

SUMMARY
The cell-cycle phase is a major determinant of repair pathway choice at DNA double strand breaks, non-homologous end joining (NHEJ), or homologous recombination (HR). Chk1 responds to genotoxic stress in S/G2 phase, but here, we report a role of Chk1 in directly promoting NHEJ repair in G1 phase. ASF1A is a histone chaperone, but it promotes NHEJ through a pathway independent of its histone-chaperone activity. Chk1 activated by ataxia telangiectasia mutated (ATM) kinase on DNA breaks in G1 promotes NHEJ through direct phosphorylation of ASF1A at Ser-166. ASF1A phosphorylated at Ser-166 interacts with the repair protein MDC1 and thus enhances MDC1’s interaction with ATM and the stable localization of ATM at DNA breaks. Chk1 deficiency suppresses all steps downstream of MDC1 following a DNA break in G1, namely histone ubiquitination, 53BP1 localization to the DNA break, and NHEJ. Thus, ASF1A phosphorylation by Chk1 is essential for DNA break repair by NHEJ in G1.
Graphical Abstract
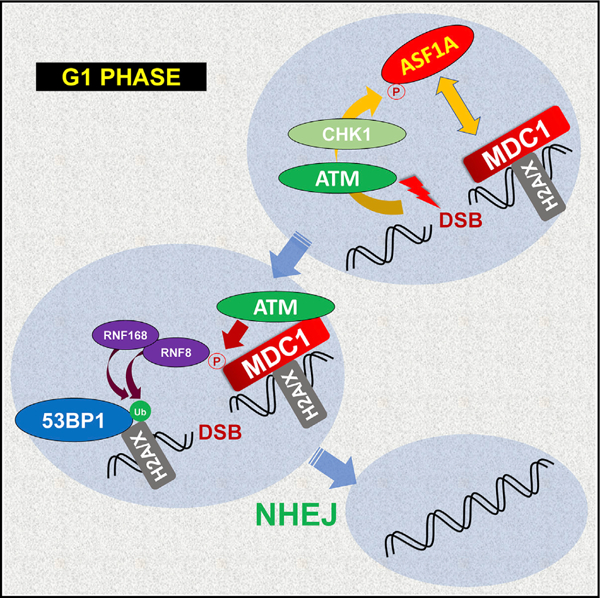
In Brief
Lee and Dutta show that ATM-dependent Chk1 requires ASF1A phosphorylation at Ser-166 to promote NHEJ repair in G1 phase at DNA double strand breaks. The phosphorylation facilitates 53BP1 localization and histone ubiquitination at the breaks by promoting its interaction with MDC1 to activate the signal cascade in the H2A/X-MDC1-ATM-RNF8 axis.
INTRODUCTION
DNA double-strand breaks (DSBs) are very toxic to cells. DSBs are generated by intrinsic and extrinsic sources, such as reactive oxygen species and ionizing radiation, and are also produced by diverse physiological events such as meiosis, telomere maintenance, V(D)J recombination, and immunoglobulin G (IgG) class switching (Pandita, 2002; Scott and Pandita, 2006; Iyama and Wilson, 2013). Unrepaired or misrepaired DSBs affect genomic integrity and have pathologic outcomes including abnormal development, premature aging, and tumorigenesis (Helleday et al., 2007; Jackson and Bartek, 2009; Li et al., 2007). In mammals, there are two major DSB repair pathways, non-homologous end joining (NHEJ) and homologous recombination (HR) (Shrivastav et al., 2008). The pathway choice between NHEJ and HR in repairing DSB depends on the cell cycle (Her and Bunting, 2018). Due to requirement of a long homology of a sister chromatid after DNA replication, HR participates in DSB repair in S/G2 phase of the cell cycle (Delacôte and Lopez, 2008; Takata et al., 1998). On the other hand, NHEJ is responsible for repairing ~75% of DSBs present in all phases of the cell cycle, and this mode of repair tends to be more mutagenic than HR (Mao et al., 2008).
Whether a DSB is repaired by HR or NHEJ is dependent on the important determinants, 53BP1 and BRCA1 (Panier and Boulton, 2014). 53BP1 recruitment at DSBs prevents end resection by antagonizing the access of BRCA1-CtIP complex, and this resection is very critical for strand-invasion in HR (Symington, 2016; Zimmermann and de Lange, 2014). In addition, cell-cycle-dependent post-translational modifications (PTMs) that occur on repair factors contribute to the pathway choice for DSB repair. In S phase, phosphorylation of CtIP by cyclin-dependent kinase (CDK) stimulates end resection by promoting the assembly of MRN-CtIP-BRCA1 complex (Chen et al., 2008; Huertas and Jackson, 2009). RNF138 promotes HR by ubiquitinating KU80 in S/G2 phase, which facilitates removal of the NHEJ-promoting complex KU70/80 from DSB ends (Ismail et al., 2015). Active CDK in S/G2 facilitates the interaction of RECQL4 with Ku70 to suppress NHEJ (Lu et al., 2017). Conversely, in G1 phase phosphorylation of 53BP1 by ataxiatelangiectasia mutated (ATM) prevents access of BRCA1 to suppress HR (Feng et al., 2015).
Chk1 is primarily activated in S phase by ataxia telangiectasia and Rad3-related (ATR), which senses single-stranded DNA produced DNA replication stress (Blackford and Jackson, 2017). However, Chk1 can also be activated by ATM: Chk1 interacts with meiotic chromosomes in an ATM-dependent manner, and ATM-dependent Chk1 activation in response to ionizing radiation suppresses tousled-like kinase 1 (TLK1) in S phase to prevent cell-cycle progression (Flaggs et al., 1997; Gatei et al., 2003; Groth et al., 2003). Although the best known function of Chk1 is cell-cycle arrest through the phosphorylation of the CDC25 family of phosphatases (Patil et al., 2013), Chk1 also promotes HR by phosphorylating Rad51 to facilitate its assembly onto single-stranded DNA (ssDNA) (Sørensen et al., 2005). Chk1 can indirectly support NHEJ by stabilizing DNA-PKcs or through the phosphorylation of a SET domain containing methyltransferase, Metnase, which methylates H3K36 near DSB and thus helps assemble the co-factor of DNA-PKcs, KU70/80 (Goudelock et al., 2003; Hromas et al., 2012). However, Chk1 substrates involved in NHEJ downstream of DNA-PK activation have not been identified. It is also not known whether Chk1, usually considered to be activated by ongoing replication in S phase, has any role in DSB repair in G1.
ASF1 plays a role in nucleosome assembly by delivering histone H3-H4 heterodimer to the histone chaperone CAF-1 (Park and Luger, 2008), and there are two paralogs of ASF1, ASF1A, and ASF1B, in higher eukaryotes (De Koning et al., 2007; Munakata et al., 2000; Silljé and Nigg, 2001). They share a common function as histone chaperones, but also have distinct functions. For example, ASF1A, but not ASF1B, promotes H3 acetylation at lys56 on newly synthesized histones and checkpoint recovery after DNA damage repair (Das et al., 2009, 2014). We and others have recently reported a role of ASF1A in NHEJ- and in HR-mediated repair independent of its histone chaperone activity: it promotes NHEJ by interacting with MDC1 and promoting the latter’s association and phosphorylation by ATM (Lee et al., 2017), while it promotes HR by facilitating Rad51 assembly onto ssDNA (Huang et al., 2018). In this study, we found that DSB-induced phosphorylation of ASF1A at S166 promotes its interaction with MDC1, which subsequently facilitates the MDC1 phosphorylation by ATM, histone ubiquitination, and 53BP1 recruitment at DSBs by activating the signal cascade in the H2A/X-MDC1-ATM-RNF8 axis. NHEJ repair in G1 phase requires ATM-dependent Chk1 activity in phosphorylating ASF1A at S166. Thus, our findings demonstrate how ATM, Chk1, and ASF1A promote NHEJ repair in G1 phase.
RESULTS
ASF1A phosphorylation at Ser-166 is required for its interaction with MDC1 on DSBs
We previously reported that ASF1A, not ASF1B, promotes NHEJ repair through its interaction with MDC1 in DSB repair, independent of its histone chaperone activity (Lee et al., 2017). ASF1A foci induced by DSBs overlap with MDC1 (Lee et al., 2017) as well as 53BP1, indicating that the interaction between ASF1A and MDC1 occurs at DSBs (Figure S1A). We wondered what made ASF1A different from ASF1B in regards to its role for NHEJ repair, and therefore checked ASF1A and ASF1B interaction with MDC1 on DSBs (Figures 1A and 1B). Although hemagglutinin HA-ASF1A or HA-ASF1B immunoprecipitates both co-precipitated histone H3 and tousled-like kinase 1 (TLK1), MDC1 was exclusively co-immunoprecipitated with ASF1A (and not ASF1B) on bleomycin treatment (Figure 1B). Because the C-terminal amino acid (aa) sequence between ASF1A and ASF1B is poorly conserved, we speculated that the C-terminal difference of ASF1A contributed to its interaction of MDC1 (Figure 1A). Co-immunoprecipitation of MDC1 with wild-type HA-ASF1A was robustly increased by bleomycin treatment, indicating that DSBs promote the ASF1A-MDC1 interaction (Figure 1C). The C-terminal deletion mutant, 1–170 amino acid (aa) of HA-ASF1A co-immunoprecipitated MDC1, but 1–160 aa of HA-ASF1A showed decreased interaction with MDC1 without decreasing interaction with histone H3 and chaperone CAF-1. This result indicates that amino acids 160–170 of ASF1A interact with MDC1 on DSB, independent of ASF1A’s histone chaperone function.
Figure 1. ASF1A phosphorylation at Ser-166 is required for its interaction with MDC1 on DSBs.
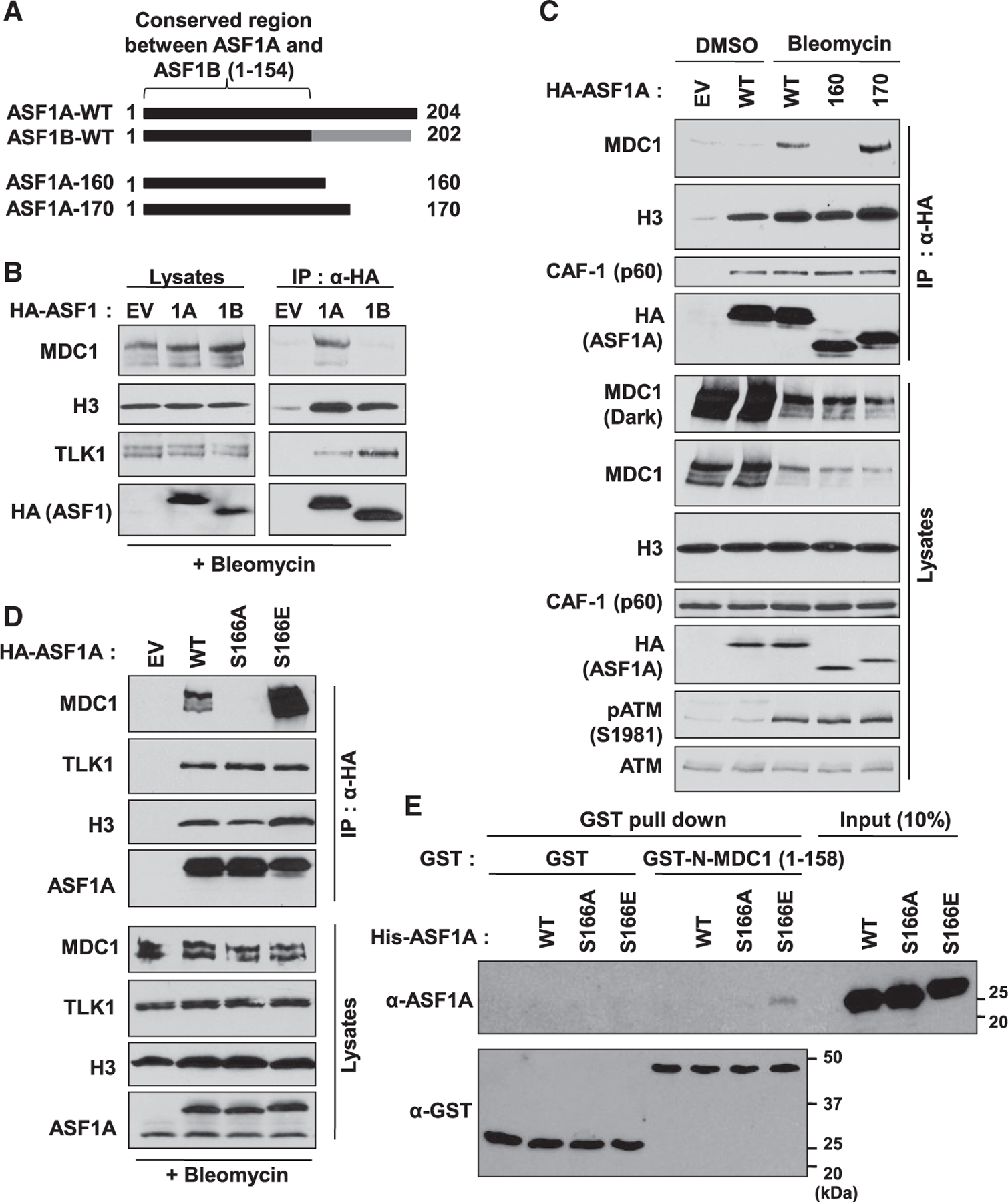
(A) Poor conservation of C-terminal ASF1A and ASF1B in humans, and C-terminal deletion mutants of ASF1A with HA tag used in (C).
(B) MDC1 interaction with ASF1A, not ASF1B, in presence of DSBs. HA-tagged ASF1A or ASF1B was immunoprecipitated with anti-HA antibody conjugated beads in HEK293T cells after 20 µg/mL bleomycin treatment for 1 h. Shown are immunoblots of immunoprecipitates and lysates. EV, empty vector; 1A, HA-ASF1A; 1B, HA-ASF1B.
(C) 160–170 amino acids of C-terminal ASF1A required for its interaction with MDC1. Immunoprecipitation (IP) was performed as described in (B).pATM, a marker for DSBs.
(D) Phospho-mimetic mutation of Ser-166 in ASF1A facilitates its interaction to MDC1. The wild-type (WT), Ser166Ala (S166A) or Ser166Glu (S166E) mutant of HA-ASF1A was applied to the IP.
(E) In vitro binding of the phospho-mimetic ASF1A to recombinant FHA domain of MDC1. Recombinant GST or GST-tagged FHA domain of MDC1 (aa 1–158) was incubated with recombinant WT or S166 mutants of his-tagged ASF1A and then immunoprecipitated by glutathione beads followed by immunoblotting against the indicated antibodies.
The FHA domain of MDC1 is required for binding to multiple phospho-proteins (Coster and Goldberg, 2010), and serine-166 (S166) of ASF1A is one of the known phosphorylation sites residing between amino acids 160–170 (Bruinsma et al., 2016; Klimovskaia et al., 2014). To test if S166 phosphorylation of ASF1A is required for its binding to MDC1, we generated phospho-defective and phospho-mimetic mutants of HA-ASF1A by substituting serine-166 to alanine (S166A) and gluta-mic acid (S166E), respectively. Although the interaction with histone H3 and TLK1 was not altered between wild-type and S166 mutants of HA-ASF1A, S166E strongly co-precipitated endogenous MDC1 even more favorably than wild-type (Figure 1D). On the other hand, S166A immunoprecipitates failed to co-precipitate MDC1. To confirm this result and avoid the possibility of any indirect interaction, we performed in vitro protein binding assay using purified recombinant GST tagged FHA domain of MDC1 (GST-N-MDC1), His6 tagged-wild-type, -S166A, and -S166E forms of ASF1A from a bacterial overexpression system (Figures 1E and S1B). Although equal amounts of recombinant GST-N-MDC1 was retrieved from co-incubation with the three forms of ASF1A, only His6-S166E was co-precipitated with the GST-N-MDC1, indicating that a phosphomimetic negative charge at Ser-166 of ASF1A promotes its interaction with the FHA domain of MDC1. Collectively, these results suggest that ASF1A interacts with MDC1 through its phosphorylation at Ser-166 on DSBs, independent of its histone chaperone activity.
Phosphomimetic S166E mutation in ASF1A promotes 53BP1 localization and NHEJ repair at DSBs
53BP1 localization at DSBs promotes NHEJ repair over HR, and we previously found that ASF1A-deficient cells were defective in DSB localization of 53BP1 and NHEJ repair (Lee et al., 2017; Panier and Boulton, 2014). Therefore, we examined whether the defects shown in ASF1A-deficient cells were recovered by the S166E mutant. U2OS or U2OS cell lines stably expressing siASF1A-resistant wild-type, S166A, or S166E of ASF1A was transfected by control small interfering RNA (siRNA) (siGL2) or siASF1A. Phosphorylation of H2AX at Ser-139 (to form γH2AX) on bleomycin treatment was not altered by ASF1A depletion and/or expression of S166 mutants, indicating ATM activation at the earliest stage of DSB repair did not require ASF1A or its phosphorylation (Figure 2A). 53BP1 focus formation was suppressed in ASF1A-depleted cells and restored in siRNA-resistant wild-type or S166E, but not in S166A-expressing cells (Figures 2B and 2C). Because MDC1 foci were not altered by ASF1A depletion and/or ASF1A mutants (Figures 2D and 2E), these results suggest that S166 phosphorylation of ASF1A plays a role in facilitating DSB localization of 53BP1 at a step after MDC1 recruitment.
Figure 2. Phosphomimetic S166E mutation in ASF1A promotes 53BP1 localization and NHEJ repair at DSBs.
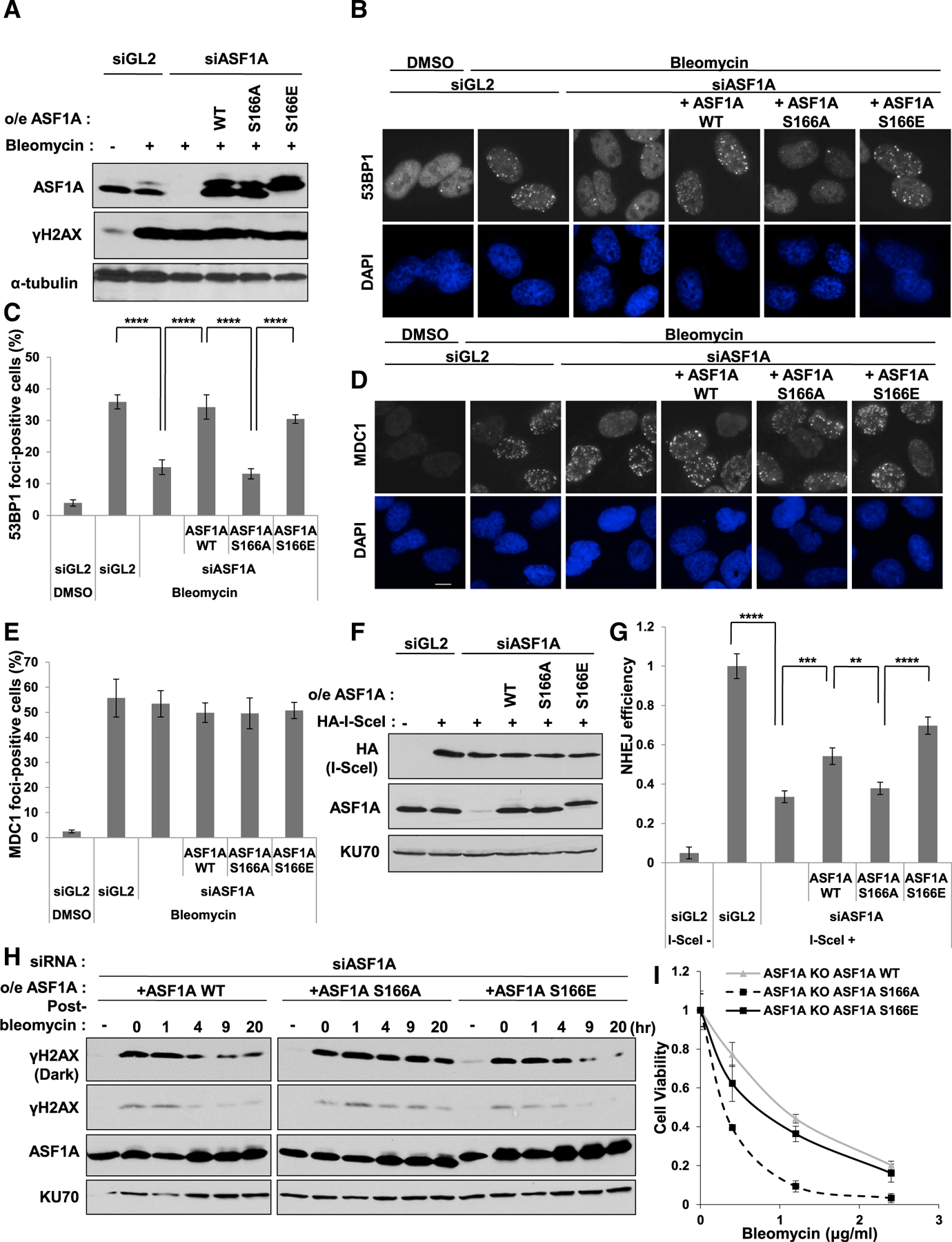
(A) γH2AX is unchanged on DSB by ectopic expression of WT or S166 mutants when endogenous ASF1A is depleted by siASF1A. Immunoblots of the whole cell extracts in the U2OS stably expressing siRNA-resistant WT or S166 mutants after transfection of control siRNA (siGL2) or siASF1A. Cells were treated with 10 µg/mL bleomycin for 1 h.
(B and C) Rescue of 53BP1 foci by siRNA-resistant WT or S166E of ASF1A in siASF1A-transfected cells. Representative images (B) and quantitation (C) are shown. Cells were treated with 10 µg/mL bleomycin for 1 h before fixation, and cells with more than 20 foci were counted. Mean ± SD of triplicates. A one-way ANOVA followed by Tukey’s post hoc test; ****p < 0.0001.
(D and E) MDC1 foci unchanged upon DSB by ectopic expression of S166 mutants of ASF1A after depletion of endogenous ASF1A. Cells with more than 10 foci were counted in (E). Mean ± SD of triplicates. Scale bar, 10 µm.
(F and G) Rescue of NHEJ efficiency by phospho-mimetic ASF1A at Ser-166 in ASF1A-depleted NHEJ/DsRed293B cells. The 293B or 293B cells stably expressing siRNA-resistant WT or S166 mutants of ASF1A were transfected with indicated siRNA and HA-I-SceI plasmids. Immunoblots of the 293B lysates are shown in (F), and NHEJ efficiency in (G) was measured as described in the STAR methods. Mean ± SD of triplicates. A one-way ANOVA followed by Tukey’s post hoc test; **p < 0.01; ***p < 0.001; ****p < 0.0001.
(H) Decrease of γH2AX after pulse-treatment of bleomycin is slowed by S166A ASF1A. The U2OS cells stably expressing siRNA-resistant WT or S166 mutants were transfected by siASF1A, then the whole cell extracts were collected at indicated time-point after pulse-treatment of 20 µg/mL bleomycin for 1 h.
(I) Cell sensitivity to bleomycin is increased by S166A ASF1A, but not by WT or S166E ASF1A. Viability measure by colony formation after 5 h treatment with bleomycin. Mean ± SD of triplicates.
NHEJ/DsRed293B cells can be used to measure cellular NHEJ efficiency (Golding et al., 2009; Mueller et al., 2013). In this system, a mutant DsRed is cut by transient expression of I-SceI endonuclease, producing two DSBs. Only intact NHEJ produces functional DsRed. Depletion of ASF1A decreased NHEJ efficiency, and siRNA-resistant ASF1A wild-type partially restored NHEJ (Figures 2F and 2G). More importantly, phosphor-mimetic S166E restored NHEJ, but S166A did not (Figure 2G). Disappearance of phospho-H2AX (γH2AX) after pulse-treatment of bleomycin is an alternative way to measure efficiency of NHEJ repair at DSBs, and was significantly retarded in S166A-expressing cells compared to ASF1A wild-type or S166E after depletion of endogenous ASF1A (Figure 2H). To determine whether the delayed repair corresponded to increased sensitivity to bleomycin, we turned to ASF1A knockout HeLa DR13–9 cells (Lee et al., 2017) expressing ASF1A-WT, -S166A, or -S166E. All cell lines had the same cell-cycle profile (data not shown). However, cells supported by ASF1A-S166A, but not ASF1A-S166E, were more sensitive to DNA damage by bleomycin (Figure 2I). These results suggest that ASF1A phosphorylation on S166 promotes the proper localization of 53BP1 as well as NHEJ repair at DSBs.
Phosphomimetic S166E mutation in ASF1A promotes histone ubiquitination by facilitating MDC1 phosphorylation by ATM and MDC1-RNF8 interaction at DSBs
53BP1 focus formation at DSBs depends on histone ubiquitination on H2A/X by a signaling cascade along the H2A/X-ATM-MDC1-RNF8 axis (Huen et al., 2007; Kolas et al., 2007; Lou et al., 2006; Mailand et al., 2007; So et al., 2009; Stucki et al., 2005). Briefly, initial activation of ATM promotes DSB localization of MDC1 by phosphorylation of H2A/X, which contributes to more stable and proper localization of ATM at DSBs. Subsequently, MDC1 phosphorylation by ATM is stimulated, and phosphorylated MDC1 recruits RNF8 ligase at DSBs through direct interaction. With the help of another E3 ligase, RNF168, histone H2A/X is ubiquitinated, which helps recruit 53BP1 to DSBs. We reported earlier that ASF1A is important for the stable association of ATM with MDC1 at DSB sites and the subsequent signaling cascade (Lee et al., 2017). Therefore, we examined whether a negative charge on S166 of ASF1A is important for the activation of this signaling cascade. Depletion of ASF1A suppressed phospho-ATM (pS1981) focus formation, which was restored by siRNA-resistant wild-type or S166E in ASF1A-depleted cells (Figures 3A and 3B). However, siRNA-resistant S166A could not rescue pATM foci suppressed by ASF1A depletion (Figures 3A and 3B). Phosphorylation of MDC1 by ATM can be measured with anti-phospho-S/TQ (Ser/Thr-Gln) antibody to recognize the phospho-residue added by ATM. Phospho-S/TQ on MDC1 was increased in ASF1A knockout cells expressing wild-type or S166E ASF1A, but not in ones expressing the S166A mutant (Figure 3C). Similarly, MDC1-RNF8 co-immunoprecipitation was decreased when S166A ASF1A replaced endogenous ASF1A, compared to the wild-type or S166E replacement (Figure 3D). These results indicate that DSB localization of ATM, phosphorylation of MDC1 by active ATM, and the MDC1-RNF8 interaction requires a negative charge on S166 of ASF1A, as can be acquired by phosphorylation.
Figure 3. Phosphomimetic S166E mutation in ASF1A promotes histone ubiquitination by facilitating MDC1 phosphorylation by ATM and MDC1-RNF8 interaction at DSBs.
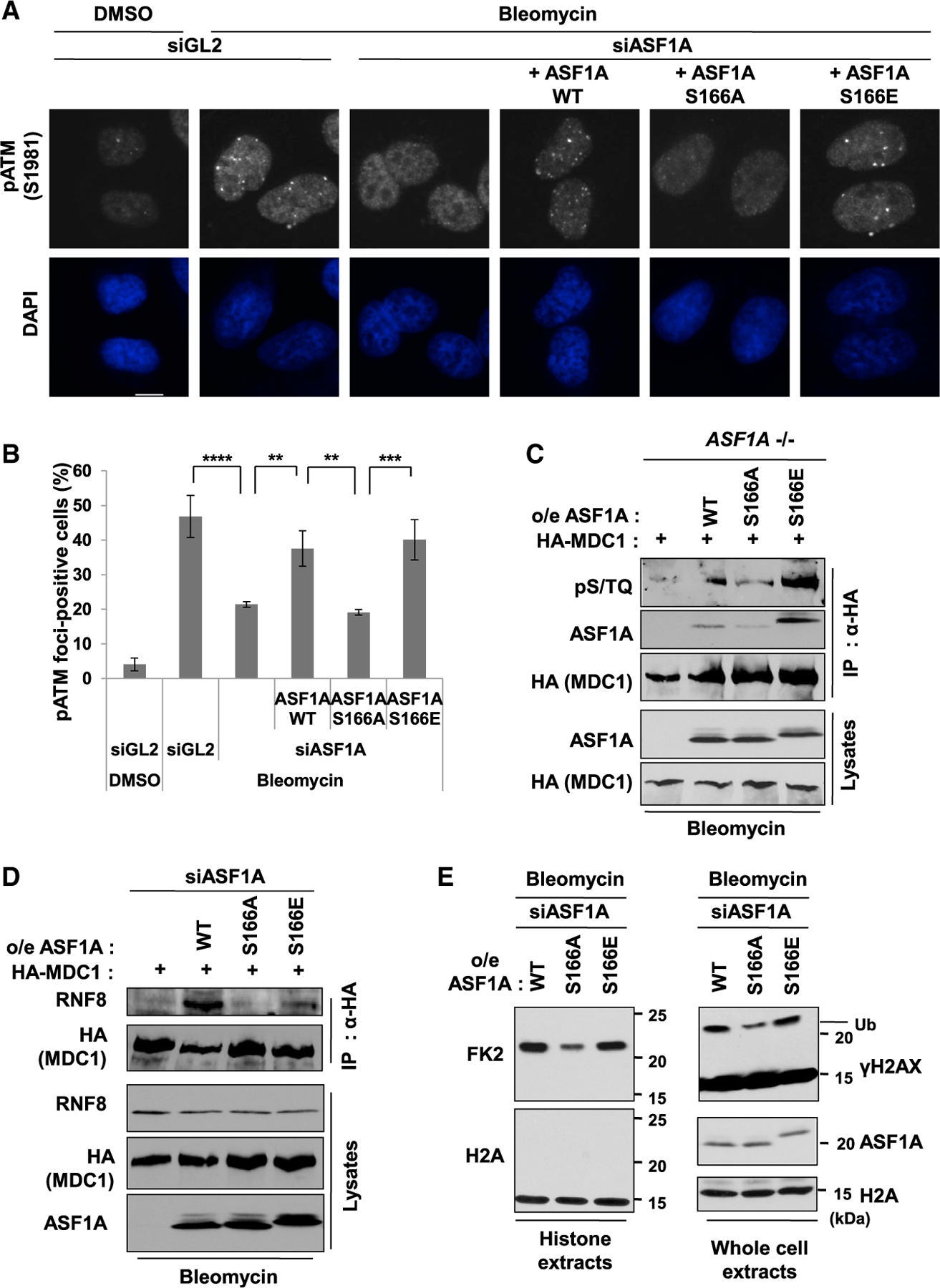
(A and B) DSB localization of phosphorylated ATM at S1981 (pS1981) promoted in presence of phosphomimetic mutation of S166 of ASF1A. Representative images (A) and quantification of cells with more than 5 foci (B). Mean ± SD of triplicates. One-way ANOVA (Tukey’s post hoc test); **p < 0.01; ***p < 0.001; ****p < 0.0001. Scale bar, 10 µm.
(C) S166E mutation of ASF1A permits ATM phosphorylation of MDC1. The ASF1A knockout 293B cells stably expressing WT or S166 mutants were transfected by HA tagged MDC1 plasmids and treated with 20 µg/mL bleomycin treatment for 2 h before harvest. Shown are immunoblots of immunoprecipitates using anti-HA antibody and lysates.
(D) S166E mutation of ASF1A promotes the MDC1-RNF8 interaction. HEK293T cells stably expressing siRNA-resistant WT or S166 mutants were transfected with HA-tagged MDC1 and siASF1A.
(E) Histone ubiquitination promoted by S166E mutation of ASF1A after DSBs. siASF1A-trans-fected U2OS stable cells with ectopic expression of siRNA-resistant WT or S166 mutants were collected, and those histone extracts (left) or whole cell extracts (right) were immunoblotted by indicated antibodies. H2A immunoblots were loading controls for both.
We next measured histone ubiquitination induced by bleomycin using FK2 antibody, which recognizes ubiquitylated proteins in histone extracts (Figure 3E, left). Alternatively, we examined mono-ubiquitination of γH2AX by the retarded mobility of the latter upon electrophoresis of whole cell extract (Figure 3E, right). Wild-type or S166E ASF1A expressing cells showed significant histone ubiquitination detected by FK2 or anti-γH2AX antibodies, but S166A ASF1A expressing cells showed significant loss of the ubiquitination. Thus, S166 phosphorylation of ASF1A is likely to enhance histone ubiquitination on H2A/X at DSBs by activating the signaling cascade along the H2A/X-ATM-MDC1-RNF8 axis culminating in 53BP1 recruitment to the DSB.
Chk1 phosphorylates Ser-166 of ASF1A on DSBs
To examine whether phosphorylation at S166 in ASF1A is upregulated by DSBs, we used a phospho-S166 specific antibody that does not recognize S166A ASF1A and found an increase in phosphorylation of S166 of wild-type ASF1A by bleomycin (Figures 4A and 4B). We wondered which kinase is responsible for this DSB-dependent phosphorylation and first focused on tousled-like kinases (TLKs) because these were already known to phosphorylate ASF1A at S166, S175, S192, and S199 even in unperturbed cells (Huang et al., 2018; Klimovskaia et al., 2014). To eliminate confusion from phosphorylation of S175/192/199, we introduced triple alanine mutations in those residues in wild-type (SAAA) or S166A (AAAA) ASF1A (Figure 4C). We then used SAAA and AAAA ASF1A as substrates in an in vitro kinase assays looking for kinases that will phosphorylate the substrate only when S166 is present. However, it is known that TLK1 is inhibited following DSB (Groth et al., 2003), and consistent with this, TLK1 purified from DNA damaged cells (Figure S2A) loses its ability to phosphorylate S166 (Figure S2B).
Figure 4. Chk1 phosphorylates Ser-166 of ASF1A on DSBs.
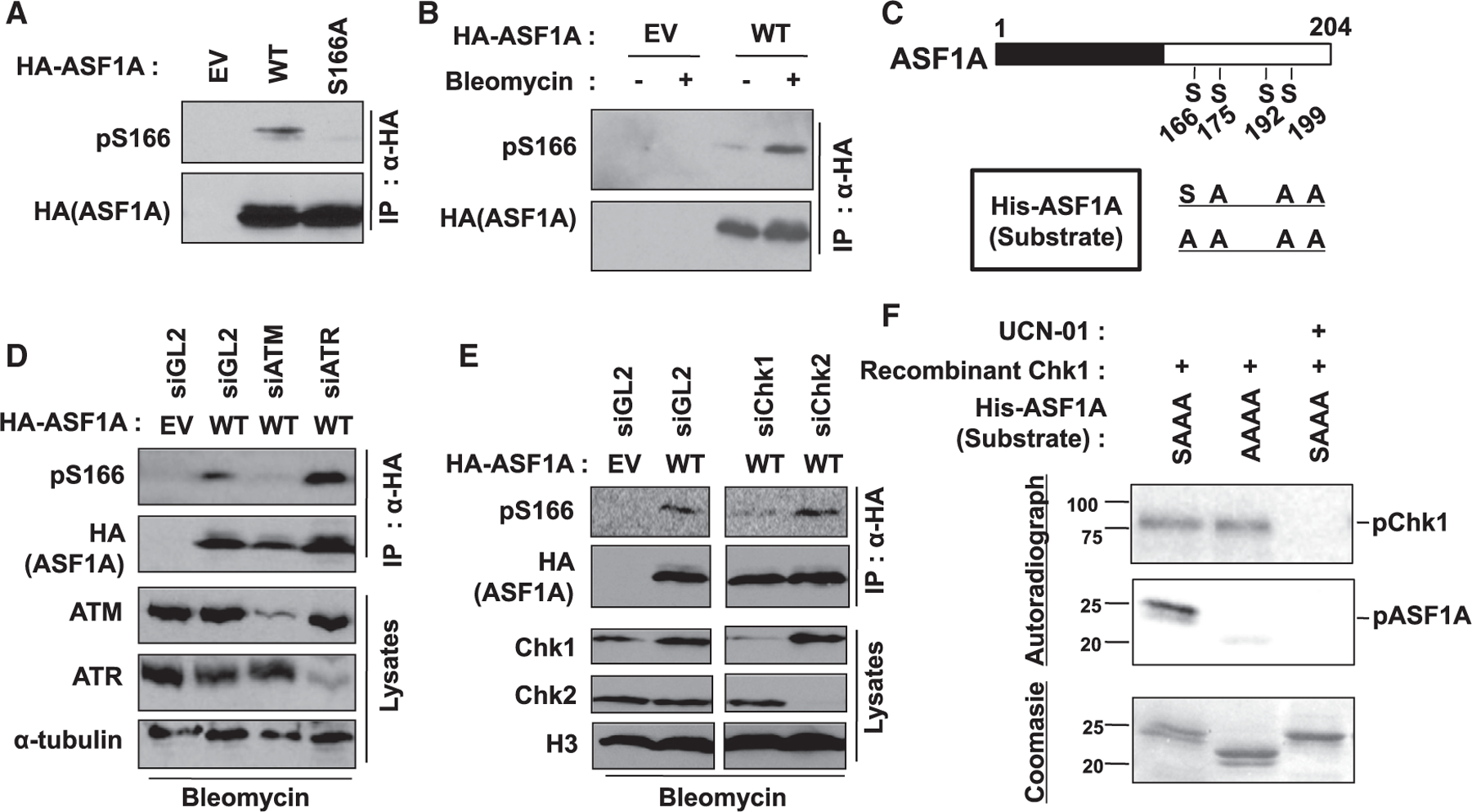
(A) Specific recognition of phosphorylated S166 in ASF1A using anti-pS166 antibody. HA-ASF1A WT or S166A mutant in HEK293T was immunoprecipitated and immunoblotted by indicated antibodies. pS166, anti-phospho-S166 antibody.
(B) Increase of phospho-S166 in ASF1A on DSBs. HEK293T cells transfected by EV or HA-ASF1A WT plasmids were incubated with or without 10 µg/mL bleomycin for 14 h.
(C) A schematic of recombinant ASF1A used as substrates in vitro kinase assays.
(D and E) ATM and Chk1 required for pS166. Indicated siRNAs were transfected twice with 24 h-interval in HEK293T cells having transient expression of HA-ASF1A WT.
(F) Chk1 directly phosphorylates S166 of ASF1A in vitro. In vitro Chk1 kinase assay was performed as described in the STAR methods. Chk1 kinase activity and S166 phosphorylation in the reactions were chased by radio-labeling of recombinant Chk1 and ASF1A, respectively. 1 µM UCN-01 was treated to inhibit Chk1 activity.
We next checked the requirement in vivo of four kinases that are known to be activated by DNA damage by siRNA knockdown of specific kinases. ATM, but not ATR, and Chk1, but not Chk2, were responsible for phosphorylation of S166 after DSB (Figures 4D and 4E). In vitro, however, S166 was not phosphorylated by ATM (Figure S2C). In contrast, recombinant Chk1 phosphorylated SAAA-ASF1A in vitro, and not the AAAA-ASF1A, even though the kinase was equally active in both lanes, as evident from the self-phosphorylation (Figure 4F). The S166 phosphorylation was repressed by UCN-01 confirming that Chk1 is directly phosphorylating the S166 of ASF1A in vitro through its catalytic activity. Therefore, we concluded that (1) ATM is the apical kinase on DSB, and (2) Chk1 is responsible for ASF1A phosphorylation at S166 on DSB.
Chk1 promotes NHEJ by facilitating histone ubiquitination on H2AX and 53BP1 localization at DSBs in G1 phase and is itself activated by ATM
Because ASF1A phosphorylation at S166 is required for NHEJ repair, and Chk1 is responsible for the phosphorylation on DSBs, we examined if depletion or inhibition of Chk1 suppressed DSB repair in an NHEJ assay (Figure 5A). The results are plotted to indicate how much a given manipulation impairs NHEJ. For example, depletion of 53BP1 impairs NHEJ by 93% (0.93), whereas depletion of BRCA1 does not decrease NHEJ. Although expression of HA-I-SceI nuclease was not suppressed by Chk1 depletion or inhibition in NHEJ/DsRed293B cells (Figures S3A and S3B), NHEJ was significantly diminished when cells were treated by siChk1 or Chk1 inhibitors, MK-8776 or UCN-01 (Figure 5A). Consistent with our previous report, depletion of ASF1A decreased NHEJ by 47% (Lee et al., 2017). Depletion of both Chk1 and ASF1A showed no further decrease of NHEJ efficiency compared to single depletion, suggesting that Chk1 and ASF1A play a role in the same pathway to promote NHEJ. Collectively, these results support that Chk1 is required for NHEJ repair through its action on ASF1A.
Figure 5. Chk1 promotes NHEJ by facilitating histone ubiquitination on H2AX and 53BP1 localization at DSBs in G1 phase and is itself activated by ATM.
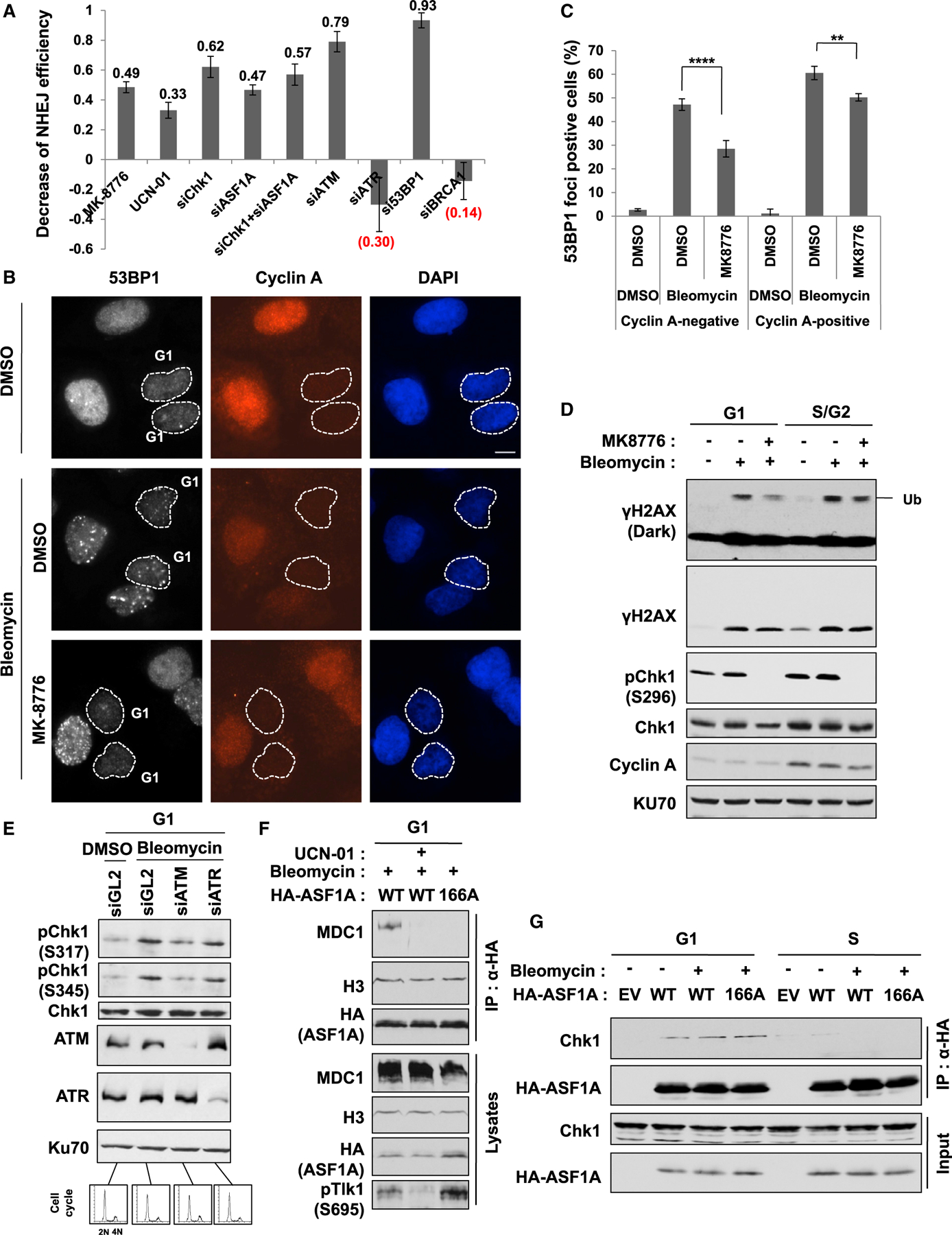
(A) Chk1 is required for NHEJ repair. NHEJ efficiency was measured as described in Figure 2G, and the decrease of NHEJ efficiency was calculated by subtracting the value in each siRNA or inhibitor treatment from controls (siGL2 or DMSO). 293B cells were transfected with siGL2 or indicated siRNA twice at 24-h intervals. 5 µM MK-8776 and 600 nM UCN-01 were added at 20 h and 5 h before harvest, respectively. Mean ± SD of triplicates.
(B and C) Chk1 inhibition decreases 53BP1 foci in G1 and S/G2 cells. U2OS cells were treated with 20 µM MK8776 for 1 h followed by 20 µg/mL bleomycin for additional 1 h. Representative images (B) and quantitation of 53BP1 foci-positive cells in cyclin A-negative (G1) and -negative (S/G2) cells (C) are shown. Cells in dashed lines indicate cyclin A-negative cells. Mean ± SD of triplicates. One-way ANOVA (Tukey’s post hoc test); **p < 0.01; ****p < 0.0001. Scale bar, 10 µm.
(D) Histone ubiquitination suppressed by Chk1 inhibition in G1, not S/G2 phase, on DSBs. U2OS cells were treated with 20 µM MK8776 for 1 h followed by 20 µg/mL bleomycin for additional 1 h in G1- or S/G2-enriched cells. Decreased autophosphorylation of Chk1 at S296 (pS296-Chk1), used as a measure of Chk1 activity.
(E) ATM-dependent phosphorylation of Chk1 in G1 phase on DSBs. HeLa DR13–9 cells were accumulated in G1 phase from nocodazole release followed by 20 µg/mL bleomycin for 1 h. Cell-cycle profiles by fluorescence-activated cell sorting (FACS) analysis at the bottom.
(F) ASF1A interaction with MDC1 requires Chk1 activity and the S166 phosphorylation in G1 phase. HEK293T cells were transfected by HA-ASF1A WT or S166A plasmids and accumulated in G1 by nocodazole release. 500 nM UCN-01 was treated for 1 h followed by bleomycin treatment. Decreased phosphorylation of TLK1 at S695 (pS695-TLK1) was used as a measure for Chk1 inactivation.
(G) Co-immunoprecipitation of ASF1A with Chk1 seen in G1 phase, but not in S phase.
We examined whether the Chk1-mediated ASF1A phosphorylation is important for DSB repair in a specific cell-cycle phase, by marking the S/G2 cells through immunostaining for cyclin A. 53BP1 foci were induced by bleomycin in both G1 (cyclin A negative) and S/G2 (cyclin A positive) cells, and the foci were significantly suppressed by MK-8776 in both phases (Figures 5B and 5C), but the inhibitory effect was more pronounced in G1. The suppression of 53BP1 foci by UCN-01 and MK-8776 in G1 phase was observed even in another cell line, HeLa DR13–9, indicating that it is not cell line-specific suppression (Figures S3C and S3D). This suggested that Chk1 contributes to 53BP1 foci formation in both G1 and S phase of the cell cycle.
Because Chk1 is known to be required for 53BP1 localization at sites of stalled replication, which is independent of γH2AX (Sengupta et al., 2004), we decided to focus on G1 cells where the Chk1-mediated activation of the ASF1A-MDC1-RNF8-histone ubiquitination pathway could be important for 53BP1 focus formation. Indeed, mono-ubiquitination on histone H2AX induced by DSBs was selectively diminished by MK8776 in G1 but not S/G2 cells (Figure 5D). The activating phosphorylation of Chk1 on S317 and S345 in G1 cells on bleomycin treatment was decreased by depletion of ATM, but not ATR, consistent with our conclusion from Figure 4D that ATM, but not ATR, is required for phosphorylation of ASF1A-S166 (Figure 5E). ASF1A interaction with MDC1 in G1 cells is also suppressed by UCN-01 or by S166A mutation of ASF1A, indicating that the MDC1-ASF1A interaction following DSB in G1 is dependent on Chk1 activity and on ASF1A phosphorylation on S166 (Figure 5F). HA-ASF1A immunoprecipitates contain endogenous Chk1 protein in G1 cells, but the interaction is not altered by DSBs or S166A mutation in HA-ASF1A (Figure 5G). It is difficult to restrict the action of I-SceI endonuclease to G1 phase or to ensure that all repairs have been completed in G1 phase, which is why we could not directly analyze the effect of Chk1 depletion of ASF1A depletion on NHEJ selectively in G1 phase. However, most NHEJ repair in asynchronous cells occurs in G1, and we showed in Figure 5A that such repair is inhibited by Chk1 inhibition or ASF1A depletion. Overall, these results suggest that Chk1 promotes NHEJ repair in G1 by phosphorylating ASF1A on S166 to facilitate the MDC1-ASF1A interaction, histone ubiquitination on H2AX, and 53BP1 localization at DSBs.
Suppression of NHEJ, histone ubiquitination on H2AX, and 53BP1 localization at DSBs in Chk1 deficiency is alleviated by phosphomimetic ASF1A at Ser-166
We next examined if Chk1 is responsible for MDC1 phosphorylation by ATM through the induction of ASF1A phosphorylation alone. Phosphorylation of endogenous MDC1 by ATM on bleomycin treatment was decreased by MK8776 in G1 cells (Figure 6A, 1st and 2nd lanes). Although the decrease of MDC1 phosphorylation on Chk1 inhibition was reversed by wild-type or S166E ASF1A expression, it was not reversed by S166A ASF1A (Figure 6A). The MDC1-RNF8 interaction induced by bleomycin treatment was reduced by MK8776, but overexpression of S166E, and not S166A, robustly rescued the interaction despite Chk1 inhibition (Figure 6B). These results indicate that phospho-mimetic mutation on S166 of ASF1A is able to rescue the signal cascade of ATM-MDC1-RNF8 axis despite Chk1 inhibition.
Figure 6. Suppression of NHEJ, histone ubiquitination on H2AX and 53BP1 localization at DSBs in Chk1 deficiency is alleviated by phosphomimetic ASF1A at Ser-166.
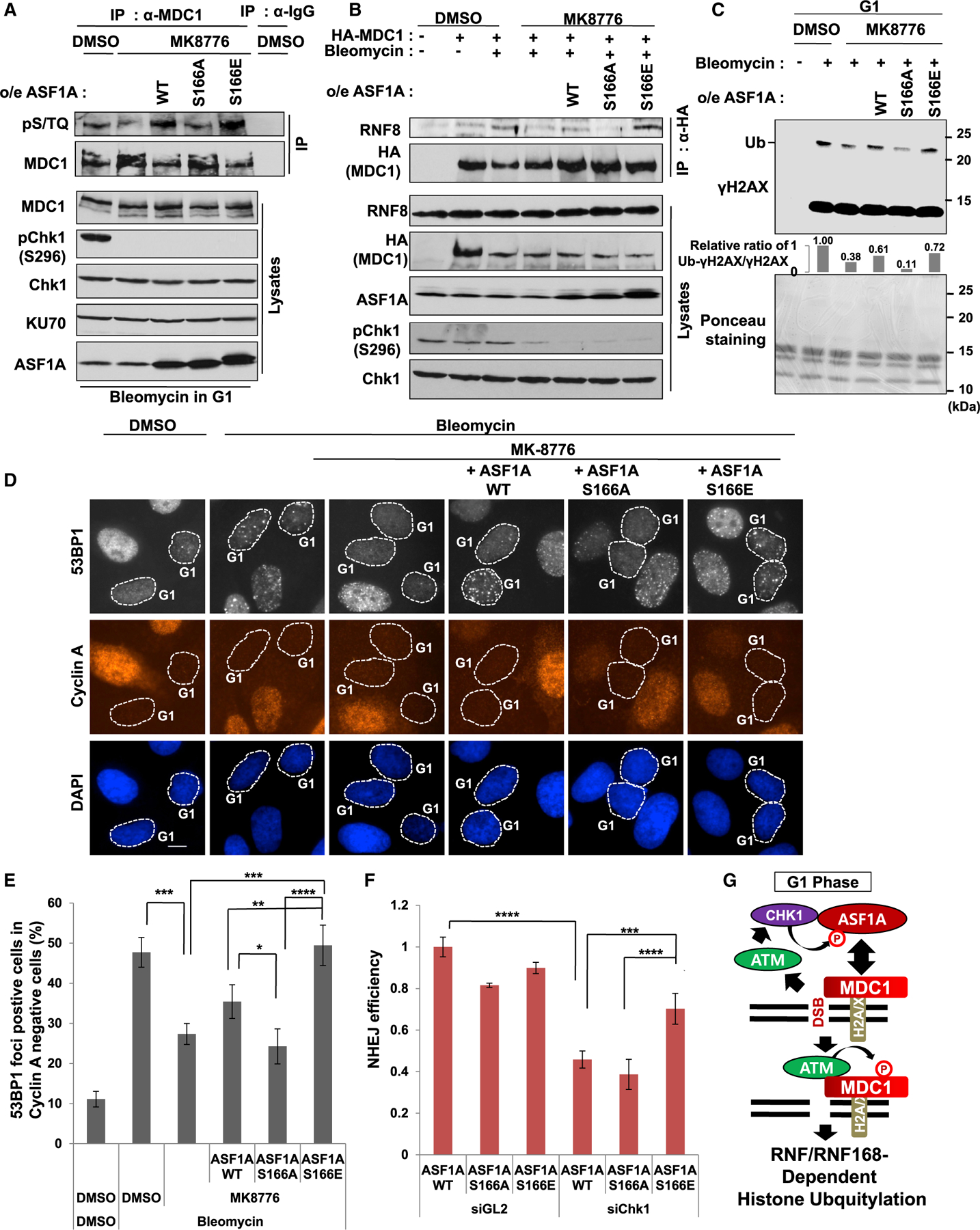
(A) MDC1 phosphorylation by ATM suppressed by Chk1 inhibition is recovered by expression of ASF1A WT or S166E in G1 cells. IPs with anti-IgG or anti-MDC1 antibodies were performed using nocodazole-released HEK293T cells. DMSO or 20 µM MK8776 was treated in G1-accumulated 293T cells. After 1 h, 20 µg/mL bleomycin was treated for 2 hr before harvest.
(B) MDC1-RNF8 interaction suppressed in Chk1 inhibition is recovered by expression of S166E. Asynchronous HEK293T with or without stable expression of WT or S166 mutants were sequentially treated with MK8776 and bleomycin with 1-h interval.
(C) Rescue of histone ubiquitination by overexpression of WT or S166E of ASF1A in G1 cells having Chk1 inhibition. Acidic histone extraction was performed using nocodazole released U2OS cells with DMSO or MK8776. Ponceau staining of the nitrocellulose membrane visualizes enriched histones as a loading control, and immunoblots for S phase histone extracts and whole cell extracts are shown in Figure S4.
(D and E) Rescue of 53BP1 foci by overexpression of ASF1A WT or S166E on Chk1 inhibition in G1 cells. Representative images (D) and quantitation of 53BP1 foci-positive cells in G1 (E) are shown. Cells with more than 10 foci were counted. Scale bar, 10 µm. Mean ± SD of triplicates. One-way ANOVA (Tukey’s post hoc test); *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
(F) Rescue of NHEJ efficiency by S166E expression of ASF1A in Chk1 deficiency. The 293B cells stably expressing ASF1A WT or S166 mutants were transfected with siChk1 and HA-I-SceI plasmids. Immunoblots of the 293B lysates are shown in Figure S4. Mean ± SD of triplicates. A one-way ANOVA followed by Tukey’s post hoc test; ***p < 0.001; ****p < 0.0001.
(G) Proposed model for a role of Chk1 in NHEJ repair through ASF1A phosphorylation in G1 phase. Details are shown in the discussion.
The decrease of mono-ubiquitination of H2AX on Chk1 inhibition was restored by overexpression of wild-type or S166E ASF1A in G1 cells, but not by S166A ASF1A (Figure 6C). The effect was specific for G1 cells, and in fact, the γH2AX monoubiquitination in S/G2 cells was not suppressed by MK8776, nor was it stimulated selectively by S166E ASF1A (Figures S4A and S4B). 53BP1 focus formation was suppressed by Chk1 inhibition in G1 (cyclin A negative) cells, and this was fully rescued by S166E ASF1A in G1 cells, but not by S166A ASF1A (Figures 6D and 6E). Thus, the phospho-mimetic mutation on S166 of ASF1A is able to rescue histone ubiquitination and 53BP1 foci formation in G1 despite Chk1 inhibition.
Finally, we examined whether suppression of NHEJ repair on Chk1 deficiency can be restored by phospho-mimetic ASF1A (Figures 6F and S4C). Compared to control siRNA, siChk1 transfection significantly suppressed NHEJ efficiency when cells expressed wild-type or S166A ASF1A, but not when they expressed S166E ASF1A (Figures 6F and S4C), indicating that S166E expression alleviates NHEJ repair suppression by Chk1 inhibition.
In all these experiments, wild-type ASF1A variably and partially rescued the MDC1–53BP1 signaling axis despite the Chk1 inhibition. This suggests that either residual Chk1 kinase activity or an alternate kinase like TLK1 can add the negative charge on S166 even in the presence of Chk1 inhibitors and partially activate the pathway when wild-type ASF1A is overexpressed.
However, it is also clear that the expression of phosphomimetic S166 mutant strongly rescued the signal cascade from MDC1 phosphorylation to NHEJ repair in cells in G1, despite Chk1 inhibition, whereas this restoration was not seen with phospho-defective mutant of ASF1A. Therefore, we conclude that Chk1 promotes NHEJ repair at DSBs in G1 phase through ASF1A phosphorylation at S166.
DISCUSSION
ASF1A promotes NHEJ repair by facilitating ubiquitin-dependent recruitment of 53BP1 to DSBs (Lee et al., 2017). The interaction of ASF1A with MDC1 enhances the latter’s interaction with ATM at DSBs followed by phosphorylation of MDC1 and activation of the signaling cascade along the MDC1-RNF8-RNF168-histone ubiquitination-53BP1 axis. An important question is how DSB is sensed by ASF1A to promote its interaction with MDC1. Our findings support the model that DSB activates Chk1 through ATM, and the ASF1A phosphorylation by Chk1 promotes NHEJ in G1 phase (Figure 6G). MDC1 is located at DSBs by recognizing γH2AX, and the phosphorylated ASF1A directly binds to the FHA domain of MDC1. Subsequently, ASF1A is replaced by ATM in its interaction with MDC1 helping the continued localization of ATM at DSB, and MDC1 is thereby phosphorylated by ATM at threonine residue T4 to recruit E3 ligase, RNF8 (Huen et al., 2007; Lee et al., 2017; Mailand et al., 2007). Finally, RNF8, with another E3 ligase RNF168, ubiquitinates histone H2A/X at DSBs through which 53BP1-Rif1 and BRCA1-RAP80 complexes are recruited to DSBs to promote NHEJ by suppressing end-resection.
ASF1A directly binds to MDC1 after the former is phosphorylated on S166. Phospho-mimetic S166E of ASF1A strongly interacted with MDC1 when compared with wild-type and phospho-defective S166A both in vivo and in vitro (Figure 1). Treatment with bleomycin increased interaction of ASF1A with MDC1 and the phosphorylation of ASF1A on S166 (Figures 1C and 4B). These data suggest that DSB-inducible phosphorylation of ASF1A at S166 facilitates its interaction with MDC1. MDC1 recruits repair proteins through its FHA domain that is a phospho-protein binding module (Coster and Goldberg, 2010). Until now, four serine residues (S166/175/192/199) in C-terminal tail of ASF1A are known to be phosphorylated by kinases such as DNA-PKcs and TLKs (Huang et al., 2018; Klimovskaia et al., 2014). We excluded the role of S175/192/199 in the interaction with MDC1 by showing that ASF1A deletion mutant, 1–170 aa, interacted with MDC1 in a manner comparable to wild-type ASF1A on DSBs. In addition, in vitro binding of recombinant FHA domain of MDC1 to S166E of ASF1A, not S166A, supported our conclusion that ASF1A directly binds to FHA domain of MDC1 after S166 phosphorylation (Figure 1E). Although ASF1A is required to promote the interaction of MDC1 with RNF8, the irreversible and stronger binding of S166E to MDC1 (Figures 1D and 1E) is accompanied by a decreased association of MDC1 with RNF8 (Figure 3D), which might suggest that irreversible association of ASF1A S166E with MDC1 partly interferes with the recruitment of RNF8. Despite the decreased recruitment, the catalytic protein RNF8 is still sufficient to permit the subsequent steps of 53BP1 recruitment and NHEJ (Figures 6E and 6F).
The FHA domain of MDC1 is also utilized for interaction with ATM (Lou et al., 2006). We previously proved that there is no tripartite complex composed of ASF1A, ATM, and MDC1, and ASF1A-MDC1interaction is functionally and temporally necessary prior to ATM-MDC1 (Lee et al., 2017). Because ATM-MDC1 interaction stabilizes the DSB localization of ATM for downstream ATM-dependent signaling events (Lou et al., 2006), the increase of ATM focus formation on expression of S166E, not S166A, supports the notion that ASF1A phosphorylation promotes ATM-MDC1 interaction by a hand-off of MDC1 to ATM (Figures 3A and 3B). Notably, this chaperone activity of ASF1A on MDC1 is independent of its chaperone function on the histone H3/H4 dimer because the interaction of histone H3 with ASF1A is not altered by S166 phosphorylation (Figure 1). Intriguingly, there is a report showing that CAF-1 interaction with H3/H4 dimer is mutually exclusive of the ASF1-H3/H4 interaction, and CAF-1 promotes the transition of H3/H4 from the ASF1-associated dimer to the DNA-associated tetramer (Liu et al., 2012). Therefore, it will be interesting to further investigate if ATM acts as CAF-1 to facilitate dissociation of ASF1A from MDC1 during DSB repair.
There is less known about the role of Chk1 in promoting NHEJ repair at DSBs. In this study, we clarify the requirement of Chk1 in NHEJ by showing that either Chk1 depletion or inhibition suppresses NHEJ efficiency (Figure 5A). Unexpectedly, we found that a role of Chk1 in promoting NHEJ through ASF1A phosphorylation is limited in G1 phase. Chk1 activates the signaling cascade along the H2A/X-ATM-MDC1-RNF8-histone ubiquitination-53BP1 localization only in G1 phase. NHEJ and HR are controlled by cell cycle, and cells may use NHEJ less in S/G2 than G1 for DSB repair (Branzei and Foiani, 2008; Heidenreich et al., 2003). Therefore, Chk1’s role in promoting ASF1A-MDC1 interaction, histone ubiquitination, and 53BP1 recruitment in G1 may contribute to the greater use of NHEJ in G1. 53BP1-Shieldin complex promotes NHEJ by protecting the DSB DNA end from strand-resection by BRCA1-CtIP, which is activated in S phase (Dev et al., 2018; Feng et al., 2015). A role of 53BP1 in promoting NHEJ in G1 thus suggests that either there is enough residual strand-resection activity in G1, or 53BP1 has an additional role independent of countering BRCA1-CtIP in facilitating NHEJ.
Chk1 can be activated primarily by ATR, but also by ATM on DSBs (Blackford and Jackson, 2017; Flaggs et al., 1997; Gatei et al., 2003; Groth et al., 2003). ATR is dispensable for NHEJ (Kim et al., 2018) and cellular phosphorylation of ASF1A at S166 on DSBs. On the other hand, ATM promotes NHEJ and the cellular phosphorylation of ASF1A (Figures 4D and 5A), through the activation of Chk1 after DSB in G1 (Figure 5E). Notably, the ATM-mediated activation of Chk1 has been reported to inhibit TLK kinases in S phase and thus suppress ASF1A phosphorylation on S166 on DSBs in S phase (Groth et al., 2003; Klimovskaia et al., 2014; Krause et al., 2003). Thus, ASF1A phosphorylation is tightly regulated by ATM-dependent Chk1 pathway and contributes to the differential function of ASF1A in G1 versus S phase. In G1 phase, DSB triggers the ATM-Chk1 pathway to promote NHEJ repair through ASF1 phosphorylation. In S/G2 phase, ASF1A phosphorylation by TLKs facilitates its histone chaperone activity required for DNA replication under unperturbed condition, but the TLK-mediated phosphorylation is suppressed after DSB by ATM-Chk1 perhaps allowing another functional phosphorylation to direct ASF1A to other pathways. For example, DNA-PKcs-dependent S192 phosphorylation of ASF1A is required for HR repair (in S/G2), or TLK2-dependent S166 phosphorylation is important for recovery from DNA damage in G2 (Bruinsma et al., 2016; Huang et al., 2018). Thus, Chk1 appears to be very important for directing ASF1A to different repair pathways dependent on the phase of the cell cycle.
Finally, S166 of ASF1A turns out to be a common phosphorylation site for Chk1 and TLKs, and therefore it is possible that other substrates are also phosphorylated by both kinases. In addition, recent studies revealed that ASF1A is increased in certain cancers such as gastrointestinal, colorectal, breast, and hepatocellular cancers, and inhibition of ASF1A sensitizes cancer cells to DNA damage (Liang et al., 2017; Wu et al., 2019; Yang et al., 2018). Now that we know that ATM and Chk1 are activators of ASF1A, ATM inhibitors (Brandsma et al., 2017) and Chk1 inhibitors (Qiu et al., 2018) may be more effective in cancers with elevated ASF1A, especially when combined with radiotherapy or chemotherapy that causes DSBs.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Anindya Dutta (ad8q@virginia.edu).
Material availability
All unique reagents generated in this study are available from the Lead Contact with a completed Material Transfer Agreement.
Data and code availability
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T, HeLa DR13–9, and NHEJ/DsRed293B cells were grown in Dulbecco’s modified Eagle’s medium (Thermo Scientific) with 10% fetal bovine serum (FBS; Sigma-Aldrich) and penicillin/streptomycin (1%, GIBCO). U2OS cells were grown in McCoy’s 5A (Corning) supplemented with 10% FBS and 1% penicillin/streptomycin. NHEJ/DsRed293B cells were obtained from Dr. J. Larner (University of Virginia) and K Valerie (Virginia Commonwealth University), and HeLa DR13–9 were gifts from J. Parvin (Ohio State University). HEK293T and U2OS cells were purchased from American Type Cell Culture (Manassas, VA). For cell cycle synchronization, cells were treated with 2 mM thymidine for 16 hr, and released in fresh medium for 6 hr. Then, cells were treated with nocodazole or thymidine for 12 hr followed by release in fresh medium for 4 to 5 hr to collect G1- or S/G2-enriched cells, respectively.
METHOD DETAILS
Generation of stably expressing cell lines of ASF1A wild-type or S166 mutants
The siRNA-resistant WT, S166A or S166E of ASF1A was inserted into retrovirus vector pLHCX (Clontech). Retroviral vectors were co-transfected with retroviral packaging vector into 293T cells using Lipofectamine2000 (Invitrogen). After 2 days, the viral supernatants were filtered by a 0.45-µm syringe filter, and poured on the U2OS, HeLa DR13–9, HEK293T or NHEJ/DsRed293B cells with fresh media at a ratio of 1:1 in the presence of 8 µg/ml polybrene. For drug selection, 100~200 µg/ml hygromycin B (Sigma Aldrich) was treated over a week.
Purification of recombinant ASF1A and FHA domain of MDC1
Human ASF1A and MDC1 FHA domain (1–158 aa) were inserted into pET28A and pGEX5X-1 vectors, respectively, and transformed in BL21(DE3) competent cells (New England Biolabs). After IPTG induction by standard protocol, the harvested cells were lysed by buffer A (50 mM Tris-HCl (pH7.5), 150 mM NaCl, 0.05% NP-40) followed by addition of 20 mg/ml lysozyme for 30 min on ice. After sonication (1 s-on/3 s-off, 90 s at 20% amplitude using Sonic Dismembrator model) followed by centrifugation at 13K for 30 min, the supernatant was obtained and incubated with glutathione-4B resins (Amersham) or Ni-NTA agarose (QIAGEN) for 1 hr at 4ºC. The Ni-NTA agarose was packed in empty PD-10 columns (GE healthcare). The columns were pre-washed and eluted by buffer B (50mM NaH2PO4, 300 mM NaCl, 10% glycerol, 0.25% Tween20, 10mM beta-mercaptoethanol, 1mM PMSF) with 50, 100, and 250 mM imidazole. The glutathione-4B resins were washed with buffer A three times by repeating 10 min rotation at 4ºC followed by centrifugation. GST-fusion proteins were eluted by buffer C (50 mM Tris-HCl (pH8.0), 5% glycerol, 10 mM Glutathione). The purity was checked by running a 12% SDS-polyacrylamide gel.
In vitro binding assay
5 ng of recombinant GST or GST-MDC1 FHA domain (1–158 aa) were incubated with 5 ng of recombinant His-ASF1A WT, S166A or S166E in binding buffer (25 mM HEPES (pH 7.5), 100 mM NaCl, 0.01% TX100, 5% glycerol, 1 mM DTT). After incubation for 3 hr at 4ºC by rotation, 20 µL of glutathione-4B resins were mixed and incubated for another 30 min. The resins were intensively washed in binding buffer for 5 times, and the bound fractions were subjected to a 12% SDS-polyacrylamide gel. His-ASF1A was detected by anti-ASF1A antibody in the immunoblots.
siRNAs and Inhibitors
The sequences of the siRNAs used in this paper are as follows: siASF1A, 5′-AAGUGAAGAAUACGAUCAAGU-3′; siATM, 5′-GCGCCTGATTCGAGATCCT-3′; siATR, 5′-GACGGTGTGCTCATGCGGC-3′; siChk1, 5′-GAAGCAGUCGCAGUGAAGA-3′; siBRCA1, 5′-CCUGUCUCCACAAAGUGUG-3′; si53BP1, 5′-GCCAGGUUCUAGAGGAUGA-3′. To maximize the knockdown efficiency, each siRNA was transfected twice at a 24 hr-interval. Lipofectamine RNAi MAX (Invitrogen) was used for transfection of siRNA following the manufacturer’s instructions. Cells were treated with UCN-01 (U6508l; Sigma-Aldrich) or MK-8776 (S2735; Selleckchem) to inhibit Chk1 kinase activity.
Primers for ASF1A deletion and mutagenesis
The following primers were used for ASF1A or MDC1 deletion constructs: 1–160 aa of pcDNA3-HA-ASF1A, forward, 5′-GGGGGGAATTCATGGCAAAGGTTCAGGTGAAC-3′, and reverse, 5′-GGGGGGGGGCGGCCGCTCACAGTTTTTCTGTGTTATCTTCCCAAT-3′; 1–170 aa of pcDNA3-HA-ASF1A, forward, 5′-GGGGGGAATTCATGGCAAAGGTTCAGGTGAAC-3′, and reverse, 5′ GGGGGGGGGC GGCCGCTCATAGATTTGGATTACTGCTCTCTGC-3′; 1–158 aa of pGEX5X-1-GST-MDC1 FHA domain, forward, 5′-GGGGGGGAT CCCCATGGAGGACACCCAGGCTATTG-3′, and reverse, 5′-GGGGGGGGGCGGCCGCTTGAGTTTCTCCCTGTACTCTGGGTG-3′; S166A, S166E, S175A, S192A, and S199A of ASF1A were made using the following primers: S166A, forward, 5′-CTGGAAGATGCAGAGAGCGCTAATCCAAATCTACAGTC-3′, and reverse, 5′-GACTGTAGATTTGGATTAGCGCTCTCTGCATCTTCCAG-3′; S166E, forward, 5′-CTGGAAGATGCAGAGAGCGAGAATCCAAATCTACAGTC-3′, and reverse, 5′-GACTGTAGATTTGGATTCTCGCTCTCTGCATCTTCCAG-3′; S175A, forward, 5′-CTACAGTCACTTCTTGCAACAGATGCATTACC-3′, and reverse, 5′-GGTAATGCATCTGTTGC AAGAAGTGACTGTAG-3′; S192A, forward, 5′-GGTCCACATCAGAAAACGCACTAAATGTCATGTTAG-3′, and reverse, 5′-CTAACAT GACATTTAGTGCGTTTTCTGATGTGGACC-3′; S199A, forward, 5′-CTAAATGTCATGTTAGAAGCCCACATGGACTGCATG-3′, and reverse, 5′-CATGCAGTCCATGTGGGCTTCTAACATGACATTTAG-3′; siRNA-resistant ASF1A was made using the following primers: siASF1A, forward, 5′-GTGGGCTCTGCAGAAAGCGAAGAATATGATCAAGTTTTAGACTC-3′, and reverse, 5′-GAGTCTAAAACTTGATCATATTCTTCGCTTTCTGCAGAGCCCAC-3′.
Immunoprecipitation, immunoblotting and antibodies
HEK293T or NHEJ/DsRed293B cells were lysed in lysis buffer (20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5 mM EDTA, 0.5 mM EGTA, 5 mM MgCl2, 10 mM NaF, 0.1 mM sodium vanadate, protease inhibitors supplemented by PhosSTOP (Roche)) for 15 min. After centrifugation at 15,000 rpm for 30 min, 1–2 mg of lysates were incubated with the indicated antibodies or 12 µL of 50% slurry of EZview Red Anti-HA Affinity beads (E6779; Sigma) for 1 hr. For immunoprecipitation of endogenous MDC1, the harvested cells were briefly sonicated after lysis, and the MDC1 immuno-complex pulled down on protein G-conjugated agarose beads. After washing for 5 times with the lysis buffer, the immunoprecipitated proteins together with lysates were subjected to SDS-polyacrylamide gel electrophoresis. For anti-pS166 immunoblots, HA-ASF1A expressing plasmids were transfected into HEK293T cells, and IP was performed with EZview Red Anti-HA Affinity beads. For other immunoblotting, total cell extracts were subjected to SDS-polyacrylamide gel electrophoresis.
Antibodies for this study were as follows: anti-ASF1A (2990S), anti-γ-H2AX (S139) (2577S), anti-H2AX (2595S), anti-pS695-TLK1 (4121S), anti-p-(S/T)-ATM/ATR substrates (2851), anti-TLK1 (4125S), anti-pS296-Chk1 (2349S), anti-pS317-Chk1 (2344S), anti-pS345-Chk1 (2341S) and anti-H2A (2578S) (Cell Signaling Technology); anti-HA (sc-805, sc-7392), anti-RNF8 (sc-271462), anti-RNF168 (sc-101125), anti-KU70 (sc-5309), anti-TLK2 (sc-393506), anti-Chk1 (sc-8408S), anti-Cyclin A (sc-751), anti-GST (sc-459), α-tubulin (sc-5286), anti-ATR (sc-1887) and anti-53BP1 (sc-22760) (Santa Cruz Biotechnology); anti-53BP1 (612522; BD Biosciences); anti-CAF-1 (p60) (NB100–57523) and anti-Chk1 (NB100–464) (Novus Biological); anti-ubiquitinylated proteins antibody, clone FK2 (04–263) (EMD Millipore); anti-pS1981-ATM (ab81292), anti-ATM (ab78), anti-H3 (ab1791), anti-MDC1 (ab11171), anti-MDC1 (ab50003), and anti-RNF8 (ab105362) (Abcam)
Immunofluorescence
Immunofluorescence was performed as previously described (Lee et al., 2015). Coverslips were washed twice with PBS, and fixed with 4% paraformaldehyde for 10 min followed by permeabilization with 0.5% Triton X-100 containing PBS for 5 min. The fixed cells were incubated in the blocking solution (10% FBS in PBST (PBS containing 0.1% Triton X-100) for 30 min at room temperature or overnight at 4ºC. After the primary antibody incubation for 1–3 hr, the antibodies were detected with Alexa Fluor 555 anti-rabbit (A21429; Life Technologies) or Alexa Fluor 488 anti-mouse (A11029; Life Technologies) immunoglobulin G secondary antibody in the blocking solution for 40 min at room temperature. Vectashield Mounting Medium containing 4’, 6’-diamidino-2-phenylindole (Vector Laboratories) mounted the coverslips and the images of stained cells were obtained by Zeiss AXIO observer A-1 equipped with Zeiss EC Plan-Apochromax 63X/1.4 oil and Zeiss AXIOCAM MRC.
Colony formation assay
Cell viability was quantified in a colony formation assay using HeLa 13–9 ASF1A KO cells stably expressing ASF1A WT or S166 mutants. About 4000 cells were plated on 6 well dishes, and the indicated concentration of bleomycin was treated for 5 hr in the next day. After one week, the cell colonies were stained with crystal violet and quantified using Gene Tools software (Syngene).
Acidic histone extraction
Acidic histone extraction was performed according to Abcam’s protocol (https://www.abcam.com/protocols/histone-extraction-protocol-for-western-blot) with slight modification. 5 × 105 U2OS cells were washed twice with ice-cold PBS containing 5 mM C4H7NaO2, and lysed in 0.5 mL of triton extraction buffer (PBS containing 0.5% Triton X-100 (v/v), 2 mM PMSF, 0.02% NaN3 (w/v)) for 10 min with gentle stirring. After centrifugation at 2000 rpm for 10 min, pellets were washed in 0.25 mL of triton extraction buffer. The pellets were resuspended in 80 µL of 0.2 N HCl overnight at 4ºC, and the supernatants were neutralized by adding 8 µL of 2 M NaOH, and applied to the Bradford assay (Bio-rad) to measure protein concentration.
In vitro kinase assays
In vitro Chk1 or TLK1 kinase assay was performed in kinase buffer D (20 mM Tris-HCl pH 7.5, 10 mM MgCl2, 1 mM DTT, 25 µM ATP, 8 µCi of [γ32P] ATP). 200 ng of recombinant Chk1 (#7735; Biovision) or immunoprecipitated HA-TLK1 was incubated with 1 µg recombinant His-ASF1A for 30 min at 30ºC and the reaction stopped by addition of Laemmli buffer. UCN-01 was directly added to the reaction tube at 100 nM concentration to inhibit Chk1. Reactions were separated on 13% SDS-polyacrylamide gels. To obtain HA-TLK1 protein, HA-TLK1 expressing plasmids were transfected into HEK293T cells, and the cells were lysed in lysis buffer (20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5 mM EDTA, 0.5 mM EGTA, 5 mM MgCl2, 10 mM NaF, 0.1 mM sodium vanadate, and protease inhibitors) after 48 hr. 150 µg of lysate was mixed and incubated with EZview Red Anti-HA Affinity beads (E6779; Sigma) for 2 hr, and the beads were washed twice with lysis buffer followed by twice with kinase buffer. In vitro ATM kinase assay was performed using 200 ng of recombinant ATM (14–933; Sigma) in kinase buffer E (10 mM Tris-HCl (pH 7.5) 50 mM KCl 10 mM MgCl2 10 mM MnCl2 1 mM DTT 200 µM ATP, 4 µCi of [γ32P] ATP).
NHEJ assay
NHEJ assays were performed as described previously (Lee et al., 2017). 3 × 105 NHEJ/DsRed293B cells (Golding et al., 2009) were plated on 6 well dishes, and transfected with indicated siRNA. On the next day, 2 µg of pCβASce plasmid was transfected using Lipofectamine2000 (Invitrogen) followed by second transfection of siRNA at a 24 hr-interval with first siRNA. After 48 hr, cells were harvested, and counted in flow cytometric analysis (BD FACS Calibur and CellQuest Pro) by the FL2 and FL1 channels for the DsRed-expressing cells to measure the NHEJ repair efficiency. The percent of DsRed-positive cells in each siRNA was normalized to that of the siGL2 transfected with pCβASce. In Figure 5A, 5 µM MK-8776 and 600 nM UCN-01 were treated for 20 hr and 5hr before harvest, and the decrease of NHEJ efficiency was calculated by subtracting the value in each siRNA or inhibitor treatment from controls (siGL2 or DMSO). Disappearance of phospho-H2AX at S139 was monitored to examine endogenous NHEJ efficiency after pulse-treatment of bleomycin as described previously (Lee et al., 2017). U2OS cells stably expressing ASF1A wild-type or S166 mutants were incubated with 20 µg/ml bleomycin for 1 hr followed by wash out, then harvested at different time points. Whole cell extracts were separated in 14% SDS-polyacrylamide gels.
QUANTIFICATION AND STATISTICAL ANALYSIS
A one-way ANOVA analysis followed by Tukey’s post hoc test was used in all the statistical analyses with a p value. *, **, *** and **** standing for p < 0.05, < 0.01, < 0.001 and < 0.0001, respectively. All data were derived from at least triplicates as indicated in figure legends. All the error bars used in the figures were obtained from three independent experiments as indicated in the relevant legends, and data were presented as mean ± standard deviation (SD).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ASF1A | Cell Signaling | Cat#2990; RRID:AB_2289918 |
| phospho-(S/T)-ATM/ATR substrates | Cell Signaling | Cat#2851; RRID:AB_330318 |
| TLK1 | Cell Signaling | Cat#4125; RRID:AB_2203885 |
| γH2AX (S139) | Cell Signaling | Cat#2577; RRID:AB_2118010 |
| H2AX | Cell Signaling | Cat#2595; RRID:AB_10694556 |
| Phospho-S695-TLK1 | Cell Signaling | Cat#4121; RRID:AB_2287401 |
| Phospho-S296-Chk1 | Cell Signaling | Cat#2349; RRID:AB_2080323 |
| Phospho-S317-Chk1 | Cell Signaling | Cat#2344; RRID:AB_331488 |
| Phospho-S345-Chk1 | Cell Signaling | Cat#2341; RRID:AB_330023 |
| H2A | Cell Signaling | Cat#2578; RRID:AB_2118804 |
| HA | Santa Cruz | Cat#sc-805; RRID:AB_631618 |
| RNF8 | Santa Cruz | Cat#sc-271462; RRID:AB_10648902 |
| RNF168 | Santa Cruz | Cat#sc-101125; RRID:AB_2180105 |
| KU70 | Santa Cruz | Cat#sc-5309; RRID:AB_628453 |
| TLK2 | Santa Cruz | Cat#sc-393506 |
| Chk1 | Santa Cruz | Cat#sc-8408; RRID:AB_627257 |
| Cyclin A | Santa Cruz | Cat#sc-751; RRID:AB_631329 |
| GST | Santa Cruz | Cat#sc-459; RRID:AB_631586 |
| α-tubulin | Santa Cruz | Cat#sc-5286; RRID:AB_628411 |
| ATR | Santa Cruz | Cat#sc-1887; RRID:AB_630893 |
| 53BP1 | Santa Cruz | Cat#sc-22760; RRID:AB_2256326 |
| 53BP1 | BD Biosciences | Cat#612522; RRID:AB_2206766 |
| CAF1 (p60) | Novus Biological | Cat#NB100–57523; RRID:AB_922168 |
| Chk1 | Novus Biological | Cat#NB100–464; RRID:AB_10002158 |
| Anti-ubiquitinylated protein antibody (clone FK2) | EMD Millipore | Cat#04–263; RRID:AB_612093 |
| Phospho-S1981-ATM | Abcam | Cat#ab81292; RRID:AB_1640207 |
| ATM | Abcam | Cat#ab78; RRID:AB_306089 |
| H3 | Abcam | Cat#ab1791; RRID:AB_302613 |
| MDC1 | Abcam | Cat#ab11171; RRID:AB_297810 |
| MDC1 | Abcam | Cat#ab50003; RRID:AB_881103 |
| RNF8 | Abcam | Cat#ab105362; RRID:AB_10711502 |
| Chemicals, peptides, and recombinant proteins | ||
| Bleomycin Sulfate | Cayman chemical | Cat#9041–93-4 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat#13778150 |
| Lipofectamine 2000 Transfection Reagent | Invitrogen | Cat#11668019 |
| EZ-View Red Anti-HA Affinity Beads | Sigma | Cat#E6779; RRID:AB_10109562 |
| UCN-01 | Sigma | Cat#U6508l |
| MK-8776 | Selleckchem | Cat#S2735 |
| Chk1 | Biovision | Cat#7735 |
| Recombinant ASF1A WT | This paper | N/A |
| Recombinant ASF1A S166A | This paper | N/A |
| Recombinant ASF1A S166E | This paper | N/A |
| Recombinant ASF1A S175/192/199A | This paper | N/A |
| Recombinant ASF1A S166/175/192/199A | This paper | N/A |
| Recombinant GST | This paper | N/A |
| Recombinant GST-MDC1 FHA (1–158a.a) | This paper | N/A |
| Experimental models: cell lines | ||
| NHEJ/DsRed293B | Golding et al., 2009; Mueller et al., 2013 | N/A |
| NHEJ/DsRed293B ASF1A knockout cell line | This paper | N/A |
| NHEJ/DsRed293B ASF1A knockout cell line stably expressing ASF1A wildtype | This paper | N/A |
| NHEJ/DsRed293B ASF1A knockout cell line stably expressing ASF1A S166A mutant | This paper | N/A |
| NHEJ/DsRed293B ASF1A knockout cell line stably expressing ASF1A S166E mutant | This paper | N/A |
| HeLa DR13–9 | Ransburgh et al., 2010 | N/A |
| HeLa DR13–9 ASF1A knockout cell line | This paper | N/A |
| HeLa DR13–9 ASF1A knockout cell line stably expressing ASF1A wildtype | This paper | N/A |
| HeLa DR13–9 ASF1A knockout cell line stably expressing ASF1A S166A mutant | This paper | N/A |
| HeLa DR13–9 ASF1A knockout cell line stably expressing ASF1A S166E mutant | This paper | N/A |
| HEK293T | ATCC | CRL-3216; RRID:CVCL_0063 |
| U2OS | ATCC | HTB-96; RRID:CVCL_0042 |
| U2OS stably expressing ASF1A wildtype | This paper | N/A |
| U2OS stably expressing ASF1A S166A | This paper | N/A |
| U2OS stably expressing ASF1A S166E | This paper | N/A |
| Oligonucleotides | ||
| siASF1a-147, 5′-AAGUGAAGAAUACGAUCAAGU-3′ | Groth et al., 2005 | N/A |
| siATM, 5′- GCGCCTGATTCGAGATCCT –3′ | Lin and Dutta, 2007 | N/A |
| siATR, 5′- GACGGTGTGCTCATGCGGC –3′ | Kedde et al., 2006 | N/A |
| siBRCA1, 5′-CCUGUCUCCACAAAGUGUG-3′ | Zhu and Dutta, 2006 | N/A |
| si53BP1, 5′-GCCAGGUUCUAGAGGAUGA-3′ | Wang et al., 2002 | N/A |
| siChk1, 5′- GAAGCAGUCGCAGUGAAGA-3′ | Nakada et al., 2012 | N/A |
| Recombinant DNA | ||
| pcDNA3-HA-ASF1A WT (siASF1a-147 resistant) | This paper | N/A |
| pcDNA3-HA-ASF1A S166A (siASF1a-147 resistant) | This paper | N/A |
| pcDNA3-HA-ASF1A S166E (siASF1a-147 resistant) | This paper | N/A |
| pLHCX-ASF1A WT (siASF1a-147 resistant) | This paper | N/A |
| pLHCX-ASF1A S166A (siASF1a-147 resistant) | This paper | N/A |
| pLHCX-ASF1A S166E (siASF1a-147 resistant) | This paper | N/A |
| pGEX5X-1-MDC1 FHA domain (1–158 aa) | This paper | N/A |
| pET28A-ASF1A WT | This paper | N/A |
| pET28A-ASF1A S166A | This paper | N/A |
| pET28A-ASF1A S166E | This paper | N/A |
| pET28A-ASF1A S175/192/199A | This paper | N/A |
| pET28A-ASF1A S166/175/192/199A | This paper | N/A |
| HA-MDC1 WT | Wu et al., 2008 | N/A |
| pCβASce (I-SceI) | Addgene | Cat#26477; RRID:Addgene_26477 |
| Software and algorithms | ||
| Photoshop 7.0 | Adobe | N/A |
| ImageJ | NIH | N/A |
| CellQuest Pro | BD Biosciences | N/A |
| Flowing software Ver. 2.5 | Flowing Software | http://flowingsoftware.btk.fi/ |
| Prism 9.0 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
ASF1A phosphorylation at S166 promotes its interaction with MDC1 and NHEJ repair at DSBs
The phosphorylation helps localize 53BP1 at DSBs by activating steps downstream of MDC1
Chk1 phosphorylates S166 of ASF1A on DSBs and is activated by ATM in G1
ATM activates Chk1 in G1 to promote NHEJ repair through this pathway
ACKNOWLEDGMENTS
We thank Dr. Anja Groth for supplying anti-pS166 ASF1A antibody. We also thank the members of the Dutta laboratory for helpful discussions and Ms. Briana Wilson for her careful reading and revision of the manuscript. This work was supported by a Farrow Fellowship and NCI Cancer Center support grants P30 CA44579 to K.Y.L. and R01 CA060499 to A.D.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108680.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Brandsma I, Fleuren EDG, Williamson CT, and Lord CJ (2017). Directing the use of DDR kinase inhibitors in cancer treatment. Expert Opin. Investig. Drugs 26, 1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, and Foiani M (2008). Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol 9, 297–308. [DOI] [PubMed] [Google Scholar]
- Bruinsma W, van den Berg J, Aprelia M, and Medema RH (2016). Tousled-like kinase 2 regulates recovery from a DNA damage-induced G2 arrest. EMBO Rep 17, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Nievera CJ, Lee AY-L, and Wu X (2008). Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem 283, 7713–7720. [DOI] [PubMed] [Google Scholar]
- Coster G, and Goldberg M (2010). The cellular response to DNA damage: a focus on MDC1 and its interacting proteins. Nucleus 1, 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Lucia MS, Hansen KC, and Tyler JK (2009). CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Roy S, Namjoshi S, Malarkey CS, Jones DNM, Kutateladze TG, Churchill MEA, and Tyler JK (2014). Binding of the histone chaperone ASF1 to the CBP bromodomain promotes histone acetylation. Proc. Natl. Acad. Sci. USA 111, E1072–E1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koning L, Corpet A, Haber JE, and Almouzni G (2007). Histone chaperones: an escort network regulating histone traffic. Nat. Struct. Mol. Biol 14, 997–1007. [DOI] [PubMed] [Google Scholar]
- Delacotô F, and Lopez BS (2008). Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: the trans-S double-strand break repair model. Cell Cycle 7, 33–38. [DOI] [PubMed] [Google Scholar]
- Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. (2018). Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol 20, 954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Li N, Li Y, Wang J, Gao M, Wang W, and Chen J (2015). Cell cycle-dependent inhibition of 53BP1 signaling by BRCA1. Cell Discov 1, 15019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaggs G, Plug AW, Dunks KM, Mundt KE, Ford JC, Quiggle MR, Taylor EM, Westphal CH, Ashley T, Hoekstra MF, and Carr AM (1997). Atm-dependent interactions of a mammalian chk1 homolog with meiotic chromosomes. Curr. Biol 7, 977–986. [DOI] [PubMed] [Google Scholar]
- Gatei M, Sloper K, Sorensen C, Syljuäsen R, Falck J, Hobson K, Savage K, Lukas J, Zhou B-B, Bartek J, and Khanna KK (2003). Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J. Biol. Chem 278, 14806–14811. [DOI] [PubMed] [Google Scholar]
- Golding SE, Morgan RN, Adams BR, Hawkins AJ, Povirk LF, and Valerie K (2009). Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther 8, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudelock DM, Jiang K, Pereira E, Russell B, and Sanchez Y (2003). Regulatory interactions between the checkpoint kinase Chk1 and the proteins of the DNA-dependent protein kinase complex. J. Biol. Chem 278, 29940–29947. [DOI] [PubMed] [Google Scholar]
- Groth A, Lukas J, Nigg EA, Silljé HHW, Wernstedt C, Bartek J, and Hansen K (2003). Human Tousled like kinases are targeted by an ATM- and Chk1-dependent DNA damage checkpoint. EMBO J 22, 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groth A, Ray-Gallet D, Quivy J-P, Lukas J, Bartek J, and Almouzni G (2005). Human Asf1 regulates the flow of S phase histones during replicational stress. Mol. Cell 17, 301–311. [DOI] [PubMed] [Google Scholar]
- Heidenreich E, Novotny R, Kneidinger B, Holzmann V, and Wintersberger U (2003). Non-homologous end joining as an important mutagenic process in cell cycle-arrested cells. EMBO J 22, 2274–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Lo J, van Gent DC, and Engelward BP (2007). DNA double-strand break repair: from mechanistic understanding to cancer treatment. DNA Repair (Amst.) 6, 923–935. [DOI] [PubMed] [Google Scholar]
- Her J, and Bunting SF (2018). How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem 293, 10502–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hromas R, Williamson E, Fnu S, Lee Y-J, Park SJ, Beck BD, You JS, Laitao A, Nickoloff JA, and Lee SH (2012). Chk1 phosphorylation of Metnase/SETMAR at Ser495 enhances DNA repair but decreases replication fork restart. Oncogene 31, 4245–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T-H, Fowler F, Chen C-C, Shen Z-J, Sleckman B, and Tyler JK (2018). The Histone Chaperones ASF1 and CAF-1 Promote MMS22L-TONSL-Mediated Rad51 Loading onto ssDNA during Homologous Recombination in Human Cells. Mol. Cell 69, 879–892.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MSY, Grant R, Manke I, Minn K, Yu X, Yaffe MB, and Chen J (2007). RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell 131, 901–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huertas P, and Jackson SP (2009). Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem 284, 9558–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail IH, Gagné J-P, Genois M-M, Strickfaden H, McDonald D, Xu Z, Poirier GG, Masson J-Y, and Hendzel MJ (2015). The RNF138 E3 ligase displaces Ku to promote DNA end resection and regulate DNA repair pathway choice. Nat. Cell Biol 17, 1446–1457. [DOI] [PubMed] [Google Scholar]
- Iyama T, and Wilson DM 3rd. (2013). DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst.) 12, 620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SP, and Bartek J (2009). The DNA-damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedde M, le Sage C, Duursma A, Zlotorynski E, van Leeuwen B, Nijkamp W, Beijersbergen R, and Agami R (2006). Telomerase-independent regulation of ATR by human telomerase RNA. J. Biol. Chem 281, 40503–40514. [DOI] [PubMed] [Google Scholar]
- Kim D, Liu Y, Oberly S, Freire R, and Smolka MB (2018). ATR-mediated proteome remodeling is a major determinant of homologous recombination capacity in cancer cells. Nucleic Acids Res 46, 8311–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimovskaia IM, Young C, Strømme CB, Menard P, Jasencakova Z, Mejlvang J, Ask K, Ploug M, Nielsen ML, Jensen ON, and Groth A (2014). Tousled-like kinases phosphorylate Asf1 to promote histone supply during DNA replication. Nat. Commun 5, 3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD, Panier S, Mendez M, Wildenhain J, Thomson TM, et al. (2007). Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science 318, 1637–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause DR, Jonnalagadda JC, Gatei MH, Sillje HHW, Zhou B-B, Nigg EA, and Khanna K (2003). Suppression of Tousled-like kinase activity after DNA damage or replication block requires ATM, NBS1 and Chk1. Oncogene 22, 5927–5937. [DOI] [PubMed] [Google Scholar]
- Lee KY, Im J-S, Shibata E, Park J, Handa N, Kowalczykowski SC, and Dutta A (2015). MCM8–9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nat. Commun 6, 7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Im J-S, Shibata E, and Dutta A (2017). ASF1a promotes non-homologous end joining repair by facilitating phosphorylation of MDC1 by ATM at double-strand breaks. Mol. Cell 68, 61–75.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Vogel H, Holcomb VB, Gu Y, and Hasty P (2007). Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol 27, 8205–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Yuan X, Yu J, Wu Y, Li K, Sun C, Li S, Shen L, Kong F, Jia J, et al. (2017). Histone Chaperone ASF1A Predicts Poor Outcomes for Patients With Gastrointestinal Cancer and Drives Cancer Progression by Stimulating Transcription of b-Catenin Target Genes. EBioMedicine 21, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JJ, and Dutta A (2007). ATR pathway is the primary pathway for activating G2/M checkpoint induction after re-replication. J. Biol. Chem 282, 30357–30362. [DOI] [PubMed] [Google Scholar]
- Liu WH, Roemer SC, Port AM, and Churchill MEA (2012). CAF-1-induced oligomerization of histones H3/H4 and mutually exclusive interactions with Asf1 guide H3/H4 transitions among histone chaperones and DNA. Nucleic Acids Res 40, 11229–11239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell 21, 187–200. [DOI] [PubMed] [Google Scholar]
- Lu H, Shamanna RA, de Freitas JK, Okur M, Khadka P, Kulikowicz T, Holland PP, Tian J, Croteau DL, Davis AJ, and Bohr VA (2017). Cell cycle-dependent phosphorylation regulates RECQL4 pathway choice and ubiquitination in DNA double-strand break repair. Nat. Commun 8, 2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, and Lukas J (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, and Gorbunova V (2008). Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst.) 7, 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller AC, Sun D, and Dutta A (2013). The miR-99 family regulates the DNA damage response through its target SNF2H. Oncogene 32, 1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munakata T, Adachi N, Yokoyama N, Kuzuhara T, and Horikoshi M (2000). A human homologue of yeast anti-silencing factor has histone chaperone activity. Genes Cells 5, 221–233. [DOI] [PubMed] [Google Scholar]
- Nakada S, Yonamine RM, and Matsuo K (2012). RNF8 regulates assembly of RAD51 at DNA double-strand breaks in the absence of BRCA1 and 53BP1. Cancer Res 72, 4974–4983. [DOI] [PubMed] [Google Scholar]
- Pandita TK (2002). ATM function and telomere stability. Oncogene 21, 611–618. [DOI] [PubMed] [Google Scholar]
- Panier S, and Boulton SJ (2014). Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol 15, 7–18. [DOI] [PubMed] [Google Scholar]
- Park Y-J, and Luger K (2008). Histone chaperones in nucleosome eviction and histone exchange. Curr. Opin. Struct. Biol 18, 282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil M, Pabla N, and Dong Z (2013). Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci 70, 4009–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Oleinick NL, and Zhang J (2018). ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol 126, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransburgh DJR, Chiba N, Ishioka C, Toland AE, and Parvin JD (2010). Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res 70, 988–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SP, and Pandita TK (2006). The cellular control of DNA double-strand breaks. J. Cell. Biochem 99, 1463–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Robles AI, Linke SP, Sinogeeva NI, Zhang R, Pedeux R, Ward IM, Celeste A, Nussenzweig A, Chen J, et al. (2004). Functional interaction between BLM helicase and 53BP1 in a Chk1-mediated pathway during S-phase arrest. J. Cell Biol 166, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastav M, De Haro LP, and Nickoloff JA (2008). Regulation of DNA double-strand break repair pathway choice. Cell Res 18, 134–147. [DOI] [PubMed] [Google Scholar]
- Silljé HH, and Nigg EA (2001). Identification of human Asf1 chromatin assembly factors as substrates of Tousled-like kinases. Curr. Biol 11, 1068–1073. [DOI] [PubMed] [Google Scholar]
- So S, Davis AJ, and Chen DJ (2009). Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol 187, 977–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, and Helleday T (2005). The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol 7, 195–201. [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, and Jackson SP (2005). MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123, 1213–1226. [DOI] [PubMed] [Google Scholar]
- Symington LS (2016). Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol 51, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, and Takeda S (1998). Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Matsuoka S, Carpenter PB, and Elledge SJ (2002). 53BP1, a mediator of the DNA damage checkpoint. Science 298, 1435–1438. [DOI] [PubMed] [Google Scholar]
- Wu L, Luo K, Lou Z, and Chen J (2008). MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 105, 11200–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Li X, Yu J, Björkholm M, and Xu D (2019). ASF1a inhibition induces p53-dependent growth arrest and senescence of cancer cells. Cell Death Dis 10, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Liu L, Cao C, Song N, Wang Y, Ma S, Zhang Q, Yu N, Ding X, Yang F, et al. (2018). USP52 acts as a deubiquitinase and promotes histone chaperone ASF1A stabilization. Nat. Commun 9, 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, and Dutta A (2006). An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication. Mol. Cell. Biol 26, 4601–4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, and de Lange T (2014). 53BP1: pro choice in DNA repair. Trends Cell Biol 24, 108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new unique reagents.