

Abstract
Cocaine is a psychostimulant with a high potential for abuse and addiction. Risk for cocaine use disorder is driven, in part, by genetic factors. Animal models of addiction-relevant behaviors have proven useful for studying both genetic and non-genetic contributions to drug response. In a previous study, we examined initial locomotor sensitivity to cocaine in genetically diverse inbred mouse strains. That work highlighted the relevance of pharmacokinetics in initial locomotor response to cocaine but was limited by a single dose and two sampling points. The objective of the present study was to characterize the pharmacokinetics and pharmacodynamics of cocaine and its metabolites (norcocaine and benzoylecgonine) in 6 inbred mouse strains (I/LnJ, C57BL/6J, FVB/NJ, BTBR T+ tf/J, LG/J, LP/J) that exhibit extreme locomotor responses to cocaine. Mice were administered cocaine at one of 4 doses and concentrations of cocaine, norcocaine, and benzoylecgonine were analyzed in both plasma and brain tissue at 5 different time points. Initial locomotor sensitivity to cocaine was used as a pharmacodynamic endpoint. We developed an empirical population PK model that simultaneously characterizes cocaine, norcocaine, and benzoylecgonine in plasma and brain tissues. We observed interstrain variability occurring in the brain compartment that may contribute to pharmacodynamic differences amongst select strains. Our current work paves the way for future studies to explore strain-specific pharmacokinetic differences and identify factors other than pharmacokinetics that are responsible for the diverse behavioral response to cocaine across these inbred mouse strains.
Keywords: pharmacokinetics, pharmacodynamics, pharmacogenetics, cocaine, inbred, mice, locomotor, norcocaine, benzoylecgonine, modeling
INTRODUCTION
Cocaine (COC) is the third most commonly used illicit drug in the United States (U.S.) after marijuana and prescription pain relievers.1 According to the most recent National Survey on Drug Use and Health, almost 1 million individuals in the U.S. meet the diagnostic criteria for COC use disorder (CUD).1 Recent data also suggests that COC use is increasing, and the number of overdose deaths involving COC has risen approximately 18% from 2011–2016.2,3 There are currently no approved treatments for CUD, due in part to large gaps in our knowledge about the underlying etiology of this devastating disorder. Studies aimed at understanding the mechanistic etiologies underlying CUD are needed in order to decrease negative clinical outcomes including accidental death, increased healthcare spending, lost productivity and increased crime rates.4–7
There is a substantial body of literature supporting the role of genetics in substance use disorders (SUD) including CUD.8,9 Human genome-wide association studies, or GWAS, have started to identify regions of the genome associated with SUDs.10 However, GWAS can be hindered by unknown environmental factors including previous and ongoing drug exposures that prevent assessment of the early stages of the addiction cycle. Some of these challenges can be overcome by using preclinical animal models, which allow for a more direct measurement of the substance’s impact on an individual. One commonly used animal model of drug sensitivity assesses initial locomotor activation following acute psychostimulant administration.11 Initial subjective responses to psychostimulants predict the progression from initial exposure to subsequent use of the psychostimulant in humans.6,12,13 Therefore, research efforts examining initial drug sensitivity can help elucidate potential mechanisms of addiction.
Individual differences in COC pharmacokinetics (PK) may be driven by inherited genetic factors, and therefore could be used for predicting the risk of developing CUD.14 However, the relationship between PK and behavioral effects of cocaine and its metabolites, as well as the impact of genetic background on this relationship, are currently underexplored in both humans and animal models.15,16 Studying panels of genetically diverse inbred mouse strains is a powerful method for probing genetic and environmental variance that contribute to complex behaviors. We previously examined initial locomotor sensitivity in response to acute cocaine administration in 45 inbred mouse strains, and observed significant phenotypic variation across strains that could be partially (~50%) attributed to genetics.11,17 Brain concentrations of COC and norcocaine (NOR) were heritable, varied across strains, and were significantly correlated with locomotor activation11. However, the level of correlation suggested that locomotor response to COC was not fully explained by pharmacokinetic (PK) parameters. This finding has been observed by others, albeit in far fewer genetic backgrounds.18–22 While our previous study utilized a diverse panel of genetic backgrounds, it was still limited to a single dose and two sampling points post exposure.11 A more comprehensive characterization of COC exposure and metabolism at multiple doses and time points in multiple genetic backgrounds is absent in the literature. More complete PK profiling is warranted in order to make definitive conclusions about the role of PK on individual differences in locomotor response to COC.
In the current study, we characterized the PK of COC, its active metabolite, NOR, and major inactive metabolite, benzoylecgonine (BZE), at four different doses and five time points across a set of six inbred mouse strains that previously showed either high- or low-locomotor response to acute COC.11,17 Our experimental design allows us to more fully investigate the relationship between genetic background, PK and behavior.
MATERIALS AND METHODS
Animals.
Male mice from six inbred mouse strains (I/LnJ, C57BL/6J, FVB/NJ, BTBR T+ Itpr3 tf/J, LG/J, LP/J) were selected based on previously reported phenotypic differences in COC-induced locomotor activation (n=395, Supplementary Table 1).11,17 C57BL/6J and I/LnJ strains were classified as high responders, while BTBR T+ Itpr3 tf/J, FVB/NJ, LG/J, and LP/J as low responders to the locomotor activating effects of an acute 20 mg/kg cocaine injection.11 Only male mice were tested for comparison to previously published data.23,24 All mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and allowed to acclimate in ventilated caging (Tecniplast, Buguggiate, Italy) located in an animal holding room in an AAALAC-accredited vivarium at the University of North Carolina for at least 10 days prior to testing. Mice were group housed (2–4 per cage) on a 12-h light/dark cycle (lights on 0700 h) and received food (PicoLab Rodent Diet 20, Purina, St. Louis, MO, USA) and water ad libitum. The mean age at the onset of testing was 67 days (± 7.3 days). All behavioral and PK experiments were conducted between 0800–1200 to minimize circadian variation. Adequate measures were taken to minimize pain or discomfort and all procedures were approved by the Institutional Animal Care and Use Committee at the University of North Carolina.
Pharmacokinetic Analysis.
Drugs.
Cocaine hydrochloride (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 0.9% saline each day prior to testing. Either cocaine or vehicle control (0.9% saline) were administered by intraperitoneal (IP) injection at a volume of 0.01 mL/g.
Pharmacokinetic Sampling
Drug naïve mice were administered a single IP injection of 5, 10, 30 or 40 mg/kg COC hydrochloride and were euthanized at 2, 5, 10, 30 or 60-min post drug administration for collection of brain and blood samples. Each strain/dose/time combination was assessed in biological triplicates (n=3 mice). Mice were anesthetized with isoflurane, and blood was collected by cardiac puncture in an EDTA-coated syringe. Whole blood was collected in EDTA-treated tubes, and the plasma separated from cells by centrifugation at 1,500 x g for 10 minutes. The right brain hemisphere was also collected, rinsed, weighed and frozen in dry ice. Brain tissue was homogenized at a ratio of 1-part tissue (mg) to 5 parts 0.5% acetic acid (μL) using a Precellys 24 bead mill homogenizer (VWR, Radnor, PA). All samples were stored at −80 °C until mass spectrometry analysis was conducted.
Sample Processing.
Cocaine and metabolites were extracted from plasma or brain homogenate by protein precipitation in 200 μL acetonitrile/0.1% formic acid containing 50 ng/mL of each deuterated internal standard. Samples were vortexed for 1 minute and centrifuged at 2,800 x g for 15 minutes. 100 μL supernatant was transferred to a sample vial containing 200 μL of 0.1% formic acid. The calibration curve (1–5,000 ng/mL) and controls (4, 40, 400, and 4,000 ng/mL) were prepared by spiking serial dilutions of the mixed analytes into untreated mouse plasma or brain homogenate.
LC-MS/MS Measurement of Drug and Metabolite Concentrations
Liquid chromatography of COC, NOR, BZE and their deuterated internal standards was accomplished using a Shimadzu LC-20AD liquid chromatograph with an Atlantis T3 (2.5 μm 2.1 × 50 mm) analytical column (Waters, Milford, MA). The mobile phase consisted of 0.1% formic acid in water (mobile A) and 0.1% formic acid in acetonitrile (mobile phase B). Analytes were measured using a Thermo TSQ Ultra triple quadrupole mass spectrometer (ThermoFisher Scientific, Waltham, MA) equipped with a heated electrospray ionization source in the positive ion mode. Multiple reaction monitoring (MRM) mode utilized the following transitions: COC (304.2->182.1), BZE (290.1->168.1), NOR (290.1->168.1), COC-d3 307.1->185.1, BZE-d3 (293.1->171.1) and NOR-d3 (293.1->171.1).
Pharmacokinetic Analyses
Plasma and brain concentrations were expressed as ng/mL and ng/g, respectively. Three mice were tested per dose and strain combination. Due to the need to euthanize individual mice at each dose and sampling time in order to collect brain samples for COC, NOR, and BZE measurements, the PK analysis was conducted as follows. Median values of plasma and brain concentrations of COC, NOR and BZE from each of the three samples per strain, dose and sampling time were calculated. For modeling and statistical analysis purposes, the aggregate of the values across time for each combination of strain and dose was considered as if coming from one mouse, which is denoted hereafter as a full profile mouse or FPM. A similar analysis method has been used in previously published research.25,26 A representative sampling map is depicted in Figure 1.
Figure 1. PK sampling schema.
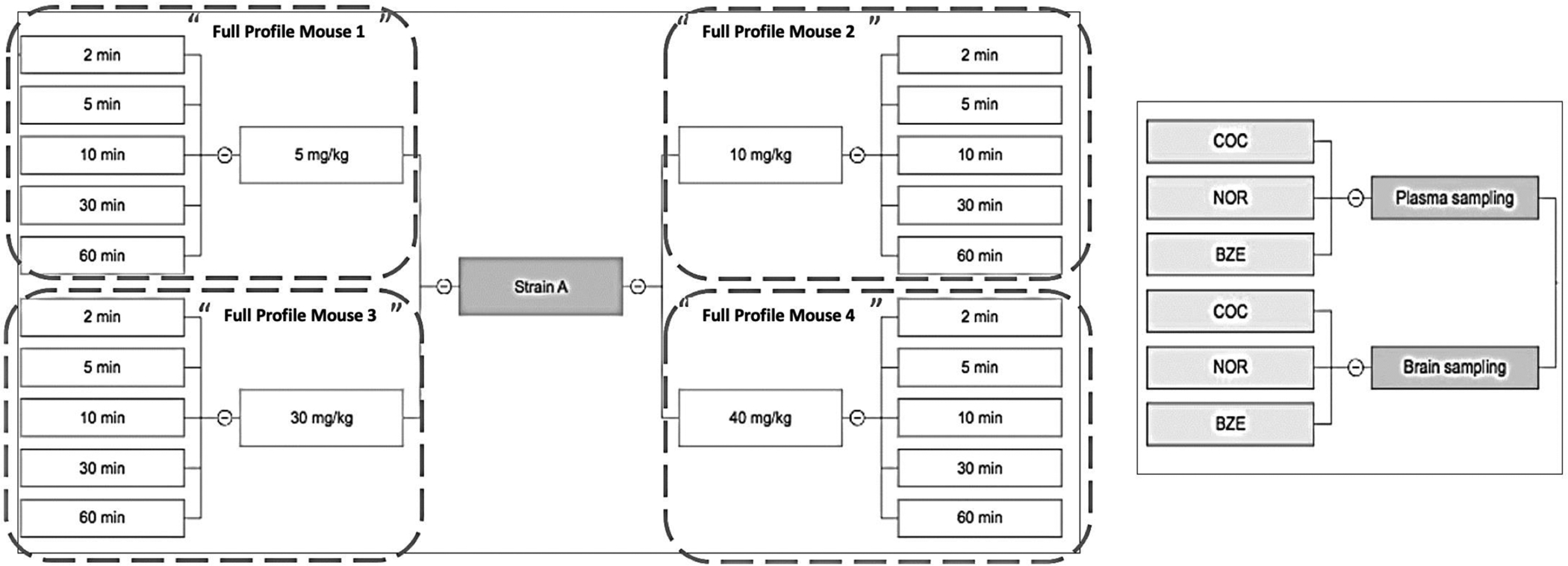
Strain A is a representative strain. A total of 4 COC doses were administered intraperitoneally (5, 10, 30, and 40 mg/kg). Mice from all dosing groups were euthanized at 5 different time points (2, 5, 10, 30, and 60 min) for quantification of COC, NOR, and BZE concentrations in plasma and brain.
All PK analyses were performed using Phoenix 8.1 (Certara, Princeton, NJ). Median concentrations of biological triplicates for COC, NOR, and BZE in plasma and brain were used for XY plot analysis. Non-compartmental analyses (NCA) for each combination of dose and analyte were conducted on data aggregated across the 6 strains. The peak plasma and brain concentrations (Cmax) of COC, NOR, and BZE, the time at which Cmax was reached (Tmax), and the observed total exposure (AUC0-last) were calculated. For the NCA analyses, we considered a CV% value greater than 40% as an indication of high interstrain PK differences. Although only one of our CV values was greater than 30%, we conducted an analysis of variance (ANOVA) for each analyte in both brain and plasma to further examine interstrain variability. Details of the ANOVA are described below in the Statistical Methods section.
Population PK modeling was performed first on the parent drug COC, and subsequently adding compartments for its metabolites, NOR and BZE.27 The absorption rate constant (Ka) was fixed at 0.71, based on previously reported COC bioavailability after intraperitoneal administration.28 One-, two-, and three-compartment PK models with linear or saturable metabolism from the plasma compartment were fit to the data. Sequentially, both linear and nonlinear formation of BZE and NOR were tested in the model in a similar manner. The Fisher’s scoring algorithm was utilized to determine the standard error of the fixed effects for all parameters.29 All dose levels were simultaneously fitted to obtain a single set of PK parameters. The final population PK model selection was made on the basis of visual inspection of the diagnostic plots, Akaike Information Criteria (AIC), and the log-likelihood (−2LL).
Pharmacodynamic Analyses
Our pharmacodynamic (PD) endpoint was defined as locomotor activation measured in centimeters travelled post drug exposure. PD data and methods for assessing locomotor response to COC are described previously.11 Briefly, mice were tested for locomotor dose response to COC using a 3-day protocol. On Days 1–2, mice received an IP injection of saline and were placed in an open-field apparatus (Med Associates ENV-515–16, Fairfax, VT) for 30 min. On day 3, mice received an IP injection of COC at one of the four doses (5, 10, 30, or 40 mg/kg) prior to being placed in the open field for 30 min. Distance travelled on each day was measured in 5 min bins and summed to represent the total distance in the 30 min interval. Locomotor response to COC was stratified by strain and calculated by subtracting the mean distance travelled after the second saline exposure (Day 2) from the mean distance travelled after COC was administered (Day 3). These measures were used for dose response analyses and NCA. The locomotor stimulation ratio was calculated by dividing distance travelled by unit time (in cm/min) for each strain at each dose. The highest ratio (Rmax), as well as the time at which Rmax was achieved (Tmax-PD), were also calculated.
Statistical Methods
Due to the study design and the need to utilize the concept of an FPM, all statistical summaries and analyses should be considered exploratory in nature. For the ANOVA, the effects in the model were strain and dose. Because there was a single FPM for each combination of strain and dose, it was not possible to include a strain by dose interaction term in the model that examined PK parameters as primary analysis. Statistical significance was reported if the P-value from the ANOVA was less than or equal to 0.05. In situations where the overall P-value for the strain effect was statistically significant, the ANOVA was followed by the testing of pairwise differences of the strain means and associated P-values. While our primary analysis is focused on the utilization of PK parameters, visual inspection of the concentration data informed differences amongst the first 3 time points (0.03, 0.08, 0.17 hours). To address these potential differences in the timing of onset of drug exposure, supplementary analyses were conducted by testing strain, time, and strain by time interaction effects for each analyte in brain and plasma. Each dose group was analyzed separately. No adjustment was made for the multiplicity of testing as we consider these results to be exploratory.
RESULTS
Non-compartmental Analysis
In plasma, both Cmax and AUC0-last increased in a dose-dependent manner for COC, NOR and BZE (Table 1), suggesting dose-dependent exposure. Mean Tmax for COC, NOR, and BZE were 2.0 min [range: 1.8 min-2.4 min], 6 min [5.4 min-7.8 min], and 34.5 min [26 min-40 min], respectively. Tmax for COC and NOR remained the same across the 4 dosing groups, suggesting that the rate of gastrointestinal absorption of COC and NOR was independent of dose. For both NOR and BZE, dose-normalized AUC0-last values demonstrated non-linear PK profiles in plasma, which informed the development of a model describing their formation. The Cmax for NOR was < 5% that of COC, whereas Cmax for BZE was < 30% that of COC. Total NOR exposure (AUC0-last) was < 12% that of COC, whereas the total BZE exposure was 33% greater than that of COC in the plasma. These data are consistent with previous studies showing that BZE has a longer plasma elimination half-life than both COC and NOR.30,31 Our data show that not only did BZE plasma concentrations decline at a later time than both COC and NOR, but BZE concentrations were still rising after 60 min (Figure 2).
Table 1. Non-compartmental analysis of plasma and brain concentrations.
Major PK parameters for COC, NOR, and BZE in plasma and brain.
| Non-compartmental Analysis | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Source | Dose (mg/kg) | AUC0-last (hr*mg/L) |
Dose Normalized AUC0-last (hr*mg/L) |
Cmax (mg/L) |
Dose Normalized Cmax (mg/L) |
Tmax (h) |
||||||
| Mean | CV% | Mean | CV% | Mean | CV% | Mean | CV% | Mean | CV% | |||
| PLASMA | COC | 5 | 0.165 | 7.78 | 0.0330 | 7.78 | 0.620 | 17.52 | 0.124 | 17.52 | 0.0300 | 0.00 |
| 10 | 0.440 | 36.77 | 0.0440 | 36.77 | 1.51 | 38.07 | 0.151 | 38.07 | 0.383 | 53.25 | ||
| 30 | 1.97 | 37.32 | 0.0658 | 37.32 | 8.76 | 66.37 | 0.292 | 66.37 | 0.0300 | 0 | ||
| 40 | 2.24 | 24.38 | 0.561 | 24.38 | 9.81 | 61.52 | 0.245 | 61.52 | 0.0300 | 0 | ||
| NOR | 5 | 0.0176 | 17.03 | 0.00351 | 17.03 | 0.0387 | 51.48 | 0.00774 | 51.48 | 0.0933 | 67.99 | |
| 10 | 0.0380 | 28.74 | 0.00380 | 28.74 | 0.0778 | 23.96 | 0.00779 | 23.96 | 0.0933 | 67.99 | ||
| 30 | 0.152 | 27.66 | 0.05008 | 27.66 | 0.314 | 36.74 | 0.0105 | 36.74 | 0.133 | 135.97 | ||
| 40 | 0.214 | 25.06 | 0.00535 | 25.06 | 0.510 | 40.51 | 0.0127 | 40.51 | 0.0867 | 52.45 | ||
| BZE | 5 | 0.393 | 28.46 | 0.0786 | 28.46 | 0.514 | 27.26 | 0.103 | 27.26 | 0.612 | 53.43 | |
| 10 | 0.837 | 26.83 | 0.0837 | 26.83 | 1.04 | 30.08 | 0.104 | 30.08 | 0.445 | 30.27 | ||
| 30 | 2.62 | 30.37 | 0.0872 | 30.37 | 3.24 | 31.76 | 0.108 | 31.76 | 0.667 | 38.73 | ||
| 40 | 2.89 | 17.14 | 0.0723 | 17.14 | 3.54 | 18.41 | 0.0884 | 18.41 | 0.583 | 34.99 | ||
| BRAIN | COC | 5 | 0.686 | 21.51 | 0.137 | 21.51 | 1.57 | 40.36 | 0.314 | 40.36 | 0.125 | 39.44 |
| 10 | 1.32 | 17.92 | 0.132 | 17.92 | 3.10 | 22.22 | 0.310 | 22.22 | 0.110 | 42.25 | ||
| 30 | 5.22 | 22.4 | 0.174 | 22.4 | 9.43 | 23.44 | 0.314 | 23.44 | 0.155 | 23.7 | ||
| 40 | 7.40 | 15.15 | 0.185 | 15.15 | 15.4 | 39.19 | 0.385 | 39.19 | 0.110 | 42.25 | ||
| NOR | 5 | 0.0366 | 42.02 | 0.00733 | 42.02 | 0.0977 | 42.96 | 0.0195 | 42.96 | 0.125 | 39.44 | |
| 10 | 0.0969 | 35.39 | 0.00969 | 35.39 | 0.205 | 36.51 | 0.0205 | 36.51 | 0.110 | 42.25 | ||
| 30 | 0.470 | 32.86 | 0.0157 | 32.86 | 0.858 | 48.30 | 0.0286 | 48.30 | 0.170 | 0.00 | ||
| 40 | 0.800 | 30.59 | 0.0200 | 30.59 | 1.48 | 40.30 | 0.0369 | 40.30 | 0.155 | 23.70 | ||
| BZE | 5 | 0.0252 | 6.14 | 0.00504 | 6.14 | 0.038 | 11.50 | 0.00760 | 11.50 | 0.75 | 38.49 | |
| 10 | 0.0506 | 21.08 | 0.00506 | 21.08 | 0.0756 | 20.46 | 0.00756 | 20.46 | 1.00 | 0.00 | ||
| 30 | 0.158 | 24.15 | 0.00528 | 24.15 | 0.253 | 22.76 | 0.00842 | 22.76 | 1.00 | 0.00 | ||
| 40 | 0.209 | 26.20 | 0.00522 | 26.20 | 0.327 | 32.62 | 0.00818 | 32.62 | 1.00 | 0.00 | ||
Figure 2. XY plots for COC, NOR and BZE in plasma.
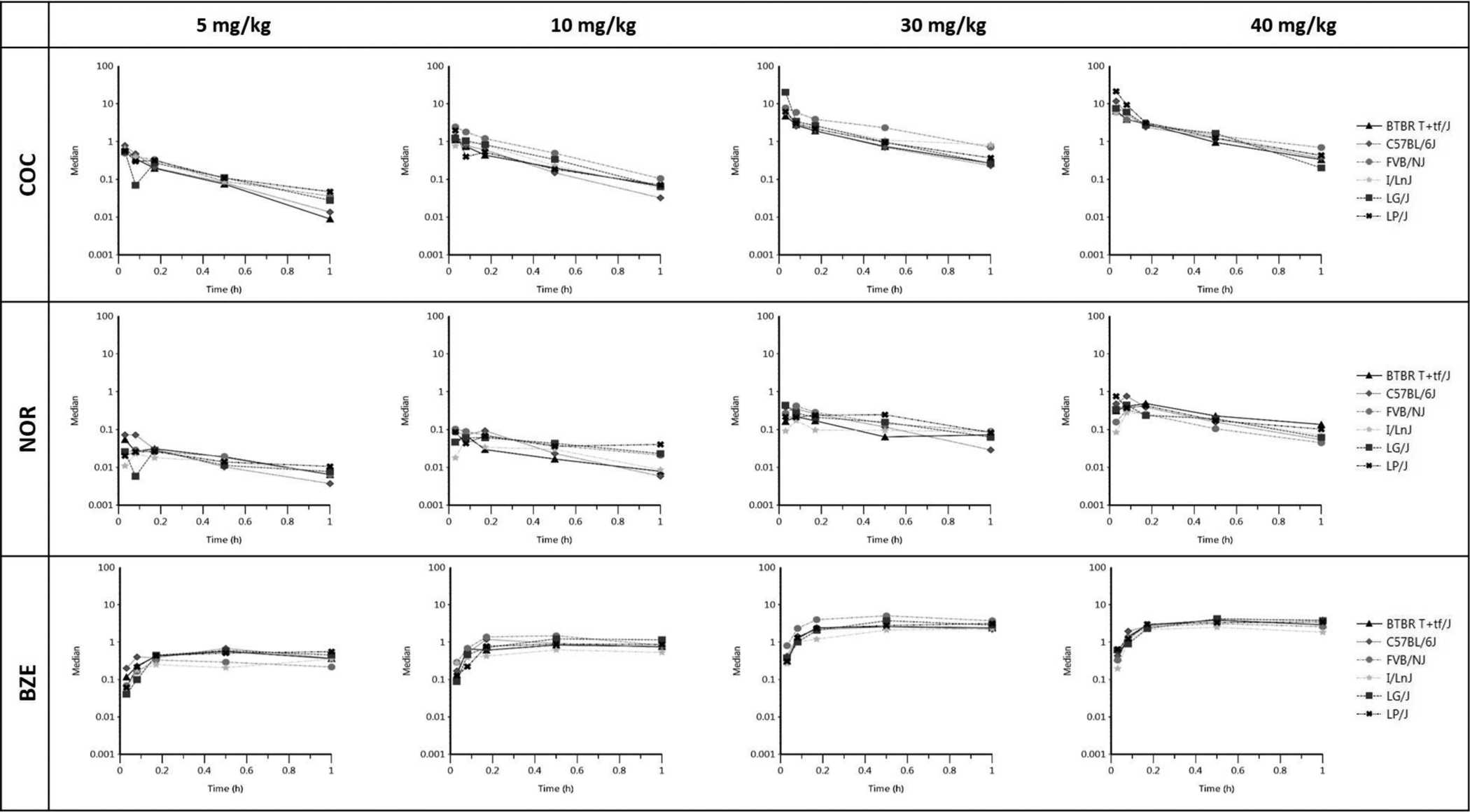
Each of the 12 graphs represents concentration versus time for the various analyte (COC, NOR, and BZE) and dose combinations. Each line on the graph corresponds to an individual FPM and each data point on the line represents the concentration of the corresponding analyte in plasma (mg/L) at the specific time (0.03, 0.08, 0.17, 0.5 and 1-hr) post COC injection at dose indicated. Median concentrations from the biological triplicate were used for each strain, dose, and time combination.
In the brain, both AUC0-last and Cmax increased in a dose-dependent manner for COC, NOR and BZE (Table 1), suggesting dose-dependent exposure. While Tmax for COC and NOR remained the same across the 4 dosing groups, Tmax for BZE was not fully captured within the experimental time at 60 min, as evidenced by the increasing concentrations of BZE in the brain at the last sampling point (Figure 3). For both NOR and BZE, dose-normalized AUC0-last values demonstrated non-linear PK profiles in the brain. These data informed the development of a model describing their formation. Additionally, total NOR exposure (AUC0-last) was < 12% that of COC and total BZE exposure was < 6% that of COC in the brain.
Figure 3. XY plots for COC, NOR and BZE in brain.
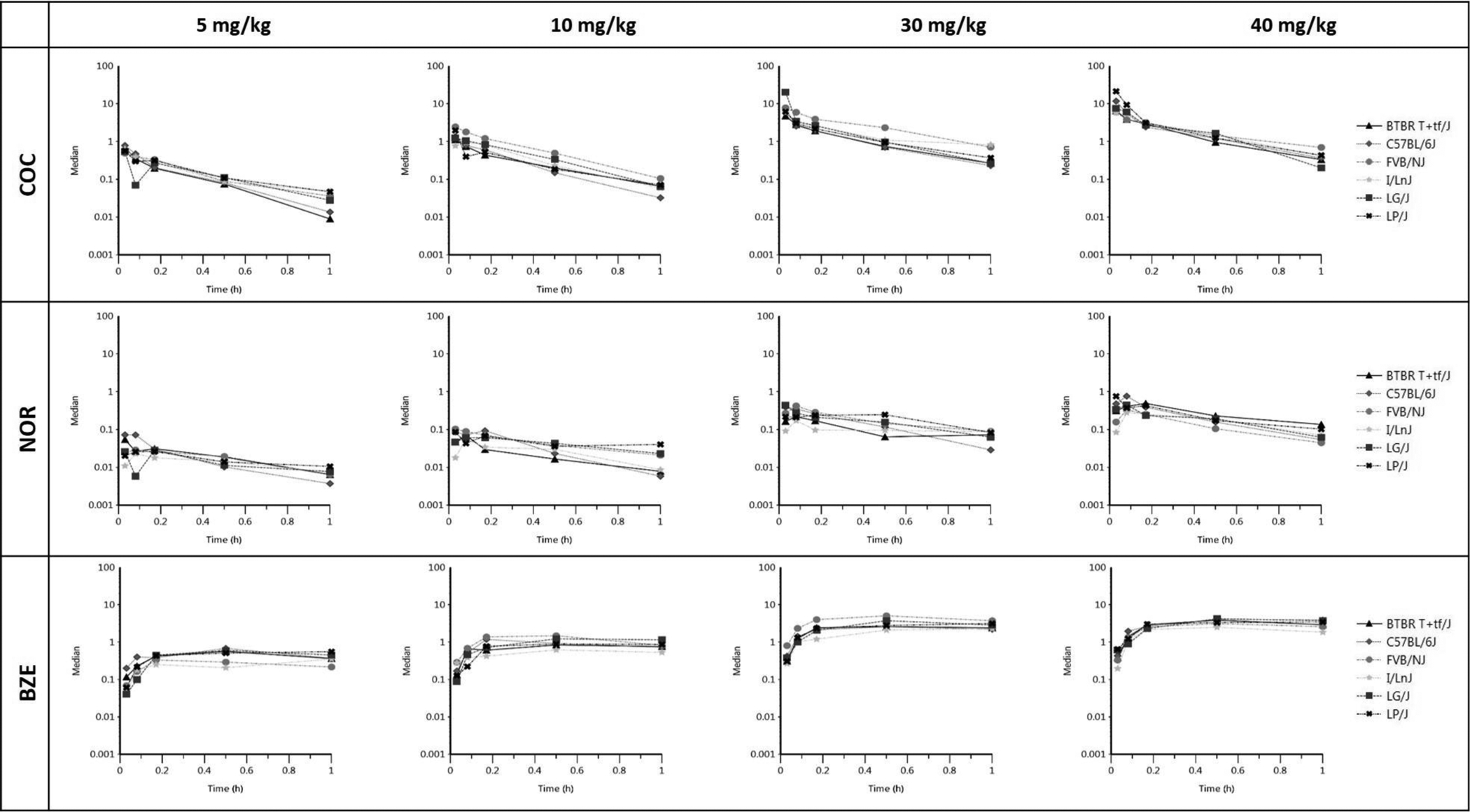
Each of the 12 graphs represents concentration versus time for the various analyte (COC, NOR, and BZE) and dose combinations. Each line on the graph corresponds to an individual FPM and each data point on the line represents the concentration of the corresponding analyte in the brain (ng/g) at the specific time (0.03, 0.08, 0.17, 0.5 and 1-hr) post COC injection at dose indicated. Median concentrations from the biological triplicate were used for each strain, dose and time combination.
Development of the population PK model
The 24 FPM mice were used as subject input for the population PK model. An open, three-compartment model was used to describe COC distribution in plasma and brain with a log-additive error term.21 The model used a first-order conditional estimation extended least squares (FOCE-ELS) approach and was constructed graphically to characterize the relationships between parent COC and metabolite formation in both the plasma and brain (Figure 4). Because our model is the first that describes all 3 analytes in both plasma and brain, the initial parameter estimates for model fitting were estimated de novo and from existing literature where possible27. The final model revealed bidirectional movement of both metabolites between the plasma and brain compartments. Notably, in the plasma, parent COC underwent Michaelis-Menten kinetics to form NOR and BZE, which were then distributed readily into the brain tissue.32,33 This finding was also supported by the observation of nonlinear formation of both NOR and BZE in NCA. Our model suggests that brain concentrations of NOR and BZE are derived from the metabolites crossing the blood brain barrier from plasma, as well as those formed from parent COC in the brain. Predicted data overlay the observed data well, indicating that the model was a reasonable fit (Figure 5). Final estimates and CV% are shown in Table 2.
Figure 4. Model used to describe COC and metabolites, NOR and BZE, in plasma (PL) and brain (BR).
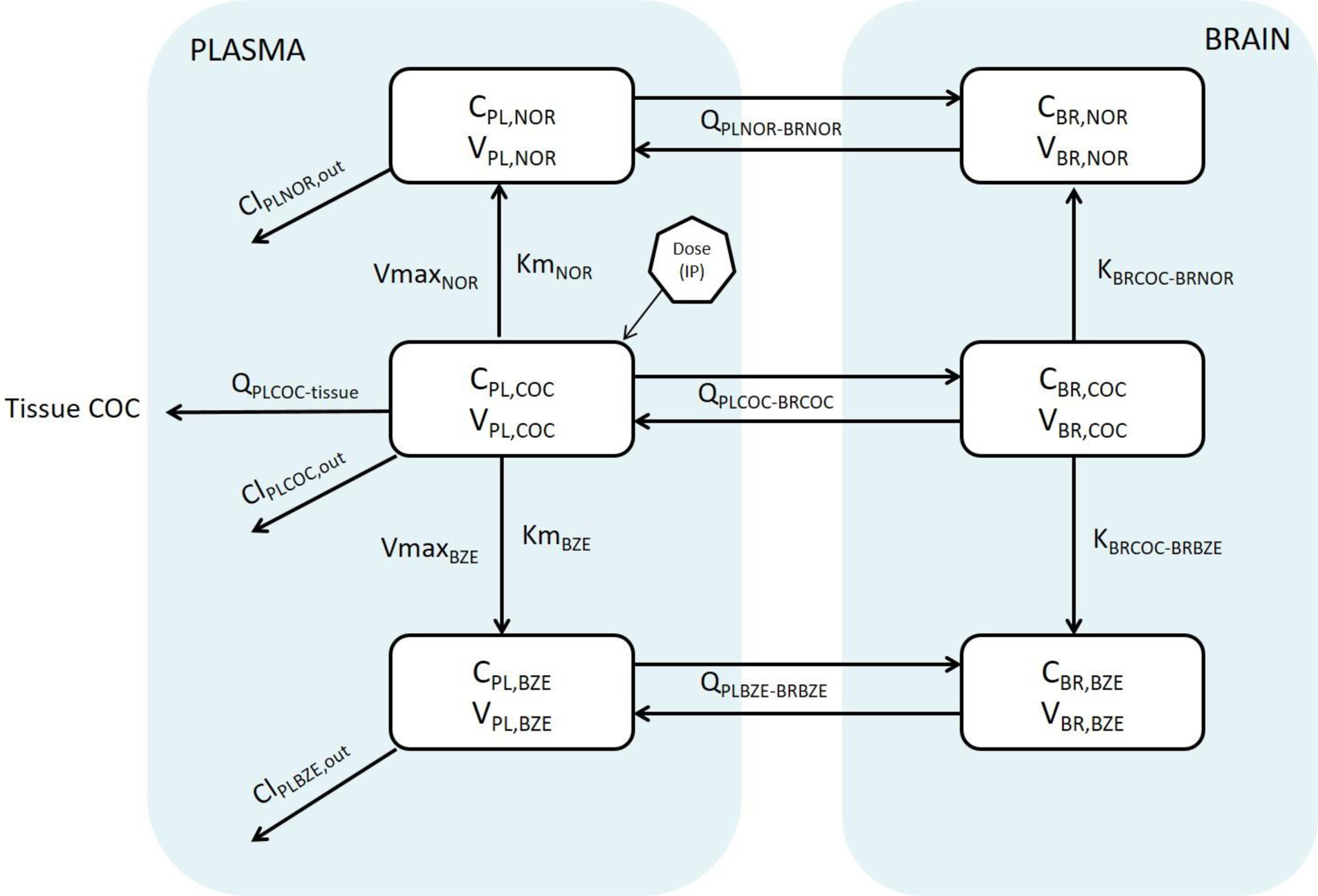
COC, NOR, and BZE were each presented in a population PK model, with COC undergoing Michaelis Menten kinetics in plasma forming NOR and BZE. All COC, NOR, and BZE undergo bidirectional transport process entering and exiting the brain. NOR and BZE are also formed in the brain from COC entered through the plasma. Abbreviations and definitions: CPL, NOR, NOR concentration in plasma; VPL, NOR, NOR volume of distribution in plasma; QPLNOR-BRNOR, NOR flow rate between plasma and brain; ClPLNOR, out, NOR clearance from plasma; VmaxNOR, maximum rate of NOR formation from COC in plasma; KmNOR, Michaelis-Menten constant for NOR formation in plasma; CBR, NOR, concentration of NOR in the brain; VBR, NOR, volume of distribution of NOR in the brain; KBRCOC-BRNOR, formation rate constant of NOR from COC in the brain; QPLCOC-tissue, flow rate of COC between plasma and tissue; ClPLCOC, out, COC clearance from plasma; CPL, COC, COC concentration in plasma; VPL, COC, COC volume of distribution in plasma; QPLCOC-BRCOC, flow rate of COC between plasma and brain; CBR, COC, COC concentration in the brain; VBR, COC, COC volume of distribution in the brain; KBRCOC-BRBZE, formation rate constant of BZE form cocaine in the brain; CBR, BZE, concentration of BZE in the brain; VBR, BZE, volume of distribution of BZE in the brain; QPLBZE-BRBZE, flow rate of BZE between plasma and brain; CPL,BZE, concentration of BZE in plasma; VPL, BZE, volume of distribution of BZE in plasma; ClPLBZE, out, plasma clearance of BZE; VmaxBZE, maximum rate of BZE formation from cocaine in plasma; KmBZE, Michaelis-Menten constant for BZE formation in plasma.
Figure 5. PK model diagnostic plots.
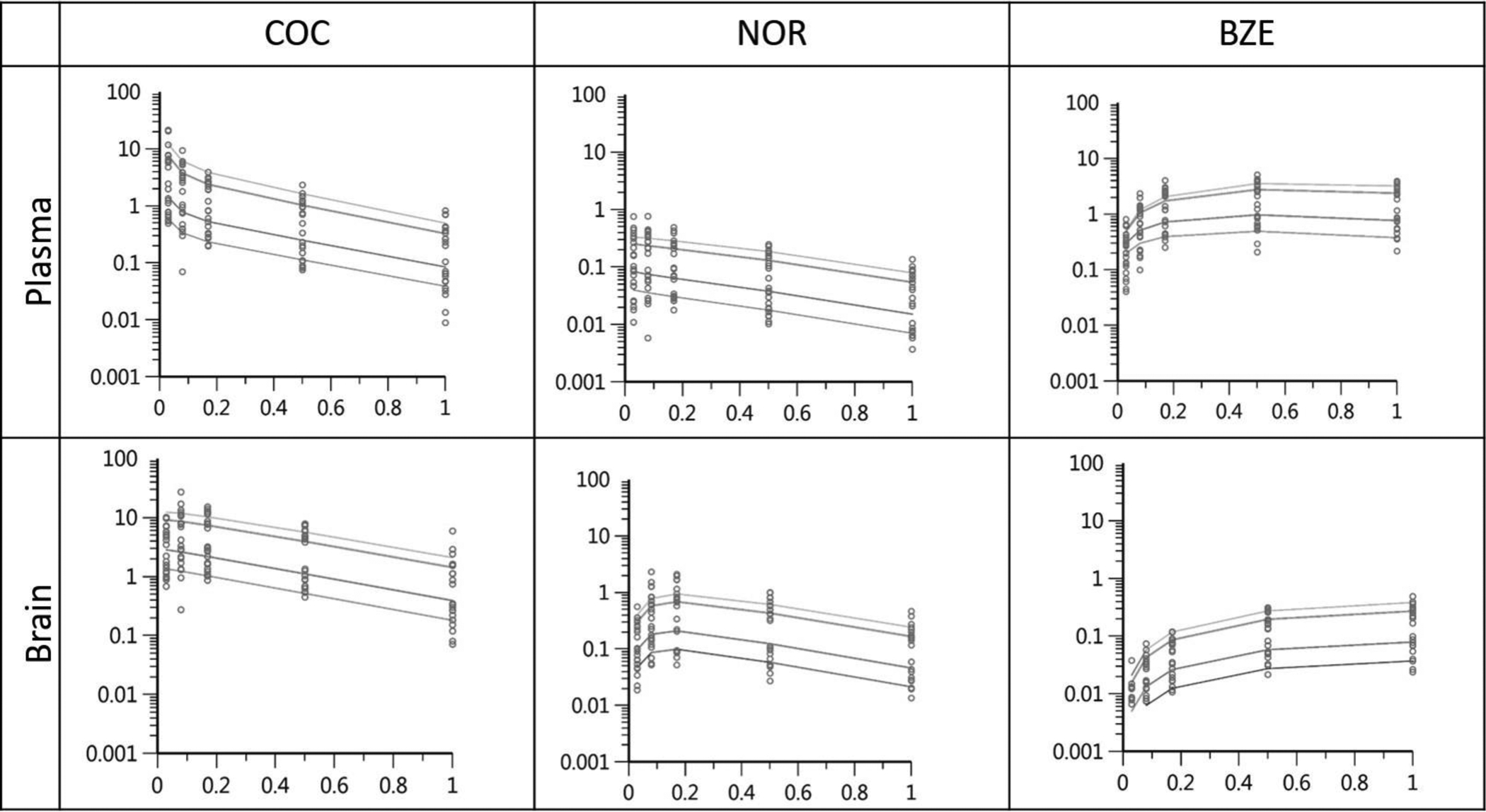
Predicted concentrations of COC, NOR and BZE in plasma and brain are depicted with a line and observed data were overlaid on top of as dots. X-axis is independent variable plotted against the Y-axis showing individually predicted dependent variables (concentrations).
Table 2.
PK model final parameter estimates
| Parameter | Definition | Without I/LnJ strain | |
|---|---|---|---|
| Estimate | CV% | ||
| CLPLCOC (mL/h) | Clearance of cocaine in plasma | 1.10 × 10−2 | 20.02 |
| CLPLBZE (mL/h) | Clearance of benzoylecgonine in plasma | 4.32 | 9.39 |
| CLPLNOR (mL/h) | Clearance of norcocaine in plasma | 2.24 | 18.67 |
| KBRCOC-BRBZE (mg/(g*h)) | Formation rate of benzoylecgonine from cocaine in the brain | 2.07×10−2 | 24.97 |
| KBRCOC-BRNOR (mg/(g*h)) | Formation rate of norcocaine from cocaine in the brain | 1.52×10−2 | 24.64 |
| KmBZE (mg/mL) | Concentration of benzoylecgonine when the rate of non-linear formation was at half its maximum value in plasma | 3.50 | 12.94 |
| KmNOR (mg/mL) | Concentration of norcocaine when the rate of non-linear formation was at half its maximum value in plasma | 1.32×104 | 20.71 |
| QPLCOC-BRCOC (mL/h) | Flow rate of cocaine from plasma to brain | 5.82 | 6.22 |
| QPLCOC-tissue (mL/h) | Flow rate of cocaine from plasma to tissue | 1.22 | 13.31 |
| QPLBZE-BRBZE (mL/h) | Flow rate of benzoylecgonine from plasma to brain | 1.40×10−2 | 68.07 |
| QPLNOR-BRNOR (mL/h) | Flow rate of norcocaine from plasma to brain | 4.02×10−1 | 25.98 |
| VPLCOC (mL) | Central volume of distribution for cocaine | 1.46×10−2 | 22.3 |
| VBRCOC (g) | Degree of penetration of cocaine in the brain | 1.93 | 4.62 |
| VBRBZE (g) | Degree of penetration of benzoylecgonine in the brain | 7.35×10−1 | 25.89 |
| VBRNOR (g) | Degree of penetration of norcocaine in the brain | 2.64×10−2 | 27.35 |
| VCOC-tissue (mL) | Volume of distribution of cocaine in the tissue | 4.11×10−2 | 22.77 |
| VmaxBZE (h−1) | Maximal saturable metabolism rate of benzoylecgonine in plasma | 6.88×10 | 9.91 |
| VmaxNOR (h−1) | Maximum saturable metabolism rate of norcocaine in plasma | 7.5×102 | 20.27 |
| VPLBZE (mL) | Volume of distribution of benzoylecgonine in plasma | 3.25 | 7.41 |
| VPLNOR (mL) | Volume of distribution of norcocaine in plasma | 5.89×10−1 | 27.49 |
Pharmacodynamic analyses
Data for Rmax and total distance travelled per strain are shown in Table 3. For the high responding I/LnJ strain, Rmax was achieved at 10 mg/kg, while total distance travelled over the 30 min interval and Rmax both decreased at 30 mg/kg. These data suggest that I/LnJ mice reached a plateau for distance travelled between the 10 and 30 mg/kg doses. The high-responder, C57BL/6J, achieved Rmax at 30 mg/kg, while the total distance travelled over the 30 min interval and Rmax both decreased at 40 mg/kg. These data suggest that response saturation for C57BL/6J was between 30 and 40 mg/kg. Among these two high responding strains, I/LnJ mice reached the maximum PD response at a lower dose than C57BL/6J mice. Similar to the high-responding C57BL/6J strains, low responding FVB/NJ mice also reached maximal locomotor response at 30 mg/kg and locomotor activity decreased at the 40 mg/kg dose. Even though these two strains both reached a plateau at 30 mg/kg in terms of locomotor activity, the data reveal a 3-fold difference in total distance travelled for the two strains. For the BTBR T+ tf/J low responding strain, Rmax increased substantially from the 10 to 30 mg/kg, and continued to increase at the 40 mg/kg dose, indicating that the dose response maximum was not achieved. For the low responding LP/J strain, both Rmax and total locomotor activity were lower in response to COC than the vehicle control. However, after LP/J mice reached peak response at dose 30 mg/kg, the total distance travelled did not increase further. This pattern was also observed in C57BL/6J and FVB/NJ stains. The LG/J strain had virtually no locomotor response at all doses up to 30 mg/kg and achieved maximum locomotor response at 40 mg/kg. However, this response was still 85% lower than the locomotor response of I/LnJ mice at 5 mg/kg.
Table 3. Descriptive results of dose response stratified by strains.
Stratified by strain, major PD parameters are summarized for ambulatory movement in relation to time from 0–30 min post cocaine injection at dose indicated. Values were adjusted for saline administration by subtracting the mean distance travelled for animals in the saline group.
| Strain | Dose (mg/kg) |
Tmax-PD (min) |
CV% | Rmax (cm/min) |
CV% | Total 30 min Distance (cm) |
CV% |
|---|---|---|---|---|---|---|---|
| I/LnJ | 5 | 10 | 36.89 | 399.23 | 26.22 | 5788.7 | 83.87 |
| 10 | 5 | 32.27 | 1211.73 | 19.05 | 19820.39 | 38.95 | |
| 30 | 10 | 35.90 | 1132.87 | 16.53 | 16277.36 | 48.32 | |
| 40 | 15 | 57.74 | 1078.3 | 19.93 | 18122.33 | 54.23 | |
| C57BL/6J | 5 | 5 | 53.56 | 143.23 | 37.98 | 3273.18 | 86.82 |
| 10 | 5 | 56.71 | 441.41 | 39.99 | 9036.48 | 56.50 | |
| 30 | 15 | 33.92 | 1236.41 | 23.81 | 28610.7 | 30.28 | |
| 40 | 15 | 39.59 | 1135.42 | 27.27 | 27040.53 | 39.05 | |
| FVB/NJ | 5 | 15 | 67.19 | 119.23 | 18.06 | 2678.24 | 64.81 |
| 10 | 30 | 56.66 | 382.95 | 31.24 | 8888.3 | 71.42 | |
| 30 | 10 | 28.75 | 594.29 | 57.16 | 12628.77 | 94.71 | |
| 40 | 30 | 40.16 | 476.75 | 44.99 | 9647.44 | 95.83 | |
| BTBR T+tf/J | 5 | 5 | 96.26 | 134.26 | 65.78 | 1664.85 | 241.18 |
| 10 | 2 | 80.55 | 232.85 | 40.29 | 2227.07 | 153.37 | |
| 30 | 20 | 49.72 | 605.26 | 54.44 | 13238.46 | 75.22 | |
| 40 | 20 | 52.56 | 673.58 | 56.10 | 15027.21 | 83.32 | |
| LG/J | 5 | 5 | 99.17 | 30.64 | 67.39 | −2364.42 | 95.44 |
| 10 | 2 | 82.65 | 146.74 | 67.01 | −432 | 1104.34 | |
| 30 | 2 | 93.60 | 145.25 | 70.96 | −600.81 | 665.26 | |
| 40 | 2 | 84.88 | 127.37 | 82.09 | 865.08 | 744.69 | |
| LP/J | 5 | 2 | 84.18 | −20.78 | 35.95 | −1657.83 | 47.12 |
| 10 | 15 | 49.31 | 103.55 | 77.40 | 476.67 | 742.99 | |
| 30 | 15 | 34.29 | 595.38 | 40.20 | 11649.02 | 61.66 | |
| 40 | 10 | 57.36 | 534.76 | 52.67 | 10829.67 | 92.31 |
PKPD analyses: Association between PK and initial locomotor activation
The PKPD relationships for COC and NOR are shown in Figure 6. Notably, C57BL/6J reached maximum PD effect at dose 30 mg/kg whereas I/LnJ reached maximum PD effect at a lower dose of 10 mg/kg in the brain. Both C57BL/6J and I/LnJ were among the high responding strains, yet exhibit different dose-response relationships. Interestingly, while the other 4 strains exhibited either a plateau or decreased locomotor activation after Cmax was reached, the locomotor activity of LG/J and BTBR T+ Itpr3 tf/J continued to increase. In comparison with COC, PKPD trends are similar for NOR, with the exception that AUC0-last and Cmax were 10-fold lower.
Figure 6. PKPD comparison across 6 strain for COC and NOR in the brain. Individual plots show.
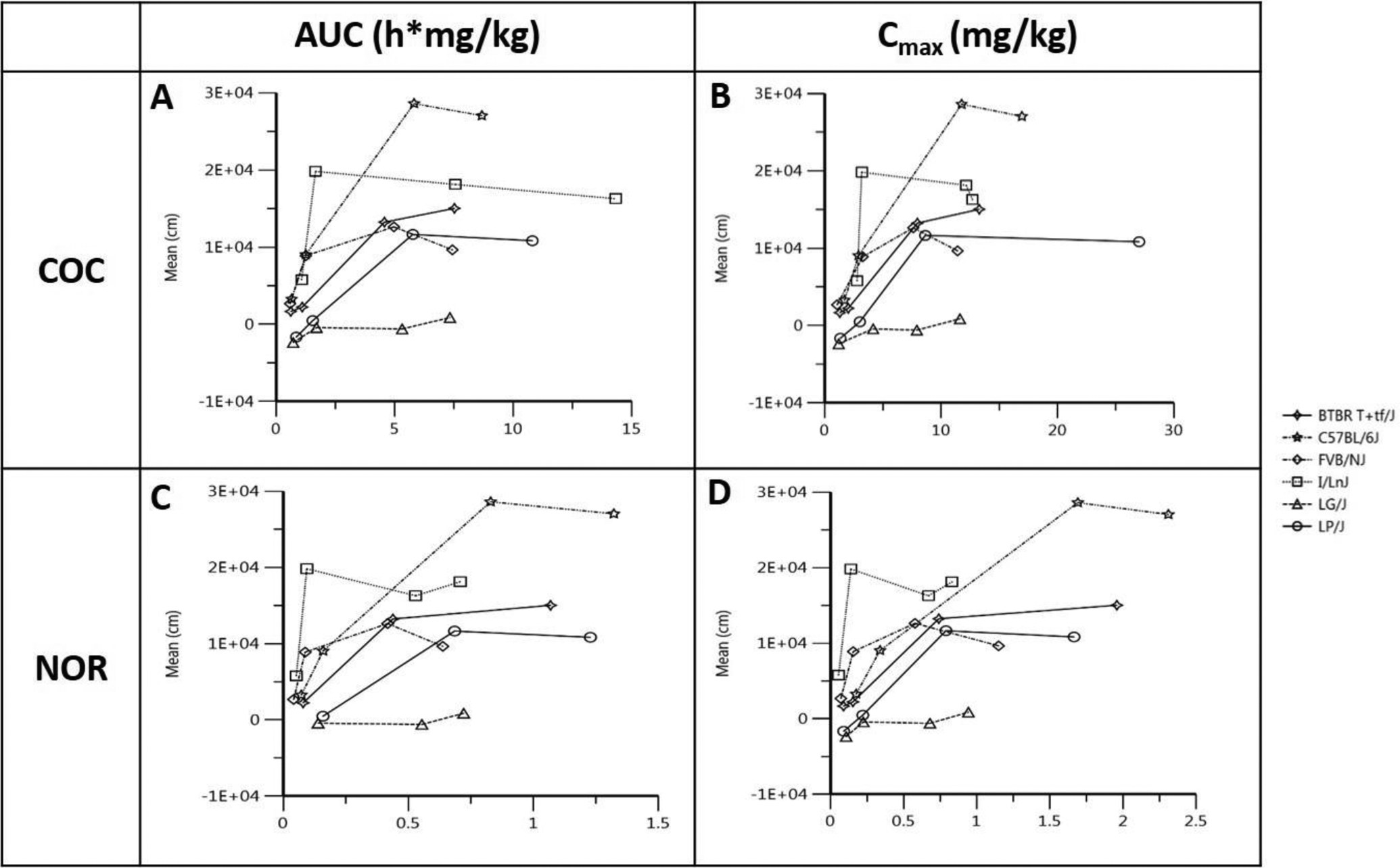
PK and PD relationships for each strain. In each panel, the PK of COC and NOR are measured as AUC and Cmax and plotted against the PD endpoint, mean distance travelled in 30 min. All 4 dosing groups are stratified by strain and represented on the curve with the first data point representing 5mg/kg and the 2nd, 3rd and 4th data point representing 10, 30 and 40 mg/kg dosing groups, respectively. A: relationship between brain AUC of COC and mean distance travelled in 30 min; B: relationship between brain Cmax of COC and mean distance travelled in 30 min; C: relationship between brain AUC or NOR and mean distance travelled in 30 min; D: relationship between brain Cmax of NOR and mean distance travelled in 30 min.
Interstrain Analysis of Variance (ANOVA)
While the majority of the PK parameters from the NCA are consistent with low interstrain differences, as evidenced by CV% values below 40% (Table 1), the inspection of plasma and brain concentrations of COC, NOR, and BZE across strains suggest interstrain differences might exist (Figures 2–3). Our ANOVA yielded a statistically significant overall strain effect for analytes in the brain compartment but not in plasma (Supplemental Table 2). We examined pairwise strain comparisons for COC or metabolites at time points for which an overall strain effect was observed and identified statistically significant individual strain differences (Table 4). Results from the supplementary ANOVA conducted on the first 3 time points are shown in Supplemental Table 3. Although specific multiple comparisons weren’t done when the effect was significant (P<0.05), differences can be viewed on the corresponding plots (Supplemental Figure 1). These results should be interpreted as descriptive in nature due to the sample size at each dose and time point.
Table 4. Overlapping PK and PD pairwise comparisons.
Significant pairwise strain comparisons for PK are shown along with significant and overlapping PD comparisons (italicized). Significant PK differences between strains in the same PD response group (i.e. high vs high, low vs low) are shaded grey.
| PK Analyte and Parameter | Strain A | Strain B | Difference | PK P-value | PD P-value |
|---|---|---|---|---|---|
| Brain NOR Cmax | C57BL/6J | FVB/NJ | 0.64 | 0.00766 | 0.02265 |
| C57BL/6J | I/LnJ | 0.71 | 0.00407 | ||
| C57BL/6J | LG/J | 0.64 | 0.00768 | 0.00010 | |
| Brain BZE Cmax | BTBR T+tf/J | LP/J | −0.09 | 0.03810 | |
| C57BL/6J | LP/J | −0.09 | 0.04600 | 0.00340 | |
| FVB/NJ | LG/J | −0.12 | 0.01070 | ||
| FVB/NJ | LP/J | −0.14 | 0.00350 | ||
| I/LnJ | LP/J | −0.10 | 0.01970 | 0.01141 | |
| Brain BZE AUC0-last | C57BL/6J | LG/J | −0.05 | 0.03920 | 0.00010 |
| FVB/NJ | LG/J | −0.07 | 0.00318 | 0.01620 | |
| FVB/NJ | LP/J | −0.07 | 0.00468 | ||
| I/LnJ | LG/J | 0.02 | 0.00765 | 0.00031 | |
| I/LnJ | LP/J | −0.06 | 0.01130 | 0.01141 |
Total distance travelled in 30 min was compared across strains and showed high interstrain variability as expected based on our previous publications.17,23 The P-value for the overall distance travelled between strains was 0.0012. We examined pairwise comparisons from the ANOVA and identified overlaps in significant interstrain differences for PK and PD (Table 4). We did not identify any overlaps corresponding to COC PK variability but several included the active metabolite NOR. All of the NOR PK/PD overlaps included the high responding C57BL/6J substrain in comparison with two low responding strains (FVB/NJ, LG/J) and another high responding strain (I/LnJ) (Table 4).
DISCUSSION
This study is the first to utilize a population PK model to characterize COC, NOR and BZE in both plasma and brain in mouse models with the goal of understanding the relationship between PK and PD in genetically and phenotypically diverse inbred mouse strains. Understanding the COC PK is important for interpreting its behavioral, or PD effects, and identifying potential mechanisms for development of therapeutics. We characterized the PK of COC across a range of doses in a series of inbred mouse strains that previously displayed either high or low locomotor response to COC administration.11,17 Ours is the first study to describe the PK of COC and its metabolites in this manner, and our experimental design allowed us to more fully investigate the relationship between genetic background, PK, and locomotor behavior of our mouse strains. We developed an empirical model that simultaneously characterizes COC, NOR and BZE in plasma and brain tissue at multiple time points and doses in six inbred mouse strains that differ for initial locomotor sensitivity to COC. Specifically, we utilized plasma and brain concentrations of COC, NOR and BZE to develop a population PK model. Existing studies have been limited to examining plasma or analyzing brain samples without population PK modeling analysis.21,34–37 We believe our model is the most comprehensive thus far, as it characterizes the metabolism of COC into NOR and BZE in the plasma compartment, and the movement of the parent drug and metabolites into the brain. Additionally, the model shows that COC also undergoes metabolism into NOR and BZE in the brain compartment.
During model development, we observed that the I/LnJ strain showed a substantial increase in the brain COC exposure at the 30 mg/kg dose compared to the other strains (Supplemental Figure 2). During model optimization, we tested our PK model with and without the I/LnJ strain data. The model without the I/LnJ strain data showed improved CV% (Table 2) and we concluded that this model might be better suited to explain the mechanisms of COC disposition. Due to our limited sample size (n=4 FPM), we could not develop a full population PK model which included the I/LnJ strain. Future experiments can be designed to include more I/LnJ mice with a wider range of COC dosing to optimize subject input for the development of the population PK model. Overall, however, the diagnostic plots (Figure 5, Supplemental Figure 3–4) show that the model is a reasonable approximation of COC distribution in plasma and brain and the processes related to metabolite formation. We acknowledge that our model was limited by the number of strains and included only responders that were representative of either high or low in the previous publication.11 Inclusion of additional strains with more diverse PD responses to COC may yield different results.
The estimated brain volume of distribution was highest for COC (Table 2), suggesting that the parent drug has the highest degree of brain penetration compared to either NOR or BZE. This observation is consistent with previous studies in both animal models and humans38,39 Our data also indicate that COC concentrations drop rapidly in the plasma, while the model also suggests that the parent drug is both readily metabolized and enters the brain. We cannot exclude the possibility that the low volume of COC in the plasma might also be attributed, in part, to unexplored mechanisms not accounted for in our model. For example, mechanisms of disposition in other tissue compartments that were not quantified in our studies. More specifically, our model shows that COC distributes into other tissue compartments in addition to the brain compartment.
Our final model establishes that BZE achieves higher plasma concentrations in mice than both COC and NOR (Table 2). The estimated parameters are consistent with the concentration versus time plots (Figure 2) showing that BZE has a greater plasma concentration than COC and a longer elimination half-life than COC and NOR. Human studies have also shown that BZE has a longer elimination half-life than both COC and NOR, hence the use of BZE as a surrogate for COC drug screening.40–42 Evidence from the literature regarding the ability of BZE to cross the blood brain barrier has been equivocal43,44 although previous studies have detected low levels of BZE in the brain.45–47 Our final model suggests that BZE can cross the blood brain barrier45–47 but whether BZE is transported or diffuses into the brain or results from cocaine metabolism in the brain is still unknown. Our data suggest that BZE in the brain results from both transport or diffusion from the periphery and COC metabolism in the brain. Future experiments utilizing peripheral administration of BZE could answer the question of whether this metabolite has the ability to cross the blood brain barrier.
One limitation for our PK model is data scarcity. Due to serial euthanization, only one FPM per dose per strain was available as individual subject data in developing the population PK model. The use of a FPM imposed limits such that only one FPM per dose and strain combination was available for analysis. Therefore, PK results should be viewed as descriptive in nature since formal dose response analyses across strains were not possible. Although data were scarce due to the experimental design, the diagnostic plots (Figure 5, Supplemental Figure 3–4) showed that the model was a reasonable approximation of COC distribution and the processes related to NOR and BZE metabolite formation. Future studies that involve more dose groups in these six strains are warranted to more accurately explain COC distribution and metabolism. Nevertheless, this PK model provides a platform for developing future PD models connecting the brain compartment directly with the PD responses.
One of the main goals of our study was to examine the role of PK on individual differences in COC-induced locomotor activation in inbred mice strains with stable genetic backgrounds. Although we observed considerable similarities in PK profiles of COC, NOR, and BZE over the course of 60 min post-COC injection (Figure 2–3), we did detect interstrain PK differences. Our observations support the effects of genetic background on COC PK as reported in previous studies.48,49 Further study is needed to determine if the differences we observed are real, or artifacts of the large number of statistical comparisons that were performed. Pairwise comparisons identified strains that exhibited significant differences in analyte concentrations in the brain. We detected statistically significant PK differences between high vs. low responders and also high vs. high and low vs. low responders (Table 4), suggesting that the relationship between PK and locomotor activation is not necessarily linear.
Comparisons for which both PK and PD were significant mostly involved the non-active metabolite, BZE (Table 4). However, several significant PK/PD pairwise comparisons were observed for the active metabolite, NOR. NOR is a lipophilic metabolite that crosses the blood brain barrier and, like COC, inhibits dopamine reuptake albeit with reduced potency.42,50,51 Previous studies have found that administration of NOR by various routes does not increase locomotor activity in rats.52,53 However, NOR has been found to decrease locomotor activity in the inbred mouse strain BALB/cBy54 most likely due to its anesthetic effects.50 We also noted that all of the statistically significant NOR comparisons involved the C57BL/6J strain (Table 4). C57BL/6J mice have higher levels of NOR compared to other inbred strains (Supplemental Figure 5). However, for all inbred strains, including C57BL/6J, the concentrations of NOR in the brain are still 6–15% that of COC (Table 1). We also noted an interesting pattern in NOR concentration in the supplementary ANOVA. At all doses, we observed a significant strain effect for NOR levels in the brain whereas COC levels were only significantly different at the 5 and 40 mg/kg doses. Regardless, we observed no strain differences greater than 10-fold (Supplemental Figure 1). These observations along with evidence from the literature suggest that strain differences in NOR are unlikely to fully explain locomotor differences between inbred strains. However, additional experiments that involve direct administration of the metabolite are warranted to accurately establish the magnitude of a PK difference that is relevant and assess the contribution of NOR in terms of locomotor activation in these genetically diverse mice.
Our data support a time-dependent relationship between brain concentrations of COC and locomotor activation. In general, locomotor activity corresponds to COC concentrations in the brain over time (Figure 6, Supplemental Figure 6–8). This relationship has also been observed in other studies.57,58 We also observed dose-dependent locomotor responses that differ across inbred strains. For example, the I/LnJ strain shows a peak locomotor response at 10 mg/kg but locomotor activity does not increase further at 30 and 40 mg/kg even though brain concentration of COC continues to increase (Figure 6). Conversely, LG/J mice show almost no locomotor response to cocaine at any dose even though their brain concentration of COC increases in a dose-dependent manner.
The rate at which psychostimulants enter the brain is an important factor in addiction liability in humans. Addiction liability is increased in individuals who take COC via routes of administration that result in a more rapid delivery of the drug to the brain.15 Thus, interstrain differences in the rate at which COC enters the brain might correspond with its behavioral effects. We observed no interstrain differences in the rate at which COC enters the brain in our population (Figure 3). We note that IP administration of COC corresponds more closely to intranasal or oral routes of administration in humans55,56 and results in slower rates of exposure and decline compared to IV drug administration.
Although our previous study concluded that high- and low-responders also show differences in IV COC self-administration,17 the role of PK in the rewarding and reinforcing effects of cocaine cannot be established based on the data presented here. The psychomotor stimulant theory of addiction proposed by Wise and Bozarth in 1987 suggested that locomotor activation and positive reinforcement induced by psychostimulants are homologous.59 However, a positive relationship between psychostimulant-induced locomotor activation and drug reinforcement and reward behaviors such as IV drug self-administration and conditioned place preference has not been supported across all studies.60 In fact, in our previously published study, we determined that inbred strain differences in cocaine-induced locomotor activation could predict some drug reinforcement behaviors but not others and that the relationship varied across different genetic backgrounds.17 For example, low responding LG/J mice acquire and maintain responses for COC in a self-administration paradigm at a level that is similar to that exhibited by high-responding strains.17 However, direct comparisons are difficult based on the different routes of administration used for locomotor activation and drug self-administration studies. Additional studies utilizing different protocols for drug delivery and different genetic backgrounds are required to more fully understand the relationship between PK and the rewarding reinforcing effects of cocaine.
Based on our previously published results and the data presented herein, we can conclude that the extreme locomotor differences in response to cocaine observed in these inbred mouse strains can be partially attributed to the PK differences of COC and NOR in the brain. However, the behavioral effects of COC likely result from other mechanisms. These mechanisms might include differences in dopaminergic transmission,61,62 changes in other monoamine neurotransmitters,63,64 or interactions between monoaminergic and other pathways.65 Genetic analyses can be used to identify mechanisms underlying strain differences. Genetic mapping studies are underway in our laboratory, and by other groups, to elucidate the biological mechanisms that bring about variation in drug response. In addition, strain differences in mechanistic pathways may also have non-genetic origins. Identifying both genetic and non-genetic mechanisms that contribute to drug response is a worthwhile endeavor that would significantly advance our understanding of CUD.
Supplementary Material
Acknowledgements
We gratefully acknowledge the reviewers for their suggestions to improve the manuscript.
Footnotes
Data Availability Statements
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Bose J, Hedden SL, Lipari RN, Park-Lee E, Tice P. Use and Mental Health Indicators in the United States: Results from the 2017 National Survey on Drug Use and Health Recommended Citation Substance Abuse and Mental Health Services Administration.; 2018.
- 2.John William S and Wu L-T. Trends and correlates of cocaine use and cocaine use disorder in the United States from 2011 to 2015. Drug Alcohol Depend. 2017;176(3):139–148. doi: 10.1016/j.physbeh.2017.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hedegaard H, Bastian BA, Trinidad JP, Spencer M, Warner M. Drugs Most Frequently Involved in Drug Overdose Deaths: United States, 2011–2016. Natl Vital Stat Rep. 2018;67(9):1–14. [PubMed] [Google Scholar]
- 4.Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33(6):267–276. doi: 10.1016/J.TINS.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spronk DB, Van Wel JHP, Ramaekers JG, Verkes RJ. Characterizing the cognitive effects of cocaine: A comprehensive review. Neurosci Biobehav Rev. 2013;37:1838–1859. doi: 10.1016/j.neubiorev.2013.07.003 [DOI] [PubMed] [Google Scholar]
- 6.Lambert NM, McLeod M, Schenk S. Subjective responses to initial experience with cocaine: an exploration of the incentive-sensitization theory of drug abuse. Addiction. 2006;101(5):713–725. doi: 10.1111/j.1360-0443.2006.01408.x [DOI] [PubMed] [Google Scholar]
- 7.Badiani A, Spagnolo P. Role of Environmental Factors in Cocaine Addiction. Curr Pharm Des. 2013;19(40):6996–7008. doi: 10.2174/1381612819999131125221238 [DOI] [PubMed] [Google Scholar]
- 8.Prom-Wormley EC, Ebejer J, Dick DM, Bowers MS. The genetic epidemiology of substance use disorder: A review. Drug Alcohol Depend. 2017;180:241–259. doi: 10.1016/j.drugalcdep.2017.06.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ducci F, Goldman D. The Genetic Basis of Addictive Disorders. Psychiatr Clin North Am. 2012;35(2):495–519. doi: 10.1016/j.psc.2012.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hancock D, Markunas C, Bierut L, Johnson E. Human Genetics of Addiction: New Insights and Future Directions. Curr Psychiatry Rep. 2017;546(7660):651–655. doi: 10.1038/nature22814.Trans-kingdom [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiltshire T, Ervin RB, Duan H, et al. Initial locomotor sensitivity to cocaine varies widely among inbred mouse strains. Genes Brain Behav. 2015;14(3):271–280. doi: 10.1111/gbb.12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidson ES, Finch JF, Schenk S. Variability in subjective responses to cocaine: Initial experiences of college students. Addict Behav. 1993;18(4):445–453. doi: 10.1016/0306-4603(93)90062-E [DOI] [PubMed] [Google Scholar]
- 13.Fergusson DM, Horwood LJ, Lynskey MT, Madden PAF. Early Reactions to Cannabis Predict Later Dependence. Arch Gen Psychiatry. 2003;60(10):1033. doi: 10.1001/archpsyc.60.10.1033 [DOI] [PubMed] [Google Scholar]
- 14.Kreek MJ, Bart G, Lilly C, Laforge KS, Nielsen DA. Pharmacogenetics and human molecular genetics of opiate and cocaine addictions and their treatments. Pharmacol Rev. 2005;57(1):1–26. doi: 10.1124/pr.57.1.1 [DOI] [PubMed] [Google Scholar]
- 15.Allain F, Minogianis E-A, Roberts DCS, Samaha A-N. How fast and how often: The pharmacokinetics of drug use are decisive in addiction. Neurosci Biobehav Rev. 2015;56:166–179. doi: 10.1016/j.neubiorev.2015.06.012 [DOI] [PubMed] [Google Scholar]
- 16.Kawa AB, Allain F, Robinson TE, Samaha AN. The transition to cocaine addiction: the importance of pharmacokinetics for preclinical models. Psychopharmacology (Berl). 2019. doi: 10.1007/s00213-019-5164-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts AJ, Casal L, Huitron-Resendiz S, Thompson T, Tarantino LM. Intravenous cocaine self-administration in a panel of inbred mouse strains differing in acute locomotor sensitivity to cocaine. Psychopharmacology (Berl). 2018;235(4):1179–1189. doi: 10.1007/s00213-018-4834-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zombeck JA, Gupta T, Rhodes JS. Evaluation of a pharmacokinetic hypothesis for reduced locomotor stimulation from methamphetamine and cocaine in adolescent versus adult male C57BL/6J mice. Psychopharmacology (Berl). 2009;201(4):589–599. doi: 10.1007/s00213-008-1327-0 [DOI] [PubMed] [Google Scholar]
- 19.Benuck M, Lajtha A, Reith M. Pharmacokinetics of systemically administered cocaine and locomotor stimulation in mice. J Pharmacol Exp Ther. 1987;243(1):144–149. [PubMed] [Google Scholar]
- 20.Wiener HL, Reith MEA. Correlation between cocaine-induced locomotion and cocaine disposition in the brain among four inbred strains of mice. Pharmacol Biochem Behav. 1990;36(3):699–701. doi: 10.1016/0091-3057(90)90277-O [DOI] [PubMed] [Google Scholar]
- 21.Sun L, Lau CE. Simultaneous pharmacokinetic modeling of cocaine and its metabolites, norcocaine and benzoylecgonine, after intravenous and oral administration in rats. Drug Metab Dispos. 2001;29(9):1183–9. [PubMed] [Google Scholar]
- 22.Benuck M, Reith ME, Lajtha A. Presence of the toxic metabolite N-hydroxy-norcocaine in brain and liver of the mouse. Biochem Pharmacol. 1988;37(6):1169–1172. http://www.ncbi.nlm.nih.gov/pubmed/3355590. Accessed September 5, 2018. [DOI] [PubMed] [Google Scholar]
- 23.Wiltshire T, Ervin RB, Duan H, et al. Initial locomotor sensitivity to cocaine varies widely among inbred mouse strains. Genes, Brain Behav. 2015;14(3):271–280. doi: 10.1111/gbb.12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts AJ, Casal L, Huitron-Resendiz S, Thompson T, Tarantino LM. Intravenous cocaine self-administration in a panel of inbred mouse strains differing in acute locomotor sensitivity to cocaine. 2019;235(4):1179–1189. doi: 10.1007/s00213-018-4834-7.Intravenous [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samtani MN, Jusko WJ. Comparison of dexamethasone pharmacokinetics in female rats after intravenous and intramuscular administration. Biopharm Drug Dispos. 2005;26(3):85–91. doi: 10.1002/bdd.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meno-Tetang GML, Lowe PJ. On the prediction of the human response: A recycled mechanistic pharmacokinetic/pharmacodynamic approach. Basic Clin Pharmacol Toxicol. 2005;96(3):182–192. doi: 10.1111/j.1742-7843.2005.pto960307.x [DOI] [PubMed] [Google Scholar]
- 27.Sun L, Lau CE. Simultaneous pharmacokinetic modeling of cocaine and its metabolites, norcocaine and benzoylecgonine, after intravenous and oral administration in rats. Drug Metab Dispos. 2001;29(9):1183–1189. [PubMed] [Google Scholar]
- 28.Mccarthy LE, Mannelli P, Niculescu M, Gingrich K, Unterwald EM, Ehrlich ME. The distribution of cocaine in mice differs by age and strain. 2004. doi: 10.1016/j.ntt.2004.07.004 [DOI] [PubMed]
- 29.NT L A fast scoring algorithm for maximum likelihood estimation in unbalanced mixed models with nested random effects. Biometrika. 1987;74:817. [Google Scholar]
- 30.Jufer RA, Wstadik A, Walsh SL, Levine BS, Cone EJ. Elimination of Cocaine and Metabolites in Plasma, Saliva, and Urine Following Repeated Oral Administration to Human Volunteers. Vol 24.; 2000. [DOI] [PubMed] [Google Scholar]
- 31.Mets B, Diaz J, Soo E, Jamdar S. Cocaine, norcocaine, ecgonine methylester and benzoylecgonine pharmacokinetics in the rat. Life Sci. 1999;65(12):1317–1328. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Zheng X, Zhan M, Zhou Z, Zhan C-G, Zheng F. Metabolic Enzymes of Cocaine Metabolite Benzoylecgonine. ACS Chem Biol. 2016;11(8):2186–2194. doi: 10.1021/acschembio.6b00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun H, Yazal J El, Lockridge O, et al. Predicted Michaelis-Menten complexes of cocaine-butyrylcholinesterase. Engineering effective butyrylcholinesterase mutants for cocaine detoxication. J Biol Chem; 2001; 276(12):9330–9336. doi: 10.1074/jbc.M006676200 [DOI] [PubMed] [Google Scholar]
- 34.Lau CE, Foster DM, Falk JL. Pharmacokinetic-pharmacodynamic modeling of the psychomotor stimulant effect of cocaine after intravenous administration: timing performance deficits. J Pharmacol Exp Ther. 1988; 288(2):535–543. [PubMed] [Google Scholar]
- 35.Parlaman JP, Thompson BL, Levitt P, Stanwood GD. Pharmacokinetic profile of cocaine following intravenous administration in the female rabbit. Eur J Pharmacol. 2007;563(1–3):124–129. doi: 10.1016/j.ejphar.2007.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Booze RM, Lehner AF, Wallace DR, et al. Dose-response cocaine pharmacokinetics and metabolite profile following intravenous administration and arterial sampling in unanesthetized, freely moving male rats. Neurotoxicol Teratol. 1997;19(1):7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimmer BA, Dobrin CV, Roberts DCS. Brain-cocaine concentrations determine the dose self-administered by rats on a novel behaviorally dependent dosing schedule. Neuropsychopharmacology. 2011;36(13):2741–2749. doi: 10.1038/npp.2011.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawks RL, Kopin IJ, Colburn RW, et al. Norcocaine : a pharmacologically active metabolite of cocaine found in brain. Life Sci. 1974; 15(12):2189–95. [DOI] [PubMed] [Google Scholar]
- 39.Morishima H, Whittington R, Iso A, et al. The Comparative Toxicity of Cocaine and Its Metabolites in Conscious Rats. Anesthesiology. 1999;(90):1684–1690. [DOI] [PubMed] [Google Scholar]
- 40.Huestis MA, Darwin WD, Shimomura E, et al. Cocaine and metabolites urinary excretion after controlled smoked administration. J Anal Toxicol. 2007;31(8):462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones AW, Holmgren A, Kugelberg FC. Concentrations of cocaine and its major metabolite benzoylecgonine in blood samples from apprehended drivers in Sweden. Forensic Sci Int. 2008;177(2–3):133–139. doi: 10.1016/j.forsciint.2007.11.009 [DOI] [PubMed] [Google Scholar]
- 42.Mule’ SJ, Casella GA, Misra AL. Intracellular disposition of [3H]-cocaine, [3H]-norcocaine, [3H]-benzoylecgonine and [3H]-benzoylnorecgonine in the brain of rats. Life Sci. 1976;19(10):1585–1596. doi: 10.1016/0024-3205(76)90105-3 [DOI] [PubMed] [Google Scholar]
- 43.Chapy H, Smirnova M, André P, et al. Carrier-mediated cocaine transport at the blood-brain barrier as a putative mechanism in addiction liability. Int J Neuropsychopharmacol. 2014;18(1). doi: 10.1093/ijnp/pyu001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spiehler V Brain concentrations of cocaine and benzoylecgonine in fatal cases. J Forensic Sci. 1985;30(4):1003–1011. [PubMed] [Google Scholar]
- 45.Dean RA, Harper ET, Dumaual N, et al. Effects of ethanol on cocaine metabolism: formation of cocaethylene and norcocaethylene. Toxicol Appl Pharmacol. 1992;117(1):1–8. [DOI] [PubMed] [Google Scholar]
- 46.Sherry Chow HH, Anavy N, Villalobos A. Direct nose-brain transport of benzoylecgonine following intranasal administration in rats. J Pharm Sci. 2001;90(11):1729–1735. doi: 10.1002/jps.1122 [DOI] [PubMed] [Google Scholar]
- 47.Misra AL, Nayak PK, Bloch R, Mulé SJ. Estimation and disposition of [3H]benzoylecgonine and pharmacological activity of some cocaine metabolites. J Pharm Pharmacol. 1975;27(10):784–786. doi: 10.1111/j.2042-7158.1975.tb09404.x [DOI] [PubMed] [Google Scholar]
- 48.Azar MR, Acar N, Erwin VG, et al. Distribution and clearance of cocaine in brain is influenced by genetics. Pharmacol Biochem Behav. 1998;59(3):637–640. doi: 10.1016/S0091-3057(97)00471-1 [DOI] [PubMed] [Google Scholar]
- 49.Mccarthy LE, Mannelli P, Niculescu M, Gingrich K, Unterwald EM, Ehrlich ME. The distribution of cocaine in mice differs by age and strain. Neurotoxicol Teratol. 2004;26:839–848. doi: 10.1016/j.ntt.2004.07.004 [DOI] [PubMed] [Google Scholar]
- 50.Reith MEA, Lajtha A. Locomotor depression in mice by norcocaine does not involve central α2-adrenergic or presynaptic dopamine receptors. Pharmacol Biochem Behav. 1986;24(2):305–307. doi: 10.1016/0091-3057(86)90355-2 [DOI] [PubMed] [Google Scholar]
- 51.Ritz MC, Cone EJ, Kuhar MJ. Cocaine inhibition of ligand binding at dopamine, norepinephrine and serotonin transporters: a structure-activity study. Life Sci. 1990;46(9):635–645. http://www.ncbi.nlm.nih.gov/pubmed/2308472. Accessed September 5, 2018. [DOI] [PubMed] [Google Scholar]
- 52.Bedford JA, Borne RF, Wilson MC. Comparative behavioral profile of cocaine and norcocaine in rats and monkeys. Pharmacol Biochem Behav. 1980;13(1):69–75. doi: 10.1016/0091-3057(80)90122-7 [DOI] [PubMed] [Google Scholar]
- 53.Elliott PJ, Rosen GM, Nemeroff CB. A comparison of cocaine and its metabolite norcocaine: effects on locomotor activity. Pharmacol Biochem Behav. 1987;26(3):573–575 [DOI] [PubMed] [Google Scholar]
- 54.Reith MEA, Meisler BE, Lajtha A. Locomotor effects of cocaine, cocaine congeners, and local anesthetics in mice. Pharmacol Biochem Behav. 1985;23(5):831–836. doi: 10.1016/0091-3057(85)90078-4 [DOI] [PubMed] [Google Scholar]
- 55.Jones RT. The pharmacology of cocaine smoking in humans. NIDA Res Monogr. 1990;99:30–41. [PubMed] [Google Scholar]
- 56.Pan H ‐ T, Menacherry S, Justice JB. Differences in the Pharmacokinetics of Cocaine in Naive and Cocaine‐Experienced Rats. J Neurochem. 1991;56(4):1299–1306. doi: 10.1111/j.1471-4159.1991.tb11425.x [DOI] [PubMed] [Google Scholar]
- 57.Benuck M, Lajtha A, Reith ME. Pharmacokinetics of systemically administered cocaine and locomotor stimulation in mice. J Pharmacol Exp Ther. 1987;243(1). [PubMed] [Google Scholar]
- 58.Unterwald EM, Ho A, Rubenfeld JM, Kreek MJ. Time course of the development of behavioral sensitization and dopamine receptor up-regulation during binge cocaine administration. J Pharmacol Exp Ther. 1994;270(3):1387–1397. [PubMed] [Google Scholar]
- 59.Ying L, Wang D. New design deforming controlling system of the active stressed lap. Adv Opt Mech Technol Telesc Instrum. 2008;7018(4):701833. doi: 10.1117/12.786465 [DOI] [Google Scholar]
- 60.Yamamoto DJ, Nelson AM, Mandt BH, et al. Rats classified as low or high cocaine locomotor responders: A unique model involving striatal dopamine transporters that predicts cocaine addiction-like behaviors. Neurosci Biobehav Rev. 2013;37(8):1738–1753. doi: 10.1016/j.neubiorev.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song R, Zhang HY, Li X, Bi GH, Gardner EL, Xi ZX. Increased vulnerability to cocaine in mice lacking dopamine D3 receptors. Proc Natl Acad Sci U S A. 2012;109(43):17675–17680. doi: 10.1073/pnas.1205297109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Merritt KE, Bachtell RK. Initial D2 dopamine receptor sensitivity predicts cocaine sensitivity and reward in rats. PLoS One. 2013;8(11). doi: 10.1371/journal.pone.0078258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaniewska M, Filip M, Przegalinski E. The Involvement of Norepinephrine in Behaviors Related to Psychostimulant Addiction. Curr Neuropharmacol. 2015;13(3):407–418. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rocha BA, Goulding EH, O’Dell LE, et al. Enhanced locomotor, reinforcing, and neurochemical effects of cocaine in serotonin 5-hydroxytryptamine 2C receptor mutant mice. J Neurosci. 2002;22(22):10039–10045. doi: 10.1523/jneurosci.22-22-10039.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drouin C, Darracq L, Trovero F, et al. α1b-Adrenergic Receptors Control Locomotor and Rewarding Effects of Psychostimulants and Opiates. J Neurosci. 2002;22(7):2873–2884. doi: 10.1523/jneurosci.22-07-02873.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.