

Abstract
Background
Multiple genetic changes, availability of cellular nutrients and metabolic alterations play a pivotal role in oncogenesis
Aims
We focus on cancer cell's metabolic properties, and we outline the cross talks between cellular oncogenic growth pathways in cancer metabolism. The review also provides a synopsis of the relevant cancer drugs targeting metabolic activities that are at various stages of clinical development.
Methods
We review literature published within the last decade to include select articles that have highlighted energy metabolism crucial to the development of cancer phenotypes.
Results
Cancer cells maintain their potent metabolism and keep a balanced redox status by enhancing glycolysis and autophagy and rerouting Krebs cycle intermediates and products of β‐oxydation.
Conclusions
The processes underlying cancer pathogenesis are extremely complex and remain elusive. The new field of systems biology provides a mathematical framework in which these homeostatic dysregulation principles may be examined for better understanding of cancer phenotypes. Knowledge of key players in cancer‐related metabolic reprograming may pave the way for new therapeutic metabolism–targeted drugs and ultimately improve patient care.
Keywords: cancer metabolism, glutaminolysis, glycolysis, growth factors, mitochondrial transmembrane potential, systems biology
Glossary
- Akt
aphakia mouse T‐cell lymphoma
- Myc
myelocytomatosis
- AMPK
AMP‐activated protein kinase
- NAD+
nicotinamide adenine dinucleotide
- CpG
cytidine phosphate guanosine
- NADP+
nicotinamide adenine dinucleotide phosphate
- CTLA4
cytotoxic T‐lymphocyte
- NFκB
nuclear factor kappa B associated protein 4
- Oxidative stress
ROS scavenging capacity
- ΔG
Gibbs free energy
- Pcr
phosphocreatine
- Δψ
trasmembrane matrix potential
- PDL1
programmed cell death ligand protein 1
- PFK
phosphofructokinase
- GLUT 1
glucose transporter 1
- Pi
inorganic phosphate
- GSH
glutathione—cysteine, glycine
- PI3k
phosphatidylinositol 3 kinase
and glutamate
- PPP
pentose phosphate pathway
- HIF1‐α
hypoxia inducible factor
- ROS
reactive oxygen species
- IDH
isocitrate dehydrogenase
- SDH
succinate dehydrogenase
- IL‐1
interleukin 1
- TCA
tricarboxylic acid
- K‐Ras
Kisten rat sarcoma
- TNF
tumor necrosis factor
- LDH
lactate dehydrogenase
- Tp53
tumor protein 53
- mTORC1
mechanistic target of rapamycine complex 1
- VEGF
vascular endothelial growth factor
1. INTRODUCTION
Malignant cells depend on the continuous activation and/or alterations of growth factors and inflammatory signals in combination with several genetic and epigenetic modifications to circumvent cell death and promote cell proliferation. This ability hinges on the constant supply of energy and recycled carbon generated through active autophagy and through complex metabolic networks rerouting.1, 2, 3, 4, 5, 6 The oncometabolic shift is coopted from the major metabolic pathways including glycolysis, the pentose phosphate pathway (PPP), the tricarboxylic acid (TCA) cycle, and the β‐oxidation of free fatty acids (FFAs).7, 8, 9, 10, 11, 12, 13 This metabolic reprogramming is controlled by a reductive and oxidative (redox) pairs such as the nicotinamide adenine dinucleotide phosphate (NADP+/NADPH) and the nicotinamide adenine dinucleotide (NAD+/NADH); the universal energy carrier adenosine triphosphate (ATP)/adenosine diphosphate (ADP); the PPP; the transmembrane mitochondrial potential (Δψ); and finally, by the intracellular pH status. Because of their central roles in global metabolism, all living organisms including cancer cells ensure the proper balance of these factors to maintain a correct thermodynamic equilibrium. Glucose, apart from generating ATP and biosynthetic intermediates, is a key nutrient that also maintains redox homeostasis. 14
Under physiological conditions, pyruvate is formed in the last irreversible step of glycolysis. Two molecules of ATP are generated in the conversion of glucose into 2 molecules of pyruvate. The fate of pyruvate is variable. When the amount of oxygen is limiting, pyruvate is reduced by NADH to form lactate. The regeneration of NAD+ in this reduction maintains glycolysis. Under aerobic condition, pyruvate enters the TCA cycle inside mitochondria to form acetyl‐CoA. Much more energy can be extracted aerobically by means of the TCA cycle (36 molecules of ATP). The third fate of pyruvate is carboxylation to maintain a minimal level of oxaloacetate to allow the TCA cycle to continue functioning. This is an example of anaplerotic reaction.2
NADH—a reducing equivalent generated during catabolic breakdown of glucose—is used in mitochondrial oxidative phosphorylation (OXPHOS) and provides energy for catabolic reactions. NADH can be either converted to NADPH within the mitochondria or is used for generation of primary metabolites from precursor molecule in the TCA cycle. NADH is used later for electron transport chain. NADPH is produced in the oxidative PPP branching off glycolysis and provides reducing equivalents critical for anabolic synthesis of FFA and nucleotides. Since the cell regulates NADH and NADPH independently, consequently, the intracellular NAD+/NADH ratio is high, while the ratio of NADP+/NADPH is kept low.4
In cancer cells, the NAD+/NADH and NADP+/NADPH redox ratios are, respectively, 5 and 10 times higher compared with normal cells.4 These high redox couple ratios may explain the anomalous characteristic of cancer cell metabolism first observed by Otto Heinrich Warburg more than 60 years ago.15, 16, 17, 18, 19 The Warburg effect established increased glycolysis as a main source of metabolic fuel (ATP) in tumor cells under hypoxic or even normoxic conditions. We will revisit the Warburg effect shortly.
Recent cancer research has shown that the high glycolytic rate converting incoming glucose to lactate (and a stoichiometric amount of H+ ions) is upregulated by hypoxia‐inducible factor (HIF1‐α), oncogenic signaling such as aphakia mouse T‐cell lymphoma (Akt), and/or by mutations of TCA cycle enzymes such as lactate dehydrogenase (LDH) or succinate dehydrogenase (SDH).20
2. MATERIALS AND METHODS
We focus on cancer cell's metabolic properties, and we outline the cross talks between cellular oncogenic growth pathways in cancer metabolism. The review also provides a synopsis of the relevant cancer drugs that are at various stages of clinical development.
3. RESULTS
3.1. Regulation of energy metabolism in the normal mammalian cell
3.1.1. Energy status
Adenosine triphosphate is an unstable molecule when it is in equilibrium with water, highly “exergonic,” and is rapidly degraded by nucleotidase and by hydrolytic activity to ADP and inorganic phosphate (Pi) (ΔG ≃ −7.3 Kcal/mol), or to adenosine monophosphate (AMP) and 2 Pi (ΔG ≃ −4.3 Kcal/mol).21, 22 It has a rapid half‐life that is on the order of seconds.1 Cellular energy depends on the ATP/ADP or ATP/AMP ratio. Adenosine triphosphate is generated in 2 ways. One takes place in mitochondria through OXPHOS (>90%—cell respiration or oxidative state), and another occurs in the cytoplasm through anaerobic glycolysis (< 10%—also known as the Embden‐Meyerhof pathway or reductive state). The smaller amount of energy that is subsequently produced in glycolysis is still enormous: gram per gram, eukaryotic cells convert 104 times more energy per second than that produced by our sun.23 Phosphofructokinase (PFK) controls the third step and the slowest reaction in glycolysis, and therefore, PFK is the rate‐limiting step in that pathway. Phosphofructokinase is allosterically inhibited by high levels of ATP, and this inhibition is reversed by high levels of AMP and thus increasing the rate of glycolysis.22 Adenosine monophosphate and not ADP is the key low energy sensing mechanism of cells for 3 reasons. First, AMP remains intracytoplasmic and cannot enter mitochondria, while ADP does. Second, ADP can be salvaged into ATP by adenylate cyclase (2 ADP = AMP + ATP). Third, any small change of ATP concentration leads to a large change in concentration of AMP due to the difference in their absolute intracellular concentrations. For example, in the cytoplasm, the ATP/ADP and ATP/AMP ratios are ≃ 500 and 80, respectively; AMP is therefore considered an amplified version of ATP.21
3.1.2. Redox status
Changes in mitochondrial ADP concentration affect ATP synthase turnover, which is integrated with NADH‐generating enzymes associated with the Krebs's cycle dehydrogenases. The free energy generated by a strong reducing agent in the reaction NADH→ NAD+ when coupled to pyruvate → lactate reaction is due to the difference in reduction potentials of the participating reactants and calculated to be +0.13 V or −6 Kcal/mol (derived from Nernst equation).22
A “plus” reduction potential means that NADH has higher affinity for electrons (e−) than does hydrogen, which is defined as 0 V. The free energy of oxidation due to the flow of e− from NADH to O2 is −52.6 Kcal/mol, an exergonic process that is used to synthesize ATP (mostly by ATP synthase), which is an endergonic process with a ΔG of ≃ +7.3 Kcal/mol (this is the reverse energy of ATP hydrolysis). The normal cytoplasmic NAD+/NADH redox ratio15, 22 is high (≃700). In any coupled reaction to ATP hydrolysis, the equilibrium constant of that reaction is converted by a factor 108 because as you recall ATP is highly exergonic.22
Despite its tremendous daily turnover (exceeding 50 kg), ATP homeostasis with a total body content of only 50 g is maintained almost constant (within 10% variation) by phosphocreatine.
3.2. Energy metabolism in tumor cells
3.2.1. Warburg effect
Even in presence of oxygen, and under the influence of improper activation of HIF1‐α, growth factors, oncogene‐orchestrated upregulation of biosynthetic enzyme expression, and under a steady flow of energy demand, cancerous cells shift their energy metabolism largely to glycolysis. The result is an increased import of glucose into the cytoplasm by glucose transporter (GLUT)‐1, while OXPHOS remains almost unchanged.2, 13, 16, 17, 24
Altered energy metabolism is therefore a common feature of cancerous tissues.5, 6 Both Warburg effect and the drafted usage of biosynthetic macromolecules are simply other traits of cancer cells that modulate rapid energy generation, biomass formation, and redox status. This cancer phenotype is viewed as an autonomous evolutionary Malthusian selection and adaptation to the dynamic tumor microenvironment, which may include limited access to oxygen and nutrients, and an acidotic milieu.13 In brief, the Warburg effect is characterized as follows:
An 18 times less ATP is produced in glycolysis than in OXPHOS (2 ATP per glucose molecule compared to 36 ATP per glucose molecule via mitochondrial respiration).
A compensatory mechanism in cancer cells—transcription factors like myelocytomatosis (Myc) and HIF1‐α that upregulate expression of glycolytic enzymes, and increase GLUT‐1.
A glucose distribution in 3 parts: 85% is converted to lactate by glycolysis, 10% for OXPHOS, and the remaining 5% is for biomass formation.20
Recently, we came to realize that under the sustained proliferation‐induced oncogenes, and to meet their incessant demand for fuels, glycolytic tumors are also capable of using OXPHOS as an alternative fuel source.25 This capacity is accomplished by dispatching the little pyruvate generated from glycolysis to the oxygen‐consuming mitochondria. 1
Besides glucose, other catabolites such as glutamine, other amino acids, FFA, and pyruvate and lactate have all been shown to be diverted within tumor cells into various biosynthetic pathways.25 Thus, cancer cells have really reengineered metabolism for efficient incorporation of carbon into biomass and have adapted to an ever‐changing cancer microenvironment. Although glycolysis is a physiologic response to hypoxia in all proliferative and embryonic tissues, cancer cells constitutively use glucose and produce lactate regardless of oxygen pressure. The rationale for this transformation is to allow a faster generation of ATP to maintain high cellular division rate.16 The synthesis of ATP is split roughly equally between glycolysis and OXPHOS (rather than generation of >90% from OXPHOS). The process is induced to satisfy demand for energy metabolism, prevent excess accumulation of NADH from oxidative respiration, and to avoid reactive oxygen species (ROS) flooding of mitochondria.24 The additional demand for energy is to support nucleotide synthesis, diversion of Krebs cycle's intermediates (that are normally in balanced flux for cataplerotic and anaplerotic reactions), biosynthesis of nucleosides, lipids for building cell membranes, and finally for the building of macromolecules required by the cancer cell descendant progeny.6, 13, 19
3.2.2. Summarized pathobiology of the Warburg effect and its consequences
Cancer cells transition to anaerobic metabolism caused by genetic alterations, inadequate oxygen delivery, and by disorganized blood supply. They activate complex and interconnected signaling pathways designed to preserve their energy and redox status. To a certain extent, OXPHOS continues. The accumulation of ROS and an altered redox status play an important part in cancer progression. The role of OXPHOS is restricted to providing TCA cycle intermediates as substrates for the de novo synthesis of lipids and nonessential amino acids.3, 16
The primary metabolic end products as a direct consequence of the Warburg effect are the generation of lactate, pyruvate, and acetyl‐coenzyme A. The latter metabolite is further shunted into the production of FFA and triglycerides.15
The cofactor necessary for the continuation of glycolysis (NAD+) is regenerated by the conversion of pyruvate to lactate.
The extracellular lactate contributes to the acidification of the extracellular matrix (ECM) that provides a shield of protection against the immune response.20
Lactic acid and ECM‐associated proteinases together breakdown the mesh of secreted proteoglycans that surround the tumor facilitating local invasion and metastases.
Finally, any excess lactate is recycled back to pyruvate supporting OXPHOS by the better oxygenated cancer cells situated at the surface of tumor.
3.2.3. Acidosis
Lactic acidosis is common to many tumors. Under glucose deprivation or hypoxia, the production of protons derived from high glycolytic rate rescues cancer cells via gene reprogramming, stimulation of autophagy, and inhibiting cellular apoptosis by enhanced Akt activation.20, 26 Additionally, as we read before, ECM acidification in the vicinity of tumor cells exerts an immune‐permissive microenvironment by attenuating T‐cell activation and monocyte migration.27 Buffering the acid milieu with bicarbonate has been shown to improve antitumor responses of anti‐CTLA4, anti‐PD1, in tumors. 28, 29
3.2.4. Free fatty acids
Citrate in the cytosol is used mainly to convert acetyl‐CoA for fatty acids and cholesterol synthesis. Mitochondrial β‐oxidation is essential for the accelerated cancer cell proliferation.30 Carnitine palmitoyltransferase (CPT1) catalyzes rate‐limiting step of FFA oxidation. CPT1 is upregulated in many types of cancer and is an important fuel source for cancer via ATP and NADPH production. NADPH as we have already seen provides redox power against ROS. In cancer, biosynthetic pathway of FFA is also affected by overall pyruvate carbon flux.31
3.2.5. Redox status
Glucose is a key nutrient that maintains redox homeostasis of NAD+/NADH and NADP+/NADPH ratio. NADPH and NADH are cofactors that carry e− and are constantly regenerated from NADP+ and NAD+ to maintain the cellular redox balance.20 High NADPH/NADP+ ratio contributes to ROS detoxification.32
The preferential conversion of glucose to pyruvate then to lactate, is to regenerate NAD+ from NADH and to maintain glycolysis. Regeneration of NAD+ is attained by reducing pyruvate to lactate coupled to the reaction NADH→NAD+.
Hypoxic cancer cells are metabolically coupled with other stroma cells; thus, lactate generated via glycolysis by hypoxic cells can be recycled as substrate for mitochondrial OXPHOS by aerobic tumor stromal cells (=lactate shuttle). The aberrant low cytoplasmic NAD+/NADH redox ratio is maintained despite recycling some of NADH (produced during high rate glycolysis and TCA cycle activity) to regenerate NAD+. This step is essential for constant ATP production, glucose oxidation, and cell survival. The low NAD+/NADH redox ratio is also critical. It facilitates increased pyruvate conversion, and the excess lactate is secreted into the extracellular milieu and reused by stabilizing HIF1‐α and by activating proangiogenic and inflammatory factors.8, 9, 33, 34
3.2.6. Mitochondrial membrane potential in cancer cells (Δψ)
The human mitochondrial DNA (mtDNA) is a 16.6 kb circular double‐stranded DNA molecule that is devoid of protective histones.5 Mitochondrial DNA comprises 37 genes that are essential for mitochondrial function. Thirteen of these genes are involved in OXPHOS. Associations of tumor‐specific mtDNA sequence variants in various human cancers have been reported.35 In cancer and normal cells alike, mtDNA is precariously located adjacent to the mitochondrial “power furnace,” ie, oxidation and local generation of ROS. Knowing the diverse roles of mitochondria in cell metabolism, apoptosis, and oxidative stress, it is not surprising that any of these genes can thus be damaged leading to mitochondrial dysfunction, inefficient e− transport culminating with a high Δψ.
Low Δψ cells (quiescent) exhibit decreased oxidative stress and DNA damage, unlike, stem‐like cancer cells (a subset of cancer cells) and cancer cells, which are characterized by a high Δψ.36, 37
The e− donated by NADH flows, at a “tunneling rate” in the range of 1015 s−1 down the mitochondrial electron transport chain, as close as 14 A0 or less spacing of redox prosthetic centers, which is then coupled to proton extrusion by chemi‐osmotic type of mechanism from enzymes complex I, III, and IV.38 This process creates the very negative mitochondrial transmembrane potential Δψ (mean approximately −140 mV in normal cells of all living organisms) for maximal ATP production and optimum ROS formation. By contrast, the mean Δψ is high approximately −220 mV in both cancer cells and in rapidly proliferating embryonic stem cells. In both scenarios, Δψ acts as a charged “battery.” The more hyperpolarized Δψ in cancer cells than normal cells is due in part to aerobic glycolysis. If Δψ value dissipates, the permeability transition pore will open, and cell death occurs by the cytoplasm release of cytochrome c, which is an apoptotic agent. The stored Gibbs free energy (which in this context acts to generate proton‐motive force) is the difference (Δψ – ΔH) between concentration gradient of Δψ (negative inside) and ΔH+ (alkali inside). This gradient is used for H+ flow reentry into the mitochondrial matrix and maximum ATP synthesis.36 Therefore, loss of Δψ inhibits the production of ATP by ATP synthase.
4. DISCUSSION
4.1. Biological processes regulating cancer metabolism
On this note, Vander Heiden and DeBerardinis5 in a recently published review explored further the complexity of cancer biology. They elucidated specific metabolic activities that participate in the reprogramming of cancer energy metabolism. In that report, the authors provided a synopsis of the multistep processes involving many signaling pathways and their interconnections in cancer. In the metabolic reprogramming‐enabling glycolysis, the impact of several environmental nutrients availability (including glucose; glutamine; serine; arginine; glycine; tryptophan; TCA cycle enzyme mutations (such as LDH, isocitrate dehydrogenase [IDH], fumarate hydratase, and SDH); TCA cycle intermediates (such as succinate, aspartate, acetate, and fumarate); oxygen; and of HIF1‐α, 5, 8, 33, 34 all support survival and improve biomass formation. We will briefly explore some of these catabolites separately.
4.1.1. Glutamine
Is the most abundant circulating amino acid in blood and in muscle. 39 Myelocytomatosis upregulation, HIF1‐α stabilization, and mTOR upregulation all promote production of glutamine.40 It is the preferred anaplerotic substrate that drives bothTCA cycle and glycolysis. Glutaminolysis is considered a hallmark of cancer metabolism because glutamine has important pleiotropic functions that are critical for cancer cell survival, proliferation, and metastasis. Glutamine acts as a carbon and amino donor for building purines and pyrimidines, and production of lipids and non‐essential amino acids leading to proliferative growth. 39 For example, human and mouse gliomas growth is supported by using glucose to synthesize glutamine, which then locks in nucleotides biosynthesis. 41
Overexpression of oncogenes enhances glutaminolysis, but when glucose is increased, slowly proliferating tumor cells rapidly switch to a glycolytic mode (=Crabtree effect).19
4.1.2. IDH, fumarate hydratase, and SDH mutations act as oncogenes
Enzymes of the TCA cycle can be severely impaired in certain human cancers and thus losing their genetically tumor‐suppressor effects. The main explanatory element is that mutations of these enzymes lead to succinate or fumarate accumulations giving rise to DNA hypermethylation and epigenetic alterations.10, 42, 43 Mutation of SDH leads to oxidative stress enhancing the Warburg effect, which is conveyed by a lack of HIF1‐α degradation.44, 45
Somatic mutation in the active site of IDH confers increased activity of this enzyme resulting in over production of 2‐hydroxyglutarate. This in turn stabilizes HIF1‐α leading to the activation of glycolysis.8
LDH is key enzyme to cancer's glycolytic phenotype irrespective of cancer cell Tp53 status.
LDH‐A Converts pyruvate to lactate allowing faster rate of glycolysis and the generation of ATP then approaches that from OXPHOS. 46
4.2. Interplay between cancer metabolism and oncogenic signaling pathways
Along those changes, cancer‐associated metabolic alterations can intersect with oncogenic signaling pathways like HIF 1‐α, ROS, VEGF, NFκB, phosphatidylinositol 3 kinase (PI3K), genetic/epigenetic instability, oncoprotein Myc, K‐Ras, AMPK, Tp53, and mechanistic target of rapamycine complex 1 (mTORC1) that control cellular processes of growth, proliferation, differentiation, and stemness.
We discuss some of these pathways (Figure 1) in the following section and also recommend articles for further reading.34, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56 In one way or another, these oncoproteins and tumor suppressors contribute to reprogramming glucose metabolism.
Figure 1.
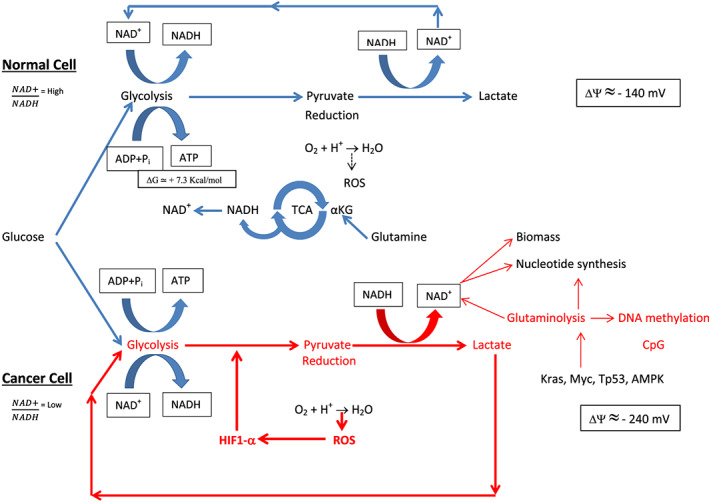
Normal and cancer cell functions. Physiologic functions are colored in blue. Upregulated functions are colored in red and are critical for cancer cell survival. Normally, the mitochondrial transmembrane potential (Δψ) is polarized (negative inside). In cancer cells, the Δψ is more hyperpolarized than normal cells. Akt, aphakia mouse T‐cell lymphoma; AMPK, adenosine monophosphate–activated protein kinase; CpG, cytidine phosphate guanosine; HIF1‐α, hypoxia inducible factor; K‐Ras, Kisten rat sarcoma (oncogene); mTOR, mammalian target of rapamycine; Myc, myelocytomatosis (oncogene); NAD+, nicotinamide adenine dinucleotide; NFκB, nuclear factor kappa B; PI3k, phosphatidylinositol 3 kinase; ROS, reactive oxygen species; SDH, succinate dehydrogenase; TCA, tricarboxylic acid; Tp‐53, tumor protein 53
4.2.1. Hypoxia inducible factor
Under normoxic conditions, HIF1‐α is a transcription regulator that remains at low‐level steady state by ubiquitination (half‐life of only 10 min). When cells experience hypoxic conditions—oxygen levels in the best perfused tissues are 9% or less—HIF1‐α degradation fails to occur, and its concentration increases rapidly causing expression of hundreds of genes with multiple targets involved in glucose metabolism. The constitutive expression of HIF1‐α—which promotes cell survival by switching to glycolytic metabolism and fast generation of ATP—is under the influence of mTORC1, Akt, and PI3K signaling. These signaling pathways allow the inhibition of the mitochondrial enzyme that converts pyruvate to acetyl‐CoA for entry into the TCA cycle. 5, 53
Investigators have reported that constitutive expression of HIF1‐α in many aggressive glycolytic tumors may also be caused by mtDNA mutations.56
4.2.2. Reactive oxygen species
By way of OXPHOS, e− generated from the respiratory complexes in mitochondria is a major source for ROS production. In cancer cells, under hypoxic microenvironment and because of insufficient e− transport, one byproduct of the metabolic shift within the mitochondria is ROS formation that is maintained at critical intensity. At this level, ROS induces DNA instability and aneuploidy, which further contribute to tumor initiation and progression. If a still higher level of ROS continues, it will serve as a crucial pro‐apoptotic factor, and the cancer cell dies. The functional significance of ROS adaptation in cancer requires further research.24
What is now known is that a moderate level of ROS reduces OXPHOS flux. By contrast in cancer‐associated fibroblasts, high expression of glycolytic transcription factor HIF1‐α induces high levels of ROS leading to suppression of OXPHOS.32
The most ROS leak comes from complex I enzyme because it has an unusually long distance of more than 14 A0 molecular spacing of redox centers, which is not optimum for a robust electron tunneling effect as discussed earlier.
Recent evidence has suggested an essential role for ROS in regulating cancer cell epithelial mesenchymal transition (EMT). Reactive oxygen species can alter redox‐sensitive proteins that possess free thiol (−SH) on cysteine residues of transcription factors involved in EMT remodeling including NFκB and HIF1‐α and promoting metastases.57 Consequently, antioxidants and flavonoids have been used as cancer drugs to retard EMT progression (Table 1).
Table 1.
List of drugs targeting metabolic activities that are undergoing therapeutic intervention or in clinical trials
| Drug/Products | Effect | Cancer Type | FDA/References |
|---|---|---|---|
| AZD3965 | CPT1 inhibitor | Lymphoma | NCT01791595 |
| Etomoxir | CPT1 inhibitor | Leukemia | 29, 30, 88 |
| Glioblastoma | |||
| Omeprazole | Proton pump inhibitor | Colorectal | NCT02518373 |
| Avastin | Anti‐VEGF A antibody | NSCLC (metastatic) | Approved/ 83, 84 |
| Hepatic | |||
| Nexavar | Anti‐VEGF receptor | Advanced/renal | Approved/ 83, 84 |
| Everlolimus | mTOR inhibitor | Multiple types | Approved/ 86 |
| Perhexilin+cisplatinum | Inhibitor β‐oxidation | Neuroblastoma | 87 |
| Metformin | Antidiabetic | Prostate | 89, 90, 91 |
| Increase NADH/NAD+ | |||
| Activates AMPK | |||
| Enhances lactate→pyruvate | |||
| L‐asparaginase | Glutamine depletion | ALL | Approved/ 92 |
| Vitamin C + doxycycline | Antioxidant/antibiotic | Cancer stem cells 95, 106 | |
| Galangin | Flavonoid | 93, 94 | |
| EMT transition | |||
| PEITC | Increase apoptosis | 58 | |
| Indoximol (NLG8189) | IDO inhibitor | 102 | |
| Ipilimumab | CTLA4 antibody | Melanoma | Approved in 2001 |
| + metastases | 103 | ||
| Duralumab | PDL1 antibody | Melanoma | Approved in 2017 |
| + metastases | 103 | ||
| Stiripentol | Antiepileptic | Compassionate use | |
| LDH inhibitor | Lymphoma | 97 | |
| Berberine | Activates AMPK | Colorectal cell line | 104 |
| Inhibits NFκB | In mice | ||
| Resveretrol | Plant phenol | CML | 105 |
| Inhibits m TOR |
Abbreviations: ALL, acute lymphocytic leukemia; CML, chronic lymphocytic leukemia; CTLA4, cytotoxic T‐lymphocyte associated protein 4; EMT, epithelial‐mesenchymal transition; NFκB, nuclear factor kappa B; NSCLC, nonsmall cell lung carcinoma; PDL1, programmed cell death protein ligand 1.
Continued ROS stress may induce the activation of NFκB leading to an increase in the expression of survival factors and ROS‐scavenging enzymes. Furthermore, cancer cells may respond to ROS stress by increasing their levels of VEGF and HIF1‐α leading to enhanced glycolysis.58 Based on this adaptation, cancer therapy aimed at increasing ROS level may thus be unsuccessful.
4.2.3. Vascular endothelial growth factor
Vascular endothelial growth factor (VEGF), originally known as vascular permeability factor, is a potent angiogenic ligand that binds to tyrosine kinase receptor. In cancer, the higher VEGF mRNA expression is NFκB dependent. In a recent series of 232 cervical adenocarcinomas, the immunohistochemical expression of metabolism related proteins, such as GLUT 1 and monocarboxylase transpoter (MCT), provided evidence for a cross‐talk between cancer metabolic remodeling and angiogenic switch.59
The expression of VEGF is inversely correlated with pH and is produced by cancer cells, myofibroblasts, and by macrophages within the cancer‐associated stroma predominantly surrounding necrotic areas.52, 59, 60 This helps to continuously induce fibrin‐rich fluid within tumor stroma, halting in their track the marching of invasive immune cells as a result of the now very dense barrier of connective tissue.52 Unlike in normal tissues, the deposition of extensive collagenous matrix by myofibroblasts may eventually replace the now dissolved fibrinous matrix by way of fibrinolysis. The consequent constitutively leaky capillaries have a major clinical implication in tumor oncopharmacology: Excessive pressure buildup in stroma due to fluid accumulation prevents efficient anticancer drug transfer from the capillary into the tumor niche.
4.2.4. Nuclear factor kappa B
Discovered 30 years ago, nuclear factor kappa B (NFκB)61 is a transcription factor that mediates inflammation. It is induced by ROS, interleukin (IL)‐1, and by tumor necrosis factor. 62, 63 Excessive and continuous—instead of brief bursts as in the case of normal wound healing—can lead tumor cells to evade the antiproliferative control mechanism without healing; hence, the metaphor that tumors are “Wounds That Do Not Heal.”64 Tumor cells thus behave as chronically wounded as a result of NFκB, matrix metalloproteinases, and a variety of activated growth factors. Those factors are released by the recruited macrophages and fibroblasts, which are somewhat resistant to hypoxia in tumor tissues.65
Recent reports have linked inflammation and metabolism and demonstrated that NFκB regulates glycolysis by upregulating GLUT3, mitochondrial respiration, and energy homeostasis.55, 63
In the absence of Tp53, NFκB suppresses OXPHOS. Nuclear factor kappa B also senses environmental nutrient levels such as glucose and amino acids and evokes gene response by varying transcriptional activity leading to an unstoppable metabolites mobilization.63 Besides NFκB, other pro‐inflammatory cytokines, including TNF‐α and IL‐1β, synergize with transcription factor such as SOX2 (sex determining region Y‐box 2). That cooperation is crucial for self‐renewal of embryonic stem cells.66 In vitro cell culture experiment of human colorectal adenocarcinoma established a signaling role for lactate modulating the endothelial cell phenotype and thereby tumor vascular morphogenesis (=tumor angiogenesis). The integrating factors between lactate and endothelial cells were ROS and NFκB. 67
4.3. Genetic instability and mutations
Oxidative stress has been linked to karyotype instability. Furthermore, in many cancers, loss of heterozygosity and chromosomal/microsatellite instability is generally recognized cancer phenotypes and is, for the most part, induced by inflammation, chronic hypoxia, and by ROS exposure of tumor tissue.68 Researchers have identified somatic mutations (genetic) due to the stochastic, ie, “probabilistic” DNA replication and to the more “deterministic” environmental signals (epigenetic). Both genetic and epigenetic transformations are responsible for deregulating cellular energetics and redox capacity.69, 70 Mitotic check‐point proteins/gene mutations cause chromosomal desegregation resulting in an aberrant chromosomal architecture or aneuploidy when expressed at supernormal levels. Aneuploidy, defined as the presence of an abnormal number of chromosomes or structure, is a widespread feature of most human cancer genome.71, 72 Under selective pressure during tumor evolution and therapy, aneuploidy may enhance adaptation and survival. Molecular abnormalities observed in cancer aneuploidy are all natural consequence of the massive overexpression and underexpression of proteins.73 For example, intratumor hypoxia induces genomic instability and leads to mutational inactivation of tumor suppressor genes Tp53 and TGF‐β, and activation of GLUT‐1, HIF1‐α, AMPK, and VEGF proteins.69, 74 These tumorgenic mutations exert metabolic autonomy and can drive metastatic phenotype observed in tumor cells.9, 69 Loss of Tp53 in fibroblasts enhances glucose transport via NFκB and accelerates glycolysis.73
4.4. Relevance of AMPK to cancer metabolism
Adenosine monophosphate–activated protein kinase (AMPK) and mTOR are concurrently activated in cancer by recycled aminoacids. AMPK is activated by AMP (sensor of low energy) and activates both OXPHOS and glycolysis, thus increasing the NAD+/NADH ratio in the cell. AMPK also functions as a metabolic tumor suppressor regulating glucose, lipids, and proteins metabolism.75
The major role of AMPK in cancer metabolism is the direct phosphorylation of Tp53 leading to its stabilization and to its transcriptional activity.76 Tp53 promotes conversion of pyruvate into acetyl‐CoA for entry into the TCA cycle generating less ATP and suppresses MCT1, thereby reducing NAD+ generation through the conversion of pyruvate to lactate.77
Activation of Tp53 also restricts cell growth at times of glucose deprivation. Tp53 is a tumor suppressor that responds to multiple cellular stresses including oncogenic activation or hypoxia and induces cell‐cycle arrest and senescence. It is well known that Tp53 is inactivated in approximately 50% of human cancers.
Free fatty acid biosynthesis is induced by phosphorylation of acetyl‐CoA carboxylase. Activation of AMPK in turn activates acetyl‐CoA carboxylase, which is a downstream target that stimulates lipid metabolism needed for building of cancer cell membrane.78
Activation of AMPK has also been found to oppose tumor progression in part by regulating inflammation and immune cells including dentritic cells, macrophages, and T‐cells. In all these cells, FFAs are used to generate ATP. AMPK is thus essential for the normal function of innate immunity. No wonder then that AMPK deficient mice have been found to produce higher level of pro‐inflammatory cytokines that correlated with tumor progression and invasion.79
To maintain protein homeostasis and supply metabolite intermediates for biosynthetic processes, even by addition of amino acids, AMPK concurrently with and independently of mTORC1 is acutely activated in an Akt‐ and insulin‐independent manner to sustain autophagy; thus, AMPK opposes mTORC1 inhibitory activity. These results have recently been reported in a murine myocytes experiment,80 Figure 2. The clinical implication of these findings is that enteral or parenteral nutritional supplement might be ineffective in slowing down muscle wasting in cancer patients. Based on the critical role of AMPK in cancer, metformin, an AMPK activator, is emerging as a possible anticancer drug.
Figure 2.
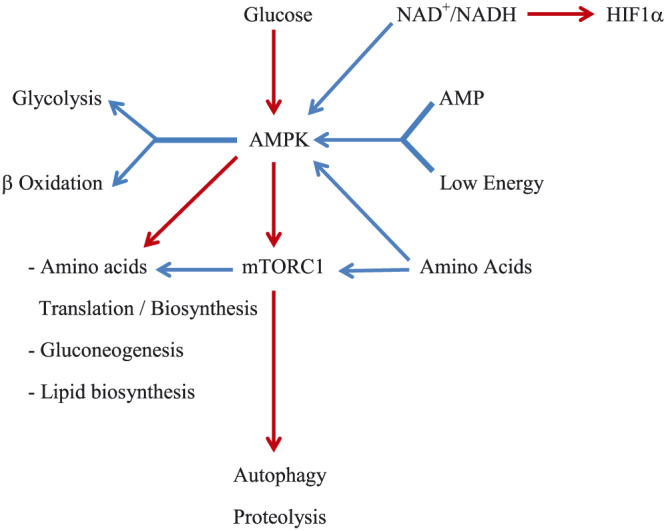
A simplified block diagram of signaling pathways by key regulators: AMPK and mTORC1. Blue arrow line means activation. Red arrow line means inhibition. These are not necessarily direct. Both, AMPK and mTORC1 are activated by the addition of amino acids. AMPK opposes mTORC1 and activates autophagy. AMP, adenosine monophosphate; AMPK, AMP‐activated protein kinase; NAD+, nicotinamide adenine dinucleotide; mTORC1, mammalian target of rapamycin complex 1; HIF1α, hypoxia inducible factor
4.5. mTORC1 and cancer
mTORC1 that was dubbed as the mammalian target of rapamycin (later changed to mechanistic) is hyperactivated in cancer. Sirolimus, a macrolide compound made by a bacterium with antifungal properties, is a rapalog, or rapamycin (named after Rapa Nui Island in the South Pacific where it was cultured from a soil sample).81 Rapamycin inhibits mTORC1 pathway. Proliferative and progrowth processes such as glycolysis, lipids, and amino acid biosynthesis, as well as inhibition of apoptosis and autophagy, are all regulated by mTORC1.5, 80 mTORC1, a serine/threonine‐specific protein kinase, is a downstream effector for multiple mutated oncogenic signals such as K‐Ras, AMPK, and PI3K/AKt pathways.75, 80, 81
Phosphatidylinositol 3 kinase—an intracytoplasmic second messenger enzyme—is one of the most common constitutively activated signal in human cancers even in the absence of growth factors. Aphakia mouse T‐cell lymphoma is also a serine/threonine kinase that reduces apoptosis by destroying Tp53 and increases protein synthesis and angiogenesis and commits to glycolysis. Phosphatidylinositol 3 kinase/AKt axis activates mTOR kinase, and both pathways represent the dominant mitogenic signals in most types of nonhematopoietic spontaneous cancer cells.52, 58 mTOR is inhibited by AMPK and switches off ATP‐consuming processes in order to save energy. Overall, therefore, mTORC1 is a highly connected regulator of many key metabolic processes.
4.6. Therapeutic targets based on oncometabolism
Overview of drugs targeting metabolic activities that are undergoing therapeutic intervention or in clinical trials is provided in Table 1.
As we read earlier, under the influence of oncogenic signals and hypoxia, cancer is characterized by a high metabolic demand with a main alteration towards glycolytic metabolism and accumulation of H+ ions. If this alteration is a common trait in human malignancies, then why not inhibit glycolysis to kill cancer cells? Actually, this strategy may probably be doomed to fail, since cancer cells circumvent any therapy designed to inhibit glycolysis by repressing respiration and exacerbating hypoxia via the Crabtree effect as mentioned above. The end result will surely increase further mutation rate.16 Efforts have thus focused on other therapeutic modalities. For example, exploiting tumor microenvironment acidification, a phase 1 trial of AZD 3965, a CPT1inhibitor, is in progress (NCT01791595) being tested for solid tumors and/or lymphoma. A phase 2 trial of Omeprazole, a proton pump inhibitor, is tested for treatment of colorectal cancer (NCT 02518373).
Other agents such as Avastin—an anti‐VEGFA monoclonal antibody (bevacizumab)—and Nexavar (Sorafenib)—a VEGF receptor antagonist—are both approved by the FDA as first line therapy for treating metastatic non‐squamous NSCLC or renal cell carcinoma, respectively.82, 83
A phase 3 randomized, double‐blind, placebo‐controlled, international, multicenter trial in patients with unresectable hepatic carcinoma showed improved survival with Avastin. Furthermore, Avastin treatment combined with other chemotherapeutic agents added an average of 4.7 months to the overall survival of patients with advanced colorectal carcinoma.83 However, the combined costs of this drug cocktail are prohibitive.84
The mTOR inhibitor everolimus, a derivative of rapamycin, has an antiangiogenic effects and has been approved by the US Food and Drug Administration in 2012 for treatment of advanced breast cancer and later approved for metastatic renal cell carcinoma.85
A clinical study in children with neuroblastoma, Perhexiline a β‐oxidation inhibitor of FFA utilization enhanced antitumor efficacy of cisplatinum cytotoxic side‐effects and improved survival. 86
Aberrant expression of CPT1A and CPT1C, regulators of FFA β‐oxidation, has been demonstrated in a cohort of 80 patients with high grade glioblastoma. 29 Carnitine palmitoyltransferase inhibitor, etomoxir impairs NADPH production and increases ROS resulting in ATP depletion and tumor cell death.30, 31 In another study, etomoxir suppressed cancer cell proliferation and sensitized human leukemic cells to apoptosis.87
Studies in mice and humans provided support for metformin—an antihypertensive agent—as an anticancer drug. Metformin increases the ratios NADH to NAD+, and lactate to pyruvate reaction in the cytoplasm and reciprocal changes in the mitochondria.88
Studies by Fendt and others showed that metformin decreases glucose oxidation and increases the dependence of prostate cancer cells on reductive glutamine metabolism.89, 90
Glutamine depletion by L‐asparaginase is an FDA‐approved cancer drug to treat acute lymphocytic leukemias.91
On the basis of the elevated ROS generation by increased intrinsic oxidative stress in cancer cell, increasing intracellular ROS above critical levels may selectively induce cancer cell death. In a preclinical study, high dose of vitamin C (ascorbic acid‐ an antioxidant) has been reported to benefit patients with colorectal cancer. Galangin, a flavonoid, can also repress mitochondria‐derived ROS and inhibits cell invasion by restraining EMT.92, 93
Although the exact mechanism by which vitamin C exerts its effect is unknown, one recently proposed mechanism is that vitamin C decreases the levels of NADPH, glutathione (GSH), and NAD+. Consequently, intracellular ROS is increased in cancer cell that carries oncogenic mutations and that expresses high levels of GLUT 1 on their plasma membrane, owing to the fact that GLUT1 is a known transporter of oxidized form of vitamin C. This is the naturally occurring form of vitamin C that acts as an extracellular e− acceptor. It maintains the NADH/NAD+ ratio by ensuring a high rate of NADH production and a continued e− flux through the plasma membrane.94
Similarly, β‐phenylethyl isothiocyanate, which is naturally present in relatively high quantities in cruciferous vegetables, increases intracellular ROS levels and apoptosis by depleting both cytoplasmic and mitochondrial GSH. Cancer cells carrying high levels of activated PI3K/AKt signaling can be selectively eradicated by exposure to β‐phenylethyl isothiocyanate. 58
Specific LDH inhibitors, such as FX11 and stiripentol, induce apoptosis that is mediated by mitochondrial ROS production in lymphoma and lung cancer cells.95, 96 The anticonvulsivant drug, Stiripentol, is not approved in the United States but is classified as an orphan drug that may be prescribed on a compassionate use basis. This drug has been shown to inhibit LDH but has not been tested as anticancer drug.
Several GLUT 1 inhibitors have been shown to kill cancer cells in vitro. But the consequence of systemic depletion of this receptor in human cancer has yet to be determined.
Multiple solid tumors overexpress specific tryptophan‐degrading dioxygenases, which catalyze tryptophan into kynurenine. This derivative promotes suppression of antitumor immune responses by promoting regulatory T (Tregs) cell phenotype. Tregs are immunosuppressive and generally suppress or down regulate the proliferation of immune effector cells. This mechanism is exploited by cancer for immune escape.97 The indoleamine 2,3‐dioxygenase (IDO) pathway regulates immune response by suppressing T‐cell function and enabling local tumor immune escape.98 A methylated tryptophan with immune checkpoint inhibitory activity, indoximod (NLG‐8189), inhibits IDO, which degrades the essential amino acid tryptophan, and may increase or maintain tryptophan levels important to T‐cell function. Tryptophan depletion is associated with immunosuppression involving T cell arrest and anergy. 99 Indoleamine 2,3‐dioxygenase is the rate‐limiting enzyme in tryptophan catabolism and is highly expressed in many cancer cells and in antigen‐presenting cells.98, 100 In vitro, IDO inhibitors significantly reverse the suppression of T cells created by IDO‐expressing dendritic cells and are being tested in phase 2 clinical trials across various cancers.101
Finally, immune check‐point blockade, by blocking intrinsic down‐regulators of immunity has increased survival in the treatment of multiple cancer types. This benefit comes at a cost of adverse events such as the occurrence of lymphomagenesis and autoimmunity.102
Examples of cancer immunotherapy that showed improved survival include Ipilimumab (CTLA4 antibody), an FDA‐approved drug in 2011 for metastatic melanoma, and Durvalumab (PDL1 antibody), an FDA‐approved drug in 2017. Both drugs can overactivate immunity so it attacks normal tissues in multiple organs.
Treatment with naturally occurring compounds such as berberine has been shown to induce apoptosis in colorectal cancer cell lines, inhibited NFκB activity, and to inhibit colon tumor formation in mouse model through activation of AMPK.103 Treatment with berberine has not been tested in human cancers.
Another compound resveratrol—a type of natural polyphenol, and a phytoalexin produced by several plants in response to injury—induces autophagy in chronic myelogenous leukemia cells by stimulating AMPK, thereby inhibiting the mTOR pathway. As we know, autophagy enables cancer cell survival by sustaining energy homeostasis and eliminating damaged organelles and proteins.104
Finally, as we have learned earlier, high oxidative stress is a characteristic of cancer stem cells.37 Therefore, pharmacologic redox modulation might be an effective therapy to eliminate those cells and hopefully abolish chemotherapy and radiotherapy resistance. A new combination drugs to eradicate cancer stem cells with doxycycline targeting mitochondrial protein translation and vitamin C targeting glycolysis has been proposed.105
4.7. Future prospects
It is noticed that cancer drug development is progressing rapidly. Yet curative cancer therapy has been limited thus far. Therefore, there is an unmet need for new anticancer drugs as we realize that the administration of agent alone could have an inadequate value. Eventually, new agent combinations with or without immunotherapy will have to be considered in the next generation of clinical investigations.
We suggest that there are, however, several challenges in this approach that researchers in the cancer field should address and solve.
First, thus far, the identification of the ultimate single molecular signal occurring in the initiation and the progression of cancer growth that might lead to targeted therapy has not had a universal success.
Second, different tumor types have different phenotypes, eroding the hope for a “magic bullet” to treating cancer.106
Third, tumor adaptability to cellular and extracellular environmental selection (Darwinian) forces conferring a competitive fitness and promoting emergence of treatment resistance. Our understanding of the mechanisms underlying such acquired resistance is still very limited.
Fourth, as we noted earlier, progressive aneuploidy may also induce cancer cells adaptation and resistance to cytotoxic drug therapy. Whether or not cancer tissue with chromosomal instability is more sensitive to further instability resulting from neoadjuvant therapy remains to be proven.107
Fifth, networks involved in tumorigenesis such as the cell‐cycle checkpoint, tumor suppressor proteins, and other oncoproteins may exhibit bistability due to positive feedback loops resulting in a robust output that is sometimes nonlinear, oscillatory, or bistable.
Examples of networks oscillation in time are NFκB and Tp53 signals. Bistability of regulatory circuits occurs when the rate of positively autoregulated gene product—by means of positive feedback loops—is strong compared with its degradation rate. AMPK is an example of a bistable signal in which the feedback loop enhances the robustness of 2 stable output states in response to a stimulus. Furthermore, bistability generally widens the distribution of proteins concentration per cell in which the distribution becomes bimodal. Subsequently, cell‐cell variability—due to different protein concentrations—determines different phenotypes of developmental tissues including cancer.108, 109
Sixth, unfortunately, as in most cases, antineoplastic compounds in preclinical trials are tested based on a single therapeutic strategy designed to hit 1 particular molecule or a metabolite. Although a single entity is easier to test pharmacologically, this approach has a potential downside owing to acquired drug resistance overtime and to the genomic instability of tumor tissue promoting increased mutagenesis and tumor‐cell transformation.108, 110
Seventh, as we learned earlier, stroma of malignant tissues prevents efficient transfer of anticancer drugs into the tumor niche.
Finally, there are concerns over a greater degree of drug toxicity, safety, and systemic side effects including immunosuppression.102
One then poses the following questions: Will targeting combinatorial networks ever decrease cancer treatment‐related resistance? And might this approach provide sustained remission? The benefits in this paradigm shift for treatment of cancer must be balanced against the potential of unforeseen pitfalls related to multiple drug side effects therapy. This concern is based upon the fact that drug can also be diverted against pathways or molecules shared by normal host cells. Moreover, evolutionary theory predicts that resistant clones would rapidly emerge.111
4.7.1. Emerging roles of cancer systems biology in developing therapeutics
Integration of complex biological processes and their network interactions as they have been revealed in cancer research requires critical analysis.112 Computational modeling to identify alternative and novel cancer therapeutic drug targets can be used by modeling dynamical molecular changes noted above.113
Here, the emerging field of “cancer systems biology analysis” through mathematical modeling might provide some of the answers.114, 115, 116, 117 By building these integrated networks, systems biology (which is defined as a process that is more than the sum of its parts) is more relevant to the natural world, and to oncogenesis. Tumor progression is described as dynamic and time‐independent—meaning current output of the collective networks does not depend on the previous input—making the process dynamic and nonlinear. This new field is still evolving, and researchers have yet to select the proper analytical and computational tools. The initial step of modeling and then integrating a wealth of high throughput data of—“omics” information—consequential to very dense and high‐dimensional biological networks is a daunting challenge. Indeed, the profile of the extensive cross‐talks and the constellation of molecular regulators critical in tumorigenesis as outlined in this review offer a critical insight into a difficult disease process.
Once signaling‐network data are obtained, data mining using systemic mathematical tools to identify their dynamics include mass action kinetics; least square regressions analyses (regular, ridge, or lasso); principal component analysis; Gaussian mixture model; and Bayesian inference to cite a few. These models need to be validated before applying them for long‐term prediction.108 As we have described earlier and because of multiple feedback loops, certain signaling networks are dynamic and nonlinear, and some are oscillatory such as Tp53, AMPK, or Akt,118 making future prediction to capture process dynamics difficult. Principal component analysis–based methods in these circumstances cannot be applied.
5. CONCLUSIONS
In this review, we outlined the transformation from the physiologic to the oncometabolic partition of metabolic energy between upregulated glycolysis and downregulated mitochondrial oxidation. We also learned about the intertwined connections between growth signals and intermediate metabolites designed to support continued tumor progression. The scientific advances from work derived over the last decade provided substantial insights into the complex tapestry of cancer pathogenesis. Improvement in our understanding of cancer pathophysiology continues, and so does the effectiveness of therapy. However, much remains to be done about both the genetic alterations and the hijacked metabolism‐dependent oncogenic state that inexorably replenishes the fuels for “this voracious beast.”
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHORS' CONTRIBUTION
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, AKA, FTC; Methodology, AKA; FTC; Investigation, AKA; FTC; Formal Analysis, AKA, FTC, BJB, YKA, KOA; Resources, AKA, FTC; Writing ‐ Original Draft, AKA, FTC; Writing ‐ Review & Editing, AKA, FTC, BJB, YKA, KOA; Visualization, AKA, BJB; Supervision, AKA.
ACKNOWLEDGMENT
No funding was received for this study.
Alameddine AK, Conlin FT, Binnall BJ, Alameddine YA, Alameddine KO. How do cancer cells replenish their fuel supply? Cancer Reports. 2018;1:e1003. 10.1002/cnr2.1003
REFERENCES
- 1. Cairns RA, Mak TW. The current state of cancer metabolism. Nat Rev Cancer. 2016;16(10):613‐614. [Google Scholar]
- 2. Ward PS, Thompson GB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11(2):85‐95. [DOI] [PubMed] [Google Scholar]
- 4. Chiarugi A, Dölle C, Felici R, Ziegler M. The NAD metabolome—a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12(11):741‐752. [DOI] [PubMed] [Google Scholar]
- 5. Vander Heiden MG, DeBerardinis RJ. Understanding the intersections between metabolism and cancer biology. Cell. 2017;68:657‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:17‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Soga T. Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 2013;104(3):275‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. DeBerardinis RJ. Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet Med. 2008;10(11):767‐777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sullivan LB, Gui DY, Vander Heiden MG. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nature. 2016;16:680‐693. [DOI] [PubMed] [Google Scholar]
- 11. Zhang S, Yang C, Yang Z, et al. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am J Cancer Res. 2015;5(3):928‐944. [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32(1):12‐19. [DOI] [PubMed] [Google Scholar]
- 13. Hanahan D, Weinberg R. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐647. [DOI] [PubMed] [Google Scholar]
- 14. Moreno‐Sánchez R, Marín‐Hernández A, Saavedra E, Pardo JP, Ralph SJ, Rodríguez‐Enríquez S. Who controls the ATP supply in cancer cells? Biochemistry lessons to understand cancer energy metabolism. Int J Biochem Cell Biol. 2014;50:10‐23. [DOI] [PubMed] [Google Scholar]
- 15. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891‐899. [DOI] [PubMed] [Google Scholar]
- 17. Warburg O. On respiratory impairment in cancer cells. Science. 1953;124:269‐270. [PubMed] [Google Scholar]
- 18. Moreira JV, Hamraz M, Abolhassani M, Bigan E, et al. The redox status of cancer cells supports mechanisms behind the Warburg effect. Metabolites. 2016;33:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hu X, Chao M, Wu H. Central role of lactate and proton in cancer cell resistance to glucose deprivation and its clinical translation. Signal Transduct Targeted Ther. 2017;2, Article number: 16047. 10.1038/sigtrans.2016.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bittar EE, Bittar N (Eds). Cellular ATP, Principles of Medical Biology. Chapter 1. 4 part II Connecticut: Elsevier; 1996:1‐47. [Google Scholar]
- 22. Stryer L (Ed). Chapter 21 Oxidative phosphorylation, Biochemistry. 4th ed. New York, NY: WH Freeman; 1995:529‐558. [Google Scholar]
- 23. Schatz G. The tragic matter. FEBS Lett. 2003;536(1‐3):1‐2. [DOI] [PubMed] [Google Scholar]
- 24. Chatterjee A, Dasgupta S, Sidransky D. Mitochondrial subversion in cancer. Cancer Prev Res. 2011;4(5):638‐654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vander Heiden MG, Locasale JW, Swanson KD, et al. Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science. 2010;329(5998):1492‐1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Corbet C, Feron O. Tumour acidosis: from the passenger to the driver's seat. Nat Rev Cancer. 2017;17(10):577‐593. [DOI] [PubMed] [Google Scholar]
- 27. Colegio OR, Chu NQ, Szabo AL, et al. Functional polarization of tumour‐associated macrophages by tumour‐derived lactic acid. Nature. 2014;513(7519):559‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pilon‐Thomas S, Kodumudi KN, El‐Kenawi AE, Russell S, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cirillo A, Di Salle A, Petillo O, Melone A, et al. High grade glioblastoma is associated with aberrant expression of ZFP57, a protein involved in gene imprinting, and of CPT1A and CPT1C that regulate fatty acid metabolism. Cancer Biol Ther. 15(6):735‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Qu Q, Zeng F, Liu X, Wang QJ, Deng F. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7(5):e2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014;71(14):2577‐2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martinez‐Outschoorn UE, Peiris‐Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14(2):113. 10.1038/nrclinonc [DOI] [PubMed] [Google Scholar]
- 33. Semenza GL. Targeting HIF‐1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721‐732. [DOI] [PubMed] [Google Scholar]
- 34. Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF‐1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012;18(20):5585‐5594. [DOI] [PubMed] [Google Scholar]
- 35. Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25(34):4647‐4662. [DOI] [PubMed] [Google Scholar]
- 36. Heerdt BG, Houston MA, Augenlicht LH. Growth properties of colonic tumor cells are a function of the intrinsic mitochondrial membrane potential. Cancer Res. 2006;66(3):1591‐1596. [DOI] [PubMed] [Google Scholar]
- 37. Sancho P, Barneda D, Heeschen C. Hallmarks of cancer stem cell metabolism. Br J Cancer. 2016;114(12):1305‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunneling in biological oxidation‐reduction. Nature. 1999;402(6757):47‐52. [DOI] [PubMed] [Google Scholar]
- 39. Yang L, Venneti S, Nagrath D. Glutaminolysis: a hallmark of cancer metabolism. Annu Rev Biomed Eng. 2017;19(1):163‐194. [DOI] [PubMed] [Google Scholar]
- 40. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marin‐Valencia I, Yang C, Mashimo T, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15(6):827‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yang M, Pollard PJ. Succinate: a new epigenetic hacker. Cancer Cell. 2013;23(6):709‐711. [DOI] [PubMed] [Google Scholar]
- 43. Wong CC, Qian Y, Yu J. Interplay between epigenetics and metabolism in oncogenesis: mechanisms and therapeutic approaches. Oncogene. 2017;1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Slane BG, Aykin‐Burns N, Smith BJ, et al. Mutation of succinate dehydrogenase subunit C results in increased O2•−, oxidative stress, and genomic instability. Cancer Res. 2006;66(15):7615‐7620. [DOI] [PubMed] [Google Scholar]
- 45. King A, Selak MA, Gottlieb E. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25(34):4675‐4682. [DOI] [PubMed] [Google Scholar]
- 46. Allison SJ, Knigh JRP, Granchi C, Rani R, et al. Identification of LDH‐A as a therapeutic target for cancer cell killing via (i) p53/NAD(H)‐dependent and (ii) p53‐independent pathways. Oncogene. 2014;3(5):e102. 10.1038/oncsis.2014.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c‐Myc and cancer metabolism. Clin Cancer Res. 2012;18:5546‐5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Choi YK, Park KG. Metabolic roles of AMPK and metformin in cancer cells. Mol Cells. 2013;36(4):279‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Flöter J, Kaymak I, Schulze A. Regulation of metabolic activity by p53. Metabolites. 2017;7(2):21. 10.3390/metabo7020021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16(10):635‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5(10):1024‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell. 2017;169(3):381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Balamurugan K. HIF‐1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer. 2016;138(5):1058‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20(1):87‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Baker RG, Hayden MS, Ghosh S. NFκB, inflammation, and metabolic disease. Cell Metab. 2011;13:11‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Weinberg RA. Tumor suppressor genes. In: Weinberg RA, ed. The Biology of Cancer. 2nd ed. New York: Garland science; 2014:231‐274. [Google Scholar]
- 57. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trachootham D, Alexandre J, Peng Huang P. Targeting cancer cells by ROS‐mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579‐591. [DOI] [PubMed] [Google Scholar]
- 59. Pinheiro C, Garcia EA, Morais‐Santos F, Moreira MA, et al. Reprogramming energy metabolism and inducing angiogenesis: co‐expression of monocarboxylate transporters with VEGF family members in cervical adenocarcinomas. BMC Cancer. 2015;15:835. 10.1186/s12885-015-1842-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shi Q, Le X, Wang B, et al. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene. 2001;20(28):3751‐3756. [DOI] [PubMed] [Google Scholar]
- 61. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF‐κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1‐2):37‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G. The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends Cell Biol. 2012;22(11):557‐566. [DOI] [PubMed] [Google Scholar]
- 64. Dvorak HF. Tumors: wounds that do not heal. N Engl J Med. 1986;315(26):1650‐1659. [DOI] [PubMed] [Google Scholar]
- 65. Schäfer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9(8):628‐638. [DOI] [PubMed] [Google Scholar]
- 66. Bass AJ, Wang TC. An inflammatory situation: SOX2 and STAT3 cooperate in squamous cell carcinoma initiation. Cell Stem Cell. 2013;12(3):266‐268. [DOI] [PubMed] [Google Scholar]
- 67. Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF‐κB/IL‐8 pathway that drives tumor angiogenesis. Cancer Res. 2011;71(7):2550‐2560. [DOI] [PubMed] [Google Scholar]
- 68. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer‐related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30(7):1073‐1081. [DOI] [PubMed] [Google Scholar]
- 69. Bristow RG, Hill RP. Hypoxia and metabolism: hypoxia, DNA repair and genetic instability. Nat Rev Cancer. 2008;8(3):180‐192. [DOI] [PubMed] [Google Scholar]
- 70. Tomasetti C, Vogelstein B. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sansregret L, Vanhaesebroeck B, Swanton C. Determinants and clinical implications of chromosomal instability in cancer. Nat Rev Clin Oncol. 10.1038/nrclinonc.2017.198. Published on Line Jan 03, 2018 [DOI] [PubMed] [Google Scholar]
- 72. Hoeijmakers JHJ. DNA damage, aging, and cancer. N Engl J Med. 2009;361:1475‐1148. [DOI] [PubMed] [Google Scholar]
- 73. Rasnick D, Duesberg PH. How aneuploidy affects metabolic control and causes cancer. Biochem J. 1999;340(3):621‐630. [PMC free article] [PubMed] [Google Scholar]
- 74. Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789‐799. [DOI] [PubMed] [Google Scholar]
- 75. Canto C, Gerhart‐Hines Z, Feige JN, Lagouge M, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jones RG, Plas DR, Kubek S, Buzzai M. AMP‐activated protein kinase induces a p53‐dependent metabolic checkpoint. Mol Cell. 2005;18(3):283‐293. [DOI] [PubMed] [Google Scholar]
- 77. Boidot R, Végran F, Meulle A, Le Breton A. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer Res. 2012;72(4):939‐948. [DOI] [PubMed] [Google Scholar]
- 78. Feng Z, Hu W, de Stanchina E, Teresky AK. The regulation of AMPK β1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF‐1‐AKT‐mTOR pathways. Cancer Res. 2007;67(7):3043‐3053. [DOI] [PubMed] [Google Scholar]
- 79. Li W, Saud SM, Young MR, Chen G, Hua B. Targeting AMPK for cancer prevention and treatment. Oncotarget. 2015;6(10):7365‐7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pezze PD, Ruf S, Sonntag AG, et al. A systems study reveals concurrent activation of AMPK and mTOR by amino acids. Nat Commun. 2016;7:13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335‐2342. [DOI] [PubMed] [Google Scholar]
- 84. Llovet JM, Ricci S, Mazzaferro V, et al. For SHARP investigators study group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378‐390. [DOI] [PubMed] [Google Scholar]
- 85. Buti S, Leonetti A, Dallatomasina A, Bersanelli M. Everolimus in the management of metastatic renal cell carcinoma: an evidence‐based review of its place in therapy. Core Evid. 2016;11:23‐36. 10.2147/CE.S98687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Vella S, Penna I, Longo L, Pioggia G, et al. Perhexiline maleate enhances antitumor efficacy of cisplatin in neuroblastoma by inducing overexpression of NDM29 ncRNA. Sci Rep. 2015;5:18144. 10.1038/srep18144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Samudio I, Harmancey R, Fiegl M, et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J Clin Invest. 2010;120(1):142‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fendt SM, Bell EL, Keibler MA, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73(14):4429‐4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Whitburn J, Edwards CM, Sooriakumaran P. Metformin and prostate cancer: a new role for an old drug. Curr Urol Rep. 2017;18:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354(2):166‐178. [DOI] [PubMed] [Google Scholar]
- 92. Shimojo Y, Akimoto M, Hisanaga T, Tanaka T. Attenuation of reactive oxygen species by antioxidants suppresses hypoxia‐induced epithelial‐mesenchymal transition and metastasis of pancreatic cancer cells. Clin Exp Metastasis. 2013;30(2):143‐154. [DOI] [PubMed] [Google Scholar]
- 93. Marcucci F, Stassi G, De Maria R. Epithelial–mesenchymal transition: a new target in anticancer drug discovery. Nat Rev Drug Discov. 2016;15(5):311‐325. [DOI] [PubMed] [Google Scholar]
- 94. Herst PM, Berridge MV. Plasma membrane electron transport: a new target for cancer drug development. Curr Mol Med. 2006;6(8):895‐904. [DOI] [PubMed] [Google Scholar]
- 95. Ooi AT, Gomperts BN. Molecular pathways: targeting cellular energy metabolism in cancer via inhibition of SLC2A1 and LDHA. Clin Cancer Res. 2015;21(11):2440‐2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Le A, Cooper CR, Gouw AM, Dinavahi R. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci U S A. 107(5):2037‐2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Newton R, Priyadharshini B, Turka LA. Immunometabolism of regulatory T cells. Nat Immunol. 2016;17(6):618‐625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18:6110‐6121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Prendergast GC. Why tumours eat tryptophan. Nature. 2011;478(7368):192‐194. [DOI] [PubMed] [Google Scholar]
- 100. Sakaguchi S, Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T. Regulatory T cells: How do they suppress immune responses? Int Immunol. 2009;21:1105‐1111. [DOI] [PubMed] [Google Scholar]
- 101. Zhai L, Spranger S, Binder DC, Gritsina G. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res. 2015;21(24):5427‐5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Postow MA, Sidlow R, Hellmann MD. Immune‐related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378(2):158‐168. [DOI] [PubMed] [Google Scholar]
- 103. Li W, Hua B, Saud SM, et al. Beriberine regulates AMP‐activated protein kinase signaling pathways and inhibits colon tumorigenesis in mice. Mol Carcinog. 2015;54(10):1096‐1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Puissant A, Robert G, Fenouille N, et al. Resveratrol promotes autophagic cell death in chronic myelogenous leukemia cells via JNK‐mediated p62/SQSTM1 expression and AMPK activation. Cancer Res. 2010;70(3):1042‐1052. [DOI] [PubMed] [Google Scholar]
- 105. De Francesco EM, Bonuccelli G Maggiolini M, Sotgia F, Lisanti MP. Vitamin C and doxycycline: a synthetic lethal combination therapy targeting metabolic flexibility in cancer stem cells (CSCs). Oncotarget. 2017;8(40):67269‐67286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Frantz S. Drug discovery: Playing dirty. Nature. 2005;437:942‐943. [DOI] [PubMed] [Google Scholar]
- 107. Zasadil LM, Andersen KA, Yeum D, Rocque GB, et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6:1‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Derbal Y. Perspective on the dynamics of cancer. Theor Biol Med Model. 2017;14:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bhalla US, Iyengar R. Emergent properties of networks of biological signaling pathways. Science. 1999;283(5400):381‐387. [DOI] [PubMed] [Google Scholar]
- 110. Sullivan LB, Gui DY, Heiden MGV. Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer. 2016;16(11):680‐693. [DOI] [PubMed] [Google Scholar]
- 111. Gillies RJ, Verduzco D, Gatenby RA. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer. 2012;12(7):487‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cava C, Bertoli G, Colaprico A, Olsen C, Bontempi G, Castiglioni I. Integration of multiple networks and pathways identifies cancer driver genes in pan‐cancer analysis. BMC Genomics. 2018;19(1):25. 10.1186/s12864-017-4423-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Barabási A‐L, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101‐113. [DOI] [PubMed] [Google Scholar]
- 114. Bizani M, Dinicola S, Manetti C, Azmi AS (Eds). Systems Biology Approach to Metabolomics in Cancer Studies. New York: Springer; 2012:3‐37. [Google Scholar]
- 115. Vera J, Wolkenhauer O. Mathematical tools in cancer signaling systems biology. In: Cesario A, Marcus F, eds. Cancer Systems Biology, Bioinformatics and Medicine Research and Clinical Applications. New York: Springer; 2011:185‐209. [Google Scholar]
- 116. Nielsen J. Systems biology of metabolism. Annu Rev Biochem. 2017;86:11.1‐11.31. [DOI] [PubMed] [Google Scholar]
- 117. Prasasya RD, Tian D, Kreeger PK. Analysis of cancer signaling networks by systems biology to develop therapies. Semin Cancer Biol. 2011;21(3):200‐206. [DOI] [PubMed] [Google Scholar]
- 118. Tsai TY, Choi YS, Ma W, Pomerening JR, Tang C, Ferrell JE. Robust, Tunable biological oscillations from interlinked positive and negative feedback loops. Science. 2008;321(5885):126‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]