

Abstract
Background
Three‐dimensional (3D) in vitro model systems can bridge the gap between regular two‐dimensional cell culture and whole‐animal studies. Analyses of cancer cell migration and invasion increasingly use differing 3D systems, which may produce conflicting findings.
Aims
We directly compared different 3D extracellular matrix systems for studying cancer cell migration/invasion by analyzing cell morphologies and quantifying aspects of cell migration including speed and directional persistence using automated computer‐based cell tracking.
Methods and results
We performed direct comparisons of five different 3D extracellular matrix cell culture systems using both HT1080 fibrosarcoma and MDA‐MB‐231 breast carcinoma cell lines. The reconstituted 3D systems included two types of collagen hydrogel and tissue matrix gel (TMG) vs cell‐derived matrices extracted from cultured primary human or cancer‐associated fibroblasts. The fibrillar matrix architecture of these systems differed. 3D rat tail collagen and TMG matrices had short, randomly oriented collagen fibrils; bovine collagen had long, larger fibril bundles; and the cell‐derived matrices were strongly oriented. HT1080 cells displayed rounded morphologies in all three reconstituted 3D matrices but became spindle shaped in the two cell‐derived matrices. MDA‐MB‐231 cell morphologies were elongated in all matrices. Quantitative measures of cell migration parameters differed markedly between the different types of 3D matrix. Comparing the reconstituted matrices, cells migrated the most rapidly and furthest in TMG. Comparing TMG with cell‐derived matrices, cells migrated more efficiently in the cell‐derived matrices. The most notable differences were in directional persistence of migration, which was greatest in the two cell‐derived matrices.
Conclusion
The morphologies of matrix fibrils and cell shape, and particularly the efficiency and directionality of cell migration, differed substantially depending on the type of 3D matrix system. We suggest that it is important to employ the 3D model system that most closely resembles the matrix environment being studied for analyses of cancer cell migration and invasion.
Keywords: cancer, cell migration, collagen, extracellular matrix, invasion, three‐dimensional culture
1. INTRODUCTION
Many important conceptual and mechanistic advances in cancer cell biology have been provided by studies of malignant cell migration using traditional two‐dimensional (2D) cell culture systems on flat tissue culture substrates of glass or plastic. Nevertheless, cells growing on flat 2D substrates can differ considerably in their morphology, cell‐cell and cell‐matrix interactions, and differentiation from those growing in more physiological three‐dimensional (3D) environments. 1 , 2 , 3 , 4 , 5 Therefore, recent research on cancer cell migration has been gradually shifting from 2D to 3D cell culture. 6 , 7 , 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15
3D matrices used recently that employ physiological extracellular matrix molecules include collagen 3D matrices, cell‐derived matrix (CDM), and tissue matrix gel (TMG). 16 , 17 , 18 , 19 , 20 Collagen 3D matrices based primarily on collagen I are used more frequently. They have the significant advantages of mimicking collagen‐rich tissues in vivo, simple molecular composition, and the ability to assemble 3D hydrogels by polymerizing collagen solutions at varying concentrations with the capacity to generate 3D matrix environments that differ in stiffness and matrix pore size for cell migration. 8 , 13 , 16
Nevertheless, collagen matrices lack other components present in the extracellular matrix (ECM) in vivo. Moreover, there can be a large range of types of collagen matrix depending on the source of the collagen (eg, rat vs bovine tissues) and the extent of endogenous covalent crosslinking, as well as matrix concentration, stiffness, and pore size of the hydrogel—all of which can alter 3D cell migration.
A new type of 3D ECM matrix is the “tissue matrix gel” from mouse or porcine tissue. Although it can also generate a polymerized matrix with pores or spaces for cell migration, TMG contains a variety of ECM molecules besides collagen that are derived from in vivo tissue, including fibronectin, laminin, and proteoglycans. 19 This major difference in molecular composition compared to purified collagen gels may affect patterns of cancer cell migration, which is a key question addressed by this study.
TMG and CDM share the feature of containing complex, physiological 3D ECM matrix molecules. However, they differ in that TMG can be polymerized from solution to generate a relatively physiological 3D matrix in terms of composition, whereas CDMs are generated directly from naturally organized 3D ECM matrices generated by fibroblastic cells without altering the original spatial organization of ECM molecules. For example, CDM systems often contain more aligned 3D fiber fibronectin and collagen than TMG, which could alter the mode and extent of directionality of tumor cell migration.
This study presents a direct comparative investigation of five different types of 3D ECM matrix for cell culture studies. We compared the organization of these matrices by confocal microscopy, as well as their effects on the cell morphology and quantifications of migration speed and directionality of two widely different types of human cancer cell arising from tissues of epithelial vs mesenchymal origin.
2. MATERIALS AND METHODS
2.1. Cell lines and tissue culture
MDA‐MB‐231 cells and hTERT PF179T cancer‐associated fibroblasts (CAFs) were authenticated and obtained directly from the American Type Culture Collection (ATCC) for this study and used immediately. Other cells were tested for mycoplasma using the MycoAlert Detection kit (Lonza). MDA‐MB‐231, HT1080, and human foreskin fibroblast (HFF) cells (a gift from Susan Yamada, NIDCR) were cultured in high glucose DMEM (Gibco) with 10% fetal bovine serum (FBS; Hyclone) at 37°C with 10% CO2, and the low‐passage primary HFF cells were used between passages 8 and 11. The hTERT PF179T CAF cells were cultured in EMEM (ATCC) with 10% FBS and 0.075% sodium bicarbonate (Gibco) at 37°C in a humidified 10% CO2 atmosphere. The media were supplemented with 100 U/mL penicillin and 100 μg/mL streptomycin.
2.2. Chemical activation of coverslips
Coverslips (Fisher Scientific) were acid‐washed with 50% nitric acid (Fisher Scientific) for 25 minutes, rinsed overnight under a continuous flow of dH2O, then dried under forced air and stored covered until needed. Triethoxysilybutraldehyde (Gelest Inc.) was diluted to 1% in 100% ethanol and then added to coverslips and incubated for 5 minutes. This silane solution was then aspirated, and the coverslip was rinsed twice with 100% ethanol and once with dH2O. Coverslip surfaces were then blown dry with forced air and cured at 65°C for 4 hours and finally stored desiccated at 4°C. 21
2.3. 3D matrices
Activated coverslips were affixed to the bottom of 24‐well glass bottom plates (Cellvis) using vacuum grease. The standard steps for preparation of each of the four general types of 3D matrix are indicated diagrammatically in Figure 1 and as described below.
FIGURE 1.

Production of the different types of 3D extracellular matrix for comparisons of cell migration/invasion. All coverslips were silanized, and each 3D matrix was generated as described in detail under section 2
2.3.1. Rat tail collagen matrix
Rat tail collagen was a kind gift from Gregory Kitten and was prepared as described. 16 Briefly, adult rat tail tendons were isolated free of blood vessels and tendon sheaths, then extracted by stirring in 0.5 M acetic acid at 4°C for 48 hours. After centrifugation at 14 000g for 1 hour, the collagen‐containing supernatant was dialyzed against 0.02 M acetic acid 4°C for 3 days and stored at 4°C. Rat tail collagen gels at a final concentration of 2 mg/mL and containing cells were produced by mixing appropriate ratios of a 6.17 mg/mL stock solution of rat tail collagen with 10x DMEM, 10x reconstitution buffer (HEPES 200 mM with 262 mM sodium bicarbonate), 1 N NaOH to adjust the pH to 7.4, and then cells in Dulbecco's phosphate‐buffered saline with calcium and magnesium (PBS++). Forty microliter of this 2 mg/mL rat tail collagen solution containing 2000 HT1080 or MDA‐MB‐231 cells was aliquoted onto the coverslip and polymerized for 30 minutes at 37°C as previously described. 16 , 22
2.3.2. Bovine collagen matrix
Bovine collagen (BC; Sigma) at a final concentration of 2 mg/mL was produced by mixing appropriate ratios of a 6.0 mg/mL stock solution of BC with 10x DMEM, 1 N NaOH to adjust the pH to 7.4, and PBS++ with or without cells. Twenty microliters of cell‐free 2 mg/mL BC was aliquoted onto the coverslip and polymerized for 5 minutes at 37°C. Deposition of this initial layer of cell‐free collagen matrix was found to be necessary in order to impede rapid cell attachment to the coverslip and to maintain cells within the hydrogel. Then, 20 μL 2 mg/mL BC containing 2000 HT1080 or MDA‐MB‐231 cells was added and polymerized for 30 minutes at 37°C. All of the BC matrices contained regular cell culture medium.
2.3.3. Tissue matrix gel
TMG was extracted from porcine breast tissue ECM as we previously reported. 19 Briefly, fresh mammary tissues from female pigs (<1 year old) were collected from a local slaughterhouse, homogenized, and decellularized; total ECM protein was then extracted and subsequently reconstituted into hydrogels at desired concentrations for the experiments. The approximate collagen concentration of TMG was determined to be 1.2 mg/mL by Sircol collagen assay. Twenty microliters of 2 mg/mL (total protein concentration) TMG without cells was aliquoted onto coverslips and polymerized for 15 minutes at 37°C; this initial hydrogel layer was used to prevent rapid cell attachment to the coverslip. An additional 20 μL of TMG containing 2000 HT1080 or MDA‐MB‐231 cells was then added and polymerized for 45 minutes at 37°C.
2.3.4. Cell‐derived matrix
CDM was produced from high‐density 14‐day cultures of HFF or hTERT PF179T CAF cells in 24 well glass bottom plates exactly as described. 23 Medium containing ascorbic acid (50 μg/mL; Sigma) was changed every 2 days. After blocking nonspecific adsorption sites with 1% heated‐denatured BSA in PBS++ at room temperature for 30 minutes, HT1080 or MDA‐MB‐231 cells (5000 per assay) were plated on top of the CDM and allowed to migrate downward into the matrix for 24 hours.
The reconstituted rat tail and BC, as well as TMG, were polymerized from a total volume of 40 μL on the coverslip in order to keep the hydrogels as thin as practical to permit high‐resolution microscopy. The thickness of each reconstituted matrix after polymerization was estimated to be 200 to 250 μm, and the CDM preparations were measured to be approximately 10 μm thick. Cells that reached the bottom of hydrogels to contact the glass coverslip were omitted from analyses because they underwent 2D spreading.
2.4. Antibodies and reagents
Atto 647 N NHS ester was obtained from Sigma‐Aldrich. Rhodamine phalloidin was from ThermoFisher Scientific. Collagen I antibody was from Novus Biologicals, and collagen I + III antibody against porcine collagen was from LSBio. Alexa Fluor 488 anti‐rabbit and Alexa Fluor 647 anti‐mouse secondary antibodies were from Jackson ImmunoResearch.
2.5. Direct labeling of 3D matrices with 647 N NHS ester fluorescent dye
Sodium bicarbonate buffer (50 mM, pH 8.5) was added to 3D matrices. This buffer was removed immediately, and 2 μg/mL Atto 647 N NHS ester in this buffer was added. Samples were incubated for 15 minutes at room temperature to permit covalent labeling. The three‐dimensional matrices were rinsed 5X with PBS++ for 15 minutes. The matrices were then rinsed with 200 mM Tris buffer (pH 7.5) to stop the coupling reaction.
2.6. Immunofluorescence staining and confocal microscopy
Fixation, permeabilization, and staining steps were performed on the coverslips in 24‐well plates. Samples were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in cytoskeletal buffer with sucrose (CBS: 10 mM MES, 138 mM KCl, 2 mM EGTA, 3 mM MgCl2 plus 320 mM sucrose) for 20 minutes at 37°C. Samples were rinsed 3X in PBS++ and permeabilized with 0.5% Triton X‐100 in CBS for 5 minutes at 37°C. Samples were then rinsed 3X over 20 minutes with PHEM + glycine buffer (60 mM PIPES, 2 mM HEPES, 10 mM EGTA, 2 mM MgCl2, 100 mM glycine, pH 6.9). Nonspecific binding sites were further blocked with 20% donkey serum (Jackson ImmunoResearch Laboratories) together with M.O.M reagent (Vector Laboratories) in PHEM + glycine buffer for 1 hour. Samples were rinsed 3X with PHEM + glycine over 20 minutes. Primary and secondary antibodies were diluted in PHEM + glycine with 10% donkey serum and incubated for 1 hour and 45 minutes, respectively.
Samples on coverslips containing RC matrix, bovine skin collagen matrix, or TMG were inverted and mounted for imaging in MatTek dishes using ProLong Gold or SlowFade Gold antifade reagent (Invitrogen). Images were collected using a spinning disk confocal microscope. The spinning disk system consisted of a Yokogawa CSU‐22 scan head (CSU‐21: modified by Spectral Applied Research, Inc.) on an automated Olympus IX‐81 microscope using a 60X SAPO‐Chromat silicone oil objective (N.A. 1.3) equipped with a custom laser launch with 488, 568, and 642 laser lines (built by Andrew Doyle). The system was controlled by MetaMorph (Molecular Devices).
2.7. Live cell imaging
Nuclei were labeled using 1 μg/mL Hoechst (Life Technologies). Each 3D matrix containing cells was cultured in Fluorobrite medium (Gibco) containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l‐glutamine (Gibco), with 10 mM dl‐lactate (Sigma) and 100x diluted OxyFluor (Oxyrase) to reduce the formation of oxygen free radicals during live imaging. Live imaging was performed using a Nikon Ti‐E microscope. A Tokai stage‐top incubator was used to maintain cells at constant 37°C with 5% CO2 and approximately 50% humidity. The system was controlled by NIS‐Elements software (Nikon). The cell migration parameters consisting of velocity, displacement, and persistence were quantified using automated computer tracking of the migration of individual cells labeled with Hoechst using the FastTracks graphical user interface developed by Brian DuChez. 10 , 24
2.8. Statistical analysis
GraphPad Prism 8 software was used for all statistical analyses. One‐way analysis of variance (ANOVA) followed by the Tukey post hoc test was used for comparisons of data sets. All error bars indicate SEM. P < .05 was considered statistically significant; n is the total number of data points per experimental condition.
3. RESULTS
We first compared three different 3D extracellular matrix model systems that can be reconstituted into a 3D matrix hydrogel by polymerizing an initially homogeneous, soluble solution of biological material. Because the goal of this study was to identify physiological matrix environments, all collagen matrix systems used were based on intact collagen extracted and purified from tissues, rather than pepsin‐digested collagen lacking endogenous telopeptide regions and crosslinking.
The most commonly used reconstituted collagen matrix in the literature is based on collagen purified from rat tails, which can be obtained commercially or purified within laboratories. Another collagen matrix with potentially different levels of endogenous covalent cross‐linking and pore size for cell migration is based on extracted, purified BC, which can also be obtained commercially. The recently described system termed “tissue matrix gel” is substantially more complex molecularly beyond its content of collagen. 19 For all three systems, solutions of soluble matrix material were polymerized under standard conditions in regular cell culture medium as detailed in section 2.
3.1. Morphologies of the fibrillar matrix and cells in 3D RC matrix, BC matrix, and TMG
As determined by direct covalent labeling by a fluorescent dye, the 3D collagen matrices generated from RC and TMG had short fibrils arranged randomly in all directions. In contrast, 3D collagen matrix from BC contained large, long bundles (Figure 2A). Thus, the fibril organization of rat and BCs differ significantly. Similar differences in fibril morphology were confirmed using anticollagen antibodies (data not shown).
FIGURE 2.
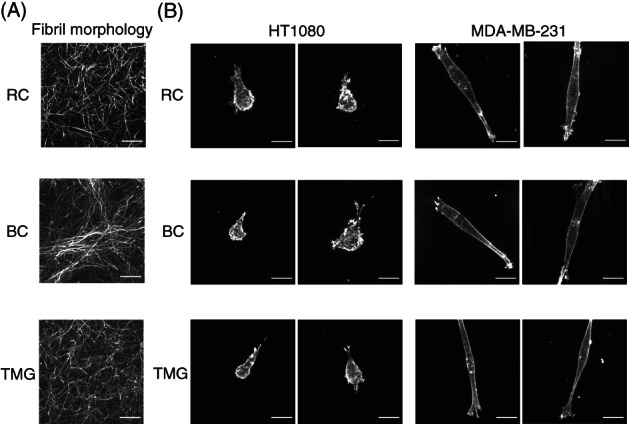
3D matrix fibril morphologies and cell morphologies of two cancer cell lines in 3D matrices reconstituted from rat tail collagen, bovine collagen, or tissue matrix gel. A, Maximum intensity projection confocal microscopy images of 10‐μm‐thick 3D matrices stained covalently using fluorescent NHS‐ester 647. B, Cellular F‐Actin labeled with rhodamine‐phalloidin. RC: Rat tail collagen matrix, BC: Bovine collagen matrix, TMG: Tissue matrix gel. Scale bars: 20 μm
The morphologies of HT1080 sarcoma cells in all three of these reconstituted 3D matrices were similar, consisting of a rounded, spherical cell shape that often protruded a single, large pseudopod terminating in a lamellipodium or lamellipodia oriented in the direction of migration, but occasionally several such processes were observed (Figure 2B). In contrast, the morphology of MDA‐MB‐231 carcinoma cells in all three of these 3D matrices was spindle‐shaped, elongated, or cylindrical with few protrusions beyond the single elongated processes extending from the end of each cell, which often contained an actin‐rich lamellipodium (Figure 2B).
3.2. Quantification of cell migration patterns in RC matrix, BC matrix, and TMG
Using automated computer‐based tracking of cell migration, we compared the three reconstituted 3D matrix systems for their effects on cancer cell migratory speed, directionality, and displacement (net distance covered per assay period). We present our results using three different methods for plotting the data (bar graphs, box‐and‐whiskers plots, and violin plots) because each presents a different viewpoint of the primary data. The cell migration velocities of both HT1080 sarcoma and MDA‐MB‐231 carcinoma cells were greater in TMG compared to both RC and BC (Figure 3A,B). Similarly, the net displacement (the distance between each cell's starting point and final end point after 16 hours) was greater in TMG than for both of the collagen matrices and for each of the two cancer cell types; the enhancement of net migratory displacement for MDA‐MB‐231 cells in TMG was particularly notable compared to the more modest increase in velocity. Thus, both mesenchymal and epithelial tumor cells migrated the most extensively in the more molecularly complex TMG microenvironment compared to the two types of collagen matrix.
FIGURE 3.
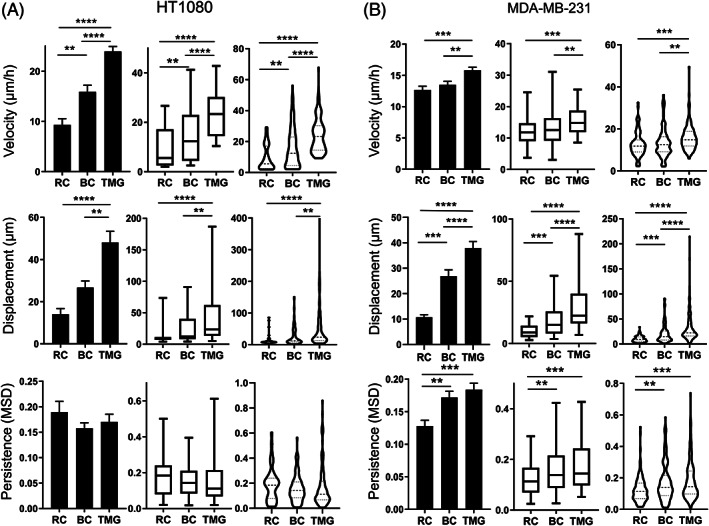
Cell migration patterns in rat tail collagen matrix, bovine collagen matrix, or tissue matrix gel. Column bar graph, box‐and‐whisker plots (5th to 95th percentile), and violin plots with the migration parameters indicated along the Y axis for HT1080 sarcoma cells, A, and MDA‐MB‐231 carcinoma cells, B, in each of the reconstituted 3D matrices. Migration parameters were quantified by automated computer tracking for velocity, displacement, and persistence. N (number of independent experiments) > 3, n > 50 cells. Error bars: SEM. MSD: mean square displacement. **P < .01, ***P < .01, ****P < .0001. RC: rat tail collagen matrix, BC: bovine collagen matrix, TMG: tissue matrix gel
On the other hand, the directional persistence of HT1080 sarcoma cells was similar within each type of matrix. In contrast, the migratory persistence of MDA‐MB‐231 carcinoma cells was significantly increased in both BC and TMG compared to classical rat collagen (RC) matrices (Figure 3A,B).
In addition, however, there were also significant differences in cell motility between the two types of collagen matrix, that is, in comparisons of RC and BC. Both cell velocity and total migratory displacement of HT1080 cells were significantly enhanced in BC compared to RC (Figure 3A). The displacement and persistence of MDA‐MB‐231 cells in BC were also higher than in RC. Although HT1080 and MDA‐MB‐231 cells both migrated in TMG with greater displacement than in RC and BC, they migrated with relatively similar low directionality in TMG, RC, and BC, though with a modest increase in directionality of MDA‐MB‐231 cells in BC and TMG compared to RC (Figure 4A,B).
FIGURE 4.
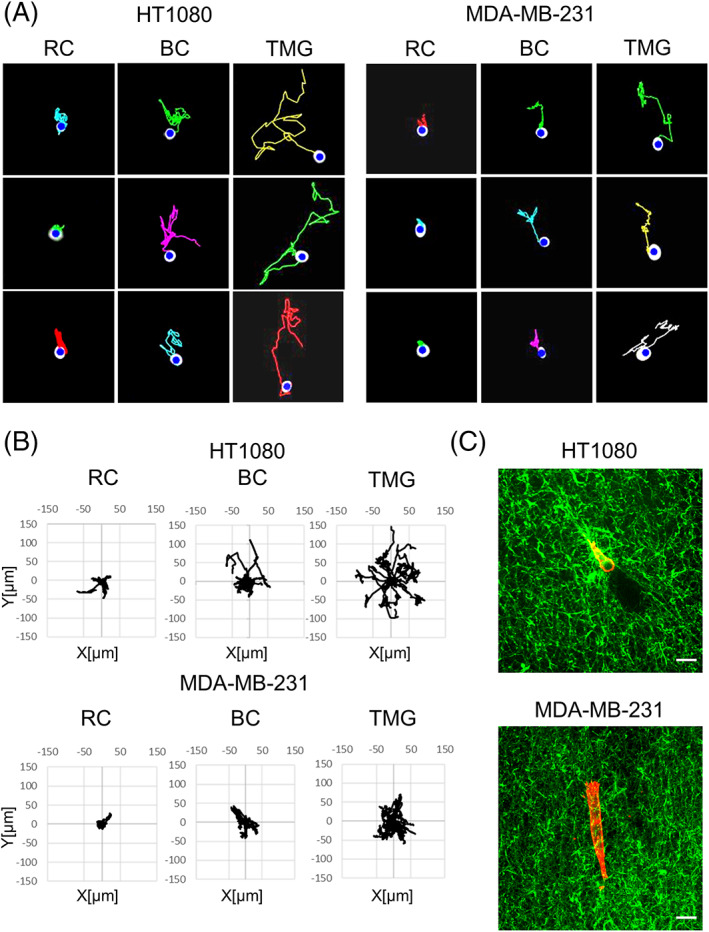
Cell migration tracks from time‐lapse video recordings of HT1080 or MDA‐MB‐231 cells in each of the three different reconstituted 3D matrices. A, Migration tracks for HT1080 cells and MDA‐MB‐231 cells in each of the reconstituted 3D matrices. B, Wind rose plots of representative HT1080 and MDA‐MB‐231 cell migration tracks in each of the three reconstituted 3D matrices. n = 20. C, Merged images of cells and collagen fibers in tissue matrix gel (TMG). Collagen in TMG was stained using anti‐pig‐collagen I + III antibody (green). HT1080 and MDA‐MB‐231 cells were stained with rhodamine‐phalloidin (red). RC: rat tail collagen matrix, BC: bovine collagen matrix, TMG: tissue matrix gel. Scale bars: 20 μm
Interestingly, there were also differences in cell degradative interactions with 3D matrix by the HT1080 and MDA‐MB‐231 cells as they migrated/invaded through collagenous TMG matrix. HT1080 sarcoma cells migrating in TMG matrix frequently displayed a trailing region lacking collagen staining, leaving behind them a local hollow tunnel devoid of 3D collagen fibers as they migrated. In contrast, MDA‐MB‐231 cells showed little evidence of such matrix degradation (Figure 4C). This difference in the pattern of matrix degradation may contribute to the observed differences between the two cell types in relative velocities in TMG environments, where HT1080 cells migrated at an average velocity of 24 μm/h compared to 16 μm/h for MDA‐MB‐231 cells.
3.3. Fibrillar matrix and cell morphology in TMG vs two types of CDM
Because TMG is molecularly complex, we next compared its biological interactions with cancer cells to the similarly complex “cell‐derived matrix” produced by tissue cultures of CAFs vs non‐malignant fibroblasts. The fibrillar matrix morphologies of CDM that had been synthesized by, and then detergent‐extracted in intact form from, hTERT PF179T CAF and human fibroblasts (HFF) were similar. Both forms of CDM were characterized by long fibrillar bundles that frequently displayed substantial local alignment (Figure 5A). There did, however, appear to be more collagen and total protein matrix in the CAF CDM than in the CDM from primary fibroblasts, consistent with a previous report (Figure 5A,B). 25 In marked contrast, TMG displayed short, randomly oriented fibrils (Figure 5A,B; see also Figure 2A).
FIGURE 5.
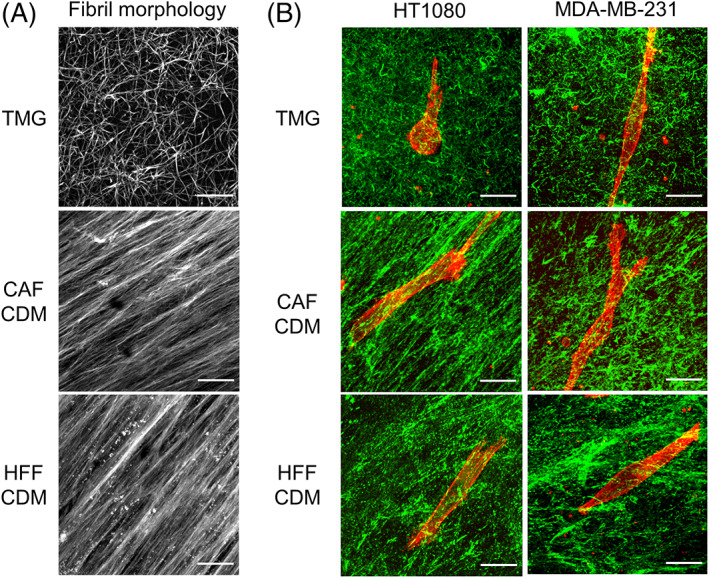
3D matrix fibril and cell morphologies in tissue matrix gel and cell‐derived matrix from cancer‐associated fibroblasts vs human foreskin fibroblasts. A, TMG and CDM were stained using fluorescent NHS‐ester 647. B, Merged images of cells and collagen fibers in TMG compared to the two CDMs. The cells were stained with rhodamine‐phalloidin (red). Collagen in TMG was stained using anti‐pig‐collagen I + III antibody (green). Collagen in the CDMs was stained with anti‐collagen I antibody (green). HT1080 and MDA‐MB‐231 cells were stained using rhodamine‐phalloidin (red). TMG: tissue matrix gel, CDM: cell‐derived matrix, CAF: cancer associated fibroblast, HFF: human foreskin fibroblast. Scale bars: 20 μm
In both types of CDM, the morphology of HT1080 cells was dramatically altered from the rounded shape characteristically observed in TMG, RC, and BC matrices to a markedly spindle‐shaped morphology with few protrusions. In contrast, the morphology of MDA‐MB‐231 cells in CDMs remained similar in all five matrices tested in this study (Figure 5B).
3.4. Cell migration patterns in TMG compared to CMDs
Both migration velocity and net displacement of both types of tumor cell tested increased significantly in both CAF CDM and HFF CDM compared to TMG (Figure 6A,B). In addition, the migratory directionality (persistence) of both cancer cells increased substantially in both CAF CDM and HFF CDM compared to TMG (Figure 6A,B), with visible straightening of their migration tracks in rose plots in which the origins of cell migration were superimposed and tracks were randomly oriented outward (Figure 7). In addition, migration differences were observed between these two types of CDM. Cell velocity, displacement, and persistence were all increased for HT1080 sarcoma cells in HFF CDM compared to CAF CDM, and velocity and displacement were also increased for MDA‐MB‐231 carcinoma cells in HFF CDM (Figure 6A,B).
FIGURE 6.
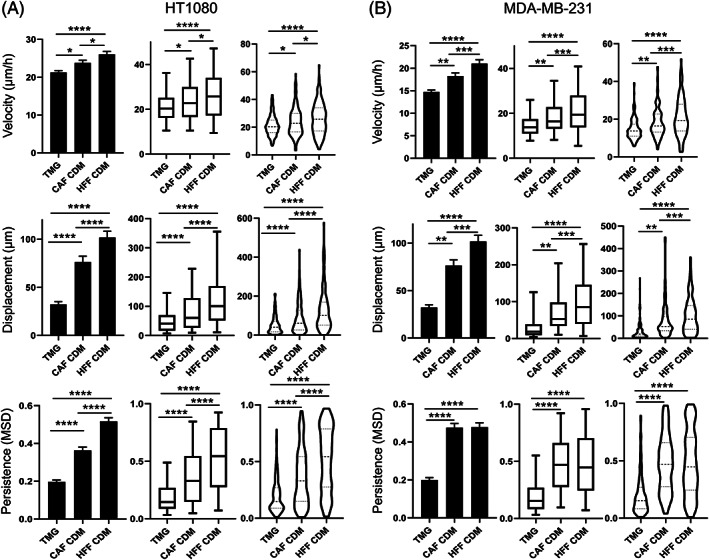
Cell migration patterns in tissue matrix gel compared to two types of cell‐derived matrix. Column bar graph, box‐and‐whisker plots (5th to 95th percentile), and violin plots with migration parameters of HT1080 sarcoma cells, A, and MDA‐MB‐231 carcinoma cells, B, in TMG and CDM. Migration patterns were quantified with respect to their velocity, displacement, and persistence. N > 3, n > 50. Error bars: SEM. MSD: mean square displacement. *P < .05, **P < .01, ***P < .01, ****P < .0001. TMG: tissue matrix gel, CDM: cell‐derived matrix, CAF: cancer associated fibroblast, HFF: human foreskin fibroblast
FIGURE 7.
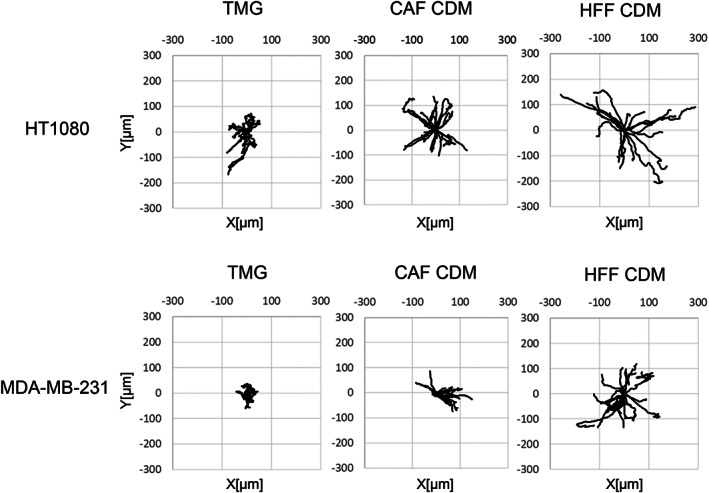
Wind rose plots of HT1080 and MDA‐MB‐231 cells migration tracks in TMG and CDM. The starting points of migration are superimposed at the origin, and the tracks are displayed as outwardly oriented tracings. n = 20. TMG: tissue matrix gel, CDM: cell‐derived matrix, CAF: cancer‐associated fibroblast, HFF: human foreskin fibroblast
4. DISCUSSION
In this study, we established that cancer cells migrate more effectively or aggressively in the more complex, arguably more physiological reconstituted 3D matrix termed TMG than in the classical 3D purified collagen matrices RC and BC from rat and bovine sources, respectively. Additionally, however, comparisons of the molecularly complex TMG with native CDMs from nonmalignant and CAF‐derived fibroblasts revealed an even higher level of cell migratory velocity, displacement, and directional persistence in the CDMs.
In addition, however, cancer cell migration patterns were found to be different even when comparing conceptually similar types of 3D matrix, that is, between the two types of purified collagen matrix (RC and BC) or between the more complex, physiological 3D CDMs from normal or CAFs.
Because rat tail collagen and BC preparations are purified collagens, they lack other ECM molecules, such as fibronectin, laminin, and proteoglycans. Such noncollagenous ECM molecules can activate a series of signal transduction pathways by interacting with various receptors, thereby regulating many cellular processes that include adhesion, migration, invasion, proliferation, differentiation, and morphogenesis—for which accumulating evidence indicates roles in carcinogenesis and malignancy for various cancers. 26 , 27 , 28 , 29 , 30 , 31 , 32 TMG contains multiple ECM molecules including fibronectin, laminin, fibrillin, proteoglycan, and so on. 19 Therefore, the enhanced cancer cell migration in TMG may be promoted by one or more of these ECM molecules.
Focusing on cell‐matrix remodeling interactions during cell migration within TMG, migrating HT1080 cells characteristically degraded the matrix to leave a collagen‐free space or tunnel behind the invading cells, whereas MDA‐MB‐231 cells could migrate without generating such tunnels. This observation would be consistent with capacity of HT1080 cells to use matrix metalloproteinases (MMPs) for more efficient migration in TMG compared to MDA‐MB‐231 cells. In fact, MMP inhibitor treatment of HT1080 cells was reported to revert their mode of cell migration back from lamellipodial to normal lobopodial migration in CDM, whereas MDA‐MB‐231 cells already use lobopodial migration with no effect of MMP inhibition on the mode of cell migration. 33
Cell morphology may also play a role in the migration pattern of HT1080 compared to MDA‐MB‐231 cells. The migratory persistence of MDA‐MB‐231 cells was increased in TMG compared to that in RC 3D matrix, whereas there were no differences in persistence when HT1080 cells migrated in TMG vs RC or BC 3D matrices. This difference correlated with differences in cell morphology. HT1080 sarcoma cells were round with some protrusions, whereas MDA‐MB‐231 cells were spindle‐shaped, elongated, or cylindrical with fewer protrusions. The protrusions of HT1080 cells extended outward nondirectionally, pulling the cell body in multiple directions, consistent with their observed more‐random patterns of migration. In contrast, the spindle‐like morphology of MDA‐MB‐231 cells with fewer protrusions in TMG may contribute to the observed greater persistence in cell migration.
Interestingly, there were also striking differences in cell motility between the complex, arguably more physiological 3D matrices, that is, TMG vs the two CDMs. One notable physical difference visible in these matrices is that the CDMs from both CAF and HFF have marked aligned collagen fibers, whereas TMG does not. Tumors have been reported to display collagen fiber alignment, termed tumor‐associated collagen signatures (TACS), at the tumor edge, and the level of TACS is considered to be a prognostic factor in cancer. 34 , 35 Riching et al reported that alignment of collagen fibers enhances the overall efficiency of migration by increasing directional persistence but does not increase the speed per se of migrating cells. 36 Our results are strikingly consistent with this finding. In our results, directionality and persistence of cell migration significantly increased in CDM from both CAF and HFF compared to migration in TMG. Our results suggest that CDM, especially from CAF, may serve as a model for the cell biological effects of TACS.
A published comparison of the nature of CDM preparations produced by non‐malignant fibroblasts compared to cancer‐associated/tumor‐associated fibroblasts revealed that the latter matrices are thicker in terms of height and numbers of fibronectin fibrils, as well as displaying a more parallel orientation of fibers. 25 Although detailed molecular analyses remain to be performed, a notable molecular difference reported by the authors was an altered ratio of total collagen to fibronectin, which was significantly increased in the tumor‐associated matrix. This increased collagen‐to‐fibronectin ratio is consistent with the latter being more mature and desmoplastic. 25 This tumor‐associated CDM can by itself induce biosynthetic changes in normal fibroblasts involving induction of desmin and α‐smooth muscle actin mimicking a cancer‐like phenotypic induction of stromagenesis. 25 Although our current study did not identify any major change in cell migratory properties in cancer‐associated CDM, dense collagen is known to induce tumor cell invadopodia implicated in local invasive behavior. 37 , 38
Although published studies of cell‐ECM interactions generally do not consider species differences in the source of matrix components utilized, such as the collagen molecules used for reconstituted matrix, there could theoretically be conditions in which the species origin of a matrix model system could become significant. For example, studies in which immune cells are added to the system could conceivably lead to differing results, especially if the 3D matrix materials are subsequently embedded in vivo. 39 Future studies using one or more of these 3D matrix systems could compare additional cell lines of cancers that are related or different from the two lines studied here, for example, other breast cancer, glioblastoma, and other cancer cell lines.
In summary, our findings have established that the migration/invasion patterns of both epithelial‐ and connective tissue‐derived cancer cells can differ markedly depending on the 3D matrix model. For example, there are differences even between simple 3D collagen model systems, as well as between them and the reconstituted more complex and physiological TMG 3D matrix. There were also major differences between the latter TMG system and CDM systems where the original matrix structure is preserved. Besides some differences in ECM content, there were differences in cancer cell motility responses to CDMs isolated from normal HFF fibroblasts compared to CAFs.
Consequently, an important point to consider is that because each tissue in vivo can have a characteristic matrix microenvironment, it is crucial to select the appropriately matched 3D in vitro matrix model for each study. The choice of model can have major effects on the results that can be obtained. The model system chosen could use a matrix matching that of the tumor cells, or it could be one with a similar molecular composition. However, particularly important elements are likely to include physical properties that include matrix fiber alignment, tissue ECM density, ECM porosity, and other physical features (eg, see Reference [8]).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ETHICAL STATEMENT
Not Applicable.
AUTHOR CONTRIBUTIONS
All authors have full access to the data in the study and take responsibility for the integrity of the data and accuracy of the data analysis. Conceptualization, Y.S. and K.M.Y.; Methodology, Y.S., K.M.Y., A.D.D., and W.L.; Investigation, Y.S., and A.D.D.; Writing – Original Draft, Y.S.; Writing – Review & Editing, K.M.Y., A.D.D., and W.L.; Funding Acquisition, K.M.Y., W.L.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the NIH, NIDCR including the NIDCR Imaging Core: ZIC DE000750‐01, as well as by startup funding and a Commercialization Gap Fund from Washington State University to W. L.
Shinsato Y, Doyle AD, Li W, Yamada KM. Direct comparison of five different 3D extracellular matrix model systems for characterization of cancer cell migration. Cancer Reports. 2020;3:e1257. 10.1002/cnr2.1257
This article has been contributed to by US Government employees and their work is in the public domain in the USA.
Funding information NIH, NIDCR, Grant/Award Numbers: ZIA DE000524, ZIA DE000718, ZIA DE000719, ZIC DE000750‐01; Washington State University, Grant/Award Numbers: Commercialization Gap Fund, Startup Funding
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Birgersdotter A, Sandberg R, Ernberg I. Gene expression perturbation in vitro ‐ a growing case for three‐dimensional (3D) culture systems. Semin Cancer Biol. 2005;15(5 SPEC. ISS):405‐412. 10.1016/j.semcancer.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 2. Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130(4):601‐610. 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 3. Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7(3):211‐224. 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 4. Nelson CM, Bissell MJ. Of extracellular matrix, scaffolds, and signaling: tissue architecture regulates development, homeostasis, and cancer. Annu Rev Cell Dev Biol. 2006;22(1):287‐309. 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ravi M, Paramesh V, Kaviya SR, Anuradha E, Solomon FDP. 3D cell culture systems: advantages and applications. J Cell Physiol. 2015;230(1):16‐26. 10.1002/jcp.24683. [DOI] [PubMed] [Google Scholar]
- 6. Pfeifer CR, Irianto J, Discher DE. Nuclear mechanics and cancer cell migration. Advances in Experimental Medicine and Biology. Vol 1146. New York LLC, NY: Springer; 2019:117‐130. 10.1007/978-3-030-17593-1_8. [DOI] [PubMed] [Google Scholar]
- 7. Hetmanski JHR, de Belly H, Busnelli I, et al. Membrane tension orchestrates rear retraction in matrix‐directed cell migration. Dev Cell. 2019;51(4):460‐475.e10. 10.1016/j.devcel.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamada KM, Sixt M. Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol. 2019;20:738‐752. 10.1038/s41580-019-0172-9. [DOI] [PubMed] [Google Scholar]
- 9. Zuela‐Sopilniak N, Lammerding J. Engineering approaches to studying cancer cell migration in three‐dimensional environments. Philos Trans R Soc Lond B Biol Sci. 2019;374(1779):20180219. 10.1098/rstb.2018.0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DuChez BJ, Doyle AD, Dimitriadis EK, Yamada KM. Durotaxis by human cancer cells. Biophys J. 2019;116(4):670‐683. 10.1016/j.bpj.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Papalazarou V, Salmeron‐Sanchez M, Machesky LM. Tissue engineering the cancer microenvironment‐challenges and opportunities. Biophys Rev. 2018;10(6):1695‐1711. 10.1007/s12551-018-0466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mohammadi H, Sahai E. Mechanisms and impact of altered tumour mechanics. Nat Cell Biol. 2018;20(7):766‐774. 10.1038/s41556-018-0131-2. [DOI] [PubMed] [Google Scholar]
- 13. van Helvert S, Storm C, Friedl P. Mechanoreciprocity in cell migration. Nat Cell Biol. 2018;20(1):8‐20. 10.1038/s41556-017-0012-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Odenthal J, Takes R, Friedl P. Plasticity of tumor cell invasion: governance by growth factors and cytokines. Carcinogenesis. 2016;37(12):1117‐1128. 10.1093/carcin/bgw098. [DOI] [PubMed] [Google Scholar]
- 15. Ji K, Sameni M, Osuala K, Moin K, Mattingly RR, Sloane BF. Spatio‐temporal modeling and live‐cell imaging of proteolysis in the 4D microenvironment of breast cancer. Cancer Metastasis Rev. 2019;38(3):445‐454. 10.1007/s10555-019-09810-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Doyle AD, Carvajal N, Jin A, Matsumoto K, Yamada KM. Local 3D matrix microenvironment regulates cell migration through spatiotemporal dynamics of contractility‐dependent adhesions. Nat Commun. 2015;6:8720. 10.1038/ncomms9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doyle AD, Yamada KM. Mechanosensing via cell‐matrix adhesions in 3D microenvironments. Exp Cell Res. 2016;343(1):60‐66. 10.1016/j.yexcr.2015.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rijal G, Li W. A versatile 3D tissue matrix scaffold system for tumor modeling and drug screening. Sci Adv. 2017;3(9):16. 10.1126/sciadv.1700764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rijal G, Wang J, Yu I, Gang DR, Chen RK, Li W. Porcine breast extracellular matrix hydrogel for spatial tissue culture. Int J Mol Sci. 2018;19(10):13. 10.3390/ijms19102912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hakkinen KM, Harunaga JS, Doyle AD, Yamada KM. Direct comparisons of the morphology, migration, cell adhesions, and Actin cytoskeleton of fibroblasts in four different three‐dimensional extracellular matrices. Tissue Eng ‐ Part A. 2011;17(5–6):713‐724. 10.1089/ten.tea.2010.0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doyle AD. Generation of micropatterned substrates using micro photopatterning. Curr Protoc cell Biol. 2009;10.15.1‐10.15.35. 10.1002/0471143030.cb1015s45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doyle AD. Generation of 3D collagen gels with controlled diverse architectures. Curr Protoc Cell Biol. 2016;72:10.20.1‐10.20.16. 10.1002/cpcb.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Beacham DA, Amatangelo MD, Cukierman E. Preparation of extracellular matrices produced by cultured and primary fibroblasts. Curr Protoc Cell Biol. 2006;33(1):10.9.1‐10.9.21. 10.1002/0471143030.cb1009s33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DuChez BJ. Automated tracking of cell migration with rapid data analysis. Curr Protoc Cell Biol. 2017;76:12.12.1‐12.12.16. 10.1002/cpcb.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Amatangelo MD, Bassi DE, Klein‐Szanto AJP, Cukierman E. Stroma‐derived three‐dimensional matrices are necessary and sufficient to promote desmoplastic differentiation of normal fibroblasts. Am J Pathol. 2005;167(2):475‐488. 10.1016/S0002-9440(10)62991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ou Y‐C, Li J‐R, Wang J‐D, et al. Fibronectin promotes cell growth and migration in human renal cell carcinoma cells. Int J Mol Sci. 2019;20(11):16. 10.3390/ijms20112792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Deep G, Kumar R, Jain AK, Agarwal C, Agarwal R. Silibinin inhibits fibronectin induced motility, invasiveness and survival in human prostate carcinoma PC3 cells via targeting integrin signaling. Mutat Res. 2014;768:35‐46. 10.1016/j.mrfmmm.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carpenter PM, Sivadas P, Hua SS, et al. Migration of breast cancer cell lines in response to pulmonary laminin 332. Cancer Med. 2017;6(1):220‐234. 10.1002/cam4.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Carpenter PM, Dao AV, Arain ZS, et al. Motility induction in breast carcinoma by mammary epithelial laminin 332 (laminin 5). Mol Cancer Res. 2009;7(4):462‐475. 10.1158/1541-7786.MCR-08-0148. [DOI] [PubMed] [Google Scholar]
- 30. Gopal S, Veracini L, Grall D, et al. Fibronectin‐guided migration of carcinoma collectives. Nat Commun. 2017;8:14105. 10.1038/ncomms14105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang W‐Y, Twu C‐W, Liu Y‐C, Lin H‐H, Chen C‐J, Lin J‐C. Fibronectin promotes nasopharyngeal cancer cell motility and proliferation. Biomed Pharmacother. 2019;109:1772‐1784. 10.1016/j.biopha.2018.11.055. [DOI] [PubMed] [Google Scholar]
- 32. Ramos G d O, Bernardi L, Lauxen I, Sant'Ana Filho M, Horwitz AR, Lamers ML. Fibronectin modulates cell adhesion and signaling to promote single cell migration of highly invasive Oral squamous cell carcinoma. PLoS One. 2016;11(3):e0151338. 10.1371/journal.pone.0151338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Petrie RJ, Harlin HM, Korsak LIT, Yamada KM. Activating the nuclear piston mechanism of 3D migration in tumor cells. J Cell Biol. 2017;216(1):93‐100. 10.1083/jcb.201605097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Provenzano PP, Inman DR, Eliceiri KW, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:11. 10.1186/1741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conklin MW, Eickhoff JC, Riching KM, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178(3):1221‐1232. 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Riching KM, Cox BL, Salick MR, et al. 3D collagen alignment limits protrusions to enhance breast cancer cell persistence. Biophys J. 2014;107(11):2546‐2558. 10.1016/j.bpj.2014.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Juin A, Billotteta C, Moreau V, et al. Physiological type I collagen organization induces the formation of a novel class of linear invadosomes. Mol Biol Cell. 2012;23(2):297‐309. 10.1091/mbc.E11-07-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Artym VV, Swatkoski S, Matsumoto K, et al. Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J Cell Biol. 2015;208(3):331‐350. 10.1083/jcb.201405099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Morris AH, Stamer DK, Kyriakides TR. The host response to naturally‐derived extracellular matrix biomaterials. Semin Immunol. 2017;29:72‐91. 10.1016/j.smim.2017.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.