

Abstract
Organic solvents are common chemicals used in industry throughout the world, however, there is evidence for adverse health effects from exposure to these compounds. Trichloroethylene (TCE) is a halogenated solvent that has been used as a degreasing agent since the early 20th century. Due to its widespread use, TCE remains one of the most significant environmental contaminants in the US, and extensive research suggests TCE is a causative factor in a number of diseases, including cancer, fetal cardiac development, and neurotoxicity. TCE has also been implicated as a possible risk factor in the development of the most common neurodegenerative movement disorder, Parkinson’s disease (PD). However, there is variable concordance across multiple occupational epidemiological studies assessing TCE (or solvent) exposure and risk for PD. In addition, there remains a degree of uncertainty about how TCE elicits toxicity to the dopaminergic system. To this end, we review the specific neurotoxic mechanisms of TCE in the context of selective vulnerability of dopaminergic neurons. In addition, we consider the complexity of combined risk factors that ultimately contribute to neurodegeneration and discuss the limitations of single-factor exposure assessments.
Introduction
Parkinson’s disease (PD) prevalence is on the rise as populations age throughout the world, with an estimated global incidence of just under 20 million by 2040.1 While aging represents the most significant risk factor for PD development, the predicted growth rate of PD incidence outpaces that of normal aging, which implies external or environmental factors are partially involved, in conjunction with genetics, sex, and race. This evidence is further supported by age-standardized prevalence rates that describe an increase in PD within populations of newly industrialized countries,2 suggesting that industrial byproducts, exposed either through occupation or as environmental contaminants, influence PD incidence.
Environmental exposures have been implicated in PD risk for decades, and renewed evidence for an environmental ‘link’ to PD has become available through improved detection methods, increased population sizes within case–control studies, and better monitoring and reporting within patient populations. Still, the integration of an individuals’ lifetime environmental exposures and PD risk remains infinitely complex, and technology to measure exposure at the individual level requires substantial engineering capabilities.3,4 Despite these challenges, there are still robust data that support significant roles of environmental exposures at the population level; for example, certain pesticides have been unequivocally linked to increased PD risk,5,6 and are now included as data in ‘Likelihood Ratios’ of prodromal PD assessment.7 In addition, the availability of genetic profiling for PD mutations and risk variants8 has improved the study of gene–environment interaction,9 providing a possible explanation for incomplete penetrance of some inherited mutations associated with PD development.10
With the rise of global industrialization, particularly across the Asian continent, concerns of environmental influence on PD risk remain, and bring to light the importance of occupational as well as long-term exposures to subthreshold levels of industrial byproducts.2 Among the most prevalent environmental contaminants from industrial origin are the organic solvent class of chemicals, such as trichloroethylene (TCE) and tetrachloroethylene (PCE), which have been ubiquitously used throughout the world, and are now significant global pollutants.12,22
A small number of studies have identified TCE as an environmental or occupational risk factor in the development of PD;12–14 perhaps the most remarkable of which is a cluster of individuals who developed PD, seemingly in relation to their proximity to the open pool of TCE within the factory where they worked.15 From this limited occupational cohort, three individuals (average age 60) were diagnosed with PD, and 27 individuals (average age 55) who worked at the facility tested positively for quantitative motor deficits, including significantly slower fine hand movement times compared to age-matched controls (p < 0.0001).15 Similarly, a study of PD-discordant twins predicted an increased odds ratio (OR) for PD development in individuals who worked with volatile organic solvents, including TCE (OR 6.1, 95% CI: 1.2–33) and PCE (OR 10.5, 95% CI: 0.97–113).14 This study only found a small number of TCE and PCE-linked PD cases (N = 9 and 5 respectively), however, the significant lag time from solvent exposure to clinical PD diagnosis (10–40 years) underscores the difficulty of assessing exposure decades before the onset of disease.14
In contrast, a population-based case–control study of occupation and risk for PD suggests no significant association between any occupation and PD.16 In this study, further evaluation of industrial toxicants revealed no elevated OR in men who self-reported workplace exposure to solvents (OR 1.0, 95% CI: 0.68, 1.34; N = 122), but did find an increased risk estimate16 in women (OR 1.7, 95% CI: 0.98, 3.04; N = 34). Likewise, a recent epidemiological study examining PD and occupation in women (N = 2590) from the Observational Study of the Women’s Health Initiative (WHI-OS), reported that no significantly elevated PD risk correlated with industrial jobs.17 Interestingly, of any occupation, the highest estimated relative risk (RR) for PD was associated with “building and grounds cleaning maintenance” (RR 1.21, 95% CI: 0.92–1.60; N = 53), which the authors identified as possible exposure to solvents, microbes, pesticides, and cleaning agents.17 Specific solvents, such as TCE or PCE, were not evaluated by Firestone et al. (2010)16 nor Burstyn et al. (2019),17 therefore correlation between occupation and PD risk should be interpreted cautiously.
Collectively, these data present an interesting contrast for occupational solvent exposure and neurodegenerative disease, however, small cohort size, recall bias, and the long time period over which idiopathic PD develops, suggests that further study is warranted. In addition, with the historical and current widespread use and environmental contamination of chemicals like TCE or PCE, occupation represents only one avenue of exposure. To this end, we review the complexity of pathogenic mechanisms resulting from volatile organic solvent exposure, predominately TCE, in relation to the selective vulnerability of dopaminergic neurons over a lifespan. Additionally, we consider the effect of solvent exposure on PD phenotype, and address the value of stratification within PD cohorts relating to exposure assessment.
TCE exposure
TCE (C2HCl3) is a halocarbon that was first synthesized in 1864 by Emil Fischer, via the reduction of hexachloroethane with hydrogen.18 Its commercial production began throughout the world during the 1920s, where its properties as a potent degreasing agent became valuable to the rising dry-cleaning industry of the era. TCE has been historically used in many industrial operations (Table 1), and is still used as a chemical feedstock or as a vapor or cold degreasing agent to remove organic material from manufactured metal parts (for detailed history, see Doherty, 2000).19 TCE is released into the environment from industrial sites; to the air from vapor release, or into soil or ground water from on-site land disposal. According to the Environmental Protection Agency (EPA) Toxics Release Inventory (TRI), 2.05 million pounds of TCE was released into the environment from industrial sites in 2017.20 TCE has an estimated half-life of approximately 7 days in the air,21 but its breakdown varies widely in soil and water depending on physical conditions, and aquifer field-tests measured its half-life at 300 days.22 Due to continuous atmospheric release and the persistence of TCE in subsurface environments, it represents one of the most significant environmental contaminants of the 20th and 21st centuries.11
Table 1.
Prevalent industrial uses of TCE
| Industrial process | Description | Major period of use | Phase |
|---|---|---|---|
| Vapor degreasing | TCE vapor condenses on object to remove organic matter | 1940s-present | Gas |
| Cold degreasing | Object is sprayed or soaked in TCE, then dried | 1940s-present | Liquid |
| Chemical feedstock | Feedstock for hydrofluorocarbon production (refrigerants) | 1930s-present | Liquid/gas |
| Textiles | Solvent for dyes, carrier solvent, waterless dying | 1900s-present | Liquid |
| Dry cleaning | Dry cleaning solvent, spot removal | 1930s-1950s, 1930s-present | Liquid |
| Military | General solvent, chemical feedstock | Reduced in 2007 | Liquid/gas |
| Anesthetic | Mostly obstetric, also general, dental, and veterinary settings | 1920s-1970s | Gas |
| Decaffeination | Extraction solvent | 1920s-1977 | Liquid |
| Animal feed processing | Soybean meal fat extraction | 1900s-1952 | Liquid |
Human exposure to TCE most commonly occurs through inhalation, as environmental partitioning of TCE is estimated to be 97.7% within the air.23 The mean concentration of TCE within the air has been steadily declining within the US, from ~1.4 μg m−3 in 1985, to 0.8 μg m−3 in 1998, though air concentrations near industrial sites where TCE is used are higher.19,21,22 TCE is a volatile compound, therefore it also poses a vapor intrusion hazard from its contamination of groundwater or soil. When released or disposed of from industrial sites into the environment, the liquid phase of TCE travels as an aqueous plume in groundwater. These “toxic plumes” of TCE eventually contaminate residential water sources, but may also gain entry into homes or offices as vapor, leading to inhalational exposure (Fig. 1). TCE was used extensively by the US military and on military bases prior to 2007 when the EPA issued a report revising the emission limits on solvents.24 In addition, TCE contaminates almost every site on the National Priorities List (Superfund sites), and therefore poses high risk of exposure to populations living near these locations. One such example, is near the Fairchild Semiconductor site in Mountain View, CA, where historical dumping of TCE from the early tech industry now contaminates the modern Silicon Valley. In addition to health concerns, the difficultly of TCE remediation from these areas has also produced a significant monetary impact; housing development within areas of Mountain View, CA have been halted until high TCE levels within the soil are remediated from the site.25
Fig. 1.

Environmental sources of trichloroethylene (TCE). TCE is predominantly released into the environment via air emissions from industrial sites (1), however, TCE waste may also contaminate groundwater and soil. TCE travels through groundwater or sewer systems in non-aqueous plumes, therefore individuals who live or work near areas of high TCE contamination may be exposed through drinking water (2), or vapor intrusion (3). Figures were created with Biorender.com.
TCE and Parkinson’s disease
Idiopathic Parkinson’s disease is the most prevalent neurodegenerative movement disorder in adults, defined by the presence of cardinal motor symptoms including, bradykinesia, rigidity, resting tremor, and postural instability.26 Underlying these motor deficits, is the progressive loss of dopaminergic neurons of the substantia nigra and their axonal projections to the caudate and putamen (striatum), where lack of the neurotransmitter dopamine results in disruption of basal ganglia signaling.27 PD is also a disease of protein mishandling and accumulation; most, but not all, individuals with PD display intracellular protein accumulations (Lewy bodies), which are highly comprised of the protein α-synuclein, within presynaptic dopaminergic neurons.28 During PD progression, α-synuclein pathology spreads from the brainstem throughout the cortices, which appears to correspond with disease severity.29,30 Neuroinflammation also plays a key role in the progression of the disease, involving resident immune cells of the brain, microglia, as well as peripheral monocytes, T-cells, and astrocytes.31,32
In addition to motor deficits, individuals with PD normally display a number of non-motor symptoms, including cognitive impairment, dementia, hallucinations, depression, sleep disturbances, decreases in gastrointestinal motility, hyposmia, and pain.33 Unlike motor symptoms, non-motor comorbidities appear to arise from a myriad of brain pathologies. For example, the accumulation and spread of α-synuclein is thought to drive cognitive impairment as the disease progresses.34,35 Similarly, symptoms of psychosis, which are reported in up to 40% of PD cases, are hypothesized to manifest from a disruption in the serotonergic system.36–38
Importantly, PD remains largely an idiopathic disease; approximately 10–15% of PD incidence is caused by heritable genetic mutations, which have been extensively studied and are outside the scope of this review (see Lill, 2016).39 A number of genetic risk factors for PD have been identified, however most display incomplete penetrance, suggesting that a combination of genetic susceptibility and environmental exposures, probably drive the majority of PD incidence.40,41
TCE was first implicated as an environmental risk factor for PD in the 1990s, when a single case study from a 47 year-old woman exposed to TCE over seven years was diagnosed with parkinsonism.42 Subsequent studies in rodents confirmed that TCE treatment caused the death of midbrain dopaminergic neurons,42 and could recapitulate other pathological hallmarks of PD, such as neuroinflammation, and α-synuclein accumulation.43,44 Like many toxicant models of PD, the dose, scheduling, and route of TCE exposure within rodents to produce dopaminergic neurodegeneration may not be directly reflective of human exposure, which is a topic that has been previously examined.45 For example, in the principal TCE exposure study, Guehl and colleagues utilized a 400 mg kg−1 intraperitoneal (i.p.) dose in OF1 (Oncins France 1) adult male mice, treated for 5 days a week over 4 weeks.42 A decade later, Liu et al. (2010)43 exposed adult male Fisher 344 rats to 200, 500, or 1000 mg kg−1 TCE in a daily oral gavage, 5 days a week over 6 weeks, and reported significant dopaminergic neuron death in the 500 and 1000 mg kg−1 dosing groups, and a corresponding loss of dopamine from the striatum. In a follow-up study, Liu et al. (2018)44 treated young adult mice (C57BL/6) with 400 mg kg−1 TCE in a daily oral gavage, 5 days a week over 8 months, which also resulted in the significant loss of dopaminergic neurons from the substantia nigra.
The United States Occupational Safety and Health Administration (OSHA) regulates workplace inhalational permissible exposure limit to TCE at 100 ppm (537.4 mg m−3) over an 8 hour workday, with short-term (5 minute) exposure at 300 ppm (1612.3 mg m−3). For liquid phase TCE, parts per million (ppm) to mg kg−1 is a 1 : 1 ratio, therefore, 100 or 300 mg kg−1 of TCE exposure in rodent models could reproduce some workplace conditions if they are at or near the OSHA limit. However, the route of administration in all animal models of PD tested to date, either i.p. injection or oral gavage, is not reflective of the majority of TCE exposure among humans, which occurs through inhalation. In addition, air sampling at industrial sites where TCE is used varies widely, therefore it is difficult to assess a true daily average occupational exposure to the solvent.46
TCE exposure is well documented from a major site of environmental contamination at the US Marine Corps Base, Camp Lejeune, North Carolina. TCE was used at Camp Lejeune – and other military bases throughout the US – along with other solvents such as PCE, vinyl chloride, and benzene.47,48 Extensive historical reconstruction estimates two water treatment plants at the base, Tarawa Terrace and Hadnot Point, were contaminated with PCE (Tarawa Terrace) and TCE (Hadnot Point) over a 30 year period between the 1950s and 1980s (Table 2), resulting in exposure to thousands of military service members, their families, and civilian employees on the base.48 Individuals who lived or worked at Camp Lejeune have increased risk for cancer, including liver, bladder, and kidney cancer, non-Hodgkin’s lymphoma, leukemia, multiple myeloma, and aplastic anemia.49 They also have increased risk for PD. In a retrospective cohort mortality study between civilian employees who worked at Camp Lejeune or Camp Pendleton, workers from Camp Lejeune had an elevated PD mortality hazard ratio (HR) of 3.13 (95% CI: 0.76, 12.81, N = 5) compared to age, sex, and race-matched individuals who lived and worked at another military base, Camp Pendleton.12
Table 2.
Camp Lejeune, North Carolina TCE contamination sitesa
| Water treatment plant | Major VOC contaminant | Peak level | Permissible water level | Date measured | Dates of plant operation |
|---|---|---|---|---|---|
| Hadnot Point | TCE | 1400 ppb | 5 ppb | May, 1982 | 1954-present |
| Tarawa Terrace | PCE | 215 ppb | 5 ppb | February, 1985 | 1952-1987 |
Table generated from reconstructed data in Maslia et al. (2016).48
There are a number of limitations to consider with the report of this elevated HR for PD from TCE exposure at Camp Lejeune; for example, there is no definitive way to measure dose or route of exposure in a retrospective study, the bulk of which likely occurred as a combination of inhalation and drinking water. In addition, individuals from Camp Lejeune were exposed to multiple chemicals at varying levels over several decades, which are difficult variables to control for. As Bove et al. (2014)12 is a mortality hazard study, the authors point out the small number of PD cases relative to cancer is likely due to the long disease progression and underreporting of PD on death certificates. Still, the Department of Veterans Affairs (VA) now officially recognizes PD as a service-connected disability associated with contamination from Camp Lejeune, and has approved VA disability compensation for individuals with PD who lived on the base for 30 days or more between 1953–1987.49 Collectively, data from occupational and environmental exposures, as well as animal models provide evidence that TCE is correlated with PD risk, and the subject warrants further investigation.
Mechanisms of dopaminergic neurotoxicity from TCE
TCE is a lipophilic compound that freely diffuses across membranes, including the blood–brain barrier (BBB).50 Once absorbed, enzymatic metabolism of TCE is extensive and wellcharacterized.51,52 TCE undergoes two types of metabolism, (1) oxidative metabolism by cytochrome P450 enzymes, predominantly Cyp2E1, or (2) direct conjugation with glutathione. While the majority of TCE biotransformation occurs via oxidative metabolism in the liver, both oxidation and conjugation of TCE can result in putative toxic metabolites, many of which are postulated to have tissue-specific effects.51,53 In addition, there is a considerable amount of evidence for species and sex-dependent differences in TCE metabolism.52,54–58 For example, oxidative metabolism kinetics of TCE are faster in mice than rats and humans,57 and human and rat liver microsomes display biphasic oxidative TCE metabolism, but is monophasic in mouse microsomes.54 The rate of glutathione conjugation of TCE is faster in male rodents (rats and mice) than female rodents, however, mice also display more rapid kinetics for glutathione conjugation than rats.54,59 TCE metabolite formation and kinetics may also vary depending on mouse strain,60–63 therefore, extrapolation of rodent-to-human TCE toxicity requires careful consideration of metabolic differences.
In humans, inhalation of TCE results in rapid distribution to multiple organs and compartments,64 including the brain.65 There is experimental evidence for TCE-induced neurotoxicity from both metabolic pathways,66–68 however, the exact mechanism by which TCE induces dopaminergic neurodegeneration remains unclear. As dopaminergic neurons of the SN are considered selectively sensitive,69–71 there may be a number of potentially convergent mechanisms that cause dopaminergic neurodegeneration from TCE exposure.
Mitochondrial disruption
TCE has been implicated as a mitochondrial toxicant since the early 1970s when inhibition of mitochondrial respiration at complex I was measured in the liver following use of TCE as an inhaled anesthetic.72 Subsequent studies have identified that mitochondrial dysfunction and a reduction of complex I activity occurs in isolated brain tissue following high oral doses of TCE (1 g kg−1).15,73 It is uncertain which metabolite of TCE is responsible for loss of complex I activity in the brain, however, at least three TCE-related metabolites have been linked to cellular mitochondrial dysfunction (Table 3, discussed below).
Table 3.
Putative mitochondrial toxicants produced by TCE metabolisma
| Structure | Metabolite name | Synonyms | Molecular formula | Mitochondrial toxicity evidence |
|---|---|---|---|---|
 |
S-(1,2-Dichlorovinyl)-L-cysteine | DCVC | C5H7Cl2NO2S | 84,86 and 145 |
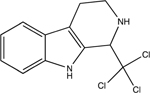 |
1-Trichloromethyl-1,2,3,4-tetrahydro-beta-carboline | TaClo | C12H11Cl3N2 | 95,96,98 and 146 |
 |
Chloral | Trichloroacetaldehyde (TCAH), trichloroethanal | C2HCl3O | 147 |
Chemical structures were created using ChemOffice Professional (Version 18, CambridgeSoft).
Dopaminergic neurons have high susceptibility to mitochondrial dysfunction, and are selectively vulnerable to environmental toxicants, such as the pesticides rotenone and paraquat, which are risk factors for PD development.5,6,74,75 In animal models, exposure to certain systemic mitochondrial toxicants (e.g. rotenone, paraquat, TCE) results in the selective loss of dopaminergic neurons from the nigrostriatal tract, while the adjacent dopaminergic neurons from the ventral tegmental area (VTA) remain largely unaffected.69,76 In addition, some mitochondrial toxicants, such as rotenone, can recapitulate other pathological hallmarks of PD, including α-synuclein accumulation, endolysosomal dysfunction, neuroinflammation, and changes within iron metabolism and handling.77–81 Similarly, rodent models of chronic, systemic TCE exposure have reported both neuroinflammation and α-synuclein accumulation in the midbrain of rats and mice.43,44 Collectively, there is evidence that systemic mitochondrial toxicant exposure produces key parkinsonian pathology extending beyond the loss of dopamine content in the striatum, or dopaminergic cell loss in the substantia nigra. This is a notable effect for mitochondrial toxicants as idiopathic PD has a long prodromal phase over which predegenerative pathology is postulated to occur (e.g. endolysosomal dysfunction leading to protein accumulation).82
Evidence for mitochondrial toxicity from TCE metabolites
TCE conjugation with glutathione produces S-(1,2-dichlorovinyl)glutathione (DCVG), which predominately occurs in the liver and kidneys. DCVG is further metabolized to S-(1,2dichlorovinyl)-L-cysteine (DCVC) by γ-glutamyltransferase (GGT) followed by cysteinylglycine dipeptidase (DP), enzymes that are highly expressed in the kidney.51 To this end, several lines of evidence show that DCVC causes mitochondrial dysfunction in proximal tubular cells of the kidney,83–86 which implicates DCVC in renal toxicity associated with TCE exposure. These data also suggest that DCVC may contribute to mitochondrial dysfunction within dopaminergic neurons, however, this mechanism of toxicity has been difficult to confirm due to the relatively low levels of glutathione-mediated biotransformation within the brain.87,88 Conversely, metabolism of TCE within the liver and kidneys to produce DCVC may be a peripheral source for the cysteine-conjugated metabolite, as DCVC appears to be a substrate for sodium-independent system L transporters (L-type amino acid transporter 1; LAT1), a major transport system across the BBB.89,90 In addition, both DCVG and DCVC were measured in brain tissue following 35S-radiolabeled uptake assays in rodents.90 Due to the relatively low antioxidant capacity of dopaminergic neurons,87,91 depletion of intracellular thiols to detoxify DCVC, could also be a source for elevated reactive oxygen species (ROS) and lipid peroxidation, both of which are mechanisms implicated in dopaminergic degeneration.92 However, the relative contribution to neurotoxicity by DCVG and DCVC remains to be measured in human TCE exposure. Of note, in a comparison between metabolic pathways of the solvents TCE and PCE in different strains of mice, PCE undergoes a greater proportion of glutathione conjugation than TCE, to produce S-(1,2,2-tri-chlorovinyl)-L-cysteine (TCVC), a potent renal toxicant, which likely causes similar mitochondrial toxicity as DCVC.93,94 As individuals are rarely exposed to TCE alone, these data high-light the importance of considering toxicokinetics of combined exposures (e.g. TCE and PCE), which could ultimately determine the fate of disease phenotype.
Another pathway involves the metabolite chloral, produced by TCE oxidative metabolism, which can interact with the neurotransmitter tryptamine to form a potent, halogenated beta-carboline toxicant, 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline (TaClo).95–97 Due to its structural similarity to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), TaClo is purported to be an inhibitor of complex I within the mitochondrial electron transport chain. Unlike MPTP, which binds to the dopamine transporter (DAT), TaClo does not appear to require a specific transporter to gain intracellular access. Treatment of rat brain homogenate with TaClo confirmed a significant reduction in mitochondrial NADH–ubiquinone reductase activity (complex I), at a concentration approximately 10-times lower than MPTP, suggesting its greater potency; TaClo IC50 ~ 250 mM; MPTP IC50 ~ 3500 μM.98 It is unknown whether TCE exposure produces high enough TaClo concentrations in the brain to cause significant damage to dopaminergic neurons,45 however, a recent study by Liu et al. (2018)44 measured ~31 ng TaClo per gram of brain tissue following an 8 month, 400 mg per kg per day oral exposure to TCE in WT mice. These data indicate that chronic exposure to TCE may be required to produce TaClo at a concentration that contributes to dopaminergic toxicity, however, more studies are needed to assess whether TaClo production is the ultimate dopaminergic toxicant following chronic TCE exposure.
Chloral, which exists in equilibrium with choral hydrate, is the major metabolite produced from the first oxidative biotransformation step of TCE with cytochrome P450 (TCE–oxygen–P450 or TCE epoxide).51,52 Chloral hydrate treatment in lymphoblastoid cells impaired ATP-linked respiration and maximal respiratory capacity, two components of mitochondrial respiration measured using the Seahorse extracellular flux assay.99 Chloral has also been shown to produce multiple mitochondrial deficits within human embryonic kidney cells (HEK293), including reducing ATP metabolism, increasing lactate dehydrogenase (LDH) release from cells, and elevating oxidative stress.100 Chloral is a CNS depressant and hypnotic, and contributes to the acute neurological effects of TCE exposure (dizziness, reduced reaction time, headache, confusion), hence the historical use of TCE as an anesthetic. It is unclear whether chloral-induced mitochondrial dysfunction could result in dopaminergic toxicity following TCE exposure, however several lines of evidence link inhaled anesthetics to general mitochondrial complex I inhibition.72,101–103 For example, sevoflurane, a common inhaled anesthetic, produces elevated ROS and depletes glutathione in a human neuroblastoma cell line (M17).104 Interestingly, this same study showed that overexpression of the PD-related protein DJ-1 (PARK7) attenuated the neurotoxic effects of sevoflurane in neuroblastoma cells. Whether TCE-derived chloral produces oxidative stress in dopaminergic neurons remains to be assessed, however, the depressive effects of TCE indicate that chloral/chloral hydrate brain levels are sufficiently high following TCE inhalation to affect global neurologic function. Taken together, this evidence suggests that TCE metabolism results in multiple putative mitochondrial toxicants, which could exert selective toxicity within dopaminergic neurons. The individual contribution or relative potency of either DCVC, TaClo, or chloral in dopaminergic neurodegeneration is difficult to predict, however, these TCE metabolites likely act in conjunction across multiple pathways to produce dopaminergic pathology.
Membrane disruption
The interaction of anesthetic compounds with biological membranes, which occurs with TCE and chloral, represents another potentially toxic mechanism in dopaminergic neurodegeneration. Inhaled anesthetics impart several changes to lipid bilayers, including physically altering bilayer structure by changing lateral pressure within the membrane, referred to as the ‘lipid hypothesis’ in their function.105 This appears to be the case for TCE, where its anesthetic effects are linked to phospholipid bilayer interaction.106 Membrane disruption and lipid raft disarrangement have been observed in tissue from PD patients.107 In addition, a well-established mechanism of protein aggregation is the interaction of α-synuclein with biological membranes.108 The majority of data for α-synuclein–membrane interactions indicate that toxic α-synuclein oligomers bind to lipid bilayers and disrupt membrane integrity, contributing to organelle and cellular dysfunction.108,109 Some evidence however, indicates solvent interaction at biological membranes may have a bidirectional effect on α-synuclein aggregation, as organic solvents, including trifluoroethanol, increased α-synuclein fibrillization in a model of biological membranes.110 Additionally, TCE treatment caused differential expression of membrane proteins in human hepatic liver cells (L-02), and is known to generally cause lipid peroxidation.111 The effect of TCE (or chloral) on dopaminergic neuron membrane integrity represents a potentially novel pathway for solvent toxicity in PD pathogenesis. In this context, PD-related pathology influenced by solvent exposure may be unique from other environmental contaminants, which could potentially result in a distinctive neurodegenerative phenotype.
Exposure windows
A key factor within epidemiological studies of TCE and PD risk is the reliance on occupational data to categorize exposure, in which adults were in contact with TCE within a defined workplace. Given the difficulty of epidemiological studies, these data are important as they represent some of the only quantifiable exposure assessments available, however, as previously discussed, there is still variability among occupation and PD risk. One potential explanation is that adult exposure to TCE does not reflect the most vulnerable window for neurotoxicity to organic solvents.
Exposure to TCE during critical periods of neuro-development (e.g. in utero, perinatal, or juvenile) causes neurotoxicity that could influence eventual risk for neurodegeneration (De Miranda and Blossom, 2020112). Due to the high susceptibility of the developing nervous system, the ability for TCE to cross the placenta,113–115 and its presence in breast milk,116 even low levels of TCE are considered potentially neurotoxic during development.66 A study of children exposed to organic solvents (N = 32) during gestation and after birth showed they have impaired motor coordination, and as well as decreases in verbal and full-scale IQ.117 Among mothers who reported occupational exposure to organic solvents, toluene was the predominant compound identified (N = 12) by Laslo-Baker et al. (2004).117 TCE (N = 2) and 1,1,1-trichloroethane (N = 2) exposures were documented, however, measurement of cognitive ability (IQ) and visual-motor coordination in children was not distinguished by specific solvent nor whether mothers were exposed to more than one solvent.117 In a larger study comparing cognitive deficits following TCE exposure from contaminated drinking water from three municipal sites in Minnesota, Ohio, and Massachusetts (total N = 54), individuals exposed as children had more severe cognitive deficits than adults, suggesting that age influenced the overall neurotoxicity potential of this solvent.118
Though data are limited, neurotoxicity resulting from perinatal exposure to low-levels of TCE, indicates that inflammation and epigenetic reprogramming may be key mechanisms of TCE-induced neuropathology. Autoimmune prone mice (MRL+/+) exposed to low-level TCE in drinking water during gestation and early life have elevated cytokine levels, reduced glutathione, and increased oxidative stress in their brain.119–121 In part, this appears to be due to altered DNA methylation (epigenetic regulation) induced by TCE exposure, which results in changes to gene expression in pathways associated with inflammation and antioxidant expression.121,122 For example, early life (6 week postnatal) TCE treatment in mice inhibited global DNA methylation in the cerebellum, which corresponded with alterations in the transmethylation and transsulfuration pathways that regulate glutathione redox potential.119,122 Long-term animal models could help determine whether early life exposure to TCE causes eventual dopaminergic neurodegeneration, however, the mechanisms governing general TCE-induced pathology in the developing brain (e.g. neuroinflammation, glutathione depletion, oxidative stress) are all mechanisms that precede dopaminergic neuron loss.31,87,123
Dose, route, and risk factors
Like all chemicals, the specific toxicity of TCE depends on its dose and route of exposure. High doses of TCE, whether inhaled or ingested, are more likely to overwhelm the P450 oxidation pathways and increase cellular antioxidant conjugation, causing depletion of glutathione and increases in oxidative damage.52 In addition, exposure to high levels of TCE produce greater levels of oxidative metabolites such as TCE oxide, which cause damage to proteins and DNA at sites close to their production (e.g. hepatocytes in the liver). In contrast, low-level TCE exposure over long periods of time may not deplete but rather consume antioxidant enzymes, leaving a reduced capacity to detoxify other reactive molecules within the cell. In this context, dopaminergic neurons of the substantia nigra would be particularly at risk, as the reactive nature of dopamine and relative paucity of antioxidant capacity within these neurons already leaves the cells vulnerable to oxidation.87,124 As previously discussed, the putative metabolites of TCE that disrupt mitochondria would also be of particular toxicity within cell types that rely on high metabolic function, such as the cells within the proximal tubule of the kidney,125 as well as dopaminergic neurons.126 To this end, short-term, high-level exposure to TCE (e.g. occupational) may be more correlative to increased cancer risk. In contrast, long-term, low-level TCE exposure (e.g. environmental) may play a stronger role in diseases with a long period of development, such as neurodegeneration.
Neither of these scenarios however, are solely reliant on exposure. Genetic risk and other disease modifying factors will ultimately determine the role for phenotypic presentation of PD. These variables are practically innumerable, however there are data to support complex interactions between genes, environment, and lifestyle in TCE-related toxicity. For example, a number of polymorphisms exist within in the P450 isoenzyme Cyp2D6, resulting in variable expression and enzymatic capacity (i.e. poor versus high metabolizers).127 Cyp2D6 polymorphisms have been linked to PD risk previously,41,127 but a lack of convincing data between certain Cyp2D6 polymorphisms and overall PD risk has limited a consensus. Recently, a study examined the role of Cyp2D6 in the bioactivation of β-carbo-lines to TaClo, and reported that Cyp2D6 was necessary to produce subsequent neurotoxicity in the adult mouse brain.128 In this context, individuals who are high Cyp2D6 metabolizers would potentially be more vulnerable to dopaminergic toxicity following TCE exposure, however, it is poor Cyp2D6 metabolizers that appear to have increased PD risk.127 Stratification of individuals with Cyp2D6 polymorphisms exposed to TCE would be necessary to quantify whether differential metabolism of solvents by P450 isoenzymes is indeed a risk factor in neurodegeneration.
The most commonly inherited genetic mutations and risk factors for PD are within the leucine-rich repeat kinase 2 (LRRK2) gene, resulting in a pathological elevation of its kinase activity.129–132 LRRK2 mutation penetrance is incomplete,133 suggesting that other risk factors, whether genetic or environmental in origin, play a role in LRRK2-mediated PD phenotype. A link between LRRK2 mutations and susceptibility to neurotoxicants has been suggested,134 and in a rat model of PD, the pesticide rotenone induced wildtype LRRK2 kinase activation within dopaminergic neurons that preceded neuron death.135 Emerging evidence shows that TCE is also capable of causing elevated LRRK2 kinase activity in dopaminergic neurons of wildtype rats, which resulted in dopaminergic neuropathology and degeneration.136 These data suggest that LRRK2 mutation carriers, who are already at increased risk for PD, may be particularly vulnerable in environments with high TCE exposure (e.g. contaminated ground water).
A number of other factors influence disease risk in TCE exposure. TCE treatment (500 μg ml−1 in drinking water) during early life is associated with changes in gut microbiome, that appear to be irreversible, even when TCE exposure has ceased.137 As the gut microbiome is now widely accepted to play a role in PD development and progression (for review see Mulak and Bonaz, 2015),138 ingestion of TCE-contaminated water sources represents a potential avenue for gut microbiota dys-regulation. In addition, high-fat diets (40% kcal fat) have been implicated as a modifier for TCE-induced toxicity,139 as TCE is a lipophilic molecule that is stored in adipose tissue.140 Similarly, alcohol can alter TCE metabolism, and may potentiate its toxicity in multiple systems.110,141
Clearly, TCE exposure and its potential for neurotoxicity on any system, including the vulnerable dopaminergic neurons of the substantia nigra, involves significant complexity of multiple factors. To study these effects is time consuming and costly, but necessary to appreciate the true risk of PD from TCE exposure; therefore, it is prudent to use caution when inter-preting data focused solely on any exposure and neurodegenerative disease.
Improving disease prediction from solvent exposure
Without a specific biomarker for PD, disease prediction in general remains limited. TCE metabolites can be measured in blood and urine for weeks following a single exposure,142 however, TCE exposure may occur years prior to the onset of PD. Recent data from a non-targeted metabolomics study of plasma from occupational TCE exposed individuals in a factory in Guangdong, China showed significant alterations in tyrosine metabolites, which was postulated to be a result of dopaminergic toxicity.143 These data indicate that alterations in dopamine metabolism within TCE-exposed individuals may be present, however, dopaminergic pathology is not to an extent where the PD-related motor symptoms were present. This type of subthreshold dopaminergic neurodegeneration likely requires other factors to ‘tip the balance’ toward a PD phenotype (e.g. genetic risk, lifestyle) and longitudinal assessment of their health would be required. Similarly, whether individuals with altered tyrosine metabolites resulting from TCE exposure would be considered in the prodromal phase of PD needs to be assessed. At present, updated criteria on PD risk from the Movement Disorders Society (MDS) suggest ‘occupational solvent exposure’ should be included in calculating Likelihood Ratio (LR) in conjunction with other environmental or lifestyle factors and prodromal markers for a positive PD diagnosis.7
Conclusions
The selective vulnerability of dopaminergic neurons to mechanisms of TCE-induced toxicity make the solvent a candidate for PD risk, but it is apparent that further research is needed on the role of TCE (and similar organic solvents) in neurodegeneration. Additionally, individuals with known PD risk factors, such as a LRRK2 mutation, may benefit from certain preventative measures (e.g. water or air filtration) if they live or work in an area with high TCE contamination. Despite efforts to reduce TCE use in the US within the last decade, it remains a relevant environmental contaminant, with evidence for moderate to high potential for neurotoxicity in vulnerable populations and life stages. In 2017, the EPA issued a report under the Toxic Substances Control Act (TSCA) proposing a ban on TCE manufacture, import, and distribution within the US for vapor degreasing.144 At present, any ban on TCE has been delayed as the EPA continues to evaluate TCE under a broader risk assessment.
Environmental significance.
Trichloroethylene (TCE) is a prototypical chemical in the category of volatile organic solvents, which includes many halogenated compounds and their structural analogs. There is renewed interest in the role of ubiquitous environmental contaminants in Parkinson’s disease (PD) risk, such as organic solvents, but precise mechanisms underlying dopaminergic pathology caused by solvent exposure remain unclear. The majority of available data regarding PD risk and solvent exposure involves TCE, however, this focused information does not preclude the potential toxicity of numerous structurally similar and abundant environmental contaminants.
Acknowledgements
Dr De Miranda is supported by the NIEHS grant K99ES029986, and the Parkinson’s Foundation. Dr Greenamyre is supported by NIH grants R01NS095387, R21ES027470, R01NS100744, the Michael J. Fox Foundation for Parkinson’s Research, American Parkinson Disease Association, and the friends and family of Sean Logan.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Dorsey ER and Bloem BR, JAMA Neurology, 2018, 75, 9–10. [DOI] [PubMed] [Google Scholar]
- 2.GBD 2016 Parkinson’s Disease Collaborators, Lancet Neurol, 2018, 17, 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loh M, Sarigiannis D, Gotti A, Karakitsios S, Pronk A, Kuijpers E, Annesi-Maesano I, Baiz N, Madureira J, Oliveira Fernandes E, Jerrett M. and Cherrie JW, Int. J. Environ. Res. Public Health, 2017, 14, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vineis P, Chadeau-Hyam M, Gmuender H, Gulliver J, Herceg Z, Kleinjans J, Kogevinas M, Kyrtopoulos S, Nieuwenhuijsen M, Phillips DH, Probst-Hensch N, Scalbert A, Vermeulen R. and Wild CP, EXPOsOMICS Consortium, Int. J. Hyg. Environ. Health, 2017, 220, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baltazar MT, Dinis-Oliveira RJ, de Lourdes Bastos M, Tsatsakis AM, Duarte JA and Carvalho F, Toxicol. Lett, 2014, 230, 85–103. [DOI] [PubMed] [Google Scholar]
- 6.Ascherio A, Chen H, Weisskopf MG, O’Reilly E, McCullough ML, Calle EE, Schwarzschild MA and Thun MJ, Ann. Neurol, 2006, 60, 197–203. [DOI] [PubMed] [Google Scholar]
- 7.Heinzel S, Berg D, Gasser T, Chen H, Yao C, Postuma RB and MDS Task Force on the Definition of Parkinson’s Disease, Mov. Disord, 2019, 30, 1600. [DOI] [PubMed] [Google Scholar]
- 8.Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, Bras J, Young E, von Coelln R, Simón-Sánchez J, Schulte C, Sharma M, Krohn L, Pihlstrøm L, Siitonen A, Iwaki H, Leonard H, Faghri F, Gibbs JR, Hernandez DG, Scholz SW, Botia JA, Martinez M, Corvol J-C, Lesage S, Jankovic J, Shulman LM, Sutherland M, Tienari P, Majamaa K, Toft M, Andreassen OA, Bangale T, Brice A, Yang J, Gan-Or Z, Gasser T, Heutink P, Shulman JM, Wood NW, Hinds DA, Hardy JA, Morris HR, Gratten J, Visscher PM, Graham RR, A. B. Singleton and 23 and Me Research Team, System Genomics of Parkinson’s Disease Consortium International Parkinson’s Disease Genomics Consortium, Lancet Neurol, 2019, 18, 1091–1102.31701892 [Google Scholar]
- 9.Klein C. and Westenberger A, Cold Spring Harbor Perspect. Med, 2012, 2, a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marras C, Canning CG and Goldman SM, Mov. Disord, 2019, 34, 801–811. [DOI] [PubMed] [Google Scholar]
- 11.Ostrowski SR, Wilbur S, Chou CHSJ, Pohl HR, Stevens Y-W, Allred PM, Roney N, Fay M. and Tylenda CA, Toxicol. Ind. Health, 2016, 15, 602–644. [DOI] [PubMed] [Google Scholar]
- 12.Bove FJ, Ruckart PZ, Maslia M. and Larson TC, Environ. Health, 2014, 13, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanner CM, Goldman SM, Ross GW and Grate SJ, Alzheimer’s Dementia, 2014, 10, S213–S225. [DOI] [PubMed] [Google Scholar]
- 14.Goldman SM, Quinlan PJ, Ross GW, Marras C, Meng C, Bhudhikanok GS, Comyns K, Korell M, Chade AR, Kasten M, Priestley B, Chou KL, Fernandez HH, Cambi F, Langston JW and Tanner CM, Ann. Neurol, 2011, 71, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gash DM, Rutland K, Hudson NL, Sullivan PG, Bing G, Cass WA, Pandya JD, Liu M, Choi D-Y,Hunter RL, Gerhardt GA, Smith CD, Slevin JT and Prince TS, Ann. Neurol, 2008, 63, 184–192. [DOI] [PubMed] [Google Scholar]
- 16.Firestone JA, Lundin JI, Powers KM, Smith-Weller T, Franklin GM, Swanson PD, Longstreth WT and Checkoway G, Am. J. Ind. Med, 2010, 53, 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burstyn I, LaCroix AZ, Litvan I, Wallace RB and Checkoway H, bioRxiv, 2019, 535732. [DOI] [PubMed] [Google Scholar]
- 18.Hardie D. and Hardie DWF, Chlorocarbons and chlorohydrocarbons. 1,1,2,2-Tetrachloroethane, in Encyclopedia of Chemical Technology, ed. Kirk RE and Othmer DF, John Wiley & Sons, New York, 1964, pp. 159–164. [Google Scholar]
- 19.Doherty RE, Environ. Forensics, 2000, 1, 69–81. [Google Scholar]
- 20.Sarkis J, Toxics Release Inventory, United States Environmental Protection Agency (EPA), 2017. [Google Scholar]
- 21.Agency for Toxic Substances and Disease Registry (ATSDR), Toxicological profile for Trichloroethylene (TCE), U.S. Department of Health and Human Services, Public Health Service, Atlanta, GA, 2019. [PubMed] [Google Scholar]
- 22.Bourg AC, Mouvet C. and Lerner DN, Q. J. Eng. Geol. Hydrogeol, 1992, 25, 359–370. [Google Scholar]
- 23.Boutonnet J-C, De Rooij C, Garny V, Lecloux A, Papp R, Thompson RS and Van Wijk D, Environ. Monit. Assess, 1998, 53, 467–487. [Google Scholar]
- 24.United States Environmental Protection Agency, Economic Impact Analysis of the Proposed Halogenated Solvent Cleaners Residual Risk Standard, 2007. [Google Scholar]
- 25.Noack M, Mountain View Voice. [Google Scholar]
- 26.Fearnley JM and Lees AJ, Brain, 1991, 114, 2283–2301. [DOI] [PubMed] [Google Scholar]
- 27.Zigmond MJ, Acheson AL, Stachowiak MK and Stricker EM, Arch. Neurol, 1984, 41, 856–861. [DOI] [PubMed] [Google Scholar]
- 28.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A. and Ghetti B, Proc. Natl. Acad. Sci. U. S. A, 2016, 95, 7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Braak H. and Braak E, Neuropathol. Appl. Neurobiol, 2015, 15, 13–26. [DOI] [PubMed] [Google Scholar]
- 30.Halliday GM, Del Tredici K. and Braak H, Parkinson’s Disease and Related Disorders, 2017, pp. 1–15. [Google Scholar]
- 31.Tansey MG and Goldberg MS, Neurobiol. Dis, 2010, 37, 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirsch EC and Hunot S, Lancet Neurol, 2009, 8, 382–397. [DOI] [PubMed] [Google Scholar]
- 33.Chaudhuri KR, Healy DG and Schapira AH, Lancet Neurol, 2006, 5, 235–245. [DOI] [PubMed] [Google Scholar]
- 34.Tsuboi Y, Uchikado H. and Dickson DW, Park. Relat. Disord, 2007, 13(suppl. 3), S221–S224. [DOI] [PubMed] [Google Scholar]
- 35.Apaydin H, Ahlskog JE, Parisi JE, Boeve BF and Dickson DW, Arch. Neurol, 2002, 59, 102–112. [DOI] [PubMed] [Google Scholar]
- 36.Politis M. and Niccolini F, Behav. Brain Res, 2015, 277, 136–145. [DOI] [PubMed] [Google Scholar]
- 37.Stahl SM, CNS Spectr, 2016, 21, 355–359. [DOI] [PubMed] [Google Scholar]
- 38.Pasquini J, Ceravolo R, Brooks DJ, Bonuccelli U. and Pavese N, Park. Relat. Disord, 2019, in press. [DOI] [PubMed] [Google Scholar]
- 39.Lill CM, Mol. Cell. Probes, 2016, 30, 386–396. [DOI] [PubMed] [Google Scholar]
- 40.Polito L, Greco A. and Seripa D, Parkinson’s Dis, 2016, 2016, 6465793–6465799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cannon JR and Greenamyre JT, Neurobiol. Dis, 2013, 57, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guehl D, Bezard E, Dovero S, Boraud T, Bioulac B. and Gross C, Eur. J. Neurol, 1999, 6, 609–611. [DOI] [PubMed] [Google Scholar]
- 43.Liu M, Choi D-Y, Hunter RL, Pandya JD, Cass WA, Sullivan PG, Kim H-C, Gash DM and Bing G, J. Neurochem, 2010, 112, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu M, Shin E-J, Dang D-K, Jin C-H, Lee PH, Jeong JH, Park S-J, Kim Y-S, Xing B, Xin T, Bing G. and Kim H-C, Mol. Neurobiol, 2018, 55, 6201–6214. [DOI] [PubMed] [Google Scholar]
- 45.Lock EA, Zhang J. and Checkoway H, Toxicol. Appl. Pharmacol, 2013, 266, 345–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonvallot N, Harrison P. and Loh M, in WHO Guidelines for Indoor Air Quality: Selected Pollutants, World Health Organization, 2010, vol. 112, pp. 1321–1329. [PubMed] [Google Scholar]
- 47.National Research Council (US) Committee on Contaminated Drinking Water at Camp Lejeune, 2009.
- 48.Maslia ML, Aral MM, Ruckart PZ and Bove FJ, Water, 2016, 8, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Department of Veterans Affairs, Fed. Regist, 2017, 82, 4173–4185. [PubMed] [Google Scholar]
- 50.Keane PC, Hanson PS, Patterson L, Blain PG, Hepplewhite P, Khundakar AA, Judge SJ, Kahle PJ, LeBeau FEN and Morris CM, Neurosci. Lett, 2019, 711, 134437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lash LH, Chiu WA, Guyton KZ and Rusyn I, Mutat. Res., Rev. Mutat. Res, 2014, 762, 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lash LH, Fisher JW, Lipscomb JC and Parker JC, Environ. Health Perspect, 2000, 108(suppl. 2), 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cichocki JA, Guyton KZ, Guha N, Chiu WA, Rusyn I. and Lash LH, J. Pharmacol. Exp. Ther, 2016, 359, 110–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elfarra AA, Krause RJ, Last AR, Lash LH and Parker JC, Drug Metab. Dispos, 1998, 26, 779–785. [PubMed] [Google Scholar]
- 55.Luo Y-S, Hsieh N-H, Soldatow VY, Chiu WA and Rusyn I, Toxicology, 2018, 409, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chiu WA, Okino MS and Evans MV, Toxicol. Appl. Pharmacol, 2009, 241, 36–60. [DOI] [PubMed] [Google Scholar]
- 57.Prout MS, Provan WM and Green T, Toxicol. Appl. Pharmacol, 1985, 79, 389–400. [DOI] [PubMed] [Google Scholar]
- 58.Luo Y-S, Furuya S, Chiu W. and Rusyn I, J. Toxicol. Environ. Health, Part A, 2018, 81, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lash LH, Xu Y, Elfarra AA, Duescher RJ and Parker JC, Drug Metab. Dispos, 1995, 23, 846–853. [PubMed] [Google Scholar]
- 60.Venkatratnam A, Furuya S, Kosyk O, Gold A, Bodnar W, Konganti K, Threadgill DW, Gillespie KM, Aylor DL, Wright FA, Chiu WA and Rusyn I, Toxicol. Sci, 2017, 158, 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yoo HS, Bradford BU, Kosyk O, Shymonyak S, Uehara T, Collins LB, Bodnar WM, Ball LM, Gold A. and Rusyn I, J. Toxicol. Environ. Health, Part A, 2015, 78, 15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yoo HS, Bradford BU, Kosyk O, Uehara T, Shymonyak S, Collins LB, Bodnar WM, Ball LM, Gold A. and Rusyn I, J. Toxicol. Environ. Health, Part A, 2015, 78, 32–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bradford BU, Lock EF, Kosyk O, Kim S, Uehara T, Harbourt D, DeSimone M, Threadgill DW, Tryndyak V, Pogribny IP, Bleyle L, Koop DR and Rusyn I, Toxicol. Sci, 2011, 120, 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fisher JW, Mahle D. and Abbas R, Toxicol. Appl. Pharmacol, 1998, 152, 339–359. [DOI] [PubMed] [Google Scholar]
- 65.Bourne JG, Anaesthesia, 1964, 19, 12–32. [DOI] [PubMed] [Google Scholar]
- 66.Salama MM, El-Naggar DA, Abdel-Rahman RH and Elhak SAG, Front. Pharmacol, 2018, 9, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Boyes WK, Bushnell PJ, Crofton KM, Evans M. and Simmons JE, Environ. Health Perspect, 2000, 108(suppl. 2), 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bale AS, Barone S, Scott CS and Cooper GS, Toxicol. Appl. Pharmacol, 2011, 255, 113–126. [DOI] [PubMed] [Google Scholar]
- 69.Brichta L. and Greengard P, Front. Neuroanat, 2014, 8, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Surmeier DJ, Lancet Neurol, 2007, 6, 933–938. [DOI] [PubMed] [Google Scholar]
- 71.Surmeier DJ, Guzman JN, Sanchez-Padilla J. and Schumacker PT, Neuroscience, 2011, 198, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hall GM, Kirtland SJ and Baum H, Br. J. Anaesth, 1973, 45, 1005–1009. [DOI] [PubMed] [Google Scholar]
- 73.Sauerbeck A, Hunter R, Bing G. and Sullivan PG, Exp. Neurol, 2012, 234, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Miranda BR, Van Houten B. and Sanders LH, in Mitochondrial Mechanisms of Degeneration and Repair in Parkinson’s Disease, Springer, Cham, Cham, 2016, vol. 11, pp. 115–137. [Google Scholar]
- 75.Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T, Matsuno-Yagi A, Miller GW and Greenamyre JT, J. Neurochem, 2007, 1469–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez TN and Greenamyre JT, Antioxid. Redox Signaling, 2012, 16, 920–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rocha EM, De Miranda BR, Castro S, Drolet R, Hatcher NG, Yao L, Smith SM, Keeney MT, Di Maio R, Kofler J, Hastings TG and Greenamyre JT, Neurobiol. Dis, 2019, 134, 104626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cannon JR, Tapias V, Na H-M, Honick AS, Drolet RE and Greenamyre JT, Neurobiol. Dis, 2009, 34, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.De Miranda BR, Fazzari M, Rocha EM, Castro S. and Greenamyre JT, Toxicol. Sci, 2019, 354, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Miranda BR, Rocha EM, Bai Q, El Ayadi A, Hinkle D, Burton EA and Timothy Greenamyre J, Neurobiol. Dis, 2018, 115, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sherer TB, Betarbet R, Kim J-H and Greenamyre JT, Neurosci. Lett, 2003, 341, 87–90. [DOI] [PubMed] [Google Scholar]
- 82.Kett LR and Dauer WT, Mov. Disord, 2016, 31, 1433–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Patel N, Birner G, Dekant W. and Anders MW, Drug Metab. Dispos, 1994, 22, 143–147. [PubMed] [Google Scholar]
- 84.Xu F, Papanayotou I, Putt DA, Wang J. and Lash LH, Biochem. Pharmacol, 2008, 76, 552–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y, Cai J, Anders MW, Stevens JL and Jones DP, Toxicol. Appl. Pharmacol, 2001, 170, 172–180. [DOI] [PubMed] [Google Scholar]
- 86.Elkin ER, Bridges D. and Loch-Caruso R, Toxicology, 2019, 427, 152283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smeyne M. and Smeyne RJ, Free Radicals Biol. Med, 2013, 62, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sian J, Dexter DT, Lees AJ, Daniel S, Jenner P. and Marsden CD, Ann. Neurol, 1994, 36, 356–361. [DOI] [PubMed] [Google Scholar]
- 89.Kanai Y. and Endou H, J. Toxicol. Sci, 2003, 28, 1–17. [DOI] [PubMed] [Google Scholar]
- 90.Patel NJ, Fullone JS and Anders MW, Mol. Brain Res, 1993, 17, 53–58. [DOI] [PubMed] [Google Scholar]
- 91.Martin HL and Teismann P, FASEB J, 2009, 23, 3263–3272. [DOI] [PubMed] [Google Scholar]
- 92.Kato Y, Asano Y. and Cooper AJ, Dev. Neurosci, 1996, 18, 505–514. [DOI] [PubMed] [Google Scholar]
- 93.Elfarra AA and Krause RJ, J. Pharmacol. Exp. Ther, 2007, 321, 1095–1101. [DOI] [PubMed] [Google Scholar]
- 94.Luo Y-S, Furuya S, Soldatov VY, Kosyk O, Yoo HS, Fukushima H, Lewis L, Iwata Y. and Rusyn I, Toxicol. Sci, 2018, 164, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bringmann G, Feineis D, God R, Peters K, Peters E-M, Scholz J, Riederer F. and Moser A, Bioorg. Med. Chem, 2002, 10, 2207–2214. [DOI] [PubMed] [Google Scholar]
- 96.Bringmann G, God R, Feineis D, Wesemann W, Riederer P, Rausch WD, Reichmann H. and Sontag KH, J. Neural Transm., Suppl, 1995, 46, 235–244. [PubMed] [Google Scholar]
- 97.Bringmann G, Feineis D, Brückner R, God R, Grote C. and Wesemann W, Eur. J. Pharm. Sci, 2006, 28, 412–422. [DOI] [PubMed] [Google Scholar]
- 98.Bringmann G, Feineis D, Brückner R, Blank M, Peters K, Peters EM, Reichmann H, Janetzky B, Grote C, Clement HW and Wesemann W, Bioorg. Med. Chem, 2000, 8, 1467–1478. [DOI] [PubMed] [Google Scholar]
- 99.Frye RE, Rose S, Wynne R, Bennuri SC, Blossom S, Gilbert KM, Heilbrun L. and Palmer RF, Sci. Rep, 2017, 7(1), 4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen Y, Xu T, Yang X, Chu W, Hu S. and Yin D, Sci. Total Environ, 2019, 664, 948–957. [DOI] [PubMed] [Google Scholar]
- 101.Cohen PJ, Anesthesiology, 1973, 39, 153–164. [DOI] [PubMed] [Google Scholar]
- 102.Kayser E-B, Suthammarak W, Morgan PG and Sedensky MM, Anesth. Analg, 2011, 112, 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zimin PI, Woods CB, Kayser EB, Ramirez JM, Morgan PG and Sedensky MM, Br. J. Anaesth, 2018, 120, 1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y, Li Y, Han X, Dong X, Yan X. and Xing Q, Cell Stress Chaperones, 2018, 23, 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cantor RS, Biochemistry, 1997, 36, 2339–2344. [DOI] [PubMed] [Google Scholar]
- 106.Huang P, Bertaccini E. and Loew GH, J. Biomol. Struct. Dyn, 1995, 12, 725–754. [DOI] [PubMed] [Google Scholar]
- 107.Marin R, Rojo JA, Fabelo N, Fernandez CE and Diaz M, Neuroscience, 2013, 245, 26–39. [DOI] [PubMed] [Google Scholar]
- 108.O’Leary EI and Lee JC, Biochim. Biophys. Acta, Proteins Proteomics, 2019, 1867, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fusco G, Chen SW, Williamson PTF, Cascella R, Perni M, Jarvis JA, Cecchi C, Vendruscolo M, Chiti F,Cremades N, Ying L, Dobson CM and De Simone A, Science, 2017, 358, 1440–1443. [DOI] [PubMed] [Google Scholar]
- 110.Munishkina LA, Phelan C, Uversky VN and Fink AL, Biochemistry, 2003, 42, 2720–2730. [DOI] [PubMed] [Google Scholar]
- 111.Tse SY, Mak IT, Weglicki WB and Dickens BF, J. Toxicol. Environ. Health, 1990, 31, 217–226. [DOI] [PubMed] [Google Scholar]
- 112.De Miranda BR and Blossom SJ, Neuroscience of Development, 2020, in press. [Google Scholar]
- 113.Beppu K, Keio J. Med, 1968, 17, 81–107. [DOI] [PubMed] [Google Scholar]
- 114.Loch-Caruso R, Hassan I, Harris SM, Kumar A, Bjork F. and Lash LH, Reprod. Toxicol, 2019, 83, 38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Elkin ER, Harris SM and Loch-Caruso R, Toxicol. Appl. Pharmacol, 2018, 338, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Beamer PI, Luik CE, Abrell L, Campos S, Martínez ME and Sáez AE, Environ. Sci. Technol, 2012, 46, 9055–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Laslo-Baker D, Barrera M, Knittel-Keren D, Kozer E, Wolpin J, Khattak S, Hackman R, Rovet J. and Koren G, Arch. Pediatr. Adolesc. Med, 2004, 158, 956–961. [DOI] [PubMed] [Google Scholar]
- 118.White RF, Feldman RG, Eviator II, Jabre JF and Niles CA, Environ. Res, 1997, 73, 113–124. [DOI] [PubMed] [Google Scholar]
- 119.Blossom SJ, Melnyk SB, Li M, Wessinger WD and Cooney CA, Neurotoxicology, 2017, 59, 164–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Meadows JR, Parker C, Gilbert KM, Blossom SJ and DeWitt JC, J. Immunotoxicol, 2017, 14, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gilbert KM, Blossom SJ, Erickson SW, Broadfoot B, West K, Bai S, Li J. and Cooney CA, Toxicol. Lett, 2016, 260, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Blossom SJ, Melnyk S, Cooney CA, Gilbert KM and James SJ, Neurotoxicology, 2012, 33, 1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blesa J, Trigo-Damas I, Quiroga-Varela A. and Jackson-Lewis VR, Front. Neuroanat, 2015, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Krüger R, Surmeier DJ and Krainc D, Science, 2017, 357, 1255–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bhargava P. and Schnellmann RG, Nat. Rev. Nephrol, 2017, 13, 629–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Harris JJ, Jolivet R. and Attwell D, Neuron, 2012, 75, 762–777. [DOI] [PubMed] [Google Scholar]
- 127.Ur Rasheed MS, Mishra AK and Singh MP, Neurochem. Res, 2017, 42, 3353–3361. [DOI] [PubMed] [Google Scholar]
- 128.Chattopadhyay M, Chowdhury AR, Feng T, Assenmacher C-A, Radaelli E, Guengerich FP and Avadhani NG, J. Biol. Chem, 2019, 294, 10336–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, Lynch J, Healy DG, Holton JL, Revesz T. and Wood NW, Lancet, 2005, 365, 415–416. [DOI] [PubMed] [Google Scholar]
- 130.Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ and Toft M, Am. J. Hum. Genet, 2005, 76, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Di Fonzo A, Rohé CF, Ferreira J, Chien HF, Vacca L, Stocchi F, Guedes L, Fabrizio E, Manfredi M, Vanacore N, Goldwurm S, Breedveld G, Sampaio C, Meco G, Barbosa E, Oostra BA and Bonifati V, Italian Parkinson Genetics Network, Lancet, 2005, 365, 412–415. [DOI] [PubMed] [Google Scholar]
- 132.Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O’Neill E, Meitinger T, Kolch W, Prokisch H. and Ueffing M, Hum. Mol. Genet, 2006, 15, 223–232. [DOI] [PubMed] [Google Scholar]
- 133.Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, Lynch T, Bhatia KP, Gasser T, Lees AJ and Wood NW, Lancet Neurol, 2008, 7, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee J-W and Cannon JR, Exp. Biol. Med, 2015, 240, 752–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Di Maio R, Hoffman EK, Rocha EM, Keeney MT, Sanders LH, De Miranda BR, Zharikov A, Van Laar A, Stepan AF, Lanz TA, Kofler JK, Burton EA, Alessi DR, Hastings TG and Greenamyre JT, Sci. Transl. Med, 2018, 10, eaar5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.De Miranda BR, Rocha EM, Castro S. and Greenamyre JT, The Toxicologist Supplement to Toxicological Sciences, 2019, vol. 168. [Google Scholar]
- 137.Khare S, Gokulan K, Williams K, Bai S, Gilbert KM and Blossom SJ, J. Appl. Toxicol, 2019, 39, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mulak A. and Bonaz B, World J. Gastroenterol, 2015, 21, 10609–10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Blossom SJ, Fernandes L, Bai S, Khare S, Gokulan K, Yuan Y, Dewall M, Simmen FA and Gilbert KM, Toxicol. Sci, 2018, 164, 313–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Albanese RA, Banks HT, Evans MV and Potter LK, Bull. Math. Biol, 2002, 64, 97–131. [DOI] [PubMed] [Google Scholar]
- 141.Larson JL and Bull RJ, J. Toxicol. Environ. Health, 1989, 28, 395–406. [DOI] [PubMed] [Google Scholar]
- 142.Bartonicek V, Br. J. Ind. Med, 1962, 19, 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walker DI, Uppal K, Zhang L, Vermeulen R, Smith M, Hu W, Purdue MP, Tang X, Reiss B, Kim S, Li L, Huang H, Pennell KD, Jones DP, Rothman N. and Lan Q, Int. J. Epidemiol, 2016, 45, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Environmental Protection Agency EPA, Fed. Regist, 2017, 1–30. [Google Scholar]
- 145.Cooper AJL, Bruschi SA and Anders MW, Biochem. Pharmacol, 2002, 64, 553–564. [DOI] [PubMed] [Google Scholar]
- 146.Kochen W, Kohlmüller D, De Biasi P. and Ramsay R, Adv. Exp. Med. Biol, 2003, 527, 253–263. [DOI] [PubMed] [Google Scholar]
- 147.Blossom SJ, Doss JC and Gilbert KM, Toxicol. Sci, 2007, 95, 401–411. [DOI] [PubMed] [Google Scholar]