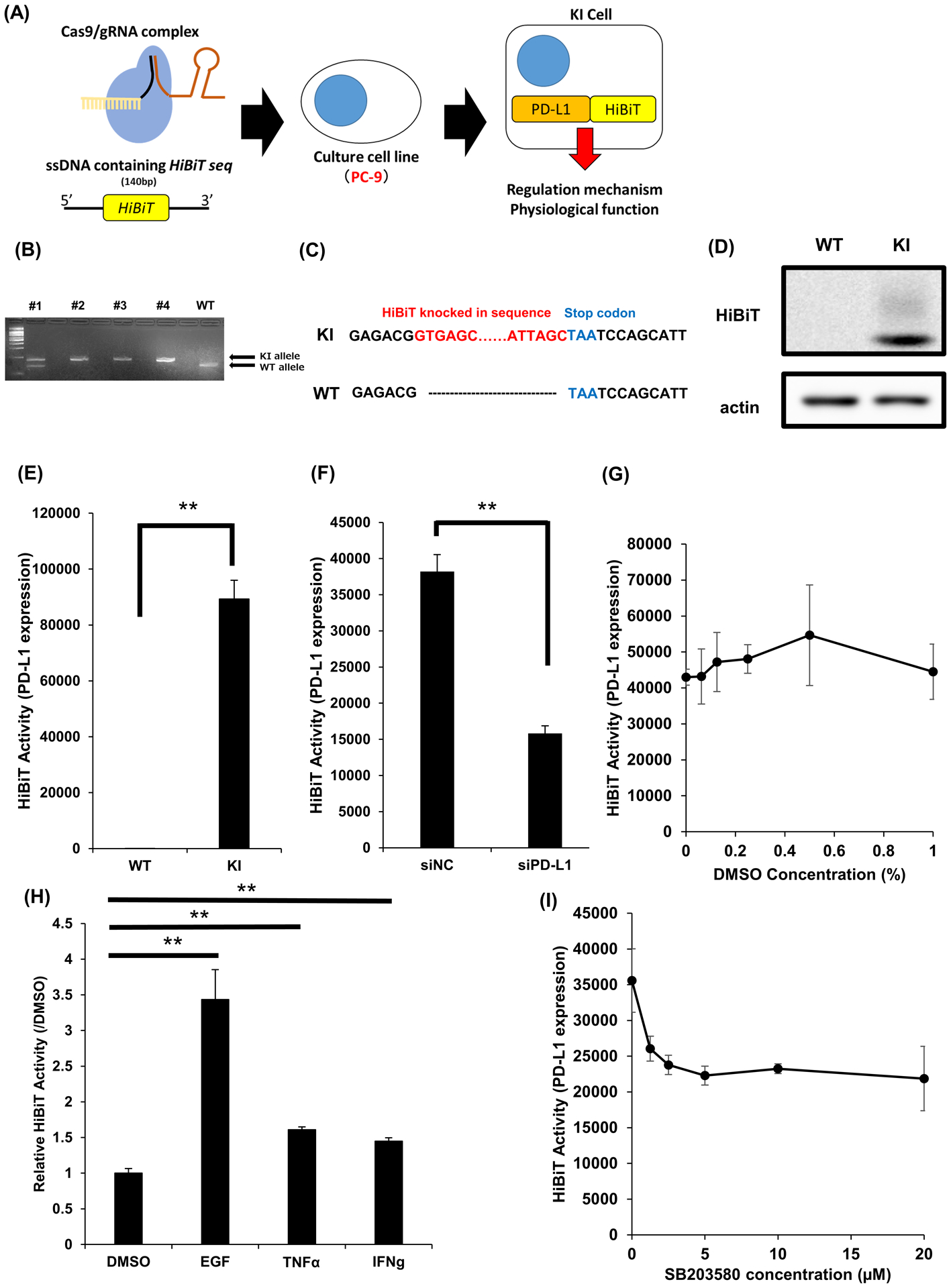
(A) Schema for constructing HiBiT knocked-in PC9 cells using CRISPR Cas9 system
(B) Results of electrophoresis for PCR products of genome extracted from picked up clones
(C) Sequencing check for HiBiT tagging revealed the targeted insertion of HiBiT tag
(D) Western Blotting confirmation for the targeted insertion of HiBiT tag in PC9-KI cells
(E) HiBiT assay for PC9 cells and PC9-KI cell to confirm the specificity of luminescence; n=3
(F) HiBiT assay for PC9-KI cells transfected with siRNA for negative control or PD-L1 for 48h; n=4
(G) HiBiT assay for PC9-KI cells with various concentration of DMSO; n=4
(H) HiBiT assay for PC9-KI cells stimulated with EGF, TNFα, and IFNγ; n=4
(I) HiBiT assay for various concentration of p38 inhibitor SB203580; n=4
Error bars represent the standard error mean (**p < 0.01, two-tailed Student’s t-test).
PD-L1, programmed death ligand 1; PCR, polymerase chain reaction; siRNA, small interfering RNA; DMSO, dimethyl sulfoxide; EGF, epidermal growth factor; TNFα, tumor necrosis factor alpha; IFNγ, interferon gamma
For the chemical screening, we seeded 4×104 PC9-KI cells on 96 well plates, incubated them for 24 h, and added 1280 chemical compounds from the LOPAC chemical compound library. At a final concentration of 1 μM, we incubated the cells for another 24 h, and then measured the HiBiT activity (Figure 2A). We defined regulators as compounds that regulated the expression of PD-L1 in the range of p <0.01. As a result, we identified thirteen upregulators and seven downregulators (Figure 2B, Table S2). Colchicine and vincristine, currently known for targeting microtubules and thapsigargin, which can increase intracellular calcium levels were newly identified as upregulators of PD-L1 (Figure 2C). We identified seven chemical compounds as candidate downregulators (Figure 2D). To exclude the possibility of overestimating the candidate downregulators due to their cytotoxicity, we performed an inhibitory concentration (IC50) assay for PC9-KI cells. As a result, brefeldin A and epidermal growth factor receptor (EGFR) inhibitor had a strong cytotoxicity at a dose of 1 μM, and their relative cell viabilities were less than 60% as compared to the treatment with DMSO. On the other hand, treatment with the p38 inhibitor and ML-240 showed milder cell toxicity, and their relative cell viabilities were more than 60% as compared to the treatment with DMSO (Supporting Figure S1). From these results, we regarded the p38 inhibitor, an already known downregulator of PD-L1, and ML-240, an AAA ATP ase p97 inhibitor, as downregulators of PD-L1.
Figure 2. Compound screening using HiBiT-KI cells.
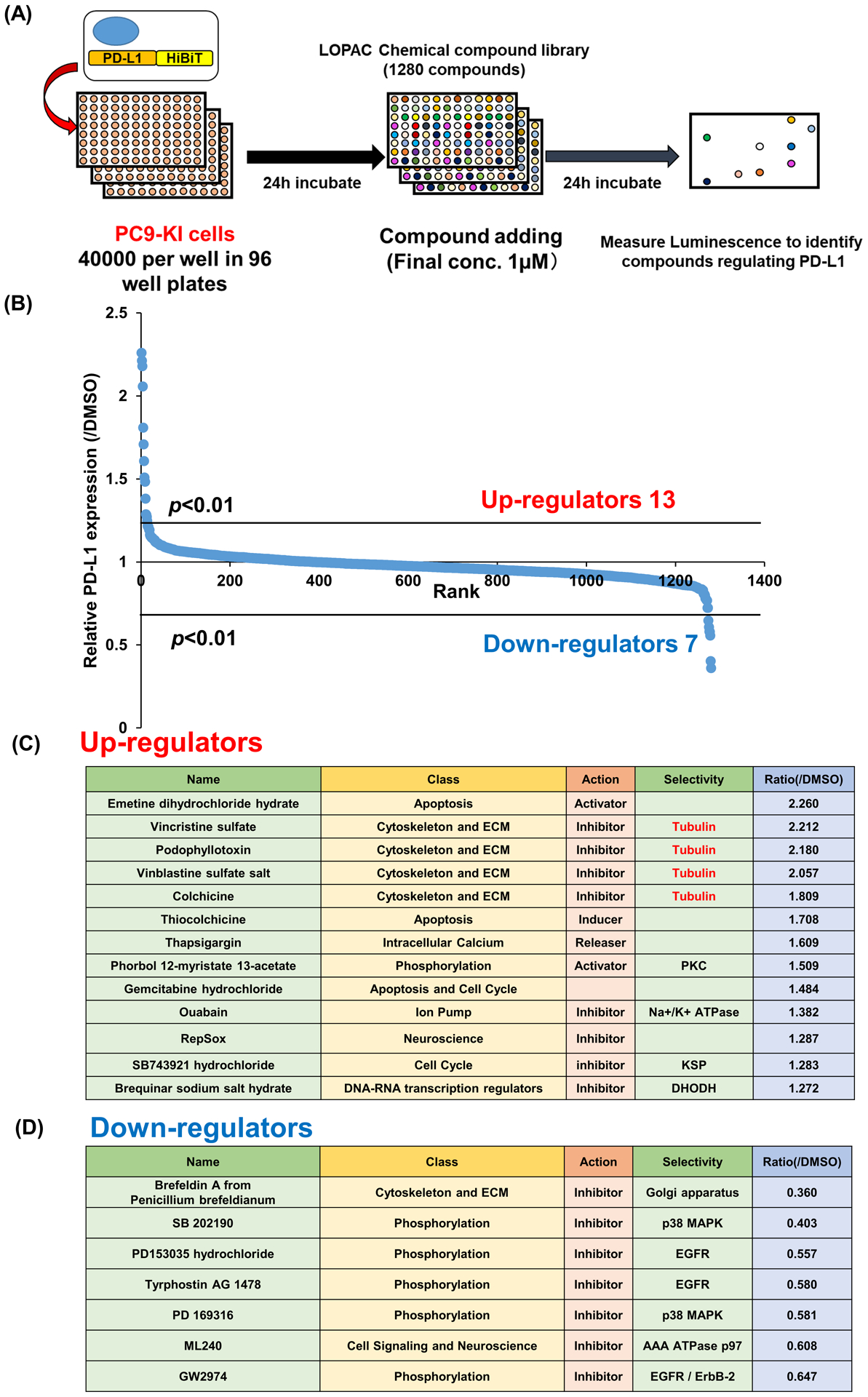
(A) Schema for chemical compounds screening with LOPAC chemical compounds library
(B) Results of chemical compounds screening
(C) Candidate upregulators in chemical compounds screening
(D) Candidate downregulators in chemical compounds screening
PD-L1: programmed death ligand 1; LOPAC, Library of pharmaceutically active compounds
To measure the effective concentration 50 (EC50) of colchicine for maximum upregulation of PD-L1, we performed a concentration dilution assay on PC9-KI cells using the HiBiT system. We determined that the EC50 of colchicine was approximately 50 nM, and it upregulated PD-L1 protein expression 1.8 times as much as DMSO (Figure 3A). The IC50 for maximum suppression of cell viabilities of PC9-KI cells by colchicine was approximately 1 nM (Figure 3B). Moreover, we used western blotting with antibodies for endogenous PD-L1 to validate the results of the HiBiT assay, and we confirmed the upregulation of PD-L1 in PC9-KI cells treated with 1 μM of colchicine overnight (Figure 3C). Furthermore, to show that the upregulation of PD-L1 by colchicine was a common phenomenon, we validated the effects of colchicine in other cell lines. By adding 1 μM of colchicine to the melanoma cell line A375 for 3h, we confirmed the upregulation of PD-L1 protein and RNA (Figure 3D, E). The IC50 for the maximum suppression of cell viability of A375 by colchicine was approximately 50 pM (Figure 3F). Adding 1 μM of colchicine to the breast cancer cell line MDAMB231 for 3 h also showed the upregulation of PD-L1 protein and RNA (Figure 3G, H). The IC50 for the maximum suppression of viability of MDAMB231 cells by colchicine was approximately 1 nM (Figure 3I).
Figure 3. Colchicine functions as a PD-L1 upregulator.
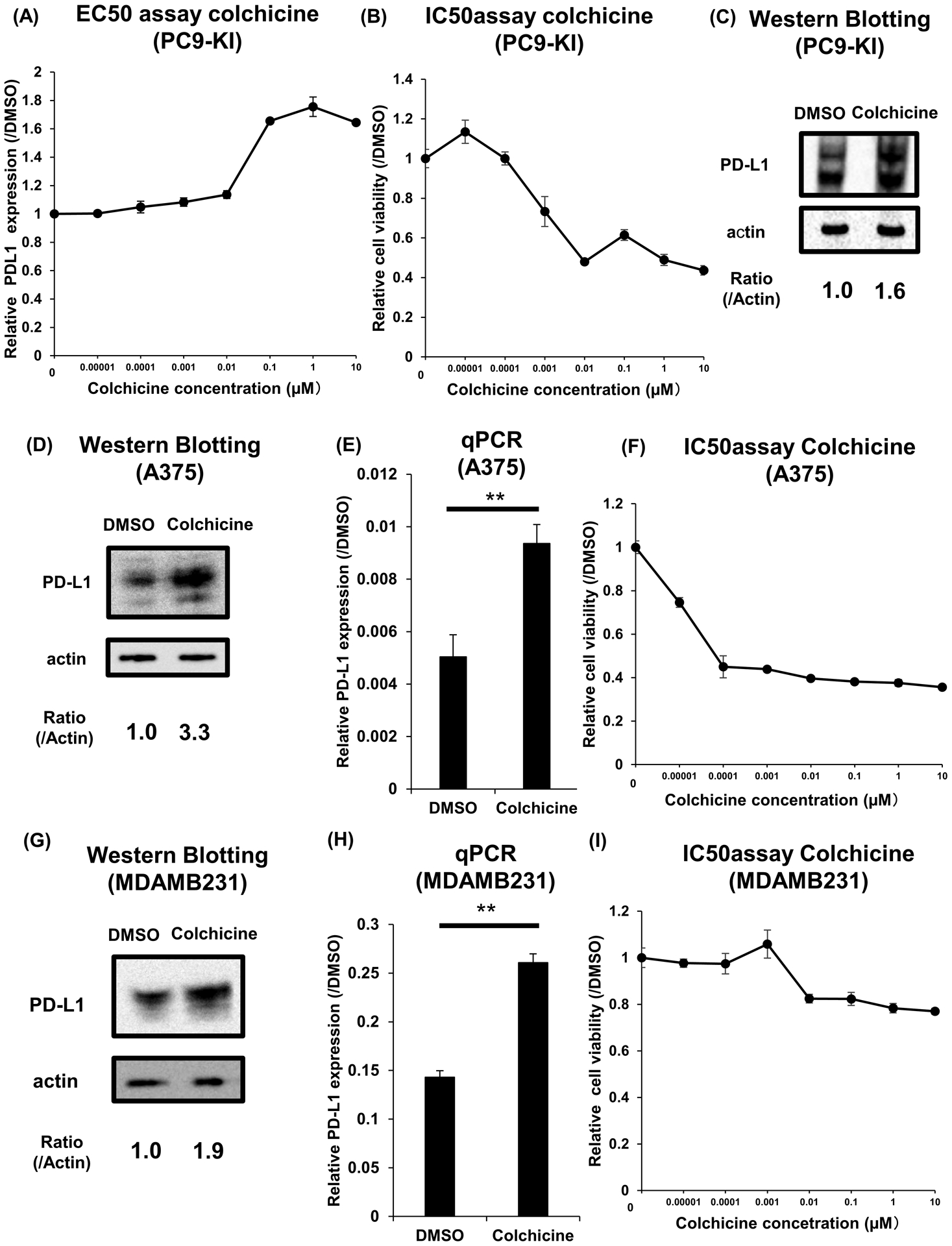
(A) EC50 assay of colchicine for maximum upregulation of PD-L1 in PC9-KI cells; n=3
(B) IC50 assay of colchicine for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(C) Western blotting for endogenous PD-L1 of PC9-KI cells treated with 1μM colchicine for overnight
(D) Western blotting for endogenous PD-L1 of A375 cells treated with 1μM colchicine for 3h
(E) qPCR of endogenous PD-L1 of A375 cells treated with 1μM colchicine for 3 h; n=3
(F) IC50 assay of colchicine for minimum downregulation of PD-L1 in A375 cells; n=3
(G) Western blotting for endogenous PD-L1 of MDAMB231 cells treated with 1μM colchicine for 3h
(H) qPCR of endogenous PD-L1 of MDAMB231 cells adding 1μM colchicine for 3h; n=3
(I) IC50 assay of colchicine for minimum downregulation of PD-L1 in MDAMB231 cells; n=3
Error bars represent the standard error mean (**p < 0.01, two-tailed Student’s t-test)
EC50, effective concentration 50; IC50, inhibitory concentration 50; PD-L1, programmed death ligand 1; qPCR, quantitative polymerase chain reaction
We also measured the EC50 of vincristine for the maximum upregulation of PD-L1 on PC9-KI cells. The EC50 of vincristine for PC9-KI cells was approximately 100nM, and the maximum upregulation of PD-L1 was twice than that with DMSO (Figure 4A). The IC50 for the maximum suppression of the viabilities of PC9-KI cells by vincristine was approximately 1 nM (Figure 4B). Furthermore, to show that the upregulation of PD-L1 by vincristine was a common phenomenon, we validated the effects of vincristine in other cell lines. By adding 1 μM of vincristine to A375 for 3h, we confirmed the upregulation of PD-L1 protein and RNA (Figure 4C, D). The IC50 for the maximum suppression of cell viability of A375 cells by vincristine was approximately 500 pM (Figure 4E). Adding 1 μM of vincristine to MDAMB231 cells for 3 h also showed moderate upregulation of PD-L1 protein and RNA (Figure 4F, G). The IC50 for the maximum suppression of cell viability of MDAMB231 cells by vincristine was approximately 500 pM (Figure 4H).
Figure 4. Vincristine functions as a PD-L1 upregulator.
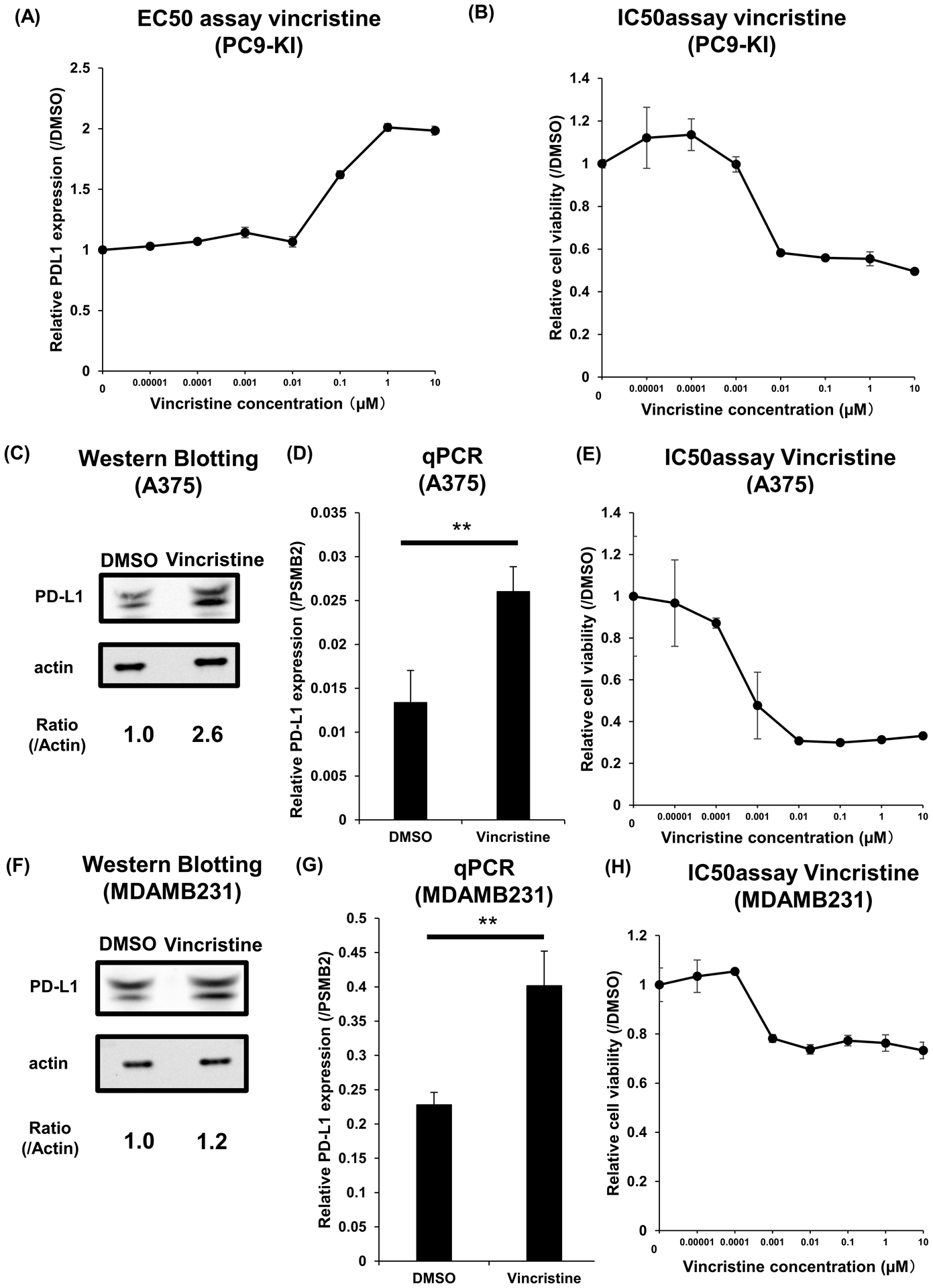
(A) EC50 assay of vincristine for maximum upregulation of PD-L1 in PC9-KI cells; n=3
(B) IC50 assay of vincristine for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(C) Western blotting for endogenous PD-L1 of A375 cells treated with 1μM vincristine for 3 h
(D) qPCR of endogenous PD-L1 of A375 cells treated with 1μM vincristine for 3h; n=3
(E) IC50 assay of vincristine for minimum downregulation of PD-L1 in A375 cells; n=3
(F) Western blotting for endogenous PD-L1 of MDAMB231 cells treated with 1μM vincristine for 3h
(G) qPCR of endogenous PD-L1 of MDAMB231 cells treated with 1μM vincristine for 3h; n=3
(H) IC50 assay of vincristine for minimum downregulation of PD-L1 in MDAMB231 cells; n=3
Error bars represent the standard error mean (**p < 0.01, two-tailed Student’s t-test).
EC50, effective concentration 50; IC50, inhibitory concentration 50; PD-L1, programmed death ligand 1; qPCR, quantitative polymerase chain reaction
Subsequently, we validated the upregulation of PD-L1 by thapsigargin. The EC50 of thapsigargin for PC9-KI cells was approximately 5 nM, and the maximum upregulation of PD-L1 was 1.5 times higher than DMSO (Figure 5A). The IC50 for maximum suppression of cell viability of PC9-KI cells by thapsigargin was approximately 5 nM (Figure 5B). Furthermore, to demonstrate that the upregulation of PD-L1 by thapsigargin was a common phenomenon in other cell lines, we validated the effects of thapsigargin in A375 cells. By adding 1 μM of thapsigargin to A375 cells for 3 h, we confirmed the upregulation of PD-L1 protein and RNA (Figure 5C, D). The IC50 for the maximum suppression of cell viability of A375 cells treated with thapsigargin was approximately 500 pM (Figure 5E).
Figure 5. Thapsigargin functions as a PD-L1 upregulator.
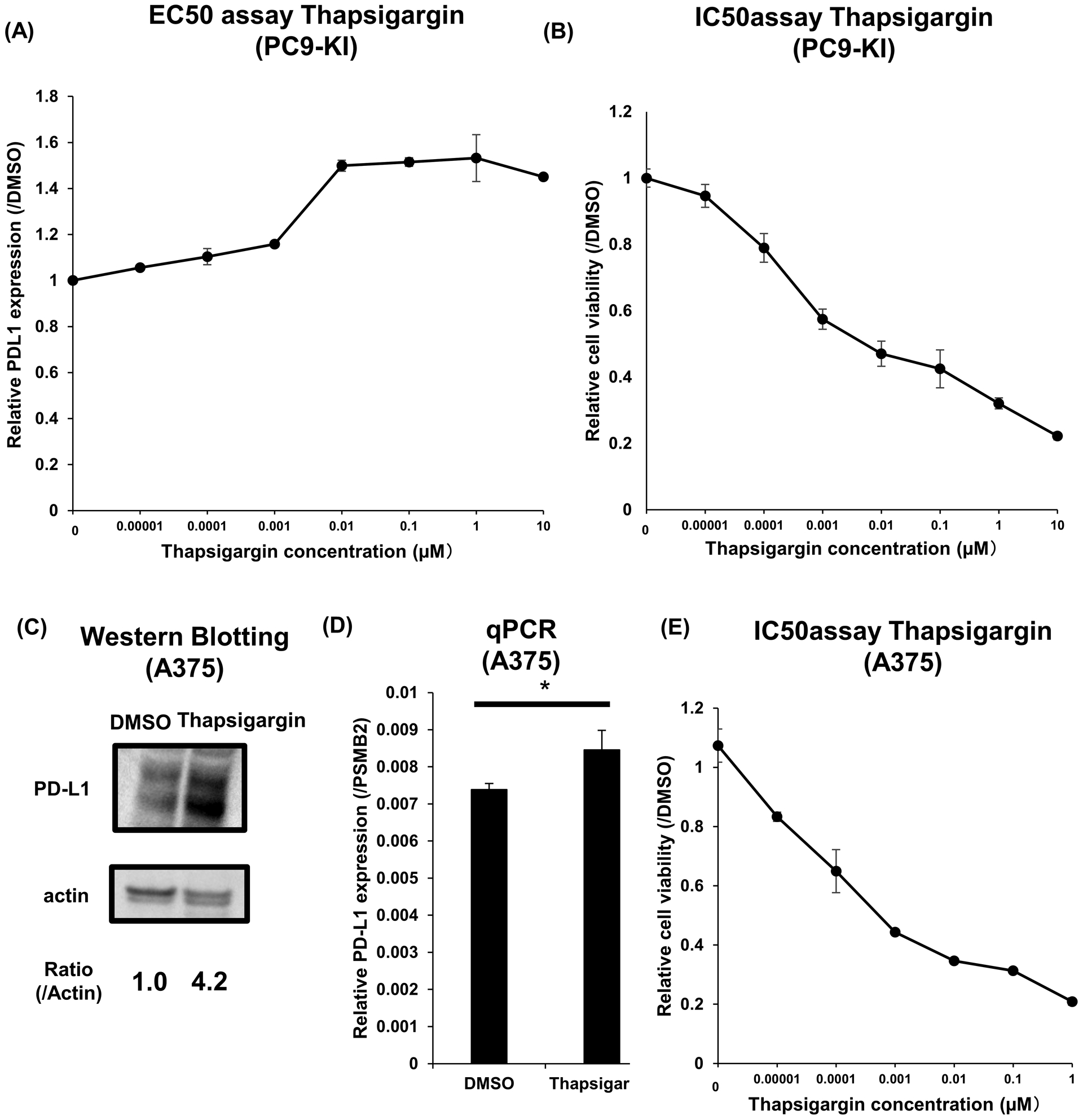
(A) EC50 assay of thapsigargin for maximum upregulation of PD-L1 in PC9-KI cells; n=3
(B) IC50 assay of thapsigargin for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(C) Western blotting for endogenous PD-L1 of A375 cells treated with 1μM thapsigargin for 3h
(D) qPCR of endogenous PD-L1 of A375 cells treated with 1μM vincristine for 3h; n=3
(E) IC50 assay of vincristine for minimum downregulation of PD-L1 in A375 cells; n=3
Error bars represent the standard error mean (*p < 0.05, two-tailed Student’s t-test).
EC50, effective concentration 50; IC50, inhibitory concentration 50; PD-L1, programmed death ligand 1; qPCR, quantitative polymerase chain reaction
Finally, we validated the downregulation of PD-L1 by the AAA ATPase p97 (VCP) inhibitor -ML-240. The EC50 of ML-240 for PC9-KI cells was approximately 1 μM, and the maximum downregulation of PD-L1 was suppressed by 10% as compared to DMSO (Figure 6A). Furthermore, to show that the downregulation of PD-L1 by ML-240 was a common phenomenon in multiple cell lines, we validated the effects of ML-240 on other cell lines. By adding 1 μM of ML-240 to A375 cells overnight, we confirmed the moderate downregulation of the PD-L1 protein, although no significant change was detected in PD-L1 RNA (Figure 6B, C). The IC50 for maximum suppression of cell viability of A375 cells by ML-240 was approximately 5 μM (Figure 6D). Adding 1 μM of ML-240 to MDAMB231 cells overnight also showed the downregulation of the PD-L1 protein and RNA (Figure 6E, F). The IC50 for the maximum suppression of cell viability of MDAMB231 cells by ML-240 was approximately 5 μM (Figure 6G).
Figure 6. ML-240 functions as a PD-L1 suppressor.
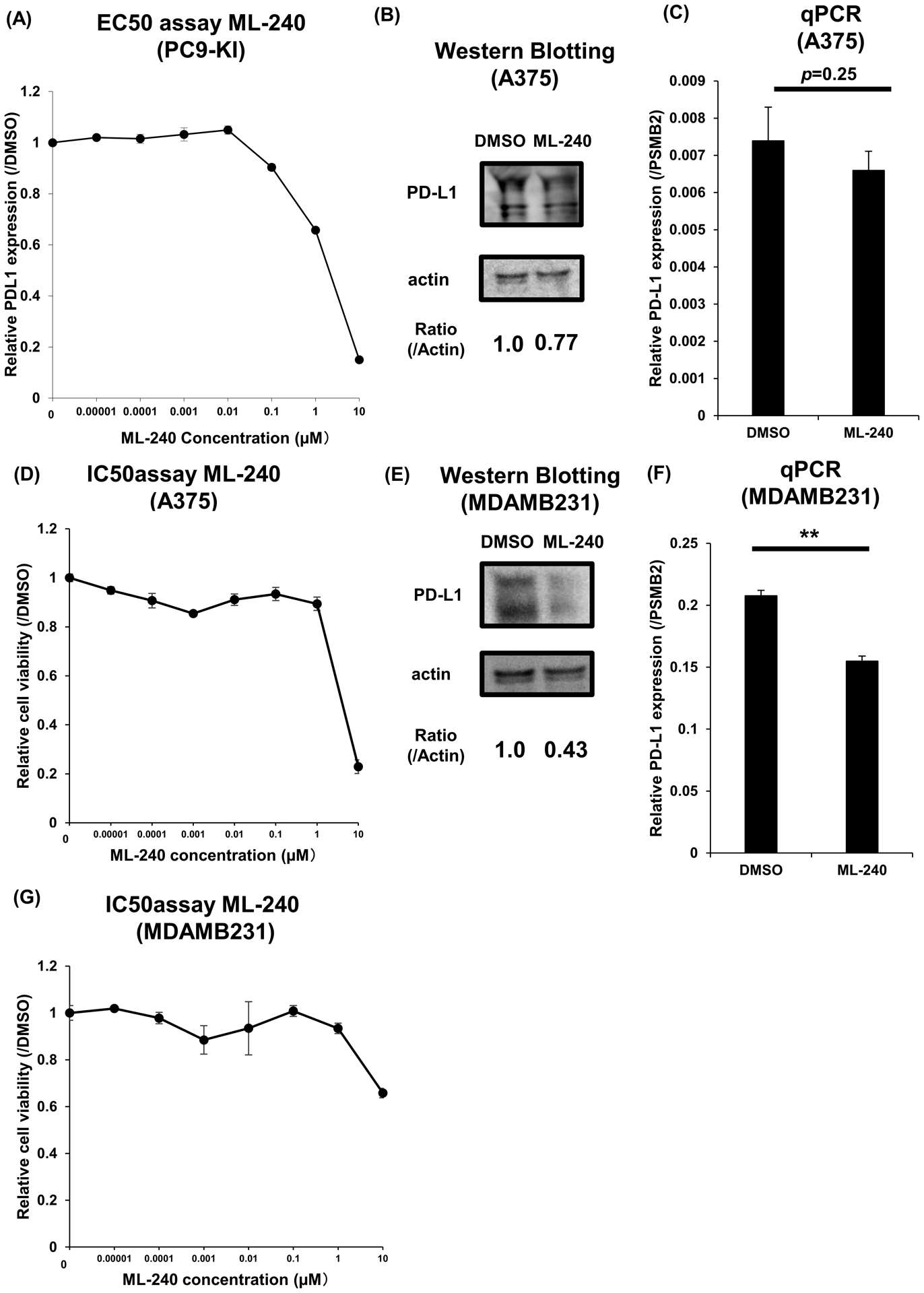
(A) EC50 assay of ML-240 for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(B) Western blotting for endogenous PD-L1 of A375 cells treated with 1μM ML-240 for 3h
(C) qPCR of endogenous PD-L1 of A375 cells treated with 1μM ML-240 for 3h; n=3
(D) IC50 assay of ML-240 for minimum downregulation of PD-L1 in A375 cells; n=3
(E) Western blotting for endogenous PD-L1 of MDAMB231 cells treated with 1μM ML-240 for 3h
(F) qPCR of endogenous PD-L1 of MDAMB231 cells treated with 1μM ML-240 for 3h; n=3
(G) IC50 assay of ML-240 for minimum downregulation of PD-L1 in MDAMB231 cells; n=3
Error bars represent the standard error mean (**p < 0.01, two-tailed Student’s t-test).
EC50, effective concentration 50; IC50, inhibitory concentration 50; PD-L1, programmed death ligand 1; qPCR, quantitative polymerase chain reaction
4. Discussion
We created the HiBiT-tagged lung adenocarcinoma cell line, PC9-KI, using CRISPR Cas9 genome editing technology, which made it possible to detect the PD-L1 protein expression changes quickly and easily. Working on PC9-KI cells, we performed chemical compound screening using the LOPAC chemical compound library, and we identified two new PD-L1 upregulators (microtubule polymerization inhibitors and the calcium releasing inducer thapsigargin) and a novel PD-L1 downregulator (a p97 inhibitor ML-240). Furthermore, we confirmed that the regulation of PD-L1 by each compound could be observed in a melanoma cell line, A375, and a breast cancer cell line MDAMB231, which suggests the potential of PD-L1 regulation in various cell lines.
4.1. Chemical screening system using a HiBiT-tagged cell line
Although chemical screening using HiBiT-tagged cells has been previously reported, our study is the first to perform chemical screening using HiBiT for detecting endogenous protein expression changes for cancer research [11–13]. We used ssDNA for homologous recombination, taking advantage of the characteristics of the HiBiT tag. Using this method, we could easily prevent off-target tagging. Compared with the classic protein detection methods such as western blotting, enzyme-linked immunosorbent assay (ELISA), and fluorescence-activated cell sorting (FACS), the HiBiT system represents a quicker and streamlined method for detecting changes in protein expression. We expect this technology to be applied to screening systems for detecting important endogenous protein expression changes in various types of cancers in the future.
We selected PC9, an EGFR-mutated lung adenocarcinoma cell line, to establish this screening system. This was because of the moderate expression of PD-L1 and the ease of applying the existing CRISPR-Cas9 gene editing technology.
Immune checkpoint inhibitors that target PD-L1 and PD-1 have been used in clinical practice in many cancer types, including melanoma and lung adenocarcinoma [3,14–17]. However, although recent studies have revealed the regulation of PD-L1 expression through signaling pathways, ubiquitination, and glycosylation, small molecule compounds that regulate PD-L1 expression have not been identified [4–9,15,18–21]. Moreover, PD-L1 expression changes based on drugs used in combination with immune checkpoint inhibitors were not defined clearly; therefore, we believe that our strategy provides a simple and quick method to confirm and validate PD-L1 protein expression changes.
Compounds that can negatively regulate PD-L1 have the potential to directly regulate tumor immunity. In addition, it has been reported that the clinical efficacy of immune checkpoint inhibitors is enhanced when PD-L1 expression increases, suggesting that compounds that positively regulate PD-L1 expression have the potential to enhance the effects of immune-checkpoint inhibitors [22]. Our study is significant because the potential of regulating PD-L1 for each drug in the chemical compound library was obtained in a cross-sectional manner by our screening system.
4.2. Newly identified compounds regulating PD-L1 from the chemical screening
Microtubule-polymerization inhibitors such as colchicine and vincristine were identified by chemical screening as compounds that specifically upregulate PD-L1 expression. The crosstalk between microtubule regulation and the immune system has been reported for its importance in the inflammasome induction [23]. On the other hand, the relationship between PD-L1 regulation and microtubules is yet to be reported, which suggests the existence of a novel microtubule-dependent mechanism for the regulation of PD-L1 expression. In a previous report, PD-L1 expression was upregulated during the M phase of the cell cycle, and CUL3-SPOP induced by Cyclin D reduced PD-L1 expression [21]. We infer from these reports that microtubule inhibitors stop the cell cycle in the M phase, which induces the upregulation of PD-L1 expression.
Although colchicine and vincristine are compounds with cytotoxic activity and anticancer potential, it is worth noting that microtubule-targeted compounds specifically upregulated PD-L1 expression, while other frequently used anticancer drugs with cytotoxic activity, such as cisplatin and fluorouracil, did not upregulate PD-L1[24–26]. This result suggests a mechanism in tumor cells that detects the cellular stress caused by inhibition of microtubule polymerization, and thereby increasing the expression of PD-L1. Moreover, according to the results of the IC50 assay in our study, vincristine and colchicine increased PD-L1 expression in the cytotoxic concentration range. These results suggest that using immune checkpoint inhibitors with microtubule inhibitors may be therapeutically useful.
Another compound identified as an upregulator was thapsigargin, a noncompetitive inhibitor of sarco/endoplasmic reticulum calcium transport ATPase (SERCA) for endoplasmic reticulum (ER) calcium uptake. Thapsigargin has the ability to increase cytosolic calcium levels and induce ER stress [27,28]. Our study showed that thapsigargin increased the expression of PD-L1 in PC9-KI and A375 cells. The results of our screening indicated that compounds that induce extracellular calcium influx did not increase PD-L1 expression; therefore, there may be a mechanism of PD-L1 expression dependent on calcium influx from the ER (Table S1).
We also identified ML-240, an inhibitor of p97, as a compound that reduces PD-L1 expression. P97 is an AAA ATPase with multiple functions including cell cycle regulation and proteasome and DNA damage repair [29,30]. Our study is the first to report possible links between PD-L1 regulation and p97. ML-240 has also been reported to have a promising potential as an anti-tumor drug [31,32]. Therefore, our reports suggest the potential of ML-240 as an anti-tumor drug, which effectively prevents tumors from evading the immune system.
4.3. Limitation.
In this study, we did not conduct in vivo experiments with immune checkpoint inhibitors. Therefore, further validation with in vivo experiments is essential for making the results clinically significant. In addition, the PC9 cell line used in this study was an EGFR-mutated cell line, which was not indicated for use with immune checkpoint inhibitors. Thus, it is necessary to evaluate its reproducibility in multiple cancer types. In our study, we confirmed the reproducibility of the breast cancer cell line MDAMB231 and the melanoma cell line A375, both of which showed similar tendencies as PC9-KI cells. Based on this information, the results of this study should be interpreted with caution with respect to clinical application.
4.4. Conclusion
We established a lung adenocarcinoma cell line (PC9-KI), with HiBiT-tagged into the C- terminus of PD-L1 by genome editing technology, and used it in chemical screening to identify compounds that regulate PD-L1 expression. As a result, we newly identified microtubule-polymerization inhibitors and thapsigargin as upregulators, and a p97 inhibitor as a downregulator of PD-L1. These compounds regulate PD-L1 not only in lung adenocarcinoma, but also in breast cancer and melanoma.
Supplementary Material
Supporting Table S1: Sequences of oligos, primers, siRNAs and guide RNAs
Supporting Table S2: Results of chemical compounds screening
Supporting Figure S1: IC50 Assay for candidate downregulators of PD-L1
(A) IC50 assay of brefeldin A for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(B) IC50 assay of p38 inhibitor for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(C) IC50 assay of EGFR inhibitor for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(D) IC50 assay of ML-240 for minimum downregulation of PD-L1 in PC9-KI cells; n=3
IC50, inhibitory concentration 50; EGFR, epidermal growth factor receptor
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science KAKENHI (Grant Nos. 20H05696, 18K19603 to H.A.), NIH grant (AR050631 to H.A.), and AMED‐CREST from AMED (Grant No. JP20gm0810008 to H.A.). We thank Dr. Hiroyuki Kagechika (Tokyo Medical and Dental University) for technical discussions, Risa Yagasaki (Tokyo Medical and Dental University) for grammar and spell check, Editage (www.editage.com) for English language editing, and Dr. Yuichi Takiguchi (Chiba University) for kindly providing PC9 cell line. We are also grateful to the entire staff of the Department of Systems BioMedicine at Tokyo Medical and Dental University (TMDU) for their support and discussion.
Abbreviations
- ACTB
Anti-beta-actin antibody
- cDNA
Complementary DNA
- crRNA
CRISPR RNA
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
Dimethyl sulfoxide
- DNA
Deoxyribonucleic acid
- EC50
Effective concentration 50
- ECL
Enhanced chemiluminescence
- EGF
Epidermal growth factor
- EGFR
Epidermal growth factor receptor
- ELISA
Enzyme-linked immunosorbent assay
- FACS
Fluorescence-activated cell sorting
- HRP
Horseradish peroxidase
- IC50
Inhibitory concentration 50
- INF
Interferon
- LOPAC
Library of pharmaceutically active compounds
- PBS
Phosphate-buffered saline
- PBST
Phosphate-buffered saline-Tween 20
- PCR
Polymerase chain reaction
- PD-1
Programmed cell death 1
- PD-L1
Programmed death-ligand 1
- PVDF
Polyvinylidene fluoride
- qPCR
Quantitative polymerase chain reaction
- RIPA
Radio-immunoprecipitation assay
- RNA
Ribonucleic acid
- RPMI
Roswell Park Memorial Institute
- SDS-PAGE
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- SERCA
Sarco/endoplasmic reticulum calcium-ATPase
- siRNA
Small interfering RNA
- ssDNA
Single-stranded DNA
- TNF
Tumor necrosis factor
- tracrRNA
Trans-activating CRISPR RNA
Footnotes
Funding sources and disclosure of conflicts of interest
The authors have no conflict of interest.
References
- [1].Zhang N and Bevan MJ (2011). CD8(+) T cells: foot soldiers of the immune system. Immunity 35, 161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Francisco LM, Sage PT and Sharpe AH (2010). The PD-1 pathway in tolerance and autoimmunity. Immunol Rev 236, 219–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y and Zang X (2015). Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med 21, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jiang X, Zhou J, Giobbie-Hurder A, Wargo J and Hodi FS (2013). The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res 19, 598–609. [DOI] [PubMed] [Google Scholar]
- [5].Atefi M et al. (2014). Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res 20, 3446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Visan I (2017). CMTM6 controls PD-L1. Nat Immunol 18, 1067. [DOI] [PubMed] [Google Scholar]
- [7].Burr ML et al. (2017). CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Castagnoli L et al. (2019). WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 38, 4047–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chan LC et al. (2019). IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest 129, 3324–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schwinn MK et al. (2018). CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem Biol 13, 467–474. [DOI] [PubMed] [Google Scholar]
- [11].Kinder TB, Dranchak PK and Inglese J (2020). High-Throughput Screening to Identify Inhibitors of the Type I Interferon-Major Histocompatibility Complex Class I Pathway in Skeletal Muscle. ACS Chem Biol 15, 1974–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kurimoto R et al. (2020). The tRNA pseudouridine synthase TruB1 regulates the maturation of let-7 miRNA. EMBO J, e104708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Oh-Hashi K, Furuta E, Fujimura K and Hirata Y (2017). Application of a novel HiBiT peptide tag for monitoring ATF4 protein expression in Neuro2a cells. Biochem Biophys Rep 12, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Robert C et al. (2015). Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372, 320–30. [DOI] [PubMed] [Google Scholar]
- [15].Borghaei H et al. (2015). Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med 373, 1627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fehrenbacher L et al. (2016). Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 387, 1837–46. [DOI] [PubMed] [Google Scholar]
- [17].Gong J, Chehrazi-Raffle A, Reddi S and Salgia R (2018). Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Marzec M et al. (2008). Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A 105, 20852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li CW et al. (2016). Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun 7, 12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mezzadra R et al. (2017). Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zhang J et al. (2018). Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lin H et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J Clin Invest 128, 1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T and Akira S (2013). Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14, 454–60. [DOI] [PubMed] [Google Scholar]
- [24].Cocco G, Chu DC and Pandolfi S (2010). Colchicine in clinical medicine. A guide for internists. Eur J Intern Med 21, 503–8. [DOI] [PubMed] [Google Scholar]
- [25].Shekelle PG et al. (2017). Management of Gout: A Systematic Review in Support of an American College of Physicians Clinical Practice Guideline. Ann Intern Med 166, 37–51. [DOI] [PubMed] [Google Scholar]
- [26].Adhikari S, Dongol RM, Hewett Y and Shah BK (2014). Vincristine-induced blindness: a case report and review of literature. Anticancer Res 34, 6731–3. [PubMed] [Google Scholar]
- [27].Rogers TB, Inesi G, Wade R and Lederer WJ (1995). Use of thapsigargin to study Ca2+ homeostasis in cardiac cells. Biosci Rep 15, 341–9. [DOI] [PubMed] [Google Scholar]
- [28].Zhou XC, Dong SH, Liu ZS, Liu S, Zhang CC and Liang XZ (2018). Regulation of gammaherpesvirus lytic replication by endoplasmic reticulum stress-induced transcription factors ATF4 and CHOP. J Biol Chem 293, 2801–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chou TF et al. (2011). Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A 108, 4834–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van den Boom J and Meyer H (2018). VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol Cell 69, 182–194. [DOI] [PubMed] [Google Scholar]
- [31].Chou TF, Li K, Frankowski KJ, Schoenen FJ and Deshaies RJ (2013). Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem 8, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Anderson DJ et al. (2015). Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell 28, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Table S1: Sequences of oligos, primers, siRNAs and guide RNAs
Supporting Table S2: Results of chemical compounds screening
Supporting Figure S1: IC50 Assay for candidate downregulators of PD-L1
(A) IC50 assay of brefeldin A for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(B) IC50 assay of p38 inhibitor for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(C) IC50 assay of EGFR inhibitor for minimum downregulation of PD-L1 in PC9-KI cells; n=3
(D) IC50 assay of ML-240 for minimum downregulation of PD-L1 in PC9-KI cells; n=3
IC50, inhibitory concentration 50; EGFR, epidermal growth factor receptor