

Abstract
Background
Atherosclerosis leads to the occurrence of cardiovascular diseases. However, the molecular mechanisms that contribute to atherosclerotic plaque rupture are incompletely characterized. We aimed to identify the genes related to atherosclerotic plaque progression that could serve as novel biomarkers and interventional targets for plaque progression.
Methods
The datasets of GSE28829 in early vs. advanced atherosclerotic plaques and those of GSE41571 in stable vs. ruptured plaques from Gene Expression Omnibus (GEO) were analyzed by using bioinformatics methods. In addition, we used quantitative reverse transcription polymerase chain reaction (qRT-PCR) to verify the expression level of core genes in a mouse atherosclerosis model.
Results
There were 29 common differentially expressed genes (DEGs) between the GSE28829 and GSE41571 datasets, and the DEGs were mainly enriched in the chemokine signaling pathway and the Staphylococcus aureus infection pathway (P<0.05). We identified 6 core genes (FPR3, CCL18, MS4A4A, CXCR4, CXCL2, and C1QB) in the protein-protein interaction (PPI) network, 3 of which (CXCR4, CXCL2, and CCL18) were markedly enriched in the chemokine signaling pathway. qRT-PCR analysis showed that the messenger RNA levels of two core genes (CXCR4 and CXCL2) increased significantly during plaque progression in the mouse atherosclerosis model.
Conclusions
In summary, bioinformatics techniques proved useful for the screening and identification of novel biomarkers of disease. A total of 29 DEGs and 6 core genes were linked to atherosclerotic plaque progression, in particular the CXCR4 and CXCL2 genes.
Keywords: Atherosclerosis, molecular mechanism, bioinformatics, core genes, differentially expressed genes (DEGs)
Introduction
Atherosclerosis, defined as the formation of fibrofatty lesions in the artery wall, causes considerable morbidity and mortality worldwide (1). Typically, atherosclerotic plaque have superficial fibrous cap composed of smooth muscle cells and relatively dense collagen. Beneath and side of cap shows more cellular area containing macrophages, T cells and smooth muscle cells. Deep to the fibrous cap is a necrotic core containing lipid debris from dead cells, foam cells fibrin, variably organized thrombus and other plasma proteins (1). Atherosclerotic plaque formation, progression, and rupture lead to later symptomatic disease such as myocardial infarction, stroke, disabling peripheral artery disease, and eventually death (2). Atherosclerosis seriously reduces people’s quality of life and life expectancy, and greatly increases health costs (3). Therefore, it is urgent to study the pathogenesis of atherosclerosis in order to find suitable targets for the prevention and treatment of this condition.
Currently, there is a lack of understanding regarding the molecular pathways of atherosclerotic plaque rupture (4). However, high-throughput gene profiling has become an effective tool for revealing the pathogenesis of cardiovascular diseases. These transcriptomics research results will provide new directions for future mechanism research of cardiovascular diseases. Using this method, researchers have already discovered several genes and pathways that are closely related to coronary artery disease and atherosclerosis (5,6). Most of these studies have focused on comparing the differential gene expression between atherosclerotic plaques and normal tissues (7-11). Fewer studies exist, however which compare the differential gene expression between early and advanced plaques, or between stable and ruptured plaques (4,12,13). In this study, we identified common differentially expressed genes (DEGs) in early and advanced plaques (GSE28829), and in stable and ruptured plaques (GSE41571), and used bioinformatics methods to determine the key pathways and core genes involved in atherosclerotic plaque progression. It is hoped our findings will provide new potential targets for the prevention, diagnosis, and treatment of atherosclerosis. We present the following article in accordance with the MDAR reporting checklist (available at http://dx.doi.org/10.21037/atm-21-193).
Methods
Microarray data information
The microarray data of the GSE28829 dataset for early and advanced plaques and the GSE41571 dataset for stable and ruptured atherosclerotic plaques, were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (NCBI-GEO) public database. The GSE28829 and GSE41571 datasets were both on the GPL570 platforms (HG-U133_Plus_2) which included 13 early plaque cases and 16 advanced plaque cases, and 6 stable plaque cases, and 5 ruptured plaque cases, respectively. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Data processing of DEGs
DEGs were identified via the GEO2R interactive web tool with |log FC| >1.5 and adjusted P value <0.05 (14). We then used Venn diagram software to check the raw data in TXT format in order to identify the common DEGs between the two sets of microarray data (14). The DEGs with log FC <0 and log FC >0 were considered downregulated and upregulated genes, respectively.
Gene ontology (GO) and pathway analysis
GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were used to study the biological characteristics of these DEGs (15). GO and KEGG pathway analyses were performed by DAVID online software (https://david.ncifcrf.gov/), which can visualize the DEG enrichment of molecular function (MF), biological process (BP), cellular component (CC), and KEGG pathways (15). A P value <0.05 in the results was considered statistically significant (15).
Protein-protein interaction (PPI) network and core gene identification
A PPI helped to clarify the regulatory functions among proteins, which were evaluated using the Search Tool for the Retrieval of Interacting Genes (STRING) (14,16). We uploaded the common DEGs obtained from the GSE28829 and GSE41571 datasets onto STRING’s website to construct a PPI network, and then used Cytoscape software (version 3.2.1) to obtain a visualized interactive network. Finally, we used Molecular Complex Detection (MCODE) application in Cytoscape to screen the core genes (16).
Animal model
Male wild-type (WT) and atherosclerosis-prone apolipoprotein E-deficient Ap(oE−/−) mice (6 weeks old, 20±2 g) with a C57BL/6J genetic background were obtained from Xipuer-Bikai Experimental Animal Co., Ltd. (Shanghai, China). The WT mice were given a normal diet and used as controls, and the ApoE−/− mice were given a Western diet (high-fat diet of 21% butter fat and 0.15% cholesterol) for 20 weeks and used as atherosclerosis models. All mice were fed in the Zhejiang University Animal Center. After the mice were euthanized, the aortas were excised and processed for histology. All animal experiments were approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine (Hangzhou, China). All animal experiments were performed in accordance with Zhejiang university guidelines for the care and use of animals.
Verification by quantitative reverse transcription quantitative polymerase chain reaction (qRT-PCR)
The extent of atherosclerosis was assessed in mice aortas by staining for lipid depositions with Oil Red O (12). The mouse aorta samples were divided into three groups: normal aortas from controls, atherosclerotic artery segments with early lesions (pathological intimal thickening and intimal xanthoma) from ApoE−/− mice, and atherosclerotic artery segments with advanced lesions (thin or thick fibrous cap atheroma) from ApoE−/− mice (12). Tri®-Reagent (Sigma-Aldrich, St. Louis, MO, USA) was used to extract total RNA from the aortic samples and PrimeScript RT reagent Kit (TaKaRa Biotechnology, Dalian, China) was used to reverse transcribe total RNA into complementary DNA (cDNA). The polymerase chain reaction (PCR) reaction was performed using SYBR Green (Thermo Fisher Scientific, USA) and specific primer pairs (sequences in Table 1). Amplification was performed in duplicate using a 7900 HT Fast Real-Time PCR system (Applied Biosystems, Thermo Fisher Scientific, USA). The experiment was repeated three times for each gene, and GAPDH was used as the endogenous reference gene.
Table 1. Primer sequences for qRT-PCR.
| Targets | Forward primer (5’-3’) | Reverse primer (3’-5’) |
|---|---|---|
| CXCR4 | GAGGCCAAGGAAACTGCTG | GCGGTCACAGATGTACCTGTC |
| CXCL2 | CTCAGACAGCGAGGCACATC | CCTCAACGGAAGAACCAAAGAG |
| GAPDH | CACCATCTTCCAGGAGCGAG | CCTTCTCCATGGTGGTGAAGAC |
qRT-PCR, quantitative reverse transcription quantitative polymerase chain reaction.
Statistical analysis
The 2−ΔCT method was used to determine the expression level of all messenger RNAs (mRNAs) (17). One-way analysis of variance was used for comparisons among three groups, and a P value <0.05 was considered statistically significant. All statistical analyses were conducted using SPSS 25.0 software (IBM Corp., Armonk, NY, USA).
Results
Identification of DEGs
There were 96 DEGs in advanced atherosclerotic plaque compared with early plaque, including 92 upregulated genes and 4 downregulated genes. In addition, there were 838 DEGs in ruptured atherosclerotic plaque compared with stable plaque, including 173 upregulated genes and 665 downregulated genes. Finally, there were 29 DEGs overlapped between GSE28829 and GSE41571 in the Venn diagram, which were all upregulated genes (log FC >0) (Table 2 and Figure 1).
Table 2. A total of 29 commonly differentially expressed genes (DEGs) identified from the GSE28829 and GSE41571 datasets.
| DEGs | Gene symbol |
|---|---|
| Upregulated | IGKV1OR2-108, CXCR4, CD37, CD36, IGHM, IGLC1, CD52, CYAT1, AQP9, CCL18, IGLJ3, FPR3, IGHV3-23, NPL, IGLL3P, RAC2, C1QB, IGKC, MS4A4A, HLA-DQB1, JCHAIN, MIR8071-2, CXCL2, IGK, SLC16A10, IGLV1-44, MLIP, IGHA2, CTSC |
| Downregulated | – |
Figure 1.
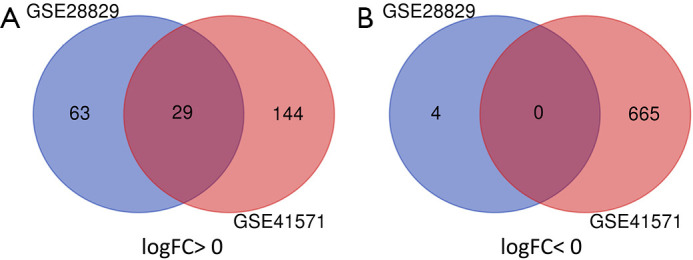
A total of 29 common DEGs in the two datasets (GSE28829 and GSE41571) were identified through Venn diagram software. (A) A total of 29 DEGs were upregulated (log FC >0), and (B) 0 DEGs were downregulated (log FC <0). DEGs, differentially expressed genes.
DEGs GO and KEGG pathway analyses
GO and KEGG pathway analyses of 29 DEGs were performed by DAVID software. GO analysis showed the following: (I) for BP, DEGs were particularly enriched in regulation of immune response, complement activation, receptor-mediated endocytosis, innate immune response, phagocytosis, and proteolysis; (II) for CC, DEGs were significantly enriched in the plasma membrane, extracellular exosome, extracellular space, extracellular region, and blood microparticle; and (III) for MF, DEGs were enriched in antigen binding, immunoglobulin receptor binding, serine-type endopeptidase activity, peptidoglycan binding, and phosphatidylcholine binding (Table 3).
Table 3. Gene ontology analysis results of differentially expressed genes during the progression of atherosclerotic plaques.
| Expression | Category | Term | Count | % | P value | FDR |
|---|---|---|---|---|---|---|
| Upregulated | GOTERM_BP_DIRECT | GO: 0006955~immune response | 12 | 57.7 | 2.96E-13 | 3.66E-10 |
| GOTERM_BP_DIRECT | GO: 0006958~complement activation, classical pathway | 7 | 33.6 | 1.31E-09 | 1.61E-06 | |
| GOTERM_BP_DIRECT | GO: 0006898~receptor-mediated endocytosis | 7 | 33.6 | 5.80E-08 | 7.16E-05 | |
| GOTERM_BP_DIRECT | GO: 0045087~innate immune response | 7 | 33.6 | 7.78E-06 | 0.009601 | |
| GOTERM_BP_DIRECT | GO: 0006910~phagocytosis, recognition | 6 | 28.8 | 1.35E-10 | 1.66E-07 | |
| GOTERM_BP_DIRECT | GO: 0006508~proteolysis | 6 | 28.8 | 2.45E-04 | 0.302523 | |
| GOTERM_CC_DIRECT | GO: 0005886~plasma membrane | 12 | 57.7 | 0.004436 | 4.37306 | |
| GOTERM_CC_DIRECT | GO: 0070062~extracellular exosome | 11 | 52.9 | 8.10E-04 | 0.812025 | |
| GOTERM_CC_DIRECT | GO: 0005615~extracellular space | 10 | 48.1 | 1.32E-05 | 0.013256 | |
| GOTERM_CC_DIRECT | GO: 0005576~extracellular region | 8 | 38.4 | 0.002154 | 2.144901 | |
| GOTERM_CC_DIRECT | GO: 0072562~blood microparticle | 7 | 33.6 | 2.04E-08 | 2.05E-05 | |
| GOTERM_MF_DIRECT | GO: 0003823~antigen binding | 7 | 33.6 | 1.13E-09 | 1.15E-06 | |
| GOTERM_MF_DIRECT | GO: 0034987~immunoglobulin receptor binding | 6 | 28.8 | 6.60E-11 | 6.69E-08 | |
| GOTERM_MF_DIRECT | GO: 0004252~serine-type endopeptidase activity | 6 | 28.8 | 7.40E-06 | 0.007488 | |
| GOTERM_MF_DIRECT | GO: 0042834~peptidoglycan binding | 2 | 9.6 | 0.013427 | 12.79255 | |
| GOTERM_MF_DIRECT | GO: 0031210~phosphatidylcholine binding | 2 | 9.6 | 0.025586 | 23.08273 | |
| Downregulated | – | – | – | – | – | – |
The results of KEGG analysis showed that 29 DEGs were particularly enriched in the chemokine signaling pathway and the staphylococcus aureus infection pathway (P<0.05) (Table 4). Four genes (RAC2, CXCR4, CXCL2, and CCL18) were markedly enriched in the chemokine signaling pathway (Table 4 and Figure 2).
Table 4. KEGG pathway analysis results of differentially expressed genes during the progression of atherosclerotic plaques.
| Pathway ID | Name | Count | % | P value | Genes |
|---|---|---|---|---|---|
| hsa04062 | Chemokine signaling pathway | 4 | 19.2 | 0.003574 | RAC2, CXCR4, CXCL2, CCL18 |
| hsa05150 | Staphylococcus aureus infection | 3 | 14.4 | 0.003796 | HLA-DQB1, C1QB, FPR3 |
Figure 2.

Four genes (RAC2, CXCR4, CXCL2, and CCL18) were significantly enriched in the chemokine signaling pathway. RAC means RAC2; Chemokine means CXCL2 and CCL18; Chemokine R means CXCR4.
PPI network and modular analysis
After the pathway analyses, 18 of the 29 DEGs were imported into the PPI network complex, and the other 11 DEGs were not contained in the PPI network complex (Figure 3A). We then used Cytoscape MCODE to screen the core genes, and identified 6 hub genes (FPR3, CCL18, MS4A4A, CXCR4, CXCL2, and C1QB) among 18 nodes (Figure 3B). Three of the six core genes (CXCR4, CXCL2, and CCL18) were markedly enriched in the chemokine signaling pathway (Table 4).
Figure 3.

The PPI network of common DEGs was constructed by the STRING online database and module analysis. (A) A total of 18 DEGs were included in the DEG PPI network; (B) core genes in the PPI network. PPI, protein-protein interaction; DEGs, differentially expressed genes.
Corroboration of two core genes using qRT-PCR
Mouse aortic samples were divided into three groups: normal aortas, early atherosclerotic plaque, and advanced atherosclerotic plaque. According to the PPI network analysis, the three core genes markedly enriched in the chemokine signaling pathway were CXCR4, CXCL2, and CCL18. qRT-PCR was performed in mouse aortic samples to confirm the expression levels of the three core genes. The results showed that the expression of two core genes (CXCR4 and CXCL2) increased significantly during the progression of atherosclerotic plaques (Figure 4). Because no homologs of CCL18 have been found in rodents (18), CCL18 was not verified by qRT-PCR.
Figure 4.

Corroboration of two core genes using qRT-PCR. (A) Oil Red O-stained mice aortas; (B) Relative expression of the CXCR4 measured by qRT-PCR; (C) Relative expression of the CXCL2 measured by qRT-PCR. qRT-PCR, quantitative reverse transcription quantitative polymerase chain reaction.
Discussion
Although great progress has been made in the prevention, diagnosis, and treatment of cardiovascular disease, it is still the leading cause of death worldwide (19). The main cause of cardiovascular disease is atherosclerosis, which is itself a chronic inflammatory disease (19). The molecular mechanisms of atherosclerotic plaque formation and development are very complex. Although some studies have found that cytokines participate in all stages of atherosclerosis, the detailed pathophysiology of the atherosclerosis is still unclear (19,20). In recent decades, the rapid development of high-throughput detection technology and the progress of bioinformatics technology have brought new methods into the study of pathophysiological mechanism of atherosclerosis.
In this study, the GSE28829 dataset that was related to early vs. advanced atherosclerotic plaques and the GSE41571 that was dataset related to stable vs. ruptured plaques from GEO were analyzed by using bioinformatics methods. We found that there were 29 common DEGs between GSE28829 and GSE41571 and that the DEGs were mainly enriched in the chemokine signaling pathway and the Staphylococcus aureus infection pathway (P<0.05). Our research suggests that the two pathways play important roles in the progression of atherosclerotic plaque and deserve further study. To identify the core genes involved in the progression of atherosclerotic plaques, we constructed a DEG PPI network and predicted the key modules by using the STRING online database and Cytoscape software. We found that FPR3, CCL18, MS4A4A, CXCR4, CXCL2, and C1QB were the top six core genes in the progression of atherosclerotic plaques. Three of the six core genes (CXCR4, CXCL2, and CCL18) were markedly enriched in the chemokine signaling pathway. Finally, we confirmed that the mRNA levels of two core genes (CXCR4 and CXCL2) were also upregulated during the progression of atherosclerotic plaques in the mouse model of atherosclerosis. These hub genes are discussed below.
Chemokines are cytokines with a molecular weight of 8–12 kDa, and mediate cell chemotaxis and stagnation by binding to their respective receptors on the cell surface (21). The C-X-C motif chemokine receptor 4 (CXCR4) belongs to a cytokine receptor and participates in a variety of biological processes, such as HIV virus invasion, the occurrence and development of autoimmune diseases, etc. (22). The ligand of CXCR4 is C-X-C motif chemokine ligand 12 (CXCL12). Together with CXCL12, CXCR4 plays a pivotal role in atherosclerosis and arterial injury (23). The expression of CXCR4 in leukocytes is closely related to the vulnerability of atherosclerosis plaque (23). Naruko et al. also reported that the proportion of CXCR4-positive cells gradually increased with the progression of atherosclerotic plaque in patients with acute coronary syndromes (24). Additionally, Hyafil et al. recently reported that CXCR4-positive macrophages also accumulated in tissue samples of human carotid plaques (22). CXCR4 was also found to be expressed in both smooth muscle cell progenitors and endothelial progenitor cells in atherosclerotic plaque progression (21). Arterial injury can induce the expression of CXCR4 in smooth muscle cells, trigger CXCR4 signaling and contribute to intimal hyperplasia (23). Our results showed increased expression of CXCR4 during the progression of atherosclerotic plaques at the mRNA level. These findings align with previous data (21-24), but the exact mechanism of the CXCR4/CXCL12 axis in atherosclerotic plaque progression is not clear and needs further study.
Chemokines induce the aggregation of inflammatory cells to the inflammatory site, which is the main factor leading to vascular inflammatory injury (25). Some chemokines, including C-C motif chemokine ligand 2 (CCL2), CCL4, CCL5, C-X-C motif chemokine ligand 10 (CXCL10), CXCL1, and CXCL16, have been identified to play an important role during the progression of atherosclerotic plaque (26-30). Inflammation within the atherosclerotic plaque is mediated by their recruitment of target inflammatory cells (25). CXCL1 and CXCL2, the common ligands of CXCR2, are the major leukocyte chemoattractants and activators (31,32). CXCL1 is a chemoattractant of neutrophils, T lymphocytes, and monocytes and is expressed in human atherosclerotic plaques (33,34). It can induce the production of free radicals and lead to the injury of vascular endothelial cells (34). CXCL2, secreted by activated monocytes, has the same activity as CXCL1 (25). It attracts neutrophil aggregation during the inflammatory process, but its role in atherosclerosis is not clear (35,36). High CXCL2 expression has been reported in patients with rheumatoid arthritis, in whom neutrophils predominate in the joints (37). Rouault et al. reported that CXCL2 contributes to the adhesion of neutrophils to endothelial cells in vitro which can then the promote inflammation and senescence of endothelial cells in turn (38). Our results showed that CXCL2 is upregulated at the mRNA level during the progression of atherosclerotic plaques, which has not been reported in previous studies. Further study of CXCL2 will help us to understand the pathophysiological mechanism of atherosclerosis and facilitate its utilization as a biomarker and intervention target for atherosclerosis.
C-C motif chemokine ligand 18 (CCL18) is also a chemokine, secreted by type 2 macrophages (39). It is a chemotactic factor for T cells and serves as a marker of anti-inflammatory activation (18). The accumulation of macrophages in the arterial intima is an important factor in promoting the progression of atherosclerosis. CCL18 may participate in human atherosclerotic plaque formation (40). CCL18 was found to be positively expressed in human atherosclerotic plaques, and its mRNA was limited to the macrophage-rich region (41). Chen et al. reported that the expression levels of CCL18 in ruptured plaques were significantly higher than that in asymptomatic stable plaques (18).
Formyl peptide receptors (FPRs) are G protein–coupled receptors and are mainly expressed in white blood cells (42). Details regarding FPR signaling are scarce (43). Three FPRs have been identified in humans, FPR1–FPR3, and putative counterparts in mice (43). FPR1 and FPR2 are positively expressed on the surfaces of leukocytes, and are closely related to the control of inflammation induction (44). However, the overall function of FPR3 remains unclear, and whether or not it is related to inflammation has not been reported (42).
The membrane spanning 4-domains A (MS4A) family of proteins includes well-known members, MS4A1-MS4A13 (45). These proteins are expressed in certain leukocyte subsets and play a key role in regulating cell activation, growth, and development (45). MS4A4A is also a member of the MS4A family and is selectively expressed by macrophage-lineage cells (46). However, our understanding of its biological roles is rather limited. MS4A4A has not been previously studied in atherosclerosis. Mattiola et al. reported that MS4A4A was induced during the differentiation of monocytes into macrophages (46), while genome-wide association studies have correlated MS4A4A with Alzheimer’s disease and cutaneous systemic sclerosis (47,48).
Finally, complement C1q B chain (C1QB) is part of the complement system, and plays an important role in the regulation of immune response (49). The activation of the complement system can lead to endothelial dysfunction, which is considered to be an important mechanism for the formation and development of atherosclerosis (50). Tian et al. reported that C1QB in particular is associated with inflammatory response (51), and Bos et al. have reported that it is significantly associated with the stage of coronary artery disease (49).
The bioinformatics analysis in our study showed that the high expression of FPR3, CCL18, MS4A4A, CXCR4, CXCL2, and C1QB had appreciable value in distinguishing plaques and even identifying advanced and ruptured stages. However, this study had several limitations. First, due to the heterogeneity of atherosclerosis and the relatively small sample size of this study, the interpretation of the findings needs should be conducted with caution. Second, this study was only a preliminary screening of core genes during the progression of atherosclerosis, and the specific pathophysiological mechanism of these core genes in the progression of atherosclerosis requires further study. In the future, we must further verify these results in a larger number of samples, and clarify the role of these core genes in atherosclerosis through in vitro and in vivo studies.
Conclusions
This study preliminarily screened and identified the key pathways and genes closely related to the progression of atherosclerotic plaques. We speculated that these core genes (FPR3, CCL18, MS4A4A, CXCR4, CXCL2, and C1QB) may play an important role during the progression of atherosclerotic plaques, influencing atherosclerosis progression through chemokine signaling pathway and Staphylococcus aureus infection pathway. These findings may provide needed insights into the pathophysiological mechanism of atherosclerosis, and offer new potential targets for the prevention, diagnosis, and treatment of atherosclerosis in the future.
Supplementary
The article’s supplementary files as
Acknowledgments
Funding: This research was funded by the Natural Science Foundation of Zhejiang Province, China (No. LQ18H180002) and the National Natural Science Foundation of China (No. 31700690).
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. This study was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine (Hangzhou, China). All animal experiments were performed in accordance with Zhejiang university guidelines for the care and use of animals. The study was conducted in accordance with the Declaration of Helsinki (as revised in 2013).
Reporting Checklist: The authors have completed the MDAR reporting checklist. Available at http://dx.doi.org/10.21037/atm-21-193
Data Sharing Statement: Available at http://dx.doi.org/10.21037/atm-21-193
Conflicts of Interest: All authors have completed the ICMJE uniform disclosure form (available at http://dx.doi.org/10.21037/atm-21-193). The authors have no conflicts of interest to declare.
(English Language Editor: J. Gray)
References
- 1.Libby P, Buring JE, Badimon L, et al. Atherosclerosis. Nat Rev Dis Primers 2019;5:56. 10.1038/s41572-019-0106-z [DOI] [PubMed] [Google Scholar]
- 2.Huang L, Chambliss KL, Gao X, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019;569:565-9. 10.1038/s41586-019-1140-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tzoulaki I, Castagne R, Boulange CL, et al. Serum metabolic signatures of coronary and carotid atherosclerosis and subsequent cardiovascular disease. Eur Heart J 2019;40:2883-96. 10.1093/eurheartj/ehz235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee K, Santibanez-Koref M, Polvikoski T, et al. Increased expression of fatty acid binding protein 4 and leptin in resident macrophages characterises atherosclerotic plaque rupture. Atherosclerosis 2013;226:74-81. 10.1016/j.atherosclerosis.2012.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puca AA, Carrizzo A, Spinelli C, et al. Single systemic transfer of a human gene associated with exceptional longevity halts the progression of atherosclerosis and inflammation in ApoE knockout mice through a CXCR4-mediated mechanism. Eur Heart J 2020;41:2487-97. 10.1093/eurheartj/ehz459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aravani D, Morris GE, Jones PD, et al. HHIPL1, a Gene at the 14q32 Coronary Artery Disease Locus, Positively Regulates Hedgehog Signaling and Promotes Atherosclerosis. Circulation 2019;140:500-13. 10.1161/CIRCULATIONAHA.119.041059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biros E, Gabel G, Moran CS, et al. Differential gene expression in human abdominal aortic aneurysm and aortic occlusive disease. Oncotarget 2015;6:12984-96. 10.18632/oncotarget.3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ayari H, Bricca G: Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J Biosci 2013;38:311-5. 10.1007/s12038-013-9310-2 [DOI] [PubMed] [Google Scholar]
- 9.Huang CC, Lloyd-Jones DM, Guo X, et al. Gene expression variation between African Americans and whites is associated with coronary artery calcification: the multiethnic study of atherosclerosis. Physiol Genomics 2011;43:836-43. 10.1152/physiolgenomics.00243.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong HS, Kim JY, Lee SH, et al. Synergy of circulating miR-212 with markers for cardiovascular risks to enhance estimation of atherosclerosis presence. PLoS One 2017;12:e0177809. 10.1371/journal.pone.0177809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hägg DA, Jernas M, Wiklund O, et al. Expression profiling of macrophages from subjects with atherosclerosis to identify novel susceptibility genes. Int J Mol Med 2008;21:697-704. 10.3892/ijmm.21.6.697 [DOI] [PubMed] [Google Scholar]
- 12.Döring Y, Manthey HD, Drechsler M, et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012;125:1673-83. 10.1161/CIRCULATIONAHA.111.046755 [DOI] [PubMed] [Google Scholar]
- 13.Nührenberg TG, Langwieser N, Binder H, et al. Transcriptome analysis in patients with progressive coronary artery disease: identification of differential gene expression in peripheral blood. J Cardiovasc Transl Res 2013;6:81-93. 10.1007/s12265-012-9420-5 [DOI] [PubMed] [Google Scholar]
- 14.Feng H, Gu ZY, Li Q, et al. Identification of significant genes with poor prognosis in ovarian cancer via bioinformatical analysis. J Ovarian Res 2019;12:35. 10.1186/s13048-019-0508-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu K, Fu Q, Liu Y, et al. An integrative bioinformatics analysis of microarray data for identifying hub genes as diagnostic biomarkers of preeclampsia. Biosci Rep 2019;39:BSR20190187. 10.1042/BSR20190187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Zhang R, Chen X, et al. Identification of hub genes and small-molecule compounds related to intracerebral hemorrhage with bioinformatics analysis. PeerJ 2019;7:e7782. 10.7717/peerj.7782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu Y, Xu J, Chen J, et al. Blocking transferrin receptor inhibits the growth of lung adenocarcinoma cells in vitro. Thorac Cancer 2018;9:253-61. 10.1111/1759-7714.12572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen P, Chen Y, Wu W, et al. Identification and validation of four hub genes involved in the plaque deterioration of atherosclerosis. Aging (Albany NY) 2019;11:6469-89. 10.18632/aging.102200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Q, Li Y, Song X, et al. Both gut microbiota and cytokines act to atherosclerosis in ApoE-/- mice. Microb Pathog 2020;138:103827. 10.1016/j.micpath.2019.103827 [DOI] [PubMed] [Google Scholar]
- 20.Ramji DP, Davies TS. Cytokines in atherosclerosis: Key players in all stages of disease and promising therapeutic targets. Cytokine Growth Factor Rev 2015;26:673-85. 10.1016/j.cytogfr.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Döring Y, Pawig L, Weber C, et al. The CXCL12/CXCR4 chemokine ligand/receptor axis in cardiovascular disease. Front Physiol 2014;5:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hyafil F, Pelisek J, Laitinen I, et al. Imaging the Cytokine Receptor CXCR4 in Atherosclerotic Plaques with the Radiotracer (68)Ga-Pentixafor for PET. J Nucl Med 2017;58:499-506. 10.2967/jnumed.116.179663 [DOI] [PubMed] [Google Scholar]
- 23.Weiberg D, Thackeray JT, Daum G, et al. Clinical Molecular Imaging of Chemokine Receptor CXCR4 Expression in Atherosclerotic Plaque Using (68)Ga-Pentixafor PET: Correlation with Cardiovascular Risk Factors and Calcified Plaque Burden. J Nucl Med 2018;59:266-72. 10.2967/jnumed.117.196485 [DOI] [PubMed] [Google Scholar]
- 24.Naruko T, Ueda M, Haze K, et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002;106:2894-900. 10.1161/01.CIR.0000042674.89762.20 [DOI] [PubMed] [Google Scholar]
- 25.Castillo L, Rohatgi A, Ayers CR, et al. Associations of four circulating chemokines with multiple atherosclerosis phenotypes in a large population-based sample: results from the dallas heart study. J Interferon Cytokine Res 2010;30:339-47. 10.1089/jir.2009.0045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deo R, Khera A, McGuire DK, et al. Association among plasma levels of monocyte chemoattractant protein-1, traditional cardiovascular risk factors, and subclinical atherosclerosis. J Am Coll Cardiol 2004;44:1812-8. 10.1016/j.jacc.2004.07.047 [DOI] [PubMed] [Google Scholar]
- 27.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med 2006;354:610-21. 10.1056/NEJMra052723 [DOI] [PubMed] [Google Scholar]
- 28.Ardigo D, Assimes TL, Fortmann SP, et al. Circulating chemokines accurately identify individuals with clinically significant atherosclerotic heart disease. Physiol Genomics 2007;31:402-9. 10.1152/physiolgenomics.00104.2007 [DOI] [PubMed] [Google Scholar]
- 29.Zernecke A, Shagdarsuren E, Weber C. Chemokines in atherosclerosis: an update. Arterioscler Thromb Vasc Biol 2008;28:1897-908. 10.1161/ATVBAHA.107.161174 [DOI] [PubMed] [Google Scholar]
- 30.de Oliveira RT, Mamoni RL, Souza JR, et al. Differential expression of cytokines, chemokines and chemokine receptors in patients with coronary artery disease. Int J Cardiol 2009;136:17-26. 10.1016/j.ijcard.2008.04.009 [DOI] [PubMed] [Google Scholar]
- 31.Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol 2011;32:452-60. 10.1016/j.it.2011.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Copin JC, da Silva RF, Fraga-Silva RA, et al. Treatment with Evasin-3 reduces atherosclerotic vulnerability for ischemic stroke, but not brain injury in mice. J Cereb Blood Flow Metab 2013;33:490-8. 10.1038/jcbfm.2012.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breland UM, Halvorsen B, Hol J, et al. A potential role of the CXC chemokine GROalpha in atherosclerosis and plaque destabilization: downregulatory effects of statins. Arterioscler Thromb Vasc Biol 2008;28:1005-11. 10.1161/ATVBAHA.108.162305 [DOI] [PubMed] [Google Scholar]
- 34.Bechara C, Wang X, Chai H, et al. Growth-related oncogene-alpha induces endothelial dysfunction through oxidative stress and downregulation of eNOS in porcine coronary arteries. Am J Physiol Heart Circ Physiol 2007;293:H3088-3095. 10.1152/ajpheart.00473.2007 [DOI] [PubMed] [Google Scholar]
- 35.Jabs A, Okamoto E, Vinten-Johansen J, et al. Sequential patterns of chemokine- and chemokine receptor-synthesis following vessel wall injury in porcine coronary arteries. Atherosclerosis 2007;192:75-84. 10.1016/j.atherosclerosis.2006.05.050 [DOI] [PubMed] [Google Scholar]
- 36.Abe Y, Yamamoto N, Nakamura K, et al. IL-13 attenuates early local CXCL2-dependent neutrophil recruitment for Candida albicans clearance during a severe murine systemic infection. Immunobiology 2019;224:15-29. 10.1016/j.imbio.2018.11.002 [DOI] [PubMed] [Google Scholar]
- 37.Inoue A, Matsumoto I, Tanaka Y, et al. TIARP attenuates autoantibody-mediated arthritis via the suppression of neutrophil migration by reducing CXCL2/CXCR2 and IL-6 expression. Sci Rep 2016;6:38684. 10.1038/srep38684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouault C, Pellegrinelli V, Schilch R, et al. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology 2013;154:1069-79. 10.1210/en.2012-1415 [DOI] [PubMed] [Google Scholar]
- 39.Luzina IG, Papadimitriou JC, Anderson R, et al. Induction of prolonged infiltration of T lymphocytes and transient T lymphocyte-dependent collagen deposition in mouse lungs following adenoviral gene transfer of CCL18. Arthritis Rheum 2006;54:2643-55. 10.1002/art.21950 [DOI] [PubMed] [Google Scholar]
- 40.Hägg DA, Olson FJ, Kjelldahl J, et al. Expression of chemokine (C-C motif) ligand 18 in human macrophages and atherosclerotic plaques. Atherosclerosis 2009;204:e15-20. 10.1016/j.atherosclerosis.2008.10.010 [DOI] [PubMed] [Google Scholar]
- 41.Reape TJ, Rayner K, Manning CD, et al. Expression and cellular localization of the CC chemokines PARC and ELC in human atherosclerotic plaques. Am J Pathol 1999;154:365-74. 10.1016/S0002-9440(10)65283-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klaver D, Posch B, Geisler A, Hermann M, et al. Peptides from allergenic lipocalins bind to formyl peptide receptor 3 in human dendritic cells to mediate TH2 immunity. J Allergy Clin Immunol 2020;145:654-65. 10.1016/j.jaci.2019.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen K, Bao Z, Gong W, et al. Regulation of inflammation by members of the formyl-peptide receptor family. J Autoimmun 2017;85:64-77. 10.1016/j.jaut.2017.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee HY, Jeong YS, Lee M, et al. Intracellular formyl peptide receptor regulates naive CD4 T cell migration. Biochem Biophys Res Commun 2018;497:226-32. 10.1016/j.bbrc.2018.02.060 [DOI] [PubMed] [Google Scholar]
- 45.Eon Kuek L, Leffler M, Mackay GA, et al. The MS4A family: counting past 1, 2 and 3. Immunol Cell Biol 2016;94:11-23. 10.1038/icb.2015.48 [DOI] [PubMed] [Google Scholar]
- 46.Mattiola I, Tomay F, De Pizzol M, et al. The macrophage tetraspan MS4A4A enhances dectin-1-dependent NK cell-mediated resistance to metastasis. Nat Immunol 2019;20:1012-22. 10.1038/s41590-019-0417-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet 2011;43:429-35. 10.1038/ng.803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deming Y, Filipello F, Cignarella F, et al. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci Transl Med 2019;11:eaau2291. 10.1126/scitranslmed.aau2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bos S, Phillips M, Watts GF, et al. Novel protein biomarkers associated with coronary artery disease in statin-treated patients with familial hypercholesterolemia. J Clin Lipidol 2017;11:682-93. 10.1016/j.jacl.2017.03.014 [DOI] [PubMed] [Google Scholar]
- 50.Hertle E, Stehouwer CD, van Greevenbroek MM. The complement system in human cardiometabolic disease. Mol Immunol 2014;61:135-48. 10.1016/j.molimm.2014.06.031 [DOI] [PubMed] [Google Scholar]
- 51.Tian ZH, Yuan C, Yang K, et al. Systematic identification of key genes and pathways in clear cell renal cell carcinoma on bioinformatics analysis. Ann Transl Med 2019;7:89. 10.21037/atm.2019.01.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as