

Abstract
Lipid droplets (LDs) are dynamic organelles that regulate the storage and homeostasis of intracellular triglycerides and other neutral lipids. Studies show that the number, morphology, and subcellular localization of LDs are altered in a growing number of diseases. As such, methodologies for imaging and quantifying LDs have become essential tools for detecting changes in cellular lipid metabolism, which could be an important indicator of disease onset or progression. We previously reported on the accumulation of LDs in podocytes of the kidney glomerulus in nephrological diseases of metabolic and non-metabolic origin. Here, we describe a high-content analysis (HCA) method for automated detection and quantification of LDs in differentiated human podocytes.
Keywords: Lipid droplets, Human podocytes, High content screening, Cell-based assay, Phenotypic drug discovery
1. Introduction
Lipid droplets (LDs), first observed by Van Leeuwenhoek in 1674, are known to be conserved from Archaea bacteria to mammals [1]. In mammals, LDs were initially thought to be confined to adipocytes but were later found to occur in almost all cell types. Analyses of the structural composition of LDs revealed the presence of a hydrophobic lipid core composed of neutral lipids, including triacylglycerols (TAG) and cholesterol esters (CE), surrounded by a phospholipid monolayer. It was originally believed that the primary function of LDs is to store lipids and provide energy in the absence of sufficient nutrients. However, recent studies have shown that LDs are complex organelles with multiple functions that include modulation of nuclear processes, protein trafficking, membrane trafficking, and phospholipid recycling [2–4]. LDs are also sites for the synthesis of eicosanoids, which have a major role in inflammation [5]. α-Synuclein, a protein linked to Parkinson’s disease, is shown to bind to LDs [6], and its overexpression promotes the LD accumulation in yeast cells [7]. In Drosophila embryos, LDs plays an important role in histone balance and metabolism in the nucleus and function as a storage site for unstable proteins [8, 9]. Abnormalities in the size or number of LDs affect the onset or progression of numerous metabolic diseases including obesity, insulin resistance and type 2 diabetes, heart disease, and nonalcoholic fatty liver disease [10–13]. As such, understanding the mechanistic pathways that contribute to changes in the morphology or number of lipid droplets is an important step toward developing therapeutic interventions for these diseases.
Accumulation of LD in podocytes has been reported by us and others in clinical and experimental diabetic kidney disease [14–16]. We recently reported glomerular accumulation of LD in kidney diseases of non-metabolic origin, such as focal and segmental glomerulosclerosis and Alport syndrome [17]. We further demonstrated that accumulation of LD in podocytes is a key predisposing factor to cell injury and kidney disease development and progression [18]. Developing strategies to reduce LD accumulation in podocytes therefor represents a novel and potentially effective approach for treating or curing kidney disease.
Visualizing LDs is, in principle, a simple technique, whereby neutral lipid dyes such as BODIPY are used to stain and image LDs within cells. However, accurate quantification of LDs, especially when dealing with a large number of cells and culture conditions, as in a screening scenario, is a complicated and laborious task. Here, we describe a method for efficiently assaying intracellular LDs. The technique utilizes digital confocal microscopy and high-content analysis (HCA) to sensitively and robustly quantify intracellular LDs in differentiated podocytes.
2. Materials
2.1. Cells and Cell Culture Reagents
Human immortalized podocytes (a kind gift from Dr. Moin Saleem [19]).
Podocyte culture media: RPMI 1640 (Sigma: R-8758) contain insulin-transferrin-selenium (Invitrogen: 414100045), 10% fetal bovine berum (Sigma-F7524), penicillin-streptomycin (optional, Sigma- P4333).
Collagen I (Corning catalog#354236).
0.02 N acetic acid.
BODIPY 493/503 stock solution (Invitrogen, 1 mg/ml in 100% ethanol).
HCS CellMask Deep Red 650/655 (Molecular Probe, USA, 10 mg/ml in DMSO).
DAPI (1 mg/ml Thermo Scientific Inc., USA).
Phosphate-buffered saline (PBS).
4% Paraformaldehyde 4% sucrose solution in 1× PBS (PFA/sucrose).
5-, 10-, and 25-ml serological pipettes.
15- and 50-ml centrifuge tubes.
T25 and T75 cell culture flasks.
Greiner Bio-One Flat Bottom 96-well plate (Catalog No. 655090).
2.2. Equipment
Biological safety cabinet.
Tissue culture incubator at 33 °C and 37 °C with humidified 5% CO2 atmosphere.
Swing bucket centrifuge for 15 and 50 ml tubes.
Water bath at 37 °C.
Hemocytometer or automated cell counter.
OPERA HCS imager (Perkin-Elmer, USA).
3. Method
All the steps should be done under a ventilated hood unless specified otherwise.
3.1. Coating 96-Well Plates
96-well plates were collagenized using 100 μl of a 0 .1mg/ml collagen I in 0.02 N acetic acid solution for at least 1 h at 37 °C.
Wash three times with 100 μl of 1× PBS before seeding podocytes.
Coated plates can be stored at 4 °C for 1 week.
3.2. Culturing and Plating of Cells
Culture human conditionally immortalized podocytes as described in [19] (see Note 1).
Seed 4000–6000 human podocytes in 150 μl of culture media in the inner 60 wells of collagenized 96-well plates and differentiate for 13 days at 37 °C (outer wells are avoided and filled with media or PBS to avoid edge well effects).
At the appropriate time point, add 50 μl of media containing treatment at 4× the desired final concentration. If serum starvation is desired, starve the cells overnight prior to treatment.
3.3. Fixation of Cells (Done on Day 14)
Discard culture medium either by gently inverting the plate or by using a multichannel suctioner, and then wash the wells three times with 100 μl of 1× PBS (see Note 2).
Fix the cells by slowly adding 100 μl of 4% PFA, 4% sucrose solution prepared in 1× PBS from the side wall of the well, and incubate the plates at room temperature for 20 min.
0Discard fixative and wash the cells three times with 100 μl of 1× PBS.
Store cells in 150 μl 1× PBS at 4 °C until ready for next step. For long-term storage, add 0.02% sodium azide to prevent microbial growth.
3.4. Staining of Lipid Droplets
Several neutral lipid staining dyes such as Nile Red, BODIPY, and LipidTox™ are commercially available. Here, we provide a lipid droplet staining protocol using BODIPY 493/503 dye.
Staining Protocol:
Prepare the staining solution by diluting DAPI (1:5000), BODIPY 493/503 (1:1000), and HCS CellMask Deep Red (1:80000) in 1× PBS (see Notes 3 and 4).
Discard PBS over cells (Subheading 3.2) and add 100 μl of the staining solution to each well, and then incubate for 30 min at room temperature.
Wash the wells three times with 1× PBS.
Proceed to image acquisition step (see Note 5).
3.5. Image Acquisition Using an Automated Confocal Microscope
Lipid droplet visualization should be done using a confocal fluorescence microscope or high-content confocal screening machine. Conventional fluorescence images will have high background, and excessive clustering of LDs will challenge quantification. Imaging a plane closer to bottom surface of the cells will result in sharper images with low background. It will also provide a section of the cell body with minimal cytoplasmic breaks, which will facilitate cell segmentation. Here, we provide a protocol for high-content image acquisition using the OPERA HCS imager.
Carry out the acquisition process using the OPERA LX software at the desired lens magnification. We recommend using μ-clear 96-well Greiner plates and visualizing the lipid droplets with the 20× magnification lens.
In the light source box, select and activate the appropriate lasers (see Note 6).
Acquire images with three channels, one channel being the UV 365 channel for the nucleus (DAPI) acquisition, second 488 channel for the BODIPY (LDs) acquisition, and third 640 for cytosol (CellMask Deep Red).
Acquire 20–30 different fields or enough fields to count at least individual 200 cells per well.
3.6. Image Analysis and Quantification of Lipid Droplets
The lipid content can be quantified either as total normalized fluorescence intensity (BODIPY intensity) or as number of spots per cells, depending on the exact biological process being interrogated. Here, we provide a workflow for image analysis using the Columbus™ software. In this example (Fig. 1a), cell nuclei are stained with DAPI (channel-1), lipid droplets with BODIPY 493/503 (channel-2), and cell cytoplasm with CellMask Deep Red (channel-3).
Fig. 1.
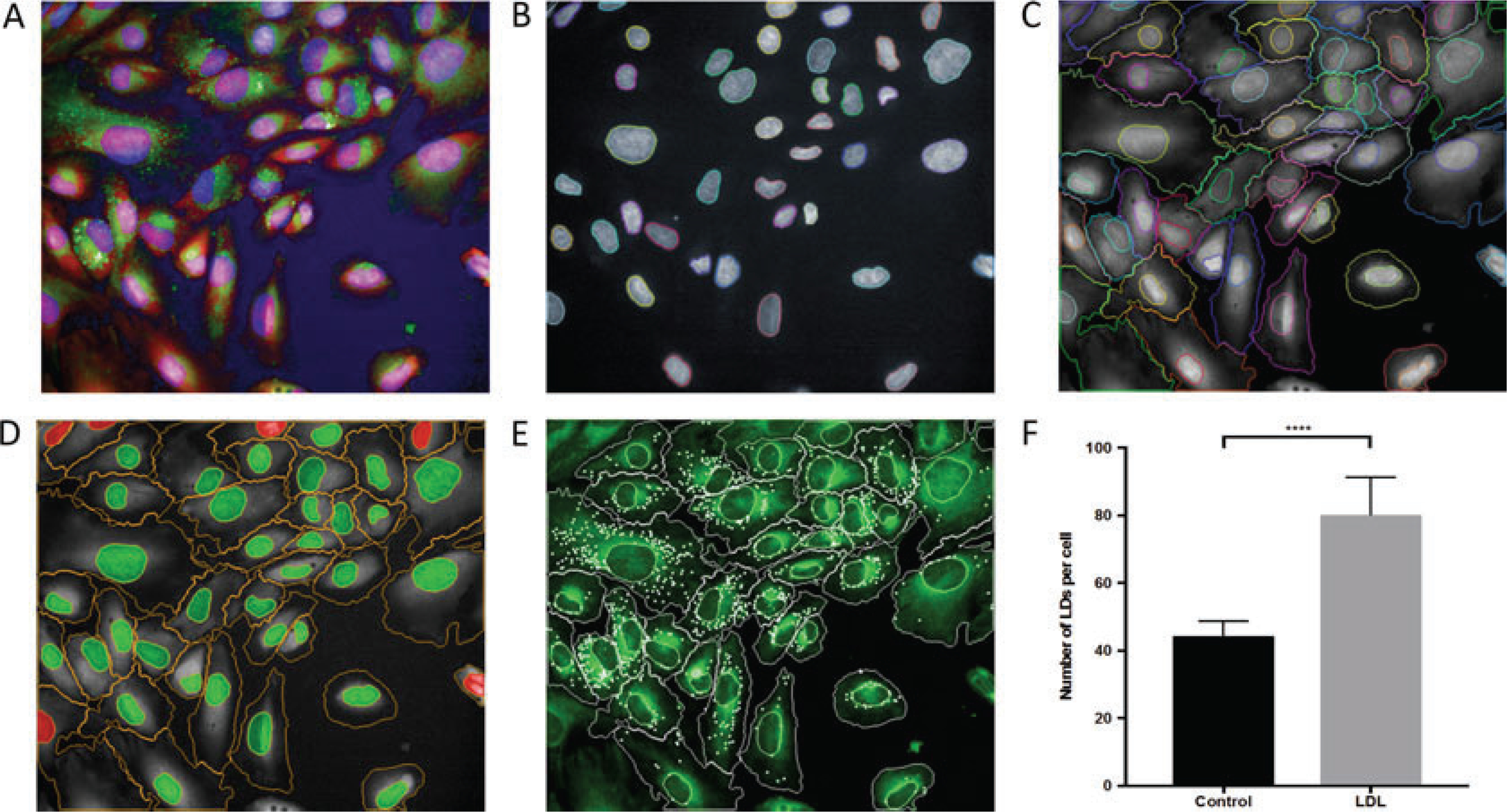
Lipid droplet quantification using the Columbus™ software. (a) Human podocytes nucleus stained with DAPI (Blue), lipid droplets with BODIPY 493/503 (green) and cytosol with CellMask Deep Red (Red); (b) identification of cell nuclei; (c) identification of the cell cytoplasm; (d) exclusion of the cells at the image border. Cells marked with green were selected for further analysis, and cells marked with red were removed from the analysis; (e) spot detection. Detected spots are marked with white. (f) LDL treatment increases LDs in cultured podocytes. Human podocytes were incubated without (control) or with 50 μg/ml acetylated low-density lipoprotein (LDL) for 24 h at 37 °C. Cells were fixed and stained as described in protocol. LDs were quantified using Columbus software
Nuclei detection: locating regions belonging to nuclei in the image is the first step in identifying and segmenting their corresponding cell bodies. For the quantification of LDs in the cells, transfer the data to Columbus software. After uploading the FLEX file, select the data from the data tree. Go to the Image analysis tab, and explore several images. If necessary, change the colors of the channels using the right hand control box. Navigate to the image analysis tab in the top menu, and select “find nuclei” from the drop-down menu to create the first step in the image analysis pipeline. After selecting the nuclear channel (in this case, channel-1), select algorithm B with common threshold 0.4, nucleus area >30 μm2, split factor 7.0, individual threshold 0.4, and contrast 0.1. Proper identification of the nuclei must be confirmed by manually checking 5–10 images (Fig. 1b). Once executed, this step will identify the nuclei within an image, which will be referred to by the default name of “total cells” (see Note 7).
Cell segmentation: in this step, cell bodies are identified by detecting areas surrounding nuclei with staining intensity in the deep red (640 nm) channel, followed by the application of a watershed algorithm to separate neighboring cells along their borders. Once nuclei are detected, add a “find cytoplasm” tab. Similar to nuclei detection, select the appropriate channel (in this case, channel-3). Select method D with an individual threshold 0.15. Method D works best when intensity decreases with the distance from nucleus and cells are in higher density. In sparse cells cultures, errors may occur with the detected cytoplasm spreading out to the background area, and a different algorithm may be required. Cell segmentation must be confirmed manually by checking 5–10 images (Fig. 1c).
Exclusion of artifacts: cells which are largely outside the border of the image may skew the analysis and must be excluded. From drop-down menu, select nuclei as the population and common filter as the method. Tick mark “remove border object.” This step will remove all cells which has nuclei on the border of the image from the analysis. The selected population will have the default name of “selected cells” (Fig. 1d) (see Note 8). Additional removal of artifacts can be accomplished in subsequent steps following the calculation of morphological properties.
Calculation of morphological properties: add a “Calculate morphology properties” step to compute the morphological properties of the selected cells. Add a “select population” tab and from drop-down menu. Set “selected cells” as the population and filter by property as the method. From the drop-down menu, select cell area and input the appropriate minimum area in μM2 (e.g., 100 μM2). Input a name for the newly selection population, e.g., “final selected cells.”
Quantification of mean BODIPY intensity: from the drop-down menu, add a “calculate intensity properties,” and tab and select “final selected cells” as the population. Select appropriate channel for the BODIPY stain (in this case, channel 2). Set the region field to “cytoplasm.” Select “mean intensity” to calculate normalized mean intensity (total cell intensity divided by cell area, averaged per well).
Spot detection: spots are defined as small regions on the image having a higher intensity than their surroundings. Add “find spot” tab from the drop-down menu, and select the appropriate channel for BODIPY (in this case, channel 2), region of interest (ROI) population as “final selected cells,” and ROI region as “cytoplasm.” Spots will be detected using a pre-defined algorithm. Select method C with radius ≤5.0, contrast <0.31, uncorrected spot to region intensity >1.3, distance between the spot 2.0 pixels, and spot peak radius 0.0 (Fig. 1e). Correct identification of the spots should be manually checked in 5–10 figures, and the method parameters tweaked as necessary (see Note 9).
Acquire results: in the “define results” section, select total number cells (final selected cells), cell area, normalized BODIPY intensity (mean per well), and spots per cell (mean per well). Perform the necessary statistical analyses to determine if significant changes in LD occur under various treatment conditions (Fig. 1f).
Footnotes
Human podocytes cultured in complete medium at 33 °C. For differentiation, cells at 40–60% confluence are thermoshifted to 37 °C and maintained for 14 days. Culture medium is changed at least three times a week.
Dispense solution along the wall of the well to avoid detaching cells. If using suction, leave some liquid over the cells.
For multiplex staining with any given combination of dyes, the choice and concentration of each individual dye must be optimized for the corresponding imaging system to minimize bleed through across different channels.
Although a single dye like CellMask Deep Red or CellMask Blue can be used for both nucleus and cytosol staining, we have found that using a different dye/channel for each result in superior nuclear detection and cell segmentation.
We recommend reading the plate on the same day it is stained. We observe that after 15 days at 4 °C, fluorescence intensity of stained cells decreases by 60–70%.
Turn on the lasers at least 15 min prior to the visualization. This will allow the signal intensity to stabilize prior to image acquisition.
It is important to fine-tune detection parameters for every preset algorithm.
In the Columbus software, it is important to remove unwanted cells after cell segmentation. Removing unwanted nuclei before defining the cytoplasm will affect your cell segmentation.
Depending on the dye, cells types, and method of staining, background intensity varies, and it is important to fine-tune the identification criteria during each experiment.
References
- 1.Murphy DJ (2012) The dynamic roles of intracellular lipid droplets: from archaea to mammals. Protoplasma 249:541–585 [DOI] [PubMed] [Google Scholar]
- 2.Liu P, Ying Y, Zhao Y, Mundy DI, Zhu M, Anderson RG (2004) Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J Biol Chem 279:3787–3792 [DOI] [PubMed] [Google Scholar]
- 3.Saka HA, Valdivia R (2012) Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu Rev Cell Dev Biol 28:411–437 [DOI] [PubMed] [Google Scholar]
- 4.Welte MA (2015) Expanding roles for lipid droplets. Curr Biol 25:R470–R481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bozza PT, Bakker-Abreu I, Navarro-Xavier RA, Bandeira-Melo C (2011) Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot Essent Fatty Acids 85:205–213 [DOI] [PubMed] [Google Scholar]
- 6.Thiam AR, Antonny B, Wang J et al. (2013) COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc Natl Acad Sci U S A 110:13244–13249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Outeiro TF, Lindquist S (2003) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302:1772–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z, Johnson MR, Ke Z, Chen L, Welte MA (2014) Drosophila lipid droplets buffer the H2Av supply to protect early embryonic development. Curr Biol 24:1485–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Z, Thiel K, Thul PJ, Beller M, Kuhnlein RP, Welte MA (2012) Lipid droplets control the maternal histone supply of drosophila embryos. Curr Biol 22:2104–2113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birch AM, Buckett LK, Turnbull AV (2010) DGAT1 inhibitors as anti-obesity and antidiabetic agents. Curr Opin Drug Discov Devel 13:489–496 [PubMed] [Google Scholar]
- 11.Greenberg AS, Coleman RA, Kraemer FB et al. (2011) The role of lipid droplets in metabolic disease in rodents and humans. J Clin Invest 121:2102–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gan L, Xiang W, Xie B, Yu L (2015) Molecular mechanisms of fatty liver in obesity. Front Med 9:275–287 [DOI] [PubMed] [Google Scholar]
- 13.Olofsson SO, Bostro m P, Andersson L et al. (2009) Lipid droplets and their role in the development of insulin resistance and diabetic dyslipidemia. Clin Lipidol 4:611–622 [Google Scholar]
- 14.Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U (2014) Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res 55:561–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M (2006) Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes 55:2502–2509 [DOI] [PubMed] [Google Scholar]
- 16.Merscher-Gomez S, Guzman J, Pedigo CE et al. (2013) Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes 62:3817–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mitrofanova A, Molina J, Varona Santos J et al. (2018) Hydroxypropyl-β-cyclodextrin protects from kidney disease in experimental Alport syndrome and focal segmental glomerulosclerosis. Kidney Int 94(6):1151–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pedigo CE, Ducasa GM, Leclercq F et al. (2016) Local TNF causes NFATc1-dependent cholesterol-mediated podocyte injury. J Clin Invest 126:3336–3350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni L, Saleem M, Mathieson PW (2012) Podocyte culture: tricks of the trade. Nephrology (Carlton) 17:525–531 [DOI] [PubMed] [Google Scholar]