

Abstract
Background
Ductal carcinoma in situ (DCIS) comprises a diverse group of preinvasive lesions in the breast and poses a considerable clinical challenge due to lack of markers of progression. Genomic alterations are to a large extent similar in DCIS and invasive carcinomas, although differences in copy number aberrations, gene expression patterns, and mutations exist. In mixed tumors with synchronous invasive breast cancer (IBC) and DCIS, it is still unclear to what extent invasive tumor cells are directly derived from the DCIS cells.
Aim
Our aim was to compare cancer‐relevant mutation profiles of different cellular compartments in mixed DCIS/IBC and pure DCIS tumors.
Methods and results
We performed targeted sequencing of 50 oncogenes in microdissected tissue from three different epithelial cell compartments (in situ, invasive, and normal adjacent epithelium) from 26 mixed breast carcinomas. In total, 44 tissue samples (19 invasive, 16 in situ, 9 normal) were subjected to sequencing using the Ion Torrent platform and the AmpliSeq Cancer Hotspot Panel v2. For comparison, 10 additional, pure DCIS lesions were sequenced.
Across all mixed samples, we detected 23 variants previously described in cancer. The most commonly affected genes were TP53, PIK3CA, and ERBB2. The PIK3CA:p.H1047R variant was found in nine samples from six patients. Most variants detected in invasive compartments were also found in the corresponding in situ cell compartment indicating a clonal relationship between the tumor stages. A lower frequency of variants were observed in pure DCIS lesions.
Conclusion
Similar mutation profiles between in situ and invasive cell compartments indicate a similar origin of the two tumor stages in mixed breast tumors. The lower number of potential driver variants found in pure DCIS compared with the in situ cell compartments of mixed tumors may imply that pure DCIS is captured earlier in the path of progression to invasive disease.
Keywords: breast tumor progression, DCIS, invasive breast cancer, mutations, targeted sequencing
List of Abbreviations
- COSMIC
catalogue of somatic mutations in cancer
- DCIS
ductal carcinoma in situ
- ER
estrogen receptor
- IBC
invasive breast cancer
- LCM
laser capture microdissection
- PALM
polyethylene membrane
- PCR
polymerase chain reaction
- PGM
personal genome machine
- PR
progesterone receptor
- qPCR
quantitative polymerase chain reaction
1. INTRODUCTION
Ductal carcinoma in situ (DCIS) is a noninvasive breast cancer. In DCIS, abnormal cells are contained within the milk ducts while the basement membrane is intact, and there is no invasion of surrounding stroma. 1 Today, DCIS comprises about 20% of all breast carcinoma diagnoses, usually detected in the context of mammography screening. 2 In situ lesions are generally accepted as nonobligate precursors to invasive breast cancer (IBC), but importantly, not all in situ lesions progress to become invasive. There is however, an increased risk of developing IBC subsequent to an in situ carcinoma if left untreated.3, 4 The clinical challenge is therefore to distinguish high risk from low risk lesions in order to offer optimal treatment to these patients. 5 Much remains to be learned about the pathogenesis of DCIS to be able to predict disease progression of these nonobligate breast cancer precursors.
Many cases of breast cancer present as mixed lesions, that is, synchronous invasive ductal carcinoma and DCIS. In such cases, the in situ lesion is thought to be the precursor of the invasive tumor and studies have reported an overall high degree of similarity of genetic aberrations between DCIS and IBC.6, 7, 8, 9, 10 Nevertheless, differences in type and frequency of mutations have also been reported. 11 It has been hypothesized that DCIS and IBC originate from the same ancestor cell, but have deviated prior to the in situ stage following separate tumor progression paths. 12 In tumors without an in situ compartment the invasive carcinoma may have arisen de novo, 13 or the preinvasive stage has been a brief, transient phase along the progression to invasive breast carcinoma. 14 More, in‐depth sequencing studies are required to investigate the intralesion heterogeneity in DCIS and whether progression to IBC is a result of clonal selection.6, 15
In this study, we have sequenced microdissected cell compartments from 26 mixed breast tumors using the Ion AmpliSeq Cancer Hotspot Panel v2. The mutation spectrum across 44 samples of carcinoma in situ, invasive carcinoma, and adjacent normal tissue showed a high degree of similarity between synchronous DCIS and IBC and a higher mutation frequency in the in situ cell compartment in mixed tumors compared with pure DCIS.
2. MATERIALS AND METHODS
2.1. Tumor tissue samples
Fresh frozen tissue from patients with mixed tumors (invasive ductal carcinoma with synchronous in situ lesion) or pure DCIS was collected at the Fresh Tissue Biobank, Department of Pathology, Uppsala University Hospital, Sweden. Histopathological evaluation of all cases was performed by a pathologist. DCIS tumor compartments were given a histopathological grade using the EORTC system 16 while invasive compartments were graded using the Elston & Ellis system. 17 Estrogen receptor (ER) and progesterone receptor (PR) status was determined by immunohistochemistry, and were previously published. 18 Samples were considered ER/PR positive if >10% of the cells showed positive nuclear staining. HER2‐status was determined using Silver‐enhanced in situ hybridization and scored as previously described. 18
2.2. Laser capture microdissection
Invasive, in situ and normal cell areas were microdissected using laser capture microdissection on a Zeiss inverted microscope PALM Laser MicroBeam System (Carl Zeiss, Germany) as previously described. 8 Frozen 14 μm‐thick sections were mounted on polyethylene membrane (PALM) covered slides and stained with hematoxylin (60 μL) mixed with RNasin for 1 minute, incubated in Zincfix (60 μL) for 30 seconds, followed by a series of 30‐seconds incubation steps in 75%, 95%, and 100% ethanol, respectively. Adjacent 4 μm‐thick sections were cut and stained by a routine hematoxilin and eosin protocol to locate the areas to be microdissected. Cells were captured into collecting caps and preserved in 50 μL Trizol at −80°C for DNA extraction. The number of cells obtained was estimated by the operator during microdissection and between 100 and 4000 cells were obtained for each sample. Pure DCIS samples were not microdissected; for these samples, whole FFPE tumor sections were used for DNA isolation.
2.3. DNA purification
DNA was isolated using Qiagen (Hilden, Germany) DNeasy Blood and Tissue Mini Kit. Samples were thawed and centrifuged at 16 000g for 15 minutes to precipitate DNA. After complete removal of Trizol, 180 μL buffer ATL and 20 μL protease was added and the tubes incubated at 56°C overnight before addition of 200 μL buffer AL. Samples were mixed well by vortexing before 200 μL ethanol was added and the samples were again mixed well by vortexing. The samples were then transferred to DNeasy Mini spin columns and further processed as per the manufacturer's instructions before DNA was eluted in 100 μL buffer AE. To improve recovery of the DNA, the elution buffer was left on the columns for 5 minutes before a final centrifugation step. For quantification and quality assessment of the DNA, quantitative polymerase chain reaction (qPCR) was performed with the KAPA hgDNA Quantification and QC Kit (KAPA Biosystems, Wilmington, MA) as per the manufacturer's instructions. Isolation of DNA from pure DCIS tumors were performed using the QIAcube system with the AllPrep DNA/RNA Universal Kit (cat.no. 80224, Qiagen, Hilden, Germany) according to protocol provided by the supplier.
2.4. Library preparation
Sequencing libraries for Ion Torrent sequencing were prepared using the Ion Torrent AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific, Waltham, MA), and the Ion AmpliSeq Cancer Hotspot Panel v2 and Sample ID Panel as per the manufacturer's instructions. Briefly, approx. 100 pg DNA was mixed with Ion AmpliSeq HiFi Master Mix and the two primer pools, and amplified for 27 cycles followed by partial digestion of the primer sequences and ligation of barcoded adapters. The libraries were purified using Agencourt AMPure XP beads (Beckman Coulter, Brea, CA) and amplified by polymerase chain reaction (PCR) for 5 cycles followed by a two‐round purification process with AMPure XP beads. The final libraries were quantified on Agilent Bioanalyzer instrument (Agilent Technologies, Santa Clara, CA) with the Agilent High Sensitivity DNA Kit and stored at −20°C. The AmpliSeq Cancer Hotspot Panel yields 207 amplicons that cover hotspot regions of 50 relevant cancer genes (Suppl. file 1).
2.5. Template preparation and sequencing
Libraries were normalized to 100 pM in Low TE and equal amounts of each library were pooled. Each pool was diluted 10 times and 20 μL were clonally amplified on the Ion OneTouch system using the Ion OneTouch 200 Template Kit v2 DL and enriched with the Ion OneTouch ES as per the manufacturer's instructions. Sequencing was carried out on the Ion Torrent Personal Genome Machine (PGM) using the Ion PGM 200 Sequencing Kit and Ion 314 or Ion 316 Chips for 400 cycles according to the manufacturer's instructions. For the microdissected samples, mean number of mapped reads was 395 584 (range 126 272‐933 608), mean read length 108 bp (range 73‐115 bp) while mean depth was 1655 (range 411‐3922). For the pure DCIS samples mean number of mapped reads was 273 769 (range 227 290‐345 207), mean read length 113 (range 112‐116 bp), and mean depth 1262 (range 1046‐1585).
2.6. Variant calling
Data was analyzed using the AmpliSeq Variant Caller plug‐in within the Ion Torrent Suite software (version 5.0.4, Thermo Fisher Scientific) with Human genome assembly build 37 (GRCh37) as reference. In total, 57 samples were sequenced. Three samples were excluded from further analysis after quality assessment and altogether were 44 microdissected samples from 26 mixed tumors and 10 pure DCIS successfully sequenced. Due to low input and varying sample quality for the microdissected samples, a strict cut‐off was applied; only variants with maximum allele frequency >10% and quality >100 across all microdissected samples were included. For the pure DCIS samples, variants with allele frequency <10% were also included since these data were of high quality. Variants were manually assessed in Integrative Genomics Viewer 19 to evaluate strand bias and potential technical artifacts. Variants were filtered against the Genome Aggregation Database version 2.1.1 20 to exclude single nucleotide polymorphisms.
2.7. Digital droplet PCR
Digital droplet PCR was performed using the RainDrop system (RainDance technologies) to validate the PIK3CA:H1047R variant found in nine samples on the Ion Torrent platform. DNA was isolated from separate FFPE tumor sections using DNeasy Mini spin columns as described above. An assay with two color fluorescent TaqMan probes was used to discriminate between droplets containing mutant and wild type alleles. A 50 μL reaction mix containing 2x KAPA Probe Force qPCR Master Mix (Sigma Aldrich, St. Louis, MO), 25x Droplet Stabilizer (RainDance Technologies), 13.3 μL nuclease free H2O, 9 μL DNA sample, and 0.7 μL primer/probe mix with 500 nM fwd/rev primer and 200 nM WT/mutant probe was made for each sample. The total reaction mix was loaded onto the RainDance Source chip for partitioning of the mix into millions of single droplets. Each droplet contains a PCR mix‐oil emulsion and a single DNA fragment (positive) or no target molecule (negative). After partitioning, a PCR amplification was performed, where each droplet acts as an individual PCR reaction. The PCR conditions were as follows: 98°C (3 minutes), 55 cycles of 95°C (10 seconds), and 60°C (1 minute) with ramp speed of 0.5°C/second, 72°C (10 minutes), 98°C (10 minutes), 12°C (10 minutes), and keep at 12°C. The samples were transferred to the RainDrop Sense instrument for automatic counting of positive and negative droplets depending on the presence or absence of a fluorescent signal enabling calculation of the absolute number of targets present in the original sample.
2.8. Statistical analysis
Fisher's exact tests were performed to test whether there was any statistically significant association between variants in genes and ER or PR status.
3. RESULTS AND DISCUSSION
In total, 19 invasive carcinoma, 16 DCIS and nine normal, microdissected tissue samples from 26 patients with mixed tumors, were subjected to targeted sequencing of 50 oncogenes and tumor suppressor genes. Amongst the samples were 13 in situ/invasive pairs and from three of these, adjacent normal epithelial cells were also sequenced. In addition, we sequenced 10 pure DCIS samples. An overview of relevant clinical information is shown in Suppl. file 2. Mean age at time of diagnosis was 52 years (range 30‐81) and mean tumor size was 23.7 mm (range 1‐80). Of all tumors, 28/36 (78%) were ER positive and 23/36 (64%) PR positive. All PR‐positive tumors were ER‐positive.
Across all samples from mixed tumors we identified 23 different, potentially pathogenic variants in seven genes (AKT1, CDH1, CDKN2A, ERBB2, MET, PIK3CA, and TP53) (Suppl. file 3). PIK3CA and TP53 were the most commonly mutated genes. Most variants were present in only one patient; but for two of the genes (AKT1 and PIK3CA), identical variants were identified in more than one patient. The number of variants in each in situ or invasive cell compartment ranged from zero to four, and most tumors (12/16 in situ, 13/19 invasive) carried only one variant. The most common variant (PIK3CA:p.H1047R) was found in nine samples from six patients. Across the samples from mixed tumors, five different PIK3CA variants were detected in 19 samples from 13 patients including one normal sample. By contrast, none of the nine TP53 variants were found in more than one patient. Among the 13 cases of pairs, for which both in situ and invasive samples were available from the same patient, we found 18 variants in six different genes (Figure 1). In six of the cases, the variant(s) were identical in both compartments.
FIGURE 1.

Genes with pathogenic variants identified in the 13 available DCIS/IBC sample pairs. DCIS (blue), IBC (red). DCIS, ductal carcinoma in situ; IBC, invasive breast cancer
Interestingly, we found five different ERBB2 variants; two (p.D769H and p.V777L) resided in both the in situ and invasive tumor compartments of the same tumor. Two other ERBB2 variants were found in a second tumor. One of these (p.D769Y) was found in both the in situ and invasive tumor compartments and the other (p.L755S) was found only in the in situ compartment. One ERBB2 variant (p.G776delinsAVGC) was found in a normal sample; however, none of the corresponding tumor cell compartments were successfully sequenced for this particular case. Altogether, these findings demonstrate large intertumor heterogeneity in mutation pattern in synchronous DCIS and IBC and indicate that ERBB2 variants also are present early in tumorigenesis.
In three tumors (UPP027, UPP208, and UPP244), normal epithelium was successfully sequenced in addition to invasive and in situ tumor tissue. Two of these tumors (UPP027 and UPP208) carried only one variant each (PIK3CA:p.H1047R and TP53:p.A84fs, respectively) and none of the corresponding normal compartments carried these variants. The last tumor with three samples (UPP244) carried two different variants; one of these (TP53:p.R175H) was found in both the in situ and invasive compartments, while the other (PIK3CA:p.H1047R) was found in the normal compartment at a frequency of 30%, absent in the in situ compartment and present at a very low frequency (3%) in the invasive compartment. Across the cohort, nine normal breast epithelium samples were sequenced, and amongst these, three carried potentially pathogenic variants (Figure 2). However, since all normal tissue samples sequenced in this study were obtained adjacent to tumor areas, they should not be considered entirely normal.
FIGURE 2.
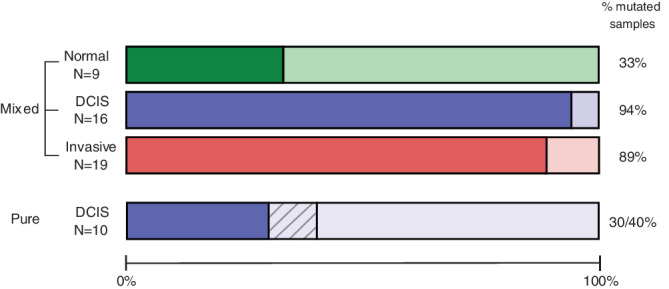
Frequency of samples with pathogenic variants. The bar plot illustrates the frequency of samples with (dark color) and without (light color) any pathogenic variant (frequency indicated at the right). The hatched area in the pure DCIS bar illustrates the frequency of variants with allele frequency below 10%. DCIS, ductal carcinoma in situ
We found a significant association between PIK3CA variants and positive PR status (P = .039, Fisher's exact test), which has been previously noted.21, 22, 23 A similar association was not seen for ER (P = .44), however; the low number of samples in this study may have prevented the identification of any such association.
In addition to microdissected tissue from mixed tumors, we sequenced 10 pure (nonmicrodissected) DCIS. Three variants in three different tumors were detected; PIK3CA:p.C420R, PIK3CA:p.E542K, and TP53:p.R213X (Suppl. file 3). Two of these were not detected in any of the microdissected samples, while the third variant (PIK3CA:p.C420R) was found in one of the invasive tumor cell compartments, but was filtered out due to allele frequency below threshold. There was a notable difference in the number of variants across the 50 genes between in situ cell compartments from mixed tumors compared with pure DCIS. Almost all in situ cell compartments from the mixed tumors, 15/16 (94%), carried at least one variant while only 3 out of 10 (30%) of the pure DCIS tumors carried any of the variants (Figure 2). However, the pure DCIS tumors were not subjected to microdissection before sequencing which may have lowered the allele frequency and compromised the detection of relevant variants. When including all variants regardless of allele frequency, three additional variants were found (Suppl. file 3). Using this cut‐off, 4/10 (40%) of the pure DCIS carry potentially pathogenic variants (Figure 2), however; comparison with the sequencing results from synchronous DCIS from mixed tumors is challenging when different thresholds are applied. Noticeably, none of the pure DCIS tumors carried the most common variant identified among the mixed tumors (PIK3CA:p.H1047R). Targeted sequencing as performed here includes only a limited number of genes and therefore we cannot exclude the possibility that mutation spectra across other putative driver genes might be similar between the two different types of in situ cancers. It also important to recognize that the pure DCIS tumors in this cohort may be earlier in the progression path to invasiveness and therefore carry fewer tumor driving mutations. Finally, heterogeneity at the DCIS stage may imply that some pure DCIS are on the verge of becoming invasive and display a mutational pattern similar to IBC whereas others will never become invasive and that these could be regarded as different entities. These findings confirm those of other studies24, 25 and highlight the importance of being conscious about distinguishing synchronous DCIS from pure DCIS lesions when studying tumor progression.
When DCIS presents synchronous with invasive disease, it is unclear whether these multiple stage‐specific cell populations have a common ancestor or develop from multiple clones. Previous sequencing studies have reported similar mutation profiles in DCIS and IBC, with PIK3CA, TP53, and GATA3 as the most commonly affected genes.8, 9, 10, 24, 26, 27, 28, 29, 30 However, different prevalence of PIK3CA variants has been observed between DCIS and IBC. One study reported PIK3CA variants restricted to the in situ compartment in two cases of synchronous DCIS and IBC, while in a third case, a reduced frequency of a specific PIK3CA variant was found in invasive cells relative to the cells from the in situ compartment. 9 In one tumor in our study, we found a PIK3CA variant in the in situ cells, and not in the invasive cell compartment, while in two tumors, we found a PIK3CA variant in the invasive cells while not in the corresponding in situ cell compartment. In our study, the sequencing panel did not include GATA3, so the high frequency of GATA3 variants previously found in DCIS could not be confirmed. 27
Ion semiconductor sequencing is a “sequencing by synthesis” method based upon detection of hydrogen ions that are released during polymerization of DNA. The technology is well suited for targeted sequencing of samples with minute amounts of DNA which is often the challenge with microdissected tissue. This has allowed us to sequence a panel of the most frequently mutated genes in cancer, in relatively few cells from stored Trizol cell fractions after microdissection. To validate our findings, we used digital droplet PCR to quantify the most frequently detected variant in this study, PIK3CA:p.H1047R and found similar frequencies as by sequencing (Table 1). Four of the samples with TP53 mutations in this study were included in a previous study of TP53 mutations in synchronous DCIS and IBC. 8 In three of these samples (UPP038, UPP208, UPP142 [pure DCIS]), we detected the same variants, while for UPP065, a 10 bp deletion of codons 106‐109, was not called by the Ion Torrent analysis pipeline. However, we identified the deletion in our data by manual inspection. This discrepancy could be due to inaccurate flow‐calls, a known artifact of PGM, which may cause homopolymers to be under‐called. 31
TABLE 1.
Validation results ddPCR
| SampleID | Input (ng DNA) | WT droplets | MUT droplets | Mutation frequency ddPCR % | Mutation frequency Ion Torrent % | Comments |
|---|---|---|---|---|---|---|
| UPP027 Normal | 0.054 | 66 | 0 | 0 | 0 | |
| UPP027 DCIS | 0.297 | 108 | 50 | 32 | 33 | |
| UPP027 Invasive | 0.01 | 58 | 20 | 26 | 30 | |
| UPP050 Normal | NA | 10 187 | 2 | 0.02 | NA | Ion Torrent sequencing failed |
| UPP050 DCIS | 0.021 | 2639 | 1279 | 33 | 43 | |
| UPP050 Invasive | 0.058 | 7330 | 20 | 0.27 | 0 | |
| UPP087 Normal | NA | 10 957 | 567 | 5 | NA | Ion Torrent sequencing failed |
| UPP087 DCIS | 0.016 | 0 | 0 | NA | 0 | ddPCR failed |
| UPP087 Invasive | 0.062 | 1102 | 272 | 20 | 40 | |
| UPP158 DCIS | 0.085 | 169 | 88 | 34 | 41 | |
| UPP158 Invasive | 0.156 | 288 | 184 | 39 | 18 | |
| UPP233 Invasive | 0.144 | 14 | 6 | 30 | 27 | |
| UPP244 Normal | 0.063 | 1 | 0 | NA | 29 | ddPCR failed |
| UPP244 DCIS | 0.144 | 2115 | 0 | 0 | 0 | |
| UPP244 Invasive | 0.081 | 43 281 | 1 | 0.002 | 3 | |
| WT‐PIK3CA_ctr | 10 | 7596 | 0 | 0 | Not applicable | Negative control |
| PIK3CA_5_ctr | 50 | 40 733 | 3091 | 7 | Not applicable | Positive control |
| PIK3CA_5_ctr | 50 | 43 064 | 3139 | 7 | Not applicable | Positive control |
Note: PIK3CA:p.H1047R.
4. CONCLUSION
In this study, we performed targeted sequencing of microdissected tissue from in situ and invasive tumor cell compartments from 26 patients with mixed DCIS/IBC tumors, in addition to 10 pure DCIS tumors. Although the number of cases included was small, we found that across the 50 cancer‐relevant genes included in the panel, the spectrum of variants was similar between synchronous DCIS and IBC supporting earlier findings of a clonal relationship between the two tumor stages and a possible selection of subclones during tumor progression. PIK3CA and TP53 were the most frequently mutated genes and alterations occurred at the DCIS stage or possibly earlier. Sequencing of 10 pure DCIS indicated a lower number of potentially pathogenic variants in these lesions compared with synchronous DCIS.
CONFLICT OF INTEREST
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
AUTHORS' CONTRIBUTIONS
All authors take responsibility for the integrity of the data and all authors approved the final manuscript. Conceptualization, V.K.,T.S.; Methodology, H.B., S.K.; Investigation, H.B., S.K.; Formal Analysis, H.B. and S.K.; Resources, F.W., V.K., T.S.; Writing ‐ Original Draft, H.B., T.S.; Writing ‐ Review & Editing, H.B.,F.W., V.K., T.S.; Visualization, H.B.; Supervision, V.K., T.S.; Funding Acquisition, V.K., T.S.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
ETHICS STATEMENT
The study complies with the Declaration of Helsinki, and was approved by the Ethics Committee at Uppsala University Hospital, Sweden (approval number 2005/118). In this retrospective cohort study informed consent from the women was not needed according to the ethical approval.
Supporting information
Suppl. file 1 Overview of the 50 genes included in the AmpliSeq Cancer Hotspot Panel (.xlsx)
Suppl. file 2 Clinical information and overview of samples analyzed (.xlsx)
Suppl. file 3 List of all pathogenic variants detected (.xlsx)
ACKNOWLEDGEMENTS
We wish to thank Simin Tahmasebpoor and Johan Botling at the Department of Pathology, Akademiska Sjukhuset, for collecting frozen tissue samples, and Inger Bergheim and Helen Vålerhaugen at Department of Cancer Genetics, Oslo University Hospital for assisting in performing sequencing and ddPCR experiments.
This work was supported by funds from the South‐Eastern Norway Regional Health Authority (2012056 to TS) and Division of Medicine, Akershus University Hospital to VK. The Cancerfonden supported part of the study (4920‐B06‐03PCD to FW) and the Fresh Tissue Biobank at the Department of Pathology, Akademiska Sjukhuset, was supported by the Swedish National Biobank Program funded by Wallenberg Consortium North and Swegene. SK was a postdoctoral fellow from the Norwegian Cancer Society.
Bergholtz H, Kumar S, Wärnberg F, Lüders T, Kristensen V, Sørlie T. Comparable cancer‐relevant mutation profiles in synchronous ductal carcinoma in situ and invasive breast cancer. Cancer Reports. 2020;3:e1248. 10.1002/cnr2.1248
Funding information Akershus Universitetssykehus, Grant/Award Number: Division of Medicine; Cancerfonden, Grant/Award Number: 4920‐B06‐03PCD; Helse Sør‐Øst RHF, Grant/Award Number: 2012056 ; Kreftforeningen
REFERENCES
- 1. Lakhani S, Ellis I, Schnitt S, Tan P, van de Vijver M. WHO Classification of Tumours of the Breast. WHO Classification of Tumours 4. 4th ed. Lyon, France: IARC Press; 2012. [Google Scholar]
- 2. Ward EM, DeSantis C, Lin CC, et al. Cancer statistics: breast cancer in situ. CA Cancer J Clin. 2015;65:481‐495. [DOI] [PubMed] [Google Scholar]
- 3. Erbas B, Provenzano E, Armes J, Gertig D. The natural history of ductal carcinoma in situ of the breast: a review. Breast Cancer Res Treat. 2006;97:135‐144. [DOI] [PubMed] [Google Scholar]
- 4. Sanders ME, Schuyler PA, Simpson JF, Page DL, Dupont WD. Continued observation of the natural history of low‐grade ductal carcinoma in situ reaffirms proclivity for local recurrence even after more than 30 years of follow‐up. Mod Pathol. 2015;28:662‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gorringe KL, Fox SB. Ductal carcinoma in situ biology, biomarkers, and diagnosis. Front Oncol. 2017;7:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cowell CF, Weigelt B, Sakr RA, et al. Progression from ductal carcinoma in situ to invasive breast cancer: revisited. Mol Oncol. 2013;7:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rane SU, Mirza H, Grigoriadis A, Pinder SE. Selection and evolution in the genomic landscape of copy number alterations in ductal carcinoma in situ (DCIS) and its progression to invasive carcinoma of ductal/no special type: a meta‐analysis. Breast Cancer Res Treat. 2015;153:101‐121. [DOI] [PubMed] [Google Scholar]
- 8. Zhou W, Muggerud AA, Vu P, et al. Full sequencing of TP53 identifies identical mutations within in situ and invasive components in breast cancer suggesting clonal evolution. Mol Oncol. 2009;3:214‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hernandez L, Wilkerson PM, Lambros MB, et al. Genomic and mutational profiling of ductal carcinomas in situ and matched adjacent invasive breast cancers reveals intra‐tumour genetic heterogeneity and clonal selection. J Pathol. 2012;227:42‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miron A, Varadi M, Carrasco D, et al. PIK3CA mutations in in situ and invasive breast carcinomas. Cancer Res. 2010;70:5674‐5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakr RA, Weigelt B, Chandarlapaty S, et al. PI3K pathway activation in high‐grade ductal carcinoma in situ‐implications for progression to invasive breast carcinoma. Clin Cancer Res. 2014;20:2326‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuerer HM, Albarracin CT, Yang WT, et al. Ductal carcinoma in situ: state of the science and roadmap to advance the field. J Clin Oncol. 2009;27:279‐288. [DOI] [PubMed] [Google Scholar]
- 13. Wong H, Lau S, Yau T, Cheung P, Epstein RJ. Presence of an in situ component is associated with reduced biological aggressiveness of size‐matched invasive breast cancer. Br J Cancer. 2010;102:1391‐1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kurbel S. In search of triple‐negative DCIS: tumor‐type dependent model of breast cancer progression from DCIS to the invasive cancer. Tumor Biol. 2013;34:1‐7. [DOI] [PubMed] [Google Scholar]
- 15. Kader T, Hill P, Zethoven M, et al. Atypical ductal hyperplasia is a multipotent precursor of breast carcinoma. J Pathol. 2019;248:326‐338. [DOI] [PubMed] [Google Scholar]
- 16. Holland R, Peterse JL, Millis RR, et al. Ductal carcinoma in situ: a proposal for a new classification. Semin Diagn Pathol. 1994;11:167‐180. [PubMed] [Google Scholar]
- 17. Elston EW, Ellis IO. Method for grading breast cancer. J Clin Pathol. 1993;46:189‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wärnberg F, Nordgren H, Bergkvist L, Holmberg L. Tumour markers in breast carcinoma correlate with grade rather than with invasiveness. Br J Cancer. 2001;85:869‐874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robinson JT, Thorvaldsdóttir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv. 2019;531210. 10.1101/531210. [DOI] [Google Scholar]
- 21. Pang B, Cheng S, Sun SP, et al. Prognostic role of PIK3CA mutations and their association with hormone receptor expression in breast cancer: a meta‐analysis. Sci Rep. 2014;4:6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nik‐Zainal S, Davies H, Staaf J, et al. Landscape of somatic mutations in 560 breast cancer whole‐genome sequences. Nature. 2016;534:47‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Saal LH, Holm K, Maurer M, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res. 2005;65:2554‐2559. [DOI] [PubMed] [Google Scholar]
- 24. Kim SY, Jung SH, Kim MS, et al. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015;6:7597‐7607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schorr MC, Pedrini JL, Savaris RF, Zettler CG. Are the pure in situ breast ductal carcinomas and those associated with invasive carcinoma the same? Appl Immunohistochem Mol Morphol. 2010;18:51‐54. [DOI] [PubMed] [Google Scholar]
- 26. Agahozo MC, Sieuwerts AM, Doebar SC, et al. PIK3CA mutations in ductal carcinoma in situ and adjacent invasive breast cancer. Endocr Relat Cancer. 2019;26:471‐482. [DOI] [PubMed] [Google Scholar]
- 27. Pang J‐MB, Savas P, Fellowes AP. Breast ductal carcinoma in situ carry mutational driver events representative of invasive breast cancer. Mod Pathol. 2017;30:952‐963. 10.1038/modpathol.2017.21. [DOI] [PubMed] [Google Scholar]
- 28. Doebar SC, Krol NM, Marion R, et al. Progression of ductal carcinoma in situ to invasive breast cancer: comparative genomic sequencing. Virchows Arch. 2019;474:247‐251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dunlap J, le C, Shukla A, et al. Phosphatidylinositol‐3‐kinase and AKT1 mutations occur early in breast carcinoma. Breast Cancer Res Treat. 2010;120:409‐418. [DOI] [PubMed] [Google Scholar]
- 30. Li H, Zhu R, Wang L, et al. PIK3CA mutations mostly begin to develop in ductal carcinoma of the breast. Exp Mol Pathol. 2010;88:150‐155. [DOI] [PubMed] [Google Scholar]
- 31. Bragg LM, Stone G, Butler MK, Hugenholtz P, Tyson GW. Shining a light on dark sequencing: characterising errors in ion torrent PGM data. PLoS Comput Biol. 2013;9:e1003031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. file 1 Overview of the 50 genes included in the AmpliSeq Cancer Hotspot Panel (.xlsx)
Suppl. file 2 Clinical information and overview of samples analyzed (.xlsx)
Suppl. file 3 List of all pathogenic variants detected (.xlsx)
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.