

Abstract
Dronedarone, an antiarrhythmic drug, has been marketed as an alternative to amiodarone. The use of dronedarone has been associated with severe liver injury; however, the mechanisms remain unclear. In this study, the possible mechanisms of dronedarone induced liver toxicity were characterized in HepG2 cells. Dronedarone decreased cells viability and induced apoptosis and DNA damage in a concentration- and time-dependent manner. Pretreatment of the HepG2 cells with apoptosis inhibitors (caspase-3, −8, and −9) or the necrosis inhibitor (Necrox-5), partially, but significantly, reduced the release of lactate dehydrogenase. Dronedarone caused the release of cytochrome c from mitochondria to cytosol, a prominent feature of apoptosis. In addition, the activation of caspase-2 was involved in dronedarone induced DNA damage and the activation of JNK and p38 signaling pathways. Inhibition of JNK and p38 by specific inhibitors attenuated dronedarone-induced cell death, apoptosis, and DNA damage. Additionally, suppression of caspase-2 decreased the activities of JNK and p38. Dronedarone triggered DNA damage was regulated by downregulation of topoisomerase IIα at both transcriptional and post-transcriptional levels. Taken together, our data show that DNA damage, apoptosis, and the activation of JNK and p38 contribute to dronedarone-induced cytotoxicity.
Keywords: dronedarone, liver toxicity, DNA damage-induced apoptosis, topoisomerase I, topoisomerase IIα, caspase-2, MAPK pathway
INTRODUCTION
Dronedarone, a noniodinated benzofuran derivative of amiodarone, has been approved by the U.S. Food and Drug Administration (FDA) since 2009 as an antiarrhythmic drug for the treatment of atrial fibrillation and atrial flutter. Dronedarone was initially developed as a safer alternative to amiodarone, which was a potent antiarrhythmic drug but caused fatal hepatitis and cirrhosis (Mason, 1987). Although dronedarone was developed to minimize amiodarone’s liver toxicity, dronedarone has been reported to cause a total of 727 cases of liver disorders, including two cases of acute liver failure leading to liver transplantation in the first two years after its launch (FDA, 2011; Joghetaei et al., 2011; De Ferrari and Dusi, 2012; Jahn et al., 2013). These reports of liver injury were followed by the warnings from U.S. FDA about possible severe hepatotoxicity in patients treated with dronedarone. To date, mechanistic studies on dronedarone induced liver toxicity are limited to only a few reports, which mainly focused on mitochondrial toxicity (Serviddio et al., 2011; Felser et al., 2013, 2014). Since dronedarone is still available on the market and used by patients, there is an unquestioned need of evaluating toxicities caused by dronedarone as well as understanding the cellular mechanisms of dronedarone-associated toxicities.
In response to damage or stress, cells exhibit multiple types of cell death, with apoptosis and necrosis being the major cell death modes (Green and Llambi, 2015; Vanden Berghe et al., 2015). Apoptosis is a caspase-dependent cell death route and is regulated by two distinct pathways: the death receptor-mediated extrinsic pathway, and the mitochondria-mediated intrinsic pathway (Taylor et al., 2008). The extrinsic pathway is triggered by binding of pro-apoptotic ligands to their corresponding receptors to form a death-inducing signaling complex (DISC), such as TNF/TNFR1 (tumor necrosis factor/tumor necrosis factor receptor 1) and Fas ligand/Fas. The DISC subsequently recruits and activates caspase-8, and then activates caspase-3, the “executioner” of apoptosis. The intrinsic pathway is characterized by mitochondrial dysfunction and the subsequent release of apoptogenic factor cytochrome c from the mitochondria into the cytosol. The Bcl-2 family, including both pro-apoptotic members (e.g., Bax and Bad) and anti-apoptotic members (e.g., Mcl-1 and Bcl-2), tightly regulates the release of cytochrome c (Chao and Korsmeyer, 1998). Cytochrome c subsequently cleaves caspase-9, which then activates caspase-3, leading to apoptotic cell death. In contrast, necrotic cell death refers to passive cell death that occurs from the loss of cell membrane integrity and does not activate any specific signaling pathways (Yuan and Kroemer, 2010; Green and Llambi, 2015).
The induction of intrinsic apoptosis has been demonstrated as an outcome of DNA damage (Norbury and Zhivotovsky, 2004; Roos and Kaina, 2006), with caspase-2 being the link between nuclear DNA damage responses and mitochondrial permeabilization (Lassus et al., 2002). Caspase-2 is the most conserved caspase and acts as both initiator and executioner during apoptosis (Vakifahmetoglu-Norberg and Zhivotovsky, 2010). In response to DNA damage, pro-caspase-2 forms an active caspase heterotetramer via autocatalytic cleavage and then promotes cytochrome c release, the key feature of apoptosis (Robertson et al., 2002; Aksenova et al., 2016).
Mitogen-activated protein kinase (MAPK) signaling pathway is involved in many cellular functions in response to various stimuli (Pearson et al., 2001). In mammalian cells, three major branches of classic MAP kinases have been well-characterized: c-jun N-terminal kinases (JNKs), p38 MAP kinases, and extracellular signal regulated kinases (ERKs). MAP kinases are catalytically inactive in their base form and require phosphorylation for their signaling cascade activation (Chang and Karin, 2001). MAP kinases play different roles in regulating cell responses to different activators. For instance, ERKs mediate cell proliferation and survival in response to growth factors, whereas p38 and JNKs are associated with apoptosis and other types of cell death in response to stressful stimuli (Xia et al., 1995). Studies, including ours, have shown that activation of JNK or JNK/p38 signaling pathway plays an important role in cellular DNA damage and apoptosis (Roos and Kaina, 2006; Chen et al., 2014a,b; Guo et al., 2015).
In the present study, we investigated whether or not DNA damage and apoptosis could be the possible mechanisms involved in the toxic effects of dronedarone. We explored the potential underlying molecular pathways in HepG2 cells using various biological markers and approaches. We demonstrated that DNA damage induced-apoptosis contributes to dronedarone-induced cytotoxicity, with the involvement of the activation of caspase-2 and JNK/p38 signaling pathway. We also studied the potential role of topoisomerase inhibition in triggering dronedarone associated DNA damage. Our results suggest that downregulation of topoisomerase IIα may be responsible for DNA damage caused by dronedarone.
MATERIALS AND METHODS
Chemicals and Reagents
Dronedarone, dimethysulfoxide (DMSO), Williams’ medium E, and MG-132 protease inhibitor were from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). Antibiotic-antimycotic was from Life Technologies (Grand Island, NY). The general caspase inhibitor (Z-VAD-FMK), the caspase-3 inhibitor (Z-DEVD-FMK), the caspase-8 inhibitor (Z-IETD-FMK), the caspase-9 inhibitor (Z-LEHD-FMK), and the caspase-2 inhibitor (Z-VAVAD-FMK) were obtained from R&D systems (Minneapolis, MN). For Western blotting assays, the primary antibodies against the caspase-3, caspase-9, cleaved caspase-8, cytochrome c, Mcl-1, Bcl-2, Bax, Bad, phospho-JNK (Thr183/Thr185), JNK, phospho-p38 (Thr180/Tyr182), p38, phospho-ERK1/2 (Thr202/Tyr204), ERK1/2, PARP-1 (poly (ADP-ribose) polymerase), phospho-Chk1(Ser345), phospho-Chk2 (Thr68), and γ-H2A.X (Ser139) were purchased from Cell Signaling Technology (Danvers, MA). Antibody for topoisomerase I was obtained from Abcam (Cambridge, MA). Antibodies for caspase-2, α-Tubulin, topoisomerase IIα, and GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture and Treatment with Dronedarone
The HepG2 human hepatoma cell line was from the American Type Culture Collection (ATCC; Manassas, VA). HepG2 cells were cultured in Williams’ medium E complete media containing 10% FBS and 1 × antibiotic antimycotic at 37°C in a humidified atmosphere with 5% CO2. The passage number did not excess 10. Unless otherwise specified, cells were seeded at a density of 5 × 105 cells/ml in a volume of 50 μl per well in 96-well plates, in a volume of 5 ml in 60 mm tissue culture dishes, or 10 ml in 100 mm tissue culture dishes. Cells were cultured for 24 hr prior to treatment with dronedarone or the DMSO vehicle control. HepaRG cells, derived from a human hepatic progenitor cell line, are terminally differentiated hepatic cells that retain many characteristics of primary human hepatocytes. HepaRG cells were purchased from Life Technologies. After thawing, HepaRG cells were seeded at 1 × 105 cells per well in 96-well plates, and maintained in Williams’ medium E supplemented with the Thaw, Plate, & General Purpose Medium Supplement (Life Technologies) for 7 days. The cells were then treated with dronedarone at different concentrations or the DMSO vehicle control for 6 hr. The final concentration of DMSO was 0.1%.
MTS Cell Viability Assay
CellTiter 96® AQueous One Solution Reagent (MTS, Promega Corp., Madison, WI) was used to detect cell viability. After treatment of dronedarone in 96-well plates, the supernatants were aspirated and then 10 μl of MTS reagent, mixed with 90 μl serum-free media, was added to each well. After incubation for 1 hr at 37°C in a humidified 5% CO2 atmosphere, the absorbance at 490 nm was measured with a Synergy 2 Microplate Reader (BioTek, Winooski, VT). Background absorbance signals, determined in a set of cell-free wells, were subtracted from sample signals. The cell viabilities were calculated by comparing the absorbance of the treated cells to that of the DMSO controls.
Lactate Dehydrogenase Assay
The cytotoxicity of dronedarone was assessed using a lactate dehydrogenase (LDH) assay. Briefly, after treatment of dronedarone in 96-well plates, 6 μl of cell-free supernatant from each well was transferred into the corresponding well of a new clear 96-well plate, and then 10 μl of 10% Triton X-100 was added to each well containing treated cells. After a 2-h lysis, 10 μl of lysates were transferred into the empty wells in each 96-well plate containing the corresponding supernatant, and 230 μl of reaction buffer (81.3 mM Tris, 203.3 mM NaCl, 0.2 mM NADH, and 1.7 mM monosodium pyruvate, pH 7.2) was added to the wells with supernatants or cell lysates. Absorption was measured at 340 nm for 5 min at 1-min intervals using a Synergy 2 Microplate Reader (BioTek, Winooski, VT). The LDH release was calculated by the following equation: percent cytotoxicity = 100 × (decrease in supernatant absorption/decrease in cell lysate absorption).
Cellular Apoptosis Assay
The treated cells undergoing early/late apoptosis were determined by FITC Annexin V Apoptosis Detection Kit with PI (Biolegend®, San Diego, CA). Briefly, HepG2 cells were seeded at a density of 1.5 × 105 cells/ml in a volume of 2 ml per well into 6-well plates and incubated overnight. The cells were exposed to various concentrations of dronedarone (6.25–25 μM) for 4 hr. After exposure, 3 × 105 cells were collected and resuspended in 100 μl annexin V binding buffer. The cell suspension was stained with 5 μl of FITC annexin V and 10 μl of propidium iodide solution for 15 min at room temperature in the dark. Before being analyzed with a CytoFLEX flow cytometer (Beckman Coulter, Indianapolis, IN), the cells were diluted with 400 μl of annexin V binding buffer. Data were acquired and analyzed using CytoFLEX CytExpert software (Beckman Coulter, Indianapolis, IN).
Caspase-3/7, Caspase-8, and Caspase-9 Activity Measurement
The enzymatic activities of caspase-3/7, −8, and −9 were assayed using luminescent assay kits (Caspase-Glo® 3/7 Assay Systems, Caspase-Glo® 8 Assay Systems, and Caspase-Glo® 9 Assay Systems, Promega) according to the manufacturer’s instructions and measured with a Synergy 2 Microplate Reader (BioTek). The caspase induction was calculated by comparing the luminescence of the treated cells to that of the DMSO controls.
Caspase-2 Activity Measurement
The enzymatic activity of caspase-2 was determined using Caspase-Glo® 2 Reagent (Promega) in combination with 1 μM of the caspase-3/7 inhibitor (Z-DEVD-FMK) and 60 μM of the MG-132 protease inhibitor and measured with a Synergy 2 Microplate Reader (BioTek). The induction of caspase-2 was calculated by comparing the luminescence of the treated cells to that of the DMSO controls.
Western Blot Analysis
Cells were plated in 60 or 100 mm tissue culture dishes and treated with dronedarone. After treating for specified times and at specified concentrations, whole-cell lysates were prepared using RIPA buffer containing Halt Protease Inhibitor Cocktail (ThermoFisher Scientific, Waltham, MA). The separation of mitochondria from cytosolic fractions was performed using a Mitochondria Isolation kit for cultured cells (ThermoFisher Scientific) according to the manufacturer’s instructions. The concentrations of the protein samples were determined using a Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA). Standard Western blots were performed. Depending on the proteins of interest, antibodies were selected against caspase-2, caspase-3, caspase-9, cleaved caspase-8, cytochrome c, Mcl-1, Bcl-2, Bax, Bad, phospho-JNK (Thr183/Thr185), JNK, phospho-p38 (Thr180/Tyr182), p38, phospho-ERK1/2 (Thr202/Tyr204), ERK1/2, PARP-1, phospho-Chk1(Ser345), phospho-Chk2 (Thr68), γ-H2A.X (Ser139), topoisomerase I, and topoisomerase IIα, followed by an incubation with secondary antibody conjugated with horseradish peroxidase (Santa Cruz Biotechnology). GAPDH or α-Tubulin was used as the internal control. The protein signals were determined and quantified with a FluroChem E System (ProteinSimple, San Jose, CA).
Assay of Topoisomerase I-Mediated Relaxation of Supercoiled Plasmid
The topoisomerase I enzyme assay was performed using a topoisomerase I drug screening kit (TopoGen, Buena Vista, CO) as described previously (Chen et al., 2013). Briefly, a 20 μl reaction mixture containing 10 mM Tris HCl (pH 7.9), 1 mM EDTA, 0.15 M NaCl, 0.1% BSA, 0.1 mM spermidine, 5% glycerol, 125 ng supercoiled plasmid DNA, and 2 units of purified human topoisomerase I was incubated at 37°C for 30 min with or without various concentrations of dronedarone. The reactions were stopped by addition of 10% SDS, and then an additional incubation at 37°C for 15 min with proteinase K was conducted to degrade denatured topoisomerase I. The relaxation of supercoiled plasmid DNA was detected using a non-EB gel and staining with EB for 15 min and imaged with a FluroChem E System (ProteinSimple).
RNA Isolation and Real-Time PCR Assay
Total RNA was isolated using the RNeasy system (Qiagen, Germantown, MD). cDNAs were generated by reverse transcription of 2 μg of total RNA using high capacity cDNA reverse transcription kits (Applied Biosystems, Foster City, CA) according to the manufacturer’s instruction. Quantitative real-time PCR for topoisomerase IIα was performed as described previously (Chen et al., 2013) to evaluate relative gene expression.
Statistical Analyses
Data are presented as the mean ± standard deviation (SD) of at least three independent experiments. Analyses were performed using Graph-Pad Prism 5 (GraphPad Software, San Diego, CA). Statistical significance was determined by one-way analysis of variance (ANOVA) followed by the Dunnett’s tests for pairwise-comparisons or two-way ANOVA followed by the Bonferroni post-test. The difference was considered statistically significant when P was less than 0.05.
RESULTS
Dronedarone Induces Cellular Damage in HepG2 and HepaRG Cells
To assess whether dronedarone induces cytotoxicity in HepG2 cells, cells were treated with dronedarone at concentrations of 6.25 to 25 μM for 2, 4, and 6 hr. Two different endpoints, MTS assay (to measure the conversion of a colored formazan product generated by NAD(P)H-dependent mitochondrial dehydrogenase activity in viable cells) and LDH release assay (to determine the damage of plasma membrane by measuring the release of the enzyme lactate dehydrogenase into the supernatants) were used to measure the general cytotoxicity of dronedarone. As shown in Figure 1A, dronedarone significantly decreased cell viability in a time- and concentration-dependent manner when measured by MTS assay. At 2 and 4 hr, 10 μM dronedarone inhibited the cell viability to about 70% of that of the DMSO control. Moreover, MTS level dropped to near zero in cells treated with 25 μM of dronedarone for 6 hr, indicating severe cell death upon dronedarone exposure.
Fig. 1.
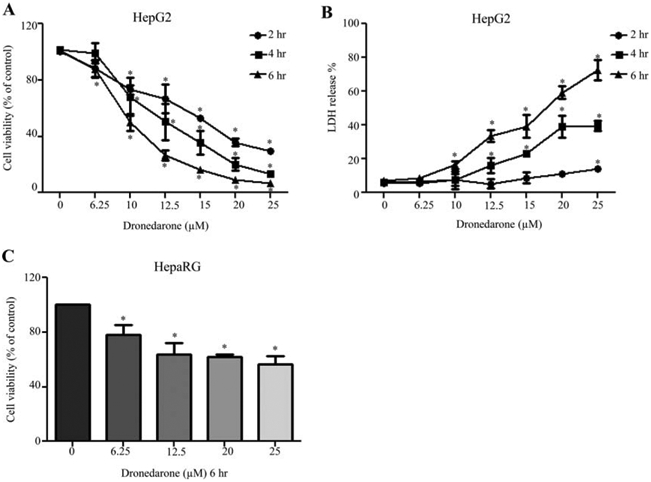
Dronedarone induces cellular damage in HepG2 and HepaRG cells. HepG2 cells were exposed to dronedarone at 6.25, 10, 12.5, 15, 20, and 25 μM for 2, 4, and 6 hr, with DMSO as the vehicle control and cytotoxicity was measured using MTS assay (A) and LDH assay (B). (C) HepaRG cells were treated with dronedarone at 6.25, 12.5, 20, and 25 μM for 6 hr and cytotoxicity was determined using MTS assay. The results shown are mean ± S.D. from three independent experiments. *, P < 0.05 compared with the control for each time point.
LDH release, an indicator of cell necrosis, was elevated in a time- and concentration-dependent manner by dronedarone treatment (Fig. 1B). At 2 hr, a significant 14% release of LDH occurred at 25 μM dronedarone exposure, while at 4 hr, a 15–40% release of LDH was observed, with the release becoming significant at 12.5 μM. At 6 hr, a 16–73% release of LDH occurred, with the release becoming significant at 10 μM, implicating more pronounced cellular damage after extended exposure to dronedarone.
HepaRG cells are terminally differentiated human hepatic progenitor cells that retain many features of primary hepatocytes, including expression of key metabolic enzymes. As shown in Figure 1C, at 6 hr, 25 μM dronedarone decreased the cell viability to about 57% of that of the DMSO control when measured by MTS assay. These data indicated that HepG2 cells have higher sensitivity although both HepG2 and HepaRG cells show significant cytotoxicity upon dronedarone exposure. Thus, the following mechanistic studies were performed in HepG2 cells.
As shown in Figure 2 by using flow cytometry analysis of Annexin V/PI staining, HepG2 cells exhibited significant increases in the percentage of early apoptotic cells and late apoptotic/necrotic cells upon dronedarone treatment. These data implied that dronedarone induced cellular damage may result from both apoptotic and necrotic cell death.
Fig. 2.

Dronedarone induces apoptotic cell death in HepG2 cells. (A) Flow cytometric analysis of Annexin V and PI staining of HepG2 cells which were exposed for 4 hr to dronedarone at indicated concentrations. Early apoptotic cells are stained only with annexin V and late apoptotic or necrotic cells are double positive for annexin V and PI staining. (B) The bar graph depicts the percentage of early or late apoptotic population. The results shown are mean ± S.D. from three independent experiments. *, P < 0.05 compared with the DMSO treated cells.
Dronedarone-Induced Cytotoxicity via Both Apoptosis and Necrosis
To determine whether apoptosis and/or necrosis are the cell death modes and to assess the involvement of intrinsic and/or extrinsic apoptosis signaling pathways, we used a number of inhibitors, including specific apoptotic and necrotic inhibitors, and tested their effects on dronedarone-induced cytotoxicity. As shown in Figure 3A, the cytosolic fraction of cytochrome c was increased, accompanied by a decreased proportion in mitochondria, indicating that dronedarone activated intrinsic apoptotic signaling pathway.
Fig. 3.
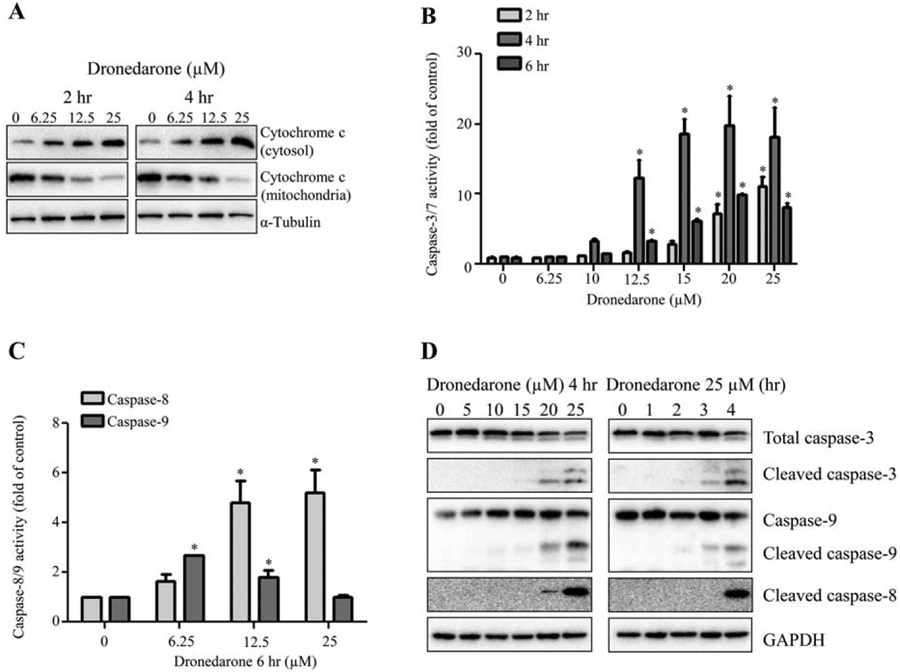
Effect of dronedarone on apoptosis related proteins in HepG2 cells. HepG2 cells were treated with the indicated concentrations of dronedarone. (A) Mitochondrial and cytosolic proteins were extracted at 2 and 4 hr after dronedarone exposure, and the expression levels of cytochrome c were examined by Western blotting with α-Tubulin being a loading control. Similar results were obtained from three independent experiments. (B, C) Cellular caspase-3/7, −8, and −9 activities were expressed as fold changes to DMSO control cells (* P < 0.05). (D) Total cellular proteins were isolated at 4 hr after dronedarone treatment with concentrations up to 25 μM or at different time points up to 4 hr with 25 μM treatment. The expression levels of caspase-3, −8, and −9 were evaluated by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments.
The enzymatic activities and protein expression levels of the key caspases in intrinsic pathway, including caspase-3/7 and caspase-9, and the major caspase family member in extrinsic pathway, caspase-8, were assessed. As indicated in Figure 3B, the peak of enzymatic activities of caspase-3/7 appeared after a 4 hr exposure to dronedarone; whereas at 6 hr, the activities were less pronounced than that at 4 hr, possibly due to the loss of cells as measured with the LDH assay (Fig. 1B). Interestingly, at 6 hr, despite severe cell death, significant increases of the activities of caspase-8 and caspase-9 were still detectable, although the activity of caspase-9 declined at 25 μM treatment (Fig. 3C). Based on the results described above, we focused our further studies on the 4-h time point where the increase of LDH release was significant but less than that at 6 hr. The cleaved forms of caspase-3, −8, and −9 representing activation were assessed using Western blot analysis. As shown in Figure 3D, dronedarone markedly increased the expression levels of cleaved forms of caspase-3, −8, and −9.
We further investigated the expression levels of Bcl-2 family members that regulate the activation of caspases and participate in the intrinsic apoptosis pathway. As shown in Figure 4A, the key anti-apoptotic members Mcl-1 and Bcl-2 were decreased in a concentration- and time-dependent manner, whereas no change was observed in the expression of pro-apoptotic members Bax and Bad, indicating that dronedarone-induced apoptosis is dependent on deactivation of anti-apoptotic Bcl-2 family members rather than activation of pro-apoptotic members.
Fig. 4.

Both apoptosis and necrosis contribute to dronedarone-induced cytotoxicity in HepG2 cells. (A) Total cellular proteins were extracted after dronedarone treatment at indicated concentrations and time-points. The levels of anti-apoptosis Bcl-2 family members, such as Mcl-1 and Bcl-2, and pro-apoptosis members, such as Bax and Bad were detected by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments. (B, C) HepG2 cells were pretreated with 10 μM of general caspase inhibitor (Z-VAD-FMK), caspase-3 inhibitor (Z-DEVD-FMK), caspase-8 inhibitor (Z-IETD-FMK), or caspase-9 inhibitor (Z-LEHD-FMK) for 1 hr prior to treatment of dronedarone for 6 hr. Cytotoxicity and apoptosis were determined by LDH assay (B) and caspase-3/7 activity (C), respectively. (D) HepG2 cells were pretreated with 5 μM of Necrox-5 (necrosis inhibitor) for 1 hr prior to treatment of dronedarone for 6 hr. Cytotoxicity was measured by LDH release assay. The bar graphs are mean ± SD of three individual experiments. *, P < 0.05 compared with treatment of dronedarone alone.
Various inhibitors have been shown to specifically block apoptotic or necrotic pathways, and we assessed their protective effects on dronedarone-induced cellular damage. As indicated in Figure 4B, pretreatment with 10 μM general caspase inhibitor (VAD-FMK), 10 μM caspase-3 inhibitor (DEVD-FMK), 10 μM caspase-8 inhibitor (IETD-FMK), or 10 μM caspase-9 inhibitor (LEHD-FMK) significantly decreased dronedarone-elevated LDH release. The inhibitory efficiency of the four inhibitors was confirmed by measuring caspase-3/7 activities (Fig. 4C). In addition, Necrox-5, a necrosis inhibitor, significantly attenuated dronedarone-induced cell death, indicating that dronedarone-induced liver toxicity is mediated by both apoptosis and necrosis (Fig. 4D).
Collectively, cytochrome c release initiated the intrinsic apoptosis pathway, caspase-8 activated the extrinsic apoptosis pathway, and necrosis, and all three pathways participated in dronedarone-induced cytotoxicity.
Dronedarone Triggers DNA Damage and Caspase-2 Activation
It is known that induction of apoptosis can be derived from DNA damage (Wyllie et al., 1980); therefore, we examined whether or not dronedarone induces DNA damage by measuring histone H2A.X phosphorylation at serine139 (γ-H2A.X), a hallmark of double-strand DNA breakage in cells (Rogakou et al., 1998). Three proteins that associate with DNA repair, including PARP-1, phosphorylated-Chk1 (p-Chk1) and phosphorylated-Chk2 (p-Chk2), were also assessed. Upon exposure to dronedarone, HepG2 exhibited concentration- and time-dependent increases in the expression of γ-H2A.X, cleavage of PARP-1, and p-Chk2, whereas p-Chk1 remained undetectable (Fig. 5A). Upregulation of γ-H2A.X and cleaved PARP-1 was observed at concentrations of 15–25 μM at the 4-h time point. At 25 μM, dronedarone-induced p-Chk2 was observed as early as 1 hr, followed by the changes in γ-H2A.X and PARP-1. These observations indicate that DNA damage was induced upon dronedarone treatment.
Fig. 5.
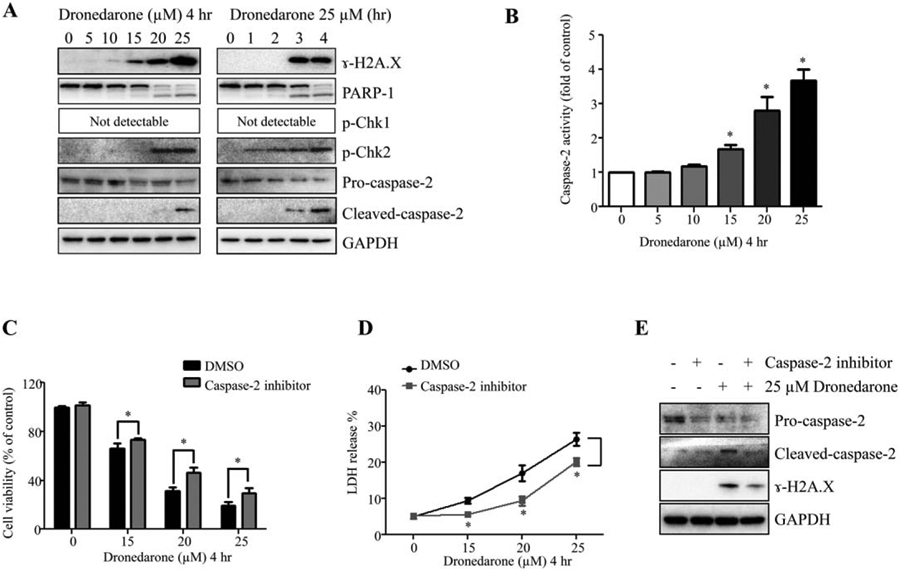
Dronedarone causes DNA damage and activates caspase-2 in HepG2 cells. (A) Total cellular proteins were extracted after dronedarone treatment at indicated concentrations and time-points. The expression levels of ɣ-H2A.X, PARP-1, p-Chk1, p-Chk2, and caspase-2 were determined by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments. (B) Caspase-2 activity was assessed as described under Materials and Methods section after various concentrations of dronedarone treatment for 4 hr. (C-E) HepG2 cells were co-treated with dronedarone at indicated concentrations and 50 μM of Z-VAVAD-FMK for 4 hr. Cytotoxicity was determined by MTS assay (C) and LDH assay (D). Results shown are mean ± SD of three individual experiments. *, P < 0.05 compared with treatment of dronedarone alone. (E) The inhibitory effect of caspase-2 inhibitor and the effect of caspase-2 inhibitor on DNA damage were assessed by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments.
Caspase-2 links DNA damage with cellular apoptosis because it is involved in the release of apoptotic effectors from mitochondria in response to DNA damage (Lassus et al., 2002; Norbury and Zhivotovsky, 2004). To determine whether or not dronedarone induces caspase-2 activation, we examined the expression level of cleaved caspase-2 and the enzymatic activity of caspase-2. As shown in Figure 5A, starting at 3 hr, 25 μM of dronedarone remarkably induced the cleavage of caspase-2, accompanied by a decrease of pro-caspase-2. In agreement with the results from Western blotting, the increase of caspase-2 enzymatic activation was observed in a concentration-dependent manner, starting at 15 μM for 4 hr (Fig. 5B).
To confirm that caspase-2 activation is involved in dronedarone-induced cytotoxicity, we applied a specific caspase-2 inhibitor Z-VAVAD-FMK. As shown in Figure 5E, 50 μM of Z-VAVAD-FMK markedly inhibited both the pro-caspase-2 and cleaved caspase-2. This suppression significantly increased cell viability compared with cells treated with dronedarone alone (Fig. 5C,D). Inhibition of caspase-2 activity decreased the extent of DNA damage caused by 25 μM dronedarone, as indicated by the less induction of ɣ-H2A.X in comparison with the treatment of dronedarone alone (Fig. 5E). Collectively, DNA damage-induced apoptosis, which is positively mediated by caspase-2, contributes to dronedarone’s cytotoxicity.
Dronedarone Activates MAPK Signaling Pathway
Many studies have shown that the MAPK signaling pathway plays an important role in apoptosis and DNA damage. Accumulating evidence has indicated that there are crosstalk between the MAPK signaling pathway and caspase-2 in stress-induced cytotoxicity (Dirsch et al., 2004; Zhivotovsky and Orrenius, 2005; Mishra and Karande, 2014). Accordingly, we examined the activation of the three branches (JNK, p38, and ERK1/2) of the MAPK signaling pathway. As shown in Figure 6A, dronedarone increased the levels of p-JNK and p-p38 in a concentration-dependent manner after a 2 hr treatment; whereas no changes of JNK, p38, p-ERK 1/2, or ERK1/2 were observed. At the 4-h time point, the maximal expression level of p-p38 occurred at 15 μM. At 20 and 25 μM, the increased expression of p-p38 declined, presumably due to cell loss as measured with LDH assay (Fig. 1B), but the expression was still upregulated when compared to the DMSO control. These results suggest that dronedarone treatment activates JNK and p38 signaling pathway but not ERK1/2 pathway.
Fig. 6.

Involvement of MAPK signaling pathway in dronedarone-induced cytotoxicity in HepG2 cells. (A) Total cellular proteins were extracted after dronedarone treatment at indicated concentrations and time-points. The expression levels of p-JNK, JNK, p-p38, p38, p-ERK1/2, and ERK1/2 were determined by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments. (B-E) After treatment with 10 μM SP600125, 10 μM SB239063, or 2 μM PD184352 for 2 hr, HepG2 cells were treated with indicated concentrations of dronedarone for 4 hr with the present of each inhibitor. Cytotoxicity was assessed by MTS assay (B) and caspase-3/7 activity was used as an indicator of apoptosis (C). The bar graphs are mean ± SD of three individual experiments. *, P < 0.05 compared with treatment of dronedarone alone. (D, E) The inhibitory effect of JNK and p38 inhibitors and the effect of JNK and p38 inhibitors on DNA damage were assessed by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments. (F) HepG2 cells were co-treated with dronedarone at indicated concentrations and 50 μM of caspase-2 inhibitor (Z-VAVAD-FMK) for 4 hr. The effects of caspase-2 inhibitor on the activation of JNK and p38 were determined by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments.
To investigate whether or not the MAPK pathway participates in dronedarone-induced cytotoxicity and apoptosis, HepG2 cells were pretreated with three specific MAPK inhibitors targeting JNK (SP600125), p38 (SB239063), and ERK1/2 (PD184352) for 2 hr, followed by a 4-h dronedarone treatment. Consistent with the results from Western blotting shown in Figure 6A, JNK and p38 but not the ERK1/2 pathway regulated dronedarone-induced cytotoxicity and apoptosis, because inhibition of JNK and p38 significantly increased cell viability and decreased caspase-3/7 activity compared with dronedarone only treated group. Inhibition of ERK1/2 showed no effect (Fig. 6B,C). The efficiency of the inhibitors was confirmed by Western blotting (Fig. 6D,E). Moreover, compared to cells treated with only dronedarone, inhibition of JNK and p38 markedly suppressed the expression of ɣ-H2A.X (Fig. 6D,E). Taken together, these data indicate that the activation of both JNK and p38 promotes dronedarone-induced apoptosis and DNA damage.
Using Western blotting, we investigated further whether or not inhibition of caspase-2 regulates the induction of p-JNK and p-p38. As shown in Figure 6F, the caspase-2 inhibitor (Z-VAVAD-FMK) markedly decreased the activation of p-JNK and slightly decreased the activation of p-p38, indicating that caspase-2 can positively regulate p-JNK and p-p38.
Dronedarone Downregulates the Expression of Topoisomerase IIα but Not Topoisomerase I
To explore further the possible mechanisms of DNA damage induced by dronedarone, we assessed the generation of reactive oxygen species (ROS), which has been recognized as a prominent trigger of DNA damage (Evans and Cooke, 2004; Chen et al., 2017). No significant alternation of intracellular ROS level was observed in dronedarone treated cells as measured by H2DCFDA staining (data not shown). In addition, the expression levels of major oxidative stress response related proteins, including nuclear factor erythroid 2-related factor 2 (Nrf2), NAD(P)H quinone oxidoreductase-1 (NQO1), γ-glutamate-cysteine synthetase (γ-GCSc), heme oxygenase-1 (HO-1), glutathione synthetase (GSS), and glutathione reductase were measured by Western blotting. The results showed no significant change upon dronedarone exposure (data not shown), implying ROS generation or oxidative stress was not involved in dronedarone induced toxicity.
DNA damage can also result from topoisomerases dysfunction, which causes single and double-strand DNA cleavage. Topoisomerase inhibitors have been reported to induce capase-2 mediated DNA damage (Perchellet et al., 2004; Hua et al., 2006; Tomicic and Kaina, 2013; Wu et al., 2015). Therefore, it was of interest to investigate the involvement of topoisomerases in dronedarone-associated DNA damage. We evaluated the expression of topoisomerase I and IIα at protein level using Western blotting. As shown in Figure 7A, a concentration-dependent decrease of topoisomerase IIα was observed at 4 hr with the decrease beginning at as low as 5 μM; a time-dependent decrease appeared as early as 1 hr with 25 μM of dronedarone treatment. Starting at 15 μM for 4 hr treatments or at 25 μM for 3 hr treatments, topoisomerase IIα protein was undetectable, indicating dronedarone caused a complete elimination of topoisomerase IIα. In contrast, topoisomerase I protein level remained approximately constant. We also examined the gene expression of topoisomerase IIα. As measured by real-time PCR, topoisomerase IIα mRNA decreased significantly in a time and concentration-dependent manner, showing the same trend as protein levels (Fig. 7B)
Fig. 7.
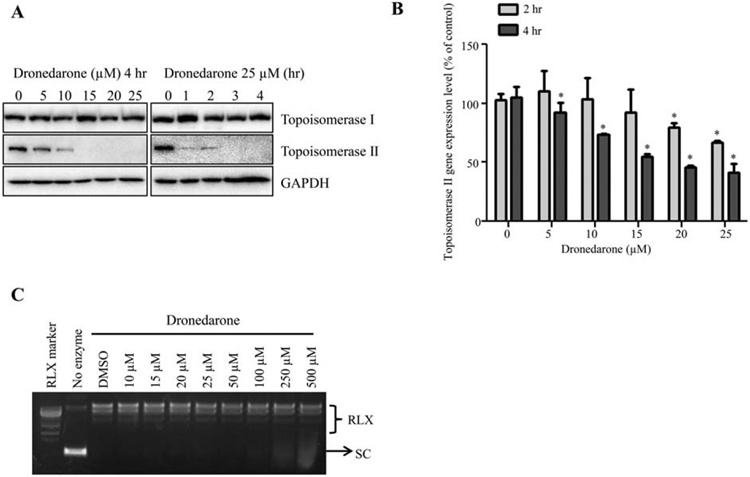
Effects of dronedarone on topoisomerase I and topoisomerase IIα. (A) Total cellular proteins were extracted after dronedarone treatment at indicated concentrations and time-points. The expression levels of topoisomerase I and topoisomerase IIα were determined by Western blotting. GAPDH was used as a loading control. Similar results were obtained from three independent experiments. (B) HepG2 cells were treated with dronedarone for 2 and 4 hr. Total RNA was isolated and real-time PCR was used to examine the expression of topoisomerase IIα at transcriptional level. The bar graph is mean α SD of three individual experiments. *, P < 0.05 compared with treatment of DMSO control. (C) Inhibitory effect of dronedarone on topoisomerase I activity was detected as described under Materials and Methods section. RLX, relaxed DNA; SC, supercoiled DNA. Representative images were from three independent experiments.
Although the protein levels of topoisomerase I were not altered significantly (Fig. 7A), there remained the possibility that topoisomerase I contributed to dronedarone-induced DNA damage by inhibiting its enzymatic activity without affecting the expression level. To rule out this possibility, we assessed the effect of dronedarone on the enzymatic activity of topoisomerase I. As indicated in Figure 7C, no inhibitory effect of topoisomerase I was observed at up to 500 μM of dronedarone exposure. These data suggest that dronedarone induces DNA damage via downregulation of a topoisomerase IIα-associated mechanism, but not a topoisomerase I-involved mechanism.
DISCUSSION
Since the approval by the U.S. FDA and the European Medicines Agency (EMA) in 2009, dronedarone has been widely used in both the U.S. and Europe (De Ferrari and Dusi, 2012). Despite the high expectations for dronedarone to be a safe and first-line treatment for atrial fibrillation, severe liver injury in humans related to the use of dronedarone has been reported. However, the mechanisms underlying dronedarone-induced hepatotoxicity are still not fully understood. An in vitro study evaluated the hepatocellular toxicity induced by dronedarone in primary human hepatocytes and the human hepatic cell line HepG2 (Felser et al., 2013) and revealed that both cell types show similar cytotoxicity upon dronedarone treatment. In this study, we assessed the cytotoxic effect of dronedarone on HepG2 and HepaRG cells. The results demonstrated that HepG2 cells exhibited more sensitivity to the insults of dronedarone (Fig. 1). Although human primary hepatocytes are considered as the most relevant in vitro cell model, their limited availability and batch-to-batch variation constrain their use in toxicity study. Thus, we used HepG2 cells to explore potential molecular mechanisms of dronedarone induced cytotoxicity. The other reasons for choosing HepG2 cells are their proven value in toxicological studies (Bova et al., 2005; Guo et al., 2006; Dykens et al., 2008; Greer et al., 2010; Felser et al., 2013; Juan-Garcia et al., 2013; Nguyen et al., 2013), and their high stability, unlimited life-span, and ready availability. We have realized that HepG2 cells have their drawbacks, such as the lack of some drug metabolizing enzymes that limits their use in drug metabolites-associated toxicity studies (Guo et al., 2011). However, our result implies that the metabolites of dronedarone contribute less than the parental drug to the toxicity because metabolism competent HepaRG cells showed a lower toxic effect comparing with HepG2 cells. In addition, using our 14 established CYPs-expressing HepG2 cell lines, a metabolism-associated toxicity screening demonstrated that the major dronedarone metabolic enzymes CYP3A4, 3A5, and 2D6 extensively detoxified cytotoxicity induced by dronedarone (unpublished data), implying that the parent form of dronedarone is the major cause for its toxicity. Thus, low expression backgroud of CYP enzymes makes HepG2 cell line an appropriate cellular model for the in-depth mechanistic studies of the toxicity of parent dronedarone at molecular level.
Clinically, the human plasma concentration of dronedarone reaches 0.28 μM in 7 days after sustained oral administration of 400 mg twice daily. In some cases, the plasma concentration can be higher than the reported concentration due to many variables such as interindividual variability in metabolic capacity and drug-drug interactions could affect metabolism of dronedarone (Hoy and Keam, 2009; Felser et al., 2013). For example, a potent CYP3A4 inhibitor could increase dronedarone exposure to as much as 25-fold (Patel et al., 2009). Studies have suggested that the cut-off concentration used for in vitro safety evaluation is 100-fold of the Cmax (maximal plasma concentration) (Lee, 2003; Dykens et al., 2008; Porceddu et al., 2012; Laifenfeld et al., 2014). The concentrations of dronedarone used were 5–25 μM, which were within the range of 100-fold of the Cmax, thus were meaningful in our in vitro mechanistic study.
In a good agreement with the results obtained from the previous study (Felser et al., 2013), we found that dronedarone caused apoptotic cell death as it increased caspase-3/7 activity and cytochrome c release, which were accompanied by downregulation of anti-apoptotic members, such as Mcl-1 and Bcl-2 (Fig. 3). Besides the caspase-3/7 activated intrinsic apoptotic pathway, we demonstrated that caspase-8 activated extrinsic apoptotic pathway and necrotic cell death also contributed to dronedarone-induced cytotoxicity (Fig. 4). Because apoptosis and necrosis are the major consequences of DNA damage (Roos and Kaina, 2006), we explored whether or not dronedarone induces DNA damage in hepatic cells, and demonstrated a concentration- and time-dependent induction of γ-H2A.X and cleavage of PARP-1, hallmarks of DNA damage (Fig. 5A). Interestingly, the induction of p-Chk2 but not p-Chk1 was observed (Fig. 5A). The Chk2 pathway regulates double-strand DNA breaks, whereas the Chk1 pathway regulates single-strand DNA breaks repair, suggesting that dronedarone may induce DNA damage via double-strand DNA breaks (Cai et al., 2009; Patil et al., 2013).
Caspase-2 is a key player in DNA damage-induced apoptosis (Lassus et al., 2002; Norbury and Zhivotovsky, 2004; Zhivotovsky and Orrenius, 2005; Vakifahmetoglu-Norberg and Zhivotovsky, 2010; Aksenova et al., 2016). Thus, we studied the role of caspase-2 in dronedarone-associated toxicity. Caspase-2 was significantly activated at both the protein expression and enzymatic activity levels in response to dronedarone treatment (Fig. 5A,B). It has been reported that caspase-2-dependent apoptosis is only observed when Chk1 is inhibited (Vakifahmetoglu-Norberg and Zhivotovsky, 2010), which is in consistent with our result that p-Chk-1 was not activated in dronedarone-treated cells (Fig. 5A). We next investigated the function of caspase-2 in dronedarone caused cytotoxicity using Z-VAVAD-FMK, a specific inhibitor of caspase-2. Application of Z-VAVAD-FMK significantly prevented cell death from dronedarone exposure (Fig. 5C,D). Interestingly, the addition of caspase-2 inhibitor also decreased the induction of γ-H2A.X. These results indicate that the activation of caspase-2 not only contributes to apoptosis and cell death but also aggravates dronedarone-induced DNA damage (Fig. 5E), indicating DNA damage and caspase-2 regulate each other and play complex roles in dronedarone induced toxicity.
The MAPK signaling pathway is another well-recognized regulator in DNA damage induced apoptosis (Roos and Kaina, 2006, 2013). We previously demonstrated that the MAPK signaling pathway is pivotal in drug-induced liver toxicity (Chen et al., 2014a,b; Guo et al., 2015), particularly, in regulating apoptosis and autophagy. In this study, the role of MAPK signaling pathway was investigated in dronedarone-caused DNA damage and apoptosis. It has been reported that among the three subgroups of MAP kinases, activation of JNK/p38 but not ERKs seems to be required for apoptosis and DNA damage (Roos and Kaina, 2006, 2013). This suggestion was supported by our findings that addition of JNK/p38 kinases inhibitors decreased cell death, caspase-3/7 activity, and the induction of γ-H2A.X (Fig. 6).
As mentioned above, caspase-2 is one of the main players in DNA damage-induced apoptosis and JNK has been recognized as a regulator of caspase-2. Since activation of both caspase-2 and JNK was observed in our study (Figs. 5A and 6A), we further explored the relationship between these two important molecules. Using a specific caspase-2 inhibitor, our results suggested that the activation of caspase-2 positively regulates the activation of JNK and p38 (Fig. 6F). It is known that the specific JNK inhibitor SP600125 was found to partially inhibit caspase-2 activation (Dirsch et al., 2004; Viana et al., 2010). Therefore, our findings suggest that the activation of caspase-2 and JNK/p38 pathways interact with each other in response to dronedarone exposure.
In this study, we also explored the possible upstream events that could trigger DNA damage in dronedarone-treated cells. First, we evaluated the ROS production and no accumulation of ROS was found in HepG2 cells treated with dronedarone, which is in consistent with the previous in vivo and in vitro reports (Serviddio et al., 2011; Felser et al., 2013). This result indicates that oxidative stress might not contribute to DNA damage caused by dronedarone. The impairment of topoisomerases can lead to DNA damage (Roos and Kaina, 2006), so next we studied the involvement of topoisomerases. Our results showed that the expression of topoisomerase IIα at both transcriptional and translational levels were significantly suppressed (Fig. 7), implying that inhibition of topoisomerase IIα may be a potential mechanism for DNA damage and a detailed mechanism worth further investigation.
In summary, we have demonstrated that dronedarone triggers DNA damage, intrinsic and extrinsic apoptosis, and necrosis in HepG2 cells. Caspase-2 and JNK/p38 activation are the linkages between DNA damage and apoptosis, and play vital roles in dronedarone-induced cytotoxicity. Inhibition of topoisomerase IIα by dronedarone could be the initial event in DNA damage and cytotoxicity. Our study not only provides new mechanistic explanations to dronedarone-induced liver toxicity but also highlights the role of DNA damage-induced apoptosis in chemical- and drug-induced liver toxicity. Due to the wide use of dronedarone in patients, awareness of the risk of dronedarone-induced liver toxicity is important for its safe application in the future.
ACKNOWLEDGMENTS
Z.R. and D.Y. were supported by appointments to the Postgraduate Research Program at the National Center for Toxicological Research administered by the Oak Ridge Institute for Science Education through an interagency agreement between the U.S. Department of Energy and the U.S. FDA. This work was supported by U.S. FDA’s intramural grant program. We appreciate Dr. Frederick Beland for his critical review of this manuscript.
Footnotes
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
DISCLAIMER
This article is not an official guidance or policy statement of the U.S. Food and Drug Administration (FDA). No official support or endorsement by the U.S. FDA is intended or should be inferred.
REFERENCES
- Aksenova VI, Kopeina GS, Zamaraev AV, Zhivotovsky BD, Lavrik IN. 2016. Mechanism of caspase-2 activation upon DNA damage. Dokl Biochem Biophys 467:132–135. [DOI] [PubMed] [Google Scholar]
- Bova MP, Tam D, McMahon G, Mattson MN. 2005. Troglitazone induces a rapid drop of mitochondrial membrane potential in liver HepG2 cells. Toxicol Lett 155:41–50. [DOI] [PubMed] [Google Scholar]
- Cai Z, Chehab NH, Pavletich NP. 2009. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol Cell 35: 818–829. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. 2001. Mammalian MAP kinase signalling cascades. Nature 410:37–40. [DOI] [PubMed] [Google Scholar]
- Chao DT, Korsmeyer SJ. 1998. BCL-2 family: Regulators of cell death. Annu Rev Immunol 16:395–419. [DOI] [PubMed] [Google Scholar]
- Chen S, Dobrovolsky VN, Liu F, Wu Y, Zhang Z, Mei N, Guo L. 2014a. The role of autophagy in usnic acid-induced toxicity in hepatic cells. Toxicol Sci 142:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wan L, Couch L, Lin H, Li Y, Dobrovolsky VN, Mei N, Guo L. 2013. Mechanism study of goldenseal-associated DNA damage. Toxicol Lett 221:64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Xuan J, Wan L, Lin H, Couch L, Mei N, Dobrovolsky VN, Guo L. 2014b. Sertraline, an antidepressant, induces apoptosis in hepatic cells through the mitogen-activated protein kinase pathway. Toxicol Sci 137:404–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhang Z, Qing T, Ren Z, Yu D, Couch L, Ning B, Mei N, Shi L, Tolleson WH, Guo L. 2017. Activation of the Nrf2 signaling pathway in usnic acid-induced toxicity in HepG2 cells. Arch Toxicol 91:1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GM, Dusi V. 2012. Drug safety evaluation of dronedarone in atrial fibrillation. Expert Opin Drug Saf 11:1023–1045. [DOI] [PubMed] [Google Scholar]
- Dirsch VM, Kirschke SO, Estermeier M, Steffan B, Vollmar AM. 2004. Apoptosis signaling triggered by the marine alkaloid ascididemin is routed via caspase-2 and JNK to mitochondria. Oncogene 23: 1586–1593. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Jamieson JD, Marroquin LD, Nadanaciva S, Xu JJ, Dunn MC, Smith AR, Will Y. 2008. In vitro assessment of mitochondrial dysfunction and cytotoxicity of nefazodone, trazodone, and buspirone. Toxicol Sci 103:335–345. [DOI] [PubMed] [Google Scholar]
- Evans MD, Cooke MS. 2004. Factors contributing to the outcome of oxidative damage to nucleic acids. Bioessays 26:533–542. [DOI] [PubMed] [Google Scholar]
- FDA. 2011. In brief: FDA warning on dronedarone (Multaq). Med Lett Drugs Ther 53:17. [PubMed] [Google Scholar]
- Felser A, Blum K, Lindinger PW, Bouitbir J, Krahenbuhl S. 2013. Mechanisms of hepatocellular toxicity associated with dronedarone–a comparison to amiodarone. Toxicol Sci 131:480–490. [DOI] [PubMed] [Google Scholar]
- Felser A, Stoller A, Morand R, Schnell D, Donzelli M, Terracciano L, Bouitbir J, Krahenbuhl S. 2014. Hepatic toxicity of dronedarone in mice: Role of mitochondrial beta-oxidation. Toxicology 323:1–9. [DOI] [PubMed] [Google Scholar]
- Green DR, Llambi F. 2015. Cell death signaling. Cold Spring Harb Perspect Biol 7: pii:a006080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer ML, Barber J, Eakins J, Kenna JG. 2010. Cell based approaches for evaluation of drug-induced liver injury. Toxicology 268:125–131. [DOI] [PubMed] [Google Scholar]
- Guo L, Dial S, Shi L, Branham W, Liu J, Fang JL, Green B, Deng H, Kaput J, Ning B. 2011. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab Dispos 39:528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Zhang L, Sun Y, Muskhelishvili L, Blann E, Dial S, Shi L, Schroth G, Dragan YP. 2006. Differences in hepatotoxicity and gene expression profiles by anti-diabetic PPAR gamma agonists on rat primary hepatocytes and human HepG2 cells. Mol Divers 10:349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Chen S, Zhang Z, Dobrovolsky VN, Dial SL, Guo L, Mei N. 2015. Reactive oxygen species and c-Jun N-terminal kinases contribute to TEMPO-induced apoptosis in L5178Y cells. Chem Biol Interact 235:27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy SM, Keam SJ. 2009. Dronedarone. Drugs 69:1647–1663. [DOI] [PubMed] [Google Scholar]
- Hua DH, Lou K, Battina SK, Zhao H, Perchellet EM, Wang Y, Perchellet JP. 2006. Syntheses, molecular targets and antitumor activities of novel triptycene bisquinones and 1,4-anthracenedione analogs. Anticancer Agents Med Chem 6:303–318. [DOI] [PubMed] [Google Scholar]
- Jahn S, Zollner G, Lackner C, Stauber RE. 2013. Severe toxic hepatitis associated with dronedarone. Curr Drug Saf 8:201–202. [DOI] [PubMed] [Google Scholar]
- Joghetaei N, Weirich G, Huber W, Buchler P, Estner H. 2011. Acute liver failure associated with dronedarone. Circ Arrhythm Electrophysiol 4:592–593. [DOI] [PubMed] [Google Scholar]
- Juan-Garcia A, Manyes L, Ruiz MJ, Font G. 2013. Involvement of enniatins-induced cytotoxicity in human HepG2 cells. Toxicol Lett 218:166–173. [DOI] [PubMed] [Google Scholar]
- Laifenfeld D, Qiu L, Swiss R, Park J, Macoritto M, Will Y, Younis HS, Lawton M. 2014. Utilization of causal reasoning of hepatic gene expression in rats to identify molecular pathways of idiosyncratic drug-induced liver injury. Toxicol Sci 137:234–248. [DOI] [PubMed] [Google Scholar]
- Lassus P, Opitz-Araya X, Lazebnik Y. 2002. Requirement for caspase-2 in stress-induced apoptosis before mitochondrial permeabilization. Science 297:1352–1354. [DOI] [PubMed] [Google Scholar]
- Lee WM. 2003. Drug-induced hepatotoxicity. N Engl J Med 349:474–485. [DOI] [PubMed] [Google Scholar]
- Mason JW. 1987. Amiodarone. N Engl J Med 316:455–466. [DOI] [PubMed] [Google Scholar]
- Mishra R, Karande AA. 2014. Endoplasmic reticulum stress-mediated activation of p38 MAPK, Caspase-2 and Caspase-8 leads to abrin-induced apoptosis. PLoS One 9:e92586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KC, Willmore WG, Tayabali AF. 2013. Cadmium telluride quantum dots cause oxidative stress leading to extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2 cells. Toxicology 306:114–123. [DOI] [PubMed] [Google Scholar]
- Norbury CJ, Zhivotovsky B. 2004. DNA damage-induced apoptosis. Oncogene 23:2797–2808. [DOI] [PubMed] [Google Scholar]
- Patel C, Yan GX, Kowey PR. 2009. Dronedarone. Circulation 120:636–644. [DOI] [PubMed] [Google Scholar]
- Patil M, Pabla N, Dong Z. 2013. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci 70:4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. 2001. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocr Rev 22:153–183. [DOI] [PubMed] [Google Scholar]
- Perchellet EM, Wang Y, Weber RL, Lou K, Hua DH, Perchellet JP. 2004. Antitumor triptycene bisquinones induce a caspase-independent release of mitochondrial cytochrome c and a caspase-2-mediated activation of initiator caspase-8 and −9 in HL-60 cells by a mechanism which does not involve Fas signaling. Anticancer Drugs 15:929–946. [DOI] [PubMed] [Google Scholar]
- Porceddu M, Buron N, Roussel C, Labbe G, Fromenty B, Borgne-Sanchez A. 2012. Prediction of liver injury induced by chemicals in human with a multiparametric assay on isolated mouse liver mitochondria. Toxicol Sci 129:332–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. 2002. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem 277:29803–29809. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- Roos WP, Kaina B. 2006. DNA damage-induced cell death by apoptosis. Trends Mol Med 12:440–450. [DOI] [PubMed] [Google Scholar]
- Roos WP, Kaina B. 2013. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett 332:237–248. [DOI] [PubMed] [Google Scholar]
- Serviddio G, Bellanti F, Giudetti AM, Gnoni GV, Capitanio N, Tamborra R, Romano AD, Quinto M, Blonda M, Vendemiale G, Altomare E. 2011. Mitochondrial oxidative stress and respiratory chain dysfunction account for liver toxicity during amiodarone but not dronedarone administration. Free Radic Biol Med 51:2234–2242. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. 2008. Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol Cell Biol 9:231–241. [DOI] [PubMed] [Google Scholar]
- Tomicic MT, Kaina B. 2013. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochim Biophys Acta 1835:11–27. [DOI] [PubMed] [Google Scholar]
- Vakifahmetoglu-Norberg H, Zhivotovsky B. 2010. The unpredictable caspase-2: What can it do? Trends Cell Biol 20:150–159. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P. 2015. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol 2:e975093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viana RJ, Ramalho RM, Nunes AF, Steer CJ, Rodrigues CM. 2010. Modulation of amyloid-beta peptide-induced toxicity through inhibition of JNK nuclear localization and caspase-2 activation. J Alzheimers Dis 22:557–568. [DOI] [PubMed] [Google Scholar]
- Wu CC, Huang KF, Yang TY, Li YL, Wen CL, Hsu SL, Chen TH. 2015. The topoisomerase 1 inhibitor austrobailignan-1 isolated from koelreuteria henryi induces a G2/M-phase arrest and cell death independently of p53 in non-small cell lung cancer cells. PLoS One 10:e0132052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie AH, Kerr JF, Currie AR. 1980. Cell death: The significance of apoptosis. Int Rev Cytol 68:251–306. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. 1995. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331. [DOI] [PubMed] [Google Scholar]
- Yuan J, Kroemer G. 2010. Alternative cell death mechanisms in development and beyond. Genes Dev 24:2592–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky B, Orrenius S. 2005. Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun 331:859–867. [DOI] [PubMed] [Google Scholar]