

Abstract
Background
Carcinogenic transformation of white blood cells during hematopoiesis leads to the development of leukemia, a cancer characterized by incompetent immune cells and a disruption of normal bone marrow function. Leukemias are diverse in type, affected population, prognosis, and treatment regimen, yet a common theme in leukemia is the dysregulated metabolism of leukemic cells and leukemic stem cells with respect to their noncancerous counterparts.
Recent findings
In this review, we highlight current findings that elucidate metabolic traits unique to the four major types of leukemia, which confer carcinogenic survival but can be potentially exploited for therapeutic intervention. These metabolic features can work in conjunction with or be independent of unique aspects of the bone marrow microenvironment that can also influence cell survival and proliferation, thus sustaining carcinogenesis.
Conclusion
Deepening our understanding of the interactions of leukemias with their niche environments in vivo will inform future treatments for leukemia, particularly for those that are refractive to tyrosine kinase inhibitors and other therapeutic mainstays.
Keywords: metabolism, leukemia, mass spectrometry, metabolomics, stable isotope tracing, tumor microenvironment
1. INTRODUCTION
Leukemias are a group of hematological malignancies caused by the rapid production of abnormal white blood cells. Such atypical white blood cells are not functional immune cells and, through space occupied, ultimately impair the ability of the bone marrow to produce sufficient numbers of red blood cells, platelets, and normal white blood cells. Several classifications exist to characterize the four major types of leukemia, each of which involves the oncogenic transformation of progenitor cells that are biased toward specific fates in the hematopoietic hierarchy. Lymphocytic leukemia refers to the transformation of marrow progenitor cells that become lymphocytes, including natural killer, T, and B cells. In myelogenous (or myeloid) leukemia, malignant transformation occurs in the marrow progenitor cells that mature into the cells of the myeloid lineage, which includes monocytes, mast cells, basophils, neutrophils, eosinophils, erythrocytes, and thrombocytes. The second classification, acute vs chronic leukemia, refers to the degree of cell maturity prior to carcinogenic transformation. Acute leukemias are malignancies of immature hematopoietic cells, and in contrast, chronic leukemias are characterized by the transformation of partially matured hematopoietic cells. In general, chronic leukemia progresses more slowly than acute cases, the latter of which, if left untreated, can result in death within weeks.
The ongoing search for effective strategies to treat hematological cancers is not unlike other cancers in that a precise and thorough understanding of their distinct genotypic and metabolic states, especially in comparison to their normal counterparts, can provide targets for selective, robust, and lasting cytoreduction of cancer cells. Investigations of hematopoiesis, particularly in the context of leukemia development, face several substantial challenges. First, although isolation of the specific progenitor populations is possible using flow cytometry, cell numbers are often low as the populations are small. A second major challenge is heterogeneity among tumors of the same leukemia type resulting from unique tumor niches, genetic variability among humans, comorbidities, and as yet unidentified disease‐related or disease‐causing mutations. The advent of systems biology‐based approaches such as omics technologies (genomics, transcriptomics, proteomics, metabolomics, among others) has transformed this research by generating complex datasets that provide a unique view into disease pathology and thus enable the development of novel therapies. Of the different tiers in the central dogma, metabolism represents that which is closest to the cellular phenotype and most responsive to environmental stimuli.1, 2, 3 As such, metabolomics (ie, the study of small molecules <1500 Da) has emerged as the approach most amenable for detecting rapid, dynamic changes in cellular state that are biologically impactful but not manifested at the genome level due to timing or magnitude of change.
Techniques for performing wide‐scale metabolomic analyses rely on nuclear magnetic resonance (NMR) spectroscopy or mass spectrometry (MS) that provide direct readouts of complex metabolite mixtures and quantify changes occurring in response to stimuli, drug treatment, time, disease, and so on. In contrast, alternative approaches to measure single or several metabolites include autofluorescence, multiphoton, hyperspectral, and fluorescence lifetime imaging microscopy (FLIM), as well as commercial kits that spectrophotometrically quantify individual metabolites based on a surrogate reading of exogenously added enzyme activities. Extracellular flux (XF) analyzers can provide more holistic views of dynamic metabolism by determining extracellular acidification rate (ie, glycolytic flux) and oxygen consumption rate (ie, mitochondrial metabolism). These alternative approaches are utilized widely and are included in studies discussed herein. However, while valuable, these approaches are sharply limited by the breadth of information obtained in comparison to NMR and, in particular, MS experiments. This review will focus on recent investigations of metabolism in the context of leukemia that serve as major efforts to identify mechanisms of drug resistance, biological phenomena underlying relapse, and new targets for leukemia therapy.
Hematopoiesis begins at the stage of hematopoietic stem cells (HSCs), which are largely quiescent and maintain a self‐renewing capacity. HSCs reside in the bone marrow, an organ marked by hypoxia and metabolic regulation by HIF‐1α.4 As a result, HSC homeostasis relies primarily on anaerobic glycolysis,5 fatty acid metabolism,6 mitophagy, and autophagy7 with little contribution of oxidative phosphorylation and high antioxidant defense systems, thus protecting this limited and precious cell population against the adverse effects of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Once activated, HSCs progress into multipotent progenitor cells (MPPs), a population devoid of self‐renewal and marked by robust proliferation (Figure 1). Through sophisticated molecular signaling that is only partially elucidated at current day, MPP subtypes (ie, MPP2, MPP3, and MPP4) are molecularly biased towards committing to specific lineages; MPP2 cells primarily form granulocyte/macrophage progenitors (GMP) and megakaryocyte‐erythroid progenitors (MEP), while MPP3 cells predominantly fuel GMP production and MPP4 cells for lymphoid lineage progenitors, both via common myeloid progenitors (CMPs), or common lymphoid progenitors (CLPs, also known as MPP4).8 Key modulators of hematopoiesis include inflammation,9, 10 the HSC microenvironment, mitochondrial dynamics,11 reactive oxygen species,12 and epigenetics,13 among other factors.
Figure 1.
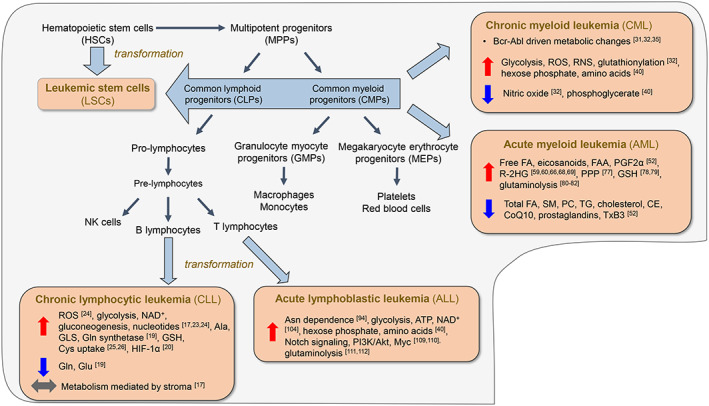
Overview of hematopoiesis and transformation. Hematopoietic stem cells (HSCs) possess self‐renewing capacity and are maintained as a small population. Generation of multipluripotent progenitor (MPP) cells devoid of self‐renewal, but with active proliferation, marks the path toward differentiation to ultimately form the cellular components of blood. Carcinogenic transformation at the HSC stage generates leukemic stem cells (LSCs); transformation of mature or immature cells at points downstream generates leukemic cells. Shown here is an overview of metabolic features of the leukemia types. Abbreviations: ATP, adenosine triphosphate; CE, cholesterol ester; CoQ10, coenzyme Q10; FA, fatty acid; FAA, fatty acid amide; GLS, glutaminase; GSH, reduced glutathione; HIF‐1α, hypoxia‐inducible factor 1α; NAD, nicotinamide adenine dinucleotide; PC, phosphatidylcholine; PGF2α, prostaglandin F2α; PPP, pentose phosphate pathway; R‐2HG, (R)‐2‐hydroxyglutarate; RNS, reactive nitrogen species; ROS, reactive oxygen species; SM, sphingomyelin; TG, triglyceride; TxB3, thromboxane B3
Carcinogenic transformation of HSCs generates leukemic stem cells (LSCs, Figure 1), which are characterized by their impaired capacity for differentiation, enhanced proliferation, and unlimited self‐renewal.14 These cells are central components of disease progression, as well as drug resistance and relapse. Contradictory to long‐term HSCs, LSCs are more dependent on oxidative respiration and have upregulated antioxidant defense systems, both adaptations which poise this cell population for rapid proliferative capacities. In addition to arising from the LSC pool,15 leukemic cells also initiate at more mature developmental stages; we discuss next the four leukemia subtypes and how their unique metabolic features underlie current treatment approaches and provide cues for the development of therapies in the future. A prominent and common feature of leukemia cell biology is the cellular interaction with the tumor microenvironment. The stroma proves to be a key regulator of cell metabolism and redox state in a manner that controls transformation and proliferation. The various roles of the tumor microenvironment are noted within each of the leukemia subtypes, but additionally, this topic is also addressed on a larger scale in the concluding section with a particular focus on extracellular matrix protein dynamics.
2. CHRONIC LYMPHOCYTIC LEUKEMIA
Chronic lymphocyc leukemia (CLL) results from an overproduction of abnormal B cell lymphocytes by the bone marrow. CLL is a slow‐progressing disease but unfortunately does not have a cure. Approximately 20 000 new cases of CLL will be diagnosed in the United States in 2018, equating to 1.2% of all newly diagnosed cancers, and 5‐year survival rates were measured at 84% from 2008 to 2014 (seer.cancer.gov, Table 1). Cells associated with CLL are often quiescent and are found in bone marrow, lymph nodes, the spleen, and circulating blood. The diverse microenvironments in which CLL cells reside require them to be highly adaptive to oxygenation status and molecular signals originating in the stroma. For example, cell signaling in non‐CLL cells in lymph nodes regulates CLL cell proliferation there as opposed to at other sites.16 Additionally, the microenvironment in the bone marrow of CLL patients is characterized by increased expression at the mRNA and protein level of the BCL‐2 family of proteins, crucial antiapoptotic factors that sustain CLL without affecting proliferation17 (and references therein). Eluding apoptosis is a hallmark of CLL cells and underlies their propensity for chemoresistance, thus serving as a promising target for therapy.18 Metabolically, CLL cells are characterized by—among other features—altered glutaminolysis, constitutive hypoxia‐inducible factor 1α (HIF‐1α) expression, and most critically widespread and continuous interactions with the stroma that function to regulate metabolic and redox homeostasis in these cells.
Table 1.
Statistics of leukemia incidence and survival in the United States
| CLL | CML | AML | ALL | ALL—Pediatric | |
|---|---|---|---|---|---|
| Est. new cases in 2018 | 20,000 | 8430 | 19,520 | 5970a | 3000a |
| Percentage of cancers diag. in 2018 | 1.2% | 0.5% | 1.1% | 0.4% | 0.2% |
| 5‐year survival rate | 84% | 67.6% | 27.4% | 71% (All)a; 40% (Adult)b | 85%‐90%c |
Statistics obtained from the National Cancer Institute's Surveillance, Epidemiology, and End Results Program (seer.cancer.gov) unless otherwise noted.
American Society for Clinical Oncology (cancer.net).
Goldstone et al. Blood. 2008, 111, 1827.
Pui et al. Leukemia. 2014, 28, 2336.
A mass spectrometry‐based metabolomic study of B lymphocytes from CLL patients (n = 4) in comparison to healthy controls (n = 5) revealed alterations in glutaminolysis in B‐CLL cells, specifically decreased levels of glutamine and glutamate and increased alanine, a product of glutaminolysis (Figure 2A).19 Further evidence supporting dysregulated glutaminolysis was found in accompanying proteomic experiments which revealed increased levels of glutaminase and glutamine synthetase in CLL cells.19 In comparison to normal lymphocytes, CLL cells have a dysregulated cell growth cycle and are primed for hypoxic response by constitutive low level expression of HIF‐1α20 even in normoxic environments such as circulating blood. Transcript‐level expression of HIF‐1α is variable among CLL patients21 and has been correlated to unfavorable prognoses in a study of 88 CLL patients.22 HIF‐1α has also been implicated as a key regulator of the interactions between CLL cells and the stroma. This molecular interfacing of CLL cells with their dynamic microenvironment confers CLL plasticity, dynamically regulating their metabolism and cell cycle. Studies of the interaction between CLL cells and the stroma often utilize coculturing of patient CLL cells with stromal cell lines. For example, a 2‐hour coculturing of patient CLL cells with NK Tert and other stromal cell lines increased oxygen consumption rate and maximum respiratory capacity and reduced glucose uptake relative to CLL cells alone.17 Several mitochondrial features in CLL cells were unaffected by coculturing, including levels of reactive oxygen species (ROS), outer mitochondrial membrane potential, mtDNA copy number, and expression levels of proteins involved in oxidative phosphorylation.17 In addition, glycolytic flux, as judged by extracellular acidification rate (ECAR), was unaltered by coculturing with NK Tert cells for 2 or 24 hours.17
Figure 2.
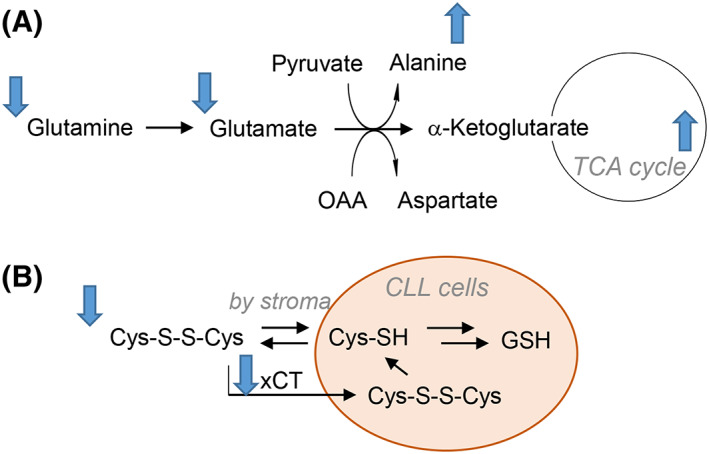
Metabolic alterations observed in CLL cells. A, Flux through glutaminolysis is increased as evidenced by decreased Gln and Glu, elevated ala, and increased expression of glutaminase and glutamine synthetase. Coculturing with stroma increases TCA cycle activity in CLL cells. B‐CLL cells have lower levels of cystine and the xCT cystine transporter; experimental evidence suggests that the stroma can regulate CLL redox homeostasis by reducing the cystine disulfide to cysteine, which enters cells and is utilized for glutathione biosynthesis
Mass spectrometry‐based metabolomics revealed that coculturing increased NAD+ along with levels of metabolites involved in the tricarboxylic acid (TCA) cycle, gluconeogenesis, and de novo synthesis of nucleotides. Furthermore, levels of nucleotide triphosphates in CLL cells were elevated at later time points such as at 24 and 48 hours of coculturing.17 Taken together, these results help to elucidate the manners by which the stroma regulates the energy demands of CLL cells and reveals that such effects are evident as quickly as 2 hours following initial interaction of these cell types. Longer term coculturing has been performed using patient CLL cells cocultured with stromal cell line HS‐5 for 6 days and revealed instead increased glucose uptake and expression of GLUT3 along with elevated glycolytic activity, as judged by increases in the expression of glycolytic enzymes, ATP levels, and ECAR relative to CLL cells alone.23
Redox homeostasis and metabolism are crucial in CLL cells, which possess higher basal levels of reactive oxygen species (ROS) than normal lymphocytes,24 thus rendering these cells more susceptible to oxidative stressors. Recent findings revealed that the synthesis of glutathione (GSH), a key tripeptide antioxidant, in CLL cells is regulated by cells in the bone marrow stroma. Coculturing did not affect protein‐level expression of a rate‐limiting enzyme of GSH synthesis, and additional experiments revealed that CLL cells have reduced cystine content and lower expression of the xCT cystine transporter compared to normal lymphocytes (Figure 2B).25 It was found in this study that the stroma reduces cystine to cysteine which is then taken up by CLL cells for GSH synthesis, revealing a mechanism by which the stroma regulates the redox environment of CLL cells.25 The reliance of CLL cells and other cancer cells on extracellular import of cysteine/cystine to sustain GSH synthesis has been exploited as a treatment option for these cancers. An engineered form of human cysteinase with two activating point mutations was recently shown to deplete the extracellular cysteine/cystine pool and kill CLL cells from patients and a mouse model.26 Cysteinase treatment reduced tumor size in breast and prostate cancer xenografts and prolonged (doubled) survival in a mouse model of CLL.26 Lastly, in patient cells and mouse splenocytes, a treatment of 0.1 μM cysteinase significantly reduced intracellular GSH and increased superoxide levels, disrupting intracellular redox homeostasis in a manner sufficient to induce cell death.26
3. CHRONIC MYELOID LEUKEMIA
Chronic myeloid leukemia (CML), also known as chronic myelogenous leukemia, is a cancer caused by transformed and growth‐unrestricted myeloid cells (Figure 1). In the United States this year, an estimated 8430 new cases of CML will be diagnosed, corresponding to 0.5% of newly diagnosed cancers (seer.cancer.gov). CML is rare in children and has a 5‐year survival rate of 67.6% in the United States (seer.cancer.gov, Table 1). CML is marked by a translocation between chromosomes 9 and 22, termed the Philadelphia chromosome, which results in the genetic fusion of the BCR and ABL genes, leading to the expression of cytosolic Bcr‐Abl kinase. Bcr‐Abl is a constitutively active tyrosine kinase whose presence leads to sustained activation of signaling involving the MAPK,27 JAK/STAT,28 MYC, RAS,29 and PI3K30 axes (reviewed in Cilloni and Saglio31). In addition, Bcr‐Abl increases the rate of cell proliferation and interferes with cell cycle signaling and quality control mechanisms, particularly those involved in DNA repair, allowing for rapid growth and propagation of mutations.32
Treatments for CML often utilize tyrosine kinase inhibitors (TKIs) to target the Abl kinase ATP‐binding domain, and although they are largely successful at inducing a remission state, TKIs are not curative for CML. Commonly used TKI therapies for CML include imatinib mesylate (marketed as Gleevec) and subsequent generation drugs nilotinib and dasatinib. The second‐generation drugs provide faster responses and are more potent but do not affect the overall survival rate for CML patients compared to first‐generation imatinib.33 Third‐generation drug ponatinib provides a treatment option for CML patients with the T315I mutation of Bcr‐Abl,34 but very likely, there are other mutations and other molecular mechanisms not yet identified that contribute to inefficacy in TKI compounds. Resistance to TKI therapy is a significant challenge in treatment of the disease; thus, it is crucial to understand the full picture of Bcr‐Abl's far‐reaching effects in order to better target future treatments.
Bcr‐Abl increases glucose transport and renders CML cells more glycolytic than their normal counterparts,35 promoting the Warburg effect that is commonly seen in many cancers. Imatinib has been shown to decrease flux through glycolysis through both reduction in glucose uptake and inactivation of glycolytic enzymes.36, 37 In one study, a continuous exposure of K562, an erythroleukemia CML cell line, to imatinib lead to inhibition of glycolysis and induction of autophagy by mechanisms involving activation of AMPK and suppression of S6K1.38
In addition to investigations of glycolysis, several studies have aimed to develop a more comprehensive cellular context for how CML cells differ metabolically from their healthy counterparts and to which degree the various TKI treatments recapitulate the phenotype of noncancerous cells. In one such study, the plasma of newly diagnosed CML patients before and after TKI treatment was compared to the plasma of age‐matched healthy control patients using gas chromatography‐mass spectrometry‐based metabolomics.39 A comparison of newly diagnosed patient (prior to TKI exposure, n = 26) plasma to the plasma of healthy controls (n = 26) revealed nine metabolites significantly altered: lactate, isoleucine, glycine, glucose, galactose, and myo‐inositol elevated in CML; glycerol, myristate, and sorbitol decreased.39 TKI therapy recipients were classified using clinical criteria as being resistant (n = 26) or sensitive (n = 26) to therapy.39 In the plasma of these patients, myristate and glycerol emerged as potential biomarkers for TKI responsiveness, both elevated in the sensitive CML group.
In a wider metabolomic approach, leukocytes and plasma of CML patients were obtained at the time of diagnosis, following hydroxyurea (HU) treatment (a common first cytoreductive step), and following subsequent TKI treatment using imatinib, nilotinib, or dasatinib.40 Principal component analysis (PCA) revealed a clustering of newly diagnosed patients with the samples obtained following HU treatment. This group was separated from a clustering of patient samples following TKI to healthy control samples, suggesting a metabolic rewiring induced by TKIs that render the cells more phenotypically similar to non‐CML leukocytes. The PCA trend existed in the patient plasma as well but was more evident in the leukocytes. A prominent theme of the findings in this study is that measurable metabolic disruptions that occur in the newly diagnosed patients in comparison to healthy controls are normalized, though not necessarily fully, by TKI treatment.40 For example, such conclusions were observed for hexose phosphate content in cells (increased in CML), hexose content in plasma (low in CML), decreased late glycolysis intermediate phosphoglycerate (decreased in CML), and leukocyte and plasma amino acid levels (increased and decreased, respectively, in CML).40 In a comparison of the three TKIs used, samples from dasatinib‐treated patients were found to cluster apart from the other TKIs, perhaps reflective of the known higher incidence this drug has for off‐target effects.
Resistance to imatinib and other TKIs, which occurs in a minority of patients, is believed to be a consequence of other mutations in the Abl domain that affect drug binding. As a result, alternative therapies for CML continue to be explored. One such effort, reported earlier this year, utilizes ivermectin, an antiparasitic compound. Ivermectin induces autophagy in breast cancer tumors by interfering with the mTOR/Akt pathway,41, 42 and has been shown to induce cell death in AML cells via chloride‐dependent membrane hyperpolarization and increased ROS.43 This study compared CML cell line K562 to primary CML cells and normal bone marrow cells, the latter two both CD‐34+ (ie, expressing a cell surface marker indicating stemness) and found that ivermectin induced caspase‐dependent apoptosis in CML but not normal hematopoietc cells via mitochondrial dysfunction and oxidative stress.44 Molecular signaling propagated by kinases in the AKT/mTOR pathway was reduced following ivermectin treatment, and increases in levels of superoxide and total intracellular ROS were both observed. Additionally, ivermectin was found to synergize with TKIs nilotinib and dasatinib to increase the amount of CML apoptosis.44 TKIs efficiently target differentiated cells, allowing leukemic stem cells to persist, thus further motivating the development of innovative treatment approaches for CML. A recent study utilized MS‐based metabolomics to profile CD‐34+ cells versus CD‐34− (differentiated) cells from four CML patients.45 CD‐34+ cells were marked by increased fatty acid oxidation (FAO) as illustrated by decreased levels of free fatty acids, increased acylcarnitine content, and increased utilization of [U‐13C] palmitate into the TCA cycle and amino acids (Figure 3).45 In addition, decreased lactate levels in CD‐34+ cells suggested a greater fraction of pyruvate carbons being utilized in the TCA cycle. Indeed, [U‐13C] glucose tracing revealed that although no changes were observed in pyruvate dehydrogenase (responsible for pyruvate decarboxylation and the formation of acetyl‐CoA), the activity of anaplerotic enzyme pyruvate carboxylase (conversion of pyruvate to oxaloacetate) was increased in CD‐34+ CML cells.45 Further experiments utilized CD‐34+ cells from control (non‐CML) patients to confirm via palmitate and glucose tracing that the observed increases in anaplerosis and oxidative metabolism in CD‐34+ CML cells are in fact a unique phenotype and are not reproducible in control CD‐34+ cells absent of CML.45 Such thorough studies of the fine details involved in how CML reprograms cell metabolism, both in stem and differentiated cells, are crucial to identify new avenues for selective and efficacious treatments, particularly for patients that are insensitive to TKI therapy.
Figure 3.
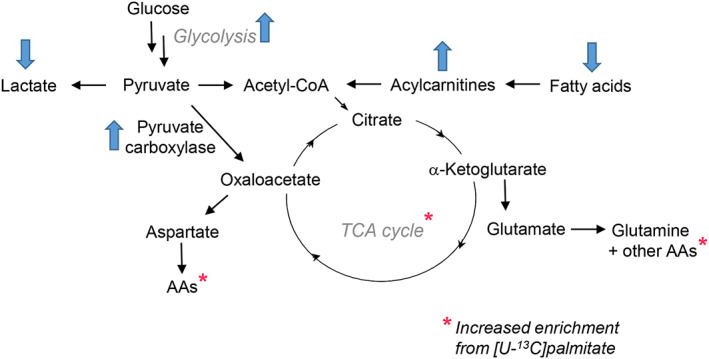
Metabolic alterations observed in CD‐34+ CML cells: Increased fatty acid oxidation leads to elevated levels of acylcarnitines and increased enrichment of TCA cycle metabolites and amino acids from stable isotope labeled palmitate. TCA flux is also increased via the enhanced activity of anaplerotic enzyme pyruvate carboxylase
4. ACUTE MYELOID LEUKEMIA
Acute myeloid leukemia (AML) is a hematological cancer that develops specifically in the myeloid lineage of the hematopoietic cellular hierarchy and results in the accumulation of immature myeloblasts that have a blockade in downstream cellular differentiation. An estimated nearly 20 000 patients were diagnosed in 2017 (seer.cancer.gov, Table 1). While great strides have been made during the last half century to treat AML thus improving survival rates, it remains an aggressive malignancy with poor prognoses of less than 30% 5‐year relative survival rates for all patients (seer.cancer.gov). While outcomes are much better for patients younger than 60 years with cure rates near 35% to 40%, prognoses for patients over the age of 60 are particularly poor in which only 5% to 15% of patients are in remission.46 These poor prognostic rates arise due to lack of response, or high rate of relapse, to the initial therapy.47
AML is heterogeneous and classified principally by cytological and genetic characteristics. While patient blood smear assessments and characterization of cell‐specific markers by flow cytometry have been historically used to diagnose AML, the advent and continuous innovation of gene and transcript sequencing technologies has expanded classification of molecular genetic subgroups.48 An analysis of 200 AML patient genomes enabled the functional categorization of several mutations thus detailing how multivariate data can be organized and potentially translated into new therapeutic strategies.49 These categories include impaired cell signaling, chromosomal dynamics, epigenetic homeostasis, and deregulation of both the transcription and splicing processes. Given the dense connectivity of biological networks, it is not surprising that the deregulated processes identified on the genetic tier also have significant metabolic components.
Metabolomic approaches have been successfully applied to elucidate metabolic programs of AML cells both basally and in response to drug treatments, as well as provide diagnostic and prognostic biomarkers (Figure 1 for summary). Wang and colleagues applied NMR to characterize features in 183 patients with AML compared to 232 age‐matched and gender‐matched healthy controls to identify a number of distinct metabolic features of disease.50 This study detailed distinct metabolic pathways pertinent to energy metabolism including glycolysis, TCA cycle, amino acid (AA), and fatty acid (FA) metabolism. The steady‐state levels of these compounds were also able to distinguish between various disease burdens, highlighting the value of metabolomics as an analytical readout. Additional work expanded on these findings with the use of GC‐MS and provided preliminary evidence that lipid levels can also potentially be used to distinguish between leukemia subtypes.51, 52 FAs are released from membrane lipids by lipases, then conjugated first to CoA by acyl‐CoA ligases, and then to carnitine by carnitine palmitoyltransferase 1 (CPT1) at the outer membrane of the mitochondria. Upon transport into the mitochondria, they undergo FAO and generate acetyl‐CoA to fuel the TCA cycle. Interestingly, the FA transporter CD36 has been suggested as a prognostic marker in AML.53 A unique population of CD36+ leukemic stem cells (LSCs) identified in murine gonadal adipose tissue was reliant on FAO and was similar to both CD36+ CML and AML cells in humans.54 In addition, coculture of AML blasts and bone marrow adipocytes increased lipolysis‐FA transport axis that was abrogated with inhibition of CPT1A.55 These metabolic adaptations protected LSCs from chemotherapy54 and are also exploited in additional models of AML drug resistance.56 Furthermore, AML cell populations that are not enriched for LSCs but are drug resistant, such as those resistant to cytarabine, also upregulate CD36, fatty acid oxidation, and overall mitochondrial activity.57 Taken together, these studies highlight a central role for mitochondria in AML development and drug resistance, further supported by the finding that inhibiting mitochondrial translation can serve as a therapeutic strategy in treating AML.58
Molecular subtype can also directly contribute to metabolic programs in AML. One such category of mutations seen in roughly 20% of AML occurs in isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2, which represent the cytosolic and mitochondrial isoforms, respectively). These NADP(+)‐dependent enzyme isoforms catalyze the oxidative decarboxylation of isocitrate to produce α‐ketoglutarate (αKG) and CO2 and play integral roles in the TCA cycle as well as the maintenance of cytosolic redox homeostasis, among other functions. Mutations in these genes result in the dysregulation of cell maintenance and proliferation, and ultimately contribute to tumorigenesis in large part due to a gain of function that enables IDH to convert αKG into (R)‐2‐hydroxyglutarate (R‐2HG),59, 60 the enantiomer of (S)‐2HG produced by lactate dehydrogenase or malate dehydrogenase under hypoxic or low pH conditions (Figure 4A).61, 62, 63 Classified as a central oncometabolite, R‐2HG is a competitive inhibitor of αKG‐dependent dioxygenases such as ten‐eleven translocation 2 protein (TET2), which converts 5‐methylcytosine to 5‐hydroxymethylcytosine in the process of DNA methylation (reviewed in Parker and Metallo64 and Sciacovelli and Frezza65). Accumulation of R‐2HG can thus destabilize the epigenetic and metabolic landscape of hematopoietic precursor cells, poising them for leukemic transformation.66, 67, 68 Given the active role IDH mutations play in leukemic development and progression, a number of small molecule inhibitors of IDH have been developed and are currently being tested or have already received FDA approval (more extensively reviewed in Ragon and DiNardo69). In addition, 2HG can alone be used as a prognostic marker in AML patients positive for IDH1/2 mutations,50 or in combination with the abundance of pyruvate, lactate, αKG, and glycerol‐3‐phosphate to predict survival outcomes.70 The implementation of high‐throughput metabolomic technologies in the future will enable the rapid screening of patient samples71, 72 and expand upon the utility of metabolomics in clinical biochemistry.73
Figure 4.
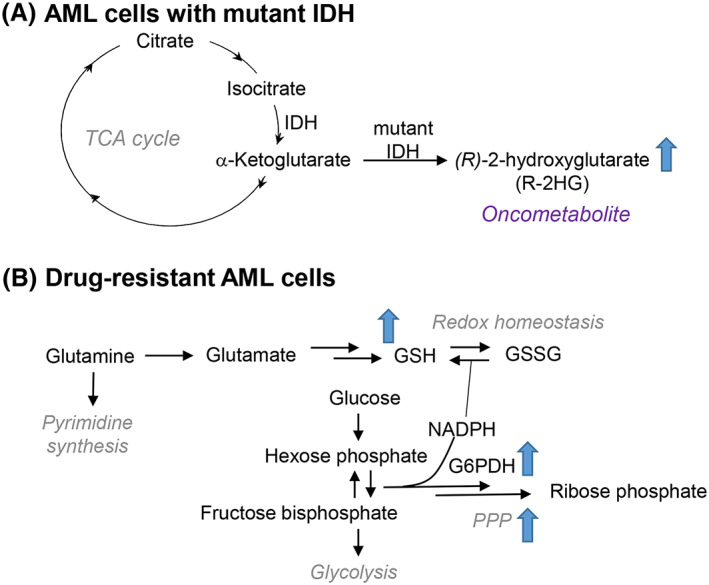
Metabolic features of AML cells. A, Mutant isocitrate dehydrogenase (IDH) generates oncometabolite (R)‐2‐hydroxyglutarate from a‐ketoglutarate. B, Hallmarks of drug‐resistant AML cells include a reliance on glutamine to fuel glutathione (GSH) biosynthesis and pyrimidine synthesis. Additionally, drug‐resistant cells have elevated pentose phosphate pathway (PPP) activity particularly of rate‐limiting enzyme glucose 6‐phosphate dehydrogenase (G6PDH)
Another common set of mutations that predict for poor outcome occur in signaling genes including activating mutations caused by internal tandem duplications (ITD) in FMS‐like tyrosine kinase 3 (FLT3).74 This mutation has become a sought‐after target in the treatment of AML due to the successful development and implementation of TK inhibitors. While inhibition of FLT3 has proven effective in treating AML,75, 76 short‐lived remission rates have prompted subsequent research into possible cellular adaptive mechanisms and have pointed to significant metabolic contributions. Studies into the downstream effects of FLT3 inhibition have determined that these cells are acutely susceptible to oxidative stress, particularly due to accumulation of mitochondrial reactive oxygen species that induce apoptosis.77 As revealed by synthetic lethal shRNA screening in combination with FLT3 inhibition, drug‐resistant cells can upregulate the activity of ataxia telangiectasia mutated (ATM) and its downstream target glucose 6‐phosphate dehydrogenase (G6PD) to activate the pentose phosphate pathway, thus enabling the cells to maintain reduced glutathione at levels sufficient to cope with oxidative stress.77 Indeed, AML cells depend strongly upon glutathione metabolism78, 79 and glutaminolysis to generate glutamate for glutathione synthesis (Figure 4B).80, 81 Glutamine utilization may also be required for pyrimidine synthesis, which represents an additional target for AML.82 Taken together, these studies have defined a viable therapeutic strategy to eliminate drug‐resistant cells by targeting FLT3 signaling in combination with glutamine metabolism and redox balance to treat AML (Gregory bioRxiv 2018).83
5. ACUTE LYMPHOBLASTIC LEUKEMIA
Acute lymphoblastic leukemia (ALL) results from the transformation of hematopoietic B cell or T cell progenitors. ALL is the most common pediatric cancer, accounting for 80% of childhood leukemias.84 Adult‐onset ALL is most prevalent in white males age >70 years (seer.cancer.gov, Table 1). For pediatric B‐ALL, the remission rate is 95% with 5‐year survival in the range of 85% to 90%.85 For adults, remission is achieved in 85% of patients but 5‐year survival is only ~40%86 and decreases to 7% if a relapse occurs.87 Independent of age, relapsed ALL is associated with poor survival rates. Known genetic alterations, including mutations, fusions, deletions, and other modifications, are associated with 50% of adult and ~70% of pediatric ALL cases.88 ALL is a heterogeneous disease marked by reprogrammed metabolism and atypical molecular signaling and, like CML, can be induced by the presence of the Philadelphia chromosome and expression of Bcr‐Abl, particularly in adult patients. We will focus here on the most recent investigations of ALL cell metabolism as fuller pictures of ALL subtypes are recently and individually reviewed (B‐ALL89, 90; T‐ALL91, 92, 93).
All cases of pediatric ALL, along with many adolescent, young adult, and adult cases, are treated with asparaginase (ASNase) therapy as a frontline approach to cytoreduction. It has been known for a half century that asparagine (Asn), normally a nonessential amino acid, is essential to ALL cells due to deficient activity of asparagine synthetase.94 A sharp reduction of the cellular Asn pool using ASNase, which hydrolyzes the Asn amide to generate aspartate and ammonia, sufficiently retards mRNA translation to ultimately induce apoptosis in ALL cells.95 Unfortunately, although ALL cells are particularly reliant on Asn, cross‐reactivity of ASNase with other cell types results in significant side effects to an extent that has limited the use of ASNase in adult ALL cases. To rectify the off‐target effects, other sources and preparations of ASNase are under investigation. For example, a recent study compared retrospectively the standard of care Escherichia coli enzyme with a polyethylene glycol conjugated (PEGylated) version in 122 ALL patients over age 14.96 Major findings included a prolonged half‐life of the PEGylated form, a 2‐month increase in relapse‐free survival time (~11 vs ~9 months with the non‐PEG‐conjugated enzyme) in patients under age 35, but no change in outcomes measured by metrics such as remission rate and survival time.96 The occurrence and the intensity of side effects measured (such as liver and kidney damage) were also comparable between the two ASNase forms.96 Resistance to ASNase therapy can occur through several mechanisms; from a metabolic standpoint, glutamine/glutamate metabolism has attracted attention as these can also serve as precursors for Asn (see Marini et al95 for a recent review on asparaginase therapy in ALL).
Within the context of longer term treatment strategies, nearly all pediatric and adult cases of ALL are treated with methotrexate (MTX). MTX is a potent inhibitor of dihydrofolate reductase, an indispensable folate pathway enzyme required to generate folate cofactors for the synthesis of nucleotides and S‐adenosylmethionine. Thus, MTX confers cytotoxicity by interfering with nucleic acid synthesis along with other molecular mechanisms. MTX is utilized in a variety of cancer types; however, adverse effects are common, and can include acute kidney injury,97 cognitive/memory impairment,98 leukoencephalopathy,99 and myelopathy. As MTX is the subject of at least hundreds of clinical trials, we will highlight here only several of the most recent developments regarding its role in the treatment of ALL with a particular focus on multiple metabolic manipulations used in conjunction.
First, a CRISPR‐Cas9 screen of the HEL (erythroblast‐like) cell line identified formimidoyltransferase cyclodeaminase (FTCD) as being linked to MTX sensitivity.100 FTCD is a crucial enzyme in the histidine degradation pathway, which utilizes tetrahydrofolate. Depletion of genes in this pathway decreased MTX sensitivity—in particular, CRISPR‐Cas9‐induced knockdown of FTCD in CML cells reduced both flux through the histidine catabolic pathway and MTX sensitivity.100 Accordingly, His supplementation was hypothesized to increase MTX sensitivity by further disrupting the folate pathway. As anticipated, tumors grown in mice treated with HEL cells were decreased in size upon combination treatment of MTX + His vs either agent alone; importantly, no adverse effects on other organs were observed up to 15 days.100 Histidine supplementation thus provides an intriguing potential route for facile dietary enhancement of MTX for leukemias.
A separate study investigated the effects of MTX when used in conjunction with mTOR inhibitors for the treatment of B‐ALL. Elevated mTOR activity is associated with poor prognoses in B‐ALL cases, but the effects of mTOR inhibitors when used alongside de novo DNA synthesis inhibitors like MTX or 6‐mercaptopurine (6‐MP) were not clear. Here, Ph+ B‐ALL cell lines BV173 and SUP‐B15 were treated for 48 hours with MTX (30 nM) or 6‐MP (10 μM) in the presence of mTORC inhibitors MLN0128 (100 nM) or rapamycin (10 nM).101 In all cases, mTORC inhibitors protected the cells from killing via MTX or 6‐MP.101 Interestingly, TKI inhibitor dasatinib also protected cells from the effects of MTX via decreased mTOR signaling.101 From a mechanistic viewpoint, inhibition of mTORC1, known to slow cell proliferation, is believed to render cells less sensitive to MTX and other agents that interfere with DNA/RNA synthesis and quality control.101
Returning to the unique features of ALL metabolism, another approach to ALL treatment utilizes glucocorticoids such as dexamethasone and prednisolone to inhibit cell proliferation by impeding glycolysis, upon which ALL cells are highly reliant to produce ATP and NAD+. In general, glucocorticoids interfere with glycolysis by mechanisms such as blocking glucose uptake and reducing the expression of glycolytic enzymes like pyruvate kinase; however, these drugs are not sufficient to induce remission when used alone. Cancer cells exposed to glucocorticoids rewire away from glycolysis toward mitochondrial metabolism and processes like autophagy in order to meet energetic demands for growth and proliferation and thus become more sensitive to drugs that target these aspects of cell metabolism.
One such recent study profiled CCRF‐CEM clones exposed to dexamethasone alone or in combination with etoposide, an inhibitor of topoisomerase 2102 that disrupts mitochondrial homeostasis,103 and other anticancer drugs. Three phenotypic subtypes emerged: (1) cells that were resistant to dexamethasone independent of etoposide treatment, (2) cells where an additive effect was observed (termed CEM‐ADD), (3) cells responsive to dexamethasone but with no observed synergy in the presence of etoposide (termed CEM‐NON).104 Reduced glycolysis was measured in both CEM‐ADD and CEM‐NON, as judged by decreased lactate levels, decreased mRNA expression of glycolytic genes hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA), and decreased protein expression of HK2.104 CEM‐ADD cells exposed to dexamethasone for 48 hours followed by treatment for 72 hours with oligomycin, an inhibitor of ATP synthase, died in greater numbers than untreated cells and cells exposed to either agent alone.
In a separate study, the effects of antibiotic tigecycline were investigated for cytotoxicity against ALL cells. Using pediatric ALL cell lines,3 primary ALL CD34+ progenitors and lymphocytes, normal bone marrow CD34+ cells, and peripheral blood mononuclear cells (PBMCs), a synergistic effect of tigecycline with doxorubin and (separately) vincristine was identified.105 In the CCRF‐CEM cell line, primary ALL 34+ progenitors, and primary ALL lymphocytes, mitochondrial respiration was adversely impacted by 2 hours following treatment with tigecycline alone. Glycolysis increased up to 6 hours, perhaps as compensation, but sharply decreased thereafter (final time point at 24 hours). All levels of tigecycline tested (5‐20 μM) for the three cell populations resulted in decreased ATP levels (luminescence assay), increased ROS (DCF assay/flow cytometry), increased content of oxidized DNA (8‐hydroxyguanine by ELISA), and increased protein oxidation (protein carbonyl content by ELISA).105 Interestingly, no difference was observed in mitochondrial membrane potential at baseline when comparing the primary ALL CD34+ progenitors, ALL lymphocytes, normal bone marrow CD34+ progenitors, and PBMCs; however, in these experiments, the primary ALL cells were found to have greater mitochondrial mass, reserve respiratory capacity, and ATP. Finally, a xenograft mouse model (CCRF‐CEM cells) found that the combinations of tigecycline with vincristine and (separately) with doxorubicin were most effective in limiting tumor size within the range of the 14 days tested.
Muschen and colleagues have recently reported new insights into the regulation of glucose metabolism in normal and malignant B cells, particularly in contrast to malignant myeloid cells. In one such study, the transcription factors PAX5 and IKZF1, which regulate genes involved in glucose uptake and metabolism, were identified as limiting ATP production via glycolysis and metabolite flow into the TCA cycle in manners that protect against transformation.106 Lesions in the PAX5 and IKZF1 genes and/or altered expression are found in the majority of adult and pediatric B‐ALL cases, leading to a loss of gatekeeper functionality that results in enhanced glucose metabolism and ATP generation (Figure 5). To illustrate, the introduction of Bcr‐Abl in mice with wild‐type PAX5 did not alter glucose uptake or utilization (as judged by ATP generation), but in mice haploinsufficient for PAX5, Bcr‐Abl increased glucose uptake 50‐fold and ATP levels 25‐fold.106 The response to glucocorticoids, which are effective for B‐lymphoid ALL but not in myeloid leukemias, was linked to glucose metabolism via the NR3C1 protein, which is increased 6‐fold to 20‐fold in B‐lymphoid cells vs myeloid leukemia cells.106 NR3C1 is positively regulated by IKZF1 and PAX5, and several experiments revealed that modulation of the levels of these transcription factors correlated with the response to glucocorticoid prednisolone.106
Figure 5.
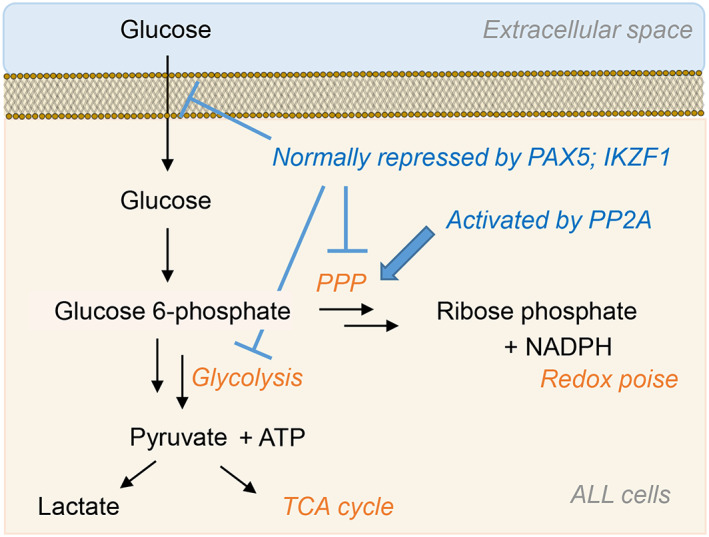
Glucose metabolism in ALL cells. Gatekeeper transcription factors PAX5 and IKZF1 limit ATP production through glycolysis to prevent transformation. Together, genetic lesions and Bcr‐Abl rewire metabolism to allow for rapid glycolytic flux. PP2A is required by ALL cells and activates the PPP to sustain redox homeostasis alongside rapid proliferation
In a subsequent report, serine/threonine phosphatase 2A (PP2A) was revealed to be essential to B cell tumors with a primary function of shifting glucose metabolism from glycolysis to the PPP to generate reducing equivalents (ie, NADPH) that counteract oxidative stress (Figure 5).107 Normal B cells possess low PPP activity due to the repressed expression of PPP enzymes by PAX5 and IKZF1 (Figure 5).107 Loss of PP2A in B cell leukemic cells increased glucose utilization and glycolysis products ATP and lactate; such changes were not observed in myeloid leukemic cells, and mitochondrial metabolism was unchanged in both cell types.107 Deletion of PP2A in B‐lymphoid cells decreased flux through the PPP, which also was not observed in myeloid cells. The PP2A deletion was found to increase phosphorylation of 6‐phosphofructokinase/fructose 2,6‐bisphosphatase 2 (Pfkfb2) at Ser483, enhancing its kinase activity (at the expense of its phosphatase activity), thereby promoting glycolysis over the PPP and illustrating a critical metabolic switch regulated by PP2A.107 The effects of this deletion were reversed by overexpression of fructose 2,6‐bisphosphate phosphatase 2 TIGAR in B‐ALL cells lacking PP2A, as judged by the restoration of NADPH/NADP ratios. Overall, these and accompanying findings identify a unique reliance of malignant B cells on the PPP and thus define a vulnerable metabolic pathway for therapeutic intervention.107
Activation of protooncogene MYC is well documented for various cancers and, through transcriptional regulation, increases metabolic activity through glycolysis and glutaminolysis (reviewed in Hsieh et al108). Notch signaling upregulates MYC, and consequently the PI3K/Akt pathway,109 and approximately half of T‐ALL patients possess activating mutations in NOTCH1.110 Recently, in the context of ALL, resistance to γ‐secretase inhibitors (GSIs), used to impede Notch signaling, was found to be associated with the loss of tumor suppressor PTEN.111 Treatment of PTEN+/+ ALL mice with GSI DBZ revealed using MS‐based metabolomics increases of glucose and its downstream metabolites, both in glycolysis and the PPP, in comparison to untreated PTEN+/+ ALL mice, and DBZ treated and untreated PTEN−/− ALL mice.111 Leukemic mice lacking PTEN had higher lactate levels and a restored glucose metabolic phenotype (in comparison to PTEN+/+ untreated) independent of DBZ exposure. Growth and proliferation of DND1, a PTEN+/+ cell line with mutated NOTCH1, was hampered by DBZ and phenotypically rescued by treatment with either methyl pyruvate or dimethyl αKG, membrane permeable sources of TCA cycle carbons.111 Finally, a comparison of stable isotope tracing using [13C6]‐glucose or [13C5]‐glutamine in NOTCH1‐induced primary ALL cells revealed that glutaminolysis is the major source of carbons for mitochondrial metabolism; thus, this pathway is a promising target for metabolic manipulation in the treatment of Notch‐implicated T‐ALL cases.111, 112 However, this reliance on glutamine‐dependent anaplerosis has been observed to decrease in cells that have acquired drug resistance.56 These metabolic changes occur concomitantly with increased glycolysis and altered FAO, further evidencing the role of metabolism in responses to environmental stimuli. Interestingly, CCRF‐CEM cells are able to acquire resistance to danorubicin in part due to increased expression of DNAJC15, which encodes for the HSP40 family member methylation‐controlled J protein (MCJ). This protein has been found to modulate mitochondrial respiration via the disruption of mitochondrial supercomplexes,113, 114 thereby decreasing mitochondrial utilization and explaining a shift towards glycolysis in drug resistance.
In another metabolic intervention approach to targeting ALL cells, thiopurine compounds, such as 6‐mercaptopurine (also termed purinethol) and 6‐thioguanine (or tioguanine), are utilized in the treatment of ALL for their ability to stall DNA replication and induce cell death. Such thiopurines have sufficient structural similarities to hypoxanthine and guanine, respectively, to be converted into nucleotides and incorporated into DNA and RNA; however, they impede protein machinery involved in processes like repair, replication, and translation, ultimately causing cytotoxicity. A recent study investigated ALL resistance to thiopurines by clonal selection of ALL cells resistant to each of the two thiopurines described and found through exome sequencing that the insensitivity of such cells to the thioguanine nucleotides occurs in the presence of mutated hypoxanthine‐guanine phosphoribosyltransferase (HGPRT), the enzyme responsible for converting hypoxanthine to inosine monophosphate and guanine to guanosine monophosphate.115
In total, ALL tumor cells are characterized by a high metabolic rate that implies a reliance on amino acids (specifically asparagine) and a robust supply and utilization of glucose through glycolysis and the PPP. Recent efforts have uncovered the metabolic rewiring that underlies transformation in these cells along with features of ALL cells that distinguish their biochemistry from their myeloid counterparts, providing new opportunities for treating this disease in a selective and effective manner. A challenging aspect of ALL treatment, not unlike for other cancers, is the potential and likely presence of as‐yet unknown genetic mutations associated with the disease etiology and/or impacting treatment response.
6. CONCLUSION—TUMOR MICROENVIRONMENT
The role of the microenvironment in harboring and promoting the growth and development of leukemic cells is becoming an increasingly appreciated aspect of hematological cancer research. The characteristics of the bone marrow depend upon a complex network of cells of hematopoietic and mesenchymal lineages, cellular signaling through chemokine receptors and adhesion molecules, vasculature, and the dense protein meshwork of the extracellular matrix (ECM) (see Tabe and Konopleva116 for a detailed review). This microenvironment provides cues for cellular homing, quiescence, self‐renewal, differentiation, and proliferation, all of which become dysregulated in LSCs and leukemia cells. One aspect of the niche with substantial metabolic implications is oxygen tension, as hypoxia is a critical regulator of HIF‐1α activity and subsequent metabolic programs. Indeed, hypoxia can contribute to chemoresistance in AML cells117, 118 and potentially serve as a prognostic marker.117 In addition, lower oxygen tension can influence interactions between transformed and stromal cells by upregulating the expression of important adhesion molecules119, 120 to attract cancer cells to hypoxic environments. As age is an important risk factor for leukemic ongogenesis (seer.cancer.gov), it is important to note the age‐associated increase in bone marrow adipocytes, which can influence oncogenesis through chemokine signaling, as well providing fatty acid substrates for FAO,121, 122 a key metabolic adaptation observed in some leukemic cells and discussed earlier in this review. Additional niche metabolites can also contribute to HSC and cancer cell behavior, with studies looking at the levels of arginine123 and asparagine124 on leukemic cell response. Valine has also been recently revealed as an important effector of the bone marrow microenvironment,125 although more work will be required to elucidate implications for oncogenesis.
As the microenvironment promotes oncogenesis, transformed cells also alter the surrounding microenvironment into a pro‐oncogenic niche and contribute to adaptive oncogenesis.126 The adaptations of LSCs enable these cells to outcompete HSCs and take control of the bone marrow niche. AML cells can transform the niche into a proleukemic environment by promoting osteogenic differentiation of mesenchymal stem cells127 or by secreting exosomes.128 Leukemic blasts can also reprogram surrounding adipocytes to upregulate lipase activity thereby providing substrates for FAO.55 Adipocytes are also a source of proinflammatory cytokines,129 some of which can alter the balance between normal and pathologic hematopoietic cell behavior.130, 131, 132
These effectors may also influence the surrounding ECM, which can harbor chemosignaling and mechanosignaling cues, the latter of which are based upon ECM stiffness and architecture.133, 134 Indeed, three‐dimensional mechanosignaling can influence leukemic cell response to therapy.135 ECM components tenascin C136 and laminins137 contribute to hematopoietic cycling and homing to the bone marrow. ECM remodeling through the activities of matrix metalloproteinases has been shown to play a role in the pathogenesis of multiple myeloma.138 The resulting architecture, as well as the individual components present in the bone marrow niche, affects tissue stiffness and mechanosignaling through axes such as YAP/TAZ.139 This transduction pathway is also tied to nutrient signaling140 and nucleotide metabolism.141 Interestingly, while YAP/TAZ is mediated by mevalonate metabolism, the cholesterol‐lowering drug lovastatin can inhibit LSCs in coculture with MSCs,142, 143 highlighting a possible connection between the ECM and LSC metabolism.
In summary, the investigations reviewed here aim not only to elucidate the fine tuning of leukemic cell metabolism particularly in contrast to each type's healthy counterpart cells but also to expand on the larger context of how the leukemia subtypes differ. Such a detailed understanding will improve treatment approaches and provide new technical approaches to investigate metabolism at the levels of genes, proteins, and metabolites. Although the ECM in particular has been challenging to study from a systems level due to inherent difficulties in quantifying insoluble proteins by liquid chromatography and mass spectrometry, innovations in sample preparation and handling144, 145 will help to reveal unique holistic changes to the bone marrow microenvironment in the future. While current therapeutic development has centered upon direct leukemic cell targeting, expanding this focus to include ECM dynamics in combination with metabolic reprogramming during oncogenesis holds promise for the development of new classes of leukemia therapies.
CONFLICT OF INTEREST
Although unrelated to the contents of the manuscript, the authors disclose that AD and TN are members of Omix Technologies, Inc.
AUTHORS' CONTRIBUTION
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, T.N. and J.A.R. F.M.L.; Writing ‐ Original Draft, T.N. and J.A.R. F.M.L.; Writing ‐ Review & Editing, T.N., A.D., and J.A.R. F.M.L.; Visualization, T.N., A.D., and J.A.R. F.M.L.; Supervision, J.A.R. F.M.L.; Funding Acquisition, T.N. and A.D. F.M.L.
ACKNOWLEDGEMENTS
AD is a recipient of the Boettcher Webb‐Waring Biomedical Research Award. TN is a recipient of US National Institutes of Health grant NIH T32 HL007171. The authors are grateful to Dr. Courtney Jones (University of Colorado Denver—Department of Hematology) for helpful discussions and feedback.
Nemkov T, D'Alessandro A, Reisz JA. Metabolic underpinnings of leukemia pathology and treatment. Cancer Reports. 2019;2:e1139. 10.1002/cnr2.1139
REFERENCES
- 1. Fiehn O. Metabolomics—the link between genotypes and phenotypes. Plant Mol Biol. 2002;48(1–2):155‐171. [PubMed] [Google Scholar]
- 2. Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17(7):451‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patti GJ, Yanes O, Siuzdak G. Innovation: metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13(4):263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takubo K, Goda N, Yamada W, et al. Regulation of the HIF‐1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391‐402. [DOI] [PubMed] [Google Scholar]
- 5. Takubo K, Nagamatsu G, Kobayashi CI, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ito K, Carracedo A, Weiss D, et al. A PML‐PPAR‐delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilkinson AC, Yamazaki S. The hematopoietic stem cell diet. Int J Hematol. 2018. [DOI] [PubMed] [Google Scholar]
- 8. Pietras EM, Reynaud D, Kang YA, et al. Functionally distinct subsets of lineage‐biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell. 2015;17(1):35‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. 2017;130(15):1693‐1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pietras EM, Mirantes‐Barbeito C, Fong S, et al. Chronic interleukin‐1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self‐renewal. Nat Cell Biol. 2016;18(6):607‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karigane D, Takubo K. Metabolic regulation of hematopoietic and leukemic stem/progenitor cells under homeostatic and stress conditions. Int J Hematol. 2017;106(1):18‐26. [DOI] [PubMed] [Google Scholar]
- 12. Testa U, Labbaye C, Castelli G, Pelosi E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp Hematol. 2016;44(7):540‐560. [DOI] [PubMed] [Google Scholar]
- 13. Yu VWC, Yusuf RZ, Oki T, et al. Epigenetic memory underlies cell‐autonomous heterogeneous behavior of hematopoietic stem cells. Cell. 2016;167(5):1310‐22.e17. [DOI] [PubMed] [Google Scholar]
- 14. Jordan CT. The leukemic stem cell. Best Pract Res Clin Haematol. 2007;20(1):13‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herault A, Binnewies M, Leong S, et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature. 2017;544(7648):53‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Herndon TM, Chen SS, Saba NS, et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia. 2017;31(6):1340‐1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vangapandu HV, Ayres ML, Bristow CA, et al. The stromal microenvironment modulates mitochondrial oxidative phosphorylation in chronic lymphocytic leukemia cells. Neoplasia. 2017;19(10):762‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Daniel C, Mato AR. BCL‐2 as a therapeutic target in chronic lymphocytic leukemia. Clin Adv Hematol Oncol. 2017;15(3):210‐218. [PubMed] [Google Scholar]
- 19. Mayer RL, Schwarzmeier JD, Gerner MC, et al. Proteomics and metabolomics identify molecular mechanisms of aging potentially predisposing for chronic lymphocytic leukemia. Mol Cell Proteomics. 2018;17:290‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koczula KM, Ludwig C, Hayden R, et al. Metabolic plasticity in CLL: adaptation to the hypoxic niche. Leukemia. 2016;30(1):65‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valsecchi R, Coltella N, Belloni D, et al. HIF‐1alpha regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood. 2016;127(16):1987‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kontos CK, Papageorgiou SG, Diamantopoulos MA, et al. mRNA overexpression of the hypoxia inducible factor 1 alpha subunit gene (HIF1A): an independent predictor of poor overall survival in chronic lymphocytic leukemia. Leuk Res. 2017;53:65‐73. [DOI] [PubMed] [Google Scholar]
- 23. Jitschin R, Braun M, Qorraj M, et al. Stromal cell‐mediated glycolytic switch in CLL cells involves Notch‐c‐Myc signaling. Blood. 2015;125(22):3432‐3436. [DOI] [PubMed] [Google Scholar]
- 24. Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS‐generating anticancer agents. Blood. 2003;101(10):4098‐4104. [DOI] [PubMed] [Google Scholar]
- 25. Zhang W, Trachootham D, Liu J, et al. Stromal control of cystine metabolism promotes cancer cell survival in chronic lymphocytic leukaemia. Nat Cell Biol. 2012;14(3):276‐286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cramer SL, Saha A, Liu J, et al. Systemic depletion of L‐cyst(e) ine with cyst(e) inase increases reactive oxygen species and suppresses tumor growth. Nat Med. 2017;23(1):120‐127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cortez D, Reuther G, Pendergast A. The Bcr‐Abl tyrosine kinase activates mitogenic signaling pathways and stimulates G1‐to‐S phase transition in hematopoietic cells. Oncogene. 1997;15:2333‐2342. [DOI] [PubMed] [Google Scholar]
- 28. Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR‐Abl‐expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol. 1997;159(10):4720‐4728. [PubMed] [Google Scholar]
- 29. Pendergast AM, Quilliam LA, Cripe LD, et al. BCR‐ABL‐induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB‐2 adaptor protein. Cell. 1993;75(1):175‐185. [PubMed] [Google Scholar]
- 30. Skorski T, Bellacosa A, Nieborowska‐Skorska M, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI‐3k/Akt‐dependent pathway. EMBO J. 1997;16(20):6151‐6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cilloni D, Saglio G. Molecular pathways: BCR‐ABL. Clin Cancer Res. 2012;18(4):930‐937. [DOI] [PubMed] [Google Scholar]
- 32. Welner RS, Amabile G, Bararia D, et al. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell. 2015;27:671‐681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Radich JP, Mauro MJ. Tyrosine kinase inhibitor treatment for newly diagnosed chronic myeloid leukemia. Hematol Oncol Clin North Am. 2017;31(4):577‐587. [DOI] [PubMed] [Google Scholar]
- 34. Breccia M, Abruzzese E, Castagnetti F, et al. Ponatinib as second‐line treatment in chronic phase chronic myeloid leukemia patients in real‐life practice. Ann Hematol. 2018;97(9):1577‐1580. [DOI] [PubMed] [Google Scholar]
- 35. Bentley J, Walker I, McIntosh E, Whetton AD, Owen‐Lynch PJ, Baldwin SA. Glucose transport regulation by p210 Bcr‐Abl in a chronic myeloid leukaemia model. Br J Haematol. 2001;112(1):212‐215. [DOI] [PubMed] [Google Scholar]
- 36. Boren J, Cascante M, Marin S, et al. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J Biol Chem. 2001;276(41):37747‐37753. [DOI] [PubMed] [Google Scholar]
- 37. Gottschalk S, Anderson N, Hainz C, Eckhardt SG, Serkova NJ. Imatinib (STI571)‐mediated changes in glucose metabolism in human leukemia BCR‐ABL‐positive cells. Clin Cancer Res. 2004;10(19):6661‐6668. [DOI] [PubMed] [Google Scholar]
- 38. Hirao T, Yamaguchi M, Kikuya M, Chibana H, Ito K, Aoki S. Altered intracellular signaling by imatinib increases the anti‐cancer effects of tyrosine kinase inhibitors in chronic myelogenous leukemia cells. Cancer Sci. 2018;109(1):121‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang B, Wang C, Xie Y, Xu L, Wu X, Wu D. Monitoring tyrosine kinase inhibitor therapeutic responses with a panel of metabolic biomarkers in chronic myeloid leukemia patients. Cancer Sci. 2018;109:777‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Karlikova R, Siroka J, Friedecky D, et al. Metabolite profiling of the plasma and leukocytes of chronic myeloid leukemia patients. J Proteome Res. 2016;15(9):3158‐3166. [DOI] [PubMed] [Google Scholar]
- 41. Wang K, Gao W, Dou Q, et al. Ivermectin induces PAK1‐mediated cytostatic autophagy in breast cancer. Autophagy. 2016;12(12):2498‐2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dou Q, Chen HN, Wang K, et al. Ivermectin induces cytostatic autophagy by blocking the PAK1/Akt Axis in breast cancer. Cancer Res. 2016;76(15):4457‐4469. [DOI] [PubMed] [Google Scholar]
- 43. Sharmeen S, Skrtic M, Sukhai MA, et al. The antiparasitic agent ivermectin induces chloride‐dependent membrane hyperpolarization and cell death in leukemia cells. Blood. 2010;116(18):3593‐3603. [DOI] [PubMed] [Google Scholar]
- 44. Wang J, Xu Y, Wan H, Hu J. Antibiotic ivermectin selectively induces apoptosis in chronic myeloid leukemia through inducing mitochondrial dysfunction and oxidative stress. Biochem Biophys Res Commun. 2018;497(1):241‐247. [DOI] [PubMed] [Google Scholar]
- 45. Kuntz EM, Baquero P, Michie AM, et al. Targeting mitochondrial oxidative phosphorylation eradicates therapy‐resistant chronic myeloid leukemia stem cells. Nat Med. 2017;23(10):1234‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. New Eng J Med. 2015;373(12):1136‐1152. [DOI] [PubMed] [Google Scholar]
- 47. Dinner SN, Giles FJ, Altman JK. New strategies for relapsed acute myeloid leukemia: fertile ground for translational research. Curr Opin Hematol. 2014;21(2):79‐86. [DOI] [PubMed] [Google Scholar]
- 48. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391‐2405. [DOI] [PubMed] [Google Scholar]
- 49. Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Eng J Med. 2013;368(22):2059‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang J‐H, Chen W‐L, Li J‐M, et al. Prognostic significance of 2‐hydroxyglutarate levels in acute myeloid leukemia in China. Proc Natl Acad Sci U S a. 2013;110(42):17017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Musharraf SG, Siddiqui AJ, Shamsi T, Choudhary MI, Rahman AU. Serum metabonomics of acute leukemia using nuclear magnetic resonance spectroscopy. Sci Rep. 2016;6:30693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pabst T, Kortz L, Fiedler GM, Ceglarek U, Idle JR, Beyoglu D. The plasma lipidome in acute myeloid leukemia at diagnosis in relation to clinical disease features. BBA Clin. 2017;7:105‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Perea G, Domingo A, Villamor N, et al. Adverse prognostic impact of CD36 and CD2 expression in adult de novo acute myeloid leukemia patients. Leuk Res. 2005;29(10):1109‐1116. [DOI] [PubMed] [Google Scholar]
- 54. Ye H, Adane B, Khan N, et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shafat MS, Oellerich T, Mohr S, et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood. 2017;129(10):1320‐1332. [DOI] [PubMed] [Google Scholar]
- 56. Staubert C, Bhuiyan H, Lindahl A, et al. Rewired metabolism in drug‐resistant leukemia cells: a metabolic switch hallmarked by reduced dependence on exogenous glutamine. J Biol Chem. 2015;290(13):8348‐8359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Farge T, Saland E, de Toni F, et al. Chemotherapy‐resistant human Acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017;7(7):716‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Skrtic M, Sriskanthadevan S, Jhas B, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20(5):674‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dang L, White DW, Gross S, et al. Cancer‐associated IDH1 mutations produce 2‐hydroxyglutarate. Nature. 2009;462:739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ward PS, Patel J, Wise DR, et al. The common feature of leukemia‐associated IDH1 and IDH2 mutations is a neomorphic enzyme activity aonverting alpha‐ketoglutarate to 2‐hydroxyglutarate. Cancer Cell. 2010;17(3):225‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Intlekofer AM, Dematteo RG, Venneti S, et al. Hypoxia induces production of L‐2‐hydroxyglutarate. Cell Metab. 2015;22(2):304‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Intlekofer AM, Wang B, Liu H, et al. L‐2‐Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol. 2017;13(5):494‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nadtochiy SM, Schafer X, Fu D, Nehrke K, Munger J, Brookes PS. Acidic pH is a metabolic switch for 2‐hydroxyglutarate generation and signaling. J Biol Chem. 2016;291(38):20188‐20197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Parker SJ, Metallo CM. Metabolic consequences of oncogenic IDH mutations. Pharmacol Ther. 2015;152:54‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sciacovelli M, Frezza C. Oncometabolites: unconventional triggers of oncogenic signalling cascades. Free Radic Biol Med. 2016;100:175‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Figueroa ME, Abdel‐Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483:474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Losman JA, Looper RE, Koivunen P, et al. (R)‐2‐hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science. 2013;339(6127):1621‐1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ragon BK, DiNardo CD. Targeting IDH1 and IDH2 mutations in acute myeloid leukemia. Curr Hematol Malig Rep. 2017;12(6):537‐546. [DOI] [PubMed] [Google Scholar]
- 70. Chen WL, Wang JH, Zhao AH, et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood. 2014;124(10):1645‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fuhrer T, Heer D, Begemann B, Zamboni N. High‐throughput, accurate mass metabolome profiling of cellular extracts by flow injection‐time‐of‐flight mass spectrometry. Anal Chem. 2011;83(18):7074‐7080. [DOI] [PubMed] [Google Scholar]
- 72. Nemkov T, Hansen KC, D'Alessandro A. A three‐minute method for high‐throughput quantitative metabolomics and quantitative tracing experiments of central carbon and nitrogen pathways. Rapid Commun Mass Spectrom. 2017;31(8):663‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. D'Alessandro A, Giardina B, Gevi F, Timperio AM, Zolla L. Clinical metabolomics: the next stage of clinical biochemistry. Blood Transfus. 2012;10(Suppl 2):s19‐s24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N‐RAS gene mutations in acute myeloid leukemia. Blood. 1999;93(9):3074‐3080. [PubMed] [Google Scholar]
- 75. Alvarez‐Calderon F, Gregory MA, Pham‐Danis C, et al. Tyrosine kinase inhibition in leukemia induces an altered metabolic state sensitive to mitochondrial perturbations. Clin Cancer Res. 2015;21(6):1360‐1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. New Eng J Med. 2017;377(5):454‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gregory MA, D'Alessandro A, Alvarez‐Calderon F, et al. ATM/G6PD‐driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc Natl Acad Sci U S a. 2016;113:E6669‐E6678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pei S, Minhajuddin M, Callahan KP, et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. 2013;288:33542‐33558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pei S, Minhajuddin M, D'Alessandro A, et al. Rational design of a parthenolide‐based drug regimen that selectively eradicates acute myelogenous leukemia stem cells. J Biol Chem. 2016;291:21984‐22000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gallipoli P, Giotopoulos G, Tzelepis K, et al. Glutaminolysis is a metabolic dependency in FLT3 ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood. 2018. blood‐2017‐12‐820035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gregory MA, Nemkov T, Reisz JA, et al. Glutaminase inhibition improves FLT3 inhibitor therapy for acute myeloid leukemia. Exp Hematol. 2018;58:52‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sykes DB, Kfoury YS, Mercier FE, et al. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167(1):171‐86.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Pollyea DA, Jordan CT. Therapeutic targeting of acute myeloid leukemia stem cells. Blood. 2017;129(12):1627‐1635. [DOI] [PubMed] [Google Scholar]
- 84. Pui C‐H, Relling MV, Downing JR. Acute lymphoblastic leukemia. New Eng J Med. 2004;350(15):1535‐1548. [DOI] [PubMed] [Google Scholar]
- 85. Pui CH, Pei D, Campana D, et al. A revised definition for cure of childhood acute lymphoblastic leukemia. Leukemia. 2014;28(12):2336‐2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard‐risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the international ALL trial (MRC UKALL XII/ECOG E2993). Blood. 2008;111(4):1827‐1833. [DOI] [PubMed] [Google Scholar]
- 87. Fielding AK, Richards SM, Chopra R, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109(3):944‐950. [DOI] [PubMed] [Google Scholar]
- 88. Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017;35(9):975‐983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. O'Dwyer KM, Liesveld JL. Philadelphia chromosome negative B‐cell acute lymphoblastic leukemia in older adults: current treatment and novel therapies. Best Pract Res Clin Haematol. 2017;30(3):184‐192. [DOI] [PubMed] [Google Scholar]
- 90. Valecha GK, Ibrahim U, Ghanem S, Asti D, Atallah JP, Terjanian T. Emerging role of immunotherapy in precursor B‐cell acute lymphoblastic leukemia. Expert Rev Hematol. 2017;10(9):783‐799. [DOI] [PubMed] [Google Scholar]
- 91. Tan SH, Bertulfo FC, Sanda T. Leukemia‐initiating cells in T‐cell acute lymphoblastic leukemia. Front Oncol. 2017;7:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bongiovanni D, Saccomani V, Piovan E. Aberrant signaling pathways in T‐cell acute lymphoblastic leukemia. Int J Mol Sci. 2017;18(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Vadillo E, Dorantes‐Acosta E, Pelayo R, Schnoor M. T cell acute lymphoblastic leukemia (T‐ALL): new insights into the cellular origins and infiltration mechanisms common and unique among hematologic malignancies. Blood Rev. 2018;32(1):36‐51. [DOI] [PubMed] [Google Scholar]
- 94. Haskell CM, Canellos GP. L‐asparaginase resistance in human leukemia—asparagine synthetase. Biochem Pharmacol. 1969;18(10):2578‐2580. [DOI] [PubMed] [Google Scholar]
- 95. Marini BL, Perissinotti AJ, Bixby DL, Brown J, Burke PW. Catalyzing improvements in ALL therapy with asparaginase. Blood Rev. 2017;31(5):328‐338. [DOI] [PubMed] [Google Scholar]
- 96. Liang J, Shi P, Guo X, et al. A retrospective comparison of Escherichia coli and polyethylene glycol‐conjugated asparaginase for the treatment of adolescents and adults with newly diagnosed acute lymphoblastic leukemia. Oncol Lett. 2018;15(1):75‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cheng DH, Lu H, Liu TT, Zou XQ, Pang HM. Identification of risk factors in high‐dose methotrexate‐induced acute kidney injury in childhood acute lymphoblastic leukemia. Chemotherapy. 2018;63(2):101‐107. [DOI] [PubMed] [Google Scholar]
- 98. Krull KR, Cheung YT, Liu W, et al. Chemotherapy pharmacodynamics and neuroimaging and neurocognitive outcomes in long‐term survivors of childhood Acute lymphoblastic leukemia. J Clin Oncol. 2016;34(22):2644‐2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Cheung YT, Sabin ND, Reddick WE, et al. Leukoencephalopathy and long‐term neurobehavioural, neurocognitive, and brain imaging outcomes in survivors of childhood acute lymphoblastic leukaemia treated with chemotherapy: a longitudinal analysis. Lancet Haematol. 2016;3(10):e456‐e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kanarek N, Keys HR, Cantor JR, et al. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature. 2018;559(7715):632‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Vo TT, Lee JS, Nguyen D, et al. mTORC1 inhibition induces resistance to methotrexate and 6‐Mercaptopurine in Ph(+) and Ph‐like B‐ALL. Mol Cancer Ther. 2017;16(9):1942‐1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Montecucco A, Zanetta F, Biamonti G. Molecular mechanisms of etoposide. EXCLI J. 2015;14:95‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yadav N, Kumar S, Marlowe T, et al. Oxidative phosphorylation‐dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015;6(11):e1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Aoki S, Morita M, Hirao T, et al. Shift in energy metabolism caused by glucocorticoids enhances the effect of cytotoxic anti‐cancer drugs against acute lymphoblastic leukemia cells. Oncotarget. 2017;8(55):94271‐94285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fu X, Liu W, Huang Q, Wang Y, Li H, Xiong Y. Targeting mitochondrial respiration selectively sensitizes pediatric acute lymphoblastic leukemia cell lines and patient samples to standard chemotherapy. Am J Cancer Res. 2017;7(12):2395‐2405. [PMC free article] [PubMed] [Google Scholar]
- 106. Chan LN, Chen Z, Braas D, et al. Metabolic gatekeeper function of B‐lymphoid transcription factors. Nature. 2017;542(7642):479‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Xiao G, Chan LN, Klemm L, et al. B‐cell‐specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell. 2018;173(2):470‐84.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hsieh AL, Walton ZE, Altman BJ, Stine ZE, Dang CV. MYC and metabolism on the path to cancer. Semin Cell Dev Biol. 2015;43:11‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Palomero T, Ferrando A. Oncogenic NOTCH1 control of MYC and PI3K: challenges and opportunities for anti‐NOTCH1 therapy in T‐ALL. Clin Cancer Res. 2008;14(17):5314‐5317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306(5694):269‐271. [DOI] [PubMed] [Google Scholar]
- 111. Herranz D, Ambesi‐Impiombato A, Sudderth J, et al. Metabolic reprogramming induces resistance to anti‐NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat Med. 2015;21(10):1182‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tzoneva G, Ferrando AA. Recent advances on NOTCH signaling in T‐ALL. Curr Top Microbiol Immunol. 2012;360:163‐182. [DOI] [PubMed] [Google Scholar]
- 113. Hatle KM, Gummadidala P, Navasa N, et al. MCJ/DnaJC15, an endogenous mitochondrial repressor of the respiratory chain that controls metabolic alterations. Mol Cell Biol. 2013;33(11):2302‐2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Champagne DP, Hatle KM, Fortner KA, et al. Fine‐tuning of CD8(+) T cell mitochondrial metabolism by the respiratory chain repressor MCJ dictates protection to influenza virus. Immunity. 2016;44(6):1299‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Yang F, Fang H, Wang D, et al. HPRT1 activity loss is associated with resistance to thiopurine in ALL. Oncotarget. 2018;9(2):2268‐2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tabe Y, Konopleva M. Leukemia stem cells microenvironment. Adv Exp Med Biol. 2017;1041:19‐32. [DOI] [PubMed] [Google Scholar]
- 117. Griessinger E, Anjos‐Afonso F, Pizzitola I, et al. A niche‐like culture system allowing the maintenance of primary human acute myeloid leukemia‐initiating cells: a new tool to decipher their chemoresistance and self‐renewal mechanisms. Stem Cells Transl Med. 2014;3(4):520‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Drolle H, Wagner M, Vasold J, et al. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk Res. 2015;39(7):779‐785. [DOI] [PubMed] [Google Scholar]
- 119. Ceradini DJ, Kulkarni AR, Callaghan MJ, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF‐1 induction of SDF‐1. Nat Med. 2004;10(8):858‐864. [DOI] [PubMed] [Google Scholar]
- 120. Fiegl M, Samudio I, Clise‐Dwyer K, Burks JK, Mnjoyan Z, Andreeff M. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood. 2009;113(7):1504‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Behan JW, Yun JP, Proektor MP, et al. Adipocytes impair leukemia treatment in mice. Cancer Res. 2009;69(19):7867‐7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. De Santo C, Booth S, Vardon A, et al. The arginine metabolome in acute lymphoblastic leukemia can be targeted by the pegylated‐recombinant arginase I BCT‐100: targeting arginine metabolism in ALL. Int J Cancer. 2018;142:1490‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Tiziani S, Kang Y, Harjanto R, et al. Metabolomics of the tumor microenvironment in pediatric acute lymphoblastic leukemia. PLoS One. 2013;8(12):e82859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Taya Y, Ota Y, Wilkinson AC, et al. Depleting dietary valine permits nonmyeloablative mouse hematopoietic stem cell transplantation. Science. 2016;354(6316):1152‐1155. [DOI] [PubMed] [Google Scholar]
- 126. Henry CJ, Marusyk A, DeGregori J. Aging‐associated changes in hematopoiesis and leukemogenesis: what's the connection? Aging (Albany NY). 2011;3(6):643‐656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Battula VL, Le PM, Sun JC, et al. AML‐induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. 2017;2(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kumar B, Garcia M, Weng L, et al. Acute myeloid leukemia transforms the bone marrow niche into a leukemia‐permissive microenvironment through exosome secretion. Leukemia. 2017;32:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hardouin P, Rharass T, Lucas S. Bone marrow adipose tissue: to be or not to be a typical adipose tissue? Front Endocrinol (Lausanne). 2016;7(85). 10.3389/fendo.2016.00085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Medyouf H, Mossner M, Jann JC, et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell. 2014;14(6):824‐837. [DOI] [PubMed] [Google Scholar]
- 131. Schepers K, Pietras EM, Reynaud D, et al. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self‐reinforcing leukemic niche. Cell Stem Cell. 2013;13(3):285‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Camacho V, McClearn V, Patel S, Welner RS. Regulation of normal and leukemic stem cells through cytokine signaling and the microenvironment. Int J Hematol. 2017;105(5):566‐577. [DOI] [PubMed] [Google Scholar]
- 133. Gattazzo F, Urciuolo A, Bonaldo P. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840(8):2506‐2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Knapik DM, Perera P, Nam J, et al. Mechanosignaling in bone health, trauma and inflammation. Antioxid Redox Signal. 2014;20(6):970‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Shin J‐W, Mooney DJ. Extracellular matrix stiffness causes systematic variations in proliferation and chemosensitivity in myeloid leukemias. Proc Natl Acad Sci U S a. 2016;113(43):12126‐12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Nakamura‐Ishizu A, Okuno Y, Omatsu Y, et al. Extracellular matrix protein tenascin‐C is required in the bone marrow microenvironment primed for hematopoietic regeneration. Blood. 2012;119(23):5429‐5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Susek KH, Korpos E, Huppert J, et al. Bone marrow laminins influence hematopoietic stem and progenitor cell cycling and homing to the bone marrow. Matrix Biol. 2018;67:47‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Panchabhai S, Kelemen K, Ahmann G, Sebastian S, Mantei J, Fonseca R. Tumor‐associated macrophages and extracellular matrix metalloproteinase inducer in prognosis of multiple myeloma. Leukemia. 2015;30:951. [DOI] [PubMed] [Google Scholar]
- 139. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179‐183. [DOI] [PubMed] [Google Scholar]
- 140. Santinon G, Pocaterra A, Dupont S. Control of YAP/TAZ activity by metabolic and nutrient‐sensing pathways. Trends Cell Biol. 2016;26(4):289‐299. [DOI] [PubMed] [Google Scholar]
- 141. Santinon G, Brian I, Pocaterra A, et al. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene‐induced senescence. EMBO J. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hartwell KA, Miller PG, Mukherjee S, et al. Niche‐based screening identifies small‐molecule inhibitors of leukemia stem cells. Nat Chem Biol. 2013;9(12):840‐848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Seton‐Rogers S. Destroying leukaemia stem cell habitats. Nat Rev Cancer. 2013;13:821. [DOI] [PubMed] [Google Scholar]
- 144. Hill RC, Calle EA, Dzieciatkowska M, Niklason LE, Hansen KC. Quantification of extracellular matrix proteins from a rat lung scaffold to provide a molecular readout for tissue engineering. Mol Cell Proteomics. 2015;14(4):961‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Barrett AS, Wither MJ, Hill RC, et al. Hydroxylamine chemical digestion for insoluble extracellular matrix characterization. J Proteome Res. 2017;16(11):4177‐4184. [DOI] [PMC free article] [PubMed] [Google Scholar]