

Abstract
Background
Glioblastoma (GBM) is a highly aggressive primary brain tumor. Currently, the suggested line of action is the surgical resection followed by radiotherapy and treatment with the adjuvant temozolomide, a DNA alkylating agent. However, the ability of tumor cells to deeply infiltrate the surrounding tissue makes complete resection quite impossible, and, in consequence, the probability of tumor recurrence is high, and the prognosis is not positive. GBM is highly heterogeneous and adapts to treatment in most individuals. Nevertheless, these mechanisms of adaption are unknown.
Recent findings
In this review, we will discuss the recent discoveries in molecular and cellular heterogeneity, mechanisms of therapeutic resistance, and new technological approaches to identify new treatments for GBM. The combination of biology and computer resources allow the use of algorithms to apply artificial intelligence and machine learning approaches to identify potential therapeutic pathways and to identify new drug candidates.
Conclusion
These new approaches will generate a better understanding of GBM pathogenesis and will result in novel treatments to reduce or block the devastating consequences of brain cancers.
Keywords: artificial intelligence, biomarkers, cancer stem cells, gap junctions, tunneling nanotubes (TNTs)
1. INTRODUCTION
Malignant tumors affecting the central nervous system are one of the most feared types of cancer. Less than 2% of all cancer aggressively affects the brain.1, 2 According to the World Health Organization (WHO), gliomas are classified in astrocytomas, oligodendrogliomas, oligo‐astrocytomas, and glioblastoma (GBM) based on the histopathological and the clinical features.3 GBM arises within the brain parenchyma, especially in the dura mater and calvarium, which is thought to seed the extracranial space with tumor cells.4 GBM is well known as one of the most aggressive, frequent, and devastating types of glioma, corresponding to 52% of all primary brain tumor cases.5 GBM incidence increases of 0.7% every 11 years,6 and it is greater in men (roughly 7.7 per 100,000) than in women (5.61 per 100,000), making this type of cancer a serious health problem.1
The most typical characteristics of GBM are robust angiogenesis, intense resistance to apoptosis, necrogenesis, genomic instability, heterogeneity, and adaptation to treatment.7, 8, 9 Formerly, GBM tumors were classified based on the morphological appearance.10 When these tumors become more aggressive, the morphology of the cells changes, and they are less differentiated under a microscope.11 Currently, GBM classification is based on both phenotype and genotype expression.12 GBM is recognized as a diffuse astrocytoma, and it could be considered as isocitrate dehydrogenase (Idh)‐wildtype type of tumor, also known as a primary GBM (90% of the cases) or Idh‐mutant tumor identified as secondary GBM (10% of the cases).3 Besides the numerous advances in biomedical research, surgical techniques, diagnosis, and treatment, both types of GBM have a poor prognosis and 14.6 months of survival rate. GBM has a dramatical annual incidence of 3.19 per 100,000.7 Currently, surgical resection combined with radiotherapy plus concomitant treatment with the adjuvant temozolomide (TMZ) is the standard care in patients younger than 70 years old with newly diagnosed GBM.13, 14, 15, 16 Extracranial metastasis in GBM patients is extremely rare (<2% of the cases) due to the presence of unique barriers such as the blood‐brain barrier (BBB), the dura mater, and the thickened basement membrane of the blood vessels, which prevent the hematogenous and lymphatic spread of intracranial tumor cells.17 Also, lack of extracellular matrix proteins prevents tumor invasion in the surrounding connective tissue and, consequently, hematogenous and lymphatic spread.2 However, genetic and molecular factors that predict extracranial invasion remain unclear and require further investigation.
Only recently, tumor heterogeneity becomes a hallmark of GBM, and it is influenced by genetic, epigenetic, and metabolic biomarkers. The unique cellular composition of these kinds of tumor gives them the capacity to become highly infiltrative and invasive, to have nuclear atypia, to increase proliferation, and to generate microvascular hyperplasia and necrotic foci.10, 18 Specifically, GBM stem cells (GBSCs) have an important role for survival and adaptation. GBSCs are one of the major contributors to the molecular and cellular heterogeneity observed in GBM. GBSCs are capable of self‐renewal and are responsible for therapeutic resistance and tumor recurrence.19, 20, 21, 22 The hierarchical GBSC cancer model proposes that tumor arises from GBSCs generated by mutations in either normal embryonic stem cells or by the dedifferentiation of already differentiated cells, resulting in uncontrolled growth and propagation.23 After surgical resection of the tumor and radio/chemotherapy therapy, the remaining GBSCs from the border of the tumor can repopulate the tumor, suggesting the presence of aggressive GBSCs at the border or infiltrated into healthy tissue.22, 24, 25 A recent study identified that both GBM and matched GBSCs have a recurrent copy number of genetic alterations in chromosome 7 polysomy, chromosome 10 monosomy, and chromosome 9p21 deletions, which are typical features of primary GBM, essential for gliomagenesis.26 These data suggests a condition of strong genomic heterogeneity in GBM as well as in GBSCs.26, 27
Genetics also play a key role in the GBM heterogeneity. Mutations can increase the resistance to standard radio and chemotherapy. One of the genetic alterations that have an important impact in GBM resistance to treatment involves the mutation of the epidermal growth factor receptor (Egfr) gene that occur in 36%–60% of primary GBM resulting in different modulation of EGFR signaling and related receptors.22, 28, 29 Also, the homozygous deletion of the cyclin‐dependent kinase inhibitor 2 A (Cdkn2 a) gene, which encodes the p16INK4a and p14ARF tumor suppressors, is more common in primary than in secondary GBM resulting in aberrant cell proliferation.30, 31, 32 Other important genetic alterations include loss of heterozygosity (LOH) of chromosome 10, which is present in up to 70% of primary GBM,31, 33 and mutations in the isocitrate dehydrogenase (Idh) gene resulting in DNA hypermethylation.22, 34 Furthermore, for primary GBM, a microRNA (miRNAs) study showed alterations in both genes: Tp53 and O6‐methylguanine DNA methyltransferase (Mgmt), which are major players in GBM pathogenesis, apoptosis, treatment resistance and survival.35 From the epigenetic point of view, tumor resistance to treatment has been studied through the expression of the DNA repair protein AlkB homolog 2 (Alkbh2) in GBM cell lines after TMZ exposure by real time polymerase chain reaction (qRT‐PCR). Alkbh2 is regulated by the p53 pathway, and it has an increased resistance to methylating agents like TMZ.36 Also, it was found that GBM cells overexpress Alkbh2 after TMZ exposure, enhancing the resistance to methylating agents, including TMZ.36 The susceptibility to cancer and treatment resistance has also been studied for the expression of the protein glutathione‐S‐transferase‐π (GSTP1) mutation in several kinds of tumors.37, 38, 39, 40 In GBM, expression of GSTP1 is highly heterogeneous however, its expression plays an important role in the protection of cells against damage from free radicals and also influences cytotoxicity to chemotherapeutics agents as TMZ.41, 42 Controversially, a study of 61 astrocytic tumor samples from grade II–IV showed that there were no differences in GSTP1 mRNA expression between diffuse astrocytomas, anaplastic astrocytomas, or GBM. Also, no difference was seen between secondary GBM before and after radio‐/chemotherapy, suggesting that glioma chemoresistance is probably multifactorial and GSTP1‐independent.38
As indicated above, the cellular heterogeneity and infiltration into healthy surrounding tissue makes complete surgical resection of the tumor almost impossible. Advances in genomic sequencing and transcriptomic profiling reveal heterogeneity of GBM, dividing it into molecular subtypes: proneural, neural, classical, and mesenchymal.14, 43 The proneural class is highly enriched with oligodendrocytes but not with astrocytes, whereas the classical GBM is strongly associated with astrocytes.43 The neural subtype is associated with oligodendrocytes and astrocytes differentiation but also is enriched with neuron products.43 The mesenchymal subtype is strongly associated with astroglial cells and tumor‐associated macrophages (TAMs).43, 44, 45 Each subtype of GBM is also characterized by different transcriptional profile including the gene expression of platelet‐derived growth factor receptor alpha (Pdgfrα), isocitrate dehydrogenase (Idh1), Egfr, and neurofibromatosis type I (Nf1).43 It was found that genes in normal cell types show a strong relationship between the GBM subtype and different neural lineages, where classical subtype showed a good response to therapy. Instead, the proneural subtype had the worst prognosis with high levels of Pdgfrα and Idh1 mutations.43, 46, 47 In the mesenchymal subtype, Nf1 expression was predominant.43, 45 Furthermore, in Idh wild‐type GBM Nf1 deficiency results in increased macrophage/microglia infiltration.48 In patients with mesenchymal GBM, it was identified that the transcriptional coactivator with PDZ‐binding motif (TAZ), is highly associated with the mesenchymal differentiation, silencing of TAZ in mesenchymal GBSCs decreased expression of mesenchymal markers, invasion, self‐renewal, and tumor formation.49, 50 The Egfr is also heterogeneously expressed in proneural, classical, and mesenchymal subtypes.51 But high levels of Egfr amplification were observed in 97% of classical GBM.43 In the case of the neural subtype of GBM, the expression of neuronal markers such as Neurofilament (NFL), Gamma‐Aminobutyric Acid Type A Receptor Alpha1 Subunit (GABRA1), Synaptotagmin 1 (SYT1), and Solute carrier family 12 member 5 (SCL12A50) were found.43 Overall, a detailed OMIC classification and consideration of tumor heterogeneity could provide new molecular targets to predict and treat GBM.
The high heterogeneity of GBM explains the poor response to treatment (radio‐ and chemotherapy), due to the presence of multiple cellular subpopulations with different sensitivities to treatment.44 On the other hand, cells from the same tumor may express different mutations or shown distinct phenotypic or epigenetic stages. For example, single cell RNA sequencing of GBMs as well as intra‐tumor analysis, shows that GBM tumors are highly heterogeneous. There was a clear association between increased heterogeneity within decrease survival.52 In agreement, large GBM tumors with high proliferation responded better to treatment than small and slower tumors, suggesting that many patients who receive standard‐of‐care treatments may get better benefit from alternative treatments that target specific tumor signatures.7
Furthermore, the high levels of the non‐neoplastic cell populations such as TAMs, endothelial cells, and fibroblast could complicate the interpretation of OMICs related approaches. Variations in the ratio among these cell types with tumor proliferating and stem cells in general correlate with different vascular density.53 Specifically, TAMs differentiate into M2 macrophages acting as protumoral macrophages, and contribute to disease progression. It was found by immunofluorescence staining an association of Nf1 deficiency with infiltration of TAMs/microglia, suggesting that Nf1 deactivation may promote macrophages/microglia recruitment in tumors.48 The altered DNA repair pathways and intratumoral heterogeneity in GBM tumors show that transcriptional heterogeneity was identified in 40% of the cases with variability in Mgmt methylation status in 14% of the cases.54 An example of intertumoral heterogeneity is the communication between glioma cells sensitive and resistant to radio and chemotherapy through long membrane extensions called tumor microtubes or tunneling nanotubes (TNTs).55, 56, 57, 58 Overall has been proposed that TNTs provide an alternative mechanism to spread chemotherapy resistance.56 However, the mechanism is unknown.
One of the major contributors to chemotherapy adaptation or resistance is the enzyme MGMT. MGMT prevents apoptosis induced by TMZ and radiation treatment by removing a methyl group into targeted guanine.59 Besides the poor knowledge of the mechanisms of tumor survival and adaptation to treatments, we recently identified an additional mechanism of MGMT mediated resistance mediated by TNTs. TNTs are long‐range communication systems between cells allowing the transfer of protective factors such as MGMT. TNTs enable resistant cells to radiotherapy and TMZ to share MGMT in order to protect tumor cells that are negative for MGMT. These novel mechanisms of tumor adaptation will be discussed below.
Only recently, several communication systems within a tumor have been discovered including, localized gap junction (GJ), hemichannels and TNTs. TNTs enable direct and targeted communication among different populations of cancer, immune, and healthy surrounding cells providing an alternative coordination and synchronization mechanism to spread disease and adaptation to treatment.55 Thus, current efforts are being directed toward personalized treatment through blocking prime signaling pathways in gliomagenesis and understanding acquired resistance. Advances in cell‐to‐cell communication are contributing in the discovery of new therapies and drugs using computational approaches such as artificial intelligence (AI) and machine learning (ML). AI and ML can analyze different database to identify new affected pathways and to design new potential drug.60
2. GLIOBLASTOMA HETEROGENEITY
GBM is a heterogeneous tumor at the histological, cellular, and molecular level (see Figure 1).9, 10, 18 Understanding tumor heterogeneity will help to design efficacious therapies for the treatment and avoid tumor regrowth. GBM tumors have distinct phenotypes within the same tumor that are critical for adaptation to fluctuations in their environment. Over the past decade, GBM tumors have increasingly recognized for their cellular complexity (see Figure 1). From this perspective, the biology of a tumor cannot be studied by analyzing one particular cell type or a particular microenviroment.44, 61 To explain the intra‐tumoral heterogeneity, two mechanisms have been proposed. One is the clonal evolution model that proposes cumulative genetic or epigenetic mutations in individual normal cells, leading the formation of cancer cells that clonally expand into cells with tumorigenic potential.23, 62, 63 The second model proposed is the cancer stem cell model that suggests that only a subset of cancer cells possess indefinite self‐renewal to initiate and maintain tumor growth.63 Therefore, the tumorigenic GBSCs differentiate into nontumorigenic GBSCs, creating a hierarchical organization. The differentiation of GBSCs provides a mechanism for generating phenotypic and functional heterogeneity that can be attributed to clonal evolution environment differences.23 This model suggests that in some cancers, only a minority of cells can proliferate extensively and some therapies that shrink tumor might not be curative because they fail to eliminate GBSCs.62
Figure 1.
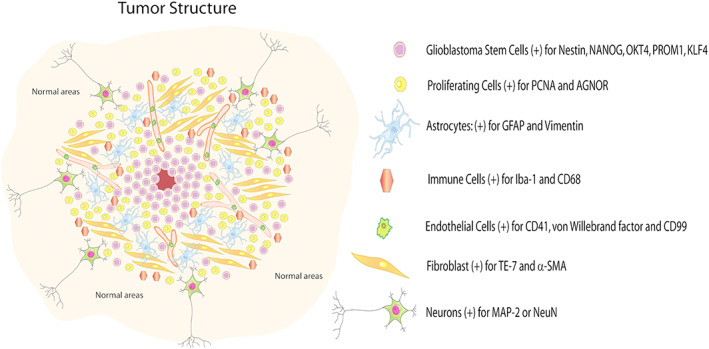
Glioblastoma tumor structure and heterogeneity. The tumor is comprised of several cell types including GBSC positive for Nestin, NANOG, OCT4, PROM1, and KLF4; proliferating cells positive for PCNA and AGNOR; astrocytes positive for GFAP and vimentin; immune cells positive for Iba‐1, CD68, and CD3; endothelial cells positive for CD31 von Willebrand factors and CD99; fibroblast positive for TE‐7 and SMA; and neurons positive for MAP‐2 or neuN. Most of these areas are repetitive and with multiple mutations. Thus, the heterogeneity of the tumor is extremely high
As shown in Figure 1, GBM intratumoral heterogeneity provides cellular niches enriched with distinct cells with different phenotypic properties, transient quiescence, self‐renewal, adaptation to hypoxia, and resistance to chemotherapy and radiation.9, 52 The tumor microenvironment in GBM is constituted by highly proliferative malignant astrocytoma cells, immune cells (lymphocytes, tumor‐associated macrophages or TAMs, and microglia), neurons, endothelial cells, and GBSC.64 The immune cells, principally macrophages, are one of the most relevant in the tumor because they constitute up to 30‐40% of the mass of the tumor,65 showing an increase in their population with the severity of the glioma.66 TAMs are highly plastic cells showing a reciprocal interaction with neoplastic tumor cells to promote growth and progression44; they are strongly correlated with intratumoral vascular density. Besides TAMs, T cells in GBM represent less than 0.25% of total tumor cells isolated from hGBM biopsy samples as examined by flow cytometry.67 Due to this intrinsic tumor heterogeneity, immune cells can be used as a potential target for new GBM drug development. However, most immune therapy approaches in GBM has fail.
GBSCs are determining factors that influence intratumoral heterogeneity, and their differentiation contributes to the response to therapy, drug resistance, and future prognosis. According to the GBSC hypothesis, tumor stem cells lose self‐renewal, and tumorigenic potential, generating a diverse progeny of the tumor bulk,68 which initiates tumor formation.69 These cells can originate phenotypically diverse cancer cells that are situated in specialized locations where the interaction with the microenvironment regulates their behavior contributes to the molecular and cellular heterogeneity in GBM tumors. GBSCs can be classified as cancer stem‐like cells (self‐renew and give rise to differentiated progeny), cancer‐initiating cells (initiate a tumor), and cancer‐propagating cells (propagate tumor).70 These cells cannot be studied in detail because most of the GBM tissues contain multiple populations of cells that express different markers. Nonetheless is possible to validate them with GBSCs enriched methods that allow separating the tumorigenic and non‐tumorigenic populations using specific GBSCs biomarkers as Sox2, Nanog, Olig2, Myc, Musashi1, Bmi1, Nestin and inhibitors of differentiation protein 1 (Id1), CD133 and, Stat3.23 GBSCs have the potential to differentiate into astrocytes, oligodendrocytes, and neurons.71
Tumorigenic GBSCs contribute to tumor initiation, infiltration, therapeutic resistance, and recurrence after surgery.72 Importantly, GBSCs behave like other kinds of stem cells but with different survival and homing capabilities.73 GBSCs could be found in both hypoxic and vascular microenvironments within tumors (perivascular niche), creating a connection between the normal neural stem/progenitors and the vasculature.74 In particular, the core region of the tumor shows high proliferation capacity and clonogenic ability, and the low expression of the differentiation markers and the genetic abnormalities are not shared with the tumor periphery. The core region also shows highly hypoxic conditions, with high enrichment of GBSCs and expression of immature markers such as CD133 and Nestin.74, 75 The intermediate layer of the tumor is hypoxic and enriched with GBSCs; it shows the expression of mixed lineage markers. The periphery of the tumor is marked by the high vascularization, the rare occurrence of GBSCs, the expression of differentiation markers, the low‐level proliferation index, and the clonogenic ability.73 This intratumoral GBSCs heterogeneity ensures metabolic adaptations to support tumor growth in diverse tumor microenvironments.76 The complex subpopulation dynamics within the heterogenous intratumoral environment was characterized by miRNA expression supporting the idea of this large variability of the tumor and particular cell populations.77 The data indicated that phenotype‐linked transcriptomics of GBSCs overlapped with anatomic tumor site, with mesenchymal‐like/nodular signatures in perinecrotic zones and with a proneural‐like/invasive signature in infiltrating areas of the tumor. GBSCs shape and adapt to microenvironmental conditions, and the complex intratumoral architecture arises from the co‐existence of diverse GBSCs within individual tumors.77
GBM intratumoral heterogeneity and treatment adaptation are one of the major barriers for the development of effective therapies. This is partially due to the tumor‐initiating cells (TICs), a cellular subset that contains highly tumorigenic GBSCs. TICs are highly resistant to conventional therapies and therefore, thought to contribute to recurrent GBM.78 Furthermore, GBSC clones from patient samples with extensive molecular and phenotypic variability among clones have a range of responses to radiation and drugs. This widespread variability was observed as a continuum of multitherapy resistance phenotypes linked to a proneural‐mesenchymal shift in the transcriptome.79 Multitherapy resistance was associated with a semi‐stable cell state that was characterized by an altered DNA methylation pattern at promoter regions of mesenchymal master regulators and enhancers. The gradient of cell states within the glioma‐initiating cell (GIC) compartment constitutes a distinct form of heterogeneity.79 A better understanding of the intratumoral heterogeneity in GBM is critical to establish faithful models and develops new therapies to treat this complex disease. We propose that one potential contributor to GBM homo‐ and heterogenecity are TNTs within the tumor.
3. MGMT TUMOR DISTRIBUTION IN GLIOBLASTOMA
The current treatments for GBM, most of time, encounter resistance or adaptation to different chemotherapeutic agents, including alkylating agents, which are highly reactive molecules that promote cell death by binding to DNA. O‐6‐methylguanine is one of the products formed in the DNA reaction of alkylating agents and plays a key role in the initiation of mutations and the cellular cytotoxic effect of these agents.80 One of the most used alkylating agents, for the treatment of GBM, is TMZ. Therapy of malignant GBM relies on treatment with O‐6‐methylating agent TMZ simultaneous with ionizing radiation. TMZ is a small lipophilic molecule which can cross the BBB81 and methylate DNA at the N‐7 (70%), N‐3 (9%) and O‐6 (6%) positions of guanine residues trigging cell death.82 MGMT normally reverses the effect of chemotherapy by restoring the guanine from O‐6‐methylguanine. Following the repair reaction, MGMT becomes inactivated, ubiquitinated, and finally proteasomal degraded. The amount of MGMT per cell is an important determinant for the ability of cancer cells to evade alkylating agent‐induced cell death, and strongly impacts the success of anticancer therapy.7 Human DNA methylation describes the covalent addition of a methyl group preferentially at 5′‐position of a cytosine or guanine nucleotide.83 When Mgmt promoter is silenced through methylation, the MGMT enzyme is reduced (<30 fmol/mg protein), and DNA cannot be repaired, increasing the sensitivity to the alkylating agent and enhancing the efficiency of therapy.7, 84 These mutations principally occur in the CpG sites, which are regions of the DNA molecule where the cytosine nucleotide is followed by a guanine nucleotide in the 5′ to 3′ direction. The hypermethylation mostly occurs at the promoter CpG island of genes that are associated with tumor suppression, DNA repair, cell cycle regulation, apoptosis, invasion, and migration.83 Overall, secondary GBMs showed a higher frequency of promoter methylation than primary GBMs; this can be caused principally for the CpG mutation sites that are more frequent in secondary (56% of the cases) than in primary (30% of the cases) GBM.30, 85 Loss of MGMT expression caused by methylation of promoter CpG islands was detected in 75% of secondary GBMs, more frequently than in primary GBMs (36%).30 The difference in frequency of Mgmt methylation between primary and secondary GBMs are clinically relevant because GBM patients with epigenetic silencing of the methylated Mgmt promoter are associated with loss of MGMT expression and diminished DNA‐repair activity generating a greater benefit from adjuvant TMZ treatment.86
Currently, MGMT is one of the most important DNA repair enzymes in GBM.80, 82, 87 This effect of MGMT causes an increase in chemoresistance87 neutralizing the cytotoxic effects of alkylating agents such as TMZ.30, 80, 88, 89 In GBM, promoter methylation of the gene encoding for MGMT is undoubtedly the genetic fingerprint with the highest impact on clinical practice. The Mgmt promoter hypermethylation is detected in approximately 32–72% of cases (35%–45% in malignant gliomas grades III and IV and 80% of grade II gliomas).85, 86, 90, 91 In long‐term survivors, the values are higher (74–83.3%).92 One of the most important genes that promote the hypermethylation is the Mgmt gene, located at chromosome 10q26.86, 87 Patients with GBM showed heterozygous deletion in the chromosome 10q26,87 suggesting that the presence of an epigenetic lesion in DNA like that can suppress the hypermethylation of tumor genes. A correlation between the presence of Tp53 mutations and Mgmt promoter methylation was found in GBM. Here, a low‐grade astrocytoma with Mgmt methylation was present in 92% of the Tp53 mutations.30 Furthermore, G:C→A:T transition mutations at CpG sites were significantly more frequent in low‐grade astrocytoma with Mgmt methylation (58%) than in those without (11%).30 These results suggest that loss of MGMT expression due to promoter methylation frequently occurs at an early stage in the pathway leading to secondary GBMs and Tp53 mutations at CpG sites in low‐grade gliomas by exogenous or endogenous factors that produce DNA adducts at the O‐6 position of guanine.
On the other hand, less than half of the Idh wild‐type of GBs have a hypermethylated Mgmt associated with the CpG island that depresses the MGMT expression and makes GBMs more sensitive to TMZ chemotherapy.84, 93 The methylation of the Mgmt promoter has been identifying as an important biomarker for GBM and is present in approximately 40% of the cases.94 The anatomic distribution of Mgmt promoter methylated in GBM tumors is proposed to occur as part of a genetic signature that develops from lower grade gliomas.
This transformation is thought to occur early in tumor development within glial cells in specific locations95, 96 supporting the hypothesis that GBM develops from neural stem cells97 and the fact that many gliomas are contiguous with the posterior subventricular zone.98 Using the analysis of differential involvement (ADIFFI) statistical mapping technique in a total of 358 patients with GBM, it was demonstrated that human GBMs occur in high frequency, contiguous with the posterior subventricular zone of the brain; Mgmt promoter‐methylated GBMs are lateralized to the left hemisphere, whereas Mgmt‐unmethylated GBMs are lateralized to the right hemisphere.83 Tumors closer to the left temporal lobe have a significantly longer overall survival compared with tumors occurring elsewhere, independent of treatment or Mgmt methylation status.98 Epigenetic silencing of the Mgmt gene by promoter methylation is associated with longer survival time and increased sensitivity to chemotherapeutic alkylating agents in GBM patients. However, patients with equivalent Mgmt promoter methylation status have variable prognoses and responses to treatment,99 suggesting that other factors are equally important in determining clinical outcome. We propose that TNT‐mediated communication and spread of MGMT contributes to the clinical outcome.
Regulation of MGMT mRNA expression is related to favorable treatment response. Mgmt promoter methylation is a powerful predictor of a survivor because hypermethylation of Mgmt is frequently expressed in long‐term survivor patients.100 Although not all patients with methylated promoter have the same response to TMZ treatment, it suggests that methylated promoter is not the only factor involved in GBM treatment resistance.99 MiRNA expression experiments classify and predict MGMT distribution in GBM samples based on mRNA expression profiles. A study in a cohort of 150 primary GBM showed that MGMT miRNAs were found to be differentially expressed between non‐tumor brain tissue.99 Furthermore, with an equivalent Mgmt promoter methylation, high‐ and low‐risk patients have distinct prognoses, with the former showing a similar survival to GBM patients with unmethylated Mgmt promoters. It was found that high‐risk patients with a methylated Mgmt promoter, who were treated with alkylating agents, had no survival advantage over low‐risk patients.99
4. GLIOBLASTOMA METABOLISM: A POTENTIAL MECHANISM OF HETEROGENEITY MEDIATED BY TNTS
Tumor metabolism is based on two major points of cell behavior: (i) the specific sourcing of metabolites; and (ii) the different cellular mechanism used to deal with different nutrients for either anabolic construction or catabolic breakdown. GBM metabolism offers new or supplementary targets for GBM therapy. Critical features of energy metabolism are related to mitochondrial genetics and apoptosis regulation in GBM. GBM functional processes are linked to mitochondrial regulation involving genomic and mitochondrial gene mutations, mitochondrial protein expression modifications, and altered metabolic regulation. Mitochondria have a crucial role because they perform numerous important cellular functions: energy generation by synthesizing ATP via oxidative phosphorylation, anabolic/catabolic reactions, metabolic regulation, signal transduction, calcium homeostasis, reactive oxygen species generation, redox control, and apoptosis. In cancer metabolism, mitochondria are indispensable for energy production, and the survival of the cells also are a crucial regulator of the apoptotic pathways. Warburg described that proliferating cancer cells preferentially convert glucose into lactate instead of pyruvate into the tricarboxylic cycle of the mitochondria, even in presence of oxygen.101, 102 This process is known as aerobic glycolysis or Warburg effect.
The metabolic signatures of cancer cells are not responses to damaged mitochondria but result from oncogene‐directed metabolic reprogramming required to support anabolic growth. Furthermore, mitochondrial DNA is a highly polymorphic molecule susceptible to a high mutational rate, which is caused by the lack of protective histones, proximity to the site of the production of (mutagenic) reactive oxygen species, and relatively limited DNA repair mechanisms. The high metabolic activity of cancer cells, impaired repair mechanisms, and increased genomic instability are typically susceptible to the accumulation of somatic DNA mutations including mtDNA mutations, which are also believed to contribute to cancer genesis and biology. Changes in mtDNA alter gene expression profiles and contribute to the compromised mitochondrial machinery of energy metabolism and apoptosis regulation. GBM tumor cells carry mtDNA mutations preferentially in the D‐loop and protein coding regions and occur in the early stage of gliomagenesis.103
Furthermore, the regulation of pro‐ and anti‐apoptotic factors is deflected in all cancers. Upregulation of the antiapoptotic Bcl‐2 and Bcl‐2‐like 2 (Bcl‐ XL) and downregulation of the proapoptotic Bcl‐2‐associated X protein (Bax) have been recurrently detected in GBM.104 Also, the energetic function of mitochondria in most malignantly transformed cells is related to the Warburg effect. It is based on mitochondrial impairment to oxidize glucose carbon to CO2. Although normal cells will largely undergo oxidative phosphorylation in the presence of glucose and oxygen, in many cancer cells, the large proportion of glucose is diverted away from mitochondrial oxidation and into glycolysis and the production of lactate by lactate dehydrogenase (LDH) even in the presence of oxygen. GBSCs have been reported to have distinctly different metabolic phenotypes compared with more differentiated tumor cells and appear to be able to easily switch between glycolytic and oxidative metabolism depending on the microenvironment.105 So GBSC population maintains a distinct metabolic phenotype compared with the tumor bulk. Glucose uptake, glycolytic enzymes, lactate, and ATP production are much higher in GBSCs compared with when they were differentiated due to diminished metabolic contribution from mitochondrial oxidation.106 Furthermore, metabolism and GBM correlation require a mention of isocitrate dehydrogenase 1/2 (Idh1/2) mutations. IDH1/2 are responsible for catalyzing the oxidative decarboxylation of isocitrate into 2‐oxoglutarate (or α‐ketoglutarate, α‐KG). α‐KG is a key molecule in the Krebs cycle. It is nitrogen scavenger and a crucial precursor of glutamine and glutamate.107 It has a potent antioxidant and immune regulation function. Mutations in the Idh1 and its homolog Idh2 gene are very common in GBM. The loss of normal enzymatic function and the abnormal production of 2‐hydroxyglutarate (2‐HG) reduce the amount of α‐KG.108 We propose that the exchange of mitochondria within the tumor via TNTs contributes to metabolic adaptation of GBM.
5. TUNNELING NANOTUBES IN GLIOBLASTOMA
Tumors are complex dynamic structures; cellular and molecular changes contribute to disease pathobiology. TNTs play a key role in cancer pathogenesis, brain invasion, proliferation, and long‐distance cell communication.109 Considering that the 10%–20% of the cells in the tumor are malignant and may not be close enough to exchange cellular information through GJ,110 TNTs become a critical cellular communication mechanism for tumor evasion and chemotherapy resistance. TNTs are long cytoplasmic F‐actin extensions of astrocytes and oligodendroglioma cells; their measures are 20–1,500 nm in length, 0.1 μm in width, and 1.57 μm2 of mean cross‐sectional area.110, 111, 112 For example, TNTs connect 10%–15% of Jurkat T cells in normal tissue culture conditions, and individual myeloid cells can support up to 75 nanotubes.113 There are different types of TNTs: an open‐ended and connexin‐containing protrusion.109, 114 GJs play a cooperative role in the communication system between the connected cells by TNTs. The presence of connexin 43 (Cx43) under HIV pathogenesis shows that the inhibition of GJ does not prevent TNT formation but interfere with the normal communication between TNT connected cells.115 TNTs are composed of Cx43. The functional role of Cx43 in astrocytoma progression of GBSC shows that Cx43 stabilizes the TNT communication. Furthermore, Cx43 deficiency results in reduced tumor size as observed by MRI and improved survival, also decreasing the radioprotective effect of TNTs in connected astrocytoma cells.112 For demonstrating tunneling microtube implication in therapy resistance, Weil and colleagues used surgical lesion experiments and implanted patient‐derived GBM stem‐like cells under a cranial window in mice using in vivo 2‐photon microscopy. They followed individual tumor regions and single glioma cells over extended periods. After the surgical removal of a cylindrical brain tissue volume colonized by GBSCs, GBM cells repopulated the lesioned area over time.56 This means that TNTs are involved in mediating the repopulation process. TNT‐connected glioma cells are more resistant to the cytotoxic effects of TMZ chemotherapy, and the microtube‐connected astrocytoma cells were protected from cell death inflicted by radiotherapy.112
In GBM, interactions and intercellular communications between malignant and non‐malignant cells are critical to improve the understanding of the disease.116 Perivascular niche plays a crucial role in many aspects of brain tumor progression. GBM stem‐like cells colonized the perivascular niche in significant numbers; it is possible to use them as a route for effective brain invasion. A subgroup of GBSCs in a perivascular position showed long‐term latency, and targeting this subpopulation of glioma cells emerges as an important task for the development of novel therapies because existing treatment modalities fall short of controlling these cells.117 It has been widely demonstrated that TNTs allow the bi‐ and unidirectional transfer of cargoes, including protein, mRNA, and organelles such as mitochondria and endoplasmic reticulum, between the connected cells.109, 111, 112, 118, 119, 120, 121
Nevertheless, the mechanisms of TNT selectivity, transport, and delivery are still unknown. The transfer of cargoes is a fast process, taking account that TNTs lifetime is less than 60 minutes.112 Several laboratories demonstrate that TNT formation is controlled by p53, Egfr, Akt, phosphoinositide‐3‐kinase (PI3K), and mTOR. p53 activation or Egfr or Akt/PI3K/mTOR induces M‐Sec overexpression, which can trigger F‐actin polymerization and contributes to TNT development from the initiating cell membrane.122 In addition, TNT formation has been studied in cancer cells, neurons, immune cells (B, T, NK cells, neutrophils, monocyte, macrophages, and dendritic cells), endothelial cells, and stem cells.55, 111, 114, 123, 124, 125 It has been demonstrated that TNT number increases when cells are under stress or pathogenic context. For example, it was demonstrated that the number of TNT between astrocytes and C6 glioma cells was increased in the presence of H2O2. 126 Also, our preliminary data show that radiation and TMZ treatment are stressful handlings that induce TNT formation.
Furthermore, in GBM, TNT formation is highly influenced by tumor type and grade, with a marked positive correlation of TNT length and unfavorable prognosis. This data suggests that cells under pathological conditions (cancer or infectious diseases) TNTs provides complex communication for cell to cell coordination and cooperation. Thus, TNT formation and function will open new therapies for the treatment of different diseases. Our group proposes that TNTs proliferate due to radiation and TMZ treatment and help tumor invasion and survival.55 We also suggest that the spread of MGMT protein into cells with insufficient or lacking MGMT occurs via TNTs to adapt the tumor to treatments and increase recurrence (see Figure 2). These mechanisms are novel. Thus, we propose that TNTs are a novel route by which MGMT and other tumor‐protective molecules could be transferred between non‐susceptible cells to treat tumor to susceptible tumor cells, preventing tumor cell death. Interestingly, TNTs are minimally expressed under physiological conditions and are only induced in cancer. Thus, TNTs are an exciting therapeutic target.
Figure 2.
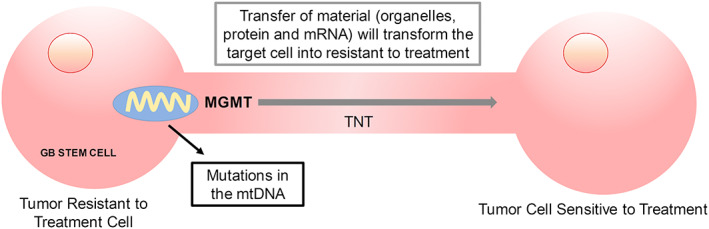
Transfer of MGMT and altered mitochondria (with mtDNA mutations) from glioblastoma tumor resistant to treatment of cancer stem cells to glioblastoma tumor sensitive to treatment stem cells, via tunneling nanotubes (TNTs)
The transfer of mitochondria via TNT induces metabolic changes in the TNT targeted cell, resulting in cell proliferation, differentiation, apoptosis, and response to therapy.55, 127 Mesenchymal stem cells (MSCs) form TNTs and transfer mitochondria to target cells (including cardiomyocytes, endothelial cells, epithelial cells, macrophages, and cancer cells) under conditions of stress or injury, leading to modifications of their functional properties.120, 128, 129 A similar process of mitochrondrial exchange by TNT like processes was observed using MSCs in co‐culture with cortical neurons or upon injection into the animal subjected to stroke. The exchange of cellular compartments improved MSCs protective abilities for better rehabilitation after stroke.130 Specifically, healthy or mutated mitochondrial DNA (mtDNA) can be shared between cells and consequently affects the metabolism of the TNT targeted cell. It has been reported that the horizontal transfer of mtDNA increases tumor‐initiating efficacy, showing that exchange of mtDNA within the tumor is essential for tumor adaptation and pathogenesis.131 Furthermore, it had been shown that mtDNA is acquired by transfer of whole mitochondria from stromal cells into the tumor, rescuing the metabolism of the tumor cell with damaged mtDNA.
The intercellular exchange of mitochondria via TNTs has been proposed as a mechanism for restoring survival and metabolism of damaged cells. The interaction between apoptotic and healthy cells connected by TNTs shows that in pheochromocytoma cells (PC12) treated with ultraviolet light were rescued when they were cocultured with untreated PC12 cells.133 Furthermore, blocking TNTs inhibited the rescue effect.133 Additionally, the TNT formation between stromal (MSCs and endothelial cells; ECs) and cancer cells showed that intercellular transfers of cytoplasmic content occurred between cancer cells and MSCs or ECs, but the exchange of mitochondria occurred preferentially between ECs and cancer cells. Cancer cells acquiring mitochondria displayed chemoresistance.127 TNTs may help drug‐sensitive cancer cells to acquire survival signals from drug‐insensitive cells and escape death during cancer treatment, suggesting that TNTs could be a novel target for the development of new cancer therapies.
6. DRUGS AND TREATMENTS
Currently, the treatment for GBM corresponds to surgical resection of the tumor, where the patient waits approximately 4 weeks for the craniotomy wound to heal before starting the therapy.134 The post‐surgery process receives radiotherapy irradiation of 2 Gy, given 5 days per week for 6 weeks, for a total of 60 Gy,13, 135 plus continuous daily TMZ, 75 mg per square meter of body‐surface area per day, 7 days per week from the first to the last day of radiotherapy, followed by six cycles of adjuvant TMZ, 150 to 200 mg per square meter for 5 days during each 28‐day cycle.13 This treatment prolongs patient survival and reduces the risk of death by 37%.13 Despite treatment, recurrences are observed within 6–7 months and occur in around 90% of the cases136, 137; may be due to the tumor adaptation, it has been found that the reappearance of GBM arise at the resection margin, wherein the highest doses are delivered and is caused by the residual GBM cells left in the surgical margins, in the peritumoral tissue between 2 cm from the tumor edge or infiltrating the normal brain parenchyma.137, 138, 139 These residual cells subsequently become exposed to standard and experimental therapy, although their study and characterization open new research lines of therapy. GBSCs have been demonstrated as being responsible for the tumor recurrence. A small population of GBSCs derived from both peritumoral tissue and GBM shows significant differences between GBM and peritumoral tissue regarding proliferation, ultrastructural peculiarities, and, at a lower extent, stemness profile.138 The residual GBM cells are difficult to image, and the maximum dose of radiotherapy cannot destroy these cells without a specific target. Chemotherapy can eliminate these cells but not completely; an in vitro drug and irradiation (5 Gy), plus concomitant treatment with TMZ (500 μM), lomustine (380 μM), and combinations, shows that, in 64% of the cases, GBM periphery cells responded dissimilar from the corresponding center cells.139 Finding new treatments to eliminate the residual GBM cells is a major challenge.
Other factors affect the progress of therapies including invasive tumor growth in a vital organ limiting the utility of local therapy, protection of tumor cells by the BBB, intrinsic resistance to the induction of cell death, and lack of dependence on single, targetable oncogenic pathways.140 Predictive molecular markers are commonly tested as part of the routine clinical interrogation of GBM patients including Mgmt, Idh, Egfr, Vegf, Tp53, phosphatase and tensin homolog (Pten), p16INK4a gene, phospholipid metabolites, cancer stem cells, and, recently, imaging biomarkers. Meta‐analyses are used to augment the validity of potential prognostic biomarkers in GBM, but significant limitations are due to GBM novel nature and incomplete understanding of GBM biology.141 Several specific biomarkers need to be investigated for a distinct prognosis, for trying to personalized therapeutic approaches and for contributing to the development of a new generation of anti‐GBM therapies.142 New progress in GBM therapy combines the current standard‐of‐care treatment and immunotherapies or alternating electric fields therapy. Central nervous system is a privileged immune organ, but microglia are the major antigen‐presenting cells in the brain tumor microenvironment,143 which could be a strategical target for immunotherapy. Escape immune system surveillance is a critical feature for GBM, and several immune suppressive mechanisms are utilized in the setting of GBM to prevent its immune detection and eradication.
The increased signal transducer initially drives immunosuppression and activator of transcription 3 (STAT3) expression that induces cell secrete immunosuppressive factors production such as TGFβ‐2, prostaglandins (PG), interleukin (IL)‐1, IL‐10 and fibrinogen‐like protein 2 (FGL2). Regulatory T cells (Tregs) are the major responsible cells that suppress immune responses by secreting cytokines (TGF‐β and IL‐10) by cell‐to‐cell–mediated contact and by cytotoxic T‐lymphocyte–associated protein 4 (CTLA‐4) activation.144 In GBM, macrophages have increased levels of programmed cell death 1 ligand 1 (PD‐L1) that binds to and activates PD1 immune‐checkpoint receptor restricting cytotoxic T cell activity. GBM vaccine therapy relies on dendritic cells (DC)‐mediated presentation of GBM‐associated peptides, multi‐peptides, cell‐activating adjuvants, antigens, or epitope derived from tumor lysates to T cells. Many clinical trials are ongoing to evaluate the efficacy of these vaccines. Genetic engineering uses oncolytic viral therapy to create viruses that selectively infect or replicate in tumor cells. The resulting tumor cell lysis not only kills the infected tumor cells directly but can also activate the immunogenic tumor cell death pathway that can stimulate antigen presentation and adaptive immune response. Also, other immunotherapy approaches are antibodies that block the inhibitory immune‐checkpoint proteins (PD‐1 and CTLA‐4) or engineered chimeric antigen receptors, but it seems more efficacious to combine the traditional standard of care with these innovative immunotherapy strategies.140 However, the effectivity of these treatments in GBM is unclear.
7. ARTIFICIAL INTELLIGENCE TO UNDERSTAND GLIOBLASTOMA HETEROGENEITY AND THE ROLE OF TNTS
Currently, advances in computational and data science have been increasing, with the purpose to find more comprehensive and coherent strategies to improve health care and medicine. AI and ML algorithms in cancer research are a powerful tool to increase the speed in the efficiency to improve the diagnoses and design new therapies, drugs, and treatments. All these new technologies have the main focus to cure or increase life expectancy in cancer patients.
In cancer imaging analyses, AI has a successful domain. The radiologist uses a collection of images (X‐ray, tomography, magnetic resonance imaging (MRI), and positron‐emission tomography) to screen and diagnose cancer.145 Furthermore, histopathological assessment to classify cancer and identify metastasis also applies ML and AI to analyze the microscopy results and improve the accuracy in the analysis. These studies employed the transfer learning technique to establish neural network connections of thousands of images database.145 To classify and identify the cancer diagnoses in GBM radionics research help to predict disease prognosis, they are providing beneficial information for personalized treatment from a variety of imaging features extracted from multiple MRI. The early diagnosis of cancer is a critical point in term of life expectation and treatment. The computational methodologies (AI or ML) to predict early stages or the detection of tumor cells are crucial. However, metastatic tumor cells are exceedingly difficult to detect from blood or biopsy samples. But it is reported that three ML algorithms combined can analyze the data from microscopy images quickly and quantify the cell morphology for instant real‐time feedback and can certainly contribute to early cancer diagnosis.146 The imaging analysis not only is useful for early detection but also the prediction of survival.
The prediction results for both 2‐class (short and long) and 3‐class (short, medium, and long) survival groups were 98.7% and 88.95%, respectively.147 In GBM, the methylation status of the promoter of the MGMT gene impacts the efficacy and sensitivity of the TMZ treatment and consequently affects patient survival. Microscopic genetic changes may manifest as macroscopic morphological changes in the brain tumors that can be detected using MRI.148 A neural network analysis of brain MRI scans of GBM patients were collected from The Cancer Imaging Archive (TCIA) combined with methylation data from The Cancer Genome Atlas (TCGA) to predict the methylation state of the MGMT regulatory regions in these patients. The results with 67% on the validation data suggest the existence of MRI features that may complement existing markers for GBM patient stratification and prognosis.148 Besides the good results in the application of the AI methodologies to the imaging analysis, there are limitations that need to be told in considerations such as the different imaging platforms, the protocols and parameter used to get the images, the criteria to classify the patients, and the demographic and treatment information of the patients.149, 150 There is extra work in the direction of strengthening the ML and AI classification models based on imaging data for reliable and clinically meaningful prediction of the assessed molecular characteristics in patients diagnosed GBM.
Despite the large amount of OMICs (metabolomic, proteomic, lipidomic, genomic, and transcriptomic) in cancer. Databases such as TCGA Research Network is collecting data from over 11,000 tumors from 33 types of cancer.151 This enormous amounts of information provide a major opportunity to develop an integrated methodology that involves statistical analysis and computational approaches, as AI and ML, to develop effective therapies for different cancer type.152 For example, an ML method is capable of identifying stemness or the potential self‐renewal and differentiation from the origin cell in a single‐cell pattern of intra‐tumor molecular heterogeneity.153 In GBM, a multigene predictor was developed using GBM microarray data from four independent data sets and is capable of identifying nine gene sets as an independent predictor of outcome in GBM survival.154 Furthermore, TCGA was used to train accurate predictors for NF1 inactivation; this gene is an important regulator of the oncogene RAS and is inactivated frequently in GBM.155 With the current database and improved classification, ML and AI models will translate into clinically relevant predictions that will guide GBM therapy. Now critical markers of TNTs can be added to these analyses to identify their role in the pathogenesis of GBM or to develop new treatment to cure GBM.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTION
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization: S.V., D.D., and E.E. Writing original draft: S.V., D.D., and E.E. Writing review and editing: S. V, D.D., and E.E.Visualization: S.V., D.D., and E.E. Supervision: S.V. and E.E. Funding acquisition: E.E.
ACKNOWLEDGEMENTS
This work was funded by the National Institute of Mental Health grant, MH09665, the National Institute of Neurological Disorders and Stroke, NS105584, and UTMB internal funding (to E.A.E).
Valdebenito S, D'Amico D, Eugenin E. Novel approaches for glioblastoma treatment: Focus on tumor heterogeneity, treatment resistance, and computational tools. Cancer Reports. 2019;2:e1220. 10.1002/cnr2.1220
REFERENCES
- 1. Butowski NA. Epidemiology and diagnosis of brain tumors. Continuum (Minneap Minn) [Review]. 2015. Apr;21(2 Neuro‐oncology):301‐313. [DOI] [PubMed] [Google Scholar]
- 2. Ray A, Manjila S, Hdeib AM, et al. Extracranial metastasis of gliobastoma: Three illustrative cases and current review of the molecular pathology and management strategies. Mol Clin Oncol. 2015. May;3(3):479‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016. Jun;131(6):803‐820. [DOI] [PubMed] [Google Scholar]
- 4. Forsyth TM, Bi WL, Abedalthagafi M, Dunn IF, Chiocca EA. Extracranial growth of glioblastoma multiforme. J Clin Neurosci. 2015. Sep;22(9):1521‐1523. [DOI] [PubMed] [Google Scholar]
- 5. Suzuki Y, Shirai K, Oka K, et al. Higher pAkt expression predicts a significant worse prognosis in glioblastomas. J Radiat Res. 2010;51(3):343‐348. [DOI] [PubMed] [Google Scholar]
- 6. Philips A, Henshaw DL, Lamburn G, O'Carroll MJ. Brain tumours: Rise in glioblastoma multiforme incidence in England 1995–2015 suggests an adverse environmental or lifestyle factor. J Environ Public Health. 2018;2018:7910754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rayfield CA, Grady F, Leon GD, et al. Distinct phenotypic clusters of glioblastoma growth and response kinetics predict survival. JCO Clinical Cancer Informatics. 2018;2:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev [Research Support, N.I.H., Extramural Research Support, Non‐U.S. Gov'tReview]. 2007. Nov 1;21(21):2683‐2710. [DOI] [PubMed] [Google Scholar]
- 9. Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res. 2011. Jun 15;71(12):4055‐4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Friedmann‐Morvinski D. Glioblastoma heterogeneity and cancer cell plasticity. Crit Rev Oncog. 2014;19(5):327‐336. [DOI] [PubMed] [Google Scholar]
- 11. Huang Q, Zhang QB, Dong J, et al. Glioma stem cells are more aggressive in recurrent tumors with malignant progression than in the primary tumor, and both can be maintained long‐term in vitro. BMC Cancer. 2008. Oct 22;8(1):304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tamimi AF, Juweid M. Epidemiology and outcome of glioblastoma. In: De Vleeschouwer S, ed. Glioblastoma. Brisbane (AU): Codon Publications; 2017. [PubMed] [Google Scholar]
- 13. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005. Mar 10;352(10):987‐996. [DOI] [PubMed] [Google Scholar]
- 14. Choi PJ, Tubbs RS, Oskouian RJ. Emerging cellular therapies for glioblastoma multiforme. Cureus. 2018. Mar 11;10(3):e2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roux A, Caire F, Guyotat J, Menei P, Metellus P, Pallud J. Carmustine wafer implantation for high‐grade gliomas: Evidence‐based safety efficacy and practical recommendations from the Neuro‐oncology Club of the French Society of Neurosurgery. Neurochirurgie. 2017. Dec;63(6):433‐443. [DOI] [PubMed] [Google Scholar]
- 16. Park JK, Hodges T, Arko L, et al. Scale to predict survival after surgery for recurrent glioblastoma multiforme. J Clin Oncol. 2010. Aug 20;28(24):3838‐3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muller C, Holtschmidt J, Auer M, et al. Hematogenous dissemination of glioblastoma multiforme. Sci Transl Med. 2014. Jul 30;6(247):247ra101. [DOI] [PubMed] [Google Scholar]
- 18. Aum DJ, Kim DH, Beaumont TL, Leuthardt EC, Dunn GP, Kim AH. Molecular and cellular heterogeneity: The hallmark of glioblastoma. Neurosurg Focus. 2014. Dec;37(6):E11. [DOI] [PubMed] [Google Scholar]
- 19. Schiera G, Di Liegro CM, Di Liegro I. Molecular determinants of malignant brain cancers: From intracellular alterations to invasion mediated by extracellular vesicles. Int J Mol Sci. 2017. Dec 20;18(12):2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Malric L, Monferran S, Gilhodes J, et al. Interest of integrins targeting in glioblastoma according to tumor heterogeneity and cancer stem cell paradigm: An update. Oncotarget. 2017. Oct 17;8(49):86947‐86968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: Origin and new perspectives on CSC targeting. BMB Rep. 2017. Mar;50(3):117‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balca‐Silva J, Matias D, Carmo AD, Sarmento‐Ribeiro AB, Lopes MC, Moura‐Neto V. Cellular and molecular mechanisms of glioblastoma malignancy: Implications in resistance and therapeutic strategies. Semin Cancer Biol. 2018. Sep 25; [Epub ahead of print]. 10.1016/j.semcancer.2018.09.007 [DOI] [PubMed] [Google Scholar]
- 23. Bradshaw A, Wickremsekera A, Tan ST, Peng L, Davis PF, Itinteang T. Cancer stem cell hierarchy in glioblastoma multiforme. Front Surg. 2016;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seymour T, Nowak A, Kakulas F. Targeting aggressive cancer stem cells in glioblastoma. Front Oncol. 2015;5:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rycaj K, Tang DG. Cancer stem cells and radioresistance. Int J Radiat Biol. 2014. Aug;90(8):615‐621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pesenti C, Navone SE, Guarnaccia L, et al. The genetic landscape of human glioblastoma and matched primary cancer stem cells reveals intratumour similarity and intertumour heterogeneity. Stem Cells Int. 2019;2019:2617030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bayin NS, Modrek AS, Placantonakis DG. Glioblastoma stem cells: Molecular characteristics and therapeutic implications. World J Stem Cells. 2014. Apr 26;6(2):230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eskilsson E, Rosland GV, Solecki G, et al. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro Oncol. 2018. May 18;20(6):743‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nadeem Abbas M, Kausar S, Wang F, Zhao Y, Cui H. Advances in targeting the epidermal growth factor receptor pathway by synthetic products and its regulation by epigenetic modulators as a therapy for glioblastoma. Cell. 2019. Apr 12;8(4):350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007. May;170(5):1445‐1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vastrad B, Vastrad C, Godavarthi A, Chandrashekar R. Molecular mechanisms underlying gliomas and glioblastoma pathogenesis revealed by bioinformatics analysis of microarray data. Med Oncol. 2017. Sep 26;34(11):182. [DOI] [PubMed] [Google Scholar]
- 32. Le Rhun E, von Achenbach C, Lohmann B, et al. Profound, durable and MGMT‐independent sensitivity of glioblastoma cells to cyclin‐dependent kinase inhibition. Int J Cancer. 2019. Jul 1;145(1):242‐253. [DOI] [PubMed] [Google Scholar]
- 33. Francis JM, Zhang CZ, Maire CL, et al. EGFR variant heterogeneity in glioblastoma resolved through single‐nucleus sequencing. Cancer Discov. 2014. Aug;4(8):956‐971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Waitkus MS, Diplas BH, Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro Oncol. 2016. Jan;18(1):16‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jesionek‐Kupnicka D, Braun M, Trabska‐Kluch B, et al. MiR‐21, miR‐34a, miR‐125b, miR‐181d and miR‐648 levels inversely correlate with MGMT and TP53 expression in primary glioblastoma patients. Arch Med Sci. 2019. Mar;15(2):504‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johannessen TC, Prestegarden L, Grudic A, Hegi ME, Tysnes BB, Bjerkvig R. The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro Oncol. 2013. Mar;15(3):269‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duran G, Aguin S, Cruz R, et al. Association of GSTP1 and ERCC1 polymorphisms with toxicity in locally advanced head and neck cancer platinum‐based chemoradiotherapy treatment. Head Neck. 2019. Apr 11;41(8):2704‐2715 [DOI] [PubMed] [Google Scholar]
- 38. Tjiong R, Stavrinou P, Rohn G, et al. Heterogeneity of human gliomas: Glutathione‐S‐transferase expression profile during disease progression and under systemic therapy. Anticancer Res. 2019. Apr;39(4):1795‐1805. [DOI] [PubMed] [Google Scholar]
- 39. Moghimi M, Sobhan MR, Jarahzadeh MH, et al. Association of GSTM1, GSTT1, GSTM3, and GSTP1 genes polymorphisms with susceptibility to osteosarcoma: A case‐control study and meta‐analysis. Asian Pac J Cancer Prev. 2019. Mar 26;20(3):675‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dong SC, Sha HH, Xu XY, et al. Glutathione S‐transferase pi: A potential role in antitumor therapy. Drug Des Devel Ther. 2018;12:3535‐3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagane M, Shibui S, Oyama H, Asai A, Kuchino Y, Nomura K. Investigation of chemoresistance‐related genes mRNA expression for selecting anticancer agents in successful adjuvant chemotherapy for a case of recurrent glioblastoma. Surg Neurol. 1995. Nov;44(5):462‐468. discussion 8‐70 [DOI] [PubMed] [Google Scholar]
- 42. Pasqualetti F, Gonnelli A, Cantarella M, et al. Association of glutathione S‐transferase P‐1 (GSTP‐1) rs1695 polymorphism with overall survival in glioblastoma patients treated with combined radio‐chemotherapy. Invest New Drugs. 2018. Apr;36(2):340‐345. [DOI] [PubMed] [Google Scholar]
- 43. Verhaak RG, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010. Jan 19;17(1):98‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. 2018;9:1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Herting CJ, Chen Z, Pitter KL, et al. Genetic driver mutations define the expression signature and microenvironmental composition of high‐grade gliomas. Glia. 2017. Dec;65(12):1914‐1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Favero F, McGranahan N, Salm M, et al. Glioblastoma adaptation traced through decline of an IDH1 clonal driver and macro‐evolution of a double‐minute chromosome. Ann Oncol. 2015. May;26(5):880‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kumar A, Boyle EA, Tokita M, et al. Deep sequencing of multiple regions of glial tumors reveals spatial heterogeneity for mutations in clinically relevant genes. Genome Biol. 2014. Dec 3;15(12):530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Q, Hu B, Hu X, et al. Tumor evolution of glioma‐intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2018. Jan 8;33(1):152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bhat KP, Salazar KL, Balasubramaniyan V, et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011. Dec 15;25(24):2594‐2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Minata M, Audia A, Shi J, et al. Phenotypic plasticity of invasive edge glioma stem‐like cells in response to ionizing radiation. Cell Rep. 2019. Feb 12;26(7):1893‐1905. e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zanca C, Villa GR, Benitez JA, et al. Glioblastoma cellular cross‐talk converges on NF‐kappaB to attenuate EGFR inhibitor sensitivity. Genes Dev. 2017. Jun 15;31(12):1212‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Patel AP, Tirosh I, Trombetta JJ, et al. Single‐cell RNA‐seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014. Jun 20;344(6190):1396‐1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013. Nov;19(11):1423‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Parker NR, Hudson AL, Khong P, et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Sci Rep. 2016. Mar 4;6(1):22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Valdebenito S, Lou E, Baldoni J, Okafo G, Eugenin E. The novel roles of connexin channels and tunneling nanotubes in cancer pathogenesis. Int J Mol Sci. 2018. Apr 24;19(5):1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weil S, Osswald M, Solecki G, et al. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017. Oct 1;19(10):1316‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sahu P, Jena SR, Samanta L. Tunneling nanotubes: A versatile target for cancer therapy. Curr Cancer Drug Targets. 2018;18(6):514‐521. [DOI] [PubMed] [Google Scholar]
- 58. Hekmatshoar Y, Nakhle J, Galloni M, Vignais ML. The role of metabolism and tunneling nanotube‐mediated intercellular mitochondria exchange in cancer drug resistance. Biochem J. 2018. Jul 31;475(14):2305‐2328. [DOI] [PubMed] [Google Scholar]
- 59. Binabaj MM, Bahrami A, ShahidSales S, et al. The prognostic value of MGMT promoter methylation in glioblastoma: A meta‐analysis of clinical trials. J Cell Physiol. 2018. Jan;233(1):378‐386. [DOI] [PubMed] [Google Scholar]
- 60. Velaga R, Sugimoto M. Future paradigm of breast cancer resistance and treatment. In: Prosperi JR, ed. Resistance to Targeted Therapies in Breast Cancer. Cham: Springer International Publishing; 2017:155‐178. [Google Scholar]
- 61. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011. Mar 4;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 62. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell. 2012. Mar 20;21(3):283‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prasetyanti PR, Medema JP. Intra‐tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017. Feb 16;16(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Manini I, Caponnetto F, Bartolini A, et al. Role of microenvironment in glioma invasion: What we learned from in vitro models. Int J Mol Sci. 2018. Jan 4;19(1):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. De Vleeschouwer S, Bergers G. Glioblastoma: To target the tumor cell or the microenvironment? In: De Vleeschouwer S, ed. Glioblastoma. Brisbane (AU): Codon Publications; 2017. [PubMed] [Google Scholar]
- 66. Korbecki J, Gutowska I, Kojder I, et al. New extracellular factors in glioblastoma multiforme development: Neurotensin, growth differentiation factor‐15, sphingosine‐1‐phosphate and cytomegalovirus infection. Oncotarget. 2018. Jan 23;9(6):7219‐7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Han S, Ma E, Wang X, et al. Rescuing defective tumor‐infiltrating T‐cell proliferation in glioblastoma patients. Oncol Lett. 2016. Oct;12(4):2924‐2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pointer KB, Clark PA, Zorniak M, Alrfaei BM, Kuo JS. Glioblastoma cancer stem cells: Biomarker and therapeutic advances. Neurochem Int. 2014. May;71:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ho IAW, Shim WSN. Contribution of the microenvironmental niche to glioblastoma heterogeneity. Biomed Res Int. 2017;2017:9634172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lathia JD, Mack SC, Mulkearns‐Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015. Jun 15;29(12):1203‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Emmenegger BA, Wechsler‐Reya RJ. Stem cells and the origin and propagation of brain tumors. J Child Neurol. 2008. Oct;23(10):1172‐1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cheray M, Begaud G, Deluche E, et al. Cancer stem‐like cells in glioblastoma. In: De Vleeschouwer S, ed. Glioblastoma. Brisbane (AU): Codon Publications; 2017. [PubMed] [Google Scholar]
- 73. Kalkan R. Glioblastoma stem cells as a new therapeutic target for glioblastoma. Clin Med Insights‐On. 2015;9:95‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Heddleston JM, Li ZZ, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009. Oct 15;8(20):3274‐3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ludwig K, Kornblum HI. Molecular markers in glioma. J Neurooncol. 2017. Sep;134(3):505‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bayin NS, Frenster JD, Sen R, et al. Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget. 2017. Sep 12;8(39):64932‐64953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Godlewski J, Ferrer‐Luna R, Rooj AK, et al. MicroRNA signatures and molecular subtypes of glioblastoma: The role of extracellular transfer. Stem Cell Reports. 2017. Jun 6;8(6):1497‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Libby CJ, Zhang S, Benavides GA, et al. Identification of compounds that decrease glioblastoma growth and glucose uptake in vitro. ACS Chem Biol. 2018. Jun 15;13(8):2048‐2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Segerman A, Niklasson M, Haglund C, et al. Clonal variation in drug and radiation response among glioma‐initiating cells is linked to proneural‐mesenchymal transition. Cell Rep. 2016. Dec 13;17(11):2994‐3009. [DOI] [PubMed] [Google Scholar]
- 80. Pegg AE. Repair of O(6)‐alkylguanine by alkyltransferases. Mutat Res. 2000. Apr;462(2‐3):83‐100. [DOI] [PubMed] [Google Scholar]
- 81. Agarwala SS, Kirkwood JM. Temozolomide, a novel alkylating agent with activity in the central nervous system, may improve the treatment of advanced metastatic melanoma. Oncologist. 2000;5(2):144‐151. [DOI] [PubMed] [Google Scholar]
- 82. Zhang J, Stevens MF, Bradshaw TD. Temozolomide: Mechanisms of action, repair and resistance. Curr Mol Pharmacol. 2012. Jan;5(1):102‐114. [DOI] [PubMed] [Google Scholar]
- 83. Thon N, Kreth S, Kreth FW. Personalized treatment strategies in glioblastoma: MGMT promoter methylation status. Onco Targets Ther. 2013. Sep 27;6:1363‐1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jovcevska I. Sequencing the next generation of glioblastomas. Crit Rev Clin Lab Sci. 2018. Jun;55(4):264‐282. [DOI] [PubMed] [Google Scholar]
- 85. Ohgaki H. Genetic pathways to glioblastomas. Neuropathology. 2005. Mar;25(1):1‐7. [DOI] [PubMed] [Google Scholar]
- 86. Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005. Mar 10;352(10):997‐1003. [DOI] [PubMed] [Google Scholar]
- 87. Inoue R, Isono M, Abe M, Abe T, Kobayashi H. A genotype of the polymorphic DNA repair gene MGMT is associated with de novo glioblastoma. Neurol Res. 2003. Dec;25(8):875‐879. [DOI] [PubMed] [Google Scholar]
- 88. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. 2015. Jun;129(6):829‐848. [DOI] [PubMed] [Google Scholar]
- 89. Zhao YH, Wang ZF, Cao CJ, et al. The clinical significance of O(6)‐methylguanine‐DNA methyltransferase promoter methylation status in adult patients with glioblastoma: A meta‐analysis. Front Neurol. 2018;9:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mellai M, Caldera V, Annovazzi L, et al. MGMT promoter hypermethylation in a series of 104 glioblastomas. Cancer Genomics Proteomics. 2009. Jul‐Aug;6(4):219‐227. [PubMed] [Google Scholar]
- 91. Hegi ME, Diserens AC, Godard S, et al. Clinical trial substantiates the predictive value of O‐6‐methylguanine‐DNA methyltransferase promoter methylation in glioblastoma patients treated with temozolomide. Clin Cancer Res. 2004. Mar 15;10(6):1871‐1874. [DOI] [PubMed] [Google Scholar]
- 92. Das P, Puri T, Jha P, et al. A clinicopathological and molecular analysis of glioblastoma multiforme with long‐term survival. J Clin Neurosci. 2011. Jan;18(1):66‐70. [DOI] [PubMed] [Google Scholar]
- 93. Madala HR, Punganuru SR, Arutla V, Misra S, Thomas TJ, Srivenugopal KS. Beyond brooding on oncometabolic havoc in IDH‐mutant gliomas and AML: Current and future therapeutic strategies. Cancers (Basel). 2018. Feb 11;10(2):49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mollemann M, Wolter M, Felsberg J, Collins VP, Reifenberger G. Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer. 2005. Jan 20;113(3):379‐385. [DOI] [PubMed] [Google Scholar]
- 95. Eoli M, Menghi F, Bruzzone MG, et al. Methylation of O6‐methylguanine DNA methyltransferase and loss of heterozygosity on 19q and/or 17p are overlapping features of secondary glioblastomas with prolonged survival. Clin Cancer Res. 2007. May 1;13(9):2606‐2613. [DOI] [PubMed] [Google Scholar]
- 96. Drabycz S, Roldan G, de Robles P, et al. An analysis of image texture, tumor location, and MGMT promoter methylation in glioblastoma using magnetic resonance imaging. Neuroimage. 2010. Jan 15;49(2):1398‐1405. [DOI] [PubMed] [Google Scholar]
- 97. Nicolis SK. Cancer stem cells and "stemness" genes in neuro‐oncology. Neurobiol Dis. 2007. Feb;25(2):217‐229. [DOI] [PubMed] [Google Scholar]
- 98. Ellingson BM, Cloughesy TF, Pope WB, et al. Anatomic localization of O6‐methylguanine DNA methyltransferase (MGMT) promoter methylated and unmethylated tumors: A radiographic study in 358 de novo human glioblastomas. Neuroimage. 2012. Jan 16;59(2):908‐916. [DOI] [PubMed] [Google Scholar]
- 99. Cheng W, Ren X, Cai J, et al. A five‐miRNA signature with prognostic and predictive value for MGMT promoter‐methylated glioblastoma patients. Oncotarget. 2015. Oct 6;6(30):29285‐29295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Gerber NK, Goenka A, Turcan S, et al. Transcriptional diversity of long‐term glioblastoma survivors. Neuro Oncol. 2014. Sep;16(9):1186‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Potter M, Newport E, Morten KJ. The Warburg effect: 80 years on. Biochem Soc Trans. 2016. Oct 15;44(5):1499‐1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ward PS, Thompson CB. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012. Mar 20;21(3):297‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002. Dec 9;1(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Krakstad C, Chekenya M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol Cancer. 2010. Jun 1;9(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Vlashi E, Lagadec C, Vergnes L, et al. Metabolic state of glioma stem cells and nontumorigenic cells. P Natl Acad Sci USA. 2011. Sep 20;108(38):16062‐16067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Zhou YF, Zhou Y, Shingu T, et al. Metabolic alterations in highly tumorigenic glioblastoma cells preference for hypoxia and high dependency on glycolysis. J Biol Chem. 2011. Sep 16;286(37):32843‐32853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wu N, Yang M, Gaur U, Xu H, Yao Y, Li D. Alpha‐ketoglutarate: Physiological functions and applications. Biomol Ther (Seoul). 2016. Jan;24(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dimitrov L, Hong CS, Yang C, Zhuang Z, Heiss JD. New developments in the pathogenesis and therapeutic targeting of the IDH1 mutation in glioma. Int J Med Sci. 2015;12(3):201‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ariazi J, Benowitz A, De Biasi V, et al. Tunneling nanotubes and gap junctions—Their role in long‐range intercellular communication during development, health, and disease conditions. Front Mol Neurosci. 2017;10:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lou E. Intercellular conduits in tumours: The new social network. Trends Cancer. 2016. Jan 1;2(1):3‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Mittal R, Karhu E, Wang JS, et al. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J Cell Physiol. 2019. Feb;234(2):1130‐1146. [DOI] [PubMed] [Google Scholar]
- 112. Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015; Dec 3;528(7580):93‐98. [DOI] [PubMed] [Google Scholar]
- 113. Davis DM, Sowinski S. Membrane nanotubes: Dynamic long‐distance connections between animal cells. Nat Rev Mol Cell Biol. 2008. Jun;9(6):431‐436. [DOI] [PubMed] [Google Scholar]
- 114. Gerdes HH, Bukoreshtliev NV, Barroso JF. Tunneling nanotubes: A new route for the exchange of components between animal cells. FEBS Lett. 2007. May 22;581(11):2194‐2201. [DOI] [PubMed] [Google Scholar]
- 115. Okafo G, Prevedel L, Eugenin E. Tunneling nanotubes (TNT) mediate long‐range gap junctional communication: Implications for HIV cell to cell spread. Sci Rep. 2017. Nov 30;7(1):16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Thome C, Blaes J, Sonner J, et al. N‐myc downstream regulated gene 1 (Ndrg1) influences the glioma tumor microenvironment. Neuro Oncol. 2016. Oct;18:2‐3. [Google Scholar]
- 117. Osswald M, Jung E, Weil S, et al. A perivascular niche for progression and resistance in glioblastoma. Neuro Oncol. 2016. Oct;18:48.26136493 [Google Scholar]
- 118. Pontes B, Viana NB, Campanati L, Farina M, Neto VM, Nussenzveig HM. Structure and elastic properties of tunneling nanotubes. Eur Biophys J. 2008. Feb;37(2):121‐129. [DOI] [PubMed] [Google Scholar]
- 119. Smith IF, Shuai J, Parker I. Active generation and propagation of Ca2+ signals within tunneling membrane nanotubes. Biophys J. 2011. Apr 20;100(8):L37‐L39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Vignais ML, Caicedo A, Brondello JM, Jorgensen C. Cell connections by tunneling nanotubes: Effects of mitochondrial trafficking on target cell metabolism, homeostasis, and response to therapy. Stem Cells Int. 2017;2017:6917941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Zhang Y. Tunneling‐nanotube: A new way of cell‐cell communication. Commun Integr Biol. 2011. May;4(3):324‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wang Y, Cui J, Sun X, Zhang Y. Tunneling‐nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011. Apr;18(4):732‐742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Jash E, Prasad P, Kumar N, Sharma T, Goldman A, Sehrawat S. Perspective on nanochannels as cellular mediators in different disease conditions. Cell Commun Signal. 2018. Nov 8;16(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hashimoto M, Bhuyan F, Hiyoshi M, et al. Potential role of the formation of tunneling nanotubes in HIV‐1 spread in macrophages. J Immunol. 2016. Feb 15;196(4):1832‐1841. [DOI] [PubMed] [Google Scholar]
- 125. Lu J, Zheng X, Li F, et al. Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget. 2017. Feb 28;8(9):15539‐15552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Zhang L, Zhang Y. Tunneling nanotubes between rat primary astrocytes and C6 glioma cells alter proliferation potential of glioma cells. Neurosci Bull. 2015. Jun;31(3):371‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Pasquier J, Guerrouahen BS, Al Thawadi H, et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med. 2013. Apr 10;11(1):94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Murray LMA, Krasnodembskaya AD. Concise review: Intercellular communication via organelle transfer in the biology and therapeutic applications of stem cells. Stem Cells. 2018. Oct 24;37(1):14‐25. [DOI] [PubMed] [Google Scholar]
- 129. Cho YM, Kim JH, Kim M, et al. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PLoS ONE. 2012;7(3):e32778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Babenko VA, Silachev DN, Zorova LD, et al. Improving the post‐stroke therapeutic potency of mesenchymal multipotent stromal cells by cocultivation with cortical neurons: The role of crosstalk between cells. Stem Cells Transl Med. 2015. Sep;4(9):1011‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Tan AS, Baty JW, Dong LF, et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015. Jan 6;21(1):81‐94. [DOI] [PubMed] [Google Scholar]
- 132. Dong LF, Kovarova J, Bajzikova M, et al. Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA‐deficient cancer cells. Elife. 2017. Feb 15;6:e22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Wang X, Gerdes HH. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015. Jul;22(7):1181‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Davis ME. Glioblastoma: Overview of disease and treatment. Clin J Oncol Nurs. 2016. Oct 1;20(5 Suppl):S2‐S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Jackson WC, Tsien CI, Junck L, et al. Standard dose and dose‐escalated radiation therapy are associated with favorable survival in select elderly patients with newly diagnosed glioblastoma. J Neurooncol. 2018. May;138(1):155‐162. [DOI] [PubMed] [Google Scholar]
- 136. Gupta K, Burns TC. Radiation‐induced alterations in the recurrent glioblastoma microenvironment: Therapeutic implications. Front Oncol. 2018;8:503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Autier L, Clavreul A, Cacicedo ML, et al. A new glioblastoma cell trap for implantation after surgical resection. Acta Biomater. 2018. Nov 19;84:268‐279. [DOI] [PubMed] [Google Scholar]
- 138. Angelucci C, D'Alessio A, Lama G, et al. Cancer stem cells from peritumoral tissue of glioblastoma multiforme: The possible missing link between tumor development and progression. Oncotarget. 2018. Jun 15;9(46):28116‐28130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Glas M, Rath BH, Simon M, et al. Residual tumor cells are unique cellular targets in glioblastoma. Ann Neurol. 2010. Aug;68(2):264‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018. Jul;15(7):422‐442. [DOI] [PubMed] [Google Scholar]
- 141. Lu VM, Phan K, Yin JXM, McDonald KL. Older studies can underestimate prognosis of glioblastoma biomarker in meta‐analyses: A meta‐epidemiological study for study‐level effect in the current literature. J Neurooncol. 2018. May 16;139(2):231‐238. [DOI] [PubMed] [Google Scholar]
- 142. Szopa W, Burley TA, Kramer‐Marek G, Kaspera W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. Biomed Res Int. 2017;2017:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Schiffer D, Mellai M, Bovio E, Annovazzi L. The neuropathological basis to the functional role of microglia/macrophages in gliomas. Neurol Sci. 2017. Sep;38(9):1571‐1577. [DOI] [PubMed] [Google Scholar]
- 144. Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015. Nov;17:9‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Yu K‐H, Beam AL, Kohane IS. Artificial intelligence in healthcare. Nature Biomedical Engineering. 2018. 2018/10/01;2(10):719‐731. [DOI] [PubMed] [Google Scholar]
- 146. Hasan MR, Hassan N, Khan R, Kim YT, Iqbal SM. Classification of cancer cells using computational analysis of dynamic morphology. Comput Methods Programs Biomed. 2018. Mar;156:105‐112. [DOI] [PubMed] [Google Scholar]
- 147. Sanghani P, Ang BT, King NKK, Ren H. Overall survival prediction in glioblastoma multiforme patients from volumetric, shape and texture features using machine learning. Surg Oncol. 2018. Dec;27(4):709‐714. [DOI] [PubMed] [Google Scholar]
- 148. Han L, Kamdar MR. MRI to MGMT: Predicting methylation status in glioblastoma patients using convolutional recurrent neural networks. Pac Symp Biocomput. 2018;23:331‐342. [PMC free article] [PubMed] [Google Scholar]
- 149. Liu Y, Xu X, Yin L, Zhang X, Li L, Lu H. Relationship between glioblastoma heterogeneity and survival time: An MR imaging texture analysis. AJNR Am J Neuroradiol. 2017. Sep;38(9):1695‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kickingereder P, Bonekamp D, Nowosielski M, et al. Radiogenomics of glioblastoma: Machine learning‐based classification of molecular characteristics by using multiparametric and multiregional MR imaging features. Radiology. 2016. Dec;281(3):907‐918. [DOI] [PubMed] [Google Scholar]
- 151. Weinstein JN, Collisson EA, Mills GB, et al. The cancer genome atlas pan‐cancer analysis project. Nat Genet. 2013. Oct;45(10):1113‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Ding L, Bailey MH, Porta‐Pardo E, et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell. 2018. Apr 5;173(2):305‐320. e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Malta TM, Sokolov A, Gentles AJ, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018. Apr 5;173(2):338‐354. e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Colman H, Zhang L, Sulman EP, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010. Jan;12(1):49‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Way GP, Allaway RJ, Bouley SJ, Fadul CE, Sanchez Y, Greene CS. A machine learning classifier trained on cancer transcriptomes detects NF1 inactivation signal in glioblastoma. BMC Genomics. 2017. Feb 6;18(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]