

Abstract
Background
The use of in vitro cell cultures is a powerful tool for obtaining key insights into the behaviour and response of cells to interventions in normal and disease situations. Unlike in vivo settings, in vitro experiments allow a fine‐tuned control of a range of microenvironmental elements independently within an isolated setting. The recent expansion in the use of three‐dimensional (3D) in vitro assays has created a number of representative tools to study cell behaviour in a more physiologically 3D relevant microenvironment. Complex 3D in vitro models that can recapitulate human tissue biology are essential for understanding the pathophysiology of disease.
Aim
The development of the 3D coculture collagen contraction and invasion assay, the “organotypic assay,” has been widely adopted as a powerful approach to bridge the gap between standard two‐dimensional tissue culture and in vivo mouse models. In the cancer setting, these assays can then be used to dissect how stromal cells, such as cancer‐associated fibroblasts (CAFs), drive extracellular matrix (ECM) remodelling to alter cancer cell behaviour and response to intervention. However, to date, many of the published organotypic protocols are low‐throughput, time‐consuming (up to several weeks), and work‐intensive with often limited scalability. Our aim was to develop a fast, high‐throughput, scalable 3D organotypic assay for use in oncology screening and drug development.
Methods and results
Here, we describe a modified 96‐well organotypic assay, the “Mini‐Organo,” which can be easily completed within 5 days. We demonstrate its application in a wide range of mouse and human cancer biology approaches including evaluation of stromal cell 3D ECM remodelling, 3D cancer cell invasion, and the assessment of efficacy of potential anticancer therapeutic targets. Furthermore, the organotypic assay described is highly amenable to customisation using different cell types under diverse experimental conditions.
Conclusions
The Mini‐Organo high‐throughput 3D organotypic assay allows the rapid screening of potential cancer therapeutics in human and mouse models in a time‐efficient manner.
Keywords: 3D model, cancer‐associated fibroblast, coculture, drug screening, extracellular matrix, organotypic
1. BACKGROUND AND RATIONALE
Mammalian tissues are highly complex microenvironments where a close interplay exists between the cellular and acellular compartments.1, 2, 3, 4 This complex and reciprocal interaction is technically challenging to reproduce in vitro as both healthy and diseased tissues co‐evolve over time. Furthermore, in vivo study of these complex tissue environments is time‐consuming and labour intensive, and experimental manipulation is typically limited.5, 6
Two‐dimensional (2D) in vitro culture models represent the mainstay of the majority of cell biology. Whilst these models have resulted in the vast knowledge accumulated over the past decades, it is now becoming increasingly apparent that data generated in these models typically do not extrapolate to the three‐dimensional (3D) setting.5 There are several reasons for this: 2D plastic surfaces are much stiffer in comparison to most tissues except bone and so do not capture the biomechanics of native tissue environments; 2D tissue culture also lacks the complex heterogeneous 3D nature of the Extracellular matrix (ECM), where extrinsic cues and clues are presented on nanoscopic, microscopic, and macroscopic scales to cells. Cell behaviour and phenotype are the complex integration of collective nonlinear interactions between cellular and environmental cues.7 Cells within 3D environments continually assimilate and process the information received leading to alterations in their intracellular signalling that governs cell fate.8 2D culture therefore cannot replicate crucial spatial compartmentalization of extrinsic signalling cues which constitutes a fundamental feature of the 3D ECM landscape and underpins the very essence of tissue and organ complexity.9
As a result, in the last few decades, there has been an explosion in the emergence of novel approaches to produce more physiologically relevant and tuneable 3D cultures, increasing not only the fidelity of these models, but also dramatically improving our ability to monitor and image cellular response enabling real‐time study.5, 10, 11, 12, 13, 14 Within these models, genetic, epigenetic, immunological, and chemical perturbation of malignant and nonmalignant cells and tissues can be tested against carefully controlled microenvironmental factors such as oxygen tension, pH, reactive oxygen species, growth factors as well as ECM biochemistry and biomechanics. These approaches are slowly bridging the gap between complex in vivo experiments and in vitro experiments. They allow us to answer not only key biological questions, but more importantly in disease models, it allows us to rapidly and reliably screen and test novel therapeutic approaches to increase the fidelity of preclinical portfolios. One caveat, however, is that many of these 3D in vitro model systems are time‐consuming and work‐intensive with often limited scalability. Thus, the miniaturisation and integration of organotypic culture models into high‐throughput assays can lead to the rapid evaluation of novel drugs in a cost‐effective and time‐effective manner.15, 16, 17, 18 From a clinical perspective, a cost‐efficient protocol to validate drug efficacy in ex vivo patient‐derived transplants could facilitate personalised decision making in oncology.19, 20, 21
Extracellular matrix deposition, contraction, and remodelling are common to diseases such as tissue fibrosis, diabetes, and tissue aging and importantly are salient features of almost all solid tumours.3, 22 Prior to and during malignant transformation and tumour expansion, the host stroma typically initiates a desmoplastic stromal response.1, 23, 24, 25, 26 This response leads to a significant deposition and remodelling of the ECM within and surrounding the tumour. Whilst still the topic of much debate as to the precise antitumourigenic and/or protumourigenic properties of this fibrotic reaction, it ultimately leads to a significantly altered tumour microenvironment both biochemically and biomechanically.27, 28, 29, 30, 31 The response is manifested by the presence of activated fibroblasts, either resident or recruited to the site of the developing tumour.1, 2, 32, 33, 34, 35, 36, 37, 38 These fibroblasts have been shown to be the major source of tumour desmoplasia, by secreting and remodelling ECM components including collagens. These cancer‐associated fibroblasts (CAFs) are now seen as critical in the progression of many solid tumours.2, 25, 32, 37, 39, 40, 41 Thus, given the critical importance of these stromal cells in tumour onset and progression, their inclusion in 3D cancer models is of utmost importance.
Here, we describe a modified organotypic assay, the “Mini‐Organo,” that adapts and improves previously published protocols10, 42, 43, 44 by significantly reducing the time for contraction and invasion to occur, so that it can be quickly scaled for large‐scale drug testing and completed within 5 days. We demonstrate its application in a wide range of mouse and human cancer biology approaches including evaluation of stromal cell (CAF) 3D ECM remodelling, 3D cancer cell (CC) invasion, and the assessment of potential anticancer therapeutic targets. Furthermore, the Mini‐Organo assay described is highly amenable to customisation using different cell types under diverse experimental conditions and so can easily be adopted to noncancer studies. An overview of the workflow for the Mini‐Organo is shown in Figure 1.
Figure 1.
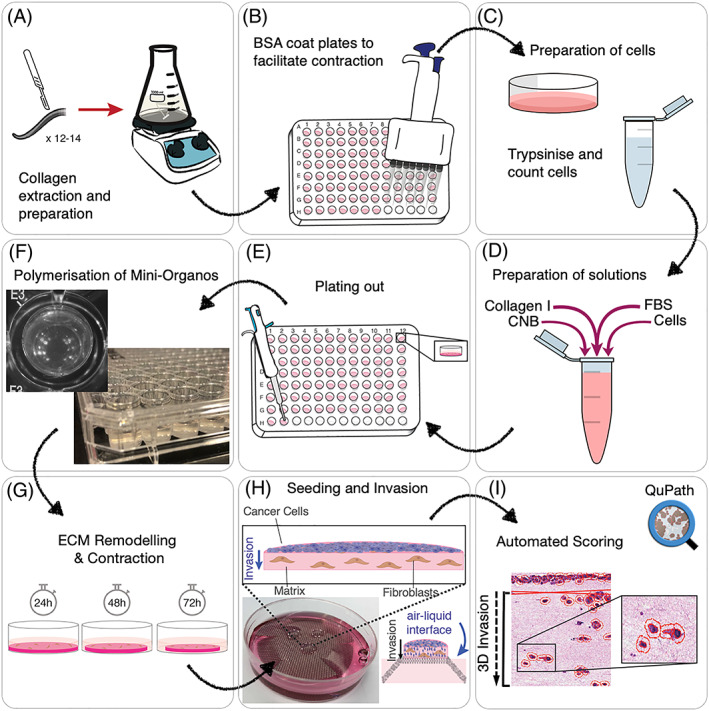
Schematic of workflow. A, Collagen extraction and preparation from rat tails for use in the Mini‐Organo. B, BSA coating of microwell plates to facilitate contraction of Mini‐Organos. C, Preparation of cells for seeding. D, Preparation of Mini‐Organo stock solution. E, Plating out of Mini‐Organos. F, Polymerisation of Mini‐Organos prior to contraction. G, Fibroblast‐driven remodelling and contraction phase. H, Transfer to air‐liquid interface and cancer cell seeding and invasion phase. I, Automated scoring
2. MATERIALS AND EQUIPMENT REQUIRED AND REAGENT SETUP
2.1. Preparation of native acid‐extracted collagen source (yields approx. 1 g of purified Collagen I)
This step details the in‐house extraction and preparation of large batches of high quality Collagen type I (hereafter “collagen I”) for use in the Mini‐Organos. It is not necessary to extract fresh collagen I for every experiment, and batches of extracted collagen can be sterilised and stored at 4°C for several months. If a suitable collagen I source is in place, steps 2a and 3a can be omitted. The “5‐day” Mini‐Organo does not include the generation of a stock collagen solution; however, for completeness, we have included this step. Here, we describe a protocol for extracting collagen I from rat tails, although other alternative methodologies and sources may be used.10, 42, 45
12 to 14 tails from rats of any age and/or strain.
Sterile (autoclaved) acetic acid solutions at 0.5 M (2 L), 0.25 M (0.5 L), and 17.4mM (~40 L) concentrations.
Filter paper (Chux, Whattman, or coffee filters—any sturdy filter paper for filtering out insoluble tail sheath).
Cooled centrifuge with ~200‐mL capacity buckets.
Toothed forceps.
Scalpels (no. 22.)
Dialysis tubing 14 000 MWCO
Sodium chloride (NaCl) salt.
Collagen quantification kit, Sircol assay (Biocolor Ltd. Cat #S1111), or similar.
2.2. Cell culture
-
Fibroblasts of interest, ie, cancer‐associated fibroblasts (CAFs) including predetermined optimal growth medium and incubation conditions. The choice of CAF used will affect the degree of contraction and also invasion. The number of CAFs and length of time for contraction of the matrices will need to be optimised for each CAF line used (described later).
-
○
In this work, we demonstrate ECM remodelling using the following CAF lines.
“KPC CAFs” derived from the autochthonous mouse model of the LSL‐KRasG12D/+, LSL‐Trp53R172H/+, Pdx1‐Cre (KPC) pancreatic ductal adenocarcinoma (PDAC).46
“KPflC CAFs” derived from the autochthonous mouse model of the LSL‐KRasG12D/+, LSL‐Trp53fl/+, and Pdx1‐Cre (KPC) PDAC.47
KPC CAFs are grown in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, penicillin‐streptomycin (10 000 U/mL penicillin G sodium and 10 000 μg/mL streptomycin sulphate). Cells are cultured at 37°C in a humidified incubator in atmospheric O2 supplemented with 5% CO2.
“KPC CCs” are derived from the autochthonous mouse model of The LSL‐KRasG12D/+, LSL‐Trp53R172H/+, and Pdx1‐Cre (KPC) PDAC.
KPC CCs are grown in DMEM containing 10% fetal bovine serum and penicillin‐streptomycin (10 000 U/mL penicillin G sodium and 10 000 μg/mL streptomycin sulphate). Cells are cultured at 37°C in a humidified incubator in atmospheric O2 supplemented with 5% CO2.
“HN‐CAFs” are derived from a human head and neck carcinoma, “V‐CAFs” are derived from a human vulval carcinoma, and “Cer‐CAFs” are derived from a human cervical carcinoma.
All human CAFs (HN‐V‐ and Cer‐CAFs) were collected from fresh patient tissue under approved ethical review and immortalised using the hTERT lentivirus. Further information and characterisation of these human CAFs have previously been published.48, 49, 50, 51
Human CAFs are grown in DMEM with high glucose, 10% fetal bovine serum, and penicillin‐streptomycin (10 000 U/mL penicillin G sodium and 10 000 μg/mL streptomycin sulphate) and supplemented with Gibco Insulin‐Transferrin‐Selenium (1×) solution. Cells are cultured at 37°C in a humidified incubator in atmospheric O2 supplemented with 5% CO2.
All cell lines were regularly tested for mycoplasma and for murine pathogens by IMPACT testing (IDEXX Laboratories, Inc.).
-
○
Reagents required
0.25%trypsin‐EDTA.
1x phosphate buffered saline (PBS).
Trypan blue for cell counting (0.4% stock solution).
Cell strainers (100, 70, and 40 micron).
2.3. Collagen I Mini‐Organo preparation and contraction
1% bovine serum albumin (BSA) in PBS.
Prepared rat tail collagen I at a minimum concentration of 2.5 mg/mL
Fetal bovine serum (FBS)
Collagen neutralisation buffer (CNB) made up of 0.5× minimal essential medium (MEM), 0.1 M HEPES buffer (pH 7.5), 238 mM NaHCO3.
Fibroblast optimised growth medium (as determined in 2b).
25 and 50‐mL plastic reservoirs.
96‐well cell culture‐treated plates
Wide, shallow ice tray for keeping plates cool during preparation of Mini‐Organos
2.4. Collagen I Mini‐Organo invasion
Stainless steel round screen mesh size 40 (sigma S0770). Each mesh is cut six times at ~25‐mm intervals, to a depth of 10 mm. This allows three arms to be bent down creating a platform approximately 3 to 4 mm high (see Figure 1H)
60 × 15‐mm cell culture dish
Sterile blunt forceps and/or micro spatula/spoon (0.5 × 0.5 × 0.7 mm) for transferring mini‐organos
Optimised growth medium.
2.5. Equipment needed
200‐μL and 1‐mL pipettes (incl. sterile tips).
Benchtop centrifuge at room temperature.
Cell counter (automated or haemocytometer).
Standard tissue culture incubator suitable for cells of interest.
Stepper pipette*.
Multichannel pipette*.
Charge‐coupled device (CCD) scanner capable of detecting a sample placed outside the ideal focal plane. Contact image sensor (CIS) scanners are not suitable due to the shallow scan depths.
* With the large amount of pipetting and smaller working volumes accompanying scaling of the Mini‐Organo, it is essential to use both a stepper pipette and multichannel pipette for accurate repeated measures to improve accuracy and reduce variability.
3. EXPERIMENTAL PROCEDURE
3.1. Preparation and quantification of high quality rat tail collagen
-
i
All work should be carried out using clean, sterile equipment. The use of a laminar flow hood is strongly advised, although not essential. Wash 12 to 14 rail tails (to give a total of 75‐100 g of tails) in 70% ethanol, and leave to soak to soften the tails but do not leave for longer than 30 minutes. To remove the collagen‐rich tendons, rotate a scalpel blade around the proximal region of the tail, carefully cut the skin of the tail along its length, and peel away the skin from the proximal cut (the skin should come off in one piece). Using toothed forceps, grip midway between the largest section of the tendon to maximise the tendon removed. Twist the tendon slightly to improve the grip and remove the tendon from the upper sheath, pulling towards the tip of the tail in a controlled movement. Take care not to remove too much accompanying sheath in the process. Once removed, collect the tendons into fresh MilliQ water. A visual demonstration of the approach has been previously published by Timpson and colleagues.42
-
ii
Solubilise the collagen I from the rat tail tendons by transferring from MilliQ to 1500 mL of 0.5 M acetic acid for at least 48 hours at 4°C. If after 48 hours large pieces of tendon are still visible, leave for another 24 hours. Use a magnetic stirrer to ensure constant mixing of the acetic acid and tendons. The solution should appear slightly viscous.
Note: Prior to solubilisation, the harvested tendons can be briefly snap frozen at −80°C for approximately 15 minutes. Once frozen, tendons can then be ground in a mortar and pestle to increase surface area and improve acetic acid dissolution of the collagen I in order to increase yield.
-
iii
Once solubilised (>48 hours), filter the collagen extract through wetted (in 0.5 M acetic acid) Whatman paper or equivalent filter to remove any remaining sheath that was transferred with the tendon. The resulting collagen I rich solution should have a thick consistency.
-
iv
Centrifuge the extracted collagen I solution at 10 000 rpm at 4°C for 1 hour. Carefully decant the supernatant and discard the pellet.
-
v
Transfer the collagen solution (supernatant) (1.5 L) to a clean 2‐L conical flask. Add NaCl salt to the supernatant to make a solution with final concentration 10% w/v NaCl solution (eg, 150 g to 1500 mL). This will precipitate out the collagen I from solution ready for collection.
Note: Be sure to add the NaCl crystals slowly, breaking up any clumps until they have fully dissolved. Do this slowly over in three to four intervals over a 30 to 60‐minute period with continuous stirring. Once all NaCl has been added and dissolved, the solution should be a homogenous, white‐opaque solution.
-
vi
Centrifuge the solution at 10 000 rpm at 4°C for 30 minutes. Carefully discard the supernatant.
-
vii
Re‐dissolve the precipitated collagen I pellet in 0.25 M sterile (autoclaved) cold (4°C) acetic acid at an approximate 1:1 volume ratio of acetic acid to precipitated collagen I for 24 hours at 4°C. This volume may be adjusted to ensure that the collagen is completely dissolved. Typically, 200 to 400 mL is required.
-
viii
Once re‐dissolved, dialyze the collagen solution against six to eight changes of 5 L of sterile 17.4 mM acetic acid (5‐mL glacial acetic acid [17.4 M] into 5 L of 4°C distilled water) for 3 to 4 days. Use a magnetic stirrer to increase dialysis efficiency. The dialysis solution should be replaced twice daily (~8‐12 hours between changes).
-
ix
Centrifuge the dialyzed collagen I at 14 000 rpm at 4°C for 1.5 hours to remove any insoluble matter. A small pellet may be visible, but this should be minimal.
-
x
Carefully decant or aliquot the collagen I solution into sterile containers.
-
xi
Collagen I concentration should be accurately determined using a kit such as the Sircol collagen quantification kit (BioColor Ltd) or a modified Lowry assay.52 With high concentrations of collagen, the higher volume of standards should be used and collagen diluted 1:10 or 1:20 to be within range.
-
xii
Adjust the collagen I concentration to 2.5 mg/mL (or desired concentration) using 0.5 mM acetic acid at 4°C.
-
xiii
Using a biological safety cabinet or equivalent UV light source, sterilise the collagen I solution for 20 minutes on ice before storing at 4°C. For long‐term storage, the solution should be lyophilized and stored at 4°C. Once this collagen stock is established, it is ready for use in the Mini‐Organo.
3.1.1. Anticipated results
A starting amount of 75 to 100 g of rat tails (representing an average of 12 rat tails weighing 6‐8 g each) should yield a final amount of approximately 1.0 to 1.5 g of purified collagen I consisting a volume of 500 mL at approximately 2.5 mg/mL collagen I. The extraction procedure above can be modified to yield higher concentrations of collagen by either increasing number of rat tails used and also by increasing the number of days dialysis (step 3a viii) is performed. However, after 4 days, the increase in yield is typically marginal. Alternatively, multiple batch extractions can be performed and pooled to create a central stock for large‐scale experiments. Contraction and/or invasion experiments described later may be affected by the batch of collagen I used and so it may be necessary to test each batch when working with multiple extractions over sustained periods of time.
It is important to keep the collagen I solution cold at all times to avoid unwanted degradation and polymerisation. A collagen I solution that is not translucent at stage viii will likely be of poor quality. Similarly, a final stock concentration of below 2.5 mg/mL will not be suitable for downstream applications described in this protocol. Higher stock concentrations can be generated, up to 4 to 5 mg/mL and may be necessary for some highly invasive CC lines (such as the fibrosarcoma cell line HT1080) or highly contractile CAFs.
The above protocol should yield approximately 1.0 to 1.5 g of purified acid‐extracted collagen I which is enough to produce approximately 30 to 50 separate 96‐well plates, equivalent to 3000 to 5000 experimental conditions.
3.2. Preparation of Mini‐Organo hydrogels
-
i
Contraction of the collagen gels is facilitated by ensuring that the gels are free from attachment to the 96‐well culture plate. To prevent the Mini‐Organo gels sticking to the well, first coat the wells with 1% BSA in sterile PBS for 1 hour in a tissue culture incubator at 37°C (or at 4°C overnight) (Figure 1B). Once incubated, remove the PBS/BSA solution and wash briefly with PBS twice before use. BSA‐coated plates can be stored in PBS in the fridge for 24 to 48 hours.
-
ii
De‐gas the collagen I solution under vacuum for 15 minutes. This step is important to prevent the formation of bubbles within the Mini‐Organos.
-
iii
Trypinise fibroblasts/CAFs according to normal passage protocol for your cell line (Figure 1C). Count the cells, resuspend in normal growth medium at the required concentration, and keep on ice. The precise cell number required per Mini‐Organo will need to be optimised prior to experimental work (optimisation described below in 3c. Anticipated Results).
-
iv
For 12 mL of collagen I Mini‐Organo mixture (enough for 1× 96‐well plate at 100 μL per well, including pipetting dead volumes), add the following components carefully and mix, taking care to avoid bubbles (Figure 1D). Ensure that the following components are kept on ice. Mix the reagents in a prechilled container on ice, adding the collagen I solution as the last step. Once the collagen I solution is neutralised, it will begin to gel unless kept at 4°C. Due to the viscosity of the collagen I Mini‐Organo solution, it is reasonable to expect to lose approximately 15% volume of the initial starting solution as dead volume during transfer to and from reservoirs. This should be taken into consideration when scaling up or down the protocol. If bubbles appear during the preparation of the gel, a brief pulse centrifugation at 4°C should remove them.
0.96‐mL CNB
1.20‐mL FBS (100%)
2.64‐mL fibroblasts/CAFs in normal growth media (or just normal growth media for acellular controls)
7.2 mL of 2.5 mg/mL extracted collagen I solution (or equivalent volume to yield a final concentration of 1.5 mg/mL collagen I [or desired concentration])
Note 1
It is important to ensure correct neutralisation of the acid‐extracted collagen at this point. The MEM within the CNB contains phenol red, and so the final gel solution should be pink/orange in colour. A yellow colour implies a pH of close to 6.0 and that the collagen I solution has not been correctly neutralised.
Note 2
It is possible to mix in additional ECM components to the Mini‐Organo mixture at this stage to test effects on fibroblast contractile behaviour. For example, the addition of ECM components such as fibronectin may increase the contraction of some fibroblast lines.
-
v
Using the stepper pipette, add 100‐μL collagen I/cell hydrogel to each well in a BSA‐coated 96‐well culture plate, taking care not to introduce bubbles (Figure 1E). Tip: drawing up and emptying the tip a couple of times will create a small dead volume (5‐10 μL). This will allow a more precise volume to be transferred to each well of the plate and, at the same time, avoid the formation of bubbles during pipetting. Pipetting the collagen against the side of the well and allowing it to run down into the well will also reduce the likelihood of generating bubbles.
-
vi
Allow gels to polymerise for 1 hour in the incubator (37°C). To maintain consistent pore sizes across different experiments, it is essential to ensure a standardised polymerisation protocol53, 54, 55 (Figure 1F).
-
vii
Once polymerised, wash the Mini‐Organos by adding 100‐μL normal growth media for 1 hour in the incubator at 37°C.
-
viii
Carefully aspirate the growth medium using a multichannel pipette (do not use vacuum aspiration).
-
ix
Add new growth media (+/− experimental interventions [growth factors/drugs] and incubate at 37°C.
-
x
Monitor and/or image contraction of Mini‐Organos using a CCD flatbed scanner capable of detecting a sample placed outside the ideal focal plane. CIS flatbed scanners are not suitable due to the shallow scan depths. The use of a flatbed scanner to image from the bottom of the plate is preferred and gives more accurate determination of degree of contraction. Images can be taken as often as necessary but should typically be carried out at least once every 24 hours.
-
xi
To obtain a quantitative measure for the level of Mini‐Organo contraction, the relative area of the well and the gel are measured using ImageJ software (as depicted in Figure 2A,B). The percentage of contraction is calculated using the following formula:
Figure 2.
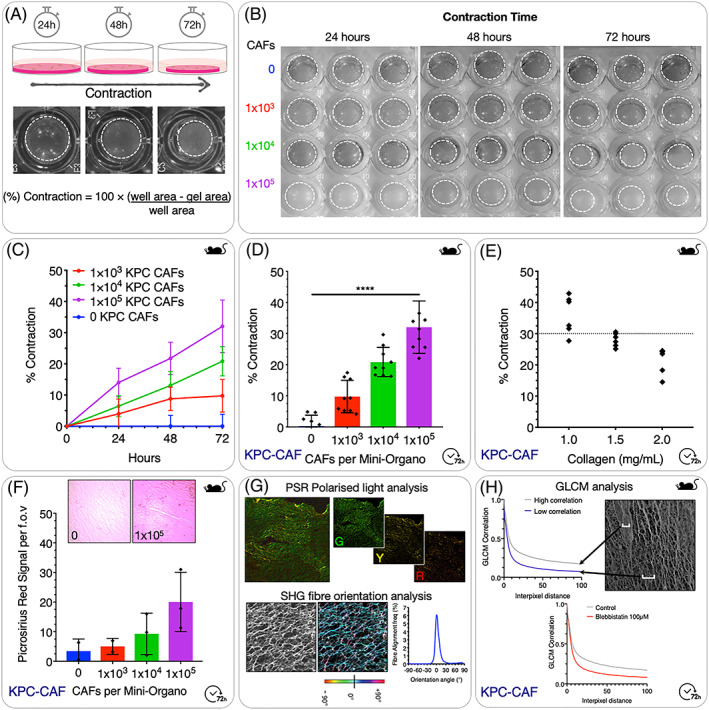
Optimisation of the Mini‐Organo contraction assay. A, Schematic for remodelling and contraction phase including representative images at 24‐hour, 48‐hour, and 72‐hour time points. B, Examples of scanned plates seeded with increasing numbers of KPC‐CAFs during contraction and remodelling phase at 24, 48, and 72 hours (Dotted lines indicate gel circumference). C, Quantification of contraction over time for different seeding densities of KPC‐CAFs. D, Quantification of final level of contraction at 72 hours is dependent on initial seeding cell number. E, Quantification of final level of contraction at 72 hours is also dependent on starting concentration of collagen I. F, Total Picrosirius Red (PSR) signal from remodelled Mini‐Organos increases as the degree of contraction increases. G, (Top) Picrosirius Red (PSR) polarised light analysis can be employed to determine density and bundling of the remodelled collagen fibres, based on birefringent properties of dye bound collagen (Red—Thick fibres; Green—thin fibres). (Bottom) Collagen fibre orientation analysis using second harmonic generation (SHG) two‐photon images can be used to quantify changes in alignment of collagen fibres during contraction. H, Grey level co‐occurrence matrix (GLCM) analysis of second harmonic generation (SHG) two‐photon images of collagen fibres can be used to map changes in fibre organisation and texture.
3.2.1. Anticipated results
The Mini‐Organos will set within 1 hour at 37°C at which time the gels should go from clear to slightly opaque (Figure 1F). It is important to ensure that the Mini‐Organos, once set, are free from bubbles. The best way to ensure this is to de‐gas the collagen I solution so that the mixed solution is free from large bubbles. Also, avoid vigorous pipetting because large bubbles will affect contraction and subsequent invasion.
The small size of the Mini‐Organo matrices requires a high degree of precision during casting as discrepancies will lead to variations in the degree of contraction. A stepper pipette will improve the accuracy of pipetting; however, a dead volume of 100 to 150 μL should be taken into consideration.
3.3. Stromal cell remodelling of the ECM: Mini‐Organo contraction assay
The first time a fibroblast/CAF line is used in the Mini‐Organo, it is essential to standardise the number of cells seeded within each hydrogel. During all optimisations, control plugs with 0 cells are necessary as a baseline for normalisation.
Once normal growth media (+/− experimental interventions [growth factors/drugs]) have been added (step 2b ix), the plates can be incubated 37°C in the requisite tissue culture incubator. Media should be changed as needed. Plugs with high numbers of cells may require media changes more frequently.
It is important to determine an optimal fibroblast/CAF seeding density that allows reproducible determination of the remodelling of the Mini‐Organos and for subsequent downstream invasion assays. This will depend on three factors: (a) initial cell seeding number, (b) length of time allowed to contract, and (c) initial starting concentration of collagen I within the plug. For short‐term experiments, such as drug screens, we recommend a scenario where the gels have contracted 30% by 72 hours. To begin with, serial dilutions of fibroblast/CAF number should be used. Because the same plate can be imaged repeatedly over several days, it is possible to determine both initial seeding number and optimal contraction time in a single experiment.
In the example illustrated in Figure 2B,C, we show the effect of changing KPC‐CAF number and incubation time on levels of contraction over 72 hours. As would be expected, higher numbers of CAFs seeded within the Mini‐Organos lead to a greater rate of contraction, which continues over the 72‐hour incubation period. Typically, depending on cell number seeded, the maximal rate of hydrogel contraction occurs within the first 72 hours, after which the rate of contraction will slow and ultimately plateau.
3.3.1. Anticipated results
The contraction of the fibroblast embedded collagen I plugs should be visible by 24 hours at 37°C after generating the matrices. The degree of contraction will be highly dependent on the initial concentration of CAFs seeded to the matrix (Figure 2B‐2D), and also the activation status of the fibroblasts/CAFs being used, as well as interventions being applied. Furthermore, the initial starting concentration of collagen will also alter the level of contraction seen at different time points (Figure 2E). This combination of factors will ultimately determine the resulting porosity of the final contracted Mini‐Organo, and as a result the ability of CCs to invade.53
If using a fibroblast/CAF line for the first time, an assessment of contractile behaviour should be undertaken and contraction curves generated (Figure 2C). This will allow determination of optimal seeding density to reach an approximate 30% to 50% contraction of the plugs prior to seeding of CCs. Typically, no significant additional contraction of the matrix is seen once the gels are transferred to the stainless steel grids at the air‐liquid interface (Section 3d: Mini‐Organo Invasion Assay).
Analysis of the extent of remodelling by embedded CAFs can be undertaken at this stage. For example, Picrosirius Red staining is a simple and effective way to determine collagen density within the plugs (Figure 2F). The specific staining of collagens by Picrosirius red is one of the most important stains to study collagen networks.56, 57 This approach also has the added advantage that under polarised light, the dye bound collagen fibre bundles appear green, red, or yellow and are easily differentiated from the black background (Figure 2G [top]). This allows for a quantitative morphometric analysis of the collagen fibres where the polarised colours reflect fibre thickness and packing (Figure 2G [top]). This approach has been applied by us and many other groups to look at collagen fibre remodelling in organotypic studies as well as in normal and diseased tissues.10, 58, 59 Finally, second harmonic generation (SHG) imaging is a label‐free imaging technique that allows imaging of noncentrosymmetric entities such as collagen fibres. The generation of an SHG signal occurs when two photons with the same wavelength fuse into a single photon with half of the original wavelength upon interaction with a non‐centrosymmetric entity. This approach allows for high resolution imaging of collagen fibres. Using SHG‐generated images, orientation of collagen fibres can be assessed (Figure 2G [bottom]).10, 58, 59, 60, 61 In addition, collagen fibre network organization can also be characterised using grey‐level co‐occurrence matrix (GLCM) analysis.10, 58, 59, 61, 62, 63 This method provides a readout of the texture of a sample by quantifying the similarity between pixels across an image64 (Figure 2H, data example shown is from blebbistatin treated CAFs [Figure 4B]).
Figure 4.

Targeting CAFs and stromal remodelling. A, Cancer associated fibroblasts derived from two matched models, the tumourigenic and highly invasive KPC (KPC‐CAFs), and the tumourigenic but poorly invasive KPflC (KPCfl‐CAFs) models of pancreatic ductal adenocarcinoma show differences in their ability to remodel the Mini‐Organos [2.5 mg/mL Col I] reflecting the differing aggressiveness of these two models. B, Treatment of KPC‐CAFs (100 000 per Mini‐Organo [2.5 mg/mL Col I]) with the nonmuscle Myosin II inhibitor Blebbistatin shows a dose‐dependent inhibition of the ability of KPC‐CAFs to remodel and contract the Mini‐Organos. C, Treatment of KPC‐CAFs (100 000 per Mini‐Organo [2.5 mg/mL Col I]) with Blebbistatin; the ROCK inhibitor Fasudil or the PHD inhibitor DMOG (which mimics hypoxia) leads to a significant reduction in the contraction of the Mini‐Organos. D‐F, Treatment of (D) human HN‐CAFs (70 000 per Mini‐Organo [3.5 mg/mL Col I + 20% Matrigel]), (E) human V‐CAFs (50 000 per Mini‐Organo [3.5 mg/mL Col I + 20% Matrigel]), and (F) human Cer‐CAFs (50 000 per Mini‐Organo [3.5 mg/mL Col I + 20% Matrigel]) with inhibitors of intracellular force generation (10‐μM Y‐27632, 10‐μM Blebbistatin, 10‐μM Mevastatin, or 10‐μM Verteporfin) show differential effects between the different tumour derived CAFs. G, Treatment of human HN‐CAFS (50 000 per Mini‐Organo [3.5 mg/mL Col I + 20% Matrigel]) and human V‐CAF (40 000 per Mini‐Organo [3.5 mg/mL Col I + 20% Matrigel]) with 2 ng/mL exogenous TGFβ leads to activation and increased contraction. H‐I, Exposure to hypoxia decreases CAF activation leading to a reduced contractile ability, which can be rescued by the addition of exogenous TGFβ in both lines.
3.4. 3D cancer cell invasion: Mini‐Organo invasion assay
Following contraction, it is then possible to use the Mini‐Organos as a physiologically relevant 3D assay to measure CC local invasion into the remodelled collagen I rich interstitial matrix. The protocol involves seeding a known number of CCs to the top of the Mini‐Organo placed at an air‐liquid interface (Figure 1H). If desired, it is possible to seed CCs on top of the gels immediately after polymerisation and washing (prior to contraction). This allows a simultaneous CAF‐CC 3D system to be exploited (simultaneous contraction/invasion) but requires a growth medium compatible with both cell types. Furthermore, it is also possible to kill the CAFs within the contracted Mini‐Organo prior to the addition of the CCs. The CAFs can be killed using either puromycin or detergent extraction48, 65 followed by extensive washing.
-
i
Prepare CCs according to standard passaging protocol to yield a cell suspension at a desired concentration. This will need to be determined in preliminary experiments. As a guide, cell numbers within the range 1 × 103 to 5 × 104 CCs per Mini‐Organo are a good starting point. Initially, seeding more CCs does not increase the absolute number of cells invading into the Mini‐Organo (Figure 3F).
Figure 3.
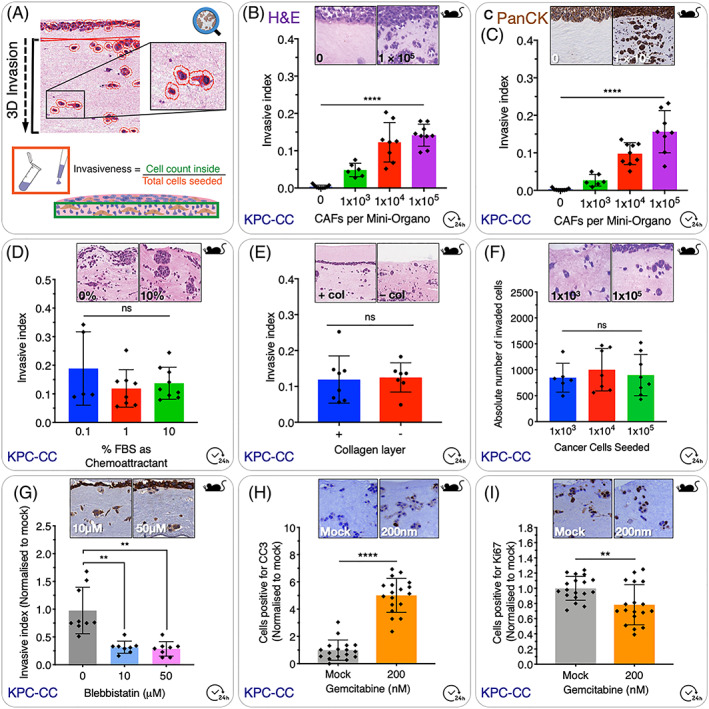
Cancer cell invasion assay. A, Schematic overview of QuPath‐based scoring system. The number of cells which have invaded into the gel is calculated as an index of the total number of cells seeded to the top of the gel. B, Increasing the number of CAFs seeded to the Mini‐Organo during the contraction phase leads to an increase in the invasion of KPC cancer cells into the Mini‐Organo at the same timepoint. C, In addition to simple haematoxylin and eosin staining (see panel B), it is possible to use immunohistochemical staining for markers of interest such as for pan‐cytokeratin (which specifically stains the epithelial tumour cells) with data recapitulating that seen in H&Es. D, The effect of chemoattractants (such as FBS) added to the media can be determined using the Mini‐Organo. E, Some (not all) cancer cell lines will invade more readily to a thin layer of 1.5 mg/mL collagen I overlaid on top following transfer to the grids (+ col vs − col). KPC‐CCs invade regardless of the presence of this additional top layer. F, Increasing the number of cancer cells seeded on top of the plug does not increase the absolute number of cancer cells invaded into the plug; however, it will decrease the invasive index accordingly (see formula in Figure 3A). G, Therapeutic targeting of cancer cell invasion can also be tested in Mini‐Organos. Treatment of KPC‐CCs with the non‐muscle Myosin II inhibitor Blebbistatin during the invasion stage (drug not present during contraction phase) and shows significant decreases in cancer cell ability to invade into the remodelled Mini‐Organos. H, Evaluation of the effects of anticancer therapies on invaded cancer cells can also be tested. Treatment of KPC‐CC with standard‐of‐care gemcitabine leads to an increase in Cleaved Caspase3 (CC3)(Apoptosis). I, decrease in Ki67 (proliferation) as measured by IHC staining
Note: It is important to ensure that a 0 CAF control is used in experiments to determine the intrinsic capacity of tumour cells to invade independently of CAFs (discussed later).
-
ii
Carefully remove media from contracted Mini‐Organos using a pipette. Avoid using a suction pump. Add 100 μL of CC suspension containing the desired number of cells.
Note: It is important to ensure an even distribution of the cell solution across the surface of the Mini‐Organo.
-
iii
Incubate for 12 to 24 hours under standard tissue culture conditions to allow cells to adhere to the contracted Mini‐Organo.
-
iv
Place sterilised stainless steel grids into a 60 × 15‐mm petri dish or six‐well plate.
-
v
Using sterilised blunt forceps, carefully transfer the Mini‐Organos from the 96wp onto the top of the stainless steel grids (Figure 1H). Multiple Mini‐Organos that are to be treated under the same conditions can be placed onto the same grid. For each separate treatment conditions, a separate dish and grid is required (Figure 1H).
-
vi
Once transferred, add complete growth media to the bottom of the dish until it just covers the base of the Mini‐Organos. The growth medium can be changed every day and will act as a chemoattractant. This growth media can also contain drugs/inhibitors/activators of interest (Figure 1H).
Optional step: The Mini‐Organos should sit at the air‐liquid interface with the top layer of CCs exposed to the air. In some studies, this may not be physiologically relevant, ie, for breast CCs, and so the top layer of CCs can be covered with an additional thin layer of collagen I (Figure 3E), to avoid direct air contact. This may not be necessary for all cell lines.
-
vii
The time taken for invasion will depend on the aggressiveness of the CC line in question and should ideally be within 72 hours. This can be optimised by varying the starting concentration of collagen I, the degree of contraction by CAFs, the number of CCs seeded, and the concentration of chemoattractant and/or inhibitors used (see examples in Figure 3). As mentioned above and discussed below, a 0 CAF control should be incorporated into each experiment.
3.4.1. Anticipated results
During the invasion step, the CCs should invade into the remodelled collagen I plug along the chemotactic gradient established by placing the Mini‐Organo on top of the grid.
Changing concentration of collagen I within the plug will alter the degree of invasion into the Mini‐Organo, as will the time allowed for invasion. If the Mini‐Organos are left too long, or the collagen concentrations are too low, CCs may invade all the way through the plug. If collagen concentrations are too high, or invasion times are too short, then the CCs may only invade a small distance into the Mini‐Organo or not at all. Altering the extent of initial contraction of the Mini‐Organos will also alter the extent to which CCs can invade into the matrix. Typically, 0 CAFs seeded to the Mini‐Organo result in no invasion by CCs, although very low concentrations of collagen or some highly aggressive CCs (such as the fibrosarcoma line HT1080) may still invade, and so a 0 CAF control is required to determine the intrinsic capacity of the tumour cells to invade independently of CAFs.
A pilot experiment to optimise the parameters that lead to approximately 50% invasion into the Mini‐Organo will allow for testing of inhibitory or stimulatory interventions. In situations where a drug, or drug(s) of interest may take more than 24 hours to act, or may need to be given sequentially on separate days, it will be necessary to optimise invasion conditions to yield a slower rate of invasion to accommodate for this.
3.5. Scoring and analysis
To determine the extent of invasion of CCs into the Mini‐Organos, they need to be fixed and processed for histological and immunohistochemical analysis. A wide range of readouts including invasion, proliferation, survival, or differentiation can be conducted (see Figure 3).
Triplicates of Mini‐Organo conditions are cut using a scalpel to generate a flat edge (approximately one‐third of the plug is removed) and placed side‐by‐side in a cassette and fixed in 10% buffered formalin or 4% paraformaldehyde plus 0.25% glutaraldehyde in PBS overnight, followed by transfer to 70% ethanol.
Once fixed, triplicate Mini‐Organos are processed and paraffin embedded flat edge down, so that when sectioned, a lateral plane through the entire Mini‐Organo is generated to provide a complete section of the Mini‐Organo.
Section the Mini‐Organos at 4 to 5 μm using a standard microtome. Three sections per plug, spaced >100 μm apart should be scored to generate an accurate invasive index.
The choice of staining of the sections will depend on the question at hand. Mini‐Organo sections can be stained with haemotoxylin and eosin (H&E) for visualising and scoring invasion (examples shown in Figure 3B and 3D‐3F). For some cell lines, specific markers can also be used such as pan‐cytokeratin (Figure 3C,G) to distinguish epithelial CCs from stromal CAFs. This may be particularly useful when using large numbers of CAFs per plug, and in particular where automated cell counting is being carried out. Additional makers such as Ki67 for proliferation, or cleaved‐caspase‐3 for apoptosis can also be used to monitor drug response of CCs that have invaded into the Mini‐Organos (Figure 3H,I).
Once stained, the whole Mini‐Organo should be imaged using either a slide scanner or tiling microscope at >20× magnification. It is recommended that scoring of the entire plug be undertaken rather than randomly chosen ROIs for an accurate quantification of invasive index. Given the smaller size of Mini‐Organos, analysis of the whole plug is readily achievable. However, care should be taken when scoring the very outer vertical edges of the gel where migration down the surface of the gel may have occurred. When classifying whether a cell has invaded, only include cells which have entered into the surface of the Mini‐Organo and travelled at least 25 μm from the plug surface. For accurate scoring, plugs should be fixed and scored whilst cells are still invading rather than left for longer at which point they may have invaded all the way through.
Analysis of cell number can be done in a number of ways using automated image analysis software, for example QuPath66 (Figures 1I and 3A) or FIJI.67 Depending on the software being used, two counting areas are outlined. The first is drawn to include all CCs within the Mini‐Organo. The upper boundary of this area is drawn approximately 25 μm from the plug surface to which CCs were seeded. The second counting area is drawn to exclude CCs still on top of the Mini‐Organo that have not invaded in. Following this, the cell detection is programmed to automatically count the cells that are within the Mini‐Organo. The automation of cell counting is advantageous in highly invasive cells lines as this enables rapid segmentation and identification of cells to determine invasive index in a time‐efficient manner.
Cell segmentation settings will be dependent on the intensity of the stain being used, the magnification of the image, and the software of choice. These should be optimised on a series of pilot Mini‐Organos prior to any unsupervised analysis.
In order to determine the invasive index, a score is calculated as the number of cells detected within the whole Mini‐organo section divided by the total number of CCs seeded (Figure 3A). An alternative and often‐used metric is to calculate the invasive index between the noninvaded upper layer of cells (on top of the gel) and the total number of invaded cells. Finally, a third approach is to calculate the area of the “noninvasive” cell layer (on top) and the “invasive area” (from the top layer down to the leading edge of the invasive cells), and normalise the invasive area with total area (noninvasive + invasive).48
Note: All slides should be stained simultaneously for comparison at the same time to avoid adjusting software parameters for each slide. Automated nucleus detection parameters are also dependent on magnification.
3.5.1. Anticipated results
The invasion of CCs into the remodelled collagen plugs can be determined by H&E staining (Figure 3B and 3D‐3F) or using a cell specific immunohistochemistry stain for a specific subset, or all CCs using markers such as pan‐cytokeratin (Figure 3C,G) as we have done previously.10, 58 Various elements can be changed to determine the effects on CC invasion. For example, standard growth serum (FBS) should drive the invasion of most CCs. Altering FBS levels may affect invasion of some CC lines. In our example, we see that the aggressive KPC CCs invade independent of FBS concentration (Figure 3D). Treatment of invaded CCs with various anticancer therapies is facilitated and allows for screening of difference combinations. Here, we show the effects of Blebbistatin on KPC CC invasion (Figure 3G) and gemcitabine, the standard‐of‐care therapy for pancreatic cancer on invaded KPC CC viability. Readouts include measurement of proliferation (Ki67 staining) (Figure 3H) and apoptosis (CC3) (Figure 3I) using our previously published staining protocols.10, 58
4. PRACTICAL APPLICATIONS OF THE MINI‐ORGANO IN CANCER BIOLOGY
The traditional organotypic contraction assays previously described10, 42 typically take 8 to 12 days to contract, and another 10 to 14 days for invasion. The Mini‐Organo has been optimised to significantly reduce the equivalent time needed, requiring only 72 hours for contraction and 24 hours for invasion representing an 80% reduction in time required. The Mini‐Organo allows for the same analysis of the extent and degree of ECM (collagen I) remodelling and organisation using both standard histological approaches such as Picrosirus red staining (Figure 2F,G) and SHG imaging for fibrillar collagen (Figure 2G,H). Here we have shown the application of Picrosirius Red staining, SHG imaging, orientation and alignment analysis,10, 60 and grey level co‐occurrence matrix (GLCM) analysis10, 58 as examples of a few of the approaches that can be applied (Figure 2F‐2H).
The contractile phase of the Mini‐Organo allows for the rapid controlled testing of interventions on fibroblast/CAF ability to remodel a collagen I rich ECM (see Figure 4). Because collagen I represents the major interstitial ECM component of most tissues, including many solid tumours, the Mini‐Organo offers a physiologically relevant and powerful approach to assessing potential stromal cell targeting in the cancer context. Targeting ECM remodelling as an approach to treat solid tumours is an exciting emerging concept that is showing promise in several settings.3, 22, 25, 41, 58, 68
4.1. Targeting and assessing stromal driven collagen remodelling
Not all CAFs should be considered equal. To highlight this, we have taken CAFs derived from two matched genetically engineered mouse models, the tumourigenic and highly invasive KPC (KPC‐CAFs), and tumourigeneic but poorly invasive KPflC (KPCfl‐CAFs) model of PDAC. CAFs isolated from these two models show differences in their ability to remodel the Mini‐Organos reflecting the differing aggressiveness of these two GEM models (Figure 4A). To further demonstrate the potential of testing antistromal cell drugs in the Mini‐Organo, we demonstrate the effects of three well‐known anticontractile agents: the nonmuscle Myosin II inhibitor Blebbistatin, the potent Rho‐associated coiled coil forming protein serine/threonine kinase (ROCK) inhibitor Fasudil58, and the competitive inhibitor of prolyl hydroxylase domain‐containing proteins (PHDs); DMOG on the KPC (murine pancreatic cancer CAFs) (Figure 4B‐4C, quantification of SHG‐GLCM analysis of blebbistatin treated Mini‐Organos also shown as example in Figure 2H). The addition of anticontractile drugs leads to a significant reduction in the contraction of the Mini‐Organos (Figure 4C) in a dose‐dependent manner (Figure 4B).
In addition, we evaluated the effects of two YAP/TAZ inhibitors, Mevastatin and Verteporfin, as well as the actomyosin inhibitor Blebbistatin, and a second highly potent selective ROCK inhibitor, Y‐27632 in 3 human patient‐derived CAF cell lines from head and neck cancer (HN‐CAF), vulval cancer (V‐CAF), and cervical cancer (Cer‐CAF).48, 49, 50, 51 Our data show that HN‐CAFs and V‐CAFs respond to YAP/TAZ, actomyosin, and ROCK inhibitors in the same manner. Interestingly, Cer‐CAFs respond to YAP/TAZ inhibitors similar to HN‐CAFs and V‐CAFS; however, they show no response to the actomyosin inhibitor Blebbistatin and the ROCK inhibitor Y‐27632 (Figure 4D‐4F). This data highlight how not all CAFs should be considered equal and reinforces the notion that cancer coculture studies should ideally be paired with suitable matching fibroblast/CAF lines that are relevant to the cancer type being studied, in order to maximise the fidelity of experimental systems.
CAFs are present at all stages of cancer and are believed to be the main cell type responsible for regulating the desmoplastic reaction in cancer. Dissecting the mechanisms underlying CAF activation has received much attention in the last decade.37, 69 As such, CAFs are believed to be promising targets in cancer. Here, we use our 3D Mini‐Organo to demonstrate that it is an excellent tool to explore the mechanisms behind CAF activation.
Transforming growth factor (TGFβ) is the most potent fibrogenic cytokine known,70, 71, 72, 73 and it is now well‐recognised that TGF‐β1 plays an important role in fibroblast activation, proliferation, and fibrotic ECM remodelling. In cancer, sustained CAF activation is partly maintained through autocrine secretion and stimulation of TGF‐β1 and SDF‐1α. As expected, exogenously added TGF‐β1 induces CAF activation and gel contraction of two distinct types of human CAFs (Figure 4G). Recent work has also demonstrated that prolonged exposure to hypoxia reduces CAF activation and ECM remodelling.51 We confirmed this finding in our model system (Figure 4H,I) and further show that hypoxic suppression of CAFs can be rescued by the addition of exogenous TGF‐β1 (Figure 4H,I). These simple 3D experiments demonstrate that the Mini‐Organo is a time‐efficient and physiologically relevant model system that can be used not only in drug screens, but also in dissecting molecular mechanisms regulating CAF activation and ECM remodelling and tumour progression.
5. CONCLUDING REMARKS
Here, we present a method for the rapid and scalable production of 3D physiologically relevant coculture assays suitable for dissecting both stromal remodelling and CC invasion. One of the major goals in designing 3D physiologically relevant in vitro approaches is to capture the complexity of intercellular communication, nutrient and oxygen gradients, as well as cell polarity and interactions with the ECM that is lacking in more traditional 2D monolayer cultures. The Mini‐Organo offers a simple, straightforward, scalable, and cost‐effective way of doing this.
Given the enormous number of new anticancer drugs and the accompanying exponentially increasing number of possible drug combinations, the use of in vivo models is prohibitively expensive. Therefore, approaches such as the “Mini‐Organo” offer a bridge between the two that allows for higher throughput screening of molecules and interventions within the standard laboratory environment for implementation into the clinic, not only in the cancer field but also many other diseases.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHORS' CONTRIBUTIONS
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, T.R.C., C.D.M.; Methodology, T.R.C., C.D.M.; Investigation, C.D.M., J.L.C., E.C.F., C.R.C., S.W., J.N.S., R.D.G., G.M., M.T., M.P., M.Y., C.V., A.Z., A.D.S.; Formal analysis, T.R.C., C.D.M., J.L.C., E.C.F., C.R.C., S.W., J.N.S.; Resources, T.R.C., C.D.M.; Writing ‐ Original Draft T.R.C., C.D.M. J.L.C.; Writing—review and editing, T.R.C., C.D.M., J.L.C., E.C.F., P.T.; Visualisation, T.R.C., C.D.M., J.L.C., E.C.F., C.R.C., S.W., J.N.S., R.D.G.; Supervision, T.R.C., C.D.M., P.T.; Funding acquisition, T.R.C., C.D.M.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request.
ACKNOWLEDGEMENTS
This work was supported by the NHMRC (T.R.C., E.C.F., J.L.C., R.D.G., and M.Y.), a Cancer Institute NSW (CINSW) fellowship (T.R.C., J.S.), Cancer Council NSW (T.R.C), and a Susan G Komen Career Catalyst (T.R.C.). T.R.C. is a recipient of an NHMRC RD Wright Biomedical Career Development Fellowship. C.D.M is supported by the Ragnar Söderberg Foundation, BioCARE, Cancerfonden, the Åke Wiberg foundation, Swedish Research Council, and the Crafoord Foundation. P.T. is a recipient of an NHMRC Senior Research Fellowship and NHMRC project grant. C.V. is a recipient of a post‐doctoral HFSP fellowship. This project was made possible by an Avner Pancreatic Cancer Foundation Grant.
Chitty JL, Skhinas JN, Filipe EC, et al. The Mini‐Organo: A rapid high‐throughput 3D coculture organotypic assay for oncology screening and drug development. Cancer Reports. 2020;3:e1209. 10.1002/cnr2.1209
The copyright line for this article was changed on 2 August 2019 after original online publication.
Contributor Information
Chris D. Madsen, Email: chris.madsen@med.lu.se.
Thomas R. Cox, Email: t.cox@garvan.org.au.
REFERENCES
- 1. Alexander J, Cukierman E. Stromal dynamic reciprocity in cancer: intricacies of fibroblastic‐ECM interactions. Curr Opin Cell Biol. 2016;42:80‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. LeBleu VS, Kalluri R. A peek into cancer‐associated fibroblasts: origins, functions and translational impact. Dis Model Mech. 2018;11(4):dmm029447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Filipe EC, Chitty JL, Cox TR. Charting the unexplored extracellular matrix in cancer. Int J Exp Pathol. 2018;99(2):58‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chitty JL, Filipe EC, Lucas MC, Herrmann D, Cox TR, Timpson P. Recent advances in understanding the complexities of metastasis. [version 2; peer review: 3 approved]. F1000Res. 2018;7:pii: F1000 Faculty Rev‐1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shamir ER, Ewald AJ. Three‐dimensional organotypic culture: experimental models of mammalian biology and disease. Nat Rev Mol Cell Biol. 2014;15(10):647‐664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Herrmann D, Conway JRW, Vennin C, et al. Three‐dimensional cancer models mimic cell‐matrix interactions in the tumour microenvironment. Carcinogenesis. 2014;35(8):1671‐1679. [DOI] [PubMed] [Google Scholar]
- 7. Cox T, Erler J. Network biology and the 3‐dimensional tumor microenvironment: personalizing medicine for the future: tumor microenvironment and therapy. Tumor Microenviron Ther. 2012;1:14‐18. [Google Scholar]
- 8. Hastings JF, Skhinas JN, Fey D, Croucher DR, Cox TR. The extracellular matrix as a key regulator of intracellular signalling networks. Br J Pharmacol. 2018;176:82‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Friedl P, Sahai E, Weiss S, Yamada KM. New dimensions in cell migration. Nat Rev Mol Cell Biol. 2012;13(11):743‐747. [DOI] [PubMed] [Google Scholar]
- 10. Conway JRW, Vennin C, Cazet AS, et al. Three‐dimensional organotypic matrices from alternative collagen sources as pre‐clinical models for cell biology. Sci Rep. 2017;7(1):16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sachs N, de Ligt J, Kopper O, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell. 2018;172(1‐2):373‐386. e10. [DOI] [PubMed] [Google Scholar]
- 12. Chonghaile TN. Patient‐derived organoids: are PDOs the new PDX? Sci Transl Med. 2018;10(451):eaau7377. [Google Scholar]
- 13. Donald E. Ingber: engineering the culture microenvironment. Trends Cell Biol. 2016;26:794‐795. [DOI] [PubMed] [Google Scholar]
- 14. Chen CS. 3D biomimetic cultures: the next platform for cell biology. Trends Cell Biol. 2016;26(11):798‐800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Puls TJ, Tan X, Husain M, Whittington CF, Fishel ML, Voytik‐Harbin SL. Development of a novel 3D tumor‐tissue invasion model for high‐throughput, High‐Content Phenotypic Drug Screening. Sci Rep. 2018;8:13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Orbach SM, Ehrich MF, Rajagopalan P. High‐throughput toxicity testing of chemicals and mixtures in organotypic multi‐cellular cultures of primary human hepatic cells. Toxicol In Vitro. 2018;51:83‐94. [DOI] [PubMed] [Google Scholar]
- 17. Kunz‐Schughart LA, Freyer JP, Hofstaedter F, Ebner R. The use of 3‐D cultures for high‐throughput screening: the multicellular spheroid model. J Biomol Screen. 2004;9(4):273‐285. [DOI] [PubMed] [Google Scholar]
- 18. Tung Y‐C, Hsiao AY, Allen SG, Torisawa YS, Ho M, Takayama S. High‐throughput 3D spheroid culture and drug testing using a 384 hanging drop array. Analyst. 2011;136(3):473‐478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carragher NO, Unciti‐Broceta A, Cameron DA. Advancing cancer drug discovery towards more agile development of targeted combination therapies. Future Med Chem. 2012;4(1):87‐105. [DOI] [PubMed] [Google Scholar]
- 20. Dawson JC, Carragher NO. Quantitative phenotypic and pathway profiling guides rational drug combination strategies. Front Pharmacol. 2014;5:118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Warchal SJ, Unciti‐Broceta A, Carragher NO. Next‐generation phenotypic screening. Future Med Chem. 2016;8(11):1331‐1347. [DOI] [PubMed] [Google Scholar]
- 22. Vennin C, Murphy KJ, Morton JP, Cox TR, Pajic M, Timpson P. Reshaping the tumor stroma for treatment of pancreatic cancer. Gastroenterology. 2018;154(4):820‐838. [DOI] [PubMed] [Google Scholar]
- 23. Gore J, Korc M. Pancreatic cancer stroma: friend or foe? Cancer Cell. 2014;25(6):711‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015;33(4):230‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cox TR, Erler JT. Fibrosis and cancer: partners in crime or opposing forces? Trends Cancer. 2016;2(6):279‐282. [DOI] [PubMed] [Google Scholar]
- 26. Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. 2018;18(6):359‐376. [DOI] [PubMed] [Google Scholar]
- 27. Weaver VM. Cell and tissue mechanics: the new cell biology frontier. Mol Biol Cell. 2017;28(14):1815‐1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oudin MJ, Weaver VM. Physical and chemical gradients in the tumor microenvironment regulate tumor cell invasion, migration, and metastasis. Cold Spring Harb Symp Quant Biol. 2016;81:189‐205. [DOI] [PubMed] [Google Scholar]
- 29. Acerbi I, Cassereau L, Dean I, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb). 2015;7(10):1120‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70(9‐10):537‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22(5):697‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Itoh G, Chida S, Yanagihara K, Yashiro M, Aiba N, Tanaka M. Cancer‐associated fibroblasts induce cancer cell apoptosis that regulates invasion mode of tumours. Oncogene. 2017;36(31):4434‐4444. [DOI] [PubMed] [Google Scholar]
- 33. Pankova D, Chen Y, Terajima M, et al. Cancer‐associated fibroblasts induce a collagen cross‐link switch in tumor stroma. Mol Cancer Res. 2016;14(3):287‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Erdogan B, Ao M, White LM, et al. Cancer‐associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216(11):3799‐3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Labernadie A, Kato T, Brugués A, et al. A mechanically active heterotypic E‐cadherin/N‐cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19(3):224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer‐associated fibroblasts are activated in incipient neoplasia to orchestrate tumor‐promoting inflammation in an NF‐kappaB‐dependent manner. Cancer Cell. 2010;17(2):135‐147. [DOI] [PubMed] [Google Scholar]
- 37. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582‐598. [DOI] [PubMed] [Google Scholar]
- 38. Prakash J. Cancer‐associated fibroblasts: perspectives in cancer therapy. Trends Cancer. 2016;2(6):277‐279. [DOI] [PubMed] [Google Scholar]
- 39. Shimoda M, Mellody KT, Orimo A. Carcinoma‐associated fibroblasts are a rate‐limiting determinant for tumour progression. Semin Cell Dev Biol. 2010;21(1):19‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cox TR, Erler JT. Molecular pathways: connecting fibrosis and solid tumor metastasis. Clin Cancer Res. 2014;20(14):3637‐3643. [DOI] [PubMed] [Google Scholar]
- 41. Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4(2):165‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Timpson P, Mcghee EJ, Erami Z, et al. Organotypic collagen I assay: a malleable platform to assess cell behaviour in a 3‐dimensional context. J Vis Exp. 2011;13(56):e3089. 10.3791/3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edward M. Effects of retinoids on glycosaminoglycan synthesis by human skin fibroblasts grown as monolayers and within contracted collagen lattices. Br J Dermatol. 1995;133(2):223‐230. [DOI] [PubMed] [Google Scholar]
- 44. Ranftl RE, Calvo F. Analysis of breast cancer cell invasion using an organotypic culture system. Methods Mol Biol. 2017;1612:199‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rajan N, Habermehl J, Coté M‐F, Doillon CJ, Mantovani D. Preparation of ready‐to‐use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat Protoc. 2006;1(6):2753‐2758. [DOI] [PubMed] [Google Scholar]
- 46. Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469‐483. [DOI] [PubMed] [Google Scholar]
- 47. Morton JP, Timpson P, Karim SA, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc Natl Acad Sci U S A. 2010;107(1):246‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gaggioli C, Hooper S, Hidalgo‐Carcedo C, et al. Fibroblast‐led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9(12):1392‐1400. [DOI] [PubMed] [Google Scholar]
- 49. Hooper S, Gaggioli C, Sahai E. A chemical biology screen reveals a role for Rab21‐mediated control of actomyosin contractility in fibroblast‐driven cancer invasion. Br J Cancer. 2010;102(2):392‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Calvo F, Ege N, Grande‐Garcia A, et al. Mechanotransduction and YAP‐dependent matrix remodelling is required for the generation and maintenance of cancer‐associated fibroblasts. Nat Cell Biol. 2013;15(6):637‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Madsen CD, Pedersen JT, Venning FA, et al. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 2015;16(10):1394‐1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Komsa‐Penkova R, Spirova R, Bechev B. Modification of Lowry's method for collagen concentration measurement. J Biochem Biophys Methods. 1996;32(1):33‐43. [DOI] [PubMed] [Google Scholar]
- 53. Wolf K, te Lindert M, Krause M, et al. Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol. 2013;201(7):1069‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yang Y, Motte S, Kaufman LJ. Pore size variable type I collagen gels and their interaction with glioma cells. Biomaterials. 2010;31(21):5678‐5688. [DOI] [PubMed] [Google Scholar]
- 55. Raub CB, Suresh V, Krasieva T, et al. Noninvasive assessment of collagen gel microstructure and mechanics using multiphoton microscopy. Biophys J. 2007;92(6):2212‐2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lattouf R, Younes R, Lutomski D, et al. Picrosirius red staining: a useful tool to appraise collagen networks in normal and pathological tissues. J Histochem Cytochem. 2014;62(10):751‐758. [DOI] [PubMed] [Google Scholar]
- 57. Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979;11(4):447‐455. [DOI] [PubMed] [Google Scholar]
- 58. Vennin C, Chin VT, Warren SC, et al. Transient tissue priming via ROCK inhibition uncouples pancreatic cancer progression, sensitivity to chemotherapy, and metastasis. Sci Transl Med. 2017;9(384):eaai8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chou A, Froio D, Nagrial AM, et al. Tailored first‐line and second‐line CDK4‐targeting treatment combinations in mouse models of pancreatic cancer. Gut. 2018;67(12):2142‐2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mayorca‐Guiliani AE, Madsen CD, Cox TR, Horton ER, Venning FA, Erler JT. ISDoT: in situ decellularization of tissues for high‐resolution imaging and proteomic analysis of native extracellular matrix. Nat Med. 2017;23(7):890‐898. [DOI] [PubMed] [Google Scholar]
- 61. Cazet AS, Hui MN, Elsworth BL, et al. Targeting stromal remodeling and cancer stem cell plasticity overcomes chemoresistance in triple negative breast cancer. Nat Commun. 2018;9(1):2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vennin C, Pajic M, Timpson P. Imaging fibrosis in pancreatic cancer using second harmonic generation. Pancreatology. 2015;15(2):200‐201. [DOI] [PubMed] [Google Scholar]
- 63. Huo CW, Chew G, Hill P, et al. High mammographic density is associated with an increase in stromal collagen and immune cells within the mammary epithelium. Breast Cancer Res. 2015;17(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cicchi R, Kapsokalyvas D, de Giorgi V, et al. Scoring of collagen organization in healthy and diseased human dermis by multiphoton microscopy. J Biophotonics. 2010;3(1‐2):34‐43. [DOI] [PubMed] [Google Scholar]
- 65. Cukierman E. Cell migration analyses within fibroblast‐derived 3‐D matrices. Methods Mol Biol. 2005;294:79‐93. [DOI] [PubMed] [Google Scholar]
- 66. Bankhead P, Loughrey MB, Fernández JA, et al. QuPath: open source software for digital pathology image analysis. Sci Rep. 2017;7(1):16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods. 2012;9(7):676‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012;12(8):540‐552. [DOI] [PubMed] [Google Scholar]
- 69. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392‐401. [DOI] [PubMed] [Google Scholar]
- 70. Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342(18):1350‐1358. [DOI] [PubMed] [Google Scholar]
- 71. Border WA, Ruoslahti E. Transforming growth factor‐beta in disease: the dark side of tissue repair. J Clin Invest. 1992;90(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jones CL, Buch S, Post M, McCulloch L, Liu E, Eddy AA. Pathogenesis of interstitial fibrosis in chronic purine aminonucleoside nephrosis. Kidney Int. 1991;40(6):1020‐1031. [DOI] [PubMed] [Google Scholar]
- 73. Westergren‐Thorsson G, Hernnäs J, Särnstrand B, Oldberg A, Heinegård D, Malmström A. Altered expression of small proteoglycans, collagen, and transforming growth factor‐beta 1 in developing bleomycin‐induced pulmonary fibrosis in rats. J Clin Invest. 1993;92(2):632‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.