

Abstract
Background
Mitochondrial dysfunction plays an important role in the development of septic cardiomyopathy. This study aimed to reveal the protective role of uncoupling protein 2 (UCP2) in mitochondria through AMP-activated protein kinase (AMPK) on autophagy during septic cardiomyopathy.
Methods
UCP2 knockout mice via a cecal ligation and puncture (CLP) model and the H9C2 cardiomyocyte cell line in response to lipopolysaccharide (LPS) in vitro were used to study the effect. The myocardial morphological alterations, indicators of mitochondrial injury and levels of autophagy-associated proteins (pAMPK, pmTOR, pULK1, pTSC2, Beclin-1, and LC3-I/II) were assessed. In addition, the mechanism of the interaction between UCP2 and AMPK was further studied through gain- and loss-of-function studies.
Results
Compared with the wild-type mice, the UCP2 knockout mice exhibited more severe cardiomyocyte injury after CLP, and the AMPK agonist AICAR protected against such injury. Consistent with this result, silencing UCP2 augmented the LPS-induced pathological damage and mitochondrial injury in the H9C2 cells, limited the upregulation of autophagy proteins and reduced AMPK phosphorylation. AICAR protected the cells from morphological changes and mitochondrial membrane potential loss and promoted autophagy. The silencing and overexpression of UCP2 led to correlated changes in the AMPK upstream kinases pLKB1 and CAMKK2.
Conclusions
UCP2 exerts cardioprotective effects on mitochondrial dysfunction during sepsis via the action of AMPK on autophagy.
Keywords: Septic cardiomyopathy, autophagy, uncoupling protein 2 (UCP2), AMPK
Introduction
Sepsis contributes to multiple organ dysfunction and is a major cause of mortality in critically ill patients (1). The cardiovascular system is an important organ system that is frequently involved in sepsis, and sepsis-induced cardiomyopathy is among the most life-threatened complications of sepsis (2,3). As one of the most critical mechanisms of septic cardiomyopathy, myocardial energy metabolism has been studied by an increasing number of studies, and mitochondrial dysfunction has been identified as the key pathological change (4,5). Cardiac mitochondria are responsible for energy generation and are essential for cardiac function. However, current clinical practice does not include mitochondria-targeted management, although both human sepsis and septic cardiomyopathy are associated with mitochondrial impairment (6). Further exploration of the mechanism of mitochondrial dysfunction may lead to future treatments for septic cardiomyopathy.
Mitochondrial uncoupling proteins, which are located in the inner membrane of mitochondria, can promote the leakage of protons and act as crucial mitochondrial membrane potential regulators (7,8). Mitochondrial uncoupling protein 2 (UCP2) in this family is the most investigated. Many studies have revealed that UCP2 has a myocardial protective effect, and UCP2 deficiency has been shown to aggravate septic cardiomyopathy in both UCP2 knockout mice and cardiomyocytes (9-11), demonstrating the crucial role of UCP2 in the mechanism of cardiovascular involvement in sepsis. Our previous study revealed that the serum UCP2 levels were markedly increased in patients with sepsis (12) and that UCP2 exerted a protective effect against lipopolysaccharide (LPS) by regulating the myocardial autophagy and apoptosis balance (13). Under a physiological status, programmed cell death processes, such as autophagy and apoptosis, play crucial roles in cell renewal and repair. However, the mechanism by which UCP2 regulates autophagy in septic cardiomyopathy is still unknown.
Therefore, based on previous studies, we hypothesized that UCP2 may exert protective effects against septic cardiomyopathy through the regulation of autophagy possibly via AMP-activated protein kinase (AMPK), which is an essential player in autophagy. Both UCP2 knockout (UCP2-KO) mice and in vitro cultures of cardiomyocytes (H9C2 cells) were used in this study to test this hypothesis. We present the following article in accordance with the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/atm-20-4819).
Methods
Animal model and treatment
Wild C57BL/6J mice (19–23 g) were obtained from Vital River Laboratory Animal Technology Company (Beijing), and all mice were 8-week-old male mice. The UCP2-KO mice were obtained from Nanjing Biomedical Research Institute of Nanjing University. The knockout mice genotypes were identified by PCR amplification of genomic DNA from the tail. The sex, age in weeks and body weight of the UCP2-KO mice did not significantly differ from those of their littermates. All wild type (WT) and UCP2-KO mice were housed in specific pathogen-free conditions under a constant humidity (40–60%) and temperature (20–24 °C) with free access to food and water under a 12/12 h light/dark cycle. The experiments were performed under a project license (NO. XHDW-2019002) granted by the Animal Care Ethics and Use Committee of Peking Union Medical College in compliance with the guidelines of the Care and Use of Laboratory Animals published by the US National Institutes of Health.
All mice (n=24) were initially housed together (5 animals/cage) for adaption for one week before randomization into the following groups of 6 mice each: (I) WT sham operation group, (II) WT + cecal ligation and puncture (CLP) group, (III) UCP2-KO + CLP group, and (IV) UCP2-KO + CLP + AICAR (AMPK agonist) group. The model of mild-grade sepsis was established through CLP (14). A midline laparotomy was performed to expose the cecum of the mice after anesthesia with 4% chloral hydrate. The cecum was punctured by a 22 G needle at a middle point. In the sham group, only the laparotomy procedures were conducted without CLP. All mice immediately received fluid resuscitation (50 mL/kg sterile saline subcutaneously) after surgery. In the treatment groups (IV), the AMPK agonist AICAR (500 mg/kg) was administered intraperitoneally. In the vehicle-treated group (III), the same volume of distilled water was administered. AICAR was purchased from Topscience Corporation, Shanghai (Cat. No. T1477). Based on a previous study (13), twelve hours after surgery, the mice were sacrificed via an injection of 10% chloral hydrate to induce overdose anesthesia, and the cardiac tissue was collected for further evaluation.
Cell culture and treatment
H9C2 cardiomyocytes were purchased from Shanghai Zhong Qiao Xin Zhou Biotechnology. The cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, USA) and penicillin/streptomycin. The cell surface area was measured in four independent high-power fields using Image Pro Plus system (Carlsbad, CA, USA).
UCP2 siRNA (siUCP2) and negative control siRNA (siNC) were purchased from Invitrogen and transiently transfected into cells using Lipofectamine 2000 (Invitrogen, USA). 24 h after the cells were transfected with siUCP2 or siNC, protein was extracted and analyzed by Western blotting. Then, the H9C2 cells were subjected to 10 µg/mL lipopolysaccharide (LPS, Sigma, USA) with or without 1 mM of the AMPK agonist AICAR (Topscience Corporation, Shanghai) as indicated below. The cells were treated with LPS 18 h after the transfection, and protein was extracted and analyzed by Western blotting 6 h later. Images of the H9C2 cells were acquired under an inverted microscope (Olympus IX-61, Tokyo, Japan). The ultrastructural changes were observed by transmission electron microscopy (JEOL, Tokyo, Japan).
Histopathological examination
Formalin-fixed, paraffin-embedded tissue specimens were stained with H&E. The morphological evaluation was performed using light microscopy by a pathologist blinded to our experimental protocol. The histopathological evaluation was scored as follows: no damage, 0; focal edema or necrosis, 1; scattered swollen cells and necrosis, 2; confluent cell necrosis with neutrophil infiltration, 3; and massive areas of cell necrosis and neutrophil infiltration, 4 (15).
Myocardial tissue inflammation was evaluated according to the percentage of infiltration of inflammatory cells and scored as follows: no lesion, 0; <25%, 1; 25–50%, 2; 50–75%, 3; and >75%, 4 (16).
Measurement of protein expression
The samples were lysed using RIPA lysis buffer (Thermo Fisher Scientific, USA); then, the total protein was quantitated using the BCA method. Primary antibodies against UCP2 (1:1,000; ab97931, Abcam), pAMPKα [1:500; 2535, Cell Signaling Technology (CST)], AMPKα (1:1,000; 2532, CST), Beclin1 (1:500; ab62557, Abcam), LC3B (1:200; NB100-2220, Novus), pLKB1 (1:500; 3482, CST), ULK1 (1:500; ab203207, Abcam), TCS2 (1:200; 23402, CST), CAMKK2 (1:200; HPA017389, Sigma), and GAPDH (1:5,000; ab181602, Abcam) were used for the protein labeling. After washing with TBST 3 times, the membranes were incubated with an HRP-conjugated secondary antibody (Applygen Gene Technology Corp). The immunoreactions were detected using an ECL kit (Thermo Fisher Scientific, USA) and exposed using Kodak 500 film (Midwest Scientific, Valley Park, MO, USA).
Formalin-fixed, paraffin-embedded sections were stained by standard immunohistochemical procedures. Briefly, 5-µm-thick sections were blocked with avidin and biotin according to the manufacturer’s instructions. The sections were incubated with the primary antibody (anti-pAMPK rabbit polyclonal antibody, 1:50; ab23875, Abcam) overnight at 4 °C. Following washes, the sections were incubated with biotinylated IgG (Zsbio Commerce Store, Beijing) for 30 min at room temperature. The peroxidase reaction was visualized using diaminobenzidine (Sigma). The images were captured by IX80 microscopy (Olympus, Tokyo, Japan) and analyzed with Image Pro Plus system (Carlsbad, CA, USA).
Detection of myocardial mitochondrial injury
Myocardial mitochondrial injury was detected by assessing the mitochondrial membrane potential and ATP concentration. A JC-1 mitochondrial membrane potential assay kit (Cat. No. 10009172, Cayman, USA) was used to detect myocardial mitochondrial injury according to the manufacturer’s instructions. A colorimetric/fluorometric assay kit (Cat. No. MAK190, Sigma, UK) was used to quantify the ATP concentration. The mitochondrial size and number were measured based on their morphology and analyzed with Image Pro Plus system (Carlsbad, CA, USA).
Statistical analysis
All data of the continuous variables are shown as the mean ± standard deviation. Statistical significance was assessed by a one-way analysis of variance (ANOVA), followed by the least significant difference (LSD) test for the comparisons between any two groups. SPSS 18.0 software (IBM Corp.) was used for all statistical calculations. P values less than 0.05 were considered statistically significant.
Results
Myocardial damage to heart tissue and H9C2 cells
Before randomization into the different groups and experiment procedures, the sex, age in weeks and body weight of all mice did not significantly differ. The mice treated with the CLP procedure began to show clinical sign of sepsis, such as chills, generalized weakness and reduced movement, approximately 8–10 h following CLP. 12 h after the surgery, the mice were sacrificed, and the cardiac tissue was collected for further evaluation.
Figure 1A,B,C,D,E,F shows the pathological changes in the myocardial tissue of the model mice. Compared with the WT + sham group, the WT + CLP group displayed marked myocardial injury with myocardial edema and interstitial inflammatory cell infiltration (Figure 1B vs. Figure 1A, Figure 1F). The hematoxylin and eosin (H&E) staining revealed marked severe necrosis and myocardial fiber rupture in the UCP2-KO + CLP mice (Figure 1C). The AICAR treatment significantly alleviated the histopathological damage to the heart tissue (Figure 1D,E).
Figure 1.
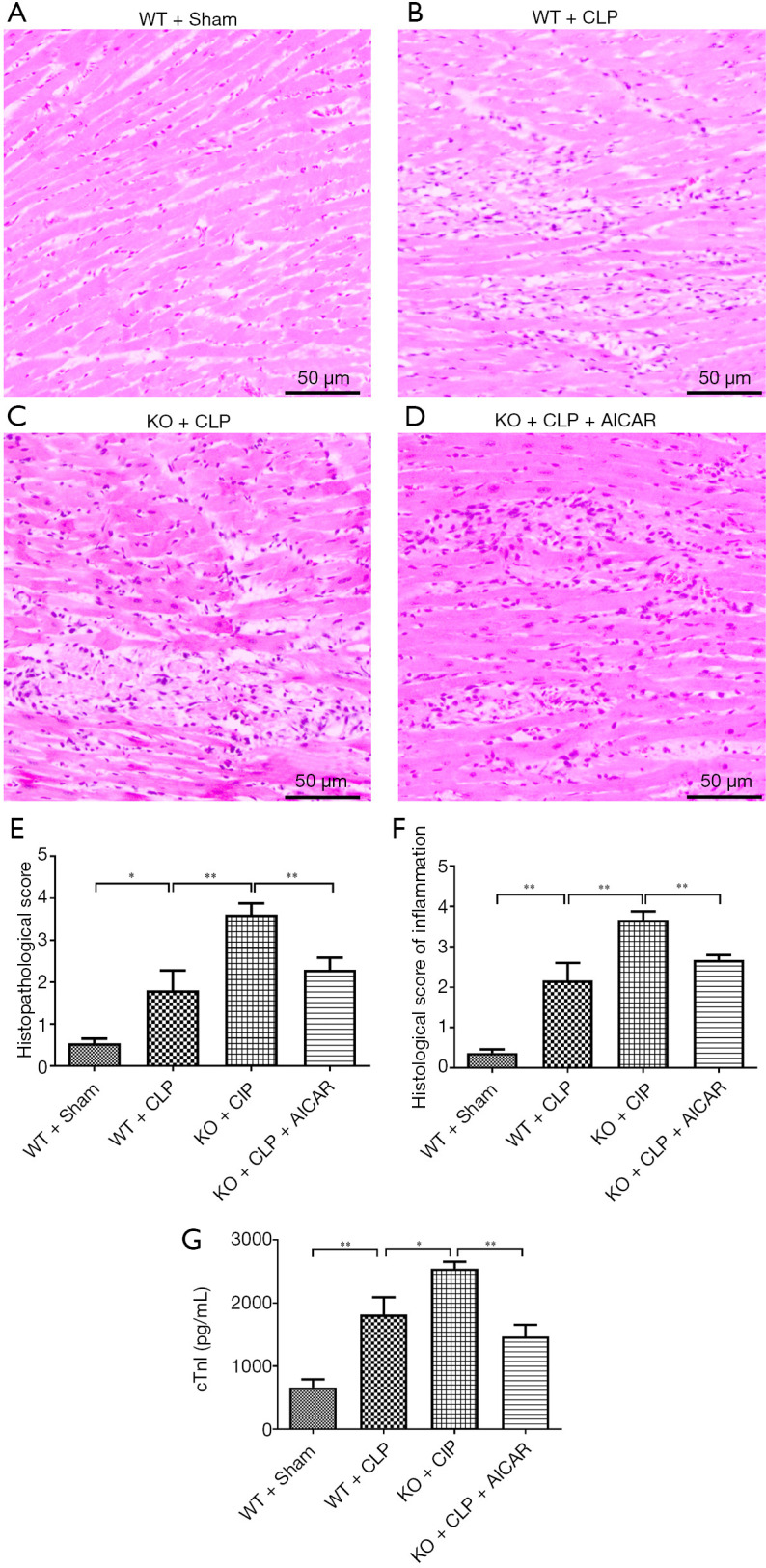
Myocardial damage to heart tissue. (A,B,C,D) H&E staining of heart tissues (×40); (E,F) histological evaluation of heart tissues; (G) cTnI levels were assessed in the mice following CLP and/or the AICAR treatment as described in the methods. *P<0.05, **P<0.01.
cTnI was quantified to assess myocardial damage after CLP. The WT + CLP group had higher cTnI levels than the WT group, and the cTnI level was increased even further in the UCP2-KO + CLP group (Figure 1G, P<0.05). The elevated cTnI level in the UCP2-KO + CLP + AICAR group was reduced after the AICAR treatment (Figure 1G, P<0.01).
Figure 2A,B,C,D,E shows the morphological alterations in the H9C2 cells in response to LPS exposure following the overexpression or silencing of UCP2 and treatment with AICAR. After the LPS exposure, the H9C2 cells became sparsely distributed and swollen and lost their original spindle shape relative to the control (non-treated) cells (Figure 2B vs. Figure 2A). The silencing of UCP2 enhanced the LPS-induced morphological alterations in the H9C2 cells (Figure 2C), but this enhancement was reduced by the AICAR treatment (Figure 2D,E).
Figure 2.

Myocardial damage to H9C2 cells. Morphological observation of H9C2 cells under phase-contrast microscopy (×200). *P<0.05.
UCP2 expression in heart tissue and myocardial cells
The UCP2 protein expression levels were detected in different groups of mice (Figure 3A,B). UCP2 protein expression was increased in the WT + CLP group. In the H9C2 cells, UCP2 protein expression was verified after overexpression or silencing (Figure 3C,D).
Figure 3.

UCP2 expression in heart tissue and myocardial cells. (A,B) UCP2 protein expression and representative image of immunoblotting of UCP2 in mouse heart tissues; (C,D) UCP2 protein expression and representative image of immunoblotting of UCP2 in cultured H9C2 cells subjected to various treatments. **P<0.01, ***P<0.001.
Mitochondrial injury in myocardial cells
Mitochondrial injury was analyzed by transmission electron microscopy, and the myocardial fibers and mitochondria were well ordered in the WT + sham group mice (Figure 4A). However, some myocardial fibers exhibited a scattered distribution and were disorganized, and the mitochondria were swollen in the WT + CLP group (Figure 4B). The above sepsis-induced morphological alterations were enhanced in the UCP2-KO + CLP group (Figure 4C) and alleviated by the AICAR treatment in the UCP2-KO + CLP + AICAR group (Figure 4D,E,F).
Figure 4.
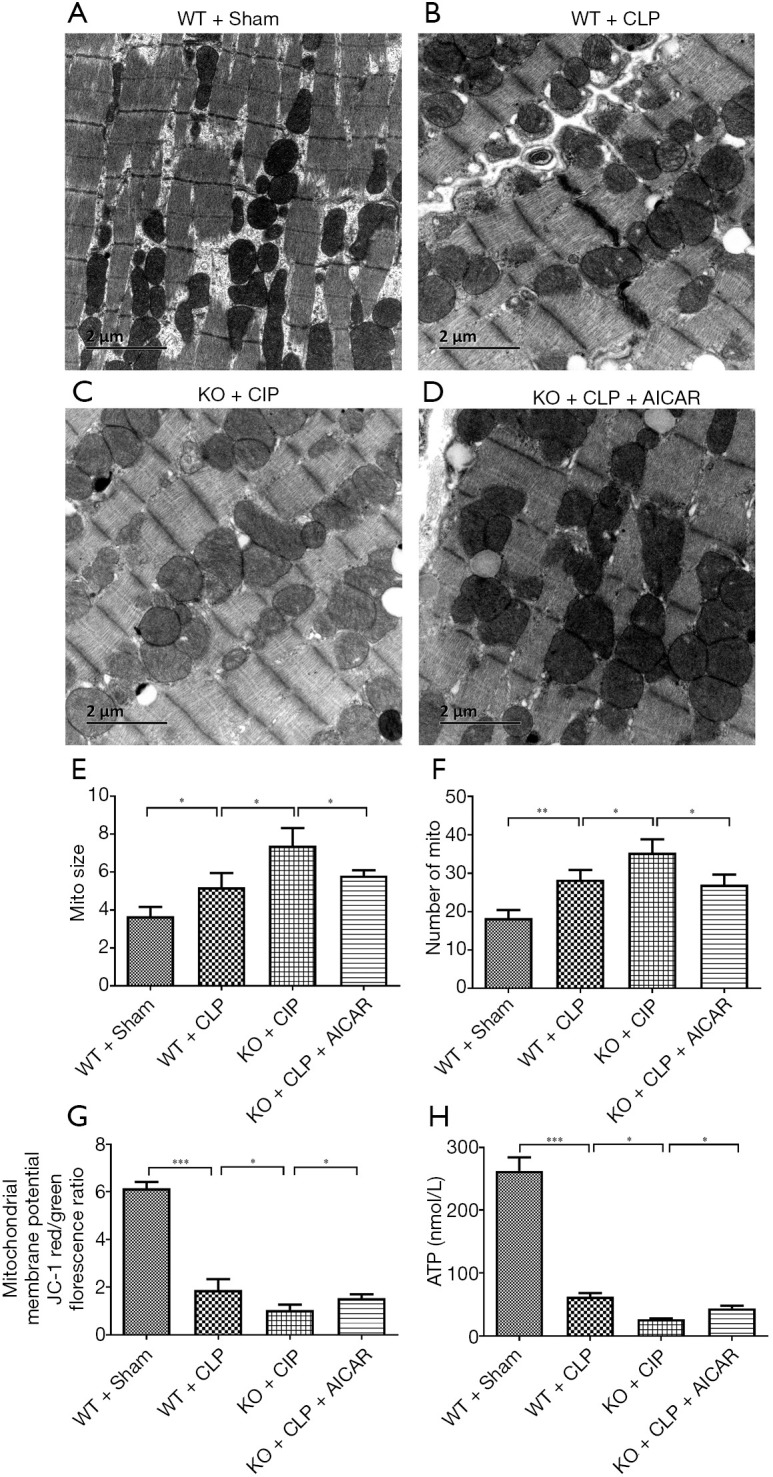
Mitochondrial injury in heart tissue. (A,B,C,D) Transmission electron microscopy observation of heart tissues (×200); (E,F) analysis of mitochondrial size and number; (G) effect on the mitochondrial membrane potential in mouse heart tissue; (H) effect on ATP synthesis in mouse heart tissue. *P<0.05; **P<0.01; ***P<0.001.
After CLP, the WT mice showed significantly decreased mitochondrial membrane potential compared with the control mice (P<0.05), which was further decreased in the UCP2-KO mice after CLP (UCP2-KO + CLP group, P<0.05) as shown in Figure 4G. However, AICAR increased the mitochondrial membrane potential in the UCP2-KO mice (UCP2-KO + CLP + AICAR group). Additionally, the ATP level was significantly reduced after CLP, and this decrease was further enhanced in the UCP2-KO group; however, the AICAR treatment significantly alleviated the CLP-induced reduction in ATP (Figure 4H, P<0.05).
The transmission electron microscopy observation of the H9C2 cells showed intact mitochondrial membranes with clear inner ridges that were neatly arranged (Figure 5A). The LPS exposure decreased the number of mitochondria and led to morphological changes with vacuolar structures, broken membranes, and ruptured or even vanished crests (Figure 5B). The cells with the UCP2 silencing exhibited further morphological and structural changes in the mitochondria (Figure 5C). In addition, the AICAR treatment protected the mitochondria in the LPS-treated cells from these subcellular structural alterations (Figure 5D).
Figure 5.

Mitochondrial injury in H9C2 cells (A,B,C,D): Transmission electron microscopy observation of heart tissues (×200).
Effect on proteins associated with autophagy
The expression of proteins associated with autophagy, including AMPK, beclin-1 and LC-3β, was assessed by immunoblotting. As shown in Figure 6A, after CLP, pAMPK/AMPK, beclin-1 and LC-3β were increased in the WT mice and decreased in the UCP2-KO CLP mice. The AICAR treatment promoted the expression of these autophagy-associated proteins. To further explore the expression of other proteins associated with autophagy, the levels of pmTOR, pULK1, pTSC2, beclin-1 and LC-3β were measured in cultured H9C2 cells. The LPS exposure significantly upregulated these proteins, except for the autophagy inhibitor pmTOR (Figure 6B). Silencing UCP2 inhibited the upregulation of pULK1, pTSC2, beclin-1 and LC-3β expression and promoted the upregulation of pmTOR, but the AICAR pretreatment reversed these effects on the regulation of these proteins (Figure 6B). Furthermore, the overexpression of UCP2 significantly increased the levels of pULK1, pTSC2, beclin-1 and LC-3β (Figure 6B).
Figure 6.

Effect on the expression of autophagy-associated proteins. (A) Expression of autophagy-associated proteins in mouse heart tissues; (B) expression of autophagy-associated proteins in cultured H9C2 cells. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Interaction between UCP2 and AMPK and the underlying mechanism
To investigate the interaction between UCP2 and AMPK, the expression level of AMPK in tissue samples from different groups was evaluated (Figure 7A-D). The phosphorylation of AMPK was significantly promoted after CLP, and the AMPK phosphorylation level was reduced in the UCP2-KO groups. The treatment with AICAR increased AMPK phosphorylation (Figure 7E). To explore the mechanism of the interaction between UCP2 and AMPK, the upstream kinases CAMKK2 and pLKB1 were analyzed. As illustrated in Figure 7F, the silencing or overexpression of UCP2 led to corresponding changes in the levels of CAMKK2 and pLKB1, which may result in AMPK activation.
Figure 7.

Interaction between UCP2 and AMPK and the underlying mechanism. (A,B,C,D) pAMPK expression in heart tissue; (E) AMPK phosphorylation levels measured in heart tissue; (F) effect on protein levels in cultured H9C2 cells treated with the indicated reagents. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Discussion
The principal findings in our study were as follows. First, the UCP2-KO mice showed more severe cardiomyocyte injury after CLP than the WT mice. AICAR, which is an agonist of AMPK, ameliorated the cardiomyocyte injury induced by CLP. The LPS exposure damaged the H9C2 cells (rat cardiomyocytes), and the cells with UCP2 silencing were more severely affected than the control cells, indicating that UCP2 can protect against sepsis. Second, the CLP- or LPS-induced mitochondrial injury and mitochondrial membrane potential loss were exacerbated by the UCP2 knockout. Moreover, both the septic animal and cell models exhibited increased levels of proteins associated with autophagy; the upregulation of autophagy-promoting proteins, such as pAMPK, pULK1, pTSC2, Beclin-1, and LC3-I/II, and the downregulation of the autophagy-inhibiting protein pmTOR were obviously limited by the UCP2 silencing. The AICAR treatment protected the cardiomyocytes, even those with UCP2 silencing, from CLP- or LPS-induced damage and attenuated the related mitochondrial injury and mitochondrial membrane potential loss. AICAR likely exerted these protective effects through AMPK activation, resulting in the promotion of autophagy. Finally, to explore the interaction between UCP2 and AMPK, we performed gain- and loss-of-function studies and found that the changes in the AMPK phosphorylation levels were correlated with UCP2 overexpression or silencing. UCP2 may regulate AMPK through its upstream kinases pLKB1 and CAMKK2. In summary, our findings reveal that UCP2 is essential for protection against septic cardiomyopathy and mitochondrial dysfunction and that these protective effects are mediated through the promotive effect of AMPK on autophagy. In addition, targeting the UCP2/AMPK axis may be a therapeutic approach to regulate mitochondrial dysfunction in septic cardiomyopathy.
Sepsis is a major public health concern worldwide (1). The function of the heart highly depends on abundant amounts of ATP, and studies have proven that mitochondrial dysfunction plays an important role in mediating organ damage in septic cardiomyopathy (2,4). UCP2, which is the most well-known isoform in the mitochondrial uncoupling protein family, is widely expressed in tissues and performs various of functions (9). Previous studies have shown that UCP2 can be upregulated in sepsis (8,11,13,17). A UCP2 knockout mouse model in vivo and cardiomyocytes transfected with siUCP2 in vitro were used to further investigate the effect of UCP2 on septic cardiomyopathy. Our studies show that UCP2 deficiency resulted in exacerbated morphologic cell damage, mitochondrial injury and mitochondrial membrane potential loss, indicating that UCP2 plays a protective role in cardiovascular disease during sepsis.
The loss of mitochondrial function is associated with different types of diseases (18). Intracellular quality control of mitochondria includes mitochondrial biogenesis, dynamic fusion and fission, and degradation. Mitochondrial degradation is executed via autophagy, which removes dysfunctional mitochondria (19,20). To date, several studies have explored UCP2 regulation of autophagy in nerve cells (21), cumulus cells (22), hepatocytes (23), and pancreatic cancer cells (24). Our previous study first confirmed that UCP2 participated in the regulation of the balance between autophagy and apoptosis in human cardiomyocytes during sepsis (13). However, the mechanism by which UCP2 regulates autophagy in septic cardiomyopathy is unknown.
Eukaryotes have evolved a very sophisticated system to sense low energy levels. AMPK is a key player in the elegant system that modulates metabolism (25). AMPK not only participates in energy switching, cell growth control, and other cellular processes, including lipid and glucose metabolism, but also is involved in various aspects of mitochondrial homeostasis, including mitophagy (26,27). The autophagy machinery comprises several multiprotein complexes that control every step, and AMPK regulates various aspects of this machinery. AMPK acts by activating ULK1, directly inactivating mTOR, or indirectly phosphorylating TSC2, all of which promote the formation of PI3K complex I to form autophagosomes. Subsequently, autophagosomes mature and fuse with lysosomes to induce the degradation of the cargo by lysosomal enzymes.
In our study, we explored the detailed changes in autophagy in both septic animal models and cell models. As a key regulator of autophagy, AMPK exhibited a significantly increased phosphorylation level but no visible change in expression. Moreover, the levels of autophagy-promoting proteins downstream of AMPK, such as pULK1 and pTSC2, were correspondingly increased, and the level of the autophagy-inhibiting protein pmTOR was decreased. Finally, the core components of the autophagosome complexes Beclin-1 and LC3-I/II were upregulated. However, with UCP2 silencing, the compensatory change in autophagy during sepsis was obviously limited, suggesting that UCP2 may contribute to cardiomyocyte activity through the regulation of the autophagy process. To further explore the mechanism by which UCP2 regulates autophagy, AICAR, which is an agonist of AMPK, was employed. As an agonist of the key regulator of autophagy, AICAR promoted the abovementioned autophagy progress even with UCP2 silencing. Moreover, AICAR protected against morphological damage to cardiomyocytes and attenuated the related mitochondrial injury and mitochondrial membrane potential loss. Therefore, we suggest that UCP2 may exert protective effects against septic cardiomyopathy through the regulation of autophagy possibly via AMPK.
Finally, we further explored the mechanism of the interaction between UCP2 and AMPK through gain- and loss-of-function studies. With UCP2 overexpression or silencing, the phosphorylation level of AMPK changed correspondingly, but AMPK expression did not noticeably change. Some previous studies identified a connection between UCP2 and AMPK in infantile hypertrophic cardiomyopathy (28) and hypertension (29). However, the specific mechanism is unclear. AMPK is a heterotrimeric complex comprising a catalytic subunit, α, and two regulatory subunits, β and γ. The α-subunit contains the kinase domain and a critical residue that is phosphorylated by an upstream kinase (25,30). One upstream kinase of AMPK is the tumor suppressor LKB1, which provides a direct connection between cancer and metabolism; LKB1 is the primary regulator of AMPK activation under energy stress in most mammalian tissues examined (31-33). AMPK can also be directly phosphorylated in response to calcium flux by the calcium-sensitive kinase CAMKK2 (also known as CAMKKβ), thus linking calcium signaling to the regulation of energy metabolism by AMPK (34,35). Our results show a similar change in pLKB1 and CAMKK2 following UCP2 overexpression or silencing. Therefore, we suggest that UCP2 may regulate AMPK through its upstream kinases pLKB1 and CAMKK2.
Conclusions
In conclusion, our study reveals that UCP2 plays a protective role in septic cardiomyopathy. Mechanistically, UCP2 may regulate mitochondrial function through AMPK-mediated promotion of autophagy, which contributes to maintaining cardiomyocyte activity. Targeting the UCP2/AMPK axis may be a therapeutic approach for the regulation of mitochondrial dysfunction in septic cardiomyopathy.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This work was supported by grants from the Beijing Municipal Natural Science Foundation Youth Program (No. 7194306).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. The experiments were performed under a project license (No. XHDW-2019002) granted by the Animal Care Ethics and Use Committee of Peking Union Medical College in compliance with the guidelines of the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Footnotes
Reporting Checklist: The authors completed the ARRIVE reporting checklist (available at http://dx.doi.org/10.21037/atm-20-4819).
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-20-4819
Conflicts of Interest: All authors completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-20-4819). JYM received support for the present research from the Beijing Municipal Natural Science Foundation of China. The other authors have no potential conflicts of interest to disclose.
References
- 1.Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016;315:801-10. 10.1001/jama.2016.0287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan P, Wang X, Liu D. The potential mechanism of mitochondrial dysfunction in septic cardiomyopathy. J Int Med Res 2018;46:2157-69. 10.1177/0300060518765896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rudiger A, Singer M. The heart in sepsis: from basic mechanisms to clinical management. Curr Vasc Pharmacol 2013;11:187-95. [PubMed] [Google Scholar]
- 4.Durand A, Duburcq T, Dekeyser T, et al. Involvement of Mitochondrial Disorders in Septic Cardiomyopathy. Oxid Med Cell Longev 2017;2017:4076348. 10.1155/2017/4076348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matkovich SJ, Al Khiami B, Efimov IR, et al. Widespread down-regulation of cardiac mitochondrial and sarcomeric genes in patients with sepsis. Crit Care Med 2017;45:407-14. 10.1097/CCM.0000000000002207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med 2013;369:840-51. 10.1056/NEJMra1208623 [DOI] [PubMed] [Google Scholar]
- 7.Ježek P, Holendova B, Garlid KD, et al. Mitochondrial uncoupling proteins: subtle regulators of cellular redox signaling. Antioxid Redox Signal 2018;29:667-714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singer M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014;5:66-72. 10.4161/viru.26907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donadelli M, Dando I, Fiorini C, et al. UCP2, a mitochondrial protein regulated at multiple levels. Cell Mol Life Sci 2014;71:1171-90. 10.1007/s00018-013-1407-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ruiz-Ramírez A, Lopez-Acosta O, Barrios-Maya MA, et al. Cell Death and Heart Failure in Obesity: Role of Uncoupling Proteins. Oxid Med Cell Longev 2016;2016:9340654. 10.1155/2016/9340654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Liu D, Chai W, et al. The Role of Uncoupling Protein 2 During Myocardial Dysfunction in a Canine Model of Endotoxin Shock. Shock 2015;43:292-7. 10.1097/SHK.0000000000000286 [DOI] [PubMed] [Google Scholar]
- 12.Huang W, Wang X, Zhang H, et al. The value of serum uncoupling protein-2 level for the patients with sepsis. Shock 2020;54:301-7. 10.1097/SHK.0000000000001523 [DOI] [PubMed] [Google Scholar]
- 13.Pan P, Zhang H, Su L, et al. Melatonin Balance the Autophagy and Apoptosis by Regulating UCP2 in the LPS-Induced Cardiomyopathy. Molecules 2018;23:675. 10.3390/molecules23030675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rittirsch D, Huber-Lang MS, Flierl MA, et al. Immunodesign of experimental sepsis by cecal ligation and puncture. Nat Protoc 2009;4:31-6. 10.1038/nprot.2008.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zingarelli B, Salzman AL, Szabó C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ Res 1998;83:85-94. 10.1161/01.RES.83.1.85 [DOI] [PubMed] [Google Scholar]
- 16.Rezkalla S, Kloner RA, Khatib G, et al. Beneficial effects of captopril in acute coxsackievirus B3 murine myocarditis. Circulation 1990;81:1039-46. 10.1161/01.CIR.81.3.1039 [DOI] [PubMed] [Google Scholar]
- 17.Jiang ZM, Yang QH, Zhu CQ. UCP2 in early diagnosis and prognosis of sepsis. Eur Rev Med Pharmacol Sci 2017;21:549-53. [PubMed] [Google Scholar]
- 18.Kiriyama Y, Nochi H. Intra- and Intercellular Quality Control Mechanisms of Mitochondria. Cells 2017;7:1. 10.3390/cells7010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 2018;19:93-108. 10.1038/nrm.2017.129 [DOI] [PubMed] [Google Scholar]
- 20.Wu Y, Yao YM, Lu ZQ. Mitochondrial quality control mechanisms as potential therapeutic targets in sepsis-induced multiple organ failure. J Mol Med (Berl) 2019;97:451-62. 10.1007/s00109-019-01756-2 [DOI] [PubMed] [Google Scholar]
- 21.Marques-Aleixo I, Santos-Alves E, Balca MM, et al. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 2015;301:480-95. 10.1016/j.neuroscience.2015.06.027 [DOI] [PubMed] [Google Scholar]
- 22.Ge H, Zhang F, Duan P, et al. Mitochondrial Uncoupling Protein 2 in human cumulus cells is associated with regulating autophagy and apoptosis, maintaining gap junction integrity and progesterone synthesis. Mol Cell Endocrinol 2017;443:128-37. 10.1016/j.mce.2017.01.020 [DOI] [PubMed] [Google Scholar]
- 23.Lou J, Wang Y, Wang X, et al. Uncoupling protein 2 regulates palmitic acid-induced hepatoma cell autophagy. Biomed Res Int 2014;2014:810401. 10.1155/2014/810401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dando I, Pacchiana R, Pozza ED, et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic Biol Med 2017;113:176-89. 10.1016/j.freeradbiomed.2017.09.022 [DOI] [PubMed] [Google Scholar]
- 25.Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 2018;19:121-35. 10.1038/nrm.2017.95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011;331:456-61. 10.1126/science.1196371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toyama EQ, Herzig S, Courchet J, et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016;351:275-81. 10.1126/science.aab4138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boutoual R, Meseguer S, Villarroya M, et al. Defects in the mitochondrial-tRNA modification enzymes MTO1 and GTPBP3 promote different metabolic reprogramming through a HIF-PPARgamma-UCP2-AMPK axis. Sci Rep 2018;8:1163. 10.1038/s41598-018-19587-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rubattu S, Bianchi F, Busceti CL, et al. Differential modulation of AMPK/PPARalpha/UCP2 axis in relation to hypertension and aging in the brain, kidneys and heart of two closely related spontaneously hypertensive rat strains. Oncotarget 2015;6:18800-18. 10.18632/oncotarget.4033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ross FA, MacKintosh C, Hardie DG. AMP-activated protein kinase: a cellular energy sensor that comes in 12 flavours. FEBS J 2016;283:2987-3001. 10.1111/febs.13698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ikeda Y, Sato K, Pimentel DR, et al. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J Biol Chem 2009;284:35839-49. 10.1074/jbc.M109.057273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jessen N, Koh HJ, Folmes CD, et al. Ablation of LKB1 in the heart leads to energy deprivation and impaired cardiac function. Biochim Biophys Acta 2010;1802:593-600. 10.1016/j.bbadis.2010.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ollila S, Makela TP. The tumor suppressor kinase LKB1: lessons from mouse models. J Mol Cell Biol 2011;3:330-40. 10.1093/jmcb/mjr016 [DOI] [PubMed] [Google Scholar]
- 34.Mungai PT, Waypa GB, Jairaman A, et al. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol 2011;31:3531-45. 10.1128/MCB.05124-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab 2005;2:21-33. 10.1016/j.cmet.2005.06.005 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as