

The paper describes the implementation of a user-friendly high-pressure small-angle X-ray scattering cell suitable for biological solutions and soft materials.
Keywords: small-angle X-ray scattering, high-pressure biology, synchrotron instrumentation
Abstract
Pressure is a fundamental thermodynamic parameter controlling the behavior of biological macromolecules. Pressure affects protein denaturation, kinetic parameters of enzymes, ligand binding, membrane permeability, ion transduction, expression of genetic information, viral infectivity, protein association and aggregation, and chemical processes. In many cases pressure alters the molecular shape. Small-angle X-ray scattering (SAXS) is a primary method to determine the shape and size of macromolecules. However, relatively few SAXS cells described in the literature are suitable for use at high pressures and with biological materials. Described here is a novel high-pressure SAXS sample cell that is suitable for general facility use by prioritization of ease of sample loading, temperature control, mechanical stability and X-ray background minimization. Cell operation at 14 keV is described, providing a q range of 0.01 < q < 0.7 Å−1, pressures of 0–400 MPa and an achievable temperature range of 0–80°C. The high-pressure SAXS cell has recently been commissioned on the ID7A beamline at the Cornell High Energy Synchrotron Source and is available to users on a peer-reviewed proposal basis.
1. Introduction
It has been established that pressure, as an analogous parameter to temperature, affects the fundamental activity and structure in biological supra-assemblies (Silva & Weber, 1993 ▸; Boonyaratanakornkit et al., 2002 ▸; Brooks et al., 2011 ▸; Czeslik et al., 2017 ▸; Winter, 2019 ▸). Pressures in the range encountered in the biosphere are known to have many effects on biomacromolecules, including alteration of protein denaturation, dynamics and kinetic constants of enzymes, ligand binding, membrane permeability, ion transduction, expression of genetic information, bacterial motility, viral infectivity, and molecular association and aggregation (Bridgman, 1914 ▸; Silva & Weber, 1993 ▸; Bartlett, 2002 ▸; Winter & Jeworrek, 2009 ▸; Brooks et al., 2011 ▸; Hnosko et al., 2012 ▸; Koo et al., 2013 ▸; Decaneto et al., 2015 ▸; Czeslik et al., 2017 ▸; Winter, 2019 ▸).
There is a growing realization that a significant part of the planetary biomass lives under high pressure in the depths of the oceans and in the Earth’s crust (Bar-On et al., 2018 ▸; Mentré & Hoa, 2001 ▸; Daniel et al., 2006 ▸). This realization is catalyzing interest in the biophysics of the large molecules of life as a function of pressure (Jenkins et al., 2018 ▸; Winter, 2019 ▸). For the purposes of this paper, we define relevant high pressures (HPs) for biophysical studies as those encountered in the known biosphere, roughly 10 MPa to somewhat below 1000 MPa (100 to 10 000 atmospheres). It is known that many living cells and cellular functions behave differently when the surrounding pressure is changed over this range (Oger & Jebbar, 2010 ▸; Winter & Jeworrek, 2009 ▸; Wang, Meng et al., 2012 ▸; Brooks et al., 2011 ▸).
Measurements of changes in biomolecular structure are key to understanding biophysical effects. Pressure alters macromolecular structure on length scales ranging from the atomic (Barstow et al., 2008 ▸), as probed by crystallography, to tens of nanometres, as probed by small-angle X-ray scattering (SAXS) (Schummel et al., 2019 ▸). However, such measurements are hampered by a paucity of high-pressure X-ray tools specific to the requirements of biological macromolecules. A goal at the Cornell High Energy Synchrotron Source (CHESS) is to remedy this situation by implementing high-pressure SAXS (HP-SAXS) and high-pressure crystallographic instrumentation specifically configured for studies of biological macromolecules in a user-friendly X-ray facility context. The purpose of this paper is to describe the HP-SAXS capabilities that have been implemented at CHESS, with the emphasis on a newly designed HP-SAXS sample cell suitable for routine studies of macromolecules in solution or dispersion. In future papers we will detail other HP cells being implemented at CHESS, including a diamond anvil cell for protein crystallographic studies and a flow-through single-crystal sapphire capillary cell for chromatography-coupled SAXS of biomolecular solutions in the range of 0.1 to 100 MPa. In the context of this paper, the difference between ‘absolute pressure’ and ‘gauge pressure’, i.e. pressure measured relative to the 0.1 MPa atmosphere, is inconsequentially small. Thus, we refer to atmospheric or ambient pressure as 0 MPa throughout.
The X-ray sample cells being implemented at CHESS are part of a larger toolkit being created by a US National Science Foundation supported Research Coordination Network (RCN) of over two dozen international collaborators interested in high-pressure biology. Other tools being worked on by RCN colleagues include HP-NMR (Peterson & Wand, 2005 ▸; Roche et al., 2019 ▸; Caro & Wand, 2018 ▸; Akasaka, 2015 ▸), optical microscopy (Hartmann et al., 2004 ▸; Vass et al., 2013 ▸; Bourges et al., 2020 ▸), methods of growing single-cell organisms in HP environments (Vezzi et al., 2005 ▸; Kato, 2006 ▸; Takai et al., 2008 ▸), tools to facilitate bioinformatic mining of extremophile genomes (Grötzinger et al., 2014 ▸; Black et al., 2013 ▸) and molecular dynamics simulations for molecules in HP environments (Ingr et al., 2016 ▸; Silva et al., 2015 ▸; Garcia & Paschek, 2008 ▸; Sarupria et al., 2010 ▸; Paschek et al., 2005 ▸; Prigozhin et al., 2019 ▸).
Several HP-SAXS cells have been described in the literature (Ando et al., 2008 ▸; Brooks et al., 2010 ▸; Pressl et al., 1997 ▸; Winter, 2002 ▸). However, in general, HP-SAXS cells are difficult for non-experts to use. Our goal was to learn from existing cells so as to devise a configuration that is well suited to an X-ray user facility context in which the users are expert in the biophysics of their samples, rather than in the arcana of HP instrumentation. Emphasis was, therefore, placed on ease and speed of changing samples, on stability and minimization of specimen cell background SAXS, and on precise and stable temperature and pressure control of the sample environment.
SAXS from biological solutions (BioSAXS) is dependent on measuring very small changes in the SAXS signals between a reference solution and one containing the macromolecules of interest, both of which scatter weakly (Skou et al., 2014 ▸). In consequence, it is of the utmost importance that all sources of background scatter be reduced as much as possible and remain reproducible when the reference solution is substituted for the sample solution (Wang et al., 2018 ▸). The need to minimize background scatter dictated a vacuum enclosure that included the downstream HP-SAXS cell window and the detector. HP cell windows are necessarily thick to withstand high pressures, yet must absorb as little of the X-ray beam as feasible. This led to the choice of windows of carefully selected synthetic single-crystal diamond, to reduce both window absorption and background SAXS. Even single-crystal diamonds are not perfect, and they exhibit SAXS that changes with the position of the beam on the crystal face, leading to a requirement that the beam hit the same place on the diamond pressure windows to within micrometre accuracy, even when the sample is changed (Wang, Tate & Gruner, 2012 ▸). This was one of the more difficult requirements to satisfy: prior cells typically relied on removal of the cell from the beamline to change the sample (Pressl et al., 1997 ▸) and/or the use of high-torque wrenches to reseal the cell (Ando et al., 2008 ▸). We required that the sample could be readily changed without moving the cell or using large forces or torques. And, of course, it is necessary that the temperature and pressure of the sample be stable, accurate and precise. All these requirements were met with the cell design described below.
2. Lessons from prior HP-SAXS cells
The literature contains descriptions of several HP-SAXS cells designed specifically for biological studies [see e.g. Winter (2002 ▸), Pressl et al. (1997 ▸), Brooks et al. (2010 ▸), Ando et al. (2008 ▸) and Heller et al. (2014 ▸), and references therein].
We chose the design of Ando et al. (2008 ▸) as a starting point. This cell is designed to withstand 400 MPa. It incorporates disposable plastic inserts to contain minimal volumes of specimen in a specimen path length optimized for ca 10 keV X-rays. The disposable inserts allow samples to be pre-loaded. The cell’s high-pressure X-ray windows are minimally thick single-crystal diamond plates. The diamonds are Bridgman/Poulter sealed (Spain & Paauwe, 1977 ▸; Poulter, 1932 ▸) onto optically smooth faces of removable steel plugs designed in such a way that the cell could be disassembled and cleaned without breaking the diamond-to-steel plug seal, i.e. the diamond windows and steel plugs can be handled as a removable unit. The steel plugs are in turn sealed into a steel pressure block via large steel closure nuts. The logic of this design is that the distance between the diamond windows is set by hard metal-to-metal contacts, and not by deformable elastic or metal seals, thereby maintaining a very reproducible window-to-window distance. Use of this cell also illustrated its drawbacks: the primary disadvantage is that the cell needs to be removed from the beamline, and the closure nuts must then be removed with a large torque wrench to change samples. This is a time-consuming process that makes it very difficult to replace the cell back on the beamline with the desired micrometre-level reproducibility. Another disadvantage is that the cell uses air paths outside the diamond windows with consequent background air scatter.
The cell developed by Brooks et al. (2010 ▸) has a dedicated side port so that samples can be loaded without requiring removal from the beamline. This allows the X-ray windows to remain in place throughout an experiment, which in turn facilitates accurate subtraction of background scattering. However, it is still necessary to use a large torque wrench to open the sample port, and this runs the danger of inadvertantly moving the cell. The two 1 mm chemical vapor deposition (CVD) diamond windows are each placed on diamond supports that use a combination of a Poulter seal and high-strength adhesives to hold the diamond in place. The cell is rated to 500 MPa and has been used for pressure jump studies.
Our intention was to draw upon the strengths of these prior cell designs so as to improve on the ease of use while minimizing the movement of cell elements in the X-ray beam path. Another consideration was to achieve a design that allowed for fast repair and changeovers if issues arose. The resultant cell is a collaboration between Cornell University and Pressure BioSciences Inc. (denoted PBI hereinafter; South Easton, Easton, Massachusetts, USA); PBI is considering selling the cell to make the design accessible to the wider community.
The following points were considered for the HP-SAXS cell detailed in the next section:
(i) A combination of a Poulter seal and retaining screw caps for the diamond windows to obtain two independent seats, each sealed by a Bridgeman seal on either end of the X-ray path.
(ii) Sample changing from a port perpendicular to the beam path using a threadless bolt-lock closure mechanism that can be opened and resealed using only hand pressure.
(iii) Multiple jackscrews to seal the diamond seating plugs, instead of closure nuts that require large torque wrenches for removal. These only need to be removed to change diamonds or make a repair.
(iv) Enhancing the cell rating to 700 MPa.
(v) Compatability with an in-line vacuum on either side of each diamond window.
(vi) Adjustability of the sample + pressurization fluid path length using metal shims. This allows for optimization of a range of X-ray energies and maximum pressures.
(vii) The ability to swap the diamond and steel seating plug assemblies quickly in case of cell failure.
3. SAXS cell design
3.1. SAXS cell body
Fig. 1 ▸ shows an overview of the SAXS cell and Table 1 ▸ sets out the specifications. The SAXS cell body (C in Fig. 1 ▸) is machined from a block of heat-treated 17-4 PH 1025 stainless steel, which is a precipitation-hardened martensitic steel. This alloy provides a good combination of high strength, corrosion resistance and machinability.
Figure 1.

A 3D half section A–A of the full cell is shown on the right. A is the internally water-cooled temperature control jacket, B the removable top pressure-seal assembly, C the stainless steel SAXS cell body, D the carbide through-rods that secure assembly B against an upward force when the cell is pressurized, E the diamond seat assembly (Fig. 2), F the specimen in a removable plastic insert suspended from the sample support column (Fig. 3) and G the diamond X-ray window. A port to the reverse side of the cell (not shown) is plumbed to the high-pressure pump to pressurize fluid in the space between the diamond windows. The assembly shown is about 15 cm tall. X-rays are incident along the arrow to G.
Table 1. HP-SAXS cell specifications.
| Cap-to-cap distance | 3.68 mm |
| Sample path length | 3–3.5 mm |
| Sample volume | 40 µl |
| Volume of pressurizing water | 2.4 cm3 |
| Aperture diameter | 1.5 mm |
| Aperture opening angle | 26° |
| Diamond diameter | 4 mm |
| Diamond thickness | 0.5 mm |
| Maximum operating pressure | 700 MPa |
| Maximum q at 14 keV (this work) | 0.7 Å−1 |
The SAXS cell body was designed for a maximum operating pressure of 700 MPa. Due to the complexity of the cell geometry and resource limitations, it was not possible to conduct destructive fatigue testing or meaningful quantitative failure calculations on the cell body, with the exception of the diamond windows (supporting information). The pressure limit was set by the manufacturer, on the basis of experience with their product line and of industry standard practice for high-pressure construction. The cell was pressurized several times to the 680 MPa limit of our available pump with no indication of failure. All the data in this paper were taken with a pressurization system capable of up to 400 MPa.
Fig. 2 ▸ details the diamond seat assembly. When the two assemblies are in place in the SAXS cell body, the inner cap-to-cap distance is 3.68 mm; this entire space is filled with water to the desired pressure. Annular metal washers placed against surface D in Fig. 2 ▸ may be used to increase this inner cap-to-cap distance. For the data described in this paper, washers were not used. The sample is held in a plastic cell (Fig. 3 ▸) suspended from the removable specimen cell and top pressure-seal assembly (B in Fig. 1 ▸). The sample thickness used in this paper was 3.2 mm, but a sample thickness of up to 3.5 mm can be accommodated without the use of annular washer shims.
Figure 2.

Detail of the diamond window seat assembly. The pusher plate, A, screws into the SAXS cell body. Six jack screws, B, are threaded through six holes in A to exert forces on the diamond seat, C. The surface, D, bottoms out against a complementary surface in the SAXS cell body as a hard metal-to-metal contact. Annular shimming washers (not shown) can be added to change this distance. The urethane O-ring, F, smaller diameter taper of C and brass retaining ring, E, form a Bridgman seal against the high-pressurization fluid in the cell. The diamond X-ray window, G, is held against the diamond seat, J, by a screw-on cap, H.
Figure 3.
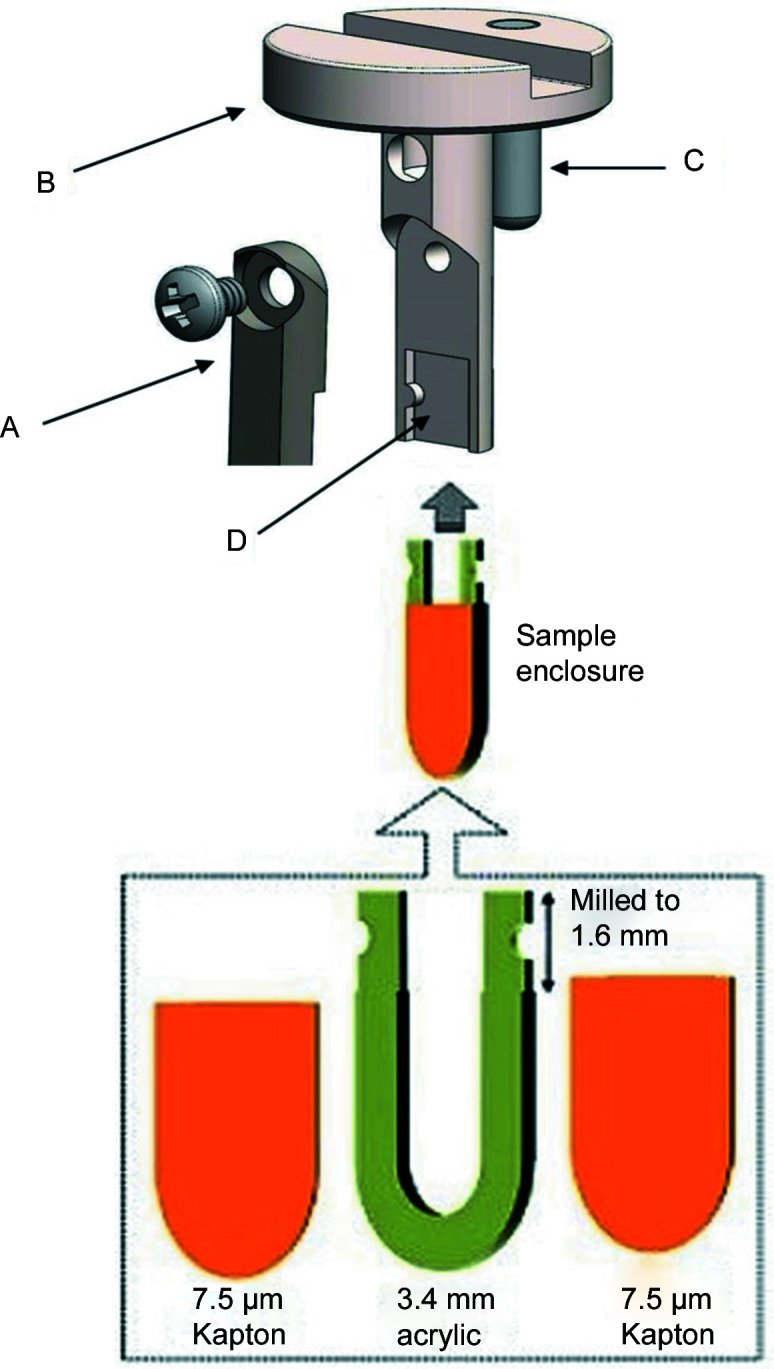
Samples are held in disposable plastic sample inserts that are suspended from a steel sample arm, B. The sample enclosure fits into a milled slot, D, in the arm and is held in place by a swinging cover, A, secured with a screw. The sample arm assembly fits into the SAXS cell body. A locating pin, C, positions the assembly so the sample enclosure face is perpendicular to the X-ray beam axis.
3.2. Sample-holder assembly
The sample is held in a disposable acrylic polymethyl methacrylate (PMMA) sample chip that is suspended from a steel sample-holder arm – see Fig. 3 ▸. Once the top pressure-seal assembly (B in Fig. 1 ▸) is removed, the sample-holder assembly is lifted out of the SAXS cell body with a magnet. The disposable plastic sample enclosure snaps into a milled indent in the suspending arm (D in Fig. 3 ▸). The plastic sample enclosure is then held firmly in place by tightening the screw of a swinging arm (A in Fig. 3 ▸).
The plastic sample enclosures used in this paper were laser cut from 3 mm thick PMMA sheet (CLAREX; Astra Products Inc., Baldwin, New York, USA), with 7.5 µm thick Kapton film windows (Chemplex Industries, Palm City, Florida, USA) being glued directly to the PMMA using cyanoacrylate glue (Brush-on Superglue; Loctite, Rocky Hill, Connecticut, USA) or five-minute two-component epoxy (Devcon; ITW Polymers Adhesives North America, Danvers, Massachusetts, USA). Care must be taken to keep the Kapton taut and the layer of glue very thin. The swinging arm/screw clamp arrangement allows a range of thicknesses of the plastic sample enclosure.
The sample is loaded into the plastic sample cell using a syringe so as to fill about half the volume of the cavity formed by the acrylic cell and Kapton windows. A typical sample load is 40 µl. The sample is then isolated from the surrounding pressure medium (water) using a plug of vacuum grease (High-Vacuum Grease; Dow Corning, Midland, Michigan, USA) or optical microscope immersion oil. The grease or oil plug acts as a freely moving piston to equilibrate the sample to the surrounding water pressure. Parafilm-M (AFNA, Oshkosh, Wisconsin, USA) has also been used successfully for this purpose in a high-pressure SAXS cell developed by the European Synchrotron Radiation Facility (ESRF, Grenoble, France) sample service (Skouri-Panet et al., 2006 ▸).
Sample loading thus involves syringing ca 30–40 µl of sample into the cell and capping with the grease or oil plug. Samples may be preloaded and stored until X-rayed.
3.3. Diamond pressure-window assemblies
The high-pressure X-ray windows are cut from single-crystal diamond. Natural, synthetic CVD (Wang, Meng et al., 2012 ▸) and high-pressure–high-temperature (HPHT) diamond windows were tested. In all cases the diamonds were cut into thin circular plates in the range of 0.5–1 mm thick. Most data in this paper, unless otherwise noted, were collected with HPHT diamonds (Almax EasyLab, Diksmuide, Belgium). Both diamond windows are type IIa (with Raman ultra-low fluorescence), oriented along the (100) axis normal to the flat face of the diamond. These circular diamond plates had acceptably consistent and small background scatter, i.e. they showed few scattering artifacts and no evidence of Kossel lines in the SAXS region. We use two pairs of windows. The data in this paper were collected with a pair of HPHT diamond plates, ca 0.5 mm thick by 4 mm diameter, and this set is intended for use up to 400 MPa. A second set of ca 1 mm thick plates cut from natural diamonds are intended for use to 700 MPa to provide sufficient safety margins (Fig. S1).
Mounting the diamonds on the flats of the conical steel plugs (Fig. 2 ▸) requires that the mating surfaces be polished to a flatness of no more than two fringes when each surface is viewed pressed against an optically flat glass plate. The polishing and mounting procedures are described in detail by Ando (2009 ▸) and Ando et al. (2008 ▸).
Data and calculations that led to the selection of these diamond thicknesses are described in the supporting information.
3.4. Pressure-control system
The pressure-control system is shown schematically in Fig. 4 ▸. The high-pressure pump is a Barocycler HUB440 Pressure Generator (PBI, South Easton, Massachusetts, USA). It is rated to 400 MPa and has been installed with standard high-pressure plumbing (High Pressure Equipment, Erie, Pennsylvania, USA). Pressure was monitored using two commercial transducers (Stellar Technology Inc., Amherst, New York, USA; Sensotec/Honeywell International Inc., Charlotte, North Carolina, USA).
Figure 4.

The pressure system controls and plumbing. The pressure generator (HUB440/880) supplies high-pressure degassed water to the pressure cell. The temperature is monitored by an RTD inserted into the block. The isolation valve permits water in the pressure generator to be recharged, when necessary, without removing pressure on the sample. The in-hutch control laptop is operated via remote desktop software.
Data from the pressure generator are acquired using the PBI HUB control software which has been developed using the LabView platform from National Instruments (Austin, Texas, USA). Connection to the HUB440 is via a USB cable to a laptop in the X-ray hutch which, in turn, is controlled from outside the hutch via a remote desktop connection.
Deionized degassed water is used as the pressurization fluid. The cell is flushed with fresh pressurization water after each experimental run. We found that the use of degassed water is essential to maintain stable backgrounds during X-ray exposure. A commercial continuous-flow degassing system (Systec Stand Alone MINI Vacuum Degassing System; Avantor Performance Materials, Center Valley, Pennsylvania, USA) was used to condition all water used for pressurization.
3.5. Temperature control system
The temperature of the SAXS cell is controlled by enveloping it in a water-cooled aluminium jacket (Fig. 1 ▸). The jacket is designed to come apart as two pieces (by the removal of four screws); the jacket may then be removed without requiring that the SAXS cell body itself be moved from its position on the X-ray beamline. The jacket encloses all four vertical sides of the cell (Fig. 5 ▸). An additional internally water-cooled aluminium plate (Fig. 5 ▸, G) is placed between the bottom of the cell and a mounting plate made of insulating fiberglass-epoxy G10 material (not visible). Thus, the SAXS cell body is covered on five sides by high-thermal-conductivity temperature-regulated aluminium. A temperature-controlled circulating water cooler is used to set the temperature. The 15 mm thick fiberglass-epoxy plate serves to thermally insulate the bottom cooling plate from the motion stages on which the SAXS cell sits (Fig. 5 ▸, G and E). The entire assembly is then encased in a plastic foam anti-condensation sleeve (Fig. 5 ▸, J). The cell is a large chunk of steel with considerable thermal mass. Hence, temperature changes may take tens of minutes to reach thermal equilibrium.
Figure 5.

Front and back views of the high-pressure sample cell. The removable pressure seal assembly (A) is held in place by carbide through-rods (D). The SAXS cell body (B) is enclosed on five sides by an aluminium water jacket (C, G) fed by chilled water via standard tube fittings (orange, shown unconnected). High-precision positioning under vacuum is accomplished using a low-profile X–Z motor stage (H, I) coupled with flexible bellows (F) coupled to KF-flange adaptors (E). The lower image shows a back view of the cell with insulating foam (J) and tube (K) to the high-pressure pump. The top foam cover is not shown.
The temperature of the SAXS cell body is monitored using an RTD-850 resistance temperature detector (RTD) sensor (Omega Engineering, Norwalk, Connecticut, USA) which comes in the form of a miniature threaded housing that fits into a No. 8-32 threaded 3/16′′ hole on the exterior of the metal block above the pressure pump inlet (Fig. 4 ▸, and Fig. 5 ▸, K). The sample cell, stage and bracket combination has been tested from 0 to 80°C under vacuum and pressurized. Temperature equilibration required approximately 20 min to warm from 25 to 40°C. Warming from a chilled state (10°C) to 25°C required only 15 min. Detailed temperature series are given in Fig. S3 of the supporting information. Temperatures above 45°C required a high-capacity chiller with a slower inherent temperature response. Once the cell body has reached thermal equilibrium, the temperature difference between the SAXS cell body and the specimen (as determined with tests of a monitoring RTD at the sample position) ranges from −0.2 to 0.8°C (supporting information).
The temperature-control jacket has integral KF25 flanges made in such a way as to press vacuum-tight O-rings against the outside flats of the SAXS cell body around the jack-screw assemblies. Thus, tightening the water-cooled jacket around the SAXS cell body automatically encases the X-ray beam path with vacuum-tight seals. The KF25 flanges mate to KF25 to KF40 conical adapters on either side of the cell (Fig. 5 ▸, E). These in turn mate to metal vacuum bellows (KF50EWB; Kurt J. Lesker Company, Jefferson Hills, Pennsylvania, USA) that are collinear with the X-ray beam axis (Fig. 5 ▸, F). The bellows allow several millimetres of motion in both directions perpendicular to the X-ray beam axis so as to position the X-ray beam on the diamond X-ray windows without moving the upstream or downstream X-ray optics. This arrangement allows vacuum X-ray beamline paths to be used both upstream and downstream of the SAXS cell to reduce air scatter, i.e. the diamond pressure windows separate the high-pressure water from vacuum. The system typically achieves a vacuum of better than 1 mTorr (1 Torr = 133.322 Pa).
4. CHESS beamline ID7A layout and characteristics
4.1. SAXS cell positioning
All diamonds exhibit some SAXS whose intensity varies with position on the diamond. A primary requirement was to obtain the lowest background SAXS from the diamond windows by scanning the X-ray beam across the windows to find the optimum spot. This was accomplished by mounting the cell on mechanical stages that allow micrometre-scale movements horizontally and vertically perpendicular to the X-ray beam over distances of several millimetres (Fig. 5 ▸, H and I). This in turn required the use of very stable low-profile translation stages that could withstand the torsional forces of the connecting pipes with minimal deviation (ATS100-50 and AVS100-13; Aerotech Inc., Pittsburgh, Pennsylvania, USA). The HP-SAXS cell is attached to the ATS100 x-stage with the G10 adaptor plate, which also serves as a thermal insulator. The low profile was a requirement of the ID7A beamline where the beam axis is only about 15 cm above the hutch optical table.
4.2. X-ray optical layout
Fig. 6 ▸ shows a schematic diagram of the essential X-ray optics of the system as installed on the CHESS ID7A beamline (a number of optical components typical of a synchrotron beamline are not shown, e.g. attenuators, beam intensity monitors etc.). A Cornell compact undulator source (manufactured by KYMA, Trieste, Italy) feeds a multilayer monochromator to produce 1.5% energy bandwidth X-rays (Temnykh et al., 2016 ▸). Table 2 ▸ gives typical X-ray beam parameters on CHESS ID7A. The hutch beam path consists of an Mo foil attenuator system (not shown), slit 1, X-ray shutter, X-ray intensity ion chamber, slit 2 and guard slit 3 prior to the SAXS cell, all in vacuum (<1 mTorr). Guard slit 3 (Advanced Design Consulting USA Inc., Lansing, New York, USA), at 36.2 m from the undulator source, serves to remove upstream parasitic scattering. The entire flight path, from just upstream of slit 2 all the way to the detector surface, is under vacuum. The KF flanges built into the SAXS cell thermal jacket mate to the upstream and downstream vacuum beam-path pipes. Operating conditions varied during the CHESS-U commissioning period: precise beam fluxes and wavelengths for particular experiments are given in the sections that follow. Generally, X-rays of 14 keV (0.88 Å) were used with flux ranging from 1.2 × 1012 to 7.8 × 1012 photons s−1 in a beam area of 250 × 250 µm. The storage ring current was 50–100 mA of positrons at 6 GeV in continuous top-up mode. CHESS is ultimately expected to reach 200 mA for routine running. Older experiments were performed using a Dectris 100KS detector (Dectris, Baden, Switzerland) with a q range of 0.01–0.3 Å−1 at 1500 mm distance. Final experiments utilized a Dectris EIGER X 4M detector with a pixel size of 75 × 75 µm, an active area of 155.2 × 162.5 mm (2070 × 2167 = 4 485 690 pixels), a distance of 1700 mm and a q range of 0.01–0.7 Å−1. The wavevector is defined as q = (4π/λ)sinθ, where 2θ is the total scattering angle and λ is the wavelength of the incident radiation.
Figure 6.

The simplified optical layout of CHESS ID7A1. Monochromatic X-rays are produced from an undulator source using a pair of multilayer mirrors (1.5% bandpass). Slit 1 effectively controls the source size while slit 2 is used to define the beam diameter. A vertical focusing mirror is located between slits 1 and 2. Slit 3, placed close to the sample cell, serves to clean up parasitic scatter from upstream components. All components downstream of slit 2 are under vacuum.
Table 2. Beamline characteristics for the CHESS ID7A station.
| Source | Cornell compact undulator |
|---|---|
| Beam size at SAXS cell | 250 × 250 µm |
| Monochromator | Dual Rh/B4C multilayers |
| Flux | 8 × 1012 photons s−1 at 0.88 Å |
| Wavelength range | 0.83–1.55 Å |
| Energy range | 8–15 keV |
| Energy bandwidth | 1.5% |
| Detector model | Dectris EIGER X 4M |
| Detector details | In vacuum |
4.3. Calibration and software integration
Optical alignment, optimization and data collection were performed using SPEC (Certified Scientific Software, Cambridge, Massachusetts, USA) macro commands on the workstation located outside the ID7A hutch. The 2D SAXS images were azimuthally integrated about the beam center and normalized by the transmitted intensities using standard image-correction procedures in the BioXTAS RAW software package (Version 1.6.4; Hopkins et al., 2017 ▸).
Silver behenate (Alpha Aesar, Ward Hill, Massachusetts, USA), C22H43AgO2, was used for calibration. It forms a lamellar phase with spacing d 001 = 58.380 Å, resulting in a series of equally spaced SAXS peaks with a first order at 0.1076 Å−1. The calibration was performed using multiple rings of the silver behenate standard, resulting in a sample-to-detector distance of 1680 mm. A CHESS-assembled molybdenum beam stop with a built-in VTS3085 photodiode (Excelitas Technologies, Waltham, Massachusetts, USA) was used to monitor the transmitted flux. This flux is automatically recorded into a data file during the X-ray exposure.
5. Performance and results
5.1. Experimental setup and protocol
Fresh degassed water is flushed through the entire pressurization fluid path before the start of the experiment and after each pressure run. The HUB440 Pressure Generator software enables water flow through the tubing at ambient pressure. This is employed along with opening of the vent button to ensure that there are no air bubbles inside the chamber before the pressure experiment begins (shown on the top of the cell in red in Figs. 1 ▸ and 4 ▸, and in white in Fig. 5 ▸). In general, to start an experiment it is advisable to first pressurize to about 5–10 MPa to allow settling of plastic cell components before making an ambient-pressure measurement (see Section 5.2 for a discussion of this phenomenon). HP-SAXS data were collected on the CHESS ID7A beamline (Acerbo et al., 2015 ▸). The samples were centrifuged at 5000 r min−1 for 10 min at 4°C before loading into the internal sample cell using a syringe or a pipette with a gel-loading tip. Sample-cell path lengths were 1.5 or 3.5 mm, as noted in the text.
The amount of equilibration time needed for any given protein to respond fully to pressure is an unknown. In these experiments we allowed 5–10 min, checking the profiles periodically for signs of change. The protein standard here, glucose isomerase, is not expected to show any significant conformational or oligomeric state changes under modest pressure (Banachowicz et al., 2009 ▸). Because radiation damage in a static sample cell is a potentially confounding factor, careful damage monitoring and the use of multiple samples may be necessary to determine a final data-collection protocol.
While BioSAXS on static samples has been performed successfully for many years prior to the use of flow cells, most current BioSAXS users may be unfamiliar with the limitations of static data collection. At the same time, new statistical tools have been developed to help detect radiation damage. For ambient BioSAXS, it is accepted practice to collect multiple sequential images and employ statistical comparisons using CORMAP (Franke et al., 2015 ▸) or a similar algorithm to determine if any damage-induced change has taken place. Systematic nonlinearity of the Guinier plot and the occurrence of upward drift in the radius of gyration are also indicators of damage. Since high-pressure BioSAXS presents special challenges beyond normal practice, we recommend that users first characterize their samples with standard ambient SAXS to estimate the maximum acceptable dose and to produce quality baseline profiles for later comparison. These undamaged profiles can then be compared with final ambient profiles collected on samples that have been exposed at higher pressures. The strategy of collecting a final ambient data set is only meaningful in cases where pressure-induced changes are reversible, and consequently, establishing reasonable dose limits is an important pre-requisite for data collection. In the tests we present here, we have also employed the practice suggested by Ando et al. (2008 ▸) of inserting waiting periods between exposures to allow damaged material to diffuse away from the beam. The precise durations of the data-acquisition and waiting times need to be determined on a case-by-case basis, which will necessarily involve expending more sample than a conventional BioSAXS experiment.
5.2. Performance
Glucose isomerase (GI), a 173 kDa tetrameric protein, has seen widespread use as a BioSAXS standard due to its availability, stability and resistance to radiation damage (Kozak, 2005 ▸; Hopkins & Thorne, 2016 ▸). Small-angle neutron scattering (SANS) measurements in solution have revealed that GI shows no observable structural changes at modest pressures of up to 150 MPa (Banachowicz et al., 2009 ▸), so we chose it as a control for assessing sample-cell performance and developing the protocol. Unlike the commercially supplied samples used in previous studies (which have become difficult to obtain), the GI used here was expressed and purified with and without the His-tag. Complete details, including buffer composition, are given in the supporting information. The radius of gyration R g measured for the His-tag-containing preparation using standard ambient size-exclusion chromatography SAXS (34.1 ± 0.1 Å) is in the upper range of typical values reported for the protein: 32–34 Å (see, for example, Small Angle Scattering Biological Data Bank entry SASDAK6; Valentini, 2012 ▸). Comparison with R g values calculated from the inverse Fourier method via P(r) gives consistently smaller values. The His-tag-free preparation shows a lower R g value (29 Å) and signs of repulsive concentration effects in the Guinier plot at the high concentration (17.9 mg ml−1) used in this study. The presence or absence of a His-tag sequence affects the net charge and may contribute to different concentration effects. The samples in this study have not been extrapolated to infinite dilution.
To assess the contribution of instrumental background, we removed the high-pressure cell and replaced it with an unobstructed vacuum path to produce a ‘windowless background’ profile. We also measured the scattering contribution from the diamond windows alone by emptying the high-pressure cell of water, drying it and measuring the window scattering in situ (Fig. 7 ▸). The diamond background rises by a factor of about 40 from the baseline to the maximum at q min = 0.0146 Å−1. Faint rings are visible starting at q min = 0.11 Å−1, which are due to some residual silver behenate standard left in the dry cell by accident. The background in the buffer rises by only a factor of 8 from the baseline over the same q-space interval and thus satisfies our normal criterion on CHESS ID7A for low background (a factor of 10). Guinier analyses of the subtracted GI profiles (4.5 and 0.9 mg ml−1) are linear all the way to q min (Fig. S4, panels A and B) and give relative errors in R g (as estimated by the BioXTAS RAW software) of 1 and 0.3%, respectively. Despite the precision of the Guinier analysis, there is little significant data beyond q = 0.1 Å−1 for any wider-angle analysis at the highest dilution. For a 173 kDa protein at the X-ray flux and exposures used here, 0.9 mg ml−1 is thus near the practical lower concentration limit for experiments that rely on shape determination.
Figure 7.

Instrumental background levels relative to the sample signal for dilutions of glucose isomerase. The high-pressure cell was replaced with a vacuum pipe to obtain a ‘windowless background’ profile. Diamond windows were measured in situ by emptying the cell of its pressurization water and drying. The artifactual ring at q = 0.11 is some residual silver behenate standard. Guinier analysis of the 4.5 mg ml−1 sample has a relative error in R g of 0.3%, while analysis of the dilute sample (0.9 mg ml−1) yields a relative error of 1%.
Previous high-pressure studies using diamond windows have occasionally reported artifacts known as Kossel lines (Kossel et al., 1935 ▸). When present, these lines can increase and move with pressure, creating potential background subtraction problems. No artifacts of this type are observable in the background images or profiles of the diamonds chosen for this study up to 400 MPa (Fig. S5).
To test the limits of data collection close to the reported damage threshold, a series of exposures of a single GI sample (4.5 mg ml−1) were taken sequentially under the following conditions: 0, 10, 100, 0, 0, 200 and 0 MPa. At each pressure, the sample was exposed for a total of 5 s (50 × 0.1 s exposures) at 13.96 keV (0.888 Å) with 1.2 × 1012 photons s−1. In between exposures, the sample was allowed to rest with no X-ray exposure for 5 min. For a 0.15 cm sample (water) path, a single 5 s exposure of a 0.025 cm diameter X-ray beam deposits a dose of 44 kGy (Meisburger et al., 2013 ▸). A single pressure measurement is therefore close to the reported damage limit for unprotected GI of 66–75 kGy (Hopkins & Thorne, 2016 ▸). For every pressure case, the 50 × 0.1 s sequential exposures passed the CORMAP test (p-value threshold of 0.01) for progressive damage. It is important to point out that this criterion only tells us that the rate of radiation damage is small enough to be ignored during the course of the exposure. The algorithm is not capable of detecting damage that has already occurred prior to exposure. A subsequent 50 × 0.1 s series of exposures on the same sample may average without apparent damage, yet comparison with the previous 50-exposure profile may show detectable progressive damage. Radiation damage is ultimately cumulative. CORMAP should be understood as a tool for gauging allowable dose, not for ascertaining damage. Once the allowable dose is known, exposure times can be chosen so as to permit a sufficient number of exposures to explore different pressure conditions on a single sample. As is the case with pressure-induced conformational changes, the effects of pressure on the rate of radiation damage are unknown a priori.
Table 3 ▸ gives the radii of gyration R g for a single sample subjected to sequential exposures under different pressure conditions. The R g values are calculated by both Guinier analysis and the inverse Fourier P(r) method. The χ2 statistic,
compares two profiles, I 1 and I 0, with known standard deviations σ0 and σ1. The profiles were scaled relative to a single reference profile so as to minimize the χ2 value. The reference profile for all calculations is the initial 0 MPa profile.
Table 3. Progressive exposure and contrast change of glucose isomerase under pressure.
| Exposure | Guinier R g 4.5 mg | P(r) R g | χ2 | Scale | Guinier R g 22 mg | P(r) R g | χ2 | Scale | Calculated contrast |
|---|---|---|---|---|---|---|---|---|---|
| 0 MPa initial | 34.5 ± 0.1 | 33.5 | 0.0 | 1.02 | 35.0 ± 0.03 | 33.3 | 0.0 | 1.02 | |
| 10 MPa | 34.7 ± 0.1 | 33.7 | 0.88 | 1.0 | 35.1 ± 0.04 | 33.4 | 1.19 | 1.0 | |
| 100 MPa | 36.0 ± 0.2 | 33.9 | 2.28 | 0.83 | 34.4 ± 0.04 | 33.1 | 3.46 | 0.82 | 0.80 |
| 0 MPa after 100 MPa | 35.3 ± 0.1 | 33.9 | 1.12 | 1.0 | |||||
| 0 MPa after 5 min | 34.9 ± 0.1 | 33.8 | 0.81 | 0.99 | |||||
| 200 MPa | 38.1 ± 0.2 | 34.6 | 7.38 | 0.67 | 33.7 ± 0.05 | 33.1 | 8.27 | 0.68 | 0.64 |
| 0 MPa after 200 MPa | 39.3 ± 0.3 | 34.8 | 4.07 | 0.98 | |||||
| 300 MPa | 34.8 ± 0.06 | 33.3 | 16.4 | 0.53 | 0.52 |
In Fig. 8 ▸, some transient settling of the cell components is evident during the initial 10 MPa pressurization. This results in an initial 2% decrease in amplitude, with subsequent profiles returning to that decreased level after pressurization. Also reported in Table 3 ▸ is the scale of the profiles relative to this ‘settled’ ambient profile (after 10 MPa). The scale changes observed here cannot be due to changes in the diamond-to-diamond distance in this cell design since the jack screws hold the diamond seats rigidly in place (Fig. 2 ▸). Further, all profiles are normalized by transmitted beam intensity, so changes in the water path length would not be evident in the amplitudes of the scattering profiles. It is most likely that this transient scale shift observed in the initial 10 MPa data comes from the plastic sample-cell windows. The scale differences between ambient and high pressure can be attributed to changes in contrast between the protein and buffer: I(0) ∝ cν2(ρprotein − ρbuffer)2, where c = sample concentration, ν = protein specific volume and ρ = bulk electron density (3.3 × 1021 cm−3 for water and 4.4 × 1021 cm−3 for protein) (Ando et al., 2008 ▸; Wang et al., 2018 ▸). Since the observed protein compressibilities are often small in comparison with water, we will assume that only the water volume changes (Grindley & Lind, 1971 ▸; Kharakoz, 2000 ▸). If rV = V ambient/V pressure = the fractional change in water volume upon compression, then
The relative scales in Table 3 ▸ derived from minimizing χ2 to superimpose the profiles agree with the scales estimated from contrast to within a few percent.
Figure 8.

Kratky plot of successive exposures of a single GI sample under alternating ambient- and high-pressure conditions. The overall lower intensity of the high-pressure profiles (100 and 200 MPa) is consistent with a change in scattering contrast which assumes that the protein is much less compressible than water. After a one-time initial transient intensity decline of 2% in going from ambient to 10 MPa, all ambient profiles overlay. Each profile was collected after waiting 5 min with no exposure to help clear potentially damaged material from the region of the beam.
Comparing ambient (0 MPa) data sets only, the Guinier plots (Fig. 9 ▸) are linear, with the exception of the final profile (labeled ‘0 MPa after 200 MPa’ in the plot) which shows significant deviation. The radius of gyration R g for those sets shows some growth at ‘0 MPa after 100 MPa’ and some recovery at ‘0 MPa after 5 min’ before finally becoming significantly larger. The χ2 statistics for those profiles also remain near unity, suggesting there is no significant change in the overall profile above experimental noise. By ‘0 MPa after 200 MPa’ there is a significant increase in both R g and χ2, indicating damage.
Figure 9.

Guinier plots of ambient-pressure GI taken before and after pressurization. A plot of the deviation from linearity (labeled ‘rel error’) shows systematic errors indicative of radiation damage only after the final exposure ‘0 MPa after 200 MPa.’
While damage is below the statistical threshold when comparing sequential 0.1 s frames, the R g for the 5 s averages suggests that subtle damage does accumulate over time even with the 5 min waiting periods. The aggregate total dose on the samples was at least 5 × 44 kGy = 220 kGy before the damage became serious in the Guinier relative error plot (Fig. 9 ▸).
Wide-angle X-ray scattering (WAXS) data are particularly sensitive to small errors in background subtraction due to the fact that the protein scattering signal falls very rapidly with increasing q. Because it is necessary to remove and re-fill the sample cell to perform buffer subtraction in these HP-SAXS experiments, interpretation of the WAXS region of the scattering profile is potentially problematic. Even without removing the cell, incomplete pressure equalization between the exterior water and the sample may alter the scattering background in subtle ways.
For better resolution of wide-angle data, we examined concentrated GI samples using an EIGER X 4M detector collecting data out to q = 0.7 Å−1. The first sample, at 22 mg ml−1 (Banachowicz et al., 2009 ▸), was protected with 0.5 mM tris(2-carboxyethyl)phosphine using beamline collection parameters that were similar to the dilute example: energy = 14.14 keV (0.8771 Å) and flux = 1.4 × 1012 photons s−1 with 10 × 1 s exposures. In Fig. S6, the ambient scattering profile was fitted to an atomic model derived from PDB entry 1mnz equilibrated using molecular dynamics (Chen & Hub, 2014 ▸) and compared with a previously published profile deposited in the Small Angle Scattering Biological Data Bank (ID SASDAK6). No attempt is made here to model HIS-tag residues.
As in the dilute case, the intensity of protein scattering curves tends to decrease under pressure due to changes in contrast resulting from differences between protein and water compressibility (Ando et al., 2008 ▸). Table 3 ▸ gives relative scale factors between the ambient profiles and the pressurized profiles up to 300 MPa using χ2 minimization to superimpose the profiles. A previous SANS study was unable to detect any structural changes in GI at pressures of up to 150 MPa and out to q = 0.15 Å−1 (Banachowicz et al., 2009 ▸). The radii of gyration (Table 3 ▸) fall slightly under pressure and progressive exposure. At wider angles, shown as aligned Kratky plots in Fig. S7, there is a systematic small upturn with pressure. This deviation is readily evident in the increasing χ2 values of Table 3 ▸. Whether this effect can be attributed to pressure or to subtle radiation damage is not clear from these simple sequential exposures.
To help separate pressure effects from damage, we collected an initial undamaged reference profile for GI using a short exposure at ambient pressure (10 × 1 s exposures at 1.6 × 1011 photons s−1; 12 kGy). As before, a high sample concentration was used (17.9 mg ml−1) to improve the WAXS signal. This GI sample contained no His-tag. The samples exhibit some concentration effect at small angles, leading to smaller R g values than are typical (29 Å−1). The focus of this final experiment is on wide-angle behavior.
We collected data at 300 MPa, then back down to 0 MPa on the same sample. A fresh sample was also taken directly at 400 MPa and then at 0 MPa for comparison. Fig. 10 ▸ shows that GI is relatively unaltered at 300 MPa except for a subtle rise in the wide-angle signal. The entire profile, including the wide-angle portion, returns to the reference profile upon depressurization. In the figure, A and B denote different fresh samples. The labels ‘after 300’ and ‘after 400’ mean that those profiles were the final measurements taken after pressurization. In all cases, there was a 5 min waiting period between exposures.
Figure 10.

GI scattering profiles (Kratky plots) before, during and after pressurization. Fresh sample, labeled A and B, was used for each pressure measurement. An initial exposure, 0 MPa A (reference), serves as a reference for undamaged ambient-pressure protein. At 300 MPa, little change is visible in the profile except for a subtle rise at wide angle. Upon depressurization, the profile returns to match the reference. At 400 MPa, significant irreversible structural change occurs at small angles. Despite the small-angle damage, the rise at wide angle still appears to be reversible upon depressurization.
At 400 MPa, the situation is radically different. The small-angle region of the profile shows irreversible change suggestive of aggregation. The total irreversible decline in intensity visible in both the pressurized and final depressurized profiles suggests sample loss due to aggregation. Similarly to the 300 MPa case, the 400 MPa profile shows a subtle rise in the wide-angle region that appears to be fully reversible upon depressurization. This supports the hypothesis that the wide-angle behavior seen here is due not to radiation damage but to pressure. An increase in WAXS intensity in a Kratky plot is often attributed to flexibility or unfolding, though further experiments are necessary to characterize the phenomenon better.
6. Discussion and conclusions
The high-pressure SAXS cell presented here introduces novel design features to improve ease of use, stability, pressure and temperature range, and general performance. This has been achieved using a combination of HPHT diamonds, in-line vacuum, a patented bolt-lock mechanism (Pressure Biosciences Inc.), a high-stability motorized positioning mechanism and a temperature-control jacket. Disposable sample cells combine with the bolt-lock mechanism to make loading and unloading samples a relatively rapid process, while helping to improve the mechanical stability that is important for the reproducibility of scattering from dilute samples. Preliminary performance tests used the protein standard glucose isomerase to evaluate possible protocols for data collection. The presence of subtle cumulative radiation damage was found to be a potentially confounding factor for interpreting pressure effects. The widely used protocol for ambient BioSAXS in which successive exposures are compared for statistical similarity to determine the onset of damage cannot be casually applied multiple times without subtle cumulative damage. A suggested strategy of applying waiting times between exposures to dissipate damage shows some effect but is not reliable on the 5 min timescale in these studies. The recommended approach here is to pre-evaluate and stay below the maximum dose limit on each sample, collecting a final ambient exposure for comparison with the undamaged (pre-evaluated) result. Glucose isomerase shows little structural change below 300 MPa but becomes irreversibly damaged by pressure at 400 MPa.
Evaluation of the equilibration times necessary for pressure-induced changes is an additional protocol step that will require more detailed future studies involving a range of protein samples. Untangling the effects of radiation damage from pressure therefore requires special consideration beyond the normal practice for BioSAXS. Flow-cell technology would greatly reduce radiation damage concerns and allow for much more precise pressure comparisons. A system under development at CHESS using conventional high-pressure chromatography equipment will be presented in a future publication, though it is unclear how such technology can yet be extended to reach the extremes of pressure possible with the static sample high-pressure system presented here.
7. Related literature
For further literature related to the supporting information, see Field (2012 ▸), Henke et al. (1993 ▸), Holzapfel & Isaacs (1997 ▸) and Nielsen et al. (2012 ▸).
Supplementary Material
Additional experimental details and extra figures. DOI: 10.1107/S1600576720014752/ei5062sup1.pdf
SAXS datafile glucose isomerase 22mg/ml 0 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup2.txt
SAXS datafile glucose isomerase 22mg/ml 100 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup3.txt
SAXS datafile glucose isomerase 22mg/ml 200 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup4.txt
SAXS datafile glucose isomerase 22mg/ml 300 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup5.txt
SAXS datafile glucose isomerase 4.5 mg/ml 0 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup6.txt
SAXS datafile glucose isomerase 0.9 mg/ml 0MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup7.txt
Acknowledgments
We thank Professor Roland Winter from TU Dortmund for valuable discussions. We also thank Martin Novak for his valuable modifications and adaptations to the apparatus.
Funding Statement
This work was funded by National Science Foundation grant DMR-1829070 to Durgesh Rai, , , and Robert Miller. National Institute of General Medical Sciences grant 1-P30-GM124166-01A1 to Richard Gillilan and Qingqiu Huang. New York State Empire Development Corporation (NYSTAR) grant none. National Nanotechnology Coordinating Office grant NNCI-1542081.
References
- Acerbo, A. S., Cook, M. J. & Gillilan, R. E. (2015). J. Synchrotron Rad. 22, 180–186. [DOI] [PMC free article] [PubMed]
- Akasaka, K. (2015). High Pressure Bioscience: Basic Concepts, Applications and Frontiers, edited by K. Akasaka & H. Matsuki, pp. 707–721. Dordrecht: Springer Netherlands.
- Ando, N. (2009). PhD thesis, Cornell University, New York, USA.
- Ando, N., Chenevier, P., Novak, M., Tate, M. W. & Gruner, S. M. (2008). J. Appl. Cryst. 41, 167–175.
- Banachowicz, E., Kozak, M., Patkowski, A., Meier, G. & Kohlbrecher, J. (2009). J. Appl. Cryst. 42, 461–468.
- Bar-On, Y. M., Phillips, R. & Milo, R. (2018). Proc. Natl Acad. Sci. USA, 115, 6506–6511. [DOI] [PMC free article] [PubMed]
- Barstow, B., Ando, N., Kim, C. U. & Gruner, S. M. (2008). Proc. Natl Acad. Sci. USA, 105, 13362–13366. [DOI] [PMC free article] [PubMed]
- Bartlett, D. (2002). Biochim. Biophys. Acta, 1595, 367–381. [DOI] [PubMed]
- Black, S. L., Dawson, A., Ward, F. B. & Allen, R. J. (2013). PLoS One, 8, e73995. [DOI] [PMC free article] [PubMed]
- Boonyaratanakornkit, B. B., Park, C. B. & Clark, D. S. (2002). Biochim. Biophys. Acta, 1595, 235–249. [DOI] [PubMed]
- Bourges, A. C., Lazarev, A., Declerck, N., Rogers, K. L. & Royer, C. A. (2020). Biophys. J. 118, 2670–2679. [DOI] [PMC free article] [PubMed]
- Bridgman, P. (1914). J. Biol. Chem. 19, 511–512.
- Brooks, N. J., Ces, O., Templer, R. H. & Seddon, J. M. (2011). Chem. Phys. Lipids, 164, 89–98. [DOI] [PubMed]
- Brooks, N. J., Gauthe, B. L., Terrill, N. J., Rogers, S. E., Templer, R. H., Ces, O. & Seddon, J. M. (2010). Rev. Sci. Instrum. 81, 064103. [DOI] [PubMed]
- Caro, J. A. & Wand, A. J. (2018). Methods, 148, 67–80. [DOI] [PMC free article] [PubMed]
- Chen, P.-C. & Hub, J. S. (2014). Biophys. J. 107, 435–447. [DOI] [PMC free article] [PubMed]
- Czeslik, C., Luong, T. Q. & Winter, R. (2017). MRS Bull. 42, 738–742.
- Daniel, I., Oger, P. & Winter, R. (2006). Chem. Soc. Rev. 35, 858–875. [DOI] [PubMed]
- Decaneto, E., Suladze, S., Rosin, C., Havenith, M., Lubitz, W. & Winter, R. (2015). Biophys. J. 109, 2371–2381. [DOI] [PMC free article] [PubMed]
- Field, J. (2012). Rep. Prog. Phys. 75, 126505. [DOI] [PubMed]
- Franke, D., Jeffries, C. & Svergun, D. (2015). Nat. Methods, 12, 419–422. [DOI] [PubMed]
- Garcia, A. E. & Paschek, D. (2008). J. Am. Chem. Soc. 130, 815–817. [DOI] [PubMed]
- Grindley, T. & Lind, J. E. Jr (1971). J. Chem. Phys. 54, 3983–3989.
- Grötzinger, S. W., Alam, I., Ba Alawi, W., Bajic, V. B., Stingl, U. & Eppinger, J. (2014). Front. Microbiol. 5, 134. [DOI] [PMC free article] [PubMed]
- Hartmann, M., Kreuss, M. & Sommer, K. (2004). Cell. Mol. Biol. 50, 479–484. [PubMed]
- Heller, W. T., Urban, V. S., Lynn, G. W., Weiss, K. L., O’Neill, H. M., Pingali, S. V., Qian, S., Littrell, K. C., Melnichenko, Y. B., Buchanan, M. V., Selby, D. L., Wignall, G. D., Butler, P. D. & Myles, D. A. (2014). J. Appl. Cryst. 47, 1238–1246.
- Henke, B. L., Gullikson, E. M. & Davis, J. C. J. (1993). At. Data Nucl. Data Tables, 54, 181–342.
- Hnosko, J., San-Martin Gonzalez, M. F. & Clark, S. (2012). J. Dairy Sci. 95, 4851–4862. [DOI] [PubMed]
- Holzapfel, W. B. & Isaacs, N. S. (1997). Editors. High Pressure Techniques in Chemistry and Physics – A Practical Approach. Oxford University Press.
- Hopkins, J. B., Gillilan, R. E. & Skou, S. (2017). J. Appl. Cryst. 50, 1545–1553. [DOI] [PMC free article] [PubMed]
- Hopkins, J. B. & Thorne, R. E. (2016). J. Appl. Cryst. 49, 880–890. [DOI] [PMC free article] [PubMed]
- Ingr, M., Kutálková, E., Hrnčiřík, J. & Lange, R. (2016). J. Theor. Biol. 411, 16–26. [DOI] [PubMed]
- Jenkins, K. A., Fossat, M. J., Zhang, S., Rai, D. K., Klein, S., Gillilan, R., White, Z., Gerlich, G., McCallum, S. A., Winter, R., Gruner, S. M., Barrick, D. & Royer, C. A. (2018). Proc. Natl Acad. Sci. USA, 115, E8153–E8161. [DOI] [PMC free article] [PubMed]
- Kato, C. (2006). Methods in Microbiology, Vol. 35, edited by F. A. Rainey & A. Oren, pp. 733–741. Amsterdam: Elsevier.
- Kharakoz, D. P. (2000). Biophys. J. 79, 511–525. [DOI] [PMC free article] [PubMed]
- Koo, J., Erlkamp, M., Grobelny, S., Steitz, R. & Czeslik, C. (2013). Langmuir, 29, 8025–8030. [DOI] [PubMed]
- Kossel, W., Loeck, V. & Voges, H. (1935). Z. Phys. 94, 139–144.
- Kozak, M. (2005). J. Appl. Cryst. 38, 555–558.
- Meisburger, S. P., Warkentin, M., Chen, H., Hopkins, J. B., Gillilan, R. E., Pollack, L. & Thorne, R. E. (2013). Biophys. J. 104, 227–236. [DOI] [PMC free article] [PubMed]
- Mentré, P. & Hui Bon Hoa, G. (2001). Int. Rev. Cytol. 201, 1–84. [DOI] [PubMed]
- Nielsen, S. S., Møller, M. & Gillilan, R. E. (2012). J. Appl. Cryst. 45, 213–223. [DOI] [PMC free article] [PubMed]
- Oger, P. M. & Jebbar, M. (2010). Res. Microbiol. 161, 799–809. [DOI] [PubMed]
- Paschek, D., Gnanakaran, S. & Garcia, A. E. (2005). Proc. Natl Acad. Sci. USA, 102, 6765–6770. [DOI] [PMC free article] [PubMed]
- Peterson, R. W. & Wand, A. J. (2005). Rev. Sci. Instrum. 76, 094101. [DOI] [PMC free article] [PubMed]
- Poulter, T. C. (1932). Phys. Rev. 40, 860–871.
- Pressl, K., Kriechbaum, M., Steinhart, M. & Laggner, P. (1997). Rev. Sci. Instrum. 68, 4588–4592.
- Prigozhin, M. B., Zhang, Y., Schulten, K., Gruebele, M. & Pogorelov, T. V. (2019). Proc. Natl Acad. Sci. USA, 116, 5356–5361. [DOI] [PMC free article] [PubMed]
- Roche, J., Royer, C. A. & Roumestand, C. (2019). Methods Enzymol. 614, 293–320. [DOI] [PubMed]
- Sarupria, S., Ghosh, T., García, A. E. & Garde, S. (2010). Proteins Struct. Funct. Bioinform. 78, 1641–1651. [DOI] [PubMed]
- Schummel, P. H., Anders, C., Jaworek, M. W. & Winter, R. (2019). ChemPhysChem, 20, 1098–1109. [DOI] [PubMed]
- Silva, E., Ricardo, Y. & Raul, G. J. (2015). RSC Adv. 5, 70005–70009.
- Silva, J. L. & Weber, G. (1993). Annu. Rev. Phys. Chem. 44, 89–113. [DOI] [PubMed]
- Skou, S., Gillilan, R. E. & Ando, N. (2014). Nat. Protoc. 9, 1727–1739. [DOI] [PMC free article] [PubMed]
- Skouri-Panet, F., Quevillon-Cheruel, S., Michiel, M., Tardieu, A. & Finet, S. (2006). Biochim. Biophys. Acta, 1764, 372–383. [DOI] [PubMed]
- Spain, I. L. & Paauwe, J. (1977). High Pressure Technology, Vol. 1, Equipment Design, Materials, and Properties. Boca Raton: CRC Press.
- Takai, K., Nakamura, K., Toki, T., Tsunogai, U., Miyazaki, M., Miyazaki, J., Hirayama, H., Nakagawa, S., Nunoura, T. & Horikoshi, K. (2008). Proc. Natl Acad. Sci. USA, 105, 10949–10954. [DOI] [PMC free article] [PubMed]
- Temnykh, A., Dale, D., Fontes, E., Lyndaker, A., Li, Y., Ruff, J., Revesz, P. & Woll, A. (2016). AIP Conf. Proc. 1741, 020003.
- Valentini, E. (2012). SASDAK6 – Glucose Isomerase, https://www.sasbdb.org/search/?q=sasdak6.
- Vass, H., Black, S. L., Flors, C., Lloyd, D., Ward, F. B. & Allen, R. J. (2013). Appl. Phys. Lett. 102, 154103.
- Vezzi, A., Campanaro, S., D’Angelo, M., Simonato, F., Vitulo, N., Lauro, F., Cestaro, A., Malacrida, G., Simionati, B., Cannata, N., Romualdi, C., Bartlett, D. H. & Valle, G. (2005). Science, 307, 1459–1461. [DOI] [PubMed]
- Wang, C., Lin, Y., Bougie, D. & Gillilan, R. E. (2018). Acta Cryst. D74, 727–738. [DOI] [PMC free article] [PubMed]
- Wang, S., Meng, Y., Ando, N., Tate, M., Krasnicki, S., Yan, C., Liang, Q., Lai, J., Mao, H., Gruner, S. M. & Hemley, R. J. (2012). J. Appl. Cryst. 45, 453–457. [DOI] [PMC free article] [PubMed]
- Wang, S. T., Tate, M. W. & Gruner, S. M. (2012). Biochim. Biophys. Acta, 1820, 957–961. [DOI] [PubMed]
- Winter, R. (2002). Biochim. Biophys. Acta, 1595, 160–184. [DOI] [PubMed]
- Winter, R. (2019). Annu. Rev. Biophys. 48, 441–463. [DOI] [PubMed]
- Winter, R. & Jeworrek, C. (2009). Soft Matter, 5, 3157–3173.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional experimental details and extra figures. DOI: 10.1107/S1600576720014752/ei5062sup1.pdf
SAXS datafile glucose isomerase 22mg/ml 0 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup2.txt
SAXS datafile glucose isomerase 22mg/ml 100 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup3.txt
SAXS datafile glucose isomerase 22mg/ml 200 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup4.txt
SAXS datafile glucose isomerase 22mg/ml 300 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup5.txt
SAXS datafile glucose isomerase 4.5 mg/ml 0 MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup6.txt
SAXS datafile glucose isomerase 0.9 mg/ml 0MPa pressure. DOI: 10.1107/S1600576720014752/ei5062sup7.txt