

Abstract
Background
Altered cell metabolism is an established hallmark of cancer. Advancement in our understanding of dysregulated cellular metabolism has aided drastically in identifying metabolic vulnerabilities that can be exploited therapeutically. Indeed, this knowledge has led to the development of a multitude of agents targeting various aspects of tumor metabolism.
Recent findings
The intent of this review is to provide insight into small molecule inhibitors that target tumor metabolism and that are currently being explored in active clinical trials as either preventive, stand‐alone, or adjuvant therapies for various malignancies. For each inhibitor, we outline the mechanism (s) of action, preclinical/clinical findings, and limitations. Sections are divided into three aspects based on the primary target of the small molecule inhibitor (s): those that impact (1) cancer cells directly, (2) immune cells present in the tumor microenvironment, or (3) both cancer cells and immune cells. We highlight small molecule targeting of metabolic pathways including de novo fatty acid synthesis, NAD+ biosynthesis, 2‐hydroxyglutarate biosynthesis, polyamine metabolism, the kynurenine pathway, as well as glutamine and arginine metabolism.
Conclusions
Use of small molecule inhibitors aimed at exploiting tumor metabolic vulnerabilities continues to be an active area of research. Identifying metabolic dependencies specific to cancer cells and/or constituents of the tumor microenvironment is a viable area of therapeutic intervention that holds considerable clinical potential.
Keywords: cancer, clinical trials, metabolic vulnerability, metabolism, small molecule inhibitors, targeted therapy
Abbreviations
- 2‐HG
2‐hydroxyglutaric acid
- ACACA
acetyl‐CoA carboxylase
- ACO
aconitase
- ACYL
ATP citrate lyase
- AH
anthranilate hydroxylase
- ALDO
aldolase
- AML
acute myeloid leukemia
- ARG
arginase
- ASS
argininosuccinate synthetase
- AST
aspartate aminotransferase
- BPTES
Bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl) ethyl sulfide
- CPS
carbomylphosphate synthase
- CS
citrate synthase
- CTCL
cutaneous T‐cell Lymphoma
- ENO
enolase
- FASN
fatty acid synthase complex
- FH
fumarase
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GDH
glutamate dehydrogenase
- GLS
glutaminase
- HAOO
3‐hydroxyanthranilic acid dioxygenase
- HK
hexokinase
- IDH
isocitrate dehydrogenase
- IDO
indolamine 2,3‐dioxygenase
- KGA
‐ kidney‐type glutaminase 1
- KMO
kynurenine 3‐monooxygenase
- KYNU
kynureninase
- LDH
lactate dehydrogenase
- MDH
malate dehydrogenase
- NA
nicotinic acid
- NAD
nicotinamide adenine dinucleotide
- NADK
NAD kinase
- NAMPT
nicotinamide phosphoribosyltransferase
- NAPRT
phosphoribosyltransferase
- NFEL2L2
nuclear factor, erythroid 2‐like 2
- NMNAT
nicotinamide mononucleotide adenylytransferase
- NOS
nitric oxide synthase
- ODC
ornithine decarboxylase
- OGDH
oxoglutarate dehydrogenase
- PAOX
polyamine oxidase
- PC
pyruvate carboxylase
- PDHA
pyruvate dehydrogenase
- Peg‐rhArg1
Pegylated Recombinant Arginase I
- PFK
phosphofructokinase
- PGAM
phosphoglycerate mutase
- PGI
glucose‐6‐phosphate isomerase
- PGK
phosphoglycerate kinase
- PK
pyruvate kinase
- PPI
proton pump inhibitors
- QPRT
quinolinate phosphoribosyltransferase
- R/R
relapsed/refractory
- SAT1
spermidine/spermine N1‐acetyltransferase
- SCS
succinate‐CoA ligase
- SDH
succinate dehydrogenase
- SLC1A5
soluble carrier family 1 member 5
- SMS
spermine synthase
- TCA
tricarboxylic acid cycle
- TDO
tryptophan 2,3‐dioxygenase
- TPI
triosephosphate isomerase
- α‐KG
alpha‐ketoglutarate
1. INTRODUCTION
Altered cell metabolism is an established hallmark of cancer.1 Fundamental differences between the metabolism of normal differentiated cells and rapidly proliferating cancer cells were first described by Otto Warburg nearly a century ago.2, 3 Warburg noted that tumors exhibit increased rates of glycolysis even in the presence of oxygen, a phenomenon now known as aerobic glycolysis or the “Warburg effect”.4 As aerobic glycolysis is an inefficient way to generate adenosine 5´‐triphosphate, Warburg postulated this observed metabolic shift was indicative of a mitochondrial defect in cancer cells. While this notion has been intensely debated, it is now recognized that cancer cells generally retain functional mitochondria capable of carrying out oxidative phosphorylation and that Warburg's observations instead reflect a reprogramming of metabolism to meet anabolic needs and provide necessary macromolecules that enable sustained proliferation.5, 6 Although the observations of Warburg and others have been fundamental to our understanding of tumor biology, they are but one aspect of adaptive tumor metabolism. In fact, alterations in cancer metabolism extend well beyond glucose metabolism and energetics to encompass a broad array of metabolic pathways that have diverse functions intrinsic to cancer cells as well as constituents of the tumor microenvironment.7, 8, 9, 10
Metabolic alterations in cancer have primarily been considered to be indirect effects of aberrantly activated cell proliferation and survival programs, rather than functionally important drivers of tumor development.1 However, findings over the last decade have greatly advanced and evolved our understanding of cancer metabolism, and it is now evident that altered metabolism is an integral effector of tumorigenesis that is intricately intertwined with cellular signaling and genetic/epigenetic regulation. This is best exemplified by gain of function mutations in the isocitrate dehydrogenase‐1/2 (IDH) enzymes that facilitate the generation of 2‐hydroxyglutarate—an onco‐metabolite that competitively inhibits the alpha‐ketoglutarate enzymes, including histone demethylases and DNA hydroxylases, resulting in distinct epigenetic phenotypes.11 Recognizing the dysregulation of cellular metabolism as an import aspect of tumorigenesis has offered the potential of clinical benefit by providing targets for the development of novel therapeutics.1
Tumors exhibit remarkable, context‐sensitive metabolic plasticity that is an essential component of their survival and progression. Metabolic fluxes are tuned according to environmental cues as well as the energy and biomass demands of cancer cells throughout tumorigenesis and in states of proliferation, nutrient attenuation, and quiescence.12 In addition to supporting primary tumors, the rewiring of cell metabolism can activate metabolic programs that confer metastatic capacity that allows cancer cells shed from primary tumors to overcome nutrient and energy deficit, survive, and initiate metastases.13 It is therefore important to elucidate and distinguish between metabolic alterations active within primary, tumor‐initiating and disseminated tumor cells. Particular attention has been drawn by Webber and colleagues14 to metabolic programs that enable metastatic cells to overcome ATP deficit, achieve anchorage independence and further tumor progression, rather than proliferation per se. Luo et al15 have explored altered lipid metabolism that is associated with metastatic disease pathogenesis and explored membrane lipid raft structures as mediators of tumor aggression and progression. This diverse metabolic landscape provides novel therapeutic potential and consideration of these multiple points of vulnerability could lead to more effective targeting of the diversity of tumor cell populations according to specific metabolic state.
The primary intent of this review is to provide an overview of small molecule inhibitors that target metabolic pathways and that are oriented towards the prevention or treatment of various malignancies. Due to the broad scope of available metabolic inhibitors, emphasis is given to those drugs in active clinical trials (Table 1). The following sections will highlight the implications, mechanism (s) of action and limitations for the inhibitors specified in Table 1 . Sections are divided into three aspects based on the primary intent of the small molecule inhibitor (s): those that impact (1) cancer cells directly, (2) immune cells present in the tumor microenvironment, or (3) both cancer cells and immune cells.
Table 1.
Small molecule inhibitors under active clinical investigation as either cancer preventive, stand‐alone, or adjuvant therapies
| Glutaminase (GLS) Inhibitors | ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Malignancy | Clinical Phase | Clinical Trial Identifier | Study Title | Status | Start Date |
| CB‐839 | Acute myeloid leukemia (AML)acute lymphocytic leukemia (ALL) | Phase I | NCT02071927 | Study of the glutaminase inhibitor CB‐839 in leukemia | Completed | 26‐Feb‐14 |
| Non‐Hodgkin's lymphoma (NHL)multiple myelomaWaldenstrom's macroglobulinemia (WM)diffuse large B‐cell lymphoma (DLBCL),other B‐cell NHL subtypes, including WMT‐cell NHL | Phase I | NCT02071888 | Study of the glutaminase inhibitor CB‐839 in hematological tumors | Completed | 26‐Feb‐14 | |
| Myelodysplastic syndrome | Phase Ib/II | NCT03047993 | CB‐839 + Azacitidine for treatment of myelodysplastic syndrome (MDS) | On‐going, not recruiting | 9‐Feb‐17 | |
| Clear cell renal cell carcinoma | Phase II | NCT03163667 | CB‐839 with Everolimus vs. placebo with Everolimus in patients with RCC | On‐going, not recruiting | 23‐May‐17 | |
| TNBC, NSCLC, renal cell carcinoma, mesothelioma, fumarate hydratase‐deficient tumors, succinate dehydrogenase (SDH)‐deficient gastrointestinal stromal tumors, SDH‐deficient non‐gastrointestinal stromal tumors, tumors harboring isocitrate dehydrogenase‐1 (IDH1) and IDH2 mutations and tumors harboring amplifications in the cMYC gene | Phase I | NCT02071862 | Study of the glutaminase inhibitor CB‐839 in solid tumors | Recruiting | 26‐Feb‐14 | |
| Clear cell renal cell carcinomamelanomanon‐small cell lung cancer | Phase I/II | NCT02771626 | Study CB‐839 in combination with nivolumab in patients with ccRCC and other solid tumors | Recruiting | 13‐May‐16 | |
| Triple negative breast cancer (TNBC) | Phase II | NCT03057600 | Study of CB‐839 in combination w/paclitaxel in patients of African ancestry and non‐African ancestry with advanced TNBC | Recruiting | 20‐Feb‐17 | |
| Colorectal cancermetastatic colorectal cancerRAS wild type colorectal cancerrefractory colorectal cancer | Phase I/II | NCT03263429 | Novel PET/CT imaging biomarkers of CB‐839 in combination with Panitumumab and irinotecan in patients with metastatic and refractory RAS wildtype colorectal cancer | Recruiting | 28‐Aug‐17 | |
| Fatty acid synthase (FASN) inhibitors | ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Malignancy | Clinical Phase | Clinical Trial Identifier | Title | Status | |
| Omeprazole | Breast cancer | Phase II | NCT02595372 | Inhibiting fatty acid synthase to improve efficacy of neoadjuvant chemotherapy | Recruiting | 3‐Nov‐15 |
| TVB‐2640 | Solid malignant tumor | Phase I | NCT02223247 | A phase 1, first‐in‐human study of escalating doses of oral TVB‐2640 in patients with solid tumors | Completed | 22‐Aug‐14 |
| Colon cancer | Phase I | NCT02980029 | Pharmacodynamic effects of fatty acid synthase (FASN) inhibition with TVB‐2640 in resectable colon cancer | Recruiting | 2‐Dec‐16 | |
| Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors | ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Malignancy | Clinical Phase | Clinical Trial Identifier | Title | Status | |
| APO866 | Cutaneous T‐cell lymphomas | Phase II | NCT00431912 | A study of APO866 for the treatment of cutaneous T‐cell lymphoma | Completed | 6‐Feb‐07 |
| B‐cell chronic lymphocytic leukemia | Phase I/II | NCT00435084 | A phase I/II study to assess the safety and tolerability of APO866 for the treatment of refractory B‐CLL | Completed | 14‐Feb‐07 | |
| Melanoma | Phase II | NCT00432107 | A study to assess APO866 for the treatment of advanced melanoma | Completed | 7‐Feb‐07 | |
| KPT‐9274 | NHLsolid tumorssarcoma | Phase I | NCT02702492 | PAK4 and NAMPT in patients with solid malignancies or NHL (PANAMA) (PANAMA) | Recruiting | 8‐Mar‐16 |
| Indoleamine 2,3‐dioxygenase inhibitors | ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Malignancy | Clinical Phase | Clinical Trial Identifier | Title | Status | |
| GDC‐0919 | Solid tumors | Phase I | NCT02048709 | Indoleamine 2,3‐dioxygenase (IDO) inhibitor in advanced solid tumors | Completed | 29‐Jan‐14 |
| INCB024360 (Epacadostat) | Myelodysplastic syndromes | Phase II | NCT01822691 | Phase II INCB024360 study for patients with myelodysplastic syndromes (MDS) | Completed | 2‐Apr‐13 |
| Ovarian cancerfallopian tube carcinomaprimary peritoneal carcinoma | Phase I | NCT02118285 | Intraperitoneal natural killer cells and INCB024360 for recurrent ovarian, fallopian tube, and primary peritoneal cancer | Completed | 21‐Apr‐14 | |
| Indoximod (1‐methyl‐D‐tryptophan) | Male breast cancer, metastatic breast cancer, recurrent breast cancer | Phase I/II | NCT01042535 | Vaccine therapy and 1‐MT in treating patients with metastatic breast cancer | On‐going, not recruiting | 5‐Jan‐10 |
| Metastatic breast cancer | Phase II | NCT01792050 | Study of chemotherapy in combination with IDO inhibitor in metastatic breast cancer | On‐going, not recruiting | 15‐Feb‐13 | |
| Solid tumors | Phase II | NCT01560923 | Phase II study of sipuleucel‐T and indoximod for patients with refractory metastatic prostate cancer | On‐going, not recruiting | 22‐Mar‐12 | |
| Melanoma | Phase II/III | NCT03301636 | A phase 2/3 study of indoximod or placebo plus pembrolizumab or nivolumab in subjects with unresectable or metastatic melanoma (NLG2107) | Not yet recruiting | 4‐Oct‐17 | |
| Epithelial ovarian, fallopian tube, or primary peritoneal cancer | Phase I | NCT02042430 | Epacadostat before surgery in treating patients with newly diagnosed stage III‐IV epithelial ovarian, fallopian tube, or primary peritoneal cancer | On‐going, not recruiting | 22‐Jan‐14 | |
| Advanced solid tumors | Phase I | NCT02559492 | Itacitinib combined with INCB024360 and/or itacitinib combined with INCB050465 in advanced solid tumors | On‐going, not recruiting | 24‐Sep‐15 | |
| Stage III‐IV melanoma | Phase II | NCT01961115 | Epacadostat and vaccine therapy in treating patients with stage III‐IV melanoma | On‐going, not recruiting | 11‐Oct‐13 | |
| Glioblastoma multiforme, glioma, gliosarcoma, malignant brain tumor, ependymoma, medulloblastoma | Phase I | NCT02502708 | Study of the IDO pathway inhibitor, indoximod, and temozolomide for pediatric patients with progressive primary malignant brain tumors | Recruiting | 20‐Jul‐15 | |
| Metastatic pancreatic cancer | Phase I/II | NCT02077881 | Study of IDO inhibitor in combination with gemcitabine and nab‐paclitaxel in patients with metastatic pancreatic cancer | Recruiting | 4‐Mar‐14 | |
| Stage III and IV melanoma | Phase I/II | NCT02073123 | Study of IDO inhibitor in combination with checkpoint inhibitors for adult patients with metastatic melanoma | Recruiting | 27‐Feb‐14 | |
| Glioblastoma multiforme, glioma, gliosarcoma, malignant brain tumor | Phase I/II | NCT02052648 | Study of IDO inhibitor and temozolomide for adult patients with primary malignant brain tumors | Recruiting | 3‐Feb‐14 | |
| Acute myeloid leukemia | Phase I/II | NCT02835729 | A study of indoximod in combination with (7 + 3) chemotherapy in patients with newly diagnosed acute myeloid leukemia | Recruiting | 18‐Jul‐16 | |
| NSCLC | Phase I/II | NCT02460367 | Immunotherapy combination study in advanced previously treated non‐small cell lung cancer | Recruiting | 2‐Jun‐15 | |
| NLG802 | Solid tumors | Phase I | NCT03164603 | NLG802 indoleamine 2,3‐dioxygenase (IDO) inhibitor in advanced solid tumors | Recruiting | 23‐May‐17 |
| Fallopian tube carcinomaovarian carcinomaprimary peritoneal carcinoma | Phase I/II | NCT02166905 | DEC‐205/NY‐ESO‐1 fusion protein CDX‐1401, poly ICLC, and IDO1 inhibitor INCB024360 in treating patients with ovarian, fallopian tube, or primary peritoneal cancer in remission | Recruiting | 18‐Jun‐14 | |
| Advanced solid tumors | Phase I/II | NCT02959437 | Azacitidine combined with pembrolizumab and epacadostat in subjects with advanced solid tumors (ECHO‐206) | Recruiting | 9‐Nov‐16 | |
| Recurrent epithelial ovarian cancerrecurrent fallopian tube cancerrecurrent peritoneal cancer | Phase I | NCT02785250 | Study of DPX‐survivac vaccine therapy and epacadostat in patients with recurrent ovarian cancer | Recruiting | 27‐May‐16 | |
| Isocitrate dehydrogenase‐2 inhibitors | ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Malignancy | Clinical Phase | Clinical Trial Identifier | Title | Status | |
| AG‐120 | Cholangiocarcinoma, chondrosarcoma, glioma, other advanced solid tumors | Phase I | NCT02073994 | Study of orally administered AG‐120 in subjects with advanced solid tumors, including glioma, with an IDH1 mutation | On‐going, not recruiting | 28‐Feb‐14 |
| Relapsed/refractory AML, untreated AML, other IHD‐1 mutated positive hematological malignancies | Phase I | NCT02074839 | Study of orally administered AG‐120 in subjects with advanced hematologic malignancies with an IDH1 mutation | On‐going, not recruiting | 28‐Feb‐14 | |
| AG‐221 (Olaparib) | Hematologic neoplasms | Phase I | NCT01915498 | Phase 1/2 study of AG‐221 in subjects with advanced hematologic malignancies with an IDH2 mutation | On‐going, not recruiting | 5‐Aug‐13 |
| Newly diagnosed AML, untreated AML, AML arising from MDS, AHD or genotoxic injury | Phase I | NCT02632708 | Safety study of AG‐120 or AG‐221 in combination with induction and consolidation therapy in patients with newly diagnosed acute myeloid leukemia with an IDH1 and/or IDH2 mutation | Recruiting | 17‐Dec‐15 | |
| AML with IDH1 or IDH2 mutations | Phase Ib/II | NCT02677922 | A safety and efficacy study of oral AG‐120 plus subcutaneous azacitidine and oral AG‐221 plus subcutaneous azacitidine in subjects with newly diagnosed acute myeloid leukemia (AML) | Recruiting | 9‐Feb‐16 | |
| Newly diagnosed AML, untreated AML, AML arising from MDS, AHD or genotoxic injury | Phase I | NCT02632708 | Safety study of AG‐120 or AG‐221 in combination with induction and consolidation therapy in patients with newly diagnosed acute myeloid leukemia with an IDH1 and/or IDH2 mutation | Recruiting | 17‐Dec‐15 | |
| AML with IDH2 mutations | Phase III | NCT02577406 | An efficacy and safety study of AG‐221 (CC‐90007) versus conventional care regimens in older subjects with late stage acute myeloid leukemia harboring an Isocitrate dehydrogenase 2 mutation (IDHENTIFY) | Recruiting | 16‐Oct‐15 | |
| Advanced glioma, cholangiocarcinoma, or solid tumors with IDH1 or IDH2 mutations | Phase II | NCT03212274 | A phase 2 study of the PARP inhibitor olaparib (AZD2281) in IDH1 and IDH2 mutant advanced solid tumors | Not yet recruiting | 11‐Jul‐17 | |
| Arginase | ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Malignancy | Clinical phase | Clinical trial identifier | Title | Status | |
| Pegylated recombinant human arginase I | Hepatocellular carcinoma | Phase II | NCT02089633 | Pegylated recombinant human arginase 1 in combination with oxaliplatin and capecitabine for the treatment of HCC (PACOX) | Completed | 18‐Mar‐14 |
| Advanced cancers | Phase I | NCT02561234 | A multiple dose, dose escalation trial of AEB1102 in patients with advanced solid tumors | Recruiting | 28‐Sep‐15 | |
| CB‐1158/INCB001158 | Advanced/metastatic solid tumors | Phase I/II | NCT02903914 | Arginase inhibitor INCB001158 as a single agent and in combination with immune checkpoint therapy in patients with advanced/metastatic solid tumors | Recruiting | 16‐Sep‐16 |
| Advanced solid tumors | Phase I/II | NCT03361228 | A study to evaluate the safety, tolerability, and antitumor activity of INCB001158 plus epacadostat, with or without pembrolizumab, in advanced solid tumors | Recruiting | 4‐Dec‐2017 |
| Inhibition of polyamine synthesis | ||||||
|---|---|---|---|---|---|---|
| Inhibitor | Malignancy | Clinical Phase | Clinical Trial Identifier | Title | Status | |
| Difluoromethylornithine (DFMO)/Eflornithine | Post‐solid organ transplantskin neoplasms | Phase II | NCT00204789 | Difluoromethylornithine (DFMO) chemoprevention of skin cancer in organ transplant recipients | Completed | 20‐Sep‐05 |
| Prostate cancer | Phase II | NCT00086736 | A randomized, placebo‐controlled phase IIb clinical trial of 2‐difluoromethylornithine (DFMO) versus bicalutamide (CASODEX) alone and in combination in patients with prostate cancer in the period prior to radical prostatectomy or brachytherapy: modulation of tissue and molecular biomarkers in human prostate tissue serum | Completed | 12‐Jul‐04 | |
| Neuroblastoma | Phase II | NCT01586260 | Preventative trial of DFMO in patients with high risk neuroblastoma in remission | On‐going, not recruiting | 26‐Apr‐12 | |
| Familial adenomatous polyposis | Phase III | NCT01483144 | Trial of eflornithine plus sulindac in patients with familial adenomatous polyposis (FAP) | On‐going, not recruiting | 1‐Dec‐11 | |
| Neuroblastoma | Phase II | NCT02395666 | Preventative trial of difluoromethylornithine (DFMO) in high risk patients with neuroblastoma that is in remission | On‐going, not recruiting | 23‐Mar‐15 | |
| Adenomatous polyp | Phase II | NCT00983580 | Acetylsalicylic acid and eflornithine in treating patients at high risk for colorectal cancer | On‐going, not recruiting | 24‐Sep‐09 | |
| Neuroblastoma | Phase I | NCT02030964 | N2012‐01: Phase 1 study of difluoromethylornithine (DFMO) and celecoxib with cyclophosphamide/topotecan (DFMO) | On‐going, not recruiting | 9‐Jan‐14 | |
| Gastric cancer and gastric intestinal metaplasia | Phase II | NCT02794428 | Chemoprevention of gastric carcinogenesis | Recruiting | 9‐Jun‐16 | |
| Colorectal neoplasms | Phase III | NCT01349881 | S0820, adenoma and second primary prevention trial (PACES) | Recruiting | 9‐May‐11 | |
| Recurrent neuroblastoma | Phase I | NCT02139397 | Study of DFMO in combination with bortezomib for relapsed or refractory neuroblastoma | Recruiting | 15‐May‐14 | |
| Non‐melanoma skin cancer | ‐ | NCT02636569 | Topical chemoprevention of skin cancer biomarkers | Recruiting | 22‐Dec‐15 | |
| Neuroblastoma | Phase II | NCT02679144 | Neuroblastoma maintenance therapy trial (NMTT) | Recruiting | 10‐Feb‐16 | |
| Anaplastic astocytoma | Phase III | NCT02796261 | Study to evaluate eflornithine + lomustine vs lomustine in recurrent anaplastic astrocytoma (AA) patients (STELLAR) | Recruiting | 10‐Jun‐16 | |
2. SMALL MOLECULE INHIBITORS: TARGETING CANCER CELLS
2.1. Fatty acid synthase inhibitors
Fatty acid synthase (FASN) is a key multi‐subunit enzyme involved in lipogenesis (Figure 1), and its overexpression is commonly observed in various malignancies; normal tissues express relatively low levels of FASN, with the exception of the liver, adipose tissue, and lactating mammary glands.16, 17 Increased expression of FASN is linked to poor prognosis and reduced disease‐free survival in numerous cancer types.18, 19, 20, 21 Fatty acid synthase facilitates the generation of long‐chain saturated fatty acids, namely palmitate, from acetyl‐CoA and malonyl‐CoA (Figure 1), thereby providing fatty acids for membrane biosynthesis and lipid‐modification of proteins.22 Regulation of FASN expression in cancer is complex; however, growth factor receptors, such as ERBB‐2 and EGFR, or loss of PTEN, can activate downstream PI3K/AKT and MAPK signaling cascades resulting in transcriptional activation of FASN expression.23 Given the importance of FASN in cancer cells and its association with poor prognosis, FASN serves as a promising therapeutic target. Ventura and colleagues recently reported that TVB‐3166, an oral FASN inhibitor, induced apoptosis, inhibited anchorage‐independent cell growth under lipid‐rich conditions, and inhibited in vivo xenograft tumor growth in a dose‐dependent manner without affecting non‐tumorous tissue.24 Ventura and colleagues demonstrated that FASN inhibition disrupted lipid raft architecture, reduced lipid biosynthesis, and inhibited PI3K/AKT/mTOR signaling and expression of oncogenic factors such as c‐Myc.24 Giro‐Perafita and colleagues illustrated that C75, a potent synthetic FASN inhibitor, reduced cell viability of triple‐negative breast cancer cell lines and sensitized doxorubicin‐resistant generated cell lines MDA‐MB‐231 and HCC1896 to doxorubicin.25 Conversely, Liu et al demonstrated that FASN upregulation confers resistance to various chemotherapeutic drugs by inhibiting drug‐induced ceramide production, caspase‐8 activation, and apoptosis in breast cancer cell lines.26 In pancreatic cancer, a positive association has been shown between FASN expression and both radiotherapy and chemotherapy. Inhibition of FASN in pancreatic cancers by siRNA against FASN or through the FASN inhibitor orlistat reduced gemcitabine resistance, whereas ectopic overexpression of FASN contributed to intrinsic resistance to gemcitabine and radiotherapy.27
Figure 1.
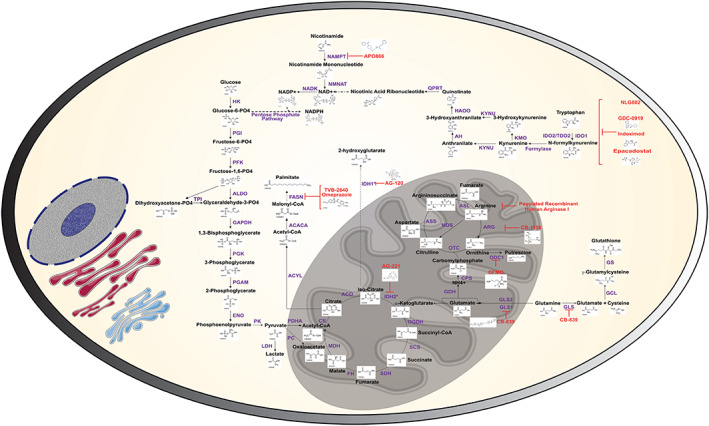
Schematic depicting metabolic pathways or proteins targeted by small molecule inhibitors. Biochemical structures for metabolites were derived from KEGG database. Chemical structures for respective inhibitors were derived from chemical‐vender websites if available. Blue text represents the enzymes, black text represents metabolites, and red text indicates the inhibitors for each step
In addition to the several outlined applications above, GlaxoSmithKline has also developed a FASN inhibitor that has been shown to induce anti‐tumor activity28, 29; however, transition of FASN inhibitors to clinical utilization remains elusive or early stage. Currently, omeprazole, an FDA‐approved proton pump inhibitor (PPI) and effective FASN inhibitor, is being actively tested in Phase 1 clinical trials in breast cancer (Table 1). Omeprazole functions by inhibiting the thioesterase domain of FASN, preventing the hydrolysis of the thioester bond between the acyl carrier protein domain and palmitate and, thereby, preventing release of free palmitate.30 Omeprazole has been shown to induce dose‐dependent reductions in cell viability in BxPC‐3 pancreatic cells30 and to inhibit breast cancer cell invasion and metastasis.31
Despite promising preclinical evidence, evaluation of the safety, tolerability, and adverse effects of FASN inhibition is warranted. Cheung and colleagues report that the use of PPIs increased risk of gastric cancer (HR: 2.44, 95% CI 1.42–4.20) in subjects treated for Helicobacter pylori.32 Moreover, this risk increased with duration of the PPI usage.32 Conversely, it has been found that the FASN inhibitor C75 readily crosses the blood‐brain barrier in rodent models, where it negatively impacts the central nervous system resulting in hypophagia and consequent weight loss. These findings underscore the importance of determining context‐specific risk versus benefit when applying FASN inhibitor therapies. Nevertheless, FASN serves as a promising target for therapeutic intervention in cancer that merits continued investigation.
2.2. Nicotinamide phosphoribosyltransferase (NAMPT) inhibitors
Nicotinamide adenine dinucleotide (NAD+) is an important metabolic reducing equivalent that is vital for bioenergetics and serves as an important cofactor for DNA maintenance and repair and SIRT‐mediated deacetylation reactions.33 NAD+ is also the precursor molecule for NADP+, an important cofactor in the pentose phosphate pathway (oxidative stress response), the citric acid cycle, and lipid biosynthesis. NAD+ is principally generated either de novo from tryptophan as part of the kynurenine pathway or recycled through the nicotinamide or nicotinic acid (NA) salvaging pathways (Figure 1).33 The nicotinamide salvaging pathway is the primary recycling pathway for NAD+ biosynthesis, for which nicotinamide phosphoribosyltransferase (NAMPT) is the rate‐limiting enzyme.33 Regulation of cell metabolism is integral to support of cell proliferation, particularly in cells under high metabolic demand. Rapid turnover of NAD+ pools by degrading enzymes such as PARPs and SIRTs render salvaging pathways as vital for restoration of NAD+ bioavailability.34 As such, it comes as no surprise that cancer cells exhibit elevated NAD+ salvaging machinery. In particular, NAMPT has been commonly observed to be elevated in various malignancies (as reviewed in Shackelford et al35). The NA salvaging pathway is frequently found to be inactive due to lack of expression of the rate‐limiting enzyme NA phosphoribosyltransferase (NAPRT).36 Conversely, de novo synthesis of NAD+ from tryptophan catabolism is minimal due to cancer cells lacking at least one enzyme in the kynurenine pathway.36 Consequently, targeting of the nicotinamide salvaging pathway via inhibiting NAMPT has received considerable attention as an intriguing target for therapeutic intervention.
NAMPT inhibition by APO866/FK866 has been shown to reduce viability of pancreatic cancer cells in vitro and to reduce pancreatic tumor growth in vivo.37 Addition of nicotinamide mononucleotide, a downstream catabolite in the nicotinamide salvaging pathway, attenuated APO866‐mediated reductions in cell viability of pancreatic cancer cell line PaTu8988T in vitro.37 Notably, the addition of NA equally prevented APO866‐induced reductions in cell viability of the PaTu8988T, suggesting activation of the NA salvaging pathway as a compensatory mechanism.37 Activation of NA salvaging pathways to counteract NAMPT inhibition is an important consideration that can have both detrimental as well as beneficial implications. Recently, O'Brien and colleagues demonstrated that treatment with the oral NAMPT‐inhibitor GNE‐617 inhibited NAD+ generation by greater than 98% and reduced tumor volume in NAPRT‐deficient PC3 and HT‐1080 xenograft models.38 However, co‐administration of NA markedly abrogated the growth‐inhibitor effects of GNE‐617.38 These findings were conserved in patient‐derived xenograft models of SAO‐737 sarcoma and STO‐399 gastric cancer.38 This would suggest a negative role of NA in antagonizing the beneficial effects of NAMPT inhibition in cancerous cells. However, activation of the NA pathway may also protect normal tissue from non‐specific toxic effects of NAMPT inhibitors. Previously, Olesen and colleagues demonstrated that co‐administration of APO866 with NA drastically reduced APO866 drug‐induced death.39 Similar to the findings by O'Brien, inclusion of NA with APO866 also negated the anti‐proliferative effects APO866.39 Thus, the use of NAMPT inhibitors comes to a juxtaposition: NAMPT inhibitors have considerable potential; however, activation of compensatory mechanisms can impede the potential benefit of such treatments. To this end, a recent open‐labeled, single‐arm, multicenter, Phase 2 clinical trial analyzed the efficacy, safety, and tolerability of APO866 in 12 patients relapsed or refractory cutaneous T‐cell lymphoma.40 Overall, APO866 demonstrated modest efficacy with one subject achieving partial response and six subjects exhibiting stable disease at 16 weeks post intervention. However, considerable adverse events were reported including pyrexia, lymphopenia, spondylitis, staphylococcal sepsis, rhabdomyloysis, and thrombocytopenia. Ultimately, the study was halted due to the lack of drug efficacy in the context of cutaneous T‐cell lymphoma.40
Currently, three Phase 2 clinical studies have been completed to explore the use of APO866, a selective NAMPT inhibitor, in both solid tumors and hematological malignancies (Table 1). Whether clinical benefit from NAMPT inhibitors will be achieved remains to be determined.
2.3. Isocitrate dehydrogenase inhibitors
In 2008, a multi‐group collaboration was set forth to sequence over 20,000 genes in 22 glioblastomas to uncover genetic alterations.41 Remarkably, one of the most common point mutations occurred in a metabolic enzyme, cytoplasmic isocitrate dehydrogenase (IDH1). Subsequent studies found that this mutation was found in ~80% of grade II‐III gliomas and secondary glioblastomas.42 Mitochondrial IDH2 has also been shown to be frequently mutated in gliomas, albeit to a lesser extent, and mutually exclusive to IDH1.42, 43 Under normal conditions, IDH1/2 generates reduced NADPH from NADP+ by catalyzing the oxidation of isocitrate to α‐ketoglutarate (α‐KG) (Figure 1).44 However, point mutations in either IDH1 or IDH2 result in the catabolism of isocitrate to the “oncometabolites” R (−)‐2‐hydroxyglutaric acid and D (−)‐2‐hydroxyglutaric acid (2‐HG), respectively, rather than oxidation to α‐ketoglutarate (Figure 1).11, 45 As expected, levels of 2‐HG are elevated in gliomas harboring mutant IDH.45 Specifically, the D‐enantiomer, but not the L‐enantiomer, of 2‐HG, has been shown to be selectively elevated in the CSF of subjects with glioma and that harbor mutated IDH genes.46 The levels of 2‐HG are so elevated in affected tumors that non‐invasive imaging modalities have proven useful in diagnosis and monitoring of patients with such glioblastomas.47 2‐HG competitively inhibits α‐KG enzymes including histone demethylases and DNA hydroxylases, resulting in distinct epigenetic phenotypes.11 Since this initial observation, IDH mutations have been observed in other tumor types, including acute myeloid leukemia (AML),48 intrahepatic cholangiocarcinoma,49 and breast adenocarcinoma.50
Cancer mutations and subsequently altered enzymatic functions make IDH unique as compared with the other metabolic targets discussed thus far, and mutated IDH enzymes have become attractive candidates for therapeutic intervention in IDH mutant malignancies. Rohle and colleagues demonstrated that IDH inhibition through a selective inhibitor, AGI‐5198, reduced 2‐HG levels in a dose‐dependent manner, induced demethylation of H3K9me3 and altered expression of genes associated with gliogenic differentiation in TS603 glioma cells.51 Rohle and colleagues further demonstrated that AG‐5198 treatment reduced tumor volume in mutant R132H‐IDH1 TS603 but not IDH1‐wild type glioma xenografts.51 Wang and colleagues demonstrated that AGI‐6780, an allosteric inhibitor of IDH2/R140Q, induced dose‐dependent reductions in 2‐HG in human glioblastoma U87 and TF‐1 cell lines expressing IDH2‐wt and IDH2/R140Q but not IDH1, indicating the selectivity of AGI‐6780 towards IDH2.52 Furthermore, AGI‐6780 reversed the IDH2/R140Q‐induced differentiation block in TF‐1 cells and induced blast differentiation in primary human IDH2/R140Q, but not IDH2‐wt, AML patient samples.52 Collectively, both studies point towards the potential clinical applications of IDH inhibition; however, they also affirm that the use of IDH inhibitors should be selective to the specific mutated form of IDH.
Currently, two IDH inhibitors are in active clinical trials, AG‐120 and AG‐221. AG‐120 is an orally available inhibitor of cytoplasmic IDH1; whereas AGI‐221 is an orally available inhibitor of mitochondrial IDH2. There are eight ongoing clinical trials exploring the use of AG‐120 and/or AG‐221 for the treatment of both hematological and solid malignancies (Table 1). Early Phase 1 studies assessing the safety and efficacy of AG‐120 in 258 subjects with mIDH1 advanced hematologic malignancies including relapsed/refractory (R/R) AML indicated acceptable tolerability with the majority of adverse events being diarrhea, leukocytosis, nausea, fatigue, febrile neutropenia, dyspnea, anemia, QT prolongation, peripheral edema, pyrexia, and decreased appetite.53 Amongst high‐risk, molecularly defined R/R AML patients, the rate of complete remission or complete remission with partial hematologic recovery was 30.4% following AG‐120 treatment, the overall response rate was 41.6%.53 Much like AG‐120, Phase 1 and 2 efficacy and tolerability studies of AG‐221 in R/R AML yielded favorable clinical outcomes including complete response rates of 19.3% and overall response rates of 40.3%.54 Adverse events associated with AG‐221 included nausea, hyperbilirubinemia, anemia, fatigue, leukocytosis, and decreased appetite.55
Whether AG‐120 or AG‐221 will provide clinical benefit or allow a sustained response in larger definitive Phase 3 trials remains to be determined; however, targeting IDH mutant tumors through the use of IDH inhibitors has shown considerable promise in early clinical trials and remains a promising avenue of therapeutic intervention.
2.4. Targeting polyamine metabolism
The polyamines spermidine and spermine and their diamine precursor putrescine are naturally occurring polycationic alkylamines essential for eukaryotic cell growth. They are directly implicated in a variety of cellular processes, including replication, transcription, translation and post‐translational modification, ion channel gating, and membrane stability.56 Not surprisingly, the requirement of polyamine metabolism is frequently dysregulated in cancer and other hyperproliferative diseases.56 The biosynthesis polyamines is regulated by ornithine decarboxylase (ODC1), the first enzyme in the polyamine pathway that mediates the decarboxylation of ornithine to generate putrescine, and adenosylmethionine decarboxylase (AMD1) which decarboxylates s‐adenosylmethionine (SAM) to provide the aminopropyl donor for the conversions to spermidine and spermine. Thus, reductions in polyamine pools lead to increases in dcSAM and corresponding reductions in SAM pools. Notably, methylation of DNA and histone tails requires the transfer of the methyl group derived from SAM, and these epigenetic changes are required for changing the pattern of peripheral tissue antigens during negative selection.57 Generation of spermidine and spermine is mediated by two sequential aminopropyl transfer reactions via spermidine synthase and spermine synthase. Intracellular polyamine levels are regulated by the enzymes spermidine/spermine N1‐acetyltransferase (SAT1), which facilitates the acetylation of spermidine/spermine mediating cellular efflux, and the oxidases polyamine oxidase (PAOX) and spermine oxidase. Transcriptional regulation of ODC1 is mediated by oncogenic c‐MYC.56 Regulation of polyamine catabolism by SAT1 has been shown to be regulated by DNA CpG hypermethylation of the SAT1 promoter region that enables elevated levels of intracellular polyamines for tumor proliferation.58 The SAT1 gene promoter also contains a polyamine‐responsive element that enables transcriptional activation through binding of nuclear factor, erythroid 2‐like 2, and polyamine‐modulating factor 1.10
Targeting of polyamine metabolism has been an active area of research for which multiple small molecule inhibitors have been developed to target various aspects of the polyamine pathway (reviewed in Casero and Marton56). Of the small molecule inhibitors, difluoromethylornithine (DFMO, elfornithine), an FDA‐approved enzyme‐activated irreversible ODC inhibitor, has been the mostly widely studied. DFMO undergoes enzymatic decarboxylation, liberating the fluoride ion and binds within the active site at lys 69 and lys 360 of ODC,59 thereby depleting intracellular polyamine pools (Figure 1). The use of DFMO in pre‐clinical settings has been widely examined.56, 60, 61 In gliomas, DFMO induces dose‐dependent cell‐cycle arrest at G0/G1 phase and intrinsic apoptosis via overexpression of Bax, Bad and reduction of bcl‐2 in vitro.60 Using a Kras‐activated p48Cre/+‐LSL‐KrasG12D/+ mouse model of pancreatic cancer, Mohammed, et al demonstrated that DFMO‐treatment induced significant reductions in PDAC incidence as compared with controls.61 Moreover, treatment with DFMO yielded a significant reduction of PanIN 3 (carcinoma in situ) lesions.61 Complementary to pre‐clinical studies, exploration of DFMO in clinical studies has been widely explored62, 63 with several clinical trials having been completed (clinicaltrials.gov). However, discretion should be taken when interpreting potential clinical benefit of DFMO. Previously, a Phase 3 study examining the efficacy of eflornithine and sulindac in preventing formation of sporadic colorectal adenomas evaluated the relationship between dietary polyamine content (sum of putrescine, spermidine, and spermine), treatment, and clinical outcome.64 When stratifying dietary polyamine content into the highest quartile versus the lower three quartiles, a significant interaction was observed between dietary polyamine levels and treatment in relation to adenoma recurrence. Moreover, there were significant reductions in risk of metachronous adenoma (risk ratio: 0.19, 95% C.I. = 0.08–0.41; P < 0.0001) in dietary polyamine quartiles 1 to 3.64 This is an important notion given that malignant cells scavenge extracellular polyamines, potentiating the possibility of reduced efficacy of ODC inhibition by DFMO.65 Consequently, dietary intervention should be monitored and controlled when examining the therapeutic potential of DFMO.
Nevertheless, there are 11 ongoing clinical trials to evaluate the clinical utility of eflornithine as a therapeutic agent (alone, or in combination with other therapies) for the treatment of cancer, namely neuroblastoma and gastrointestinal cancers (Table 1), including a randomized, double‐blind, Phase 3 trial oriented at evaluating the efficacy and safety of elfornithine/sulindac as a combination therapy compared with monotherapies in patients with familial adenomatous polyposis.66 Interim analyses of this Phase 3 study identified eight serious adverse events including depression, deep vein thrombosis, seasonal migraine, post‐polypectomy bleed, adhesive small bowl obstruction, lung adenocarcinoma, small bowel ileus, and pancreatitis; however, the authors note that these adverse events were not related to study treatment.66
Whether DFMO alone or in combination with other therapies such as sulindac is an effective preventive therapy remains to be determined; however, preclinical and clinical findings to date support the continued exploration of targeting polyamine metabolism.
3. SMALL MOLECULE INHIBITORS: TARGETING IMMUNE CELLS
3.1. Indoleamine 2, 3‐dioxygenase inhibitors
The essential amino acid tryptophan is an important precursor for protein synthesis, neurotransmitters, and de novo nicotinamide adenine‐dinucleotide biosynthesis.67 Tryptophan depletion and accumulation of its downstream catabolites, such as kynurenine (Figure 1), elicit potent immunosuppressive effects.9 Given the direct implications of immunosuppression on anti‐tumor interventions, considerable focus has been given to the development IDO inhibitors. Holmgaard and colleagues demonstrated that anti‐CTLA‐4 treatment in IDO‐deficient B16F10 melanoma xenograft mice significantly impeded tumor development and prolonged tumor‐free survival compared with IDO‐wt B16F10 xenograft mice.68 Furthermore, anti‐CTLA‐4 treatment in IDO‐deficient mice compared with control resulted in enhanced accumulation of tumor‐infiltrating CD4+ and CD8+ effector T cells and reduced the population of CD4 + FOX3+ Treg cells.68 Co‐treatment of wt B16F10 xenograft mice with 1‐methyltryptophan, a competitive inhibitor of IDO1, and anti‐CTLA‐4 equally resulted in prolonged survival and reduced tumor burden in addition to elevated infiltration of CD8+ T cells and reduced CD4 + FOX3+ Treg cell prevalence compared with controls.68 Ninomiya and colleagues illustrated that only IDO‐positive Raji lymphoma cells significantly abrogated the anti‐tumor growth effects of CD19‐CART cells in SCID‐Beige opposite flank engraftment xenograft mice.69 Importantly, the addition of co‐stimulatory domains CD19.ζ (CD3ζ chain alone), CD19.28.ζ (CD3ζ chain and CD28 endodomain), or CD19.28.4‐1BBζ (CD3z, CD28, and 4‐1BB endodomains) was not able to attenuate the inhibitory effects of the IDO‐downstream tryptophan catabolite 3‐hydroxykynurenine (Figure 1) on CD19‐CART proliferation.69 Furthermore, the addition of 1‐methyltryophan prior to CD19‐CART treatment improved the efficacy of CD19‐CART mediated suppression of tumor growth in IDO‐positive Raji tumors.69 Both of these studies demonstrate the value of IDO inhibition, particularly as a means of attenuating an immunosuppressive milieu, and, perhaps more importantly, emphasize the use of IDO inhibitors as adjuvant therapies to immuno‐therapy approaches such as anti‐tumor CAR‐T cells.
As a caveat, a recent survey of cancer cell lines demonstrated considerable variability in the expression pattern of IDO1 and tryptophan 2,3‐dioxygenase (TDO), with 16% of analyzed cancer cells lines being positive for IDO1, 19% being positive for TDO and 15% being positive for both.70 Current IDO1 inhibitors do not cross inhibit TDO.71 Thus the use of IDO1 inhibitors in subjects that do not exhibit elevated tumor IDO1 expression but instead are TDO and/or IDO2 positive would likely have minimal to no clinical benefit. Concurrently, immune‐suppression is multi‐factorial and not solely mediated by tryptophan depletion and kynurenine accumulation. For instance, is has been shown that prostaglandin E2 (PGE2) generation in cancer cells can induced Treg differentiation and promote T‐cell anergy through both direct effects on T‐cells and indirect effects on APCs.72 This would insinuate a potential synergistic benefit from using both cyclooxygenase‐2 and IDO inhibitors.
Currently, there are 10 ongoing clinical trials to assess the efficacy of the IDO1 inhibitor indoximab (1‐methyl‐d‐tryptophan) as both a mono‐ and adjuvant therapy for various malignancies in addition to several other clinical trials exploring the use of other IDO1‐inhibitors (GDC‐0919, Epacadostat, NLG802) (Table 1). Early efficacy and safety trials of epacadostat plus pembrolizumab, a PD‐1 checkpoint inhibitor, in patients with advanced lung cancers demonstrated favorable clinical outcomes including overall response rates and disease control response rates of 43% and 57%, respectively.73 Major adverse events related to epacadostat plus pembrolizumab treatment included fatigue, arthralgia, increased aspartate aminotransferase (AST), and increased lipase activity.73 Concurrently, early Phase 1 and 2 trials of epacadostat plus pembrolizumab in advanced melanoma have also shown promising results.74 Despite the encouraging results from the mentioned above Phase 1 and 2 trials, recent findings from the Pase 3 ECHO‐301/KEYNOTE‐252 trial concluded that the combination of pembrolizumab and epacadostat failed to improved progression‐free survival versus single‐agent pembrolizumab in patients with unresectable or metastatic melanoma.
IDO targeting offers considerable clinical promise; however, it is clear that its context appropriate application and combination with other therapies will be paramount to truly obtaining optimal clinical benefit.
4. SMALL MOLECULE INHIBITORS: TARGETING BOTH CANCER AND IMMUNE CELLS
4.1. Glutaminase inhibitors
Several lines of evidence have demonstrated that tumor cells exhibit a “glutamine addiction” to support anabolic metabolism.75 Extracellular glutamine can donate carbon and nitrogen to supply anabolic pathways, resulting in replenishment of tricarboxylic acid cycle (TCA) cycle intermediates and promoting synthesis of nucleotides, proteins, and lipids.6 Glutamine uptake is principally mediated by members of four amino acid transporter families, for which solute carrier family 1 member 5 has the highest affinity and is frequently upregulated in human cancer cell lines.76 While intracellular glutamine can be utilized to supply amino groups for the hexosamine biosynthetic pathway or as an exchange for the import of other amino acids such as arginine, cysteine and leucine, its primary metabolic fate in cancer is often deamination to glutamate through kidney‐type glutaminase 1 (KGA) and GAC, a splice variant encoded by GLS1.75 The contribution of GLS2‐encoding glutaminase enzymes is not a major factor in most cancers.77, 78, 79 Glutamate, in turn serves as a carbon donor for the TCA cycle via deamination to α‐ketoglutarate by glutamate dehydrogenase (Figure 1), or as a substrate for glutathione biosynthesis.75 Currently, there are three well‐studied inhibitors of KGA/GAC: Bis‐2‐(5‐phenylacetamido‐1,3,4‐thiadiazol‐2‐yl) ethyl sulfide (BPTES), (5‐(3‐Bromo‐4‐(dimethylamino)phenyl)‐2,2‐dimethyl‐2,3,5,6‐tetrahydrobenzo [a]phenanthridin‐4(1H)‐one) (968), and CB‐839. Both BPTES and CB‐839 are non‐competitive selective inhibitors of GAC and KGA,78, 80 whereas 968 is an allosteric inhibitor of GAC/KGA.81 In triple‐negative breast cancer, CB‐839, as compared with BPTES, was shown to be the superior inhibitor with an IC50 ~13‐fold lower than that of BPTES.78 In this study, CB‐839 elicited significant anti‐tumor activity in both a patient‐derived xenograft (PDX) model and in a JIMT‐1 implanted CB.17 SCID mouse model, resulting in respective tumor growth suppression by 61% and 54%, relative to vehicle control.78 Tumor abundances of glutamine increased 4 hours post CB‐839 treatment, whereas glutamate and aspartate levels decreased, consistent with GLS activity inhibition.78
While GLS inhibitors may serve as monotherapies, their combination with other concurrent therapies such as radiotherapy, chemotherapy or immunotherapy is equally intriguing. With respect to radio‐ and chemotherapy, studies in PDAC have demonstrated that GLS inhibition leads to increased radio‐sensitivity, largely due to increased ROS generation,82 an aspect likely linked to alterations in glutathione biosynthesis, a well‐documented contributor to drug resistance.83 Indeed, studies in triple‐negative breast cancer have demonstrated considerable effects of CB‐839 on reducing glutathione levels in a cell line‐dependent manner.78 Additionally, glutaminolysis, a biochemical reaction by glutamine is lysed to glutamate, aspartate, CO2, pyruvate, lactate, alanine, and citrate, has been linked to cisplatin resistance in gastric cancers.28 With respect to immunotherapy, Calithera has initiated a Phase 2 study aimed at exploring the safety, tolerability, and efficacy of CB‐839 in combination with Nivolumab, a PD‐1/PD‐L1 check point inhibitor, in patients with renal cell carcinoma, melanoma, or non‐small cell lung cancer. Interim results indicated tolerable toxicity with mild to moderate adverse events, namely fatigue, nausea, and photophobia. Positive responses were obtained in melanoma patients with overall response rates of 19% and an overall disease control rate of 44% (Calithera; Society for Immunotherapy of Cancer Meeting).
Currently, the use of CB‐839 in combination with chemotherapy or immunotherapies is being explored in multiple Phase 1 and 2 studies on solid tumors (Table 1). However, the broad applicability of CB‐839 as an anti‐cancer agent is tentative. A recent study by Davidson et al84 found that in vivo glutaminase inhibition exhibited markedly lower efficacy in KRAS driven lung cancer mouse model as compared with in vitro studies. The authors attribute this discrepancy to differences in the tumor microenvironment and nutrient utilization.84 Conversely, studies by Christen and colleagues demonstrated preferential utilization of pyruvate rather than glutamine for PC‐dependent anaplerosis in breast‐derived lung metastases and that this was sufficient to render cells insensitive to glutamine anaplerosis inhibition.85 These findings necessitate the importance of identifying the correct target population and of understanding the interaction between cancer metabolism, the tumor microenvironment, and nutrient availability.
Regardless, the ultimate clinical utility of glutaminase inhibitors as single or combinatorial agents remains to be determined; however, preclinical and preliminary clinical results support the value of targeting glutaminase as a therapy for cancer.
4.2. Targeting arginine metabolism
Whereas the abovementioned inhibitors aim to directly target enzymes that support tumor‐associated metabolic processes, one could in principle achieve similar effects using an inverse approach wherein recombinant enzymes are applied therapeutically to reduce the availability of key nutrients required for tumor growth within the tumor microenvironment. This concept has been introduced through the use of recombinant arginase I that aims to deplete the microenvironment of the amino acid arginine. Arginine is versatile amino acid that serves as an important metabolic precursor for protein biosynthesis, nitric oxide production, polyamine biosynthesis, and nitrogen disposal.86 It has been well documented that malignant cells are particularly sensitive to arginine depletion both in vitro and in vivo.87, 88, 89 Normal cells have the ability to replenish arginine from citrulline through a two‐step process involving the conversion of citrulline to argininosuccinate to arginine by argininosuccinate synthase (ASS) and argininosuccinate lyase, respectively (Figure 1). However, malignant cells often do not express ASS, thereby impeding their ability to replenish arginine.89 Correspondingly, tumor cells may have adequate levels of ASS but lack expression of ornithine transcarbamylase, the enzyme that mediates the conversion of arginine to ornithine (Figure 1),88 thereby limiting their capacity to regenerate arginine. Consequently, tumor cells are auxotrophic for arginine, and attenuating arginine availability is a plausible therapeutic strategy. This approach has led the development of Pegylated Recombinant Arginase I (Peg‐rhArg1) that is currently being evaluated in multiple Phase 1 and Phase 2 clinical trials (Table 1). A Phase 1 study lead by Poon and colleagues examined the pharmacokinetics and pharmacodynamics of Peg‐rhArg1 in advanced hepatocellular carcinoma subjects.90 A total of 15 patients were enrolled with patients being split among weekly doses of 500 U/kg (n = 3), 1000 U/kg (n = 3), 1600 U/kg (n = 3), or 2500 U/kg (n = 6).90 Plasma arginine depletion was observed in a dose‐dependent manner.90 The most common drug‐associated non‐hematological adverse events were diarrhea, abdominal discomfort, and nausea; no hematological adverse events were observed.90 The best overall response was stable disease for >8 weeks in 4 subjects (26.7%).90 Izzo and colleagues examined the effects of polyethyelene glycol‐conjugated arginine deaminase (ADI‐SS PEG) in 19 patients with unresectable hepatocellular carcinoma.91 An ADI‐SS PEG dose of 160 U/m2 sufficiently lowered plasma arginine from a baseline ~130 μM to below levels of detection (<2 μM) for more than 7 days.91 ADI‐SS PEG was well tolerated with no serious adverse events.91 Of the 19 subjects on ADI‐SS PEG, two (10.5%) had complete responses, seven (36.8%) had partial responses, seven (36.8%) had stable disease, and three (15.9%) exhibited progressive disease.91 Whereas clinical trials have been focused primarily on hepatocellular carcinomas, the application of Peg‐rhArg1 has also been tested in preclinical models of other malignancies including AML,92 prostate cancer,88 and non‐Hodgkin's lymphoma93 indicating broad potential application. The application of Peg‐rhArg1 as a monotherapy or adjuvant therapy holds promise and provides proof of concept for similar therapies that would aim to exploit the auxotrophic vulnerabilities of various malignancies.
However, it is important to note that arginine reliance is not exclusive to cancer cells. In fact, arginine metabolism is also intimately linked to T cell fate and function. In particular, arginine has been shown to play a key role in the activation of T cells and in generation of central memory‐like cells endowed with a higher survival capacity, a function that is linked to a shift in activated T cells away from glycolysis and towards oxidative phosphorylation.94 To this end, studies by Geiger and colleagues demonstrated that arginine promotes T cell survival through interaction with the transcriptional regulators BAZ1B, PSIP1, and TSN.94 Moreover, Geiger and colleagues show that stimulation of TCR transgenic CD8+ OT‐I T cells specific for the OVA257–264 peptide arginine‐supplemented medium for 4 days endowed OT‐I T cells with a higher survival capacity as compared with control when transferred into lymphopenic Cd3e −/− mice.94 Importantly, arginine‐treated OT‐I T cells adoptively transferred into wild‐type mice bearing B16 melanoma tumors expressing the OVA antigen yielded superior anti‐tumor responses, marked by significant reductions in tumor size and improved overall survival.94 Collectively, this implies that elevated arginine levels promote the survival capacity of CD8+ T cells and their anti‐tumor activity in vivo. Keeping this notion in mind, Calithera has developed the arginase inhibitor CB‐1158 (Figure 1) with the overall intent of preventing arginine depletion in the local tumor microenvironment. Pre‐clinical data has demonstrated that CB‐1158 reverses the capacity of polymorphonuclear cells and myeloid‐derived suppressor cells to inhibit T‐cell activation and proliferation ex‐vivo by preventing arginine depletion.95 Moreover, pre‐clinical data has demonstrated that CB‐1158 increases plasma and tumor arginine levels in mouse syngeneic tumor models, resulting in increased pro‐inflammatory markers and activated CD8 T‐cells in the tumor.95 The pharmacokinetics and pharmacodynamics of CB‐1158 are currently being explored in a Phase 1 study of solid tumors (Table 1). To this end, preliminary findings from the Phase 1 study evaluating safety and tolerability of CB‐1158 as a monotherapy and in combination with anti‐PD1 have found that CB‐1158 is well tolerated with no dose‐limiting toxicities or drug‐related grade 3 adverse events.95 CB‐1158 was found to be rapidly absorbed (Tmax = 4 hours); doses of 50 and 100 mg resulted in stead‐state plasma trough levels of 1.6 and 4.5 μM which was sufficient to achieve >90% arginase inhibition and increase plasma arginine levels by 2.4‐ and 4‐fold, respectively.95 Dose escalations studies are currently on‐going.
Although both Peg‐rhArg1 and CB‐1158 have shown promising preclinical and early clinical success, the two strategies are contradictory in nature; one favoring the depletion of arginine (Peg‐rhArg1) while the other favors its accumulation (CB‐1158). As such, the efficacy of either strategy as a successful therapy for cancer will be of particular interest and likely context dependent.
5. CONCLUSION
In the preceding overview, we have summarized the use of small molecule inhibitors currently in active clinical trials that are aimed at targeting both aberrant metabolic pathways in tumor cells as well as in the surrounding tumor microenvironment. Preclinical and early clinical results have shown promise and warrant close evaluation. Moreover, the use of small molecule inhibitors aimed at exploiting tumor metabolic vulnerabilities continues to be an active area of research, extending far beyond the targets mentioned in these sections. Yet, the one unifying consensus is that identifying metabolic dependencies specific to cancer cells and/or constituents of the tumor microenvironment is a viable area of therapeutic intervention that holds considerable clinical potential.
CONFLICT OF INTEREST/FINANCIAL DISCLOSURE STATEMENT
The authors certify that they have NO affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers' bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent‐licensing arrangements), or non‐financial interest (such as personal or professional relationships, affiliations, knowledge, or beliefs) in the subject matter or materials discussed in this manuscript.
FUNDING INFORMATION
J.F.F. is a recipient of junior mentored faculty fellowship by Duncan Family Institute for Cancer Prevention and Risk Assessment, The University of Texas MD Anderson Cancer Center, Houston, TX.
AUTHOR'S CONTRIBUTIONS
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, S.C.T. and J.F.F.; Methodology, S.C.T. and J.F.F. (if even applicable); Investigation, S.C.T. and J.F.F.; Formal Analysis, S.C.T. and J.F.F.; Resources, S.H.; Writing‐Original Draft, S.C.T. and J.F.F.; Writing‐Review and Editing, S.C.T., J.F.F., J.V.V., J.B.D., S.H.; Visualization, S.C.T. and J.F.F.; Supervision, S.H.; Funding Acquisition, Not applicable.
Tripathi SC, Fahrmann JF, Vykoukal JV, Dennison JB, Hanash SM. Targeting metabolic vulnerabilities of cancer: Small molecule inhibitors in clinic. Cancer Reports. 2019;2:e1131. 10.1002/cnr2.1131
Satyendra C. Tripathi and Johannes F. Fahrmann contributed equally to the manuscript.
REFERENCES
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646‐674. [DOI] [PubMed] [Google Scholar]
- 2. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Otto AM. Warburg effect (s)‐a biographical sketch of Otto Warburg and his impacts on tumor metabolism. Cancer Metab. 2016;4(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY). 2009;324:1029‐1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7(1):11‐20. [DOI] [PubMed] [Google Scholar]
- 6. DeBerardinis RJ, Mancuso A, Daikhin E, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104(49):19345‐19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jeon SM, Hay N. Expanding the concepts of cancer metabolism. Exp Mol Med. 2018;50(4):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang F, Du G. Dysregulated lipid metabolism in cancer. World J Biol Chem. 2012;3(8):167‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435‐5440. [DOI] [PubMed] [Google Scholar]
- 10. Casero RA, Pegg AE. Polyamine catabolism and disease. Biochem J. 2009;421(3):323‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Borodovsky A, Seltzer MJ, Riggins GJ. Altered cancer cell metabolism in gliomas with mutant IDH1 or IDH2. Curr Opin Oncol. 2012;24(1):83‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Teoh ST, Lunt SY. Metabolism in cancer metastasis: bioenergetics, biosynthesis, and beyond. Wiley Interdiscip Rev Syst Biol Med. 2018;10(2). [DOI] [PubMed] [Google Scholar]
- 14. Weber GF. Metabolism in cancer metastasis. Int J Cancer. 2016;138(9):2061‐2066. [DOI] [PubMed] [Google Scholar]
- 15. Luo X, Cheng C, Tan Z, et al. Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer. 2017;16(1):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Flavin R, Peluso S, Nguyen PL, Loda M. Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol (London, England). 2010;6:551‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sul HS, Wang D. Nutritional and hormonal regulation of enzymes in fat synthesis: studies of fatty acid synthase and mitochondrial glycerol‐3‐phosphate acyltransferase gene transcription. Annu Rev Nutr. 1998;18(1):331‐351. [DOI] [PubMed] [Google Scholar]
- 18. Ogino S, Nosho K, Meyerhardt JA, et al. Cohort study of fatty acid synthase expression and patient survival in colon cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2008;26(35):5713‐5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shurbaji MS, Kalbfleisch JH, Thurmond TS. Immunohistochemical detection of a fatty acid synthase (OA‐519) as a predictor of progression of prostate cancer. Hum Pathol. 1996;27(9):917‐921. [DOI] [PubMed] [Google Scholar]
- 20. Takahiro T, Shinichi K, Toshimitsu S. Expression of fatty acid synthase as a prognostic indicator in soft tissue sarcomas. Clin Cancer Res. 2003;9(6):2204‐2212. [PubMed] [Google Scholar]
- 21. Visca P, Sebastiani V, Botti C, et al. Fatty acid synthase (FAS) is a marker of increased risk of recurrence in lung carcinoma. Anticancer Res. 2004;24(6):4169‐4173. [PubMed] [Google Scholar]
- 22. Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763‐777. [DOI] [PubMed] [Google Scholar]
- 23. Bandyopadhyay S, Pai SK, Watabe M, et al. FAS expression inversely correlates with PTEN level in prostate cancer and a PI 3‐kinase inhibitor synergizes with FAS siRNA to induce apoptosis. Oncogene. 2005;24(34):5389‐5395. [DOI] [PubMed] [Google Scholar]
- 24. Ventura R, Mordec K, Waszczuk J, et al. Inhibition of de novo palmitate synthesis by fatty acid synthase induces apoptosis in tumor cells by remodeling cell membranes, inhibiting signaling pathways, and reprogramming gene expression. EBioMedicine. 2015;2:806‐822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Giro‐Perafita A, Palomeras S, Lum D, et al. Preclinical evaluation of fatty acid synthase and EGFR inhibition in triple negative breast cancer. Clin Cancer Res. 2016;22(18):4687‐4697. [DOI] [PubMed] [Google Scholar]
- 26. Liu H, Wu X, Dong Z, et al. Fatty acid synthase causes drug resistance by inhibiting TNF‐alpha and ceramide production. J Lipid Res. 2013;54(3):776‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yang Y, Liu H, Li Z, et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int J Biochem Mol Biol. 2011;2(1):89‐98. [PMC free article] [PubMed] [Google Scholar]
- 28. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4(3):e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hardwicke MA, Rendina AR, Williams SP, et al. A human fatty acid synthase inhibitor binds beta‐ketoacyl reductase in the keto‐substrate site. Nat Chem Biol. 2014;10(9):774‐779. [DOI] [PubMed] [Google Scholar]
- 30. Fako VE, Wu X, Pflug B, Liu JY, Zhang JT. Repositioning proton pump inhibitors as anticancer drugs by targeting the thioesterase domain of human fatty acid synthase. J Med Chem. 2015;58(2):778‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jin UH, Lee SO, Pfent C, Safe S. The aryl hydrocarbon receptor ligand omeprazole inhibits breast cancer cell invasion and metastasis. BMC Cancer. 2014;14(1):498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheung KS, Chan EW, Wong AYS, Chen L, Wong ICK, Leung WK. Long‐term proton pump inhibitors and risk of gastric cancer development after treatment for Helicobacter pylori: a population‐based study. Gut. 2018;67(1):28‐35. [DOI] [PubMed] [Google Scholar]
- 33. Sampath D, Zabka TS, Misner DL, O'Brien T, Dragovich PS. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol Ther. 2015;151:16‐31. [DOI] [PubMed] [Google Scholar]
- 34. Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly (ADP‐ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517‐528. [DOI] [PubMed] [Google Scholar]
- 35. Shackelford RE, Mayhall K, Maxwell NM, Kandil E, Coppola D. Nicotinamide phosphoribosyltransferase in malignancy: a review. Genes Cancer. 2013;4(11‐12):447‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xiao Y, Elkins K, Durieux JK, et al. Dependence of tumor cell lines and patient‐derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE‐618. Neoplasia (New York, NY). 2013;15:1151‐1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chini CC, Guerrico AM, Nin V, et al. Targeting of NAD metabolism in pancreatic cancer cells: potential novel therapy for pancreatic tumors. Clin Cancer Res. 2014;20(1):120‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. O'Brien T, Oeh J, Xiao Y, et al. Supplementation of nicotinic acid with NAMPT inhibitors results in loss of in vivo efficacy in NAPRT1‐deficient tumor models. Neoplasia (New York, NY). 2013;15:1314‐1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Olesen UH, Thougaard AV, Jensen PB, Sehested M. A preclinical study on the rescue of normal tissue by nicotinic acid in high‐dose treatment with APO866, a specific nicotinamide phosphoribosyltransferase inhibitor. Mol Cancer Ther. 2010;9(6):1609‐1617. [DOI] [PubMed] [Google Scholar]
- 40. Goldinger SM, Gobbi Bischof S, Fink‐Puches R, et al. Efficacy and safety of APO866 in patients with refractory or relapsed cutaneous T‐cell lymphoma: a phase 2 clinical trial. JAMA Dermatol. 2016;152(7):837‐839. [DOI] [PubMed] [Google Scholar]
- 41. Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science (New York, NY). 2008;321:1807‐1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469‐474. [DOI] [PubMed] [Google Scholar]
- 43. Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pollard PJ, Ratcliffe PJ. Cancer. Puzzling patterns of predisposition. Science (New York, NY). 2009;324:192‐194. [DOI] [PubMed] [Google Scholar]
- 45. Dang L, White DW, Gross S, et al. Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature. 2009;462(7274):739‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kalinina J, Ahn J, Devi NS, et al. Selective detection of the D‐enantiomer of 2‐hydroxyglutarate in the CSF of glioma patients with mutated Isocitrate dehydrogenase. Clin Cancer Res. 2016;22(24):6256‐6265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choi C, Ganji SK, DeBerardinis RJ, et al. 2‐hydroxyglutarate detection by magnetic resonance spectroscopy in IDH‐mutated patients with gliomas. Nat Med. 2012;18(4):624‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mardis ER, Ding L, Dooling DJ, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361(11):1058‐1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saha SK, Parachoniak CA, Bardeesy N. IDH mutations in liver cell plasticity and biliary cancer. Cell Cycle (Georgetown, Tex). 2014;13:3176‐3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fathi AT, Sadrzadeh H, Comander AH, et al. Isocitrate dehydrogenase 1 (IDH1) mutation in breast adenocarcinoma is associated with elevated levels of serum and urine 2‐hydroxyglutarate. Oncologist. 2014;19(6):602‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rohle D, Popovici‐Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science (New York, NY). 2013;340:626‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang F, Travins J, DeLaBarre B, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science (New York, NY). 2013;340:622‐626. [DOI] [PubMed] [Google Scholar]
- 53. DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with Ivosidenib in IDH1‐mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386‐2398. [DOI] [PubMed] [Google Scholar]
- 54. Cohen JD, Javed AA, Thoburn C, et al. Combined circulating tumor DNA and protein biomarker‐based liquid biopsy for the earlier detection of pancreatic cancers. Proc Natl Acad Sci U S A. 2017;114(38):10202‐10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722‐731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Casero RA Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6(5):373‐390. [DOI] [PubMed] [Google Scholar]
- 57. Herzig Y, Nevo S, Bornstein C, et al. Transcriptional programs that control expression of the autoimmune regulator gene Aire. Nat Immunol. 2017;18(2):161‐172. [DOI] [PubMed] [Google Scholar]
- 58. Mank‐Seymour AR, Murray TR, Berkey KA, Xiao L, Kern S, Casero RA Jr. Two active copies of the X‐linked gene spermidine/spermine N1‐acetyltransferase (SSAT) in a female lung cancer cell line are associated with an increase in sensitivity to an antitumor polyamine analogue. Clin Cancer Res. 1998;4(8):2003‐2008. [PubMed] [Google Scholar]
- 59. Alexiou GA, Lianos GD, Ragos V, Galani V, Kyritsis AP. Difluoromethylornithine in cancer: new advances. Future Oncol (London, England). 2017;13:809‐819. [DOI] [PubMed] [Google Scholar]
- 60. Alexiou GA, Tsamis KI, Vartholomatos E, et al. Combination treatment of TRAIL, DFMO and radiation for malignant glioma cells. J Neurooncol. 2015;123(2):217‐224. [DOI] [PubMed] [Google Scholar]
- 61. Mohammed A, Janakiram NB, Madka V, et al. Eflornithine (DFMO) prevents progression of pancreatic cancer by modulating ornithine decarboxylase signaling. Cancer Prev Res (Philadelphia, Pa). 2014;7:1198‐1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Laukaitis CM, Gerner EW. DFMO: targeted risk reduction therapy for colorectal neoplasia. Best Pract Res Clin Gastroenterol. 2011;25(4‐5):495‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Saulnier Sholler GL, Gerner EW, Bergendahl G, et al. A phase I trial of DFMO targeting polyamine addiction in patients with relapsed/refractory neuroblastoma. PLoS One. 2015;10(5):e0127246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Raj KP, Zell JA, Rock CL, et al. Role of dietary polyamines in a phase III clinical trial of difluoromethylornithine (DFMO) and sulindac for prevention of sporadic colorectal adenomas. Br J Cancer. 2013;108(3):512‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Soda K. The mechanisms by which polyamines accelerate tumor spread. J Exp Clin Cancer Res: CR. 2011;30(1):95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Burke CA, Dekker E, Samadder NJ, Stoffel E, Cohen A. Efficacy and safety of eflornithine (CPP‐1X)/sulindac combination therapy versus each as monotherapy in patients with familial adenomatous polyposis (FAP): design and rationale of a randomized, double‐blind, Phase III trial. BMC Gastroenterol. 2016;16(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol. 2003;81(4):247‐265. [DOI] [PubMed] [Google Scholar]
- 68. Holmgaard RB, Zamarin D, Munn DH, Wolchok JD, Allison JP. Indoleamine 2,3‐dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA‐4. J Exp Med. 2013;210(7):1389‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ninomiya S, Narala N, Huye L, et al. Tumor indoleamine 2,3‐dioxygenase (IDO) inhibits CD19‐CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125(25):3905‐3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pilotte L, Larrieu P, Stroobant V, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3‐dioxygenase. Proc Natl Acad Sci U S A. 2012;109(7):2497‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Platten M, von Knebel DN, Oezen I, Wick W, Ochs K. Cancer immunotherapy by targeting IDO1/TDO and their downstream effectors. Front Immunol. 2014;5:673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol (Baltimore, Md: 1950). 2012;188:21‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gangadhar TC, Schneider BJ, Bauer TM, et al. Efficacy and safety of epacadostat plus pembrolizumab treatment of NSCLC: Preliminary phase I/II results of ECHO‐202/KEYNOTE‐037. J Clin Oncol. 2017;35:9014. [Google Scholar]
- 74. Hamid O, Bauer TM, Spira AI, et al. Safety of epacadostat 100 mg bid plus pembrolizumab 200 mg Q3W in advanced solid tumors: Phase 2 data from ECHO‐202/KEYNOTE‐037. J Clin Oncol. 2017;35:3012. [Google Scholar]
- 75. De Vitto H, Perez‐Valencia J, Radosevich JA. Glutamine at focus: versatile roles in cancer. Tumour Biol: The Journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37(2):1541‐1558. [DOI] [PubMed] [Google Scholar]
- 76. Hassanein M, Hoeksema MD, Shiota M, et al. SLC1A5 mediates glutamine transport required for lung cancer cell growth and survival. Clin Cancer Res. 2013;19(3):560‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Le A, Lane AN, Hamaker M, et al. Glucose‐independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gross MI, Demo SD, Dennison JB, et al. Antitumor activity of the glutaminase inhibitor CB‐839 in triple‐negative breast cancer. Mol Cancer Ther. 2014;13(4):890‐901. [DOI] [PubMed] [Google Scholar]
- 79. Seltzer MJ, Bennett BD, Joshi AD, et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. 2010;70(22):8981‐8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. DeLaBarre B, Gross S, Fang C, et al. Full‐length human glutaminase in complex with an allosteric inhibitor. Biochemistry. 2011;50(50):10764‐10770. [DOI] [PubMed] [Google Scholar]
- 81. Wang JB, Erickson JW, Fuji R, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18(3):207‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Li D, Fu Z, Chen R, et al. Inhibition of glutamine metabolism counteracts pancreatic cancer stem cell features and sensitizes cells to radiotherapy. Oncotarget. 2015;6(31):31151‐31163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Traverso N, Ricciarelli R, Nitti M, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longev. 2013;2013:972913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Davidson SM, Papagiannakopoulos T, Olenchock BA, et al. Environment impacts the metabolic dependencies of Ras‐driven non‐small cell lung cancer. Cell Metab. 2016;23(3):517‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Christen S, Lorendeau D, Schmieder R, et al. Breast cancer‐derived lung metastases show increased pyruvate carboxylase‐dependent anaplerosis. Cell Rep. 2016;17(3):837‐848. [DOI] [PubMed] [Google Scholar]
- 86. Morris SM Jr. Arginine metabolism: boundaries of our knowledge. J Nutr. 2007;137:1602s‐1609s. [DOI] [PubMed] [Google Scholar]
- 87. Wheatley DN. Arginine deprivation and metabolomics: important aspects of intermediary metabolism in relation to the differential sensitivity of normal and tumour cells. Semin Cancer Biol. 2005;15(4):247‐253. [DOI] [PubMed] [Google Scholar]
- 88. Hsueh EC, Knebel SM, Lo WH, Leung YC, Cheng PN, Hsueh CT. Deprivation of arginine by recombinant human arginase in prostate cancer cells. J Hematol Oncol. 2012;5(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Pegylated arginine deiminase (ADI‐SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res. 2002;62(19):5443‐5450. [PubMed] [Google Scholar]
- 90. Yau T, Cheng PN, Chan P, et al. A phase 1 dose‐escalating study of pegylated recombinant human arginase 1 (Peg‐rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2013;31(1):99‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Izzo F, Marra P, Beneduce G, et al. Pegylated arginine deiminase treatment of patients with unresectable hepatocellular carcinoma: results from phase I/II studies. J Clin Oncol Off J Am Soc Clin Oncol. 2004;22(10):1815‐1822. [DOI] [PubMed] [Google Scholar]
- 92. Mussai F, Egan S, Higginbotham‐Jones J, et al. Arginine dependence of acute myeloid leukemia blast proliferation: a novel therapeutic target. Blood. 2015;125(15):2386‐2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zeng X, Li Y, Fan J, et al. Recombinant human arginase induced caspase‐dependent apoptosis and autophagy in non‐Hodgkin's lymphoma cells. Cell Death Dis. 2013;4(10):e840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Geiger R, Rieckmann JC, Wolf T, et al. L‐Arginine modulates T cell metabolism and enhances survival and anti‐tumor activity. Cell. 2016;167:829‐42.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Papadopoulos KP, Tsai FY‐C, Bauer TM, et al. CX‐1158‐101: a first‐in‐human phase 1 study of CB‐1158, a small molecule inhibitor of arginase, as monotherapy and in combination with an anti‐PD‐1 checkpoint inhibitor in patients (pts) with solid tumors. J Clin Oncol. 2017;35:3005. [Google Scholar]