

Abstract
Atrial fibrillation (AF) is the most frequent arrhythmia in adults. The prevalence and incidence of AF is going to increase substantially over the next a few decades. Because AF increases the risk of stroke, heart failure, dementia, and others, it severely impacts the quality of life, morbidity and mortality. Although the pathogenesis of AF is multifaceted and complex, focal ectopic activity and reentry are considered as the fundamental proarrhythmic mechanisms underlying AF development. Over the past 2 decades, large amount of evidence points to the key role of intracellular Ca2+ dysregulation in both initiation and maintenance of AF. More recently, emerging evidence reveal that NLRP3 (NACHT, LRR, PYD domain-containing 3) inflammasome pathways contribute to the substrate of both triggered activity and reentry, ultimately promoting AF. In this article, we review the current state of knowledge on Ca2+ signaling and NLRP3 inflammasome activity in AF. We also discuss the potential crosstalk between these two quintessential contributors to AF promotion.
Keywords: atrial fibrillation, calcium, NLRP3 inflammasome, delayed afterdepolarization, ryanodine receptor type-2, SERCA, sodium-calcium exchanger
Introduction
Atrial fibrillation (AF) is the most common sustained arrhythmia. It is associated with an increased risk of stroke, heart failure, dementia and death, thereby substantially influencing morbidity and mortality [81]. Current strategies for AF treatment include rate control by reducing the activation rate of the ventricles and rhythm control by converting the rapid and irregular atrial activation into sinus rhythm [123]. AF ablation is commonly used as a rhythm control strategy; however, the recurrence rate of AF is high in patients after successful AF ablation [52,113]. On the other hand, the conventional anti-arrhythmic drugs targeting cardiac ion channels yield modest benefits in converting AF to sinus rhythm and are often proarrhythmic [32,36]. The translational challenges to prevent AF are largely attributed to our limited ability to detect AF, the complex pathophysiology of the underlying AF-promoting atrial cardiomyopathy [53], and the progressive nature of AF leading to arrhythmia persistence and therapy resistance [66,68,157]. Mounting evidence demonstrate that aberrant calcium (Ca2+) handling in cardiac cells plays a central role in the development of AF [69]. Moreover, recent work also points to the involvement of sterile inflammatory signaling in the pathogenesis of AF [129]. In this article, we first review the pathophysiology of AF; then, we assess the specific roles of abnormal Ca2+ homeostasis and inflammatory signaling in AF paradigms; finally, we elaborate on the potential crosstalk between these two pathways that may support the progression of AF to more persistent forms.
1. Pathophysiology of AF
In a healthy heart, the electrical impulses are generated by a cluster of pacemaker cells located in the sinoatrial node (SAN). These electrical impulses travel through the cardiac conduction system and cause the sequential depolarization of atria and ventricles [29]. To date, two types of arrhythmic events are being recognized as main determinants of AF - ectopic (triggered) firing and reentry [110]. Ectopic firing refers to the state where the SAN is no longer the sole source for initiating electrical impulses, which could be attributed to enhanced automaticity or focal triggered activity. Reentry refers to events where the impulse waves activate a rotating entry path, forming a circus movement or a rotor generates spiral waves [114].
1.1). Enhanced Automaticity
Haissaguerre et al. first discovered that focal triggers at the base of the pulmonary veins (PVs), near the superior vena cava and the posterior wall of the left atrium, could fire spontaneously and initiate AF [60]. These triggers from PVs are now widely accepted as a major source of ectopic firing in AF (almost 90% cases) [44]. This report laid foundation and rationale for the modern AF ablation technology by applying either high energy (radiofrequency ablation) or cold temperature (cryoablation) to destroy the tissues surrounding PVs and isolating the spontaneous firing from PVs, thereby eliminating fibrillatory conduction to the atria [3]. However, recent studies revealed that approximately 1/3 of ectopic firings could come from the right atrium, suggesting that other regions also could initiate and maintain AF [128].
1.2). Triggered activity
Triggered activity (TA) refers to spontaneous membrane voltage oscillations during or after an action potential (AP). TAs could be induced by either early afterdepolarizations (EADs) or delayed afterdepolarizations (DADs). Both events are heavily influenced by the altered functions of Ca2+-handling proteins in cardiomyocytes. EADs are generated as a result of prolongation of the AP duration (APD). When APD is abnormally prolonged, it allows the reactivation of voltage dependent inward Ca2+ currents during phase 2 of the AP [83,110]. EADs can be also linked with APD shortening, occurring late in phase-3 of the AP. During the late phase-3 of APD, Na+/Ca2+ exchanger (NCX) can be activated by large amplitude Ca2+ transients from sarcoplasmic reticulum (SR), particularly when the APD is abbreviated, which produces a depolarizing transient inward current (Iti) that could cause late phase-3 EADs [18,83]. In contrast, DADs occur after the AP repolarization is completed and are primarily associated with the enhanced activation of the NCX due to increases in spontaneous Ca2+ release from SR [110,152].
1.3). Reentry
Ectopic firing is a frequent initiator of AF-maintaining reentry, but it could also sustain AF by itself by producing fibrillatory conduction even in the absence of a proarrhythmic atrial substrate. Reentry is considered the main mechanism that maintains AF. Circus movement reentry was found in jellyfish for the first time in 1906 [145]. This concept was then translated to arrhythmias by studies on cardiac tissue that exhibited circular electrical pathways. The formation of reentry can be a consequence of anatomical and functional substrates. The anatomical substrate has a prominent anatomical structure where the non-excitable scars (e.g. necrotic tissue or fibrosis) are surrounded by a circular pathway. A functional substrate could develop due to heterogeneities in excitability or conduction, whereby abbreviated refractoriness and slow conduction both promote reentry [153]. The effective refractory period (ERP) is determined by the cardiomyocyte APD, and conduction velocity (CV) is determined by the cellular excitability that is influenced by the amplitude of Na+ current (INa), the state of cell-to-cell communication via gap junctions, and the heterogeneity of tissue composition. Conceptually, the product of ERP and CV determines the wavelength (WL = ERP x CV) of a circuit. When ERP is abbreviated or CV is slowed, often referred as electrical remodeling, the WL of a circuit decreases [31]. The smaller the circuit is, the more circuits the atria can accommodate. ERP shortening, as a consequence of APD abbreviation, is associated with the increases in repolarizing K+ currents carried by the slow delayed rectifier K+ currents IKs [19] inward rectifier K+ current IK1 , the constitutively active G-protein coupled inward rectifying K+ current IK,ACh [35,151], and 2-pore-domain K+ currents IK2P [126], and the reduction in depolarizing L-type Ca2+ current (ICa,L) [28]. Reduced CV could be the result of impaired INa[135], reduced expression or altered distribution of connexins, and increased local extracellular volume due to interstitial fibrosis and local inflammation [73]. Atrial enlargement or hypertrophy also support the formation of an increased number of wavelets. Thus, shortened ERP, reduced CV, and atrial enlargement are the best-established factors supporting the evolution of proarrhythmic substrates for AF.
2. Ca2+ dysregulation and AF development
Ca2+ is a second messenger and plays an essential role in several fundamental biological processes, including muscle contraction, synaptic transmission, membrane trafficking, cardiac contractility, gene transcription, and cell division [21,27,49,51,61]. The association between the increased frequency of spontaneous SR Ca2+ release events (SR Ca2+ leak), which frequently underlie triggered activity, and AF was first reported by Hove-Madsen and colleagues in 2004 [72]. Since then, substantial amount of evidence collectively supports the notion that enhanced SR Ca2+ leak likely plays a causative role in AF development. In this section, we focus our discussion on 1) the role of Ca2+ homoeostasis in normal cardiac electrophysiology, and 2) the role of abnormal function of Ca2+-handling proteins in the promotion of triggered activity and in the evolution of the reentrant substrate associated with AF development (Figure 1).
Figure 1. Putative molecular mechanisms contributing to abnormal Ca2+ handling associated with atrial fibrillation (AF).
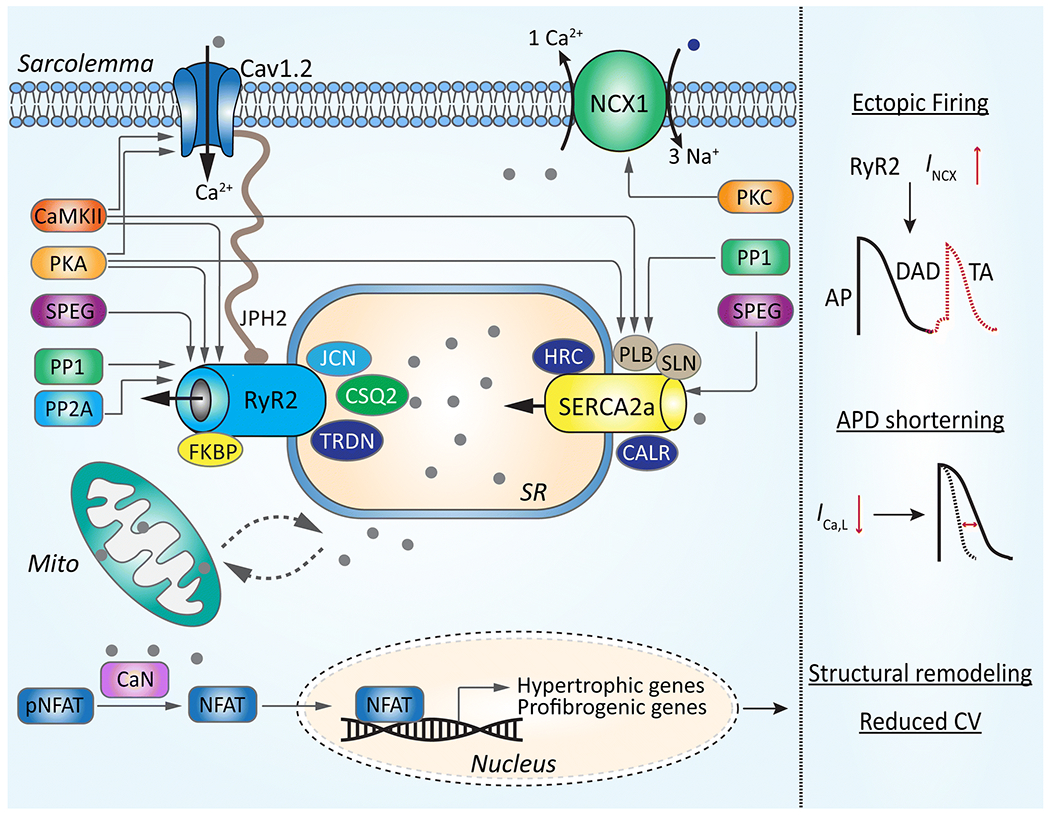
Focal ectopic firing due to the delayed afterdepolarization (DAD)-induced triggered activity (TA), APD shortening, and atrial enlargement as a result of the upregulation of hypertrophic and profibrogenic genes are key events associated with dysregulated Ca2+ handling in atrial cardiomyocytes during AF. DAD-inducing sarcoplasmic reticulum (SR) Ca2+ release (SR Ca2+ leak) is higher due to the enhanced activity of ryanodine receptor type-2 (RyR2). The latter could be a consequence of an impaired interaction with FKBP (FK506-binding protein 12.6) or junctophilin-2 (JPH2), altered phosphorylation by CaMKII, PKA, and SPEG, or a combination of both. The SERCA2a-mediated SR Ca2+ uptake could be increased in pAF because of a reduction of sarcolipin (SLN) or a hyperphosphorylation of phospholamban (PLB). In cAF, the hyperphosphorylation of PLB is associated with the enhanced Inhibitor-I (I-1) mediated inhibition of protein phosphatase type-1 (PP1). These alterations lead to the enhanced SR Ca2+ leak, which can activate the Na+/Ca2+ exchanger 1 (NCX1) and promote DADs. The shortening of APD is partially due to the reduced level of Cav1.2 (α-subunit of L-type Ca2+ channel, LTCC) or increased LTCC dephosphorylation due to enhanced activity of PP1 and PP2A. Together with structural remodeling involving Ca2+-induced calcineurin (CaN)-mediated activation of NFAT (nuclear factor of activated T-cell), a transcription factor associated with the hypertrophy and fibrosis, AP shortening promote AF-maintaining reentry.
2.1). Ca2+ homeostasis in cardiomyocytes
Ca2+ is a key contributor of excitation–contraction coupling (ECC) in cardiomyocytes [9]. When an AP starts due to the rapid activation of voltage-gated Na+ channel (Nav1.5), the related membrane depolarization activates the voltage-dependent Ca2+ channel (also known as L-type Ca2+ channel, LTCC) located on the sarcolemma, allowing a small influx of Ca2+ into the cytosol. The Ca2+ entry via LTCC triggers the intracellular Ca2+ channel located on the SR – ryanodine receptor type-2 (RyR2) to open and release a much larger amount of Ca2+ from SR into the cytosol, a process known as Ca2+-induced Ca2+release (CICR). Since activated RyR2 channels can activate neighboring RyR2 channels, the rapid rise of intracellular Ca2+ concentration from about 100 nM during diastole to about 1 μM during systole creates the systolic Ca2+ transient. Ca2+ binds to troponin C, leading to crossbridge of the myofilaments and cardiomyocyte contraction [9,46]. Following contraction, Ca2+ dissociates from the myofilaments, and cytosolic Ca2+ begins to decrease due to removal from cytosol via several mechanisms, thereby initiating relaxation. The decline in cytosolic Ca2+ concentration during diastole is primarily due to the SR Ca2+ uptake carried out by sarcoplasmic reticulum Ca2+/ATPase type-2a (SERCA2a), and Ca2+ removal from cytosol via NCX type-1 (NCX1) and plasma membrane Ca2+ ATPase (PMCA). Recent studies suggest that cytosolic Ca2+ can also be taken up into the mitochondria by the mitochondria Ca2+ uniporter (MCU) located on the outer membrane of mitochondria [40]. However, the relative contribution of mitochondrial Ca2+ transporters to the overall Ca2+ homeostasis remains to be quantitatively determined. The complex interplay between Ca2+ signaling from the SR and mitochondria has been discussed in details elsewhere [40].
2.2). Abnormal Ca2+ handling promotes triggered activity
Ca2+-handling abnormalities and cellular DAD-mediated triggered activity are critical events in atrial cardiomyocytes of patients with paroxysmal (pAF) and long-standing persistent (chronic) AF (cAF), as well as patients developing postoperative AF (poAF) [37,67,111,148–150]. Abnormal Ca2+ release can be estimated as alteration in frequency of Ca2+ sparks and spontaneous Ca2+ waves (SCaWs) [156]. Ca2+ spark is generated by the spontaneous Ca2+ release via a cluster of RyR2 channels located within 2-4 μm vicinity [23]. Increased Ca2+ spark frequency (CaSF) and SCaWs activate NCX1 [11], which is electrogenic. In its forward mode, NCX1 brings 3 Na+ ion into the cell for each Ca2+ ion pumped out of cell, which produces a transient depolarizing inward current that can potentially cause a DAD. Development of DADs increases the propensity for TAs. Simultaneous recording of membrane APs and intracellular Ca2+ signals of cardiomyocytes by combined patch clamping and Ca2+ imaging techniques have demonstrated that the SCaWs can directly evoke DADs and TAs in atrial cardiomyocytes [11,50,91,149]. The potential specific contributions of individual Ca2+-handling proteins to TA are discussed below.
RyR2
RyR2 is a tetrameric channel, first cloned from rabbit cardiac muscle in 1990 [108,112]. Since then, multiple mechanisms have been identified that can regulate the opening of this main intracellular Ca2+-release channel [39]. The stoichiometry of the RyR2 macromolecular complex is a key to maintain the normal RyR2 activity. Several binding partners of RyR2 affect the opening and closing states of this macromolecular complex. The most known RyR2-binding partner is FK506-binding protein 12.6 (FKBP12.6), and the lack of FKBP12.6 enhances the opening of RyR2 channel, increases CaSF, and promotes AF inducibility in FKBP12.6−/− knockout mice [91,134]. Similarly, junctophilin-2 (JPH2), a tethering protein that can help to maintain the optimal distance between sarcolemma and SR membrane at the junctional SR, is another important binding partner of RyR2. Beavers et al. showed a reduction of JPH2 levels in pAF patients and a mutation of JPH2 that reduces binding of JPH2 to RyR2 channels enhances SR Ca2+ leak via RyR2 and the susceptibility to inducible AF in the JPH2E169K knockin mince [8]. Conversely, RyR2 protein levels are strongly increased in pAF patients, which likely results from an impaired posttranscriptional regulation of RYR2 due to the reduced level of the microRNA (miR)-106-25 cluster [25]. Genetic ablation of miRNA-106b-25 in mice recapitulated the increase of RyR2 protein seen in pAF patients, and increased CaSF and SCaWs [25]. Interestingly, the amount of RyR2-bound JPH2 was reduced in the miR-106-25−/− mice, suggesting that JPH2-deficient RyR2 channels might be overactive, contributing to the SR Ca2+ leak.
Post-translational modifications (PTMs) are known to affect activity of RyR2 channels. Despite many controversies, PTMs of RyR2 appears to play a critical role for RyR2 dysfunction in AF [38]. Phosphorylation, oxidation, and nitrosylation are the best known PTMs associated with RyR2 activity. RyR2 channels can be phosphorylated by protein kinase A (PKA) at Serine-2808 (S2808), Ca2+/calmodulin kinase II (CaMKII) at Serine-2814 (S2814), and striated muscled preferentially expressed protein kinase (SPEG) at Serine-2367 (S2367) [20,148,155]. Some studies also suggest that PKA phosphorylates RyR2 at Serine-2030 (S2030) [75]. Phosphorylation of S2808 or S2814 enhances open probability of RyR2, thereby increasing Ca2+ release from SR [21,148,155]. Vest et al. demonstrated that the increase in S2808 phosphorylation of RyR2 enhances AF susceptibility [148]. Following this, several studies from different groups revealed that levels of Threonine-287 autophosphorylated (active) CaMKII and S2814-phosphorylated RyR2 are unaltered in pAF patients, but both are increased in cAF and poAF patients [21,92,111,149,150]. Disease modeling with a phosphomimic mutation of RyR2-S2814 (S2814D) in a knockin mouse model recapitulated the enhancement of SR Ca2+ leak and resulted in increased AF inducibility [149]. In addition to the canonical activation mechanism, oxidation of CaMKII at Methionine-281/282 in mice infused with angiotensin II enhances its activity and triggers AF, mediated by the hyperphosphorylated and hyperactive RyR2 [117]. Opposite to the effects of PKA and CaMKII, SPEG-mediated phosphorylation of RyR2-S2367 stabilizes RyR2 channel [20]. A recent study showed that the level of SPEG and RyR2-S2367 phosphorylation was reduced in pAF patients and the loss of SPEG inhibitory modulation of RyR2 increases CaSF and AF susceptibility in mice [20]. However, the steady-state phosphorylation of the RyR2 channels is also affected by channel dephosphorylating protein phosphatases [43,65]. Defective regulations via protein phosphatase type-1 (PP1) and PP2A might contribute to the enhanced phosphorylation of RyR2 at both S2808 and S2814, respectively [43,141,173]. Additionally, spinophilin-1 (Sp1), functioning as a regulatory subunit of PP1 holoenzyme, facilitates the PP1-mediated regulation of RyR2. The loss of Sp1 in mice (Sp1−/−) leads to hyperphosphorylation of RyR2 at S2814, promoting atrial ectopy and pacing-induced AF [26]. A recent study by Alsina et al. revealed that PPP1R3A, a newly discovered PP1-regulatory subunit, also regulates RyR2 channel and is downregulated in AF patients. The reduced PPP1R3A levels impairs PP1 targeting to both RyR2 and PLB, causing hyperphosphorylation of both proteins and the loss of PPP1R3A in mice enhances SR Ca2+ leak and increases AF susceptibility [2].
RyR2 is also regulated by luminal Ca2+ levels of the SR. Calsequestrin type-2 (CSQ2) is a Ca2+-buffering protein located inside the SR. When SR Ca2+ is low, CSQ2 becomes monomeric Ca2+-free form, and interacts with the accessory proteins such as triadin (TRDN) and junctin (JCN) to prevent RyR2 from opening. As the SR luminal Ca2+ increases, CSQ2 form dimers and polymers, dissociate from RyR2 complex, relieving the RyR2 inhibition [22,145]. Csq2 loss-of-function mouse model displays SCaWs and DADs, and are more susceptible to inducible AF [45,169]. Collectively, these studies demonstrate multiple mechanisms of altered RyR2 function that are all potential contributors to AF development.
SERCA2a
The activity of the SERCA2a pump is tightly regulated by the micropeptides phospholamban (PLB) and sarcolipin (SLN) [5,161]. PLB and SLN directly bind to and serve as endogeneous inhibitors of SERCA. Phosphorylation of PLB by PKA at Serine-16 (S16) or CaMKII at Threonine-17 (T17) relieves SERCA2a inhibition, allowing larger SR Ca2+ reuptake, which increase SR Ca2+ content [76,82]. SPEG may also regulate atrial SERCA2a function, with loss of SPEG resulting in reduced SR Ca2+ reuptake [121]; however, the latter is not a consistent finding [20] and requires further validation. In pAF patients and cAF patients with heart failure, although the expression of SERCA2a is reduced in pAF patients, the overall SERCA2a function increases in both AF populations due to the enhanced phosphorylation of PLB by PKA and CaMKII or reduced SLN level [111,130,144,150]. In cAF patients with preserved ejection fraction, the hyperphosphorylation of PLB is also attributed to the enhanced inhibition of PP1 by hyperphosphorylated (hyperactive) Inhibitor-1 (I-1) [43]. Genetic ablation of SLN in mice (SLN1−/−) enhances SERCA2a activity and promotes DADs [5,161]. Because both SR Ca2+ release and Ca2+ uptake are augmented, AF patients likely experience a faster Ca2+ cycling. The latter could more frequently activate NCX1, thereby causing membrane depolarizations that ultimately lower the threshold for DADs and related TAs in AF patients. As mentioned above, the CaMKII expression and activity are increased in cAF patients and promote arrhythmogenesis through phosphorylation of multiple targets (RyR2, PLB, and LTCC), proving CaMKII as a critical nodal point in AF pathogenesis. For more detailed overview on the CaMKII-mediated regulation of Ca2+-handling proteins and its relationship with AF development, we refer to a recent review article [69].
NCX1
The most known pathological consequence of augmented NCX1 function is the promotion of DADs and TAs. As a result of enhanced RyR2 activity or SR Ca2+ overload due to increased SERCA2a activity, SR Ca2+ leak can over-activate the forward mode NCX1 and generate the transient inward current INCX, which causes a membrane depolarization potentially producing DADs. Once the depolarization reaches the threshold to activate the fast sodium channel, a premature AP will be triggered [50,115,147]. Increased NCX1 function has also been linked to the development of late phase-3 EADs [18,42,139]. The level of NCX1 has an impact on the APD. NCX1 overexpression in transgenic mice prolongs APD, and knockout NCX1 in mice shortens APD [116]. Of note the enhanced NCX1 function in cAF patients appears to involve an upregulation of protein expression, which should amplify the consequences of increased SR Ca2+ leak [150]. However, the molecular mechanism underlying the upregulation of NCX1 in AF remains elusive. Because NCX1 is a membrane protein, the enhanced function of NCX1 in cAF could also be associated with an enhanced trafficking. Under normal condition, a considerable amount of NCX1 is present in the cytoplasm [59]. Thus, it raises the possibilities that the fast trafficking of NCX1 from cytoplasm to the plasma membrane might be enhanced in the context of cAF, which warrants direct demonstration. In ventricular myocytes, EHD3 (Eps15-homology-domain-containing gene produce 3) modulates NCX1 trafficking [58]. Whether this mechanism holds true in atrial myocytes remains to be determined. Moreover, some studies also suggest that NCX1 can be phosphorylated by either PKA or PKC [171]. Further studies are needed to elucidate the relationship between the phosphorylation of NCX1 and AF. Of note, the intracellular Na+ level ([Na+]i) appears to be reduced in a rabbit model of AF induced by rapid atrial pacing. Greiser et al. reported that the resting [Na+]i in rabbit atrial myocytes might decrease as a consequence of the rapid-pacing induced reduction of ICa,L and APD shortening [56]. The reduced [Na+]i level may favor the forward-mode NCX activation in atrial myocytes, which requires further validation and detailed analysis.
2.3). Abnormal Ca2+ handling promotes a reentrant substrate
Although large amount of data pointing to the causative role of abnormal Ca2+ handling in the formation of TA, there are also evidence that abnormal Ca2+ handling can promote reentry by a reduction of APD/ERP, a slowing in atrial conduction, or promoting atrial structural remodeling (hypertrophy and fibrosis), which is detailed below.
2.3.1). LTCC & reduced ERP
Decreased function of LTCC is a hallmark of cAF. In cAF patients and the rabbit model of rapid atrial pacing, mimicking the rapid atrial rhythm during AF, amplitude of ICa,L is reduced due to the downregulation of Cav1.2, the α-subunit of LTCC [10,28,119]. Increased LTCC dephosphorylation due to enhanced activity of PP1 and PP2A also contributes to the lower ICa,L [10,28,119]. The reduction in ICa,L shortens the APD, and thus ERP, allowing the formation of AF-maintaining reentry in patients with cAF [28].
The downregulation of Cav1.2 is attributed to a number of mechanisms. First, Luo et al. reveal that the enhanced microRNA (miR)-328 mediated posttranscriptional regulation of CACNA1C (encoding Cav1.2) is associated with cAF in patients and in a canine model of AF [98]. They also showed that overexpression of miR-328 through adenovirus infection in canines and transgenic mice decreases ICa,L and APD in atrial myocytes, increasing the vulnerability to AF. Conversely, inhibition of miR-328 by antagomiR reversed these alterations [98], suggesting that miR-328 could be a target to prevent AF progression [62]. A loss of transvers tubule (T-tubules) can also lead to a loss-of-function of LTCC channel [86]. The autophagosome-mediated degradation of Cav1.2 has been linked to the increased incidence and burden of AF in cAF patients and rabbit model of atrial rapid pacing [167]. Overexpression of autophagy gene 7 (Atg7) facilitates targeting of the ubiquitin-binding proteins RFP2 and p62 to Cav1.2, accelerating Cav1.2 degradation [167].
In addition to reduced expression of Cav1.2 protein, the open probability of LTCC is directly regulated by the cytosolic Ca2+ concentration. When the level of intracellular Ca2+ increases in the microdomain of Cav1.2, Ca2+-bound calmodulin interacts with Cav1.2 and inactivates the channel [170]. The enhanced SR Ca2+ leak in AF might cause a stronger inactivation of LTCC, a hypothesis that needs experimental verification. Notably, the calmodulin-mediated inactivation of Cav1.2 serves as a self-protecting mechanism that can prevent cells from Ca2+ overload in the early stage of AF and is usually reversable. When the arrhythmia transitions into more persistent forms of AF, additional mechanisms involving transcriptional and posttranscriptional regulations and protein-degradation process as described above become more important causing a downregulation of Cav1.2, often associated with electrical remodeling in cAF patients.
2.3.2). Abnormal Ca2+ handling and structural remodeling
Structural remodeling is a key factor for the maintenance of AF. Ca2+ signaling also plays a role in structural remodeling. When cytosolic Ca2+ levels rise, this can activate the Ca2+-sensitive phosphatase calcineurin (CaN) [17,33,140]. Active CaN dephosphorylates nuclear factor of activated T-cell (NFAT)-c3 and NFAT-c4, and subsequently promotes the translocation of these transcription factors into the nucleus, thereby initiating the transcription of multiple hypertrophic and profibrogenic genes associated with structural remodeling (Figure 1). The enhanced activity of CaN-NFAT pathway correlates with both the enlargement of the left atrium and the maintenance of AF in cAF patients and the canine model of AF [93,100]. Fibrosis, another important feature of structural remodeling associated with AF, could also be affected by the dysregulated Ca2+ signaling in cardiac fibroblasts. For example, the expression of transient receptor potential canonical type-3 (TRPC3) channels, which permeate Ca2+, was increased in AF patients. The increased Ca2+ entry via TRPC3 channels promotes fibroblast proliferation and their differentiation to collagen-secreting myofibroblasts by activating the extracellular signal-regulated kinase (ERK) signaling [63]. For a detailed review on the diverse Ca2+-handling systems in fibroblast and their potential roles in fibrosis, please refer to recent review articles [68,109].
Another important aspect of Ca2+-related structural remodeling is occurring within the specialized t-tubule system [143]. Although earlier studies suggested that the t-tubule system is rudimentary in small animals like rats and mice [80,158], there are t-tubules in the atria of large animals and the human [122]. Of note, recent studies with improved methodology in sample preparation and imaging system have revealed that large networks of axial tubule (AT) structures consistently exist in atrial myocytes of the human, large and small animals, with ATs facilitating rapid Ca2+ release at axial junctions [13,14]. While the t-tubule system tends to run perpendicular to the long axis in ventricular myocytes, ATs predominantly run along the long-axis in atrial myocytes. Lenaerts et al. first reported the loss of atrial tubular structure in a sheep model of persistent AF [86]. The decreased atrial tubular density is associated with increased cell volume and hypertrophy, decreased ICa,L, reduced coupling between LTCC and RyR2, and increased NCX1 activity in atrial myocytes of sheep with persistent AF [86,143]. However, whether AF-related remodeling of the atrial t-tubule system is a cause or bystander of AF development requires further extensive examination. One possibility is that the remodeled atrial t-tubule system could promote AF via re-distribution of NCX1 thereby increasing the propensity for DADs. Alternatively the related decrease in ICa,L causes an abbreviation of APD and ERP, promoting reentry. Given the presence of ATs across species, it would be important to identify the mechanisms of AT development and study how dysfunction in AF formation could promote AF in animal models and perhaps patients with AF.
3. Inflammasome and AF development
Inflammasome refers to the multimeric molecular platform responsible for the activation of pro-inflammatory caspases, which in turn lead to the processing and secretion of pro-inflammatory cytokines [15,127,129]. Over the past 20 years, inflammasomes have been recognized for their roles not only in the host defense against invading pathogens but also in the development of auto-inflammatory, cancer, and metabolic diseases [34,103]. In recent years, inflammasome, particularly the NLRP3 (NACHT, LRR, PYD domain-containing 3) inflammasome, has been linked to the pathological progression of several cardiovascular diseases including atherosclerosis, hypertension, cardiomyopathy, ischemic heart disease, and arrhythmias [89,94]. Inflammatory cytokines have been associated for a long time with the onset and maintenance of AF, as well as the outcome of AF ablation [89], and recent studies suggest that inflammasome activation may play a causative role in AF development, partially via the dysregulation of Ca2+ handling.
3.1). NLRP3 inflammasome
Among various forms of inflammasomes identified so far, the NLRP3 inflammasome is the best characterized and most extensively studied inflammasome complex [127,129]. NLRP3 inflammasome is unique in a way that it responds to a diverse stimuli [57]. The NLRP3 inflammasome is composed of the sensing subunit NLRP3, the adaptor protein ASC, and the effector subunit pro-caspase-1 [127]. NLRP3 protein contains a N-terminus pyrin domain responsible for the recruitment of ASC, a central nucleotide-binding oligomerization domain that enables the activation of the inflammasome signaling platform, and a C-terminus leucine-rich repeat (LRR) functioning in ligand sensing and autoregulation [142]. Basal level of NLRP3 is relatively low and insufficient for active inflammasome formation [7]. Meanwhile, NLRP3 is kept in an inactive ubiquitinated state until a priming signal evokes de-ubiquitination [118]. It is well-known that the activation of the NLRP3 inflammasome requires two processes: priming and triggering (Figure 2). The priming stimuli, such as ligands of toll-like receptors (TLRs), NLRs and cytokine receptors, can induce the transcription factor NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)-mediated transcription of the inflammasome component genes Nlrp3, Asc, pro-caspse-1, as well as the effector genes pro-Il-1b and pro-Il-18 [96,165] . Activation of NF-κB also leads to de-ubiquitination of NLRP3 through the deubiquitinating enzyme BRCC3 [118,120]. As the second step to the NLRP3 inflammasome, triggering stimuli promotes the assembly of NLRP3, ASC, and pro-caspase-1 proteins, facilitating the autocleavage of caspase-1 [138]. To date, a wide range of trigging stimuli have been identified, including ATP, nigericin, silica, uric acid, ionic flux (i.e. K+ efflux, Ca2+ flux, Na+ influx, and Cl− efflux), mitochondrial dysfunction, the production of reactive oxygen species (ROS), and cathepsin B released from the damaged lysosome [54,71,105,172]. In addition to the triggering stimuli, the activation of NLRP3 requires spatial arrangement driven by the microtubule network [101,106]. Active caspase-1 is an aspartate-specific cysteine protease that proteolytically cleave the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18. Furthermore, active caspase-1 also cleaves gasdermin D (GSDMD), which results in fragmentation of GSDMD. The N-terminus fragment of GSDMD (Nt-GSDMD) form pores on the plasma membrane, which facilitates the release of IL-1β and IL-18. In some cases, pore-forming action by Nt-GSDMD may also trigger a lytic, pro-inflammatory form of cell death, known as pyroptosis [48,77,97,131] (Figure 2).
Figure 2. Potential mechanisms underlying NLRP3 inflammasome activation.

Activation of NLRP3 involves two major processes: priming and triggering. Damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) activate toll-like receptor (TLR), and subsequently induce NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)-mediated transcription of inflammasome components (NLRP3, ASC, and pro-Caspase-1) and effectors (pro-IL-1β and pro-IL-18). A wide array of triggering signals may promote the inflammasome assembly. To date, the most established triggering stimuli include 1) K+ efflux via the purinergic receptor P2X7R, 2) increased intracellular Ca2+ levels, 3) ER stress, 4) enhanced cathepsin-B release by lysosome rapture, and 5) increased ROS generation. Spatial arrangement organized by the microtubule network is also essential for the inflammasome assembly. The activation of inflammasome promotes the autocleavage of caspase-1. Mature caspase-1 activates IL-1β, IL-18, and gasdermin-D (GSDMD). N-fragmented GSDMD (Nt-GSDMD) creates membrane pores and facilitate the release of mature IL-1β and IL-18.
3.2.). Overactive NLRP3 inflammasome promotes AF
Clinically, inflammation is frequently detected in patients with pAF, cAF and poAF. Several inflammatory markers including IL-1β and IL-18 correlate with the progression of AF, and can also predict the recurrence rate of AF after ablation [47,88,89,99,159]. Because the production of IL-1β and IL-18 is partially attributed to the activation of NLRP3 inflammasome, it suggests that NLRP3 inflammasome may play a role in AF pathogenesis. Recent studies have demonstrated that the NLRP3 inflammasome activity is increased in atrial samples of pAF, cAF, and poAF patients, as well as in diabetic patients who are at increased risk for AF [47,67,164]. An increased activity of the NLRP3 inflammasome was also noted in animal models of AF including the dogs with atrial tachypacing-induced AF and the CREM transgenic mouse model with spontaneous AF development [79,90,164]. Although NLRP3 inflammasomes exist in multiple cell types including cardiomyocytes, cardiac fibroblasts, macrophages, and adipocytes, etc.[129,146], direct comparison of the NLRP3 inflammasome components in human atrial myocytes versus human atrial fibroblasts revealed that cardiomyocyte NLRP3 inflammasome activation strongly contributes to the overall increase of NLRP3 activity in atrial tissues of AF patients [164]. Additionally, in the macrophage specific Atg7 knockout mice, the increased activity of macrophage NLRP3 inflammasome exerts minimum impact on AF susceptibility [132,164]. These data underscore that cardiomyocytes are key drivers of increased inflammasome activity in atria. However, it is currently unknown whether and how the NLRP3 inflammasome activation is altered in atrial adipocytes, particularly in the epicardial adipose tissue (EAT) of AF patients. Since obesity is a known risk factor of AF and EAT is considered as an important local source for the paracrine modulation of cardiomyocytes [6,55,102], subsequent work should specifically address the role of adipocyte NLRP3 inflammasome in EAT and its potential involvement in AF pathogenesis.
To demonstrate the potential causality between the cardiomyocyte NLRP3 activity and AF pathology, a mouse model with cardiomyocyte-restricted expression of a constitutively activated NLRP3 was recently developed (αMHC:NLRP3A350V/+). The A350V mutation can facilitate the inter-domain interaction between NLRP3 and other inflammasome components, thereby promoting its activation [16]. αMHC:NLRP3A350V/+ mice exhibit atrial ectopic activity, shorter atrial ERP, enlarged atria, and atrial hypertrophy and fibrosis, all of which predispose to AF development. Despite the lack of spontaneous AF, αMHC:NLRP3A350V/+ mice are very susceptible for rapid pacing-induced AF. Selective blockade of NLRP3 by the inhibitor MCC950 or a short-hairpin RNA-mediated knockdown of Nlrp3, both prevent the AF induction in αMHC:NLRP3A350V/+ mice. Consistently, the genetic ablation of NLRP3 in CREM transgenic mice prevented the development of spontaneous AF [164]. Combined these findings support a causative role for NLRP3 in AF development. Nevertheless, many important questions that need clarification remain: 1) how is the inflammasome activated in AF; 2) what are the specific effects of IL-1β, caspase-1, and GSDMD on atrial function in AF; and 3) whether NLRP3 inflammasome has IL-1β independent functions in cardiac cells.
3.3). Inflammasome activation amplifies other inflammatory signaling pathways associated with AF
Activation of NLRP3 inflammasome can potentially boost the activity of other prominent inflammatory cytokines linked to AF development, such as IL-6 and tumor necrosis factor alpha (TNFα). IL-1β, the effector of NLRP3 inflammasome activation, can trigger the expression of IL-6 and TNFα via IL-1 receptor (IL-1R) activation and the formation of active NF-κB. Clininical studies have shown that the increased levels of IL-6 and TNFα associate with AF development in patients and with adverse outcome of AF ablation [1,24,87,136]. In a rat model of sterile pericarditis, the development of poAF is accompanied with increases in IL-6 and TNFα, and atrial fibrosis [74], and the anti-AF effect of colchicine in this model is partially attributed to inhibition of IL-1β-induced expression of IL-6 [160]. The AF-promoting effects of IL-6 and TNFα are largely associated with their ability to promote hypertrophy and fibrosis in AF patients and mouse models [73]. Studies in cardiomyocytes also reveal that TNFα directly reduces ICa,L, CaTs, and SERCA2a and can induce DADs perhaps because of increases in INCX [85,124]. Cardiomyocyte-specific overexpression of TNFα in mice also lead to downregulation of Cx40 and enhances AF susceptibility [125]. Since NF-κB is a master transcription factor controlling the expression of many inflammatory cytokines, and NF-κB activation is likely the consequence of the several cytokine receptors including IL-1R and TNFα receptor (TNFR), a deleterious inflammatory signaling circle involving multiple cytokines may exist in atrial tissue supporting the development of AF substrates.
4. Crosstalk signaling linking altered Ca2+ handling and NLRP3 inflammasome activation
Although previous studies have dealt with the role of altered Ca2+ signaling and NLRP3 inflammasome activity in AF development, emerging recent work suggest these two systems may have common nodal points of mutual regulation. Here, we discuss some potential crosstalk possibilities between Ca2+ handling and NLRP3 inflammasome that could exist in the context of AF (Figure 3).
Figure 3. Crosstalk signaling between altered Ca2+ handling and NLRP3 inflammasome signaling in atrial fibrillation (AF).
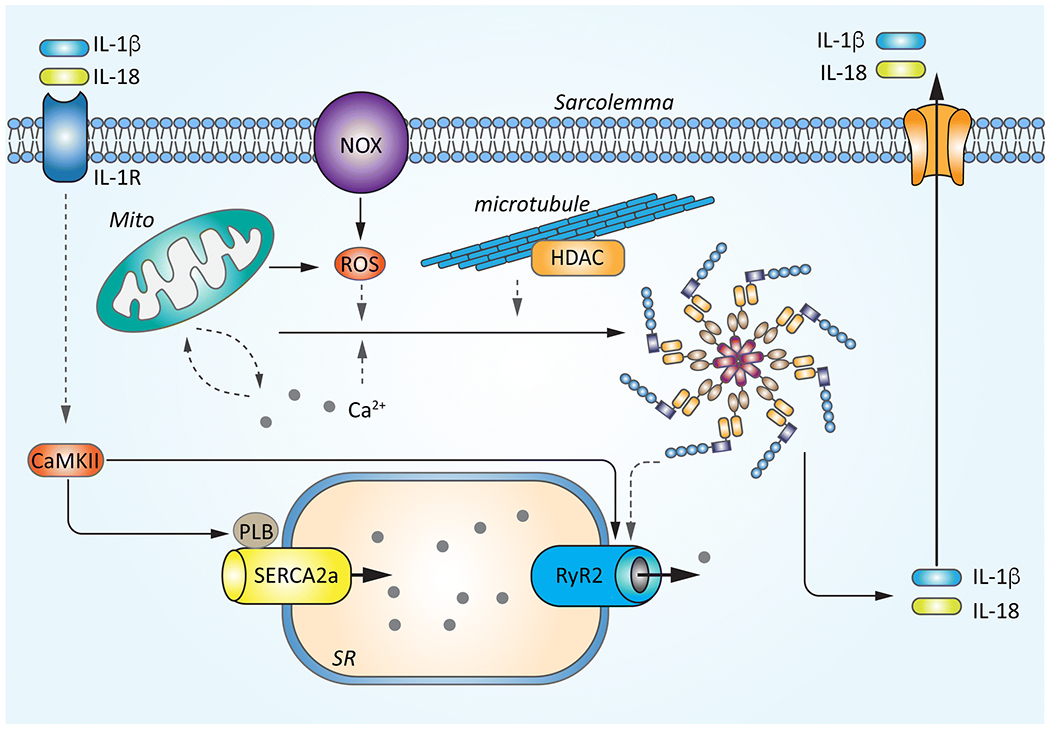
The increase in sarcoplasmic reticulum (SR) Ca2+ release (SR Ca2+ leak) in cardiomyocytes may directly activate the NLRP3 inflammasome by facilitating the inter-domain interactions between inflammasome components or may have indirect effects by promoting the mitochondria (mito)-derived ROS production. Increased levels of ROS due to the enhanced function of mitochondria or NADPH oxidase type-2 (NOX2) can activate CaMKII and RyR2, perpetuating the Ca2+-induced activation of the inflammasome. Abnormal Ca2+ signaling might also trigger the HDAC6-mediated inflammasome activation. Conversely, the enhancement of the NLRP3 inflammasome could amplify the CaMKII-mediated augmentation in SR Ca2+ handling via abnormal IL-1β signaling or elevate RyR2 protein level and RyR2-mediated Ca2+ release via IL-1β or caspase-1 independent mechanisms. Solid lines indicate established regulation patterns, dash lines indicate putative mechanisms that require direct demonstration.
4.1). Intracellular Ca2+ may activate the NLRP3 inflammasome
Ca2+ has multiple and indispensable roles in cellular life [30]. Earlier studies revealed the mobilization of intracellular Ca2+ during the process of NLRP3 inflammasome activation in macrophages and that Ca2+ signaling is required for NLRP3 inflammasome activation [107]. Multiple sources of Ca2+ may lead to the increase of intracellular Ca2+ that contributes to NLRP3-inflammasome activation. In immune cells, where the Ca2+ sensing receptor (CaSR) exists, CaSR can be sensitized by elevated extracellular Ca2+ to cause influx of Ca2+, which activates the NLRP3 inflammasome [84]. In non-excitable cells, the IP3 receptor (IP3R) is the main Ca2+ release channel from the endoplasmic reticulum (ER). Inhibition of IP3R by 2-aminoethyl diphenylborinate (2-APB) reduces intracellular Ca2+ and prevents IL-1β secretion in macrophages [84,154]. Although there is a correlation between intracellular Ca2+ and inflammasome activation, the precise mechanisms linking Ca2+ fluxes with NLRP3-inflammasome activation remain elusive. In macrophages Ca2+ could facilitate the interaction between NLRP3 and ASC as recently suggested [84]. Alternatively, the elevation in cytosolic Ca2+ can lead to mitochondrial Ca2+ overload, generation of mitochondria-derived reactive oxygen species (ROS), and subsequent activation of the NLRP3 inflammasome [70,95,107]. ER stress might also activate the NLRP3 inflammasome, and in many occasions, ER stress is influenced by dysregulations of the Ca2+ homeostasis [104]. Furthermore, the Ca2+-sensitive phosphatase CaN is also involved in the inflammasome activation. In a mouse model with cardiac-specific heterozygous overexpression of CaN, increased inflammation is attributed to the upregulation of Nlrp3, increased mature Casp-1, and elevated serum level of IL-1β [12]. Conversely, the CaN inhibitor cyclosporine reduced IL-1β production due to the decreased mRNA and protein levels of IL-1β in macrophages, positioning active CaN as an important contributor to both priming and triggering of the NLRP3-inflammasome [154]. Since increased SR Ca2+ release and elevated diastolic Ca2+ are common in AF, it would be interesting to elucidate whether and how the abnormal SR Ca2+ release events and mitochondrial Ca2+ signaling contribute to NLRP3-inflammasome activation in atrial cardiomyocytes of AF patients.
It is worth highlighting that ROS and reactive nitrogen species (RNS) signaling modulates the activity of CaMKII and several Ca2+ handling proteins in cardiomyocyte independently [166]. It is well documented that the elevated level of atrial ROS/RNS is associated with AF [4,41,78,133]. The sources for the increased ROS/RNS production in atria has been linked to the increased expression of NADPH oxidase 2 (NOX2) and the enhanced mitochondrial function [4,41,78]. The increased ROS level can activate CaMKII by oxidation at Methionine-281/282, which should phosphorylate RyR2 and PLB, as discussed above [117,166]. In parallel, ROS/RNS can directly cause the oxidation or s-nitrosylation of RyR2, which enhances the RyR2-mediated SR Ca2+ leak [162]. All of these actions lead to an elevated intracellular Ca2+ level, which could contribute to the activation of NLRP3 inflammasome [163].
Recent studies demonstrate that histone deacetylase 6 (HDAC6) plays an indispensable role for the microtubule transport and assembely of inflammasome in macrophages [101,106]. Coincidentally, earlier work have shown that increased HDAC6 activity is a key component of AF-related atrial remodeling in both patients and HL-1 cells subjected to high frequency in vitro pacing [89]. HDAC6 activity contribibutes to atrial tachycardia remodeling-induced decreases in LTCC current and Ca2+ transient amplitudes, and sacromere contractility [168]. Subsequent work should address whether HDAC6-mediated inflammasome activation requires activation of Ca2+ signaling and delinieate the underlying mechanisms.
4.2). NLRP3 inflammasome activation enhances SR Ca2+ leak
Because the enhanced NLRP3 inflammasome activity in cardiomyocytes promotes aberrant SR Ca2+ leak, a crosstalk between these two systems likely occurs. In the αMHC:NLRP3A350V/+ mouse model, where NLRP3 was specifically activated in cardiomyocytes only, CaSF was increased and RyR2 protein was upregulated, which was associated with enhanced atrial ectopic firing [164]. In atrial cardiomyocytes of poAF patients, the more frequent occurrence of SCaWs was associated with a pre-existing low-grade local inflammatory state [67]. Acute application of IL-1β to mouse atrial-cardiomyocytes (HL-1 cells) significantly increased phosphorylation of RyR2-S2814 and PLB-T17, suggesting that the increases of CaMKII and RyR2 activities seen in atria of poAF patients might be a direct consequence of IL-1β signaling. Indeed, inhibition of CaMKII by the inhibitor KN-93 reversed the IL-1β induced hyperphosphorylation of RyR2 and PLB [67]. Similarly, in a heart failure model induced by transverse aortic constriction, the activation of NLRP3 inflammasome and inflammation in ventricular tissue could be attenuated by cardiomyocyte-specific knockout of CaMKII [137]. Together, these recent studies suggest that a self-amplifying feed-forward loop via a NLRP3/CaMKII nexus exists and could play an important role in the pathophysiology of AF and heart failure. Clearly further work is needed to dissect the precise interaction patterns between Ca2+ signaling and NLRP3-inflammasome activation in cardiomyocytes, which is expected to lead to the discovery of novel potential drugs targets to treat cardiovascular diseases.
Conclusion
Aberrant Ca2+ homeostasis due to versatile mechanisms is essential in promoting the formation of ectopic (triggered) activity and a reentrant substrate for AF promotion. NLRP3 inflammasome has emerged as a novel mechanism associated with AF pathogenesis. Dysregulated Ca2+ and NLRP3-inflammasome activation may crosstalk creating a feed-forward loop of joint amplification in AF. Basic science discoveries are key guides for the development of novel anti-AF therapeutics [64]. Thus, identifying the nodal points of crosstalk signaling between these two systems may allow to develop strategies to halt their deleterious interaction potentially preventing AF induction and its perpetuation.
Acknowledgments
Funding source
This study is supported by grants from the National Institutes of Health (R01HL136389 to N.L. and D.D., R01HL147108 to N.L., and R01HL131517 and R01HL089598 to D.D.), the European Union (H2020, MAESTRIA to D.D.), the German Research Foundation (DFG, Do 769/4-1 to D.D.), and Baylor College of Medicine (CVRI pilot grant to N.L.)
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Reference
- 1.Abe I, Teshima Y, Kondo H, Kaku H, Kira S, Ikebe Y, Saito S, Fukui A, Shinohara T, Yufu K, Nakagawa M, Hijiya N, Moriyama M, Shimada T, Miyamoto S, Takahashi N (2018) Association of fibrotic remodeling and cytokines/chemokines content in epicardial adipose tissue with atrial myocardial fibrosis in patients with atrial fibrillation. Heart rhythm 15:1717–1727. doi: 10.1016/j.hrthm.2018.06.025 [DOI] [PubMed] [Google Scholar]
- 2.Alsina KM, Hulsurkar M, Brandenburg S, Kownatzki-Danger D, Lenz C, Urlaub H, Abu-Taha I, Kamler M, Chiang DY, Lahiri SK, Reynolds JO, Quick AP, Scott L Jr., Word TA, Gelves MD, Heck AJR, Li N, Dobrev D, Lehnart SE, Wehrens XHT (2019) Loss of Protein Phosphatase 1 Regulatory Subunit PPP1R3A Promotes Atrial Fibrillation. Circulation 140:681–693. doi: 10.1161/CIRCULATIONAHA.119.039642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrade JG, Champagne J, Dubuc M, Deyell MW, Verma A, Macle L, Leong-Sit P, Novak P, Badra-Verdu M, Sapp J, Mangat I, Khoo C, Steinberg C, Bennett MT, Tang ASL, Khairy P, Investigators C-DS (2019) Cryoballoon or Radiofrequency Ablation for Atrial Fibrillation Assessed by Continuous Monitoring: A Randomized Clinical Trial. Circulation 140:1779–1788. doi: 10.1161/CIRCULATIONAHA.119.042622 [DOI] [PubMed] [Google Scholar]
- 4.Antoniades C, Demosthenous M, Reilly S, Margaritis M, Zhang MH, Antonopoulos A, Marinou K, Nahar K, Jayaram R, Tousoulis D, Bakogiannis C, Sayeed R, Triantafyllou C, Koumallos N, Psarros C, Miliou A, Stefanadis C, Channon KM, Casadei B (2012) Myocardial redox state predicts in-hospital clinical outcome after cardiac surgery effects of short-term pre-operative statin treatment. Journal of the American College of Cardiology 59:60–70. doi: 10.1016/j.jacc.2011.08.062 [DOI] [PubMed] [Google Scholar]
- 5.Babu GJ, Bhupathy P, Timofeyev V, Petrashevskaya NN, Reiser PJ, Chiamvimonvat N, Periasamy M (2007) Ablation of sarcolipin enhances sarcoplasmic reticulum calcium transport and atrial contractility. Proceedings of the National Academy of Sciences of the United States of America 104:17867–17872. doi: 10.1073/pnas.0707722104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batal O, Schoenhagen P, Shao M, Ayyad AE, Van Wagoner DR, Halliburton SS, Tchou PJ, Chung MK (2010) Left atrial epicardial adiposity and atrial fibrillation. Circulation Arrhythmia and electrophysiology 3:230–236. doi: 10.1161/CIRCEP.110.957241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes-Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E (2009) Cutting Edge: NF-κB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. The Journal of Immunology 183:787. doi: 10.4049/jimmunol.0901363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, Dobrev D, Ackerman MJ, Wehrens XHT (2013) Mutation E169K in junctophilin-2 causes atrial fibrillation due to impaired RyR2 stabilization. Journal of the American College of Cardiology 62:2010–2019. doi: 10.1016/j.jacc.2013.06.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415:198–205. doi: 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- 10.Bosch RF, Scherer CR, Rüb N, Wöhrl S, Steinmeyer K, Haase H, Busch AE, Seipel L, Kühlkamp V (2003) Molecular mechanisms of early electrical remodeling: transcriptional downregulation of ion channel subunits reduces ICa,Land Itoin rapid atrial pacing in rabbits. Journal of the American College of Cardiology 41:858–869. doi: 10.1016/s0735-1097(02)02922-4 [DOI] [PubMed] [Google Scholar]
- 11.Boyden PA, Dun W, Stuyvers BD (2015) What is a Ca(2+) wave? Is it like an Electrical Wave? Arrhythm Electrophysiol Rev 4:35–39. doi: 10.15420/aer.2015.4.1.35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, Jabagi H, Wright JR Jr, MacDonald JA, Lees-Miller JP, Roach D, Semeniuk LM, Duff HJ (2013) The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1β. Experimental Physiology 98:462–472. doi: 10.1113/expphysiol.2012.068338 [DOI] [PubMed] [Google Scholar]
- 13.Brandenburg S, Kohl T, Williams GS, Gusev K, Wagner E, Rog-Zielinska EA, Hebisch E, Dura M, Didie M, Gotthardt M, Nikolaev VO, Hasenfuss G, Kohl P, Ward CW, Lederer WJ, Lehnart SE (2016) Axial tubule junctions control rapid calcium signaling in atria. The Journal of clinical investigation 126:3999–4015. doi: 10.1172/JCI88241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandenburg S, Pawlowitz J, Fakuade FE, Kownatzki-Danger D, Kohl T, Mitronova GY, Scardigli M, Neef J, Schmidt C, Wiedmann F, Pavone FS, Sacconi L, Kutschka I, Sossalla S, Moser T, Voigt N, Lehnart SE (2018) Axial Tubule Junctions Activate Atrial Ca(2+) Release Across Species. Front Physiol 9:1227. doi: 10.3389/fphys.2018.01227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nature Reviews Immunology 16:407–420. doi: 10.1038/nri.2016.58 [DOI] [PubMed] [Google Scholar]
- 16.Brydges SD, Mueller JL, McGeough MD, Pena CA, Misaghi A, Gandhi C, Putnam CD, Boyle DL, Firestein GS, Horner AA, Soroosh P, Watford WT, O’Shea JJ, Kastner DL, Hoffman HM (2009) Inflammasome-mediated disease animal models reveal roles for innate but not adaptive immunity. Immunity 30:875–887. doi: 10.1016/j.immuni.2009.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bukowska A, Lendeckel U, Hirte D, Wolke C, Striggow F, Rohnert P, Huth C, Klein HU, Goette A (2006) Activation of the calcineurin signaling pathway induces atrial hypertrophy during atrial fibrillation. Cellular and molecular life sciences : CMLS 63:333–342. doi: 10.1007/s00018-005-5353-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burashnikov A, Antzelevitch C (2006) Late-phase 3 EAD. A unique mechanism contributing to initiation of atrial fibrillation. Pacing Clin Electrophysiol 29:290–295. doi: 10.1111/j.1540-8159.2006.00336.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caballero R, de la Fuente MG, Gomez R, Barana A, Amoros I, Dolz-Gaiton P, Osuna L, Almendral J, Atienza F, Fernandez-Aviles F, Pita A, Rodriguez-Roda J, Pinto A, Tamargo J, Delpon E (2010) In humans, chronic atrial fibrillation decreases the transient outward current and ultrarapid component of the delayed rectifier current differentially on each atria and increases the slow component of the delayed rectifier current in both. Journal of the American College of Cardiology 55:2346–2354. doi: 10.1016/j.jacc.2010.02.028 [DOI] [PubMed] [Google Scholar]
- 20.Campbell HM, Quick AP, Abu-Taha I, Chiang DY, Kramm CF, Word TA, Brandenburg S, Hulsurkar M, Alsina KM, Liu HB, Martin B, Uhlenkamp D, Moore OM, Lahiri SK, Corradini E, Kamler M, Heck AJR, Lehnart SE, Dobrev D, Wehrens XHT (2020) Loss of SPEG Inhibitory Phosphorylation of Ryanodine Receptor Type-2 Promotes Atrial Fibrillation. Circulation 142:1159–1172. doi: 10.1161/CIRCULATIONAHA.120.045791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chelu MG, Sarma S, Sood S, Wang S, van Oort RJ, Skapura DG, Li N, Santonastasi M, Muller FU, Schmitz W, Schotten U, Anderson ME, Valderrabano M, Dobrev D, Wehrens XH (2009) Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. The Journal of clinical investigation 119:1940–1951. doi: 10.1172/jci37059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen H, Valle G, Furlan S, Nani A, Gyorke S, Fill M, Volpe P (2013) Mechanism of calsequestrin regulation of single cardiac ryanodine receptor in normal and pathological conditions. The Journal of general physiology 142:127–136. doi: 10.1085/jgp.201311022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng H, Lederer MR, Xiao RP, Gomez AM, Zhou YY, Ziman B, Spurgeon H, Lakatta EG, Lederer WJ (1996) Excitation-contraction coupling in heart: new insights from Ca2+ sparks. Cell Calcium 20:129–140. doi: 10.1016/s0143-4160(96)90102-5 [DOI] [PubMed] [Google Scholar]
- 24.Cheng T, Wang XF, Hou YT, Zhang L (2012) Correlation between atrial fibrillation, serum amyloid protein A and other inflammatory cytokines. Mol Med Rep 6:581–584. doi: 10.3892/mmr.2012.934 [DOI] [PubMed] [Google Scholar]
- 25.Chiang DY, Kongchan N, Beavers DL, Alsina KM, Voigt N, Neilson JR, Jakob H, Martin JF, Dobrev D, Wehrens XH, Li N (2014) Loss of microRNA-106b-25 cluster promotes atrial fibrillation by enhancing ryanodine receptor type-2 expression and calcium release. Circulation Arrhythmia and electrophysiology 7:1214–1222. doi: 10.1161/CIRCEP.114.001973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiang DY, Li N, Wang Q, Alsina KM, Quick AP, Reynolds JO, Wang G, Skapura D, Voigt N, Dobrev D, Wehrens XH (2014) Impaired local regulation of ryanodine receptor type 2 by protein phosphatase 1 promotes atrial fibrillation. Cardiovascular research 103:178–187. doi: 10.1093/cvr/cvu123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho CH, Woo JS, Perez CF, Lee EH (2017) A focus on extracellular Ca(2+) entry into skeletal muscle. Exp Mol Med 49:e378. doi: 10.1038/emm.2017.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christ T, Boknik P, Wohrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, Dobrev D (2004) L-type Ca2+ current downregulation in chronic human atrial fibrillation is associated with increased activity of protein phosphatases. Circulation 110:2651–2657. doi: 10.1161/01.CIR.0000145659.80212.6A [DOI] [PubMed] [Google Scholar]
- 29.Christoffels VM, Smits GJ, Kispert A, Moorman AF (2010) Development of the pacemaker tissues of the heart. Circulation research 106:240–254. doi: 10.1161/CIRCRESAHA.109.205419 [DOI] [PubMed] [Google Scholar]
- 30.Clapham DE (2007) Calcium signaling. Cell 131:1047–1058. doi: 10.1016/j.cell.2007.11.028 [DOI] [PubMed] [Google Scholar]
- 31.Comtois P, Kneller J, Nattel S (2005) Of circles and spirals: bridging the gap between the leading circle and spiral wave concepts of cardiac reentry. Europace : European pacing, arrhythmias, and cardiac electrophysiology : journal of the working groups on cardiac pacing, arrhythmias, and cardiac cellular electrophysiology of the European Society of Cardiology 7 Suppl 2:10–20. doi: 10.1016/j.eupc.2005.05.011 [DOI] [PubMed] [Google Scholar]
- 32.Dan GA, Dobrev D (2018) Antiarrhythmic drugs for atrial fibrillation: Imminent impulses are emerging. Int J Cardiol Heart Vasc 21:11–15. doi: 10.1016/j.ijcha.2018.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De A (2011) Wnt/Ca2+ signaling pathway: a brief overview. Acta biochimica et biophysica Sinica 43:745–756. doi: 10.1093/abbs/gmr079 [DOI] [PubMed] [Google Scholar]
- 34.De Nardo D, Latz E (2011) NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol 32:373–379. doi: 10.1016/j.it.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dobrev D, Friedrich A, Voigt N, Jost N, Wettwer E, Christ T, Knaut M, Ravens U (2005) The G protein-gated potassium current I(K,ACh) is constitutively active in patients with chronic atrial fibrillation. Circulation 112:3697–3706. doi: 10.1161/CIRCULATIONAHA.105.575332 [DOI] [PubMed] [Google Scholar]
- 36.Dobrev D, Nattel S (2010) New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet 375:1212–1223. doi: 10.1016/S0140-6736(10)60096-7 [DOI] [PubMed] [Google Scholar]
- 37.Dobrev D, Voigt N, Wehrens XH (2011) The ryanodine receptor channel as a molecular motif in atrial fibrillation: pathophysiological and therapeutic implications. Cardiovascular research 89:734–743. doi: 10.1093/cvr/cvq324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobrev D, Wehrens XH (2014) Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circulation research 114:1311–1319; discussion 1319. doi: 10.1161/CIRCRESAHA.114.300568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobrev D, Wehrens XHT (2017) Calcium-mediated cellular triggered activity in atrial fibrillation. J Physiol 595:4001–4008. doi: 10.1113/JP273048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorn GW 2nd, Maack C (2013) SR and mitochondria: calcium cross-talk between kissing cousins. Journal of molecular and cellular cardiology 55:42–49. doi: 10.1016/j.yjmcc.2012.07.015 [DOI] [PubMed] [Google Scholar]
- 41.Dudley SC Jr., Hoch NE, McCann LA, Honeycutt C, Diamandopoulos L, Fukai T, Harrison DG, Dikalov SI, Langberg J (2005) Atrial fibrillation increases production of superoxide by the left atrium and left atrial appendage: role of the NADPH and xanthine oxidases. Circulation 112:1266–1273. doi: 10.1161/CIRCULATIONAHA.105.538108 [DOI] [PubMed] [Google Scholar]
- 42.Edwards AG, Grandi E, Hake JE, Patel S, Li P, Miyamoto S, Omens JH, Heller Brown J, Bers DM, McCulloch AD (2014) Nonequilibrium reactivation of Na+ current drives early afterdepolarizations in mouse ventricle. Circulation Arrhythmia and electrophysiology 7:1205–1213. doi: 10.1161/CIRCEP.113.001666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U, Dobrev D (2006) Molecular determinants of altered Ca2+ handling in human chronic atrial fibrillation. Circulation 114:670–680. doi: 10.1161/CIRCULATIONAHA.106.636845 [DOI] [PubMed] [Google Scholar]
- 44.European Heart Rhythm A, European Cardiac Arrhythmia S, American College of C, American Heart A, Society of Thoracic S, Calkins H, Brugada J, Packer DL, Cappato R, Chen SA, Crijns HJ, Damiano RJ Jr., Davies DW, Haines DE, Haissaguerre M, Iesaka Y, Jackman W, Jais P, Kottkamp H, Kuck KH, Lindsay BD, Marchlinski FE, McCarthy PM, Mont JL, Morady F, Nademanee K, Natale A, Pappone C, Prystowsky E, Raviele A, Ruskin JN, Shemin RJ (2007) HRS/EHRA/ECAS expert Consensus Statement on catheter and surgical ablation of atrial fibrillation: recommendations for personnel, policy, procedures and follow-up. A report of the Heart Rhythm Society (HRS) Task Force on catheter and surgical ablation of atrial fibrillation. Heart rhythm 4:816–861. doi: 10.1016/j.hrthm.2007.04.005 [DOI] [PubMed] [Google Scholar]
- 45.Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D, Knollmann BC (2014) Suppression of spontaneous ca elevations prevents atrial fibrillation in calsequestrin 2-null hearts. Circulation Arrhythmia and electrophysiology 7:313–320. doi: 10.1161/CIRCEP.113.000994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fearnley CJ, Roderick HL, Bootman MD (2011) Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol 3:a004242. doi: 10.1101/cshperspect.a004242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fender AC, Kleeschulte S, Stolte S, Leineweber K, Kamler M, Bode J, Li N, Dobrev D (2020) Thrombin receptor PAR4 drives canonical NLRP3 inflammasome signaling in the heart. Basic Res Cardiol 115:10. doi: 10.1007/s00395-019-0771-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fink SL, Cookson BT (2006) Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cellular Microbiology 8:1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x [DOI] [PubMed] [Google Scholar]
- 49.Foskett JK, White C, Cheung K-H, Mak D-OD (2007) Inositol Trisphosphate Receptor Ca2+ Release Channels. Physiological Reviews 87:593–658. doi: 10.1152/physrev.00035.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fujiwara K, Tanaka H, Mani H, Nakagami T, Takamatsu T (2008) Burst emergence of intracellular Ca2+ waves evokes arrhythmogenic oscillatory depolarization via the Na+-Ca2+ exchanger: simultaneous confocal recording of membrane potential and intracellular Ca2+ in the heart. Circulation research 103:509–518. doi: 10.1161/CIRCRESAHA.108.176677 [DOI] [PubMed] [Google Scholar]
- 51.Gallo EM, Canté-Barrett K, Crabtree GR (2006) Lymphocyte calcium signaling from membrane to nucleus. Nat Immunol 7:25–32. doi: 10.1038/ni1295 [DOI] [PubMed] [Google Scholar]
- 52.Ganesan AN, Shipp NJ, Brooks AG, Kuklik P, Lau DH, Lim HS, Sullivan T, Roberts-Thomson KC, Sanders P (2013) Long-term outcomes of catheter ablation of atrial fibrillation: a systematic review and meta-analysis. J Am Heart Assoc 2:e004549. doi: 10.1161/JAHA.112.004549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goette A, Kalman JM, Aguinaga L, Akar J, Cabrera JA, Chen SA, Chugh SS, Corradi D, D’Avila A, Dobrev D, Fenelon G, Gonzalez M, Hatem SN, Helm R, Hindricks G, Ho Sy , Hoit B, Jalife J, Kim YH, Lip GY, Ma CS, Marcus GM, Murray K, Nogami A, Sanders P, Uribe W, Van Wagoner DR, Nattel S (2017) EHRA/HRS/APHRS/SOLAECE expert consensus on atrial cardiomyopathies: Definition, characterization, and clinical implication. Heart rhythm 14:e3–e40. doi: 10.1016/j.hrthm.2016.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gong T, Yang Y, Jin T, Jiang W, Zhou R (2018) Orchestration of NLRP3 Inflammasome Activation by Ion Fluxes. Trends in Immunology 39:393–406. doi: 10.1016/j.it.2018.01.009 [DOI] [PubMed] [Google Scholar]
- 55.Goudis CA, Korantzopoulos P, Ntalas IV, Kallergis EM, Ketikoglou DG (2015) Obesity and atrial fibrillation: A comprehensive review of the pathophysiological mechanisms and links. Journal of cardiology 66:361–369. doi: 10.1016/j.jjcc.2015.04.002 [DOI] [PubMed] [Google Scholar]
- 56.Greiser M, Kerfant BG, Williams GS, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, van der Velden J, Lederer WJ, Dobrev D, Schotten U (2014) Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. The Journal of clinical investigation 124:4759–4772. doi: 10.1172/JCI70102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gross O, Thomas CJ, Guarda G, Tschopp J (2011) The inflammasome: an integrated view. Immunological Reviews 243:136–151. doi: 10.1111/j.1600-065X.2011.01046.x [DOI] [PubMed] [Google Scholar]
- 58.Gudmundsson H, Curran J, Kashef F, Snyder JS, Smith SA, Vargas-Pinto P, Bonilla IM, Weiss RM, Anderson ME, Binkley P, Felder RB, Carnes CA, Band H, Hund TJ, Mohler PJ (2012) Differential regulation of EHD3 in human and mammalian heart failure. Journal of molecular and cellular cardiology 52:1183–1190. doi: 10.1016/j.yjmcc.2012.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gudmundsson H, Hund TJ, Wright PJ, Kline CF, Snyder JS, Qian L, Koval OM, Cunha SR, George M, Rainey MA, Kashef FE, Dun W, Boyden PA, Anderson ME, Band H, Mohler PJ (2010) EH domain proteins regulate cardiac membrane protein targeting. Circulation research 107:84–95. doi: 10.1161/CIRCRESAHA.110.216713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, Garrigue S, Le Mouroux A, Le Metayer P, Clementy J (1998) Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. The New England journal of medicine 339:659–666. doi: 10.1056/NEJM199809033391003 [DOI] [PubMed] [Google Scholar]
- 61.Hamilton NB, Kolodziejczyk K, Kougioumtzidou E, Attwell D (2016) Proton-gated Ca2+-permeable TRP channels damage myelin in conditions mimicking ischaemia. Nature 529:523–527. doi: 10.1038/nature16519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harada M, Luo X, Murohara T, Yang B, Dobrev D, Nattel S (2014) MicroRNA regulation and cardiac calcium signaling: role in cardiac disease and therapeutic potential. Circulation research 114:689–705. doi: 10.1161/CIRCRESAHA.114.301798 [DOI] [PubMed] [Google Scholar]
- 63.Harada M, Luo X, Qi XY, Tadevosyan A, Maguy A, Ordog B, Ledoux J, Kato T, Naud P, Voigt N, Shi Y, Kamiya K, Murohara T, Kodama I, Tardif JC, Schotten U, Van Wagoner DR, Dobrev D, Nattel S (2012) Transient receptor potential canonical-3 channel-dependent fibroblast regulation in atrial fibrillation. Circulation 126:2051–2064. doi: 10.1161/CIRCULATIONAHA.112.121830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heijman J, Algalarrondo V, Voigt N, Melka J, Wehrens XH, Dobrev D, Nattel S (2016) The value of basic research insights into atrial fibrillation mechanisms as a guide to therapeutic innovation: a critical analysis. Cardiovascular research 109:467–479. doi: 10.1093/cvr/cvv275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heijman J, Dewenter M, El-Armouche A, Dobrev D (2013) Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. Journal of molecular and cellular cardiology 64:90–98. doi: 10.1016/j.yjmcc.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 66.Heijman J, Guichard JB, Dobrev D, Nattel S (2018) Translational Challenges in Atrial Fibrillation. Circulation research 122:752–773. doi: 10.1161/CIRCRESAHA.117.311081 [DOI] [PubMed] [Google Scholar]
- 67.Heijman J, Muna AP, Veleva T, Molina CE, Sutanto H, Tekook MA, Wang Q, Abu-Taha I, Gorka M, Kunzel S, El-Armouche A, Reichenspurner H, Kamler M, Nikolaev VO, Ravens U, Li N, Nattel S, Wehrens XH, Dobrev D (2020) Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Post-Operative Atrial Fibrillation. Circulation research 127:1036–1055. doi: 10.1161/CIRCRESAHA.120.316710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heijman J, Voigt N, Nattel S, Dobrev D (2014) Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circulation research 114:1483–1499. doi: 10.1161/CIRCRESAHA.114.302226 [DOI] [PubMed] [Google Scholar]
- 69.Heijman J, Voigt N, Wehrens XH, Dobrev D (2014) Calcium dysregulation in atrial fibrillation: the role of CaMKII. Frontiers in pharmacology 5:30. doi: 10.3389/fphar.2014.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horng T (2014) Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends in Immunology 35:253–261. doi: 10.1016/j.it.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hornung V, Latz E (2010) Critical functions of priming and lysosomal damage for NLRP3 activation. European Journal of Immunology 40:620–623. doi: 10.1002/eji.200940185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J (2004) Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87 [DOI] [PubMed] [Google Scholar]
- 73.Hu YF, Chen YJ, Lin YJ, Chen SA (2015) Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol 12:230–243. doi: 10.1038/nrcardio.2015.2 [DOI] [PubMed] [Google Scholar]
- 74.Huang Z, Chen XJ, Qian C, Dong Q, Ding D, Wu QF, Li J, Wang HF, Li WH, Xie Q, Cheng X, Zhao N, Du YM, Liao YH (2016) Signal Transducer and Activator of Transcription 3/MicroRNA-21 Feedback Loop Contributes to Atrial Fibrillation by Promoting Atrial Fibrosis in a Rat Sterile Pericarditis Model. Circulation Arrhythmia and electrophysiology 9. doi: 10.1161/CIRCEP.115.003396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jones PP, Meng X, Xiao B, Cai S, Bolstad J, Wagenknecht T, Liu Z, Chen SR (2008) Localization of PKA phosphorylation site, Ser(2030), in the three-dimensional structure of cardiac ryanodine receptor. Biochem J 410:261–270. doi: 10.1042/BJ20071257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kasamatsu J, Oshiumi H, Matsumoto M, Kasahara M, Seya T (2010) Phylogenetic and expression analysis of lamprey toll-like receptors. Developmental & Comparative Immunology 34:855–865. doi: 10.1016/j.dci.2010.03.004 [DOI] [PubMed] [Google Scholar]
- 77.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015) Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526:666–671. doi: 10.1038/naturel5541 [DOI] [PubMed] [Google Scholar]
- 78.Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, Pillai R, Channon KM, Casadei B (2005) A myocardial Nox2 containing NAD(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circulation research 97:629–636. doi: 10.1161/01.RES.0000183735.09871.61 [DOI] [PubMed] [Google Scholar]
- 79.Kirchhof P, Marijon E, Fabritz L, Li N, Wang W, Wang T, Schulte K, Hanstein J, Schulte JS, Vogel M, Mougenot N, Laakmann S, Fortmueller L, Eckstein J, Verheule S, Kaese S, Staab A, Grote-Wessels S, Schotten U, Moubarak G, Wehrens XH, Schmitz W, Hatem S, Muller FU (2013) Overexpression of cAMP-response element modulator causes abnormal growth and development of the atrial myocardium resulting in a substrate for sustained atrial fibrillation in mice. Int J Cardiol 166:366–374. doi: 10.1016/j.ijcard.2011.10.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kirk MM, Izu LT, Chen-Izu Y, McCulle SL, Wier WG, Balke CW, Shorofsky SR (2003) Role of the transverse-axial tubule system in generating calcium sparks and calcium transients in rat atrial myocytes. J Physiol 547:441–451. doi: 10.1113/jphysiol.2002.034355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kornej J, Borschel CS, Benjamin EJ, Schnabel RB (2020) Epidemiology of Atrial Fibrillation in the 21st Century: Novel Methods and New Insights. Circulation research 127:4–20. doi: 10.1161/CIRCRESAHA.120.316340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kranias EG, Hajjar RJ (2012) Modulation of cardiac contractility by the phospholamban/SERCA2a regulatome. Circulation research 110:1646–1660. doi: 10.1161/CIRCRESAHA.111.259754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Landstrom AP, Dobrev D, Wehrens XHT (2017) Calcium Signaling and Cardiac Arrhythmias. Circulation research 120:1969–1993. doi: 10.1161/CIRCRESAHA.117.310083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee G-S, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ (2012) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492:123–127. doi: 10.1038/nature11588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee SH, Chen YC, Chen YJ, Chang SL, Tai CT, Wongcharoen W, Yeh HI, Lin CI, Chen SA (2007) Tumor necrosis factor-alpha alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci 80:1806–1815. doi: 10.1016/j.lfs.2007.02.029 [DOI] [PubMed] [Google Scholar]
- 86.Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D’Hooge J, Heidbuchel H, Sipido KR, Willems R (2009) Ultrastructural and functional remodeling of the coupling between Ca2+ influx and sarcoplasmic reticulum Ca2+ release in right atrial myocytes from experimental persistent atrial fibrillation. Circulation research 105:876–885. doi: 10.1161/CIRCRESAHA.109.206276 [DOI] [PubMed] [Google Scholar]
- 87.Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, Darbar D (2010) Role of inflammation and oxidative stress in atrial fibrillation. Heart rhythm 7:438–444. doi: 10.1016/j.hrthm.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, Darbar D (2010) Role of inflammation and oxidative stress in atrial fibrillation. Heart rhythm 7:438–444. doi: 10.1016/j.hrthm.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li N, Brundel B (2020) Inflammasomes and Proteostasis Novel Molecular Mechanisms Associated With Atrial Fibrillation. Circulation research 127:73–90. doi: 10.1161/CIRCRESAHA.119.316364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, Respress JL, Ather S, Skapura DG, Jordan VK, Horrigan FT, Schmitz W, Muller FU, Valderrabano M, Nattel S, Dobrev D, Wehrens XHT (2014) Ryanodine receptor-mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation 129:1276–1285. doi: 10.1161/CIRCULATIONAHA.113.006611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li N, Wang T, Wang W, Cutler MJ, Wang Q, Voigt N, Rosenbaum DS, Dobrev D, Wehrens XH (2012) Inhibition of CaMKII phosphorylation of RyR2 prevents induction of atrial fibrillation in FKBP12.6 knockout mice. Circulation research 110:465–470. doi: 10.1161/CIRCRESAHA.111.253229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Liang X, Xie H, Zhu PH, Hu J, Zhao Q, Wang CS, Yang C (2009) Enhanced activity of inositol-1,4,5-trisphosphate receptors in atrial myocytes of atrial fibrillation patients. Cardiology 114:180–191. doi: 10.1159/000228584 [DOI] [PubMed] [Google Scholar]
- 93.Lin CC, Lin JL, Lin CS, Tsai MC, Su MJ, Lai LP, Huang SK (2004) Activation of the calcineurin-nuclear factor of activated T-cell signal transduction pathway in atrial fibrillation. Chest 126:1926–1932. doi: 10.1378/chest.126.6.1926 [DOI] [PubMed] [Google Scholar]
- 94.Liu D, Zeng X, Li X, Mehta JL, Wang X (2017) Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Research in Cardiology 113:5. doi: 10.1007/s00395-017-0663-9 [DOI] [PubMed] [Google Scholar]
- 95.Liu Q, Zhang D, Hu D, Zhou X, Zhou Y (2018) The role of mitochondria in NLRP3 inflammasome activation. Molecular Immunology 103:115–124. doi: 10.1016/j.molimm.2018.09.010 [DOI] [PubMed] [Google Scholar]
- 96.Liu T, Zhang L, Joo D, Sun SC (2017) NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2. doi: 10.1038/sigtrans.2017.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535:153–158. doi: 10.1038/nature18629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F, Zhang Y, Shan H, Luo X, Bai Y, Sun L, Song W, Xu C, Wang Z, Yang B (2010) MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation 122:2378–2387. doi: 10.1161/CIRCULATIONAHA.110.958967 [DOI] [PubMed] [Google Scholar]
- 99.Luan Y, Guo Y, Li S, Yu B, Zhu S, Li S, Li N, Tian Z, Peng C, Cheng J, Li Q, Cui J, Tian Y (2010) Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. EP Europace 12:1713–1718. doi: 10.1093/europace/euq321 [DOI] [PubMed] [Google Scholar]
- 100.Luo X, Pan Z, Shan H, Xiao J, Sun X, Wang N, Lin H, Xiao L, Maguy A, Qi XY, Li Y, Gao X, Dong D, Zhang Y, Bai Y, Ai J, Sun L, Lu H, Luo XY, Wang Z, Lu Y, Yang B, Nattel S (2013) MicroRNA-26 governs profibrillatory inward-rectifier potassium current changes in atrial fibrillation. The Journal of clinical investigation 123:1939–1951. doi: 10.1172/JCI62185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, Deng Q, Orning P, Alam HB, Maliga Z, Sharif H, Hu JJ, Evavold CL, Kagan JC, Schmidt FI, Fitzgerald KA, Kirchhausen T, Li Y, Wu H (2020) HDAC6 mediates an aggresome-like mechanism for NLRP3 and pyrin inflammasome activation. Science 369. doi: 10.1126/science.aas8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mahajan R, Lau DH, Brooks AG, Shipp NJ, Manavis J, Wood JP, Finnie JW, Samuel CS, Royce SG, Twomey DJ, Thanigaimani S, Kalman JM, Sanders P (2015) Electrophysiological, Electroanatomical, and Structural Remodeling of the Atria as Consequences of Sustained Obesity. Journal of the American College of Cardiology 66:1–11. doi: 10.1016/j.jacc.2015.04.058 [DOI] [PubMed] [Google Scholar]
- 103.Man SM, Kanneganti T-D (2015) Regulation of inflammasome activation. Immunological Reviews 265:6–21. doi: 10.1111/imr.12296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Menu P, Mayor A, Zhou R, Tardivel A, Ichijo H, Mori K, Tschopp J (2012) ER stress activates the NLRP3 inflammasome via an UPR-independent pathway. Cell Death Dis 3:e261. doi: 10.1038/cddis.2011.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature Immunology 14:454–460. doi: 10.1038/ni.2550 [DOI] [PubMed] [Google Scholar]
- 106.Misawa T, Takahama M, Kozaki T, Lee H, Zou J, Saitoh T, Akira S (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14:454–460. doi: 10.1038/ni.2550 [DOI] [PubMed] [Google Scholar]
- 107.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, Horng T (2012) Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proceedings of the National Academy of Sciences 109:11282. doi: 10.1073/pnas.1117765109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nakai J, Imagawa T, Hakamat Y, Shigekawa M, Takeshima H, Numa S (1990) Primary structure and functional expression from cDNA of the cardiac ryanodine receptor/calcium release channel. FEBS Lett 271:169–177. doi: 10.1016/0014-5793(90)80399-4 [DOI] [PubMed] [Google Scholar]
- 109.Nattel S (2017) Molecular and Cellular Mechanisms of Atrial Fibrosis in Atrial Fibrillation. JACC Clin Electrophysiol 3:425–435. doi: 10.1016/j.jacep.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 110.Nattel S, Heijman J, Zhou L, Dobrev D (2020) Molecular Basis of Atrial Fibrillation Pathophysiology and Therapy: A Translational Perspective. Circulation research 127:51–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schondube FA, Hasenfuss G, Maier LS (2010) CaMKII-dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circulation research 106:1134–1144. doi: 10.1161/CIRCRESAHA.109.203836 [DOI] [PubMed] [Google Scholar]
- 112.Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, MacLennan DH (1990) Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem 265:13472–13483 [PubMed] [Google Scholar]
- 113.Packer DL, Mark DB, Robb RA, Monahan KH, Bahnson TD, Poole JE, Noseworthy PA, Rosenberg YD, Jeffries N, Mitchell LB, Flaker GC, Pokushalov E, Romanov A, Bunch TJ, Noelker G, Ardashev A, Revishvili A, Wilber DJ, Cappato R, Kuck KH, Hindricks G, Davies DW, Kowey PR, Naccarelli GV, Reiffel JA, Piccini JP, Silverstein AP, Al-Khalidi HR, Lee KL, Investigators C (2019) Effect of Catheter Ablation vs Antiarrhythmic Drug Therapy on Mortality, Stroke, Bleeding, and Cardiac Arrest Among Patients With Atrial Fibrillation: The CABANA Randomized Clinical Trial. JAMA 321:1261–1274. doi: 10.1001/jama.2019.0693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pandit SV, Jalife J (2013) Rotors and the dynamics of cardiac fibrillation. Circulation research 112:849–862. doi: 10.1161/CIRCRESAHA.111.300158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pogwizd SM, Bers DM (2004) Cellular basis of triggered arrhythmias in heart failure. Trends in cardiovascular medicine 14:61–66. doi: 10.1016/j.tcm.2003.12.002 [DOI] [PubMed] [Google Scholar]
- 116.Pott C, Philipson KD, Goldhaber JI (2005) Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux. Circulation research 97:1288–1295. doi: 10.1161/01.RES.0000196563.84231.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Purohit A, Rokita AG, Guan X, Chen B, Koval OM, Voigt N, Neef S, Sowa T, Gao Z, Luczak ED, Stefansdottir H, Behunin AC, Li N, El-Accaoui RN, Yang B, Swaminathan PD, Weiss RM, Wehrens XH, Song LS, Dobrev D, Maier LS, Anderson ME (2013) Oxidized Ca(2+)/calmodulin-dependent protein kinase II triggers atrial fibrillation. Circulation 128:1748–1757. doi: 10.1161/CIRCULATIONAHA.113.003313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Py Bénédicte F, Kim M-S, Vakifahmetoglu-Norberg H, Yuan J (2013) Deubiquitination of NLRP3 by BRCC3 Critically Regulates Inflammasome Activity. Molecular Cell 49:331–338. doi: 10.1016/j.molcel.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 119.Qi XY, Yeh YH, Xiao L, Burstein B, Maguy A, Chartier D, Villeneuve LR, Brundel BJ, Dobrev D, Nattel S (2008) Cellular signaling underlying atrial tachycardia remodeling of L-type calcium current. Circulation research 103:845–854. doi: 10.1161/CIRCRESAHA.108.175463 [DOI] [PubMed] [Google Scholar]
- 120.Qiao Y, Wang P, Qi J, Zhang L, Gao C (2012) TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Letters 586:1022–1026. doi: 10.1016/j.febslet.2012.02.045 [DOI] [PubMed] [Google Scholar]
- 121.Quan C, Li M, Du Q, Chen Q, Wang H, Campbell D, Fang L, Xue B, MacKintosh C, Gao X, Ouyang K, Wang HY, Chen S (2019) SPEG Controls Calcium Reuptake Into the Sarcoplasmic Reticulum Through Regulating SERCA2a by Its Second Kinase-Domain. Circulation research 124:712–726. doi: 10.1161/CIRCRESAHA.118.313916 [DOI] [PubMed] [Google Scholar]
- 122.Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW, Dibb KM (2011) Transverse tubules are a common feature in large mammalian atrial myocytes including human. American journal of physiology Heart and circulatory physiology 301:H1996–2005. doi: 10.1152/ajpheart.00284.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Roy D, Talajic M, Nattel S, Wyse DG, Dorian P, Lee KL, Bourassa MG, Arnold JM, Buxton AE, Camm AJ, Connolly SJ, Dubuc M, Ducharme A, Guerra PG, Hohnloser SH, Lambert J, Le Heuzey JY, O’Hara G, Pedersen OD, Rouleau JL, Singh BN, Stevenson LW, Stevenson WG, Thibault B, Waldo AL, Atrial F, Congestive Heart Failure I (2008) Rhythm control versus rate control for atrial fibrillation and heart failure. The New England journal of medicine 358:2667–2677. doi: 10.1056/NEJMoa0708789 [DOI] [PubMed] [Google Scholar]
- 124.Saba S, Janczewski AM, Baker LC, Shusterman V, Gursoy EC, Feldman AM, Salama G, McTiernan CF, London B (2005) Atrial contractile dysfunction, fibrosis, and arrhythmias in a mouse model of cardiomyopathy secondary to cardiac-specific overexpression of tumor necrosis factor-{alpha}. American journal of physiology Heart and circulatory physiology 289:H1456–1467. doi: 10.1152/ajpheart.00733.2004 [DOI] [PubMed] [Google Scholar]
- 125.Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, Mann DL, Khoury DS (2007) Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac-restricted overexpression of tumor necrosis factor. American journal of physiology Heart and circulatory physiology 292:H1561–1567. doi: 10.1152/ajpheart.00285.2006 [DOI] [PubMed] [Google Scholar]
- 126.Schmidt C, Wiedmann F, Voigt N, Zhou XB, Heijman J, Lang S, Albert V, Kallenberger S, Ruhparwar A, Szabo G, Kallenbach K, Karck M, Borggrefe M, Biliczki P, Ehrlich JR, Baczko I, Lugenbiel P, Schweizer PA, Donner BC, Katus HA, Dobrev D, Thomas D (2015) Upregulation of K(2P)3.1 K+ Current Causes Action Potential Shortening in Patients With Chronic Atrial Fibrillation. Circulation 132:82–92. doi: 10.1161/CIRCULATIONAHA.114.012657 [DOI] [PubMed] [Google Scholar]
- 127.Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832. doi: 10.1016/j.cell.2010.01.040 [DOI] [PubMed] [Google Scholar]
- 128.Schweikert RA, Perez Lugones A, Kanagaratnam L, Tomassoni G, Beheiry S, Bash D, Pisano E, Saliba W, Tchou PJ, Natale A (2001) A simple method of mapping atrial premature depolarizations triggering atrial fibrillation. Pacing Clin Electrophysiol 24:22–27. [DOI] [PubMed] [Google Scholar]
- 129.Scott L Jr., Li N, Dobrev D (2019) Role of inflammatory signaling in atrial fibrillation. Int J Cardiol 287:195–200. doi: 10.1016/j.ijcard.2018.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shanmugam M, Molina CE, Gao S, Severac-Bastide R, Fischmeister R, Babu GJ (2011) Decreased sarcolipin protein expression and enhanced sarco(endo)plasmic reticulum Ca2+ uptake in human atrial fibrillation. Biochemical and biophysical research communications 410:97–101. doi: 10.1016/j.bbrc.2011.05.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526:660–665. doi: 10.1038/nature15514 [DOI] [PubMed] [Google Scholar]
- 132.Shin JN, Fattah EA, Bhattacharya A, Ko S, Eissa NT (2013) Inflammasome activation by altered proteostasis. J Biol Chem 288:35886–35895. doi: 10.1074/jbc.M113.514919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Simon JN, Ziberna K, Casadei B (2016) Compromised redox homeostasis, altered nitroso-redox balance, and therapeutic possibilities in atrial fibrillation. Cardiovascular research 109:510–518. doi: 10.1093/cvr/cvw012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sood S, Chelu MG, van Oort RJ, Skapura D, Santonastasi M, Dobrev D, Wehrens XH (2008) Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart rhythm 5:1047–1054. doi: 10.1016/j.hrthm.2008.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sossalla S, Kallmeyer B, Wagner S, Mazur M, Maurer U, Toischer K, Schmitto JD, Seipelt R, Schondube FA, Hasenfuss G, Belardinelli L, Maier LS (2010) Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. Journal of the American College of Cardiology 55:2330–2342. doi: 10.1016/j.jacc.2009.12.055 [DOI] [PubMed] [Google Scholar]
- 136.Stanciu AE, Vatasescu RG, Stanciu MM, Serdarevic N, Dorobantu M (2018) The role of pro-fibrotic biomarkers in paroxysmal and persistent atrial fibrillation. Cytokine 103:63–68. [DOI] [PubMed] [Google Scholar]
- 137.Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, Brown JH (2018) Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca(2+)/Calmodulin-Dependent Protein Kinase II delta Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 138:2530–2544. doi: 10.1161/CIRCULATIONAHA.118.034621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Swanson KV, Deng M, Ting JPY (2019) The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nature Reviews Immunology 19:477–489. doi: 10.1038/s41577-019-0165-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Szabo B, Sweidan R, Rajagopalan CV, Lazzara R (1994) Role of Na+:Ca2+ exchange current in Cs(+)-induced early afterdepolarizations in Purkinje fibers. J Cardiovasc Electrophysiol 5:933–944. doi: 10.1111/j.1540-8167.1994.tb01133.x [DOI] [PubMed] [Google Scholar]
- 140.Tao H, Yang JJ, Shi KH, Li J (2016) Wnt signaling pathway in cardiac fibrosis: New insights and directions. Metabolism: clinical and experimental 65:30–40. doi: 10.1016/j.metabol.2015.10.013 [DOI] [PubMed] [Google Scholar]
- 141.Terentyev D, Hamilton S (2016) Regulation of sarcoplasmic reticulum Ca(2+) release by serine-threonine phosphatases in the heart. Journal of molecular and cellular cardiology 101:156–164. doi: 10.1016/j.yjmcc.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ting JPY, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot J-P, Inohara N, Mackenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, Reith W, Schreiber S, Steimle V, Ward PA (2008) The NLR gene family: a standard nomenclature. Immunity 28:285–287. doi: 10.1016/j.immuni.2008.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Trafford AW, Clarke JD, Richards MA, Eisner DA, Dibb KM (2013) Calcium signaling microdomains and the t-tubular system in atrial mycoytes: potential roles in cardiac disease and arrhythmias. Cardiovascular research 98:192–203. doi: 10.1093/cvr/cvt018 [DOI] [PubMed] [Google Scholar]
- 144.Uemura N, Ohkusa T, Hamano K, Nakagome M, Hori H, Shimizu M, Matsuzaki M, Mochizuki S, Minamisawa S, Ishikawa Y (2004) Down-regulation of sarcolipin mRNA expression in chronic atrial fibrillation. Eur J Clin Invest 34:723–730. doi: 10.1111/j.1365-2362.2004.01422.x [DOI] [PubMed] [Google Scholar]
- 145.van Ooijen G, van den Burg HA, Cornelissen BJ, Takken FL (2007) Structure and function of resistance proteins in solanaceous plants. Annual review of phytopathology 45:43–72. doi: 10.1146/annurev.phyto.45.062806.094430 [DOI] [PubMed] [Google Scholar]
- 146.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17:179–188. doi: 10.1038/nm.2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Venetucci LA, Trafford AW, O’Neill SC, Eisner DA (2008) The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovascular research 77:285–292. doi: 10.1093/cvr/cvm009 [DOI] [PubMed] [Google Scholar]
- 148.Vest JA, Wehrens XH, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR, Marks AR (2005) Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C [DOI] [PubMed] [Google Scholar]
- 149.Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S, Dobrev D (2014) Cellular and molecular mechanisms of atrial arrhythmogenesis in patients with paroxysmal atrial fibrillation. Circulation 129:145–156. doi: 10.1161/CIRCULATIONAHA.113.006641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XH, Dobrev D (2012) Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 125:2059–2070. doi: 10.1161/CIRCULATIONAHA.111.067306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Voigt N, Trausch A, Knaut M, Matschke K, Varro A, Van Wagoner DR, Nattel S, Ravens U, Dobrev D (2010) Left-to-right atrial inward rectifier potassium current gradients in patients with paroxysmal versus chronic atrial fibrillation. Circulation Arrhythmia and electrophysiology 3:472–480. doi: 10.1161/CIRCEP.110.954636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wakili R, Voigt N, Kaab S, Dobrev D, Nattel S (2011) Recent advances in the molecular pathophysiology of atrial fibrillation. The Journal of clinical investigation 121:2955–2968. doi: 10.1172/JCI46315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Waks JW, Josephson ME (2014) Mechanisms of Atrial Fibrillation - Reentry, Rotors and Reality. Arrhythm Electrophysiol Rev 3:90–100. doi: 10.15420/aer.2014.3.2.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weber K, Schilling JD (2014) Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem 289:9158–9171. doi: 10.1074/jbc.M113.531202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wehrens XH, Lehnart SE, Reiken SR, Marks AR (2004) Ca2+/calmodulin-dependent protein kinase II phosphorylation regulates the cardiac ryanodine receptor. Circulation research 94:e61–70. doi: 10.1161/01.RES.0000125626.33738.E2 [DOI] [PubMed] [Google Scholar]
- 156.Wier WG, Cannell MB, Berlin JR, Marban E, Lederer WJ (1987) Cellular and subcellular heterogeneity of [Ca2+]i in single heart cells revealed by fura-2. Science 235:325–328. doi: 10.1126/science.3798114 [DOI] [PubMed] [Google Scholar]
- 157.Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA (1995) Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 92:1954–1968. doi: 10.1161/01.cir.92.7.1954 [DOI] [PubMed] [Google Scholar]
- 158.Woo SH, Cleemann L, Morad M (2005) Diversity of atrial local Ca2+ signalling: evidence from 2-D confocal imaging in Ca2+-buffered rat atrial myocytes. J Physiol 567:905–921. doi: 10.1113/jphysiol.2005.092270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Wu N, Xu B, Xiang Y, Wu L, Zhang Y, Ma X, Tong S, Shu M, Song Z, Li Y, Zhong L (2013) Association of inflammatory factors with occurrence and recurrence of atrial fibrillation: A meta-analysis. International Journal of Cardiology 169:62–72. doi: 10.1016/j.ijcard.2013.08.078 [DOI] [PubMed] [Google Scholar]
- 160.Wu Q, Liu H, Liao J, Zhao N, Tse G, Han B, Chen L, Huang Z, Du Y (2020) Colchicine prevents atrial fibrillation promotion by inhibiting IL-1beta-induced IL-6 release and atrial fibrosis in the rat sterile pericarditis model. Biomed Pharmacother 129:110384. doi: 10.1016/j.biopha.2020.110384 [DOI] [PubMed] [Google Scholar]
- 161.Xie LH, Shanmugam M, Park JY, Zhao Z, Wen H, Tian B, Periasamy M, Babu GJ (2012) Ablation of sarcolipin results in atrial remodeling. American journal of physiology Cell physiology 302:C1762–1771. doi: 10.1152/ajpcell.00425.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR (2015) Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep 5:11427. doi: 10.1038/srep11427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yang X, An N, Zhong C, Guan M, Jiang Y, Li X, Zhang H, Wang L, Ruan Y, Gao Y, Liu N, Shang H, Xing Y (2020) Enhanced cardiomyocyte reactive oxygen species signaling promotes ibrutinib-induced atrial fibrillation. Redox Biol 30:101432. doi: 10.1016/j.redox.2020.101432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yao C, Veleva T, Scott L Jr., Cao S, Li L, Chen G, Jeyabal P, Pan X, Alsina KM, Abu-Taha ID, Ghezelbash S, Reynolds CL, Shen YH, LeMaire Sa , Schmitz W, Muller FU, El-Armouche A, Tony Eissa N, Beeton C, Nattel S, Wehrens XHT, Dobrev D, Li N (2018) Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 138:2227–2242. doi: 10.1161/CIRCULATIONAHA.118.035202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ylikoski J, Pirvola U, Happola O, Panula P, Virtanen I (1989) Immunohistochemical demonstration of neuroactive substances in the inner ear of rat and guinea pig. Acta Otolaryngol 107:417–423. doi: 10.3109/00016488909127533 [DOI] [PubMed] [Google Scholar]
- 166.Yoo S, Aistrup G, Shiferaw Y, Ng J, Mohler PJ, Hund TJ, Waugh T, Browne S, Gussak G, Gilani M, Knight BP, Passman R, Goldberger JJ, Wasserstrom JA, Arora R (2018) Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 3. doi: 10.1172/jci.insight.120728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yuan Y, Zhao J, Gong Y, Wang D, Wang X, Yun F, Liu Z, Zhang S, Li W, Zhao X, Sun L, Sheng L, Pan Z, Li Y (2018) Autophagy exacerbates electrical remodeling in atrial fibrillation by ubiquitin-dependent degradation of L-type calcium channel. Cell Death Dis 9:873. doi: 10.1038/s41419-018-0860-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, Cubukcuoglu Deniz G, Durdu S, Akar AR, Sibon OC, Nattel S, Henning RH, Brundel BJ (2014) Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 129:346–358. doi: 10.1161/CIRCULATIONAHA.113.005300 [DOI] [PubMed] [Google Scholar]
- 169.Zhang JC, Wu HL, Chen Q, Xie XT, Zou T, Zhu C, Dong Y, Xiang GJ, Ye L, Li Y, Zhu PL (2018) Calcium-Mediated Oscillation in Membrane Potentials and Atrial-Triggered Activity in Atrial Cells of Casq2(R33Q/R33Q) Mutation Mice. Front Physiol 9:1447. doi: 10.3389/fphys.2018.01447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang T, Miyamoto S, Brown JH (2004) Cardiomyocyte calcium and calcium/calmodulin-dependent protein kinase II: friends or foes? Recent Prog Horm Res 59:141–168. doi: 10.1210/rp.59.1.141 [DOI] [PubMed] [Google Scholar]
- 171.Zhang YH, Hancox JC (2009) Regulation of cardiac Na+-Ca2+ exchanger activity by protein kinase phosphorylation--still a paradox? Cell Calcium 45:1–10. doi: 10.1016/j.ceca.2008.05.005 [DOI] [PubMed] [Google Scholar]
- 172.Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–225. doi: 10.1038/nature09663 [DOI] [PubMed] [Google Scholar]
- 173.Zhu W, Wang C, Hu J, Wan R, Yu J, Xie J, Ma J, Guo L, Ge J, Qiu Y, Chen L, Liu H, Yan X, Liu X, Ye J, He W, Shen Y, Wang C, Mohler PJ, Hong K (2018) Ankyrin-B Q1283H Variant Linked to Arrhythmias Via Loss of Local Protein Phosphatase 2A Activity Causes Ryanodine Receptor Hyperphosphorylation. Circulation 138:2682–2697. doi: 10.1161/CIRCULATIONAHA.118.034541 [DOI] [PMC free article] [PubMed] [Google Scholar]