

Summary
The accumulation of proteins into insoluble aggregates is a common feature of several neurodegenerative diseases. Aggregated α-synuclein is a major component of Lewy bodies that pathologically define Parkinson's disease (PD). Here, we present methods for the detection of pathogenic conformations of α-synuclein in induced pluripotent stem cell (iPSC) patient-derived neuron models and brain tissue. These methods can be applied to studies of PD pathogenesis and the discovery of novel therapeutics that restore physiological α-synuclein.
For complete details on the use and execution of this protocol, please refer to Cuddy et al. (2019) and Zunke et al. (2018).
Subject areas: Molecular biology, Neuroscience, Protein biochemistry
Graphical abstract
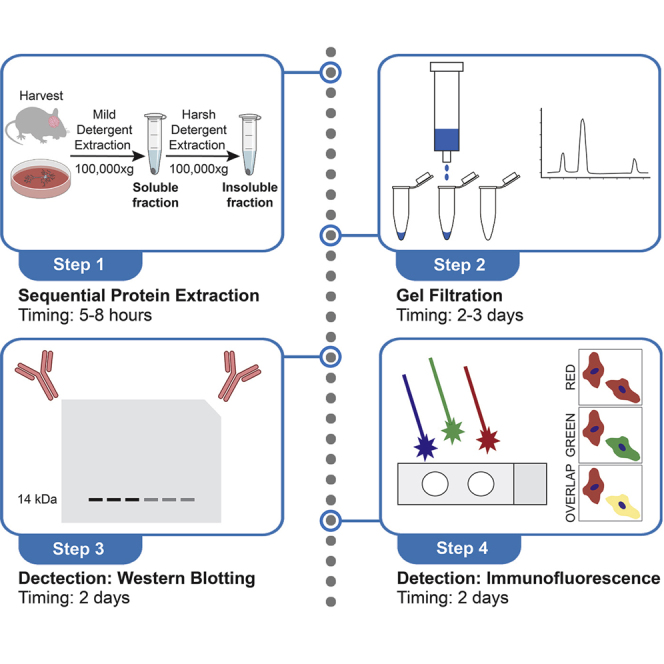
Highlights
-
•
α-Synuclein aggregates can be characterized by sequential protein extraction
-
•
Protocol is optimized for detecting α-synuclein in iPSC-derived neurons or brain tissue
-
•
Gel filtration is a useful method for the detection of oligomeric intermediates
-
•
Inclusions can be further analyzed by immunofluorescence and Thioflavin S staining
The accumulation of proteins into insoluble aggregates is a common feature of several neurodegenerative diseases. Aggregated α-synuclein is a major component of Lewy bodies that pathologically define Parkinson's disease (PD). Here, we present methods for the detection of pathogenic conformations of α-synuclein in induced pluripotent stem cell (iPSC) patient-derived neuron models and brain tissue. These methods can be applied to studies of PD pathogenesis and the discovery of novel therapeutics that restore physiological α-synuclein.
Before you begin
Prepare protein extraction buffers
Timing: 10 min
-
1.To prepare 1% Triton extraction buffer, add the appropriate amounts of protease inhibitor cocktail (PIC), sodium fluoride (NaF), sodium orthovanadate (Na3VO4), and phenylmethylsulfonyl fluoride (PMSF) to 1% Triton base buffer (described under recipe details listed under “materials”).
-
a.Vortex the solution, and keep on ice.
-
a.
-
2.To prepare 2% SDS extraction buffer, add the appropriate amount of protease inhibitor cocktail (PIC) to 2% SDS base buffer. Refer to recipe details listed under “materials.”
-
a.Vortex the solution, and keep at 20°C–22°C.
-
a.
CRITICAL: The completed Triton and SDS extraction buffers with PIC should be prepared fresh each time.
Prepare mobile phase for gel filtration
-
3.
To prepare the mobile phase, dilute 10× PBS without Ca2+/Mg2+ (Thermo Fisher Scientific #70011-044) in Milli-Q H2O to 1× PBS.
-
4.
Filter the solution using a Steritop 0.22 μm filter.
Prepare buffers for western blot
-
5.
To prepare 1× running buffer: dilute the 10× stock (Boston BioProducts #BP-150) with Milli-Q H2O. Mix well.
-
6.
To prepare 1× transfer buffer: dilute the 10× stock (Boston BioProducts #BP-190) with Milli-Q H2O and add 20% MeOH. Mix well.
-
7.
To prepare 5× Laemmli SDS sample buffer, refer to recipe details listed under “materials.”
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti alpha-synuclein LB509 (WB 1:500; IF 1:100) | Abcam | Cat# ab27766; RRID: AB_727020 |
| Rabbit polyclonal anti alpha-synuclein C20 (WB 1:2,000; IF 1:300) | Santa Cruz Biotechnology | Cat# SC-7011-R; RRID: AB_2192953 |
| Mouse monoclonal anti alpha-synuclein syn211 (WB 1:500–1,000; IF 1:100) | Thermo Fisher Scientific | Cat# 32-8100; RRID: AB_2533094 |
| Mouse monoclonal anti alpha-synuclein syn303 (WB 1:500–1,000) | BioLegend | Cat# 824301; RRID: AB_2564879 |
| Mouse monoclonal anti alpha-synuclein syn505 (WB 1:500–1,000) | Thermo Fisher Scientific | Cat# 32-8300; RRID: AB_2533225 |
| Alexa Fluor 680 goat anti-mouse IgG secondary (WB 1:10,000) | Invitrogen | Cat# A21058; RRID: AB_2535724 |
| IRdye 800CW goat anti-rabbit IgG secondary (WB 1:5,000) | Li-Cor Biosciences | Cat# 92632211; RRID AB_621843 |
| Alexa Fluor 568 goat anti-mouse IgG secondary (IF 1:400) | Invitrogen | Cat# A11031; RRID: AB_144696 |
| Alexa Fluor 647 goat anti-rabbit IgG secondary (IF 1:400) | Invitrogen | Cat# A21245; RRID: AB_2535813 |
| Chemicals, peptides, and recombinant proteins | ||
| Triton X-100 | Sigma | Cat# T8787 |
| NaCl | Sigma | Cat# S3014 |
| HEPES | Sigma | Cat# H4034 |
| EDTA | Sigma | Cat# E1644 |
| MgCl2 | Sigma | Cat# M9272 |
| Glycerol | Sigma | Cat# G5516 |
| SDS | Sigma | Cat# L4509 |
| Tris base | Sigma | Cat# T1503 |
| Tris acid | Sigma | Cat# T5941 |
| Protease Inhibitor Cocktail (PIC) | Roche | Cat# 11 836 170 001 |
| Sodium fluoride (NaF) | Sigma | Cat# 201154 |
| Sodium orthovanadate (Na3VO4) | Sigma | Cat# 450243 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma | Cat# 78830 |
| 1× phosphate buffered saline (PBS), pH 7.4 | Thermo Fisher Scientific | Cat# 10010-023 |
| 10× phosphate buffered saline (PBS), pH 7.4 | Thermo Fisher Scientific | Cat# 70011-044 |
| 10× Tris-Glycine-SDS Running Buffer | Boston BioProducts | Cat# BP-150 |
| 10× Transfer Buffer | Boston BioProducts | Cat# BP-190 |
| 10× Tris-buffered saline (TBS) | Boston BioProducts | Cat# BM-301 |
| Methanol (MeOH) | Fisher | Cat# A412P-4 |
| 10% Formaldehyde (PFA), ultrapure, EM grade | Polysciences | Cat# 04018-1 |
| 2-Mercaptoethanol (BME) | Thermo Fisher Scientific | Cat# 21985023 |
| Bromophenol blue | Sigma | Cat# 114391 |
| Simply Blue Safe Stain | Novex | Cat# LC6065 |
| Intercept Blocking Buffer | Li-Cor Biosciences | Cat# 927-70001 |
| Tween 20 | Sigma | Cat# P1379 |
| Bovine serum albumin (BSA), heat shock, fatty acid free | Roche | Cat# 03117057001 |
| Normal goat serum (NGS) | Jackson ImmunoResearch | Cat# 005-000-121 |
| Ethanol (200 proof, molecular biology grade) | Fisher | Cat# BP2818-500 |
| DAPI Fluoromount mounting media | Southern Biotech | Cat# 0100-20 |
| Thioflavin S | Sigma | Cat# T1892 |
| Critical commercial assays | ||
| Micro BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23235 |
| Gel Filtration Calibration Kit | Cytiva (formerly GE Healthcare) | Cat# 28403842 |
| Other | ||
| Cell lifters | Fisher | Cat# 08-100-240 |
| 1.5 mL microcentrifuge tubes | Fisher | Cat# 02-681-320 |
| Ultracentrifugation tubes | Beckmann Coulter | Cat# 344062 |
| PVDF immobilon FL transfer membrane, 0.45 μm pore size | Millipore | Cat# IPFL00010 |
| Steritop bottle top filters, 0.22 μm | Millipore | Cat# S2GPT10RE |
| Mini gel tank | Thermo Fisher Scientific | Cat# A25977 |
| Mini blot module (Bolt semi-wet transfer system) | Thermo Fisher Scientific | Cat# B1000 |
| Superdex 200 Increase 10/300 gel filtration column | Cytiva (formerly GE Healthcare) | Cat# 28990944 |
| Amicon ultracentrifugal protein concentrators (10 kDa MWCO) | Millipore | Cat# UFC5010BK |
| Assistant microscope coverglass, 12 mm, No. 0 | Carolina Biological | Cat# 92100100030 |
| Microscope slides | Fisher | Cat# 12-550-15 |
| Microcentrifuge (e.g., Sorvall Legend Micro 21R) | N/A | N/A |
| Homogenizer (e.g., Glas-Col model #099C K54, 333–4,000 rpm) | N/A | N/A |
| Teflon pestle and 1 mL capacity conical glass vessel (e.g., DWK Life Sciences Kimble Kontes Duall Tissue Grinders, Cat# K885480-0020) | N/A | N/A |
| Ultracentrifuge (e.g., Beckmann Coulter Optima MAX-TL) and TLA100.1 type rotor | N/A | N/A |
| Probe or water-based sonicator (e.g., Qsonica Q800R3 system) | N/A | N/A |
| Western blot imaging system (e.g., Li-Cor Biosciences Odyssey, Azure Sapphire) | N/A | N/A |
| HPLC system (e.g., Agilent 1260 Infinity II) | N/A | N/A |
| Confocal microscope (e.g., Leica TCS SPE laser with CTR4000/DMI4000B) | N/A | N/A |
Materials and equipment
Materials
| 1% Triton Base Buffer | Final concentration | Amount |
|---|---|---|
| Triton X-100 | 1% | 0.5 mL |
| 5 M NaCl | 150 mM | 1.5 mL |
| 1 M HEPES pH 7.4 | 20 mM | 1 mL |
| 0.5 M EDTA | 1 mM | 100 μL |
| 1 M MgCl2 | 1.5 mM | 75 μL |
| 100% glycerol | 10% | 5 mL |
| Milli-Q H2O | n/a | 41.825 mL |
| Total | 50 mL |
Check that the pH is ~7.4. The Triton base buffer can be stored at 4°C for 2–3 years.
| Triton extraction buffer | Final concentration | Amount |
|---|---|---|
| 1% Triton Base Buffer | n/a | 4.425 mL |
| PIC | n/a | ½ tablet |
| 500 mM NaF | 50 mM | 500 μL |
| 200 mM Na3VO4 | 2 mM | 50 μL |
| 0.1 M PMSF | 0.5 mM | 25 μL |
| Total | 5 mL |
For smaller volumes of extraction buffer, you may need to prepare 10× PIC. To do this, dissolve ¼ PIC tablet in 250 μL 1% Triton base buffer. Store 10× PIC-Triton at −20°C.
| 2% SDS base buffer | Final concentration | Amount |
|---|---|---|
| 10% SDS | 2% | 10 mL |
| 1 M Tris pH 7.4 | 50 mM | 2.5 mL |
| Milli-Q H2O | n/a | 37.5 mL |
| Total | 50 mL |
The SDS base buffer can be stored at 20°C–22°C for 2–3 years.
| SDS extraction buffer | Final concentration | Amount |
|---|---|---|
| 2% SDS Base Buffer | n/a | 5 mL |
| PIC | n/a | ½ tablet |
| Total | 5 mL |
For smaller volumes of extraction buffer, you may need to prepare 10× PIC. To do this, dissolve ¼ PIC tablet in 250 μL 1% SDS base buffer. Store 10× PIC-SDS at −20°C.
| PFA fixative solution | Final concentration | Amount | Final concentration | Amount |
|---|---|---|---|---|
| 10% paraformaldehyde (PFA) | 0.4% | 2 mL | 4% | 4 mL |
| 10× PBS | 1× | 5 mL | 1× | 1 mL |
| Milli-Q H2O | n/a | 43 mL | n/a | 5 mL |
| Total | 50 mL | 10 mL |
| 5× Laemmli SDS sample buffer | Final concentration | Amount |
|---|---|---|
| 0.5 M Tris-HCl pH 6.8 | 100 mM | 2 mL |
| 14.3 M 2-mercaptoethanol (BME) | 900 mM | 630 μL |
| Glycerol | 20% | 2 mL |
| SDS | 10% | 1 g |
| Bromophenol blue | 0.015% | 1.5 mg |
| Total | 10 mL |
Equipment
-
(1)
A motor-driven homogenization apparatus is recommended for the initial extraction steps. We use a Glas-Col model 099C K54 that can achieve up to 4,000 rpm, with a Teflon pestle and conical glass vessel such as DWK Life Sciences #K885480-0020.
-
(2)
To help solubilize detergent insoluble α-synuclein aggregates, we recommend an indirect sonication apparatus using a cup horn submerged in a temperature-controlled water bath. A relatively powerful cup horn is required for this step similar to what is used for DNA fragmentation assays, such as the Qsonica system Q800R3 or similar system. Note that a direct probe sonication method can also be used, although it requires a more careful technique (equal pulse time, probe placement in middle of liquid sample, temperature control, limitation of frothing) to limit extraction variability between samples.
-
(3)For the gel filtration step, a liquid chromatography (LC) system is required that can achieve at least 50 bar constant pressure. We use an Agilent 1260 Infinity system with a quaternary pump, thermo-cooled autoinjector, diode array detector, and thermo-cooled fraction collector. With this system, we use a Superdex 200 Increase 10/300 Increase column from Cytiva (formerly GE Healthcare; formerly Pharmacia). Other LC systems can be used that are capable of achieving the same parameters.
-
(a)Another option for gel filtration is to use a TOSOH SuperSW 3000 column with a TSK gel. This contains 4 μm resin in a 4.6 mm ID × 30 cm long stainless-steel-encased column that can withstand >120 bar of pressure. The SuperSW3000 column can be used when sample is limited. For example, results can be achieved when only 50 μg of total lysate is injected on the column. However, the fractions should be concentrated to <100 μL and analyzed by an ELISA method, since there will be insufficient protein for western blot detection.
-
(a)
-
(4)
For immunofluorescence analysis of α-synuclein puncta, we use a Leica TCS SPE CTR4000/DMI4000B confocal microscope with laser excitation. Confocal microscopy facilitates imaging of iPSC-derived neurons since they often culture in clumps, making single focal plane analysis difficult.
-
(5)
For imaging thioflavin S aggregates, a microscope with either a laser excitation at 405 nm or an excitation filter that can achieve 400–440 nm is optimal, such as an Olympus BP400-440 (wide band, cube U-MWBV) or BP420-440 (narrow band, cube U-MNBV) exciter filter, with BA475 barrier filter. Although not optimal, thioflavin S can also be detected on a more common filter capable of detecting GFP or FITC, but only if the bandwidth is wide enough to cover an excitation range of 450 nm and below such as Olympus filter BP420-480, cube U-MSWB. This cube has a wide bandwidth ranging from 420–480 nm. For simultaneous imaging with α-synuclein, using immunofluorescent secondary antibodies that emit red visible color (e.g., Alexa Fluor 568) is required to prevent signal crossover. This can be excited with a 561 nm laser line, or exciter filters (Olympus BP510-550, cube U-MWG; or BP530-550, cube U-MNG). Note that a barrier filter on the Olympus cube will be required to prevent crossover (e.g., BA475 for U-MWBV or U-MNBV, or BA515 for U-MSWB). Far-red fluorophores such as Alexa Fluor 647 will also work, as their emission-excitation spectrum is even farther away from thioflavin S. DAPI or Hoechst fluorescence for nuclear visualization is very close to Thioflavin S, but could be excited using an Olympus BP360-370 narrow band exciter filter with barrier filter BA420 (Olympus cube U-MNU). Note that BP360-370 may partially excite thioflavin S but not to the same extent as DAPI, and therefore crossover is usually not an issue. However, this depends on the abundance of aggregates in the sample. The user should optimize the settings prior to performing an experiment, as microscopy equipment varies greatly between laboratories. Performing the proper positive and negative controls (discussed below) will help to determine optimal settings.
Step-by-step method details
Sequential protein extraction of α-synuclein from harvested cells or tissues
To model PD, we generate midbrain dopaminergic neurons from PD patient-derived iPSCs using a well-established differentiation protocol that is initiated by dual SMAD inhibition (Kriks et al., 2011). The concepts of midbrain and other types of neural differentiation, as well as other technical considerations of culture modeling, have been discussed in detail previously (Zunke and Mazzulli, 2019). To obtain suitable starting material from iPSC-derived neurons, we typically plate 0.5 × 106 cells per well in a 24-well plate. If more material is needed, plate 1 × 106 cells per well in a 12-well plate or 2 × 106 cells per well in a 6-well plate. For brain tissue, we recommend using at least 10 mg per sample. Our culture studies have focused on the analysis of familial forms of PD, such as patients that express the SNCA triplication or A53T α-synuclein, or Gaucher disease patients with GBA1 mutations. These lines will often accumulate detectable levels of α-synuclein pathology between 60-to-90 days post-differentiation. However, extending the culture to 120 days may facilitate detection of insoluble α-synuclein, since it allows aggregates more time to grow and mature into stable structures. It is important to characterize the neuron viability of the line to be analyzed, so that these biochemical assays can be performed prior to the onset of neurotoxicity.
Soluble and insoluble α-synuclein protein is sequentially extracted from cells and frozen tissues using extraction buffers containing detergents of increasing strength to differentiate the physiological conformations from pathogenic forms. Under physiological conditions, α-synuclein is primarily extracted in mild, non-ionic detergents such as Triton X-100, therefore it is necessary to use an extraction buffer that will effectively disrupt the cell membrane while keeping the integrity of the soluble protein structure intact. We recommend using an extraction buffer containing 1% vol/vol Triton X-100. Since neurons are relatively fragile, using a gentle method of extraction such as homogenization is recommended for this step. For extraction of insoluble α-synuclein, a harsher detergent, such as the anionic surfactant sodium dodecyl sulfate (SDS), and a vigorous method of extraction such as ultrasonication is recommended.
Harvest cells
-
1.
Rinse the cells with cold 1× PBS while on ice and aspirate.
-
2.Add fresh cold PBS and scrape off the cells using a cell scraper, collecting into 1.5 mL microcentrifuge tubes kept on ice.
-
a.Collect each well from a plate into an individual microcentrifuge tube.
-
a.
-
3.
Spin down the collected cells in a tabletop centrifuge at 400 × g for 5 min at 4°C. Carefully aspirate off and discard the supernatant. It is critical that no liquid is left on the cell pellet prior to freezing or starting the next lysis step.
-
4.
The pellet can be stored at −80°C until analysis.
Making an extract
-
5.To lyse frozen cells or tissues, briefly thaw samples on ice and then add 1% Triton X-100-containing extraction buffer that is supplemented with a cocktail of protease inhibitors directly to the sample. Refer to recipe details listed under “materials.”
-
a.Use 50–100 μL per sample for a 24-well sized plate; 100–200 μL per sample for a 12-well plate; 200–300 μL per sample for a 6-well plate.
-
b.For frozen brain tissue, add 10 volumes of lysis buffer per milligram of tissue, and incubate on ice for 10 min to soften the tissue and prepare for homogenization.
-
a.
Note: For cell cultures, the volume will depend on the cell confluency. The indicated volumes are for iPSC-derived midbrain neurons which are cultured at relatively high density, so lower extraction volumes may be required for other cell types.
-
6.
Transfer the sample into a clean 1 mL capacity conical glass vessel.
-
7.
Homogenize the sample by slowly moving the Teflon pestle up and down 20-to-40 times. Place the homogenized lysates back in the original microcentrifuge tubes.
Note: The suspension should be homogeneous and without any clumps. The length of this step can vary depending on the rigidity of the culture or tissue. If clumps are present, the homogenization step should be repeated until a smooth, even homogenate is achieved. The homogenate should be translucent. A pipet tip should be barely visible through the homogenate when held up to a light. If the tip is not at all visible, then more lysis buffer should be added.
-
8.
Incubate the lysate in an ice-water slurry for at least 30 min, mixing slowly (e.g., 60 rpm) on a platform rotator.
-
9.
To fully disrupt lipid bilayers, perform three freeze/thaw cycles by placing the lysates in a 80°C ethanol bath for 2 min followed by a 37°C water bath for 30 s to 1 min. The thawing time will depend on the lysis volume, but should be done just until a small piece of ice is left. A white precipitate containing Triton-insoluble materials might be visible.
Note: Normally, freeze/thaw cycles can be disruptive to native protein structure. However, glycerol is included in the lysis buffer to limit this. The freeze/thaw step can be skipped if disrupting native structure is a concern. However, we have carefully tested the effect of this extraction procedure on native dimers and multimers such as neural specific enolase and GAPDH, and have not found any evidence of native structure breakdown (Zunke et al., 2018).
-
10.
Transfer the lysates, including any insoluble material, to ultracentrifugation tubes.
Note: Occasionally, the insoluble material can stick to the pipet tips. If this occurs, simply move the solution up and down several times until the particles are released.
-
11.
Clarify the lysates in an ultracentrifuge at 100,000 × g for 30 min at 4°C.
Note: Ultracentrifugation is required here to efficiently separate soluble α-synuclein from fibrillar and amorphous polymers.
-
12.Carefully transfer the supernatant to a new 1.5 mL microcentrifuge tube without disturbing the pellet. The supernatant is labeled as the Triton-soluble fraction.
-
a.Determine Triton-soluble protein concentrations using a micro BCA protein assay kit. Between 2–10 μg/μL is expected.
-
b.If extracting brain tissue, repeat steps 5–12. The total volume added will be 20 volumes of lysis buffer. This step is required to prevent carryover between the fractions.
-
a.
-
13.
Wash the pellet with 50 μL of Triton-containing extraction buffer by gently pipetting up and down with a p-200 tip to break up the pellet. It is normally not necessary to completely dissolve the pellet again, since the goal here is to wash away any soluble liquid that could have been trapped in the pellet during centrifugation. Repeat step 11 and remove the wash buffer carefully.
Note: Depending on the amount of the starting material and the nature of the sample, the pellet may not fully dissolve during this step.
Pause point: The Triton-insoluble pellet can be stored at −80°C until further extraction with SDS lysis buffer.
-
14.Resuspend the Triton-insoluble pellet in extraction buffer containing 2% SDS and supplemented with protease inhibitors. Refer to recipe details listed under “materials.”
-
a.For cell cultures using 1 X 106 cells per sample, use a minimum of 30 μL SDS lysis buffer per pellet, as a few μL will likely be lost when transferring among various tubes. Larger pellet sizes will require more lysis buffer volume (e.g., 50–70 μL) for an efficient extraction.
-
b.For brain tissue, use 5 to 10 volumes of SDS lysis buffer compared to the starting tissue weight (e.g., 100 μL volume for a 10 mg sample). If the sample appears viscous and hard to pipet, add more lysis buffer until a pipetable, partially translucent homogenate is obtained.
-
a.
Note: The pellet may not fully dissolve during this step, and particles may remain. This is normal.
-
15.
Without delay, boil the samples at 100°C for 10 min.
Note: We prefer to use a water bath for this step as opposed to a dry block, since water more efficiently surrounds the microcentrifuge tube and quickly raises sample temperature.
-
16.Sonicate the samples using either:
-
a.Water-bath sonicator with a cup horn, such as the Qsonica system Q800R3.
-
i.Sonicate samples for 10 min at 30% power, 20°C. The parameters and instructions of the Qsonica machine should be strictly followed, including the use of thin-wall tubes, water levels adjusted to cover the sample inside the tubes, and appropriate distance between the sample and the cup horn. If the sample is not solubilized after 10 min, then repeat this step until the pellet is completely dissolved.Note: We prefer this method since it better controls temperature and is more consistent between samples, compared to probe sonicators that can only do one sample at a time.
-
i.
-
b.Direct probe sonicator
-
i.Perform three each 3–5-s-long pulses per sample at 30% amplitude. Be sure to be extremely consistent with the pulse times between each sample and avoid excessive heat. Avoid touching the side walls of the microcentrifuge tubes.Note: This step should be done in a safety chamber to prevent contact with aerosols and with ear protection.
-
i.
-
a.
-
17.
Transfer the sonicated samples to a microcentrifuge tube and boil once again for 10 min. It is safe to keep samples at 20°C–22°C for short periods, which avoids precipitation of SDS.
Note: The boiling and sonication steps should dissolve pellets from cell cultures completely. However, if brain tissue is used, there may be some insoluble material remaining.
-
18.
Spin the lysates in an ultracentrifuge at 100,000 × g for 30 min at 20°C–22°C. Here again, 20°C–22°C is used (not 4°C) to prevent precipitation of SDS.
Note: Depending on the cell material starting size, a pellet may or may not be present after this step. If tissue is used, a pellet will likely be present, which can be further extracted in 70% formic acid/water by incubation at 37°C and sonication, as described in step 16.
-
19.Carefully transfer the supernatant to a new 1.5 mL microcentrifuge tube without disturbing the pellet. The supernatant is labeled as the SDS-soluble fraction.
-
a.Determine SDS protein concentrations using a BCA protein assay kit. BCA can tolerate only low levels of SDS so it is optimal to dilute the sample 1:200 to 1:1,000 to limit interference with the assay.
-
a.
Note: SDS interferes with some protein assay reagents such as Coomassie blue used in the Bradford assay.
-
20.Prepare the Triton and SDS samples for western blot analysis, as described in section under “Detection of α-synuclein via western blot.”
-
a.If analysis of soluble monomers and oligomers is desired, prepare the Triton-soluble extract as described in section "Gel filtration analysis of α-synuclein".
-
a.
Gel filtration analysis of α-synuclein
High and low molecular weight conformers of α-synuclein can be separated and reliably detected via gel filtration followed by western blot analysis. The Triton-soluble fraction is most often analyzed by this method, since it can separate monomers from soluble oligomeric intermediates that are en route to forming higher order, insoluble polymers. However, SDS fractions can be used for this process if desired. There are other methods capable of separating monomers from oligomers such as native gel electrophoresis using Coomassie brilliant blue in the sample buffer to help resolve protein bands (native blue gels). However, this method does not have the same sensitivity at detecting oligomeric intermediates as compared to gel filtration. Optimal sensitivity is critical since in some models, intermediates can be in low abundance and difficult to detect. The gel filtration/western blot method described here can be used with any antibody that is suitable for western blot analysis. This is because after separation, the gel filtration fractions are then boiled in SDS and analyzed by PAGE. This exposes the epitopes efficiently so that the antibody of choice will have a greater capacity to bind to its epitope. This is in contrast to native blue gels where proteins are not denatured, thus leaving some epitopes involved in protein-protein interactions and within the NAC region of self-associated α-synuclein buried and undetectable.
-
21.
Remove the Superdex 200 Increase 10/300 gel filtration column’s storage buffer with 0.22 μm sterile-filtered Milli-Q H2O. We use a flow rate of 0.5 mL/min.
-
22.
Equilibrate the column using 2 column volumes of 0.22 μm sterile-filtered 1× PBS, pH 7.4 as the mobile phase. Refer to recipe details listed under “before you begin.”
-
23.Calibrate the column with high molecular weight standards according to the manufacturer's instructions (e.g., Cat# 28403842 from Cytiva, formerly GE Healthcare).
-
a.It is important to use the same injection volume as the sample that will be analyzed. We use a 250 μL injection volume, which is 1% of the column volume. Note that it is not necessary to calibrate the column before every run. This is usually done after 10–20 runs, after deep cleaning the column in NaOH, or if retention times and column pressures change significantly.
-
a.
-
24.
Prepare the samples for loading. Dilute ~1 mg of the Triton-soluble lysate into a final volume of 250 μL using the PBS mobile phase. Centrifuge the sample at 20,000 × g, 4°C, for 10 min to remove any particulates in the sample. This will prevent the injection syringe from clogging.
-
25.
Set up the software for the following sequence: Set the pump flow to 0.5 mL/min (isocratic), injection volume of 250 μL, data collection in the diode array detector at 210 and 280 nm, and the fraction collector to collect every minute. The fraction collection start time should be set to collect just before the void volume of the column.
Note: For the Superdex 200 Increase column, we typically calculate a void volume of 8 mL and start fraction collection at 16 min. We end the collection at 40 min, which is sufficient time for small proteins (5 kDa) to elute from the column. Be sure to set the autoinjector and fraction collector at 4°C. If this option is not available, the system should run in a chromatography refrigerator or cold room.
-
26.
Load the sample into the injection vial or manually inject the sample, and start the sequence through the software.
-
27.After 40 min, collect the fractions and concentrate to a suitable volume for loading on SDS-PAGE gels (typically around 20–30 μL per sample). We use Amicon 10 kDa MWCO concentrators according to the manufacturer’s instructions.
-
a.We typically combine fractions in the following manner to enhance the signal and conserve gel space: Fractions 1–5, 6–9, 10–12, 13–17, 18–20, and 21–24. Oligomers of α-synuclein normally elute between fractions 2–8, while monomers elute at fraction 15 corresponding to a 35 Å-sized protein.
-
a.
Note: Fraction concentration could take up to 4 h.
-
28.
Clean the column with 2 column volumes of PBS, followed by 2 column volumes of filtered Milli-Q H2O, then store the column in 20% ethanol. Note that the sample injector and fraction collector should be cleaned in the same way to prevent salt precipitation in these components while the machine is stationary. This cleaning protocol is used for regular cleaning after each sample is run. The column should undergo an additional deep cleaning using one column volume of 0.1 M NaOH followed by 2 column volumes of water after every 10-20 separations, or if column back pressure increases.
-
29.
Once the samples have been concentrated, prepare them for western blot analysis, as described in section "Detection of α-synuclein via western blot".
Detection of α-synuclein via western blot
Below is a standard western blotting protocol that we use for the detection of soluble and insoluble α-synuclein. We recommend using at least two α-synuclein antibodies that detect distinct epitopes to confirm results. Antibody specificity should be validated by analyzing SNCA knock-out cell lines.
-
30.Prepare 50 μg per lane of both Triton and SDS extracted protein lysates to load on a 12% SDS-PAGE gel.
-
a.To prepare a sample, combine the appropriate volume of lysate with 4 μL of 5× Laemmli SDS sample buffer (for a final 1×) and supplement with Milli-Q H2O for a total volume of 20 μL. Refer to recipe details listed under “materials.”
-
b.Boil the samples for 10 min and load on an SDS-PAGE gel set up in 1× running buffer (refer to recipe details listed under “before you begin”).
-
a.
Note: For gel filtration fractions, simply load the entire concentrated fraction volume since the protein amount has already been normalized during the injection.
Note: Gradient gels can also be used, such as 4%–12% acrylamide. However we have found that gradients are not necessary.
-
31.
Run the gel at a maximum 150 V for 1–1.5 h just until the dye front runs off the gel. α-Synuclein has a calculated MW of 14,460 Da based on its calculated amino acid sequence, but migrates at approximately 18 kDa likely due to its highly acidic C-terminus (Weinreb et al., 1996).
-
32.Transfer the gel to a PVDF membrane using 1× Tris-glycine-based transfer buffer containing 20% MeOH (refer to recipe details listed under “before you begin”).
-
a.To activate the PVDF membrane, pre-wet it in 100% MeOH, and then equilibrate it in transfer buffer for 2–5 min until it sinks. We use low autofluorescent immobilon FL 0.45 μm membranes since we use fluorescent secondary detection.Note: PVDF is recommended for α-synuclein detection as it allows for higher signal detection compared to nitrocellulose membranes. Nitrocellulose membranes can also be used if they have low autofluorescent properties or if other non-fluorescent detection is used (e.g., radioactivity or HRP-conjugated secondary antibodies).
-
b.Transfer at constant 30 mV for 1 h.Note: This condition will vary depending on the transferring apparatus. We use the Bolt semi-wet transfer system from Thermo Fisher. In our experience, wet or semi-wet transfers are best for this application in order to efficiently elute the protein from the gel and onto the membrane. Other systems may work but should be tested and optimized first.
-
a.
-
33.
Place the membrane into 0.4% PFA fixative solution (refer to recipe details listed under “materials”) for 30 min at 20°C–22°C. Slowly shake the membrane on a rotator (e.g., 60 rpm) to ensure full coverage.
Note: This step should be done in a fume hood to prevent PFA inhalation.
-
34.
Wash off the PFA with Milli-Q H2O, 3× for 5 min each. Discard the PFA solution and washes into a designated PFA waste container.
-
35.
Stain the leftover gel in Simply Blue Safe Stain (or Coomassie brilliant blue) on a shaker for 1–2 h or for 12–16 h, then wash/destain in water for 12–16 h, and image on an Odyssey Imaging System, Azure Sapphire, or similar system. The stained gel can be used as a loading control during the analysis stage.
-
36.
Block the membrane for 1 h. We use a 1:1 mixture of 1× Tris-buffered saline pH 7.4 (TBS) and Li-Cor Odyssey blocking buffer. If using chemiluminescent detection, the blots can be blocked in 1× TBS containing 5% non-fat milk. Milk cannot be used with fluorescent detection systems because it contains autofluorescent substances.
-
37.Dilute anti-α-synuclein antibody in the blocking buffer. Incubate the blots with the antibody mix for 12–16 h at 4°C while shaking.
-
a.We typically use C20 or syn211 to detect all forms of α-synuclein, while LB509, syn303, and syn505 are better at detecting pathogenic aggregates and oligomers. Most of the antibodies are used at a 1:500 dilution (refer to “key resources table”), but the end concentration varies depending on the antibody batch and should be determined by the user.
-
a.
-
38.
The following day, wash the blots 3× for 5 min each with 1× TBS-0.2% Tween.
-
39.Dilute secondary antibodies in blocking buffer. Incubate the blots for 1 h at 4°C while shaking.
-
a.We typically use Alexa Fluor 680 goat anti-mouse IgG or IR dye 800CW goat anti-rabbit IgG secondary antibodies (refer to “key resources table”).
-
a.
-
40.
Wash the blots 3× for 5 min each with 1× TBS-0.2% Tween, followed by a 10min wash with 1× TBS (no tween).
-
41.
Image the blots on an Odyssey Imaging System, Azure Sapphire, or similar system. Blots should always be first probed with secondary alone and developed without the primary, to determine the background signal.
Note: Sequential extraction/western blots should be sequentially probed using different α-synuclein antibodies. Monocolonal and polycolonal antibodies can be added simultaneously if using a two-color fluorescent detection system (e.g., LB509 with C20, or syn211 with C20). Other antibodies such as syn303 and syn505 can also be used, as they can detect additional α-synuclein cross-linked bands that are associated with pathological inclusions. Blotting for reliable loading controls can then be done to assess both loading, extraction efficiency, and carry over between the fractions. For soluble fractions, we use established soluble proteins such as GAPDH or neuron specific enolase (NSE) as loading controls. For insoluble fractions, we use vimentin. Tubulin can be used as well, however it is normally found in both soluble and insoluble fractions at approximately equal levels. Total protein levels can be used to control for loading, by staining the gel after transfer or by using a total protein stain on the blot prior to the blocking step (see step 42).
Note: Gel filtration/western blots should be sequentially probed as above, followed by either GAPDH, tubulin, or NSE as loading controls and to monitor the column performance. For example, NSE is a known globular dimer under native extraction conditions, and therefore should elute off the column in a fraction that corresponds to ~90 kDa, however will migrate by SDS-PAGE at ~47 kDa. Technical issues during the extraction procedure may result in the breakdown of native structure, which should be evident from the analysis of these control proteins. Total protein levels can be used to control for loading in this step as well, by staining the gel after transfer or by using a total protein stain on the blot prior to the blocking step (see step 42).
-
42.Analysis:
-
a.For sequential extraction/western blots, quantify the α-synuclein reactive species and normalize to total protein (from the Coomassie-stained gel) or a reliable loading control as mentioned above. As an alternative to using specific proteins for loading controls, the membrane can be stained with a total protein dye prior to the blocking step such as AzureRed Fluorescent Total Protein Satin from Azure biosystems (SKU: AC2124). The dye can then be imaged by either UV or green light (e.g., using the 520 nm channel of an Azure Sapphire imager). Li-Cor also makes a total protein stain called Revert 700 (Part No. 926-11011) that can be detected on the 700 nm channel, however this may interfere with subsequent antibody detection on this channel. Total protein normalization is often more accurate than using one particular protein, since the protein levels of housekeeping genes may change depending on the experimental conditions. α-Synuclein bands that migrate above 18 kDa monomer may be present, if pathogenic aggregates are cross-linked and stable to SDS and heat. In this case, these bands should be validated to react with other α-synuclein antibodies, and compared to an SNCA knock-out lysate to determine specificity.
-
b.For gel filtration/western blots, we determine α-synuclein oligomer-to-monomer ratios. Elevation in the proportion of oligomers normally indicates that pathogenic aggregation has occurred, however this requires further analysis of the oligomeric fraction (Zunke et al., 2018). Additionally, oligomers and monomers can be analyzed separately and normalized to total protein obtained from the Coomassie-stained gel or total protein stain on the membrane. Note that although control proteins such as GAPDH and NSE are analyzed, they should not be used for normalizing α-synuclein levels in particular fractions since they do not elute equally in each fraction.
-
a.
Detection of α-synuclein aggregates via immunofluorescence imaging
Below we describe a protocol for the immunofluorescence detection of α-synuclein. This step is complementary to those described above, and necessary to validate results obtained from biochemical extractions. The analysis of aggregates in fixed cells eliminates some of the confounds that might occur during a cell lysis procedure, including aggregation events that could occur post-lysis. Furthermore, more detailed information can be gained regarding the type of pathological aggregate that is formed, based on the subcellular location (i.e., juxtanuclear vs. within neurites) and reactivity with thioflavin dyes that bind to amyloidogenic structures.
Note: A positive control where α-synuclein aggregation is induced in cells should be included. The most robust method to induce aggregation is to add pre-formed α-synuclein fibrils (PFFs) made from recombinant α-synuclein to the culture media, followed by 2–3 days of incubation. Detailed protocols for the generation of PFFs and addition to cultures have been described in detail elsewhere (Volpicelli-Daley et al., 2014).
-
43.
Plate cells on glass coverslips and culture until the time of the experiment. For iPSC-derived neurons, we typically use 12 mm diameter cover glass placed in a 24-well culture cluster, seeded at 0.4 × 106 cells per well.
Note: The coverglass used here is from Carolina Biological Supply, Assistant Glass cover slips, 12 mm (Cat# 633009). Before coating with PDL/laminin, the glass is pretreated with 70% nitric acid/water for 30 min, followed by three 1-h washes in Milli-Q water, using a volume of water that is 10 times that of the nitric acid.
-
44.
Remove the culture media and immediately add 4% PFA fixative solution. Refer to recipe details listed under “materials.” Incubate for 15 min at 20°C–22°C in a fume hood.
-
45.Carefully remove the PFA and add double the volume of water to wash the cells. Repeat 3× for 5 min each.
-
a.Discard the PFA solution and washes into a designated PFA waste container.
-
a.
-
46.
Permeabilize the cells with 0.1% to 0.3% Triton X-100 diluted in 1× PBS for 30 min at 4°C. The final Triton concentration must be determined by the end user.
Note: Typically, 0.1% Triton is better to visualize physiological α-synuclein found within synapses. 0.3% Triton better reveals pathogenic inclusions since a portion of the physiological species are extracted out of cells with the higher detergent percentage.
-
47.
To prevent non-specific antibody binding, block the cells by adding 300 μL of 2% bovine serum albumin (BSA) and 5% normal goat serum (NGS) diluted in the optimized concentration of Triton-PBS for 30 min at 20°C–22°C . Note that the same concentration of Triton X-100 should be used here as determined in step 46.
Note: The cells are cultured on cover glass within a 24-well-sized culture plate. We perform steps 44–47 within the plate.
-
48.
Incubate cells with an anti-α-synuclein primary antibody diluted in blocking buffer for 12–16 h at 4°C. See the “key resources table” for a list of suitable antibodies.
Note: To conserve antibody, we remove the cover glass from the well during this step and invert it onto a 50 μL droplet of primary antibody solution placed on parafilm. This is incubated inside a humidified chamber for 12–16 h. Keep the 24-well plate to use for the subsequent wash and secondary incubation steps.
-
49.Place the cover glass back into the 24-well plate. Carefully wash the cells 3× with Triton-PBS for 10 min each using 500 μL per well, then add secondary antibodies (300 μL of a 1:400 dilution) for 2 h at 20°C–22°C.
-
a.We typically use Alexa Fluor 568 goat anti-mouse IgG or Alexa Fluor 647 goat anti-rabbit IgG secondary antibodies.
-
a.
Note: Alexa Fluor conjugated antibodies may aggregate over time. The secondary antibody solution should be centrifuged at 20,000 × g for 10 min at 4°C to remove any particulates.
Note: The samples should be protected from light from this step forward.
-
50.
Wash the cells 3× again with Triton-PBS for 10 min each, using 500 μL per well.
Note: If thioflavin staining is not required, proceed to step 56.
-
51.
Dissolve 0.05% thioflavin S w/v in 50% ethanol/water and vortex well to dissolve. Filter the solution through a 0.22 μm filter to remove particulate matter.
-
52.
Aspirate off the Triton-PBS wash solution and directly add thioflavin S. Incubate cells with thioflavin S solution for 15 min at 20°C–22°C protected from light.
-
53.
Aspirate the solution and wash the cells twice with 50% ethanol in water for 20 min each.
-
54.
Wash the cells once with 80% ethanol in water for 20 min.
-
55.
Remove the ethanol and wash with Triton-PBS for 5 min.
-
56.
Just prior to mounting, remove the Triton-PBS and briefly rinse one time with water. Immediately mount cover glass slips onto microscope slides with 10 μL of DAPI mounting media, and allow to dry for 10 min in the dark. Finally, the edges of the cover glass should be sealed with a light layer of clear nail polish.
Note: If co-staining with thioflavin S, first perform a comparison of mounting media with and without DAPI to determine if crossover occurs in the user’s particular microscopy setup. Mounting media without DAPI is available from Southern Biotech (Cat# 0100-01).
-
57.
Visualize results using a confocal microscope for α-synuclein and DAPI. Using the confocal setup described in point 4 under “equipment,” the excitation/emission settings are as follows: DAPI, excitation= 405 nm laser line, emission = 410–450 nm; α-Synuclein-Alexa Fluor 568, excitation = 561 nm laser line, emission at 610–700 nm.
-
58.
For α-synuclein/thioflavin S colocalization, use a microscopy setup with exciter filters described above in point 5 under the “equipment” section. For thioflavin S, excitation = 400–450 nm, emission = 450–550 nm; α-synuclein-Alexa Fluor 568, excitation = 510–550 nm, emission at 610–700 nm.
Figure 1.

Representative example of α-synuclein and thioflavin S staining in cultured cells
(A) Transfected SH-SY5Y cells expressing A53T α-synuclein (α-syn) were cultured as described (Mazzulli et al., 2006), fixed, and stained using α-syn syn211 (red) and Thio S (green). The microscopy setup included an Olympus IX-FLA using BP360-370 (DAPI), BP420-480 (Thio S), and BP510-550 (α-syn) exciter filters.
(B) iPSC-derived midbrain neurons from controls or patients harboring the A53T α-synuclein mutation were cultured for 90 days, fixed, and stained using LB509 for α-syn (red) and Thio S (green), and nuclei were detected with DAPI (blue), using a similar setup as described in (A).
(C) Representative images of Thio S stained α-synuclein positive neurites from A53T iPSC-derived neurons as described in (B). The scale bars are 10 μm in all images. Arrowheads indicate large juxtanuclear inclusions, while arrows indicate cells with smaller punctated aggregates and diffuse Thio S staining. Asterisks show examples of background/non-specific staining of dead cells.
-
59.
Collect between 5 and 10 image fields per cover glass, typically using a 40× or 63× oil objective lens. The number of images to collect depends on the cell confluency. Ideally, between 10 and 20 cells per field should be obtained, with 100–200 cells total to use for quantification.
Note: If the aim of the experiment is to determine a change in pathological aggregates between cell lines, genotypes, or drug treatments, it is important to blind-code the samples and seek optimal fields of view based on the quality of the cells.
-
60.Parameters for identifying amyloidogenic α-synuclein aggregates:
-
a.Identify the size and shape of the inclusion: Usually juxtanuclear inclusion bodies are around 1/16-th to 1/8-th of the size of the nucleus (arrow heads in Figures 1A and 1B). Smaller puncta may be visible in the cytoplasm (arrow in Figure 1A). Large, elongated inclusions may fill the entire diameter of neuronal extensions (see Figure 1C). Occasionally, a diffuse thioflavin S signal may be present, however they are most often punctate. The appearance of the signal will depend on the resolution capabilities of the microscope. Some super-resolution microscopes may reveal more fine, punctated structures.
-
b.Location of the inclusion: Amyloidogenic aggregates tend to collect near the nucleus at the microtubule-organizing center. Aggregates in aged iPSC-derived midbrain neurons also occur in neurites and elongated structures that tend to expand a portion of the neuronal extension (Figure 1C).
-
a.
Expected outcomes
In this protocol, we describe biochemical and immunocytological staining methods for detection of pathological forms of α-synuclein within models of Parkinson’s disease and related synucleinopathies. Most of the procedures described here can also be applied to frozen brain tissue from human or mouse models. Our recently published work demonstrates how these techniques can be applied to determine mechanisms of aggregation and discovery of novel therapeutic pathways (Cuddy et al., 2019; Mazzulli et al., 2016a; Mazzulli et al., 2016b; Zunke et al., 2018). While each individual method we describe here is suitable for detection of pathological forms of α-synuclein, it is best to use a combination of methods to reach conclusive results.
The protocol described here is optimized for detection of α-synuclein in iPSC-derived neurons or brain tissue. Other dividing cell lines may be used, however it should be noted that some rapidly dividing cell lines have a very different metabolic rate compared to post-mitotic neurons. Therefore, even though dramatic levels of α-synuclein can be overexpressed in cell lines such as 293T cells, it can be difficult to detect insoluble aggregates, since aggregates require time to polymerize, mature, and undergo post-aggregation stabilization such as oxidative cross links. If cells are dividing too rapidly, there will be insufficient time for insoluble polymers to form, and instead they will be diluted out during cell division. Therefore, the cell model used to study protein aggregation should be carefully considered. These issues have been discussed in detail in a recent review (Zunke and Mazzulli, 2019).
Sequential protein extraction followed by western blot analysis can distinguish between physiological and pathological forms of α-synuclein. It is important to note that α-synuclein solubility is operationally defined based on decades of work from labs that have previously characterized the biophysical and biochemical properties of pathogenic aggregates. The homogenization step in 1% Triton X-100 buffer will efficiently disrupt cell membranes and subcellular organelles, releasing soluble proteins into solution while larger polymers remain as particulates. The homogenate containing a mix of soluble and insoluble materials may therefore appear turbid. Clarification of the homogenate is done by ultracentrifugation at 100,000 × g for 30 min, which previous studies have found is sufficient to pellet pathogenic aggregates of α-synuclein in vivo (Duda et al., 2002) as well as purified synthetic α-synuclein fibrils (Giasson et al., 1999). In addition to pathogenic aggregates, other large physiological polymers will also be present in the pellet fraction. These are most often structural proteins such as the intermediate filament protein vimentin, which can be used as a loading control.
Imaging inclusions in fixed cells is an important method to confirm biochemical assays. This can be achieved by staining for α-synuclein alone, or by co-staining with thioflavin S to detect amyloidogenic aggregates. Expectations for this analysis have been discussed above under step 60. Using a positive control, such as PFF-treated cultures, will help to train the experimenter’s eye for identifying aggregates, as well as to determine the optimal microscopy setup. Figure 1 shows representative examples of α-synuclein/thioflavin S colocalization in cultures that express A53T α-synuclein.
Since α-synuclein is normally a highly soluble protein under physiological conditions, increased detection of α-synuclein in the Triton-insoluble fraction would suggest that the protein is misfolded and aggregated into either amyloid-like fibrils or amorphous structures. Representative examples of soluble and insoluble α-synuclein western blots can be found in Figure 1A of Zunke et al. (2018). Amyloidogenic inclusions can be confirmed by thioflavin S staining described above, or by immuno-electron microscopy (not described here, but utilized in Zunke et al. (2018). Gel filtration analysis of soluble lysates can offer further insight into the aggregation state of the protein. Our previous studies indicated that physiological α-synuclein exists in equilibrium as a soluble monomer, as well as a soluble oligomer in human iPSC-derived midbrain neurons (Zunke et al., 2018). An elevation of the oligomer/monomer ratio usually indicates that the protein is forming pathogenic oligomers that are intermediates of amyloid fibrils. For a representative example of a gel filtration analysis of α-synuclein, please see Figure S2 of Zunke et al. (2018). The oligomeric fraction can be further analyzed by a battery of assays that are described in detail elsewhere (Zunke et al., 2018).
For all of the assays described above, we use multiple antibodies against α-synuclein that have been extensively characterized by us and other labs. Using a combination of antibodies that detect certain post-translational modifications associated with disease (e.g., nitration, oxidization, or phosphorylation) and epitopes that span the entire protein sequence can provide a more comprehensive view and insight into the mechanism of aggregation.
Limitations
A limitation of the sequential extraction protocol will likely be sensitivity at detecting insoluble, pathogenic α-synuclein in culture models. In brains of some transgenic mouse models that express aggressive α-synuclein mutations (e.g., Giasson et al., 2002; Tsika et al., 2010), it should be relatively easy to detect insoluble α-synuclein if starting with >20 mg of tissue. However, tissue culture models do not accumulate the same levels of insoluble α-synuclein as what is typically found in vivo. This is likely due to the incubation/maturation time and different environment that occurs in vitro. For example, cultures undergo regular media changes which replenishes essential co-factors, vitamins, and other supplements that are found in neural basal/B27 media. Detection of aggregates is even more challenging in dividing cells lines as discussed above and elsewhere (Zunke and Mazzulli, 2019). However, some detection issues can be overcome by simply increasing the starting culture material and using a more sensitive antibody that preferentially detects pathological α-synuclein. It may be required to harvest a confluent 6-well or 10 cm-sized dish, and use a very small volume of SDS extraction buffer to the Triton-insoluble pellet (e.g., 50–100 μL). Loading up to 80 μg per gel lane is possible, if using a 10-well, 1.5 mm thick, tris-glycine system.
For gel filtration analysis, it is important to keep in mind that the protein size is an estimate that is based on the elution profile of protein standards that are all globular with well-defined structures. The size exclusion technique reports on a particular proteins’ adsorption and elution behavior in solution through the gel matrix. Monomers of α-synuclein are not globular in structure (Weinreb et al., 1996) but are an extended, rod-like shape. Therefore, although α-synuclein monomers are predicted to weigh 14,460 Da based on amino acid sequence and mass spectrometry, the extended shape causes it to elute off of a gel filtration column much earlier than expected, at around a MW of a 60 kDa globular protein. Therefore, it is more accurate to report the Stoke’s radius or elution volume instead of the MW, since the latter is an interpretation. In this case, the radius indicates how much space a particle or protein occupies in the gel matrix.
Another issue in gel chromatography of oligomeric species in complex cell lysates is the possibility of protein-protein interactions that may alter the elution profile. It may be necessary to perform further studies to characterize the protein. If α-synuclein extracted from a lysate elutes off of the column very early (e.g., 100 Å), it could be either a self-associated oligomer, or a monomer that is in a large complex with other proteins. We have previously used immuno-FRET assays, or dot-blots using antibodies that selectively bind oligomers, to help distinguish between these possibilities (Zunke et al., 2018).
Thioflavin S is a commonly used dye for the detection of amyloidogenic inclusions because of its high binding affinity for the β-pleated sheet contents of proteins. However, one drawback of thioflavin S is that it is not protein specific and will detect any protein with a β-pleated sheet. Therefore, careful colocalization analysis needs to be performed when examining whether a protein has formed amyloidogenic inclusions. As with most fluorescence techniques, a significant limitation to immunofluorescence is sample photobleaching. Thioflavin S is not as robust as some fluorophores such as Alexa dyes, and rapid fading of thioflavin fluorescence intensity may occur. Therefore, using optimal concentrations of the dye, and rapidly imaging the samples may help in preventing signal fade. It should be noted that thioflavin T can also be used for staining cultures, which shifts its excitation maximum from 385 to 450 nm only upon binding to β-pleated sheets, and can be detected at 482 nm (FITC channel) (LeVine, 1999). Most labs use thioflavin S for histology or cell staining, however the reasons for using this dye over thioflavin T are not clear and likely to be for historical reasons.
Troubleshooting
Problem 1
In section 1, the Triton-insoluble α-synuclein is not detectable.
Potential solution
Repeat the experiment by increasing the amount of starting material and dissolve the insoluble pellet in a smaller extraction volume to concentrate the sample. Increase the amount of protein loaded per lane. Also, increasing the concentration of the α-synuclein antibody and incubating the blots for several days should increase sensitivity. Some ECL-based reagents may also provide greater sensitivity, but should be used with caution since it is easy to obtain saturated, non-quantifiable signals.
Problem 2
In section 1 or 3, there is protein carryover within the SDS fraction.
Potential solution
Excessive carry over of soluble α-synuclein into the insoluble pellet can generate misleading results and often occurs from inefficient removal of the supernatant fractions after ultracentrifugation. Carryover is detected by probing insoluble fractions with loading control proteins that are mainly soluble (GAPDH or NSE). Performing at least one pellet wash with Triton-containing extraction buffer is important prior to doing an SDS extraction. If carryover is significant, it may be necessary to wash and spin down the pellet 2–3 times.
Problem 3
In section 3, there is very poor or no detection of α-synuclein via western blot analysis.
Potential solution
α-Synuclein tends to easily detach from western blotting membranes. A mild fixation of the membrane with 0.4% PFA is necessary to prevent this detachment. Alternatively, increasing the concentration of the primary antibody or incubating the membranes with the primary for an additional night may be helpful.
Problem 4
In section 4, there is high background when performing thioflavin S immunofluorescence imaging.
Potential solution
Be sure to extensively wash out the thioflavin S after removing it from cells. It is also critical to capture images at the appropriate exposure and to adjust the imaging scaling so that non-specific background signal is eliminated. Some images can appear positive when in fact they have simply been overexposed. Background levels are established by staining SNCA knock-out cells, control cells that do not demonstrate pathology, or disease cells with the thioflavin S dye omitted. In the case of amyloidogenic α-synuclein aggregates, it is required to co-stain the thioflavin signal by colocalization with α-synuclein antibodies and follow the guidelines noted above for identifying aggregates such as size and location within the cell. The addition of negative controls (e.g., SNCA knock-out) and positive controls (cultures where pre-formed fibrils are added) are essential in setting up the technique and training the observer’s eye so that an authentic signal can be identified with confidence.
Problem 5
In section 4, there is sample bleaching when performing thioflavin S immunofluorescence imaging.
Potential solution
Thioflavin S is not as robust as some fluorophores used in immunofluorescence protocols such as Alexa dyes, and can bleach quickly. Therefore limiting the time of light exposure time is important. Additionally, images should be taken as quickly as possible to avoid loss of fluorescence intensity.
Problem 6
In section 3, high back pressure in the gel filtration column occurs, or low detection of α-synuclein in fractions.
Potential solution
High back pressures can be observed if particulates or insoluble materials are accidentally injected in the LC system or get into the column. This occurs more often in older, re-used columns that are not well maintained. It is critical to clean the gel filtration column every 10-to-20 separations with 1 column volume 0.1 M NaOH followed by water, to remove any aggregates that have retained in the column. Aggregates that are stuck in the column will not only increase the back pressure, but also will reduce the amount of α-synuclein that elutes from the column. It is also critical to change the top filter on the bed of the resin as required, usually when the pressure increases by 1–2 bar from its normal baseline value.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Joseph R. Mazzulli, jmazzulli@northwestern.edu
Materials availability
All of the reagents described here are commercially available through the indicated vendors.
Data and code availability
This study did not generate nor analyze any datasets or codes.
Acknowledgments
This work was supported by grants RF1NS109157 and R01NS092823 from the NINDS/NIA awarded to J.R.M.
Author contributions
The protocols were written by J.R.M. and I.S.
Declaration of interests
J.R.M. has received consulting fees from SK Biopharmaceuticals, Neuron23, Sanofi-Genzyme, and Lysosomal Therapeutics. J.R.M. has ownership/investment interests in Lysosomal Therapeutics.
References
- Cuddy L.K., Wani W.Y., Morella M.L., Pitcairn C., Tsutsumi K., Fredriksen K., Justman C.J., Grammatopoulos T.N., Belur N.R., Zunke F. Stress-induced cellular clearance is mediated by the SNARE protein ykt6 and disrupted by alpha-synuclein. Neuron. 2019;104:869–884.e11. doi: 10.1016/j.neuron.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda J.E., Giasson B.I., Mabon M.E., Lee V.M., Trojanowski J.Q. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann. Neurol. 2002;52:205–210. doi: 10.1002/ana.10279. [DOI] [PubMed] [Google Scholar]
- Giasson B.I., Duda J.E., Quinn S.M., Zhang B., Trojanowski J.Q., Lee V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Giasson B.I., Uryu K., Trojanowski J.Q., Lee V.M. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999;274:7619–7622. doi: 10.1074/jbc.274.12.7619. [DOI] [PubMed] [Google Scholar]
- Kriks S., Shim J.W., Piao J., Ganat Y.M., Wakeman D.R., Xie Z., Carrillo-Reid L., Auyeung G., Antonacci C., Buch A. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson's disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.R., Kamitani T. Improved immunodetection of endogenous alpha-synuclein. PLoS One. 2011;6:e23939. doi: 10.1371/journal.pone.0023939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine H., 3rd Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Mazzulli J.R., Mishizen A.J., Giasson B.I., Lynch D.R., Thomas S.A., Nakashima A., Nagatsu T., Ota A., Ischiropoulos H. Cytosolic catechols inhibit alpha-synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J. Neurosci. 2006;26:10068–10078. doi: 10.1523/JNEUROSCI.0896-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli J.R., Zunke F., Isacson O., Studer L., Krainc D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. U S A. 2016;113:1931–1936. doi: 10.1073/pnas.1520335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli J.R., Zunke F., Tsunemi T., Toker N.J., Jeon S., Burbulla L.F., Patnaik S., Sidransky E., Marugan J.J., Sue C.M. Activation of beta-glucocerebrosidase reduces pathological alpha-synuclein and restores lysosomal function in Parkinson's patient midbrain neurons. J. Neurosci. 2016;36:7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsika E., Moysidou M., Guo J., Cushman M., Gannon P., Sandaltzopoulos R., Giasson B.I., Krainc D., Ischiropoulos H., Mazzulli J.R. Distinct region-specific alpha-synuclein oligomers in A53T transgenic mice: implications for neurodegeneration. J. Neurosci. 2010;30:3409–3418. doi: 10.1523/JNEUROSCI.4977-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley L.A., Luk K.C., Lee V.M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014;9:2135–2146. doi: 10.1038/nprot.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinreb P.H., Zhen W., Poon A.W., Conway K.A., Lansbury P.T., Jr. NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded. Biochemistry. 1996;35:13709–13715. doi: 10.1021/bi961799n. [DOI] [PubMed] [Google Scholar]
- Zunke F., Mazzulli J.R. Modeling neuronopathic storage diseases with patient-derived culture systems. Neurobiol. Dis. 2019;127:147–162. doi: 10.1016/j.nbd.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunke F., Moise A.C., Belur N.R., Gelyana E., Stojkovska I., Dzaferbegovic H., Toker N.J., Jeon S., Fredriksen K., Mazzulli J.R. Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron. 2018;97:92–107.e10. doi: 10.1016/j.neuron.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate nor analyze any datasets or codes.