

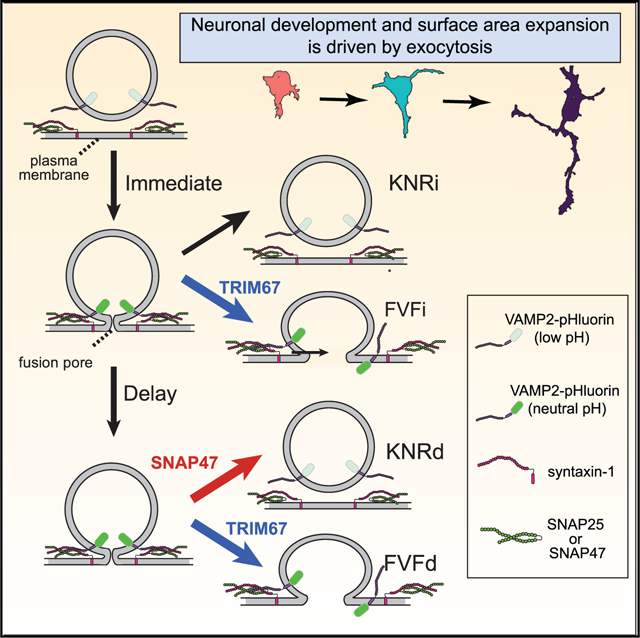
SUMMARY
Neuronal morphogenesis involves dramatic plasma membrane expansion, fueled by soluble N-ethylmaleimide-sensitive factor attachment protein eceptors (SNARE)-mediated exocytosis. Distinct fusion modes described at synapses include full-vesicle fusion (FVF) and kiss-and-run fusion (KNR). During FVF, lumenal cargo is secreted and vesicle membrane incorporates into the plasma membrane. During KNR, a transient fusion pore secretes cargo but closes without membrane addition. In contrast, fusion modes are not described in developing neurons. Here, we resolve individual exocytic events in developing murine cortical neurons and use classification tools to identify four distinguishable fusion modes: two FVF-like modes that insert membrane material and two KNR-like modes that do not. Discrete fluorescence profiles suggest distinct behavior of the fusion pore. Simulations and experiments agree that FVF-like exocytosis provides sufficient membrane material for morphogenesis. We find the E3 ubiquitin ligase TRIM67 promotes FVF-like exocytosis in part by limiting incorporation of the Qb/Qc SNARE SNAP47 into SNARE complexes and, thus, SNAP47 involvement in exocytosis.
Graphical Abstract
In Brief
Urbina et al. identify four exocytic modes in developing neurons: KNRd, KNRi, FVFd, FVFi. Simulations and experiments suggest that FVFi and FVFd provide material for plasma membrane expansion. Deletion of Trim67 decreases FVFi and FVFd while reducing surface area. SNAP47 incorporation into SNARE complexes alters fusion pore behavior, increasing KNRd.
INTRODUCTION
Membrane-bound compartments define eukaryotic cells. The transfer of material between compartments requires fusion of lipid bilayers. A fusion pore between bilayers is initiated by assembly of SNARE proteins into a complex (Figure 1A). Specific SNAREs mediate fusion between compartments; for example, vesicle-associated VAMP2 and plasma membrane (PM)-associated syntaxin-1 and SNAP25 facilitate docking and fusion of synaptic vesicles (Sudhof and Rothman, 2009; Schoch, 2001; Söllner et al., 1993). Evoked exocytosis of neurotransmitters involves multiple fusion events clustered at the synapse, a diffraction-limited area. This restriction renders visualization of individual fusion events difficult. During neuronal development, however, constitutive exocytosis mediated by the same SNAREs is not spatially restricted and is sufficiently infrequent to resolve individual fusion events (Bello et al., 2016; Urbina et al., 2018).
Figure 1. Heterogeneity in single-vesicle exocytic fusion events.
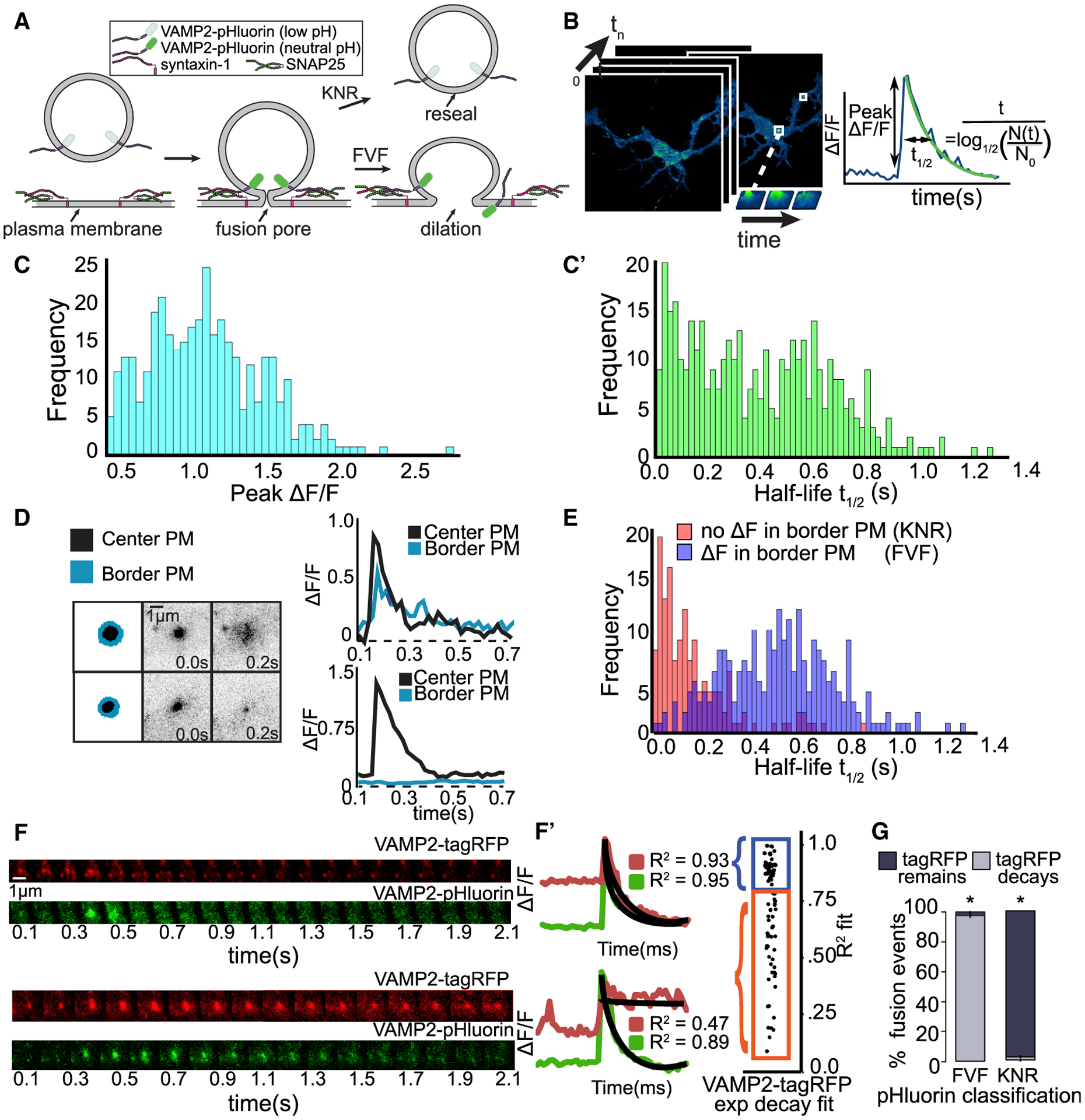
(A) Schematic of exocytic fusion depicts quenching and fluorescence of VAMP2-pHluorin during full-vesicle fusion (FVF) and kiss-and-run fusion (KNR).
(B) Automated detection of exocytic events (green) over time. Features are extracted, including the peak ΔF/F and half-life of fluorescence.
(C) Frequency of peak ΔF/F (C) and fluorescence half-life (t1/2) (C’) of individual events.
(D) Schematic of region of exocytic event (black) and bordering plasma membrane (PM; blue). Plots of ΔF/F over time demonstrate an event with spreading of fluorescence into bordering PM (top, FVF) and an event without spreading of fluorescence (bottom).
(E) Frequency of t1/2 categorized by ΔF in bordering PM. Spreading fluorescence events (ΔF, presumably FVF) have slower fluorescence decay than those without (no ΔF, presumably KNR).
(F) Dual-color imaging of VAMP2-pHluorin and VAMP2-tagRFP (red fluorescence protein) reveals vesicle fusion and vesicle fate, respectively. Example images and ΔF/F curves show that VAMP2-tagRFP either exponentially decays after ΔF/F (top, FVF) or remains in vesicle (bottom, KNR).
(G) Classification of fusion events based on VAMP2-pHluorin behavior and VAMP2-tagRFP behavior agree. Error bars represent standard error of the percent. *p < 0.05, based on a chi-square analysis of expected ratios if classes were assigned randomly.
Prior to synaptogenesis, VAMP2-mediated fusion inserts membrane material into the PM, contributing to neuronal morphogenesis (Urbina et al., 2018). However, potentially not all exocytic events add membrane. At the synapse, two modes of exocytosis are described and yet their contribution remains controversial (Alabi and Tsien, 2013; Albillos et al., 1997; He and Wu, 2007; Elhamdani et al., 2006). During full-vesicle fusion (FVF), the fusion pore dilates and the vesicle membrane incorporates into the PM. In contrast, during kiss-and-run fusion (KNR), the fusion pore opens transiently, secretes lumenal cargo, and reseals, retaining vesicular identity (Alabi and Tsien, 2013; Albillos et al., 1997; Bowser and Khakh, 2007; Holroyd et al., 2002; Wang et al., 2003). As presumably only FVF donates membrane, regulating the mode of exocytosis may tune PM expansion during development.
Fusion pore size and kinetics vary with fusion mode, yet how fusion mode is regulated is debated. In vitro, the number of SNARE complexes regulate pore dilation (Bao et al., 2018; Bello et al., 2016). The composition of SNARE complexes also regulates the fusion pore. The SNARE family contains more than 60 members (Burri and Lithgow, 2004). SNAP47 is an atypical SNAP25 family member that can substitute for SNAP25 in complexes with VAMP2 and syntaxin-1 in vitro. SNARE-interacting proteins, such as TRIM9, synaptotagmins, complexins, and α-synuclein, may also regulate fusion pores (Archer et al., 2002; Logan et al., 2017; Wang et al., 2003; Winkle et al., 2014). We found that an interaction between SNAP25 and the brain-enriched E3 ubiquitin ligase TRIM9 prevents SNARE complex formation and attenuates the exocytosis and axon branching in developing neurons (Winkle et al., 2014). Like SNAREs, the mammalian TRIM family of ubiquitin ligases contains ~70 members (Napolitano and Meroni, 2012). The TRIM9 paralog TRIM67 is enriched in the developing cortex and regulates axonal projections, filopodia, spatial learning, and memory (Boyer et al., 2018, 2020) but has not been implicated in exocytosis.
We use VAMP2-pHluorin expression to explore modes of single exocytic events in developing neurons. We develop computer vision classifiers to reveal four distinct modes of fusion, which exhibit distinct fluorescence behavior following fusion pore opening. Experimental manipulations and simulations agree that membrane provided by FVF-like modes of VAMP2-mediated exocytosis approximate PM expansion of developing neurons. The E3 ubiquitin ligase TRIM67 regulates exocytic mode by reducing SNAP47 protein and limiting SNAP47 incorporation into SNARE complexes. SNAP47 colocalizes with a subset of VAMP2-mediated exocytic events and alters fusion mode.
RESULTS
Heterogeneity in vesicle fusion
VAMP2 is the most highly expressed vesicle (v)-SNARE during neuronal development (Cardoso-Moreira et al., 2019; La Manno et al., 2016; Urbina and Gupton, 2020) and is present on recycling vesicles and post-Golgi vesicles (Figures S1A and S1B). VAMP2-pHluorin-labeled exocytic events in embryonic murine cortical neurons were imaged at 2 days in vitro (2 DIV) by total internal reflection fluorescence (TIRF) microscopy. pHluorin is a pH-sensitive variant of GFP that is quenched in the vesicular lumen and fluorescent at extracellular pH (Miesenböck et al., 1998; Figure 1A). Fusion events were characterized by a rapid fluorescence increase when the fusion pores open (Figure 1B) and slower fluorescent decay. Exocytic events were automatically detected (Urbina et al., 2018). To evaluate heterogeneity, we examined the normalized peak change in fluorescence per event (peak ΔF/F; Figures 1B and 1C), an estimate of relative VAMP2-pHluorin per vesicle, and the event half-life (t1/2; Figures 1B and 1C’), which describes fluorescence decay. Unlike peak ΔF/F, t1/2 exhibited a bimodal distribution, suggesting two populations of events. We hypothesized that these peaks could represent FVF and KNR (Figure 1A; Alabi and Tsien, 2013; Holroyd et al., 2002; Wang et al., 2003). FVF in astrocytes exhibits diffusion of VAMP2-pHluorin from the fusion site, whereas KNR remains fluorescent until pore closure and re-acidification (Bowser and Khakh, 2007). Events with longer t1/2 exhibited fluorescence spreading, consistent with diffusion, whereas events with a rapid t1/2 did not (Figure 1D). These responses aligned with the bimodal distribution of t1/2 (Figure 1E). Thus, there is sufficient information to differentiate VAMP2-pHluorin fusion events.
Vesicles that fuse by KNR retain their identity; yet, re-acidification prevents visualization. We imaged neurons expressing both VAMP2-pHluorin and VAMP2-tagRFP (Figure 1F). Exponential decay of both markers confirmed diffusion of v-SNAREs during putative FVF (Figure 1F’, top, blue). Events without VAMP2-pHluorin diffusion maintained VAMP2-tagRFP fluorescence (Figure 1F’, bottom, orange), indicating vesicles persisted following putative KNR. The agreement between VAMP2-tagRFP fluorescence behavior and pHluorin-based classification (Figure 1G) indicates that FVF and KNR were discernable in the pHluorin-based assay.
Multiple unbiased classifiers converge on four exocytic modes
To explore fusion heterogeneity in an unbiased fashion, we turned to classification and clustering approaches. Most clustering algorithms rely on a single method. In contrast, we used a three-pronged approach. Detected events were first temporally aligned to peak ΔF/F (Figure 2A, red dots). As a ground truth method, we selected features that discern differences between KNR and FVF, including ΔF/F and t1/2 (Figure 2B). Principal-component analysis (PCA) was performed, and five principal components (PCs) capturing 85% of the variance were kept (Figure 2B’). Second, we used hierarchical cluster analysis (Figure 2C). Hierarchical clustering summated a Euclidean distance cost (Σ) between each pair of events (Figure 2Ci) and iteratively merged lowest-cost events (agglomerative) or divided events (divisive, Figure 2Cii). Euclidean distance matrices demonstrated groupings of events (Figure 2Ciii). Third, we used dynamic time warping (DTW; Figure 2D) to measure spatiotemporal dissimilarity between all event pairs. DTW allowed non-linear temporal matching between events (Figure 2Di) by creating a time warp matrix, for which each square was the Euclidean distance between respective time points of two ΔF/F time series (Figure 2Dii). The optimal warp path for a pair of events was the lowest cost path through the matrix, which may be distinct from the Σ Euclidean distance (Figure 2Dii). The sum of the warp path (DTW cost) plotted on a distance matrix demonstrated grouping of events (Figure 2Diii).
Figure 2. Multiple unbiased classifiers converge on four exocytic modes.
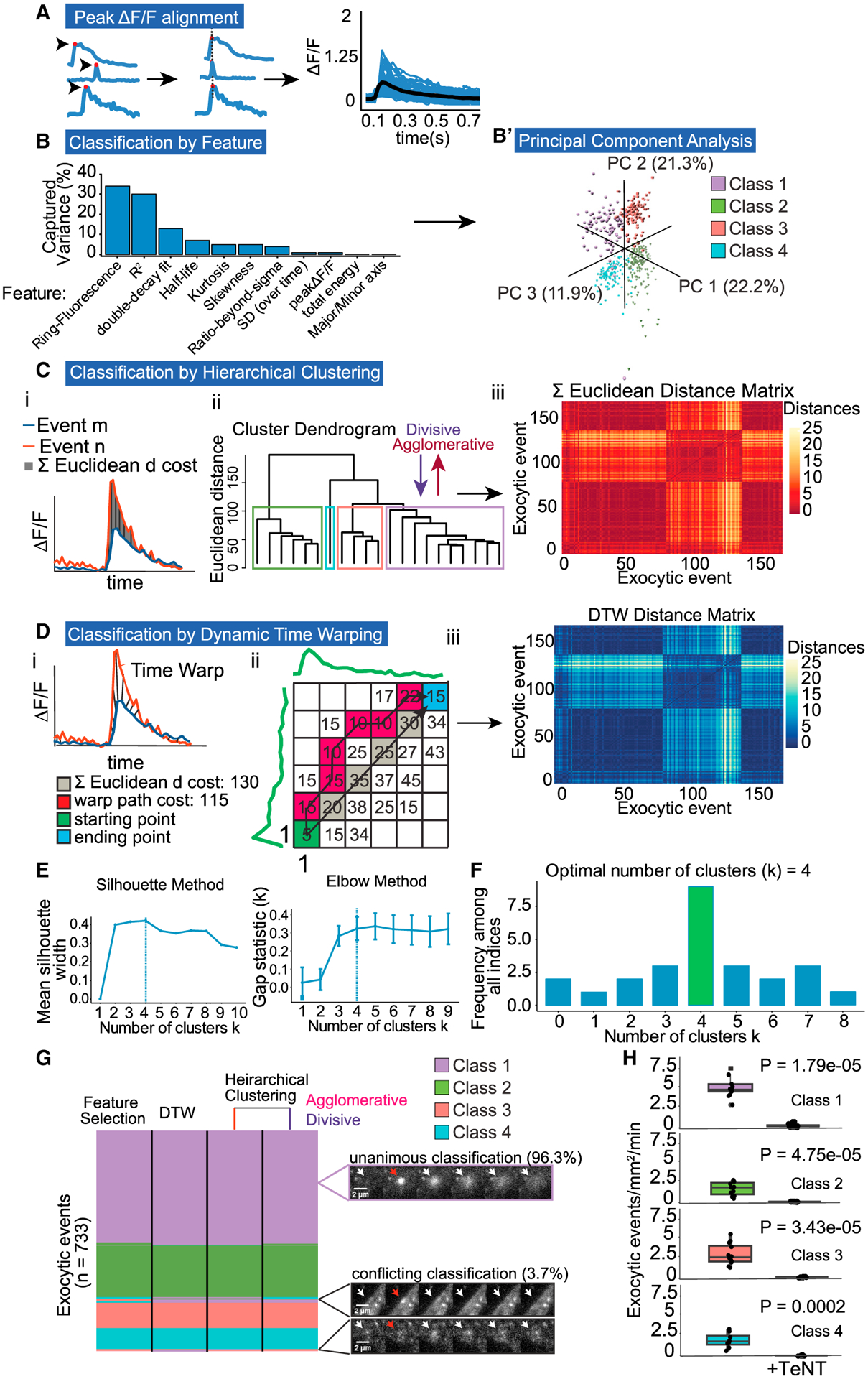
(A) ΔF/F curves are temporally registered to peak ΔF/F (red dot). Registered ΔF/F plot showing average (black) and individual events (blue).
(B) Features that represent % of captured variance from PCA.
(B’) Three principal components plotted reveal four exocytic classes.
(C) Event classification by hierarchical clustering. (i) ΔF/F plot depicting the summed (Σ) Euclidean distance between two exocytic events, m and n. (ii) Dendrogram of Σ Euclidean distances between multiple events, linked by agglomerative or divisive hierarchical clustering reveal four classes. (iii) Each pixel in distance matrix is the Σ Euclidean distance between two ΔF/F curves (n = 733).
(D) Event classification by dynamic time warping (DTW). (i) ΔF/F plot depicting time warped distance between two events, m and n. (ii) A matrix of Euclidean distances measured at all time points comparing a pair of events, starting at peak ΔF/F (green square, coordinates 1,1). Unlike the Σ Euclidean distance (gray), DTW uses the warp path with the lowest cost (red squares) to reach the ending point (blue square). The sum of the total warp path is the DTW cost per pair of events. (iii) Each pixel in distance matrix is the DTW cost between two ΔF/F curves (n = 733).
(E) Two example indices, silhouette and elbow methods, used in the plurality rules committee suggest four exocytic classes. Error bars report standard error.
(F) Decision histogram of the committee of indices, with the plurality of indices choosing four exocytic classes (8/20).
(G) Classification comparison of each clustering method. A total of 96.3% of 733 events were classified unanimously. Purple inset: representative unanimously classified exocytic event. Black inset: representative events with conflicting classification. Cell edge effects (top black inset) or low signal-to-noise ratio (bottom black inset) account for the majority of conflict. White arrows denote exocytic events. Red arrows denote peak ΔF/F.
(H) Frequency of all event classes drops to near zero with tetanus toxin (TeNT) treatment (Welch’s t test, n = 12 cells per experiment).
Using the output of each classifier, we took the plurality rules decision from a committee of the most common and best-performing clustering indices (Charrad et al., 2014) to confidently determine the number of exocytic clusters (k). Among the indices used were the gap statistic and the elbow method (Figure 2E). The committee converged on a k of 4 classes for each classification method instead of the 2 predicted by the literature, with 8 of 20 indices selecting 4 classes (Figure 2F). A total of 96.3% of 733 exocytic events were classified unanimously by different methods, indicating variations between events, and clusters were sufficiently robust to be discerned by multiple methods (Figure 2G). Inspection of conflicting classifications revealed edge cases, with low signal to noise or occurrence close to or on the cell periphery (Figure 2G, inset). To confirm that unexpected classes were not an artifact of false-positive identification of exocytic events, we treated neurons with tetanus neurotoxin (TeNT; Figure 2H), which cleaves VAMP2 and blocks fusion (Link et al., 1992), including VAMP2-pHluorin-mediated events (Urbina et al., 2018). All four classes were sensitive to TeNT and thus are bona fide exocytic events.
Distinguishing features of exocytic modes
Class averages and representative image sequences revealed distinct fluorescence profiles (Figure 3A; Video S1). Class 1 and 2 exhibited an instantaneous fluorescence decay after peak ΔF/F (fusion pore opening). In contrast, class 3 and 4 exhibited a delay between peak ΔF/F and fluorescent decay onset, observed as a plateau in ΔF/F. A single exponential decay fit well to class 1 and 2 (Figure 3B) but was a poor fit for class 3 and 4 (Figure 3C). Sequential exponentials resulted in a higher R2 (Figure 3D). The t1/2s for class 3 and 4 were calculated from the second exponential, which revealed no differences in t1/2 between class 1 and 3 or class 2 and 4 (Figure 3E).
Figure 3. Distinguishing features of four modes of exocytosis.
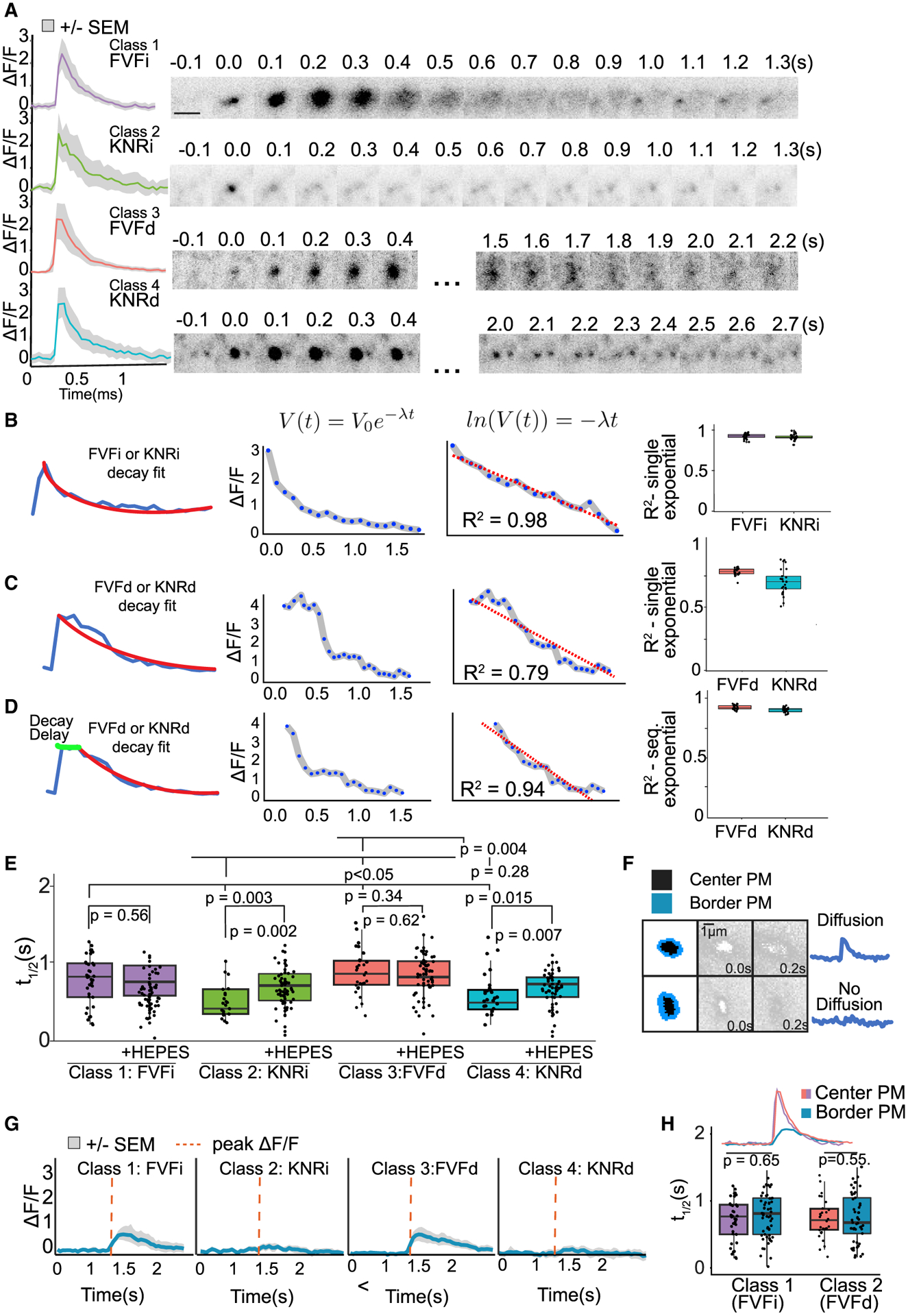
(A) Mean fluorescent curves ± SEM (gray) and representative images from each class (right).
(B and C) Example single exponential decay fit (V(t) = V0e−λt) to class 1 or 2 instantaneous (i) events (B) or to class 3 or 4 delay (d) events (C). The log of an exponential decay curve (ln(V(t)) = −λt) is linear, and the linear regression fit (R2) is an indicator of how good a fit the exponential decay curve is (center graph). Boxplots (right) of R2 of class 1 (FVFi) and class 2 (KNRi) events show a good fit (B), whereas fit to class 3 (FVFd) and class 4 (KNRd) is poor.
(D) Sequential exponential decay curves better fit to FVFd and KNRd.
(E) t1/2 of each class untreated or treated with HEPES. Class 2 and 4 are HEPES sensitive (n = 14 cells per condition; Welch’s t test followed by Benjamini-Hochberg correction).
(F) Schematic of region of exocytic event (black) and bordering PM (blue). Plots of border PM ΔF/F over time demonstrate an event with fluorescence spreading (top, FVF) and an event without (bottom).
(G) Class means ± SEM of ΔF/F in PM bordering exocytic events. Red dotted line indicates peak ΔF/F of center of event.
(H) t1/2 of fluorescence decay in PM bordering FVFi or FVFd events (border pixels) is not different from the t1/2 of the exocytic event (center pixels) (n = 14 cells per condition; Welch’s t test).
Fluorescence decay following FVF is due to VAMP2-pHluorin diffusion into the bordering PM (Figure 3F). The PM surrounding class 1 and 3 events exhibited a fluorescent peak followed by an exponential decay with the same t1/2 as center pixels (Figures 3G and 3H; Video S1), consistent with diffusion and FVF. In contrast, VAMP2-pHluorin fluorescence did not increase in the PM surrounding class 2 and 4 events (Figures 3F and 3G), suggesting they may be KNR. Loss of fluorescence following KNR is due to either vesicle reacidification or vesicle retreat from the evanescent wave. The t1/2 of class 2 and 4 did not change after increasing evanescent wave depth (Figure S1C), indicating fluorescence loss was not due to vesicle retreat. In contrast, pH buffering by cell-impermeable HEPES slowed fluorescence decay of class 2 and 4 but not class 1 and 3 (Figure 3E). This finding suggested that HEPES entered the fusion pore of class 2 and 4 vesicles and retarded reacidification, consistent with KNR. The t1/2 of HEPES-sensitive events were similar to single-vesicle KNR events measured in astrocytes (Bowser and Khakh, 2007) but faster than those reported at synapses (Atluri and Ryan, 2006). Based on the mechanisms of fluorescence decay and either the instantaneous (i) or delayed (d) onset of fluorescence decay after peak ΔF/F, we named the classes FVFi, (class 1), KNRi (class 2), FVFd (class 3), and KNRd (class 4).
Both VSV-G and FM4–64 localized to vesicles that fused by different modes, indicating fusion mode did not correspond to vesicle class (Figures S1A and S1B). Surface levels of VAMP2-pHluorin varied between cells and experiments but did not alter exocytic mode distribution (Figures S1D and S1E). This result suggested that cellular VAMP2 levels did not alter exocytic modes. There was a stratification of peak ΔF/F between modes (Figure S1F), suggesting vesicular VAMP2 levels may influence fusion mode.
To ensure the exocytic modes were not an artifact of VAMP2-pHluorin, we used alternative fusion reporters. Transferrin receptor (TfR) attached to pHuji, a pH sensitive red fluorescent protein (Shen et al., 2014), showed 100% overlap with VAMP2-pHluorin fusion events (Figure S2A). Using all three classifiers with the committee of indices, we detected four classes of exocytosis for TfR-pHuji with identical ratios to VAMP2-pHluorin (Figures S2B and S2C). The t1/2 of KNRi and KNRd for TfR-pHuji was the same as for VAMP2-pHluorin (Figures S2D and S2E), consistent with the similar pH sensitivity of the fluorophores (Martineau et al., 2017), supporting that decay was due to reacidification. In contrast, the t1/2 of FVFi and FVFd were different (Figure S2D and S2E), consistent with distinct size and diffusion behaviors of VAMP2 and TfR (Di Rienzo et al., 2013; Fujiwara et al., 2002, 2016; Jaqaman and Grinstein, 2012; Lenne et al., 2006).
As an additional marker of exocytosis, we turned to VAMP7, which mediates exocytosis of a discrete population of vesicles in developing neurons, albeit at lower frequencies than VAMP2 (Gupton and Gertler, 2010; Urbina et al.,2018). VAMP7-mediated fusion is autoinhibited by its longin domain (Burgo et al., 2013; Martinez-Arca et al., 2001); a VAMP7-pHluorin construct lacking the longin domain (VAMP7ΔLD) increased the frequency of exocytosis (Urbina et al., 2018) sufficiently to identify exocytic modes. Four modes of VAMP7ΔLD exocytosis were detected by the three classifiers with distinct relative abundance from VAMP2-pHuorin (Figure S2F). The VAMP7ΔLD and TfR data confirm the existence of four exocytic modes.
A truncated VAMP2 alters exocytic mode
The VAMP2 transmembrane domain catalyzes fusion initiation and pore expansion (Dhara et al., 2016). In astrocytes, expression of a mutant lacking the majority of the transmembrane domain (VAMP21–96; Figure S3A) in the presence of endogenous VAMP2 was shown by capacitance measurements to stabilize narrow fusion pores and block full fusion but not transient KNR (Guček et al., 2016). Although lacking the majority of the transmembrane domain, VAMP21–96 continues to fractionate and associate with membranes (Caccin et al., 2003; Wang et al., 2020). To validate classification and determine if fusion pore behavior distinguished modes, we expressed tagRFP-VAMP21–96 with full-length VAMP2-pHluorin, which is capable of and reports fusion. VAMP21–96 reduced exocytic frequency (Figure S3B; Video S2), by specifically reducing FVFi and FVFd events (Figure S3C) and shifting exocytic distribution toward KNRd and KNRi (Figure S3D). This shift aligns with capacitance measurements (Guček et al., 2016) and suggests that the FVF-like fusion is mechanistically distinct from KNR. These data suggest endogenous VAMP2 and VAMP2-pHluorin still mediate fusion, but the mode is affected by the presence of VAMP21–96.
TRIM67 biases exocytic mode toward FVF
Although each class harbors bona fide and distinct exocytic events, the molecular mechanisms influencing exocytic modes remained unclear. Our work identified the E3 ubiquitin ligase TRIM9 as a regulator of exocytosis (Urbina et al., 2018; Winkle et al., 2014). Deletion of Trim9 elevated exocytic frequency, but the ratio of classes was not affected (Figure 4A; Video S3). TRIM67 is a TRIM9 paralog enriched in developing neurons (Boyer et al., 2018). Deletion of Trim67 altered exocytic mode but not frequency; KNRi and KNRd increased 2-fold, whereas FVFi and FVFd decreased (Figures 4A and 4B). Thus, exocytic frequency and mode are independently regulated by E3 ligases. FVFd events were enriched in neurites of both Trim67−/− and Trim67+/+ neurons, and the distribution of exocytic classes within the soma was not distinct from the whole neuron (Figure 4C).
Figure 4. TRIM67 biases exocytic mode toward full-vesicle fusion.
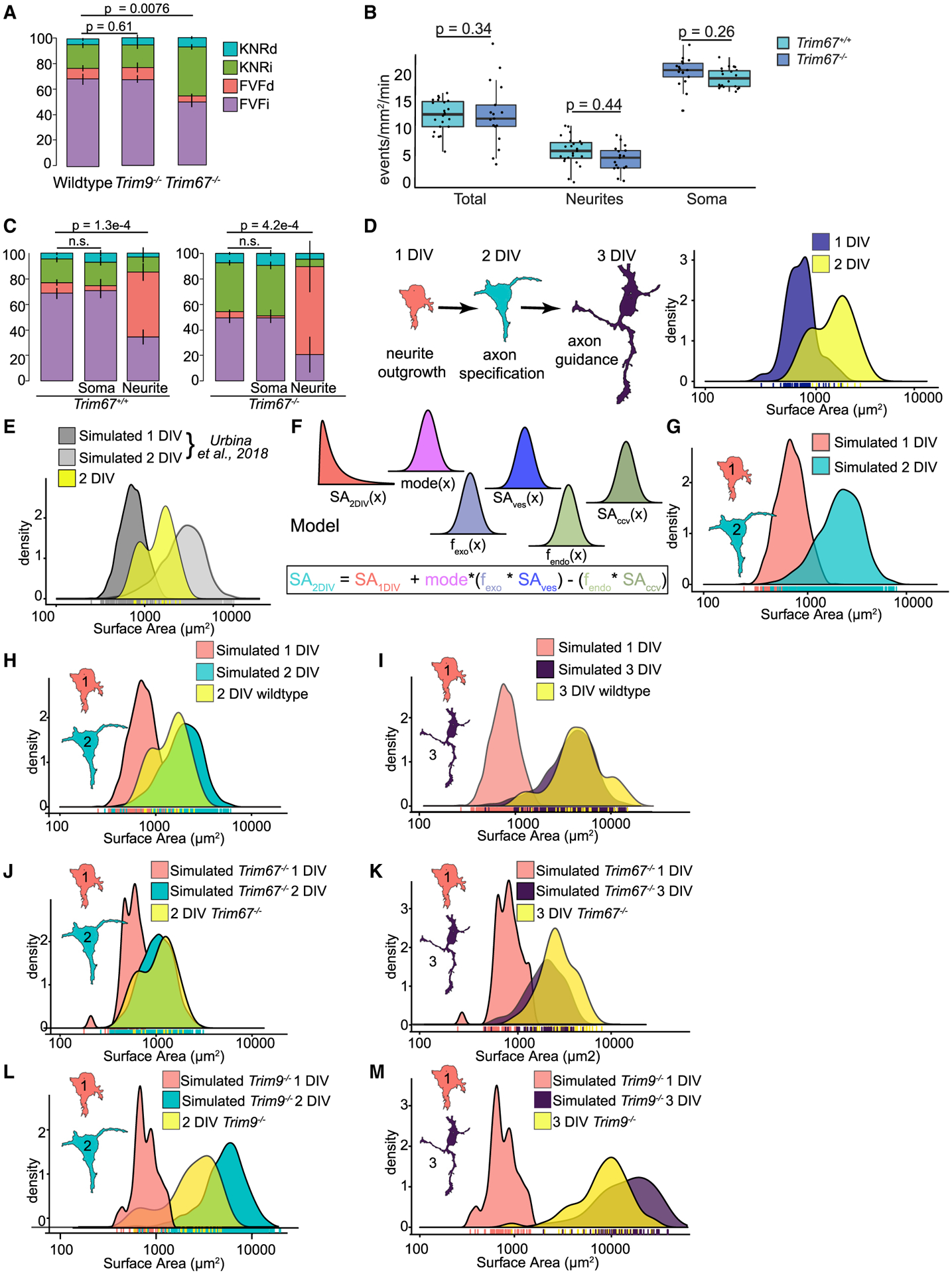
(A) Proportion of KNRd, KNRi, FVFd, and FVFi in wild-type, Trim9−/−, or Trim67−/− neurons (n = 15 neurons per condition; n = 4 biological replicates; multivariate linear regression, error bars report standard error of the percent).
(B) Frequency of exocytic events in the whole neuron, soma, and neurites of Trim67+/+ or Trim67−/− neurons (n = 15 neurons per condition; n = 3 biological replicates; Welch’s t test followed by Benjamini Hochberg correction).
(C) Proportion of KNRd, KNRi, FVFd, and FVFi VAMP2-pHluorin exocytic events in the whole neuron, soma, and neurites of Trim67+/+ or Trim67−/− neurons (n = 15 neurons per condition; multivariate linear regression, error bars report standard error of the percent).
(D) Schematic of neuronal morphogenesis in vitro (left) and empirically measured surface areas from GFP-CAAX expressing wild-type neurons at indicated time points (right). Surface area calculations were performed as described in Urbina et al. (2018).
(E) Simulated 1 and 2 DIV neurons using the model from Urbina et al. (2018) that considers all exocytic events FVF. Empirically measured surface area of cortical neurons in vitro plotted in yellow.
(F) Overview of model that includes exocytic mode. Surface area at 2 DIV (SA2DIV) was estimated by first generating a population of neurons at 1 DIV (SA1DIV) based on empirical measurements of neurons at 1 DIV. The distribution of modes (mode), frequency of exocytosis (fexo), surface area of vesicles (SAves), frequency of endocytosis (fendo), and surface area of clathrin-coated vesicles (SAccv) were incorporated into estimating the surface area expansion.
(G) Simulated surface areas at 1 and 2 DIV. Surface areas at 1 DIV are simulated based on empirical measurements of surface area at that time point. Surface areas at 2 DIV use the 1 DIV simulation and model described in (F).
(H) Simulated surface areas from (G), with empirically measured surface areas at 2 DIV overlaid in yellow demonstrating a better fit than the model that only considers FVF in (E).
(I) Simulated neurons at 3 DIV (purple) with measured surface area at neurons 3 DIV overlaid in yellow.
(J and K) Predicted PM growth simulated at 2 DIV (J) and 3 DIV (K) using the relative proportion of modes from Trim67−/− neurons with empirically measured Trim67−/− neurons overlaid in yellow.
(L and M) Simulations of PM growth at 2 DIV (L) and 3 DIV (M) using the distribution of frequencies from Trim9−/− cortical neurons compared with empirically measured Trim9−/− neurons (yellow overlay).
During morphogenesis, neuronal surface area increases (Figure 4D). Our previous experimental and modeling work suggested that VAMP2-mediated exocytosis provided excess material for PM expansion, which was partially balanced by clathrin-mediated endocytosis (Urbina et al., 2018) (Figure 4). The model used empirically measured surface areas of neurons and non-coated and clathrin-coated vesicles, along with the frequencies of exocytosis and endocytosis to predict PM expansion. A caveat was the assumption that all exocytic events supplied PM material. Upon considering that KNRi and KNRd comprise ~22% of events (Figure 4A), we updated the model with exocytic modes (Figure 4F). Accounting for KNRi and KNRd improved the similarity of predictions to measured values of surface area (Figures 4H and 4E). We extended the simulation to 3 DIV (Figure 4I). The modeled neurons matched the measured surface area well; however, a population of neurons with larger surface areas arose that were not reflected in simulations.
We next simulated growth of Trim67−/− neurons (Figures 4J and 4K). Incorporation of increased KNR and decreased FVF predicted a reduction in surface area expansion in Trim67−/− neurons at both 2 and 3 DIV. Experimentally measured surface areas agreed (Figures 4J and 4K). These data suggested a bias in exocytic mode was sufficient to alter surface area. Consistent with decelerated morphogenesis, callosal axons in Trim67−/− mice were delayed in midline crossing (Boyer et al., 2020). Simulations using the increased exocytic frequency of Trim9−/− neurons predicted increased neuronal surfaces areas, which was confirmed empirically at 2 and 3 DIV (Figure 4L). The remarkable fit between simulated and empirically measured data suggested that we captured relevant factors responsible for developmental neuronal growth.
The t-SNARE SNAP47 interacts with TRIM67 and localizes to VAMP2-mediated exocytic events
TRIM67 did not localize to VAMP2-mediated exocytic events (Figure 5A; Video S4). However, expression of RFP-tagged TRIM67 in Trim67−/− neurons (Figure S4A) rescued the ratio of exocytic modes (Figure S4B) without changing the frequency of exocytosis (Figure S4C). These data confirmed that TRIM67 biased the mode of exocytosis toward FVFi and FVFd and away from KNRi and KNRd and likely did so remotely.
Figure 5. The t-SNARE SNAP47 interacts with TRIM67 and localizes to VAMP2-mediated exocytic events.
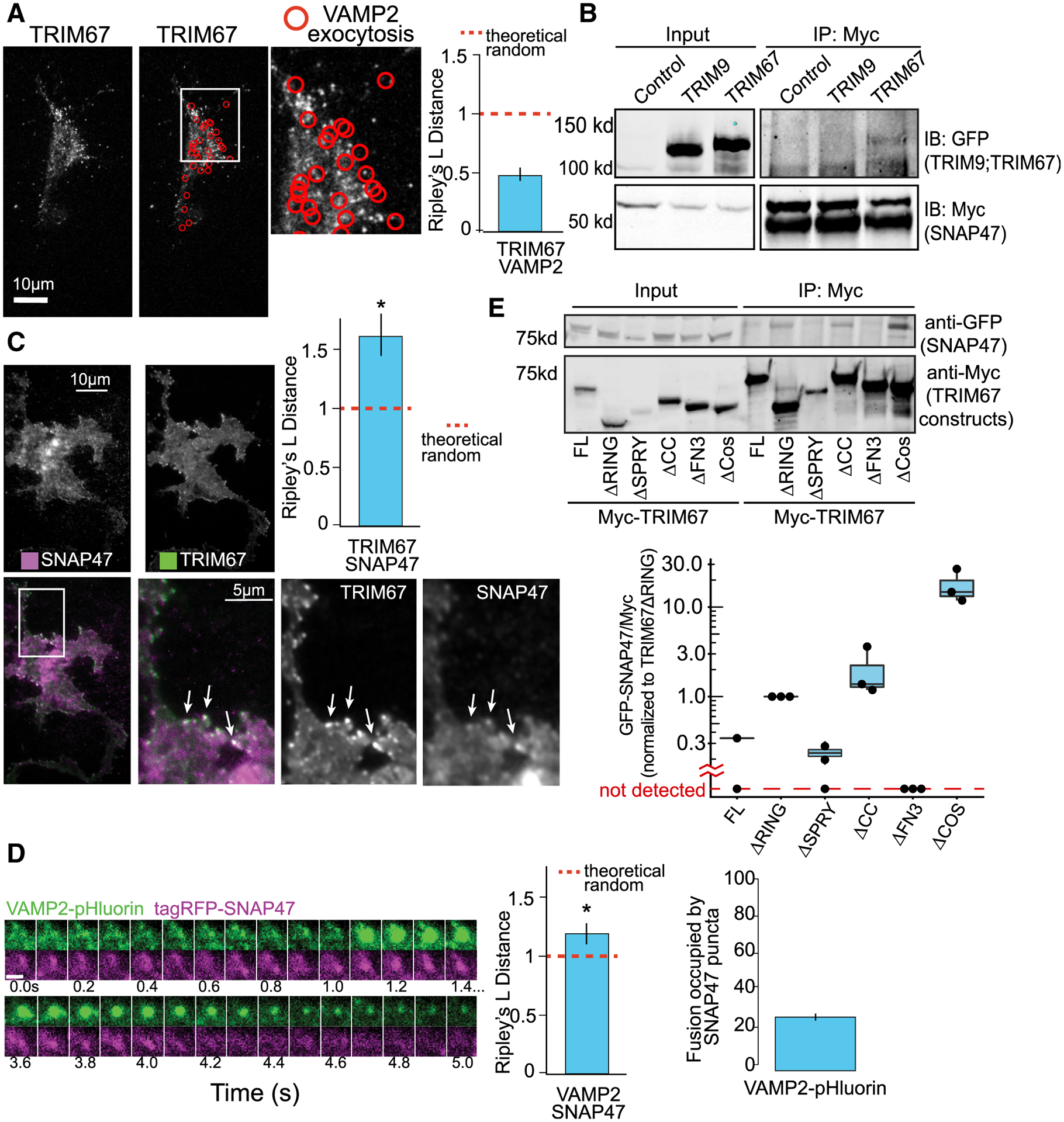
(A) Representative images and Ripley’s L distance +/− standard error colocalization analysis of TRIM67-tagRFP and VAMP2-pHluorin events (red circles) n = 15 cells; n = 4 biological replicates; value lower than theoretical random suggests no colocalization.
(B) Immunoprecipitation (IP:Myc) of Myc-SNAP47 from HEK293 cells expressing Myc-SNAP47 and either GFP-TRIM67 or GFP-TRIM9, blotted for GFP and Myc.
(C) Average of 10 frames of images and colocalization analysis of TagRFP-SNAP47 and TRIM67-GFP in cortical neurons at 2 DIV (n = 14 cells; n = 3 biological replicates; values above theoretical line are colocalized (*). Error bars report standard error.
(D) Example of VAMP2-pHluorin and TagRFP-SNAP47 colocalization during an exocytic event. n = 16 cells; values above theoretical line are colocalized (*). Error bars report standard error. Scale bar, 1 μm.
(E) Structure-function coimmunoprecipitation (coIP) assays. MycTRIM67 construct IP blotted for GFP-SNAP47 and Myc. Graph represents relative GFP-SNAP47/Myc-TRIM67 constructs normalized to Myc-TRIM67ΔRING; blots without a detectable band of GFP-SNAP47 represented below graph break.
Because few interaction partners of TRIM67 are known, we used the BioID method (Roux et al., 2012) to identify candidate TRIM67-interacting partners that may regulate exocytosis (Menon et al., 2020). One interesting candidate and the only SNARE protein significantly enriched (~4-fold) over the negative control (Myc-BirA*; Table S1) was SNAP47, a t-SNARE in the SNAP25 family. SNAP47 competes with SNAP25 and forms thermally stable, fusion-competent SNARE complexes (Holt et al., 2006). However, SNAP47-containing SNARE complexes are less efficient in liposomal fusion (Holt et al., 2006). Immunoprecipitation of Myc-SNAP47 coprecipitated GFP-TRIM67 but not GFP-TRIM9 (Figure 5B), confirming that TRIM67 and SNAP47 interact. TIRF microscopy revealed that TRIM67 and SNAP47 colocalized at the periphery of neurons (Figure 5C, arrows; Video S5). SNAP47 also colocalized with a subset (28%) of VAMP2 exocytic events (Figure 5D; Video S6). These data suggested that SNAP47 and TRIM67 interacted at sites distal from exocytic events.
Multiple domains of TRIM67 modulate the interaction with SNAP47 and exocytic mode
To determine domains of TRIM67 important for interacting with SNAP47, we performed coimmunoprecipitation assays by using domain mutants expressed in Trim67−/− HEK293 cells (Boyer et al., 2018; Figure 5E). Similar experiments were performed in Trim67−/− neurons to examine exocytic mode (Figures S4B and S4C). TRIM67ΔFN3 failed to precipitate detectable levels of SNAP47 (Figure 5E) or rescue exocytic modes (Figure S4B). TRIM67ΔSPRY precipitated similar amounts of SNAP47 compared to TRIM67 and rescued exocytic mode (Figures 5E and S4B). In contrast, more SNAP47 was detected in precipitates of TRIM67ΔRING or TRIM67ΔCC, yet these mutants failed to rescue the ratio of exocytic modes. Mutation of conserved Zn-coordinating cysteines required for ligase activity (TRIM67LD; Boyer et al., 2020) also failed to rescue exocytic mode (Figure S4B). None of these mutants altered the frequency of exocytosis (Figure S4C). TRIM67ΔCOS, however, decreased the frequency of exocytosis but did not rescue the ratio of exocytic modes (Figures S4B and S4C). Interestingly, TRIM67ΔCOS precipitated more SNAP47 (Figure 5E). These data fit a Goldilocks principle, in which the interaction strength between TRIM67 and SNAP47 must fall within certain margins to rescue exocytic mode, i.e., mutants that showed a stronger interaction than full-length TRIM67 or mutants that did not bind TRIM67 were unable to rescue exocytic mode.
TRIM67 alters exocytosis by modulated SNAP47 protein levels
Analysis of cell lysates revealed a 1.5-fold increase in SNAP47 protein in Trim67−/− neurons (Figure 6A), suggesting that TRIM67 regulated SNAP47 expression or stability. A cycloheximide chase revealed that SNAP47 was degraded by the proteasome at the same rate in Trim67+/+ and Trim67−/− cortical neurons (Figure S5A). The levels of ubiquitinated SNAP47 were also not altered by the absence of TRIM67 (Figure S5B). We hypothesized that elevated SNAP47 in Trim67−/− neurons may alter exocytic mode. Like deletion of Trim67 (Figure 4B), overexpression of SNAP47 in Trim67+/+ neurons did not alter the frequency of exocytosis (Figure 6B). However, it increased KNRd and decreased FVFi (Figure 6C). This change was more pronounced in events with detectable tagRFP-SNAP47. SNAP47 fluorescence intensity was highest at FVFd and KNRd events (Figure 6D), suggesting SNAP47 influences mode. Strikingly, the delay time before the onset of fluorescent decay increased for KNRd and FVFd (Figure 6E). Once decay initiated, overexpression of SNAP47 did not affect t1/2 (Figure 6F). These findings indicated increased SNAP47 promoted KNRd and was sufficient to shift exocytic mode. Cell surface area was reduced at 3 DIV after SNAP47 overexpression, suggesting reduced membrane delivery by exocytosis affects PM expansion (Figure 6G). Small interfering RNA (siRNA) reduced SNAP47 levels by ~80% in Trim67−/− neurons (Figures 6H and 6H’). This did not alter exocytic frequency (Figure 6I) but decreased KNRd and KNRi and increased FVFi and FVFd (Figure 6J). In contrast, knockdown of SNAP47 in Trim67+/+ neurons did not alter exocytic frequency or mode (Figures 6I and 6J). These data suggest that physiological levels of SNAP47 were insufficient to alter exocytic mode at this developmental stage, whereas increasing SNAP47 levels by overexpression or deletion of TRIM67 revealed a role for SNAP47 in exocytic mode regulation.
Figure 6. TRIM67 alters exocytosis by modulating SNAP47 protein levels.
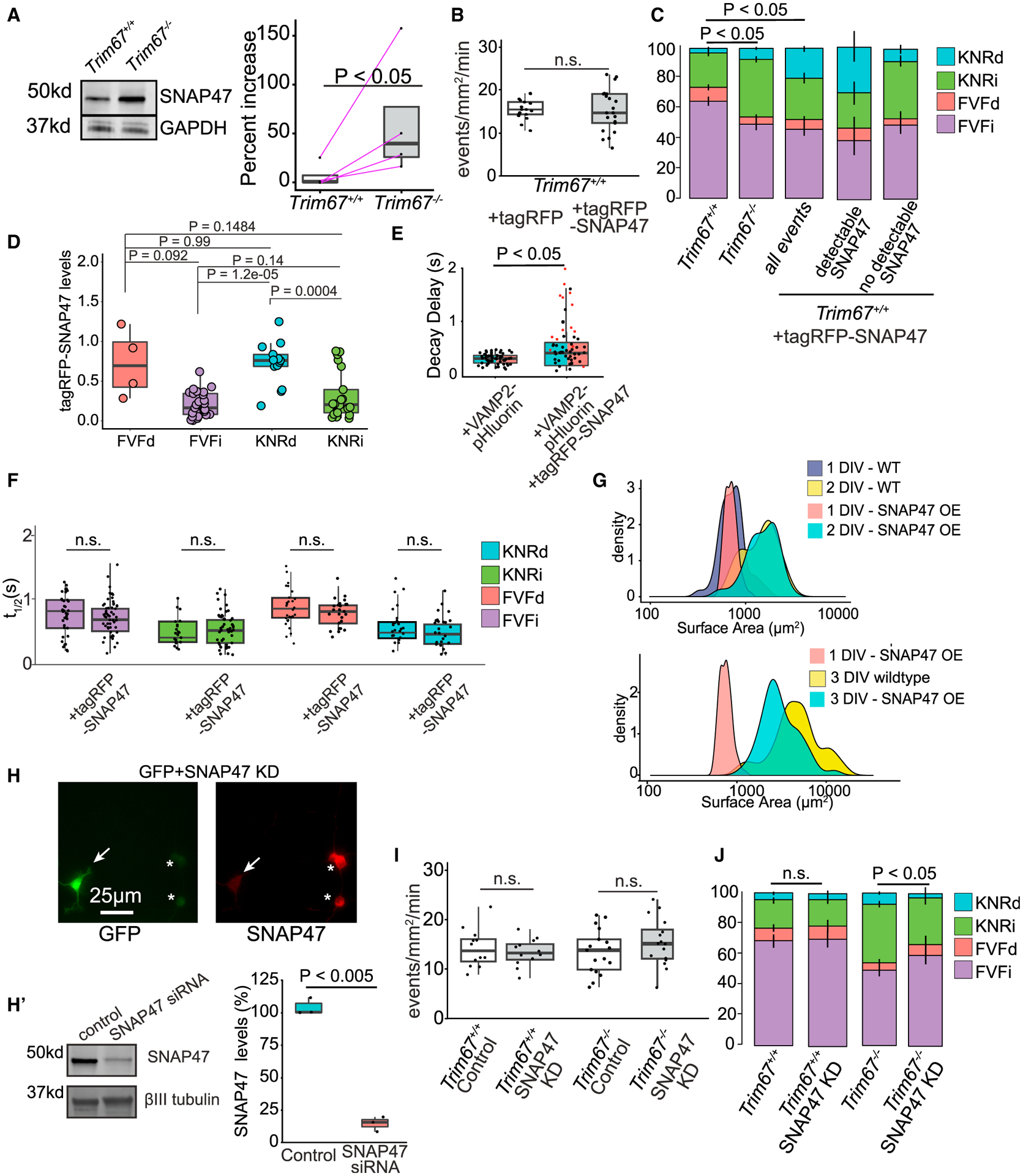
(A) Immunoblot of SNAP47 and GAPDH from lysate of Trim67+/+ and Trim67−/− cortical neurons at 2 DIV. Quantification of percent increase of SNAP47/GAPDH from 4 blots (right; log-paired t test, analysis in STAR methods).
(B) Frequency of VAMP2-pHluorin exocytosis ± tagRFP or tagRFP-SNAP47 (n = 16 neurons per condition, n = 3 biological experiments, Welch’s t test).
(C) Relative proportions of exocytic modes in Trim67+/+ neurons, Trim67−/− neurons, or Trim67+/+ neurons expressing tagRFP-SNAP47 (n = 16 neurons per condition; n = 3 biological replicates; multivariate linear regression, error bars report standard error of the percent).
(D) tagRFP-SNAP47-normalized fluorescence intensity at fusion sites (n = 16 neurons, n = 3 biological replicates; Welch’s t test with Benjamini-Hochberg correction).
(E) Quantification of time after peak ΔF/F before fluorescence decay onset of KNRd and FVFd events (n = 17 neurons; n = 4 biological replicates; Welch’s t test).
(F) t1/2 of each exocytic class ± TagRFP-SNAP47 (n = 17 neurons per condition; Welch’s t test with Benjamini-Hochberg correction).
(G) Measured neuronal surface areas at indicated times and conditions.
(H) Example SNAP47 immunocytochemistry images in neurons expressing siRNA for SNAP47 knockdown (KD) (GFP+, arrow) or without (*). Cells were transfected with an eGFP expression plasmid and SNAP47 siRNA, followed by immunostaining of SNAP47; GFP-positive neurons did not stain for SNAP47, suggesting KD of SNAP47. (H’) Immunoblot and quantification of SNAP47 and βIII tubulin protein levels in SNAP47 KD compared with control (n = 3 experiments; Welch’s t test).
(I) Frequency of VAMP2-pHluorin exocytosis ± SNAP47 siRNA (n = 12 neurons per condition; Welch’s t test).
(J) Relative proportions of exocytic classes in Trim67+/+ neurons and Trim67−/− neurons, with SNAP47 KD (n = 12 neurons per condition, n = 3 biological replicates; multivariate linear regression, error bars report standard error of the percent).
SNAP47 forms more SNARE complexes in Trim67−/−neurons
The increased SNAP47 protein in Trim67−/− neurons altered the ratio of SNAP47:SNAP25 (Figure 7A). Immunoprecipitation of SNAP47 showed that SNAP47 interacted with syntaxin-1 and VAMP2 (Figure 7B). We hypothesized that SNAP47 altered exocytosis by forming more SNARE complexes in Trim67−/− neurons. VAMP2, SNAP25, and syntaxin-1 exhibited similar proportions of complex:monomer between genotypes. In contrast, SNAP47 exhibited increased amounts of complex:monomer in Trim67−/− neurons (Figure 7C). This finding suggested that TRIM67 inhibited the incorporation of SNAP47 into SNARE complexes and, thus, SNAP47-mediated fusion events. Therefore, in the absence of TRIM67, increased SNAP47 protein levels may compete with SNAP25 to alter SNARE complex composition and exocytic mode.
Figure 7. TRIM67 reduces SNAP47 incorporation into SNARE complexes.
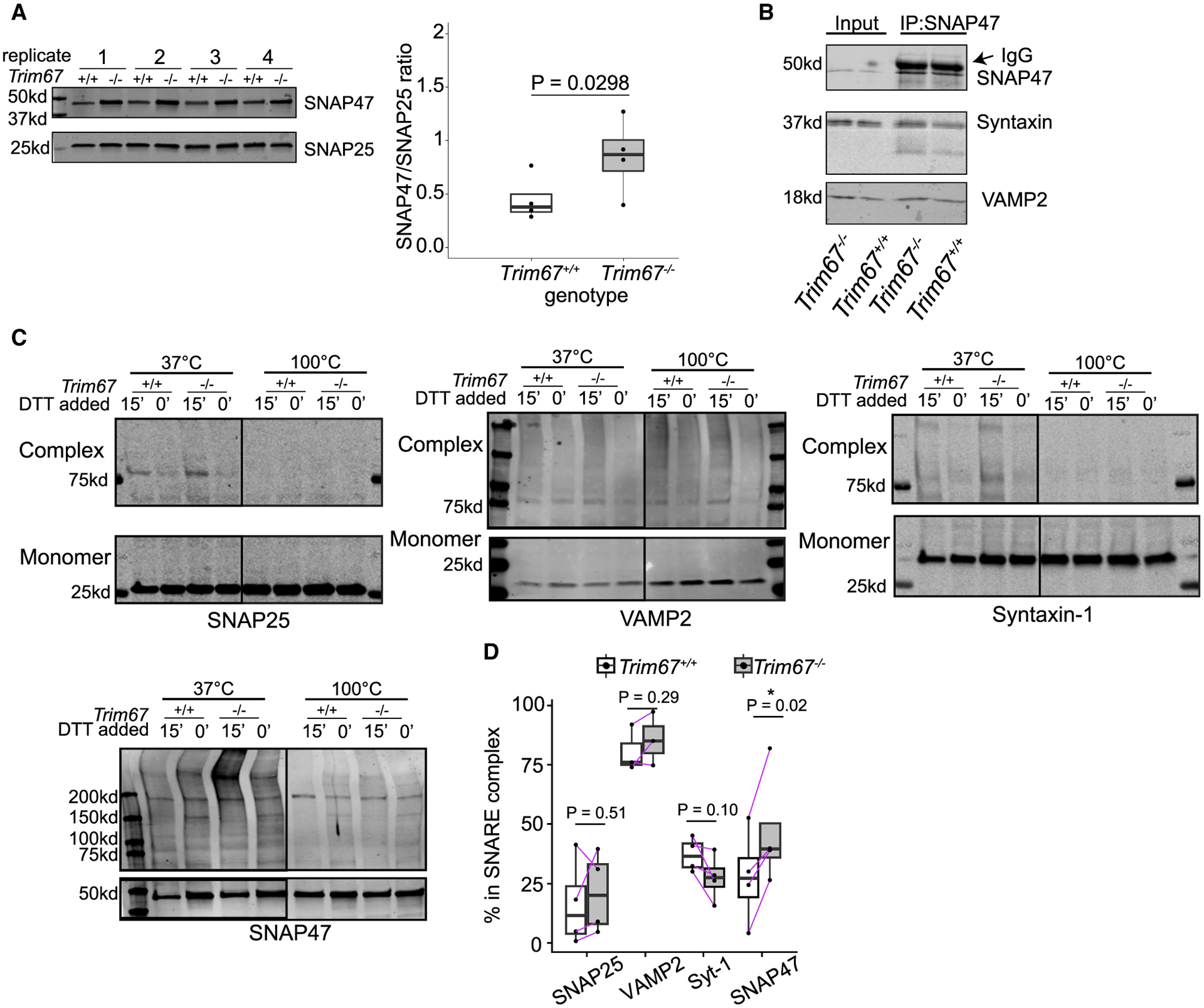
(A) Immunoblot and quantification of SNAP47 and SNAP25 from Trim67+/+ and Trim67−/− neuronal lysates at 2 DIV (four biological replicates). Quantified is the ratio of SNAP47/SNAP25 intensity, log-ratio paired t test.
(B) IP of endogenous SNAP47 in neuronal membrane fractions at 2 DIV immunoblotted for SNAP47, syntaxin-1, and VAMP2.
(C) Immunoblot of monomeric and high-molecular-weight SNARE proteins, interpreted as SNARE complexes. Cortical neurons were treated with N-ethylamaleimide (NEM, 15 min) to preserve SNARE complexes followed by DTT (15 min) to quench NEM, or NEM+DTT as a negative control (30 min). Increased intensity of bands above 37 kD (50 kD for SNAP47) in the presence of NEM only at 37°C indicates formed SNARE complexes.
(D) Quantification of % of each SNARE protein in complex (intensity of lane above 37 kD or 50 kD/(monomer + intensity of lane); n = 4 blots, log-ratio paired t test).
DISCUSSION
Here, we developed unsupervised classifiers based on VAMP2-pHluorin fluorescence to identify four exocytic classes in developing neurons. These classes were independent of cellular VAMP2 levels, occurred with both post-Golgi vesicles and recycling vesicles, and were detected with other markers and vesicle types. Distinguishable mechanisms of VAMP2-pHluorin fluorescence decay that were either pH sensitive or diffusion dependent suggested that two classes were KNR-like and two classes were FVF-like. The onset of fluorescence decay further distinguished exocytic classes. The alignment of our experiments using VAMP21–96 with capacitance measures supports the hypothesis that the delay in fluorescence decay is either a delay in fusion pore dilation (FVFd) or closure (KNRd). Our data support the hypotheses that TRIM67 altered exocytic mode in part by regulating SNAP47 incorporation into SNARE complexes and that SNAP47 pauses the fusion pore in an open state.
Distinguishing exocytic modes with classifiers and protein perturbation
Distinguishing exocytic modes is complicated by the fast and diffraction-limited nature of vesicle fusion (Albillos et al., 1997; Bao et al., 2018) and remains controversial at the synapse (He and Wu, 2007; Alabi and Tsien, 2013; Karatekin, 2018). Using developing neurons and their unique spatial distribution and temporal frequency of exocytosis, we resolved individual events. In contrast to semi-automated, supervised classification algorithms (Díaz et al., 2010; Yuan et al., 2015), our classifier requires no a priori information. Instead, automated exocytic detection coupled with three classifiers and clustering indices repeatedly delivered the surprising finding that four classes of exocytic events exist in developing neurons. This approach was robust: altering t1/2 of a subset of events with HEPES did not alter classification. Furthermore, classification was identical between VAMP2 and TfR at fusion events, even though TfR exhibited distinct diffusion dynamics in FVFi and FVFd. Some classes exhibited a delay after fusion, in which fluorescence does not decay instantaneously. This delay may occur if the fusion pore stabilized in an open state, which could facilitate the release of larger cargo. This is supported by the expression of a VAMP2 peptide known to stabilize the fusion pore (Guček et al., 2016). Alternatively, the delay may represent a decision point, prior to FVF or KNR. Finally, the delay could reveal inefficiency in fusion, as the opening and dilation of the fusion pore represent an energy barrier that SNARE machinery must overcome (Chernomordik and Kozlov, 2008).
Work in mature neurons support the existence of FVF and KNR, which influenced our interpretation (Alabi and Tsien, 2013; Albillos et al., 1997; He and Wu, 2007; Elhamdani et al., 2006). Previous work in chromaffin cells suggested additional exocytic modes differing from FVF and KNR (Shin et al., 2018, 2020; Wu et al., 2014). Exocytic events previously interpreted as KNR were argued to be omega-shrink fusion, in which a vesicle fuses and adds up to 80% of its membrane before retreating from the PM, with slowed or absent diffusion of VAMP2-pHluorin (Chiang et al., 2014). Vesicles in chromaffin cells are larger and fluorescent decay an order of magnitude slower than those of developing neurons. Therefore, fusion events in these two systems may not be directly comparable.
Exocytic mode and morphogenesis
Prior to synaptogenesis, exocytosis provides membrane to the expanding PM (Pfenninger, 2009; Urbina et al., 2018), which would suggest FVF predominates in developing neurons. In mature neurons, secretion by KNR may be more relevant to maintain surface area. Supporting a role for KNR here, protein levels of TRIM67 drop in the adult cortex (Boyer et al., 2018). Whether KNR-like modes of exocytosis carry cargo that contribute to morphogenesis remains to be determined. Although vesicular cargo is unidentified, consistent with other studies, VAMP2 may be common exocytic machinery for both post-Golgi and recycling vesicles, (Kubo et al., 2015; Oishi et al., 2006). Whether these VAMP2-containing vesicles represent a shared sorting compartment with both newly synthesized cargo and recycled cargo is unknown, but such a compartment occurs in neuroendocrine cells (Park et al., 2011). In developing neurons, FVFi events comprised most soma events, whereas FVFd predominated in neurites, with overall events more frequent in soma. Both FVFi and FVFd provide membrane material; functional differences between these modes of fusion are not known. Membrane curvatures and tension or membrane composition may trigger spatial differences in fusion mode. However, whether these modes represent distinct vesicles and cargo remains a possibility. The increased KNRi and KNRd along with decreased FVFi in the soma of Trim67−/− neurons were associated with reduced neuronal surface area, supporting a role for FVFi events as a mechanism for providing new membrane required for morphogenic PM expansion. The fluid nature of the PM may account for how changes in the soma affect morphogenesis. Membrane remodeling may also be affected by actin dynamics in the axonal growth cone. Because TRIM67 regulates filopodia in the growth cone (Boyer et al., 2020), TRIM67-mediated coordination of both exocytosis and actin dynamics likely contributes to morphogenesis, yet relative contributions are not known. The reduction in PM surface area associated with SNAP47 overexpression, however, suggests that altering exocytic mode is sufficient to alter PM expansion.
Our simulations that included exocytic mode remarkably recapitulated developmental increases in neuronal surface area. Although the simple model makes a number of assumptions, including that vesicles fusing by different modes are the same size and that vesicles sizes and rates of endocytosis are not altered by deletion of Trim9 or Trim67, the accurate fit to empirical data suggests that our model captures relevant parameters to accurately predict surface area expansion. However, simulations in Trim9−/− neurons overestimated expansion, suggesting possibly that endocytosis was also elevated in these neurons. Consistent with this idea, we recently identified several candidate TRIM9 interactors associated with clathrin coats (Menon et al., 2020). We conclude that VAMP2-mediated fusion is the primary source of membrane addition in developing neurons and that manipulating exocytic frequency or mode alters neuronal growth. Perhaps analogously, in vivo Vamp2−/− neurons have a largely normal morphology, suggesting compensatory mechanisms such as membrane added by endoplasmic reticulum (ER)-PM contact sites or exocytosis mediated by other SNARE proteins like VAMP7 (Fuschini et al., 2018; Galli et al., 1998) participate in remodeling.
Regulation of exocytic mode
Regulation of fusion pore kinetics and exocytic mode by regulatory proteins and a number of SNAREs is well documented (Bao et al., 2018; Bretou et al., 2014; Gauthier et al., 2011; Wen et al., 2016; Logan et al., 2017; Segovia et al., 2010; Archer et al., 2002). Effects of VAMP21–96 expression suggest that altering the number of SNARE complexes with fusion-competent endogenous VAMP2 and VAMP2-pHluorin affects exocytic mode. Analogously, exocytic mode was altered by increased incorporation of SNAP47, a Qb/Qc t-SNARE, into SNARE complexes. SNAP47 is capable of substituting for SNAP25 to form SNARE complexes with VAMP2 and syntaxin-1. These complexes are inefficient in liposomal fusion assays compared with SNAP25-containing complexes (Holt et al., 2006). This is consistent with our finding of increased SNAP47 at FVFd and KNRd events and in increased delay time between fusion pore opening and fluorescence decay. We hypothesize that the SNAP47-containing complexes may be the primary SNARE complex involved in KNRd. Interestingly SNAP47 has many unique features that may alter fusion kinetics; it lacks cysteine palmitoylation-mediated membrane targeting, has a long N-terminal extension, and has a linker region between the Qb and Qc SNARE domains (Holt et al., 2006). Structure-function experiments and observations that TRIM67 interacted with and colocalized with SNAP47 distal to exocytic sites and that SNAP47 colocalized with a subset of VAMP2-pHluorin events with distinct fusion behavior fit with a hypothesis that TRIM67 regulated exocytic mode by a weak interaction with SNAP47. Future studies will examine the mechanisms by which TRIM67 regulates SNAP47 protein level and incorporation into SNARE complexes, and how they may be related. SNAP47 also localizes to the ER, Golgi, and post-Golgi compartments (Kuster et. al, 2015), suggesting that TRIM67 and SNAP47 potentially mark secretory vesicle release sites in addition to regulating modes of exocytosis.
SNAP47 knockdown did not completely rescue the distribution in modes of exocytosis in Trim67−/− neurons, suggesting that TRIM67 regulates exocytic fusion by additional mechanisms. Myosin II, actin remodeling at the PM, and membrane tension are all suggested to alter exocytosis (Aoki et al., 2010; Wen et al., 2016). TRIM67 interacts with the actin-polymerase VASP at filopodia tips in the growth cone, a highly dynamic region with local changes in membrane tension and actin remodeling (Boyer et al., 2020; Staykova et al., 2011; Wen et al., 2016), but whether VASP alters exocytosis is not known. Proximity biotinylation approaches revealed several candidate TRIM67 interactors that are poised to regulate exocytosis, including the exocyst complex, tomosyn, and Munc18 (Menon et al., 2020). Separate pools of vesicles with distinct fusion characteristics and cargo (Hua et al., 2011; Rizzoli and Jahn, 2007) may fuse with different modes of fusion. Future work must define how TRIM67 interfaces with these vesicle populations and distinct mechanisms regulating fusion and also whether this unique regulation occurs at the synapse or is specific to earlier developmental time points.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephanie Gupton (sgupton@unc.edu).
Materials availability
The plasmid generated in this study is available upon request from the Gupton lab.
Data and code availability
All image analysis and classification code generated and detailed in this study are deposited at https://github.com/GuptonLab. Specifically, the exocytosis classifier is available in the repository https://github.com/GuptonLab/Exocytosis-Classification.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD021758 and DOI 10.6019/PXD021758.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mouse lines were on a C57BL/6J background and bred at UNC with approval from the Institutional Animal Care and Use Committee. Mouse colonies were maintained in specific pathogen-free environment with 12–12 hr light and dark cycles. Timed pregnant females were obtained by placing male and female mice together in a cage overnight; the following day was designated as embryonic day 0.5 (E0.5) if the female had a vaginal plug. Trim9−/− mice were described in Winkle et al., 2014 and Trim67−/− mice were described in Boyer et al. (2018). Mice were genotyped using standard procedures. For all culture experiments, embryos from time-matched homozygous WT, homozygous Trim9−/−, or homozygous Trim67−/− were used.
Culture, of primary neurons and HEK293 cell lines
Male and female mouse embryos were used to generate primary cortical neuron. Cortical neuron cultures were prepared from day E15.5 embryos as previously described (Viesselmann et al., 2011). Briefly, cortices were micro-dissected and neurons were dissociated with 0.25% trypsin for 15 min at 37°C followed by quenching with neurobasal media supplemented with 10% FBS and 0.5 mM glutamine. Cortices were gently triturated 15x and cells were counted by hemocytometer. Cells were spun at 0.1xg for 7 min at room temperature. Pelleted neurons were resuspended in neurobasal media supplemented with B27 (1:50 of manufacturer stock (Invitrogen)) and plated on cover glass, Mattek dishes, or tissue culture plastic coated with 1 mg/ml poly-D-lysine (Sigma-Aldrich). This same media was used for all time-lapse experiments.
HEK293 cells (female) were obtained from Rothenfußer (Klinikum der Universität München, München, Germany). HEK293 cells were maintained at 5% CO2 at 37°C in DMEM with glutamine (Invitrogen) supplemented with 10% FBS (Hyclone). Trim67−/− HEK293 cells were generated from this line by CRISPR/Cas9 editing as described in Boyer et al. (2020).
METHOD DETAILS
Transfection and treatment of primary neurons and HEK293 cell lines
Male and female mouse embryos were used to generate cultures and were randomly allocated to experimental groups. Cortical neuron cultures were prepared as described above. For transfection of plasmids and siRNA, neurons were resuspended after disassociation in Lonza Nucleofector solution (VPG-1001) and electroporated with Amaxa Nucleofector according to manufacturer protocol. A pool of 4 siRNAs targeting mouse SNAP-47 and control Luciferase siRNA (Target Sequence: 5′-CGTACGCGGAATACTTCGA-3′) (Dharmacon, Thermofisher Scientific) were electroporated along with GFP into E15.5 cortical neurons (2 μg GFP + 50 pmol siRNAs). Neurons were fixed and immunostained at 3 DIV. For TeNT treatment of VAMP2-pHluoring expressing neurons, 50 nM of TeNT (Sigma) was added to the neuronal media 30 min prior to imaging. For HEPES treatment, VAMP2-pHluorin exocytic events were imaged immediately after addition of 60 mM HEPES to the imaging media. For the TeNT and HEPES treatment, neurons were electroporated with VAMP2-pHluorin and imaged at 2 DIV. as described in “Live imaging and image analysis.” For assays investigating SNAP47 protein stability, 2 DIV neurons were treated with cycloheximide (50 μg/μl) for 0, 2, 4, 8 hr or cycloheximide (50 μg/μl) and bortezomib (200 nM) for 8 hr or cycloheximide (50 μg/μl) and chloroquine (50 μM) for 8 hr prior to lysis.
HEK293 cells were transfected with Lipofectamine 2000 (Invitrogen) or Polyplus jetPRIME® reagent according to manufacturer protocol.
Imaging
Live and fixed cell imaging
All live cell time-lapse imaging was performed on an inverted Olympus microscope (IX81-ZDC2) with MetaMorph acquisition software, an Andor electron-multiplying charge-coupled device (iXon), and a live cell imaging chamber (Tokai Hit Stage Top Incubator INUG2-FSX). A UAPON 100x/1.49 NA DIC TIRF objective (Olympus) was used for all live cell TIRF imaging assays. GFP-CAAX images of basal surface area were acquired with an Olympus 40x, 1.4 NA Plan Apo DIC objective. The live cell imaging chamber-maintained humidity, temperature at 37°C, and CO2 at 5%. Fixed cells were imaged using Zeiss LSM 780 confocal laser scanning microscope using a 20x/0.75 U-Plan S-Apo objective at room temperature in glycerol and n-propyl-gallate-based mounting medium.
For all exocytosis assays, E15.5 murine cortical neurons expressing VAMP2-pHluorin, VAMP7ΔLD-pHluorin, VAMP2-pHuji, FM4–64 dye, or Tfr-pHuji and/or tagRFP expressing constructs were imaged at the 2 DIV with a 100× 1.49 NA TIRF objective and a solid-state 491-nm laser illumination and a solid-state 561-nm laser, both at 35% power at 110-nm penetration depth, unless indicated otherwise. Images were acquired using stream acquisition, imaging at 10 Hz for 2 min with 100 ms exposure time. For all colocalization experiments and dual-color live cell imaging, a Hamamatsu W-VIEW GEMINI image splitter optic (A12801–01) was used for simultaneous imaging of both lasers by splitting and projecting the beams, by wavelength, side-by-side onto an electron-multiplying charge-coupled device (iXon).
For spontaneous FM4–64 dye loading, neurons were incubated in a 10 μM solution of FM4–64 dye in Hank’s Balanced Salt Solution (HBSS) for 30 min at 37°C. The media was then replaced with non-dye containing HBSS for 15 min to wash the cells. This washing step was performed 3 times. After the 3rd wash, cells are re-incubated in Neurobasal media and imaged.
Image analysis
ImageJ software was used for viewing of images and frames.
Detection of exocytic events
To identify the soma and neurites, the soma was segmented from the neurites as a rough ellipsoid encompassing the body of the neuron. All VAMP2-pHluorin, tfR-pHuji, and VAMP7ΔLD-pHluorin exocytic events were detected using the automated detection software as described (Urbina et al., 2018) in MATLAB and R with Rstudio. Briefly, the algorithm defines exocytic events as transient, non-motile (mean-square displacement < 0.2 μm2), Gaussian shaped objects that reached peak fluorescent intensity four deviations above the local background intensity.
Analysis of exocytic events
Once events were detected, a region of interest (ROI) of 25×25 pixels surrounding the Gaussian-shaped exocytic event and the bordering plasma membrane 10 frames before (1 s) and 100 frames following (10 s) was used for subsequent event analysis. The border plasma membrane was defined as follows: A Difference-of-Gaussian (Marr and Hildreth, 1980) filter was performed on each ROI around an exocytic event to segment the Gaussian shaped fluorescent signal at time point 1. The border pixels of exocytic events were defined as the average fluorescence of a 15 pixel-wide perimeter surrounding the segmented Gaussian shaped fluorescence.
Several parameters were measured within this ROI (Table S2). This includes the normalized change in fluorescence (ΔF/F) of the Gaussian, defined as the (average intensity of an ROI - background fluorescence)/background fluorescence. The background fluorescence was calculated as the average of the region of the Gaussian in the first 10 frames (1 s) before an exocytic event is detected. To ensure all relevant fusion information was obtained, fluorescence was tracked for 10 s following an exocytic event because a subset of exocytic events have an extended decay; this empirically determined time-frame ensured fluorescence returned to baseline (Urbina et al., 2018).
To determine the rate of decay of fluorescence, an exponential decay model was used for FVFi and KNRi: First we estimate the decay constant, λ, from the data:
In which V(t) is the fluorescence intensity at each time point and t is time in seconds. To ensure that the exponential decay constant is a good fit, we take the natural log of the data to estimate the R2 value:
After ensuring a good fit, we determine the half-life (t1/2) from the decay constant.
To estimate the rate of decay of FVFd and KNRd, two sequential exponential decay models were fit to the Gaussian. The log of fluorescence from peak ΔF/F until the end of decay were fit using segmented regression, with the breakpoint between the two regressions decided by iteratively moving the breakpoint one time-step, fitting segmented regression, and minimizing the sum of squared error. The breakpoint that has the lowest total error was considered the true breakpoint. The first regression represents the “delay” before decay starts, and second regression’s t1/2 represents the fluorescence decay.
Classification of exocytosis
Exocytic events were classified using three independent methods of classification: Feature extraction and principle component analysis, hierarchical clustering, and dynamic time warping (DTW).
Feature selection and principal component analysis
Feature selection was performed using both MATLAB and R. In MATLAB, image features were extracted from the ROI. In R, features were extracted from the the Gaussian shaped exocytic event, and the bordering plasma membrane (example in Figure 1B). A full list of features extracted is appended in Table S2. After features were extracted from all exocytic events, principal component analysis was performed using the R package “FactoMineR” (Lê et al., 2008) using the singular value decomposition approach. The principal components that captured the top 85% of variance were kept and used for the committee of indices.
Hierarchical clustering
Hierarchical clustering was performed using R’s base “stats” package and the “cluster” package.
Agglomerative
This function performed a hierarchical cluster analysis using a set of dissimilarities for the n objects being clustered. Initially, each object was assigned to its own cluster and then the algorithm proceeds iteratively, at each stage joining the two most similar clusters, continuing until there was a single cluster. At each stage distances between clusters were recomputed by the Lance-Williams dissimilarity update formula. In hierarchical cluster displays, a decision was needed at each merge to specify which subtree should go on the left and which on the right. Since, for n observations there are n−1 merges, there are 2(n−1) possible orderings for the leaves in a cluster tree, or dendrogram. The algorithm iordered the subtree so that the tighter cluster was on the left (the last, i.e., most recent, merge of the left subtree was at a lower value than the last merge of the right subtree). Single observations were the tightest clusters possible, and merges involving two observations were ordered by observation sequence number.
Divisive
fully described in chapter 6 of Kaufman and Rousseeuw (1990). The algorithm constructed a hierarchy of clusterings, starting with one large cluster containing all n observations. Clusters were divided until each cluster contains only a single observation. At each stage, the cluster with the largest diameter were selected. (The diameter of a cluster was the largest dissimilarity between any two observations.) To divide the selected cluster, the algorithm first selected the most disparate observation (i.e., the largest average dissimilarity to the other observations of the selected cluster). This observation initiated the “splinter group.” In subsequent steps, the algorithm reassigned observations that were closer to the “splinter group” than to the “old party.” The result was a division of the selected cluster into two new clusters.
Dynamic Time Warp
Dynamic time warping was implemented using the ‘dtw’ package in R (Giorgino, 2009) to compare two time series, X = (x1,…, xN) and Y = (y1,…, yM). DTW assumed a nonnegative, local dissimilarity function f between any pair of elements xi and yj, with the shortcut:
d, the cross-distance matrix between vectors X and Y, was the only input to the DTW algorithm. The warping functions φx and φy remapped the time indices of X and Y respectively. Given φ, the average accumulated distortion between the warped time series X and Y was computed:
where mφ(k) was a per-step weighting coefficient and Mφ was the corresponding normalization constant, which ensured that the accumulated distortions were comparable along different paths. To ensure reasonable warps, constraints were imposed on φ. Monotonicity was imposed to preserve their time ordering and avoid meaningless loops:
The goal of DTW was to find the optimal alignment φ such that
Which represented the warp path cost of X and Y. The warp path cost for each exocytic event pairing was built into a matrix.
Clustering
Each of the classifiers were independently clustered by a committee of indices. Numerous clustering validity algorithms have been proposed (Rousseeuw, 1987; Pelleg et al., 2000; Sugar and James, 2003). These algorithms combine information about intracluster compactness and intercluster isolation, as well as other factors, such as geometric or statistical properties of the data, the number of data objects, and the dissimilarity or similarity measurements. However, different algorithms lead to different clusters, and even in a single algorithm, choice of parameters can lead to different clusters. To perform classification, a plurality-rules decision of 20 well-used algorithms for determining clustering was used. This committee of indices includedA full list of the 20 methods used in this committee of indices can be found in Charrad et al. (2014).
The analysis described above was developed for VAMP2-pHluorin, and exploited for VAMP7ΔLD-pHluorin and TfR-pHuji. Adjustments were made in TfR-pHuji analysis because it was larger (~980 amino acids) than VAMP2-pHluorin (~350 amino acids) and had distinct diffusion characteristics (Di Rienzo et al., 2013; Fujiwara et al., 2002, 2016) TfR undergoes hop diffusion, a model of transmembrane protein diffusion characterized by various transmembrane proteins anchored to the actin-based membrane skeleton meshwork acting as rows of pickets that temporarily confine diffusion. To capture this information, an additional parameter measuring goodness-of-fit to linear or exponential decay was added. The goodness-of-fit was defined as the R2 of fitting a linear regression to the fluorescent decay divided by the R2 of fitting an exponential curve for the half-life as described under “Analysis of exocytic events.” A linear fit will have a high linear goodness-of-fit, while an exponential curve will have a lower value.
Colocalization
To perform co-localization analysis, TRIM67tagRFP and SNAP47tagRFP puncta were identified by difference-of-Gaussian filter. The x,y centroids of VAMP2-pHluorin events, TagRFP-SNAP47 puncta, or TRIM67-tagRFP puncta were fit to a marked point process model, which allowed comparisons of groups of points, and Ripley’s L value distances were measured.(Grantham, 2012). Ripley’s L value measures the average distance between points normalized to the number of puncta and total cellular area. This average distance was statistically compared to what distances expected if points were “randomly” localized, allowing a measure of colocalization. Ripley’s L values here were compared to theoretically random marked points, setting a threshold of 0.05% chance to fall below the theoretical Ripley’s L value. All values were normalized to the theoretical Ripley’s L value for comparisons.
Surface area measurements and modeling predictions
Empirical Surface Area Estimation
To estimate the surface area of neurons during DIV 1–3, TIRF images of GFP-CAAX expressing cells at indicated time points were used. The soma was identified and segmented from the neurites, and the basal surface area of each were calculated separately. Total neurite area was estimated by doubling the measured basal membrane area. The average height of the soma (~11 μm) was obtained from confocal image z stacks through cortical neurons at 2 DIV. The major and minor axis of the soma were measured, and used with the average height of the soma to calculate the surface area using the following calculation for a truncated ellipsoid:
Where a,b, and c represent the 3 axes of the soma (length, width, and height, respectively). The surface area of soma and neurites were summated to obtain the final surface area estimate of the neuron.
Model Selection
A Bayesian linear model based on Urbina et al. (2018) was adapted to predict neuronal surface area increases from DIV1 to DIV 2 and 3. Surface area at 2 and 3 DIV, the predicted and independent variables, are a positive, continuous, right skewed distribution, which has non-uniform standard deviation over time. Several models were constructed, selecting for probability distributions that would fit the criteria of positive, continuous, and right-skewed data: log-normal, Skew-normal, and Gamma. Models using the t-distribution and the normal distribution as a base model were also constructed, based on their well-characterized distributions. Previously published data were used as priors. Namely, the surface area of neurons at 1 DIV, the surface area of vesicles, the frequency of exocytosis, the frequency of clathrin-mediated endocytosis, and the size of clathrin-coated vesicles were used. This was combined with newly measured surface area of neurons at 1 DIV (described under “Empirical surface area estimation”) to model membrane addition. This provided confidently constructed priors that were estimated from previously measured neuronal surface area, instead of subjectively chosen priors. From this, four hierarchical models of surface area expansion were constructed: Gamma with a log-link, log-normal, skewed-normal, and log-normal with a lower bound truncation of 0. Given the difference in the standard deviation between the observed surface area at 1 and 2 DIV (Urbina et al., 2018), a fifth model was added, in which the standard deviation was also modeled based on the measured standard deviation at 1 and 2 DIV. Four chains were run with 100,000 samples after a burn-in period of 10,000 steps for a total of 110,000 steps to ensure chain convergence and enough coverage of the posterior distribution. Model evaluation using Rhat and ESS suggested the chains were well-mixed and converged properly. Each of these models were then compared by computing Watanabe-Akaike information criteria and using leave-one-out validation to compute the expected log pointwise predictive distribution for the difference in each of the models. Model selection using these criteria indicate that using the log-normal family results in the best fit for these data, followed by the gamma distribution. Taking all of these terms into account, an equation for the hierarchical Bayesian linear model was created, using the log-normal distribution:
Western blot
Analysis of protein lysates
To investigate SNAP47, SNAP25, and GAPDH protein levels, cultured E15.5 cortical neurons of indicated genotypes were lysed in immunoprecipitation (IP) buffer (10% glycerol, 1% NP-40, 50 mM Tris pH 7.5, 200 mM NaCl, 2 mM MgCl2, 1 mM NEM, and protease and phosphatase inhibitors at 2 DIV. Lysates were collected and centrifuged at 19,745 xg for 10 min. Supernatants were diluted in 4x sample buffer (200 mM Tris-Cl, pH 6.8, 8% SDS, 0.4% Bromophenol Blue, 40% glycerol, 20% 2-Mercaptoethanol), boiled for 10 min at 100°C, and resolved by SDS-PAGE. For SNAP47 half-life measurements, cycloheximide treated cortical neurons were lysed (300 mM sucrose, 1% NP-40, 50 mM Tris pH 7.5, 200 mM NaCl, 2 mM MgCl2 and protease, phosphatase and deubiquitinase inhibitors). Lysates were collected and centrifuged at 19,745 xg for 10 min. Protein concentrations were estimated, and lysates were boiled with 2x sample buffer and resolved by SDS-PAGE.
Co-immunoprecipitation assay
For coimmunoprecipitation assays in HEK293 cells expressing GFP-SNAP47 and either MycTRIM9, MycTRIM67, or MycTRIM67 domain deletion mutants. were lysed in IP buffer and lysates collected and centrifuged at 19745xg for 10 min. 500–1,000 μg of protein per sample was used per IP (the same amount of protein was maintained across all conditions per experiment). Myc-tagged proteins were precipitated with anti-Myc antibody (9E10) overnight, followed by Protein A coupled agarose beads. Beads were washed twice with IP buffer, pelleted, and boiled with 2X sample buffer. For endogenous SNAP47 immunoprecipitation from cultured cortical neurons, E15.5 cortical neurons at 2 DIV were treated with 1mM NEM (15 min) prior to lysis. Membrane proteins were enriched through ultra-centrifugation of neuronal lysates, as described in Shimojo et al. (2015). Briefly, cortices of wild-type mice were homogenized in extraction buffer (20 mM HEPES-NaOH (pH 7.4), 320 mM sucrose, 5 μg/ml leupeptin, 2 μg/ml aprotinin, and 1 μg/ml pepstatin) post NEM treatment. Membrane fractions were isolated by two sequential steps of centrifugation at 3,000 and 100,000 3 g and solubilized for 30 min in 20 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, and 1% Triton X-100. Protein extracts were cleared by centrifugation at 100,000 3 g and incubated overnight with Sepharose beads coated with anti-SNAP47, or control IgG antibodies. Attached complexes were then washed five times with extraction buffer, eluted from the beads with an SDS gel loading buffer, and resolved by SDS-PAGE.
SNARE complex assay
SNARE complex assays were performed as described in Hayashi et al. (1994) with modification. E15.5 cortical neurons at 2 DIV were treated with either 1mM NEM (15 min) to prevent SNARE disassembly or 1mM NEM+ 2mM DTT (15 min) to allow disassembly, prior to lysis with homogenization buffer (10mM HEPES-NaOH, pH 7.4, 150mM NaCl, 1mM EGTA, 1mM NEM, and protease inhibitors). After collection of lysates, Triton X-100 was added to a final composition of 1% Triton x-100 followed by trituration 10x and solubilization for 2 min on ice. Lysates were then centrifuged at 10min, 6000 g. Samples were diluted in 4x sample buffer (200mM Tris-Cl, pH 6.8, 8% SDS, 0.4% Bromophenol Blue, 40% glycerol, 20% 2-Mercaptoethanol) into two replicates, one replicate incubated at 100°C for 10 min and the second incubated at 37°C for 10 min. SNARE complexes and monomers were then resolved by SDS-PAGE. SDS-resistant SNARE complexes at 37°C run at higher molecular weights than their monomeric constituents. These high molecular weight SNARE proteins were not present upon simultaneous NEM,DTT treatment or at 100°C incubation, which allow and promote disassembly, respectively (Banerjee et al., 1996).
Ubiquitination experiment
For ubiquitination assay, Trim67−/− or rescued (transfected with Myc or Myc-TRIM67 respectively) HEK cells were co-transfected with HA-tagged Ubiquitin and control GFP or GFP-SNAP47 using Lipofectamine 2000. GFP-Trap beads (Chromotek) were used to enrich GFP-SNAP47 and GFP according to manufacturer protocol. Briefly, cells were treated with MG132 for 4 h then lysed in ubiquitination immunoprecipitation (Ub-IP) buffer (50 mM Tris-Cl, 150 mM NaCl, 1 mM EDTA, 0.5% Triton X, 0.7% N-ethylmaleimide, and protease and phosphatase inhibitors, pH 7.3–7.4). Lysates were collected and centrifuged at 19745 xg for 10 min 4°C. 500–1,000 μg of protein per sample was used in each assay (the same amount of protein was maintained across all conditions per experiment). 15 μl GFP-TRAP beads were incubated with the lysate for 2.5 hr followed by three washes. The first wash was performed using 10mM Tris-Cl, pH7.5, 150 mM NaCl, 0.5 mM EDTA, 0.7% NEM. This was followed by two washes using first a stringent buffer (8 M Urea, 1% SDS) and then SDS wash buffer (1% SDS). The beads were then boiled with 2X sample buffer. HA-ubiquitin that co-immunoprecipitated with GFP-SNAP47 and migrated at a higher apparent molecular weight was interpreted as ubiquitinated SNAP47 (SNAP47-Ub)
Immunoblotting
All protein samples were resolved by SDS-PAGE followed by transfer onto 0.45 μM (0.22 μM for the SNARE complex assay) nitrocellulose paper and analyzed by immunoblotting. Blots were probed with indicated primary antibodies, followed by IRDye® 680LT Goat anti-Rabbit Secondary Antibody and/or IRDye® 800CW Donkey anti-Mouse IgG Secondary Antibody and imaged on an Odyssey Li-Cor. Western blots were analyzed using Fiji (ImageJ) or Li-Cor Image Studio Lite. Total fluorescence of labeled bands representing relative protein were normalized to control conditions for each experiment as indicated. For all western blot comparisons, the relative band intensity or relative ratios (indicated for each blot) were log-transformed followed by paired-t test..
Mass Spectrometry
To identify potential interacting partners for TRIM67, we performed three biological replicates using the Proximity-Dependent Biotin Identification (BioID) approach (Roux et al., 2012). Briefly, the negative control Myc-BirA* and Myc-BirA* TRIM67ΔRING were packaged into Short Term Herpes Simplex Virus (HSV)(MGH) and driven under a IE 4/5 promoter. GFP expression is driven in tandem downstream of a mCMV promoter. Cortical neurons from wild-type and Trim67−/− E15.5 litters were dissociated and plated on PDL coated tissue culture dishes. Approximately 40 hours post-plating the neurons were transfected with HSV (MOI = 1.0) carrying either the negative control Myc-BirA* or Myc-BirA* TRIM67ΔRING. 6 hr post-infection neurons were treated with 50 μM Biotin for 24 hr. After incubation the cells were lysed using RIPA buffer (150 mM NaCl, 25 mM Tris-HCl, pH 7.5, 0.1% SDS, 1.0% NP-40, 0.25% Deoxycholic acid, 2 mM EDTA, 10% glycerol, protease and phosphatase inhibitors). Biotinylated proteins were then enriched from the lysate using streptavidin-conjugated Sepharose beads. The enriched proteins were digested with trypsin and eluted using the RapiGEST SF Surfactant protocol (Waters). C18 column and Ethyl acetate extraction protocols were employed to prepare the peptides for mass spectrometry.
Reverse-phase nano-high-performance liquid chromatography (nano-HPLC) coupled with a nanoACQUITY ultraperformance liquid chromatography (UPLC) system (Waters Corporation; Milford, MA) was used to separate trypsinized peptides. Trapping and separation of peptides were performed in a 2 cm column (Pepmap 100; 3-m particle size and 100-Å pore size), and a 25-cm EASYspray analytical column (75-m inside diameter [i.d.], 2.0-m C18 particle size, and 100-Å pore size) at 300 nL/min and 35°C, respectively. Analysis of a 150 min gradient of 2% to 25% buffer B (0.1% formic acid in acetonitrile) was performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific). The ion source was operated at 2.4kV and the ion transfer tube was set to 300°C. Full MS scans (350–2000 m/z) were analyzed in the Orbitrap at a resolution of 120,000 and 1e6 AGC target. The MS2 spectra were collected using a 1.6 m/z isolation width and were analyzed either by the Orbitrap or the linear ion trap depending on peak charge and intensity using a 3 s TopSpeed CHOPIN method.32 Orbitrap MS2 scans were acquired at 7500 resolution, with a 5e4 AGC, and 22 ms maximum injection time after HCD fragmentation with a normalized energy of 30%. Rapid linear ion trap MS2 scans were acquired using an 4e3 AGC, 250 ms maximum injection time after CID 30 fragmentation. Precursor ions were chosen based on intensity thresholds (> 1e3) from the full scan as well as on charge states (2–7) with a 30 s dynamic exclusion window. Polysiloxane 371.10124 was used as the lock mass. All raw mass spectrometry data were searched using MaxQuant version 1.5.7.4. Search parameters were as follows: UniProtKB/Swiss-Prot human canonical sequence database (downloaded 1 Feb 2017), static carbamidomethyl cysteine modification, specific trypsin digestion with up to two missed cleavages, variable protein N-terminal acetylation and methionine oxidation, match between runs, and label-free quantification (LFQ) with a minimum ratio count of 2. To rank candidate protein-protein interactions by likelihood of interaction, LFQ values for proteins identified in control and pulldown experiments from three biological replicates were input into SAINTq (version 0.0.4). Interactions were sorted by SAINT’s interaction probability and a false discovery rate threshold of 25% was employed. A full list of identified proteins are reported elsewhere (Menon et al., 2020).
QUANTIFICATION AND STATISTICAL ANALYSIS
The software package R was used for statistical analysis of data. Both R and Adobe Illustrator were used for the generation of figures. At least three independent biological replicates were performed for each assay, often more. Sample sizes for all experiments measuring frequency were estimated using power analysis (Power = 0.8) and the estimated effect size based off of previous work in Urbina et al. (2018). For two-sample comparisons of normally distributed data, Welch’s t test was used for two independent samples, or paired t test for paired samples. For multiple comparisons, Welch’s or paired t tests corrected using Benjamini-Hochberg method. For comparison of ratios for the four classes, multivariate linear regression was used, with the expected ratio of the four classes as the dependent variables and the conditions as the independent variables. For analysis of Figure 1G, a chi-square expected ratio test was performed to determine if the pHluorin classification assigned FVF or KNR classes different than randomly. For analysis of non-normally distributed data, the was used to determine significance followed by the method described above.
Plots
All boxplots were graphed as follows: the box represents the median value (middle of boxplot) and the interquartile range, which is the distance between the first and third quartile of the data (with the first quartile being the middle value between smallest number and median of the dataset, and the third quartile representing the middle value between the largest number and median of the dataset). The whiskers extend out to the last data point within 1.5x the interquartile range.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-TRIM67 | Boyer et al., 2018 | N/A |
| Mouse monoclonal anti-TRIM9 | Abnova | H00114088-M01; RRID:AB_509096 |
| Mouse monoclonal anti-Myc | Abcam | 9E10; RRID:AB_10573245 |
| Mouse monoclonal anti-B-III Tubilin | Thermofisher | MA1-9587; RRID:AB_2241264 |
| Mouse monoclonal anti-ubiquitin | SCBT | Sc-8017; RRID:AB_2762364 |
| Mouse monoclonal anti-GAPDH | SCBT | Sc-166545; RRID:AB_2107299 |
| GFP-Trap Agarose | Chromotek | gta-10 |
| Rabbit monoclonal anti-VAMP2 | Cell Signaling | #13508; RRID:AB_2798240 |
| Rabbit monoclonal anti-Syntaxin-1 | Santa Cruz | Sc-12736; RRID:AB_2271330 |
| Mouse monoclonal anti-SNAP25 | Synaptic Systems | 111011; RRID:AB_887794 |
| Rabbit polyclonal anti-SNAP47 | Synaptic Systems | 111403; RRID:AB_887899 |
| Mouse monoclonal anti-HA | Laboratory of Patrick Brennwald | 12CA5 |
| IRDye 800CW Donkey anti-mouse IgG | Li-Cor | 926–32212; RRID:AB_621847 |
| IRDye 680LT Goat anti-mouse IgG | Li-Cor | 926–68020; RRID:AB_10706161 |
| Rabbit polyclonal anti-HA | ThermoFisher | 71–5500; RRID:AB_2533988 |
| Chemicals, peptides, and recombinant proteins | ||
| TeNT | Sigma Aldrich | T3194-25UG |
| HEPES Buffer | Fisher Scientific | MT25060Cl |
| Cyclohexamide | Fisher Scientific | AC357420010 |
| Bortezomib | Sigma Aldrich | 5043140001 |
| Chloroquine | Sigma Aldrich | C6628 |
| Glutamine | Invitrogen | 25030149 |
| FBS | Hyclone | SH3039603 |
| PDL | Sigma-Aldrich | P7886-500MG |
| Critical commercial assays | ||
| Amaxa Nucleofector Kit | Lonza | vvpg-1001 |
| jetPRIME® DNA and siRNA Transfection Reagent | Genessee | 55–132 |
| Lipofectamine 2000 | Thermo Fisher | 11668030 |
| Deposited data | ||
| Mass spectrometry and proteomics | This Paper | PRIDE identifier PXD021758 and DOI 10.6019/PXD021758 |
| Experimental models: cell lines | ||
| HEK293 | Klinikum der Universität München | N/A |
| Experimental models: organisms/strains | ||
| Mouse: C57B176J | Jackson Laboratory | 000664 |
| Mouse: C57BL76J Trim67−/− | Boyer et al., 2018 | N/A |
| Mouse: C57BLV6J Trim9−/− | Winkle etal., 2014 | N/A |
| Oligonucleotides | ||
| siRNA: Pool targeting SNAP47 CGTACGCGGAATACTTCGA | Dharmacon/Horizon Discovery | M-051677-01-0005 |
| Recombinant DNA | ||
| Plasmid: VAMP2-pHluorin | Laboratory of James Rothman | N/A |
| Plasmid: VAMP2-tagRFP | This paper | N/A |
| Plasmid: tagRFP | Clontech | N/A |
| Plasmid: VAMP21−96-tagRFP | This paper | N/A |
| Plasmid VAMP7ΔLD-GFP | Laboratory of Thierry Galli | N/A |
| Plasmid: Myc-SNAP47 | Laboratory of Thierry Galli | N/A |
| Plasmid: VAMP7ΔLD-pHluorin | N/A | |
| Plasmid: tagRFP-SNAP47 | This paper | N/A |
| Plasmid: VAMP2-pHuji | This paper | N/A |
| Plasmid: TRIM67 | Boyer et al., 2020 | N/A |
| Plasmid: TRIM67ΔCC | Boyer et al., 2020 | N/A |
| Plasmid: TRIM67ΔCOS | Boyer et al., 2020 | N/A |
| Plasmid: TRIM67ΔRING | Boyer et al., 2020 | N/A |
| Plasmid: TRIM67ΔSPRY | Boyer et al., 2020 | N/A |
| Plasmid: TRIM67ΔFN3 | Boyer et al., 2020 | N/A |
| Plasmid: Flag-Ubiquitin | Laboratory of Ben Philpot | N/A |
| Plasmid: GFP-CAAX | Laboratory of Richard Cheney | N/A |
| Plasmid: TfR-pHuji | Laboratory of David Perrais | Addgene #61505; RRID:Addgene_61505 |
| Software and algorithms | ||
| R | R Core Team, 2020 | https://www.r-project.org/ |
| RStudio | RStudio Team, 2020 | https://rstudio.com/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| MATLAB | MATLAB | https://www.mathworks.com/products/matlab.html |
| Automated Detection of Exocytosis software | Urbina et al., 2018 | https://github.com/GuptonLab |
| Classification of Exocytosis software | This paper | https://github.com/GuptonLab |
| Other | ||
| Sepharose beads | Sigma | N/A |
| FM4–64 Dye | ThermoFisher | T13320 |
Highlights.
Unsupervised classifiers identify four exocytic classes in developing neurons
FVFi and FVFd modes insert membrane material; KNRi and KNRd do not insert membrane
TRIM67 limits SNAP47 SNARE complexes, promoting FVFi and membrane expansion
SNAP47 incorporation into SNARE complexes delays fusion pore expansion or closure
ACKNOWLEDGMENTS
Shawn Gomez provided critical suggestions and comments on DTW. Caroline Monkiewicz, Vong Thoong, and Chris Hardie performed mouse husbandry. We thank Michelle Itano and the UNC Neuroscience Imaging Core for training and access to the Zeiss 780 laser scanning confocal microscope, funded in part by P30 NS045892 and U54 HD079124. Funding from the National Institutes of Health supported this research, including R01NS112326 (S.L.G.), R35GM135160 (S.L.G.), and F31NS103586 (F.L.U.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108743.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alabi AA, and Tsien RW (2013). Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu. Rev. Physiol 75, 393–422. [DOI] [PubMed] [Google Scholar]
- Albillos A, Dernick G, Horstmann H, Almers W, Alvarez de Toledo G, and Lindau M (1997). The exocytotic event in chromaffin cells revealed by patch amperometry. Nature 389, 509–512. [DOI] [PubMed] [Google Scholar]
- Aoki R, Kitaguchi T, Oya M, Yanagihara Y, Sato M, Miyawaki A, and Tsuboi T (2010). Duration of fusion pore opening and the amount of hormone released are regulated by myosin II during kiss-and-run exocytosis. Biochem. J 429, 497–504. [DOI] [PubMed] [Google Scholar]
- Archer DA, Graham ME, and Burgoyne RD (2002). Complexin regulates the closure of the fusion pore during regulated vesicle exocytosis. J. Biol. Chem 277, 18249–18252. [DOI] [PubMed] [Google Scholar]
- Atluri P, and Ryan T (2006). The Kinetics of Synaptic Vesicle Reacidification at Hippocampal Nerve Terminals. J. Neurosci 26, 2313–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Barry VA, DasGupta BR, and Martin TFJ (1996). N-Ethylmaleimide-sensitive factor acts at a prefusion ATP-dependent step in Ca2+-activated exocytosis. J. Biol. Chem 271, 20223–20226. [DOI] [PubMed] [Google Scholar]
- Bao H, Das D, Courtney NA, Jiang Y, Briguglio JS, Lou X, Roston D, Cui Q, Chanda B, and Chapman ER (2018). Dynamics and number of trans-SNARE complexes determine nascent fusion pore properties. Nature 554, 260–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello OD, Auclair SM, Rothman JE, and Krishnakumar SS (2016). Using ApoE Nanolipoprotein Particles To Analyze SNARE-Induced Fusion Pores. Langmuir 32, 3015–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowser DN, and Khakh BS (2007). Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc. Natl. Acad. Sci. USA 104, 4212–4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer NP, Monkiewicz C, Menon S, Moy SS, and Gupton SL (2018). Mammalian TRIM67 Functions in Brain Development and Behavior. eNeuro 5, ENEURO.0186–18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer NP, McCormick LE, Menon S, Urbina FL, and Gupton SL (2020). A pair of E3 ubiquitin ligases compete to regulate filopodial dynamics and axon guidance. J. Cell Biol 219, e201902088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretou M, Jouannot O, Fanget I, Pierobon P, Larochette N, Gestraud P, Guillon M, Emiliani V, Gasman S, Desnos C, Lennon-Duménil A-M, and Darchen F (2014). Cdc42 controls the dilation of the exocytotic fusion pore by regulating membrane tension 25, 3195–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgo A, Casano AM, Kuster A, Arold ST, Wang G, Nola S, Verraes A, Dingli F, Loew D, and Galli T (2013). Increased activity of the vesicular soluble N-ethylmaleimide-sensitive factor attachment protein receptor TIVAMP/VAMP7 by tyrosine phosphorylation in the Longin domain. J. Biol. Chem 288, 11960–11972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burri L, and Lithgow T (2004). A complete set of SNAREs in yeast. Traffic 5, 45–52. [DOI] [PubMed] [Google Scholar]
- Caccin P, Rossetto O, Rigoni M, Johnson E, Schiavo G, and Montecucco C (2003). VAMP/synaptobrevin cleavage by tetanus and botulinum neurotoxins is strongly enhanced by acidic liposomes. FEBS Lett. 542, 132–136. [DOI] [PubMed] [Google Scholar]
- Cardoso-Moreira M, Halbert J, Valloton D, Velton B, Chen C, Shao Y, et al. (2019). Gene expression across mammalian organ development. Nature 571, 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrad M, Ghazzali N, Boiteau V, and Niknafs A (2014). NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw 61, 1–36. [Google Scholar]
- Chernomordik LV, and Kozlov MM (2008). Mechanics of membrane fusion. Nat. Struct. Mol. Biol 15, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhara M, Yarzagaray A, Makke M, Schindeldecker B, Schwarz y., Shaaban A, Sharma S, Böckmann RA, Lindau M, Mohrmann R, et al. (2016). v-SNARE transmembrane domains function as catalysts for vesicle fusion. Elife 5, e17571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rienzo C, Gratton E, Beltram F, and Cardarelli F (2013). Fast spatiotemporal correlation spectroscopy to determine protein lateral diffusion laws in live cell membranes. Proc. Natl. Acad. Sci. USA 110, 12307–12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz E, Ayala G, Díaz ME, Gong L-W, and Toomre D (2010). Automatic detection of large dense-core vesicles in secretory cells and statistical analysis of their intracellular distribution. IEEE/ACM Trans. Comput. Biol. Bioinformatics 7, 2–11. [DOI] [PubMed] [Google Scholar]
- Elhamdani A, Azizi F, and Artalejo CR (2006). Double patch clamp reveals that transient fusion (kiss-and-run) is a major mechanism of secretion in calf adrenal chromaffin cells: high calcium shifts the mechanism from kiss-and-run to complete fusion. J. Neurosci 26, 3030–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Ritchie K, Murakoshi H, Jacobson K, and Kusumi A (2002). Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol 157, 1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara TK, Iwasawa K, Kalay Z, Tsunoyama TA, Watanabe Y, Umemura YM, Murakoshi H, Suzuki KGN, Nemoto YL, Morone N, and Kusumi A (2016). Confined diffusion of transmembrane proteins and lipids induced by the same actin meshwork lining the plasma membrane. Mol. Biol. Cell 27, 1101–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuschini G, Cotrufo T, Ros O, Muhaisen A, Andrés R, Comella JX, and Soriano E (2018). Syntaxin-1/TI-VAMP SNAREs interact with Trk receptors and are required for neurotrophin-dependent outgrowth. Oncotarget 9, 35922–35940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli T, Zahraoui A, Vaidyanathan VV, Raposo G, Tian JM, Karin M, Niemann H, and Louvard D (1998). A novel tetanus neurotoxin-insensitive vesicle-associated membrane protein in SNARE complexes of the apical plasma membrane of epithelial cells. Mol. Biol. Cell 9, 1437–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier NC, Fardin MA, Roca-Cusachs P, and Sheetz MP (2011). Temporary increase in plasma membrane tension coordinates the activation of exocytosis and contraction during cell spreading. Proc. Natl. Acad. Sci. USA 108, 14467–14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgino T (2009). Computing and Visualizing Dynamic Time Warping Alignments inR: The dtw Package. J. Stat. Softw 31. [Google Scholar]
- Grantham N (2012). Analyzing multiple independent spatial point processes. BS Thesis (California Polytechnic State University). [Google Scholar]
- Guček A, Jorgačevski J, Singh P, Geisler C, Lisjak M, Vardjan N, Kreft M, Egner A, and Zorec R (2016). Dominant negative SNARE peptides stabilize the fusion pore in a narrow, release-unproductive state. Cell. Mol. Life Sci 73, 3719–3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton SL, and Gertler FB (2010). Integrin signaling switches the cytoskeletal and exocytic machinery that drives neuritogenesis. Dev. Cell 18, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, McMahon H, Yamasaki S, Binz T, Hata Y, Südhof TC, and Niemann H (1994). Synaptic vesicle membrane fusion complex: action of clostridial neurotoxins on assembly. EMBO J. 13, 5051–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, and Wu L-G (2007). The debate on the kiss-and-run fusion at synapses. Trends Neurosci. 30, 447–455. [DOI] [PubMed] [Google Scholar]
- Holroyd P, Lang T, Wenzel D, De Camilli P, and Jahn R (2002). Imaging direct, dynamin-dependent recapture of fusing secretory granules on plasma membrane lawns from PC12 cells. Proc. Natl. Acad. Sci. USA 99, 16806–16811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt M, Varoqueaux F, Wiederhold K, Takamori S, Urlaub H, Fasshauer D, and Jahn R (2006). Identification of SNAP-47, a novel Qbc-SNARE with ubiquitous expression. J. Biol. Chem 281, 17076–17083. [DOI] [PubMed] [Google Scholar]
- Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, and Edwards RH (2011). v-SNARE composition distinguishes synaptic vesicle pools. Neuron 71, 474–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaqaman K, and Grinstein S (2012). Regulation from within: the cytoskeleton in transmembrane signaling. Trends Cell Biol. 22, 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatekin E (2018). Toward a unified picture of the exocytotic fusion pore. FEBS Lett. 592, 3563–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman L, and Rousseeuw PJ (1990). Finding Groups in Data: An Introduction to Cluster Analysis (Wiley). [Google Scholar]
- Kubo K, Kobayashi M, Nozaki S, Yagi C, Hatsuzawa K, Katoh Y, Shin H-W, Takahashi S, and Nakayama K (2015). SNAP23/25 and VAMP2 mediate exocytic event of transferrin receptor-containing recycling vesicles. Biol. Open 4, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuster A, Nola S, Dingli F, Vacca B, Gauchy C, Beaujouan J-C, Nunez M, Moncion T, Loew D, Formstecher E, et al. (2015). The Q-soluble N-Ethylmaleimide-sensitive Factor Attachment Protein Receptor (Q-SNARE) SNAP-47 Regulates Trafficking of Selected Vesicle-associated Membrane Proteins (VAMPs). J. Biol. Chem 290, 28056–28069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Gyllborg D, Codeluppi S, Nishimura K, Salto C, and Zeisel A (2016). Molecular diversity of midbrain development in mouse, human and stem cells. Cell 167, 566.el9–580.el9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê S, Josse J, and Husson F (2008). FactoMineR: An R Package for Multivariate Analysis. J. Stat. Softw 25, 1–18. [Google Scholar]
- Lenne P-F, Wawrezinieck L, Conchonaud F, Wurtz O, Boned A, Guo X-J, Rigneault H, He H-T, and Marguet D (2006). Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 25, 3245–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link E, Edelmann L, Chou JH, Binz T, Yamasaki S, Eisel U, Baumert M, Südhof TC, Niemann H, and Jahn R (1992). Tetanus toxin action: inhibition of neurotransmitter release linked to synaptobrevin proteolysis. Biochem. Biophys. Res. Commun 189, 1017–1023. [DOI] [PubMed] [Google Scholar]
- Logan T, Bendor J, Toupin C, Thorn K, and Edwards RH (2017). α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci 20, 681–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr D, and Hildreth E (1980). Theory of edge detection. Proc. R. Soc. Lond. B Biol. Sci 207, 187–217. [DOI] [PubMed] [Google Scholar]
- Martineau M, Somasundaram A, Grimm JB, Gruber TD, Choquet D, Taraska JW, Lavis LD, and Perrais D (2017). Semisynthetic fluorescent pH sensors for imaging exocytosis and endocytosis. Nat. Commun 8, 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Arca S, Coco S, Mainguy G, Schenk U, Alberts P, Bouillé P, Mezzina M, Prochiantz A, Matteoli M, Louvard D, and Galli T (2001). A common exocytotic mechanism mediates axonal and dendritic outgrowth. J. Neurosci 21, 3830–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon S, Goldfarb D, Ho CT, Cloer EW, Boyer NP, Hardie C, Bock AJ, Johnson EC, Anil J, Ben Major M, and Gupton SL (2020). The TRIM9/TRIM67 neuronal interactome reveals novel activators of morphogenesis. Mol. Biol. Cell. mbc E20-10-0622. 10.1091/mbc.e20-10-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, and Rothman JE (1998). Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature 394, 192–195. [DOI] [PubMed] [Google Scholar]
- Napolitano LM, and Meroni G (2012). TRIM family: Pleiotropy and diversification through homomultimer and heteromultimer formation. IUBMB Life 64, 64–71. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Arakawa T, Tanimura A, Itakura M, Takahashi M, Tajima Y, Mizoguchi I, and Takuma T (2006). Role of VAMP-2, VAMP-7, and VAMP-8 in constitutive exocytosis from HSY cells. Histochem. Cell Biol 125, 273–281. [DOI] [PubMed] [Google Scholar]
- Park JJ, Gondré-Lewis MC, Eiden LE, and Loh YP (2011). A distinct trans-Golgi network subcompartment for sorting of synaptic and granule proteins in neurons and neuroendocrine cells. J. Cell Sci 124, 735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelleg D, Pelleg D, and Moore A (2000). X-means: Extending K-means with Efficient Estimation of the Number of Clusters. Proceedings of the 17th International Conf. on Machine Learning, 727–734. [Google Scholar]
- Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfenninger KH (2009). Plasma membrane expansion: a neuron’s Herculean task. Nat. Rev. Neurosci 10, 251–261. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, and Jahn R (2007). Kiss-and-run, collapse and ‘readily retrievable’ vesicles. Traffic 8, 1137–1144. [DOI] [PubMed] [Google Scholar]
- Rousseeuw PJ (1987). Silhouettes: A graphical aid to the interpretation and validation of cluster analysis. J. Comput. Appl. Math 20, 53–65. [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch S (2001). SNARE Function Analyzed in Synaptobrevin/VAMP Knockout Mice. Science 294, 1117–1122. [DOI] [PubMed] [Google Scholar]
- Segovia M, Alés E, Montes MA, Bonifas I, Jemal I, Lindau M, Maximov A, Südhof TC, and Alvarez de Toledo G (2010). Push-and-pull regulation of the fusion pore by synaptotagmin-7. Proc. Natl. Acad. Sci. USA 107, 19032–19037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Rosendale M, Campbell RE, and Perrais D (2014). pHuji, a pH-sensitive red fluorescent protein for imaging of exo- and endocytosis. J. Cell Biol 207, 419–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo M, Courchet J, Pieraut S, Torabi-Rander N, Sando Iii R, Polleux F, Maximov A, Sando R, Polleux F, Maximov A, and Maximov A (2015). SNAREs Controlling Vesicular Release of BDNF and Development of Callosal Axons. Cell Rep 11, 1054–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W, Arpino G, Thiyagarajan S, Su R, Ge L, McDargh Z, Guo X, Wei L, Shupliakov O, Jin A, O’Shaughnessy B, and Wu LG (2020). Vesicle Shrinking and Enlargement Play Opposing Roles in the Release of Exocytotic Contents. Cell Rep. 30, 421–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin W, Ge L, Arpino G, Villarreal SA, Hamid E, Liu H, Zhao WD, Wen PJ, Chiang HC, and Wu LG (2018). Visualization of Membrane Pore in Live Cells Reveals a Dynamic-Pore Theory Governing Fusion and Endocytosis. Cell 173, 934–945.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, and Rothman JE (1993). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324. [DOI] [PubMed] [Google Scholar]
- Staykova M, Holmes DP, Read C, and Stone HA (2011). Mechanics of surface area regulation in cells examined with confined lipid membranes. Proc. Natl. Acad. Sci. USA 108, 9084–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, and Rothman JE (2009). Membrane Fusion: Grappling with SNARE and SM Proteins. Science 323, 474–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugar CA, and James GM (2003). Finding the Number of Clusters in a Data-set. J. Am. Stat. Assoc 98, 750–763. [Google Scholar]
- Urbina FL, and Gupton SL (2020). SNARE-Mediated Exocytosis in Neuronal Development. Front. Mol. Neurosci 13, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbina FL, Gomez SM, and Gupton SL (2018). Spatiotemporal organization of exocytosis emerges during neuronal shape change. J. Cell Biol 217, 1113–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viesselmann C, Ballweg J, Lumbard D, and Dent EW (2011). Nucleofection and primary culture of embryonic mouse hippocampal and cortical neurons. J. Vis. Exp, 2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-T, Lu J-C, Bai J, Chang PY, Martin TFJJ, Chapman ER, and Jackson MB (2003). Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature 424, 943–947. [DOI] [PubMed] [Google Scholar]
- Wang C, Tu J, Zhang S, Cai B, Liu Z, Hou S, Zhong Q, Hu X, Liu W, Li G, et al. (2020). Different regions of synaptic vesicle membrane regulate VAMP2 conformation for the SNARE assembly. Nat. Commun 11, 1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen PJ, Grenklo S, Arpino G, Tan X, Liao H-S, Heureaux J, Peng S-Y, Chiang H-C, Hamid E, Zhao W-D, et al. (2016). Actin dynamics provides membrane tension to merge fusing vesicles into the plasma membrane. Nat. Commun 7, 12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkle CC, McClain LM, Valtschanoff JG, Park CS, Maglione C, and Gupton SL (2014). A novel Netrin-1-sensitive mechanism promotes local SNARE-mediated exocytosis during axon branching. J. Cell Biol 205, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Hamid E, Shin W, and Chiang H (2014). Exocytosis and Endocytosis: Modes, Functions, and Coupling Mechanisms. Annu. Rev. Physiol 76, 301–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan T, Lu J, Zhang J, Zhang Y, and Chen L (2015). Spatiotemporal detection and analysis of exocytosis reveal fusion “hotspots” organized by the cytoskeleton in endocrine cells. Biophys. J 108, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All image analysis and classification code generated and detailed in this study are deposited at https://github.com/GuptonLab. Specifically, the exocytosis classifier is available in the repository https://github.com/GuptonLab/Exocytosis-Classification.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (Perez-Riverol et al., 2019) with the dataset identifier PXD021758 and DOI 10.6019/PXD021758.