

Abstract
Osteoarthritis (OA) is a degenerative disease of the joints and a leading cause of physical disability in adults. Intra-articular (IA) therapy is a popular treatment strategy for localized, single-joint OA; however, small-molecule drugs such as corticosteroids do not provide prolonged relief. One possible reason for their lack of efficacy is high clearance rates from the joint through constant lymphatic drainage of the synovial tissues and synovial fluid and also by their exchange via the synovial vasculature. Advanced drug delivery strategies for extended release of therapeutic agents in the joint space is a promising approach to improve outcomes for OA patients. Broadly, the basic principle behind this strategy is to encapsulate therapeutic agents in a polymeric drug delivery system (DDS) for diffusion- and/or degradation-controlled release, whereby degradation can occur by hydrolysis or tied to relevant microenvironmental cues such as pH, reactive oxygen species (ROS), and protease activity. In this review, we highlight the development of clinically tested IA therapies for OA and highlight recent systems which have been investigated preclinically. DDS strategies including hydrogels, liposomes, polymeric microparticles (MPs) and nanoparticles (NPs), drug conjugates, and combination systems are introduced and evaluated for clinical translational potential.
Keywords: osteoarthritis, clinical trials, corticosteroid drug delivery, intra-articular therapies, sustained drug release
Duvall - Table of Contents
Osteoarthritis is a degenerative disease of the joints and a leading cause of physical disability in adults. Intra-articular therapy is a common treatment strategy; however, therapeutics are susceptible to rapid clearance from the joint. Drug delivery strategies for extended residence time of therapeutic agents is a promising approach to address this issue. In this review, we highlight clinically and preclinically tested systems for osteoarthritis therapy.
1. Introduction
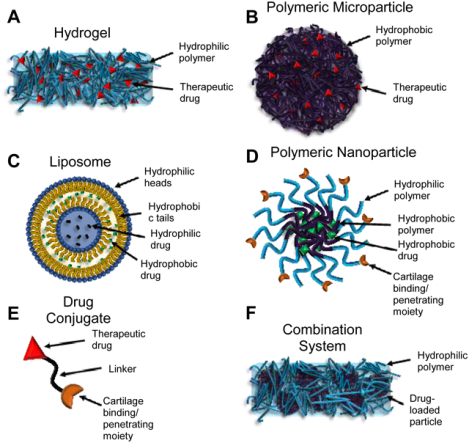
Osteoarthritis (OA) is the most common joint disorder, affecting 9.6% of men and 18% of women over 60 [1]. Primary (idiopathic) OA occurs in the majority of patients and develops slowly over time, presenting with progressive degradation of cartilage, joint inflammation, osteophytes, synovial thickening, and articular calcium crystal deposition (Figure 1) [2,3]. In contrast, post-traumatic OA (PTOA), typically develops after a traumatic injury to the joint and can cause earlier onset cartilage degeneration in younger patients. The molecular events that promote progression of disease for both of these types of OA are still largely unknown. The primary symptom of OA is joint pain, leading to loss of function and progressively worsening disability. Known risk factors for the disease include age, obesity, and gender [2]. There are no clinical disease-modifying OA drugs (DMOADs) which halt or reverse the progression of OA; the current standard of care focuses on pain management [4,5].
Figure 1.
Schematic representation of (A) a healthy knee joint and (B) a knee joint afflicted with osteoarthritis (OA). Pathologically, OA presents with cartilage destruction, synovial thickening, joint space narrowing, osteophytes, and calcium crystal deposition.
1.1. Clinical Treatment of OA
Early-stage OA patients are advised to attempt lifestyle changes, if applicable (usually for primary OA patients), such as exercise and weight loss to slow disease. Additionally, these patients are advised to use topical analgesics (over the counter gel formulations such as salicylates [6]), topical non-steroidal anti-inflammatory drugs (NSAIDs) (i.e. diclofenac (DF) [7]), or oral analgesics (i.e. acetaminophen [8]) to manage pain. If these conservative treatments are unsuccessful, oral NSAIDs are typically the next treatment option. Despite their efficacy, oral NSAIDs are associated with side effects, particularly in the gastrointestinal (GI) system, with one study finding 86.6% of a cohort of OA patients experiencing increased GI risk with NSAID treatment [9]. These complications can render oral NSAIDs unusable for many OA patients.
As disease progresses, intra-articular (IA) corticosteroids are commonly implemented to further manage pain and reduce inflammation. These molecules act by binding intracellular glucocorticoid receptors and influencing gene expression, particularly by upregulating production of anti-inflammatory cytokines and downregulating production of inflammatory cytokines and cyclooxygenase-2 [10–14]. Additionally, there is more recent evidence for disease-modifying effects of corticosteroids, including reducing glycosaminoglycan (GAG) loss [15,16] and reducing interleukin-1β (IL-1β) and matrix metalloproteinase (MMP) levels [17]. However, these effects have not been demonstrated clinically, where the major clinical benefit remains pain relief (4–8 week duration [18]). Many corticosteroids utilized for IA injection are non-water-soluble corticosteroid ester prodrugs formulated as crystal suspensions, which provide prolonged retention and activity in the joint because the active drug must be released by cellular esterases [19,20]. Despite their effects on pain, IA corticosteroids can cause significant side effects, including occasional occurrence of a “post-injection flare” (redness and swelling 3–48 hours after injection) and adrenal suppression. Additionally, there is evidence that frequent corticosteroid injections can cause cartilaginous arthropathy; for this reason, IA corticosteroids have typically been limited to 1 injection every 3 months [18,21]. Notably, a recent study has found that even this dosing regimen could be detrimental to cartilage volume: patients who received IA triamcinolone acetonide (TCA) every 3 months exhibited significantly greater cartilage volume loss compared to saline after 2 years, with no significant improvement in pain [22]. This finding is in contrast with the above preclinical findings and suggests that the effect of corticosteroids on cartilage could be dose-dependent. In light of this study and others which have found questionable efficacy of IA corticosteroids, these agents are typically administered cautiously and at best provide transient relief to patients.
Another IA treatment alternative is viscosupplementation, which refers to the technique of injecting a viscoelastic material into the joint space to provide lubrication and mechanical cushioning in the joint to reinforce the degenerated articular cartilage surface. The material used is hyaluronic acid (HA), which has been approved by the Food and Drug Administration (FDA) for this purpose in the U.S. since 1997 [23]. Current products on the market range from 500–6,000 kDa HA, which are produced by bacterial fermentation or isolated from avian sources (chicken combs). Approved formulations contain between 1–2.2% HA and require 1–5 injections per treatment course [24]. The incidence of side effects is low (IA HA can also cause post-injection flare); however, the effectiveness of this intervention is disputed, with conflicting evidence on improved outcomes over placebo [25,26]. Additionally, comparison of the efficacy of HA injections compared to corticosteroid injections have not concluded the superiority of one over the other, and patients often respond differently [27].
There are also surgical options, typically reserved as a last resort for treating OA. For patients with moderate disease, techniques including arthroscopic lavage and microfracture can be effective. The former is meant to clear debris from the joint, and the latter forms a defect in the subchondral bone, allowing egress of blood and stem cells from the bone marrow into the synovial cavity to promote formation of fibrocartilage on the articular surface [5]. As patients reach late-stage disease, total joint arthroplasty (replacement) is performed, whereby the damaged articular surfaces are removed and replaced with an implanted mechanical joint. Despite their highly invasive nature, these procedures are relatively low-risk and provide 80–90% patient satisfaction [28]. However, these prosthetic joints may require replacement if utilized in younger patients (such as PTOA patients), as they are estimated to last approximately 20 years [28]. Additionally, the financial burden of joint arthroplasty is significantly higher than that of more conservative treatments. A 2019 study found that over a 2-year period, knee arthroplasty was performed on 3.2% of knee OA patients but contributed 69% of the total cost ($3.44 billion) [29]. The extremely disproportionate cost of these procedures is a major motivation to develop more effective IA therapies.
1.2. Translation of DDSs for OA
Because of the localized nature of OA, injectable IA therapies are an attractive strategy to increase target site exposure and reduce potential side effects. A study by Furman et al. demonstrates this point with the IL-1 receptor agonist protein (IL-1Ra; anakinra) [30]. In a mouse model of PTOA, local IA therapy with 1 injection of IL-1Ra following articular fracture significantly prevented cartilage degeneration and synovial inflammation while systemic IL-1Ra administered daily or by continuous infusion over 4 weeks did not. However, efficacy of IA injected molecules, such as corticosteroids and proteins, is limited by the high clearance rate from lymphatic drainage and synovial vascular absorption [4]. Advanced drug delivery systems (DDSs) have been investigated as a strategy to extend drug residence time in the joint. By encapsulating active molecules into an exogenous, injectable carrier, or by direct conjugation of the drug to a cartilage-penetrating moiety, controlled release and slower clearance can be achieved for IA delivered therapies. Because OA is chronic in nature, it is an ideal candidate disease for treatment with drug delivery sustained release technologies. Additionally, local administration of a high drug dose in controlled-release systems is expected to reduce the incidence of systemic exposure and consequent side effects such as adrenal suppression by corticosteroids [4].
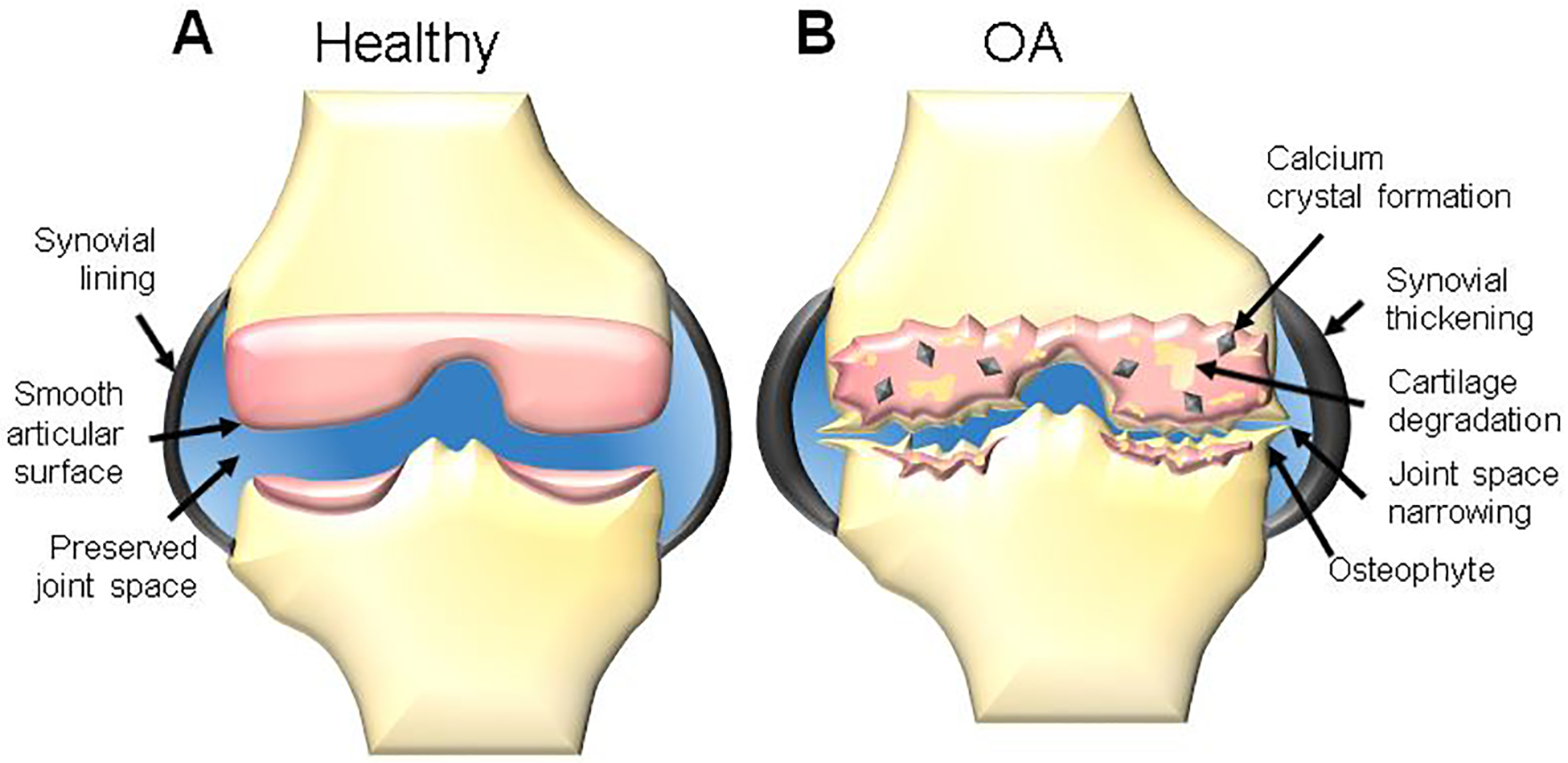
Despite many promising preclinical studies, there are few clinically approved IA therapies based on sustained release strategies. Clinical investigation of DDSs for OA has steadily increased over the past 10 years (Figure 2A–B), with 6 clinical products currently in development. These products include 3 HA hydrogels, 2 polymeric microparticles (MPs), and 1 liposome (Figure 2C). The purpose of this review is to critically evaluate the existing clinical trials for injectable controlled-release systems in OA and draw conclusions about characteristics which promote clinical translation. For hydrogel, MP, and liposomal systems (Figure 3A–C), clinically-tested products are presented first (Table 1), and preclinical systems which have shown efficacy in animal models of OA follow. Based on our review of clinical products, we evaluate preclinical studies for translational potential based on the following 3 criteria:
Frequency of dosing: Unlike orally administered or subcutaneously injected drugs, IA therapies must be administered by a health professional. Therefore, retention time in the joint is critical to clinical effectiveness due to low patient compliance with frequent doctors’ visits [31].
Therapeutic efficacy: A “successful” preclinical study should demonstrate significant therapeutic efficacy through histological evaluation, gait analysis, reduction of OA biomarkers, and pain reduction. These metrics are particularly important for DMOAD candidates, which should halt or reverse disease progression and whose action is expected to be enhanced with improved retention. The ideal DMOAD would provide both structural protection of joint tissues and pain relief in the clinical setting to restore function and reduce/prevent disability in patients.
Complexity: Increasingly complex technologies are more difficult to reliably and reproducibly manufacture at scale for human use. Laboratory settings allow great freedom for innovation in new polymers, drugs, and interesting combinations, which is evident in preclinical studies. In contrast, relative simplicity is a common thread among the products that have advanced into clinical testing and approval.
Figure 2.

Analysis of clinical trials for IA therapy of OA. (A) Number of IA DDS undergoing clinical trials from 2010–2020 based on year posted on ClinicalTrials.gov. Pie charts show proportions of (B) phases of clinical trials pursued, (C) classes of DDS investigated, and (D) types of drug investigated.
Figure 3.
Schematic representation of the major classes of technologies used to extend drug residence time in the joint for osteoarthritis: (A) hydrogels, (B) polymeric microparticles, (C) liposomes, (D) polymeric nanoparticles, (E) drug conjugates, and (F) hybrid hydrogel/particle systems.
Table 1:
Summary of Clinical Findings for Intra-Articular Therapies for Osteoarthritis
| Product | Polymer/ Drug/ Formulation | Major Clinical Findings | Reference(s) |
|---|---|---|---|
| FX006 (Zilretta™) | PLGA/ TCA/ MP |
PK: FX006 increased synovial TCA levels and decreased systemic TCA levels. Efficacy: FX006 significantly improved pain outcomes over ~3 months compared to saline but not TCA suspension. |
[102], [105], [106], [112], [114], [119] |
| TLC599 | Mixed lipids/ DSP/ Liposome |
PK: No clinical results reported (DSP detected in joint at 120 days in beagle dogs). Efficacy: TLC599 significantly improved pain outcomes over ~6 months compared to saline. |
[164], [167] |
| EP-104IAR | PVA/ FP/ MP |
PK: No clinical results reported (FP detected in joint at 60 days in beagle dogs). Efficacy: No clinical results reported |
[135] |
| Cingal | Cross-linked HA/ TH/ Hydrogel |
PK: No clinical results reported Efficacy: Cingal significantly improved pain outcomes over ~6 months compared to saline but not free TH. |
[47], [54] |
| SI-613 | HA/ DF/ Hydrogel |
PK: No clinical results reported (DF t1/2 of 24 hours in rabbits). Efficacy: SI-613 significantly improved pain outcomes compared to saline. |
[60], [61] |
| Condrotide Plus | HA/ PN/ Hydrogel |
PK: No clinical results reported Efficacy: Condrotide Plus significantly improved KSS over HA; pain relief was extended with Condrotide Plus. |
[66] |
Abbreviations: DF=diclofenac; DSP=dexamethasone sodium phosphate; FP=fluticasone propionate; HA=hyaluronic acid; KSS=Knee Society Score; MP=microparticle; PK=pharmacokinetics; PLGA=poly(lactic-co-glycolic acid); PN=polynucleotides; PVA=poly(vinyl alcohol); t1/2=half-life; TCA=triamcinolone acetonide; TH=triamcinolone hexacetonide
Another notable criterion that should be mentioned is safety, which is crucial to approval of any drug meant for use in humans. However, preclinical studies will not be explicitly evaluated for safety as this metric is not often rigorously studied in the preclinical setting.
It is notable that the clinically tested products (with the potential exception of Condrotide Plus) have so far not fulfilled the goal of DMOAD development, as they deliver pain-relieving agents including corticosteroids and NSAIDs (Figure 2D). Therefore, we extend this review to also highlight preclinical studies of polymeric nanoparticles (NPs), cartilage-penetrating drug conjugates, and combination systems which have shown DMOAD potential (Figure 3D–F). Though this review is written from the perspective of DDS technologies and a detailed discussion of drug candidates is outside the scope, Table 2 summarizes small molecule and biologic therapeutics which have been clinically tested for disease-modifying effects. Therapeutics with DMOAD potential have been reviewed in detail by others [32,33].
Table 2:
Potential DMOADs Delivered IA in Clinical Trials
| Potential DMOAD | Mechanism of Action | Phase of Clinical Testing |
|---|---|---|
| KA34 | KGN analog | Phase 1 |
| Sc-rAAV2.5IL-1Ra | Viral genome delivery of IL-1Ra | Phase 1 |
| Anakinra | IL-1Ra agonist | Phase 2 |
| Sprifermin | RhFGF-18 | Phase 2 |
| UBX0101 | MDM2/p53 interaction inhibitor (senolytic) | Phase 2 |
| ADSC/ BMDSC/ MSC | Stem cells | Phase 3 |
| PRP | Growth factor cocktail | Phase 3 |
| SM04690 | Wnt inhibitor | Phase 3 |
| TissueGene-C | Chondrocytes genetically modified to express TGF-1β | Phase 3 |
Abbreviations: ADSC=adipose-derived stem cells; BMDSC=bone-marrow derived stem cells; DMOADs=disease-modifying osteoarthritis drugs; IL-1Ra=interleukin-1 receptor agonist; KGN=kartogenin; MDM2=mouse double minute 2 protein; MSC=mesenchymal stem cells; PRP=platelet-rich plasma; RhFGF-18=recombinant human fibroblast growth factor-18; TGF-1β=transforming growth factor-1β; Wnt=Wingless and Int-1 signaling
2. Hydrogels
Hydrogels are hydrophilic polymer networks that swell in the presence of water, absorbing up to thousands of times their dry polymer mass [34] (Figure 3A). Depending upon the polymer composition, functionalization with cell adhesive or degradable moieties, and crosslinking density, hydrogels can be tuned to have different mechanical properties, degradation behavior, and biological responses. Hydrogels are attractive for OA therapy because they can fill the narrowed joint space to potentially provide cushioning and lubrication that mimics the native cartilage extracellular matrix (ECM) [35,36], though pain relief following viscosupplementation is still debated [26]. In addition to mechanical function, hydrogels can also be loaded with and serve as a depot for a variety of agents, including cells and drugs [37]. Hyalgan (Fidia SPA), Orthovisc (Anika Therapeutics, Inc.), and Synvisc (Genzyme) are all commercial HA products used in the clinic [38]. Additionally, synthetic polymers such as polyacrylamide and poly(vinyl alcohol) (PVA) have been used to formulate injectable and implantable hydrogels for OA. Cartiva (Cartiva, Inc.) is an implantable PVA hydrogel which is FDA-approved for treatment of OA in the great toe, and Aquamid (A2 Reumatologi Idraetsmedicin A/S) is an injectable polyacrylamide hydrogel being tested in patients with knee OA (NCT03060421) [39].
While all FDA-approved hydrogel OA products are designed purely for mechanical supplementation, many groups are interested in using hydrogels as drug-delivery vehicles to combine their mechanical benefits with their potential as drug release depots. Recent reviews on this topic are available that focus on tissue engineering for focal chondral defects [35]; delivery of biologics including HA, platelet-rich plasma (PRP), and stem cells [40]; and polymeric hydrogels for drug delivery [36]. The following discussion focuses on recent hydrogel-based systems for drug delivery which have been studied clinically and preclinically (Table 3) for OA therapy.
Table 3:
Preclinical Hydrogel Systems
| Polymer/ Drug/ Formulation | Model | Dosing Scheme (Dose) | Outcome | Ref. |
|---|---|---|---|---|
| HA/ TCA/ Hydrogel | Mouse MMx model | Single injection (100 μg TCA) | HA-TCA improved withdrawal force over 28 days | [75] |
| cHA/ Dex/ Hydrogel | Rat ACLT model | Single injection (10 or 25 μg Dex) | cHA-Dex improved osteophyte and synovial inflammation scores 8 weeks post-injection | [76] |
| HA/ sCT/ Hydrogel | Rabbit ACLT model | Three injections once per week (25, 100, or 400 IU sCT) | HA-sCT extended release of sCT and improved histological score with 3 weekly injections | [77] |
| GLT-HA-FD/ PRP/ Hydrogel | Rabbit ACLT model | Single injection (not stated) | Hydrogel reduced cartilage damage and increased proteoglycans at 8 weeks | [80] |
| HA-PX/ DK/ Hydrogel | Rat MIA model | Injections twice per week (500 μg DK) | HA-PX + DK reduced joint swelling, serum cytokines, and joint damage scores at 2 weeks | [86] |
| PCLA-PEG-PCLA/ CXB/ Hydrogel | Horse LPS-induced synovitis model | Single injection (300 mg CXB) | CXB-gel was detectable for 2 weeks and reduced prostgalndin-E2 4 hours post-injection | [87] |
Abbreviations: ACLT=acterior cruciate ligament transection; cHA=cross-linked hyaluronic acid; CXB=celecoxib; Dex=dexamethasone; DK=Diclofenac potassium; FD=fucoidan; GLt=gelatin; HA=hyaluronic acid; LPS=lipopolysaccharide; MIA=monoiodoacetate; MMx=medial meniscectomy; PCLA=poly(ε-caprolactone-co-lactide); PEG=poly(ethylene glycol); PRP=platelet-rich plasma; PX=Poloxamer 407; sCT=salmon calcitonin; TCA=triamcinolone acetonide.
2.1. Clinical Hydrogel Products
2.1.1. Cingal
Anika Therapeutics is developing an injectable, cross-linked HA gel loaded with triamcinolone hexacetonide (TH) for IA therapy of OA (Cingal). This product is the next generation of their currently FDA-approved non-drug-loaded HA viscosupplementation product, Monovisc. Monovisc is formulated from HA (1–3 MDa) cross-linked via p-phenylene-bis(ethylcarbodiimide) chemistry, and the FDA-approved formulation is a 4 ml injection containing 88 mg HA [41,42]. Cingal consists of the same formulation with TH mixed into the HA matrix (0.204 mg TH per mg HA), and is already approved for OA therapy in Canada and the European Union [43,44].
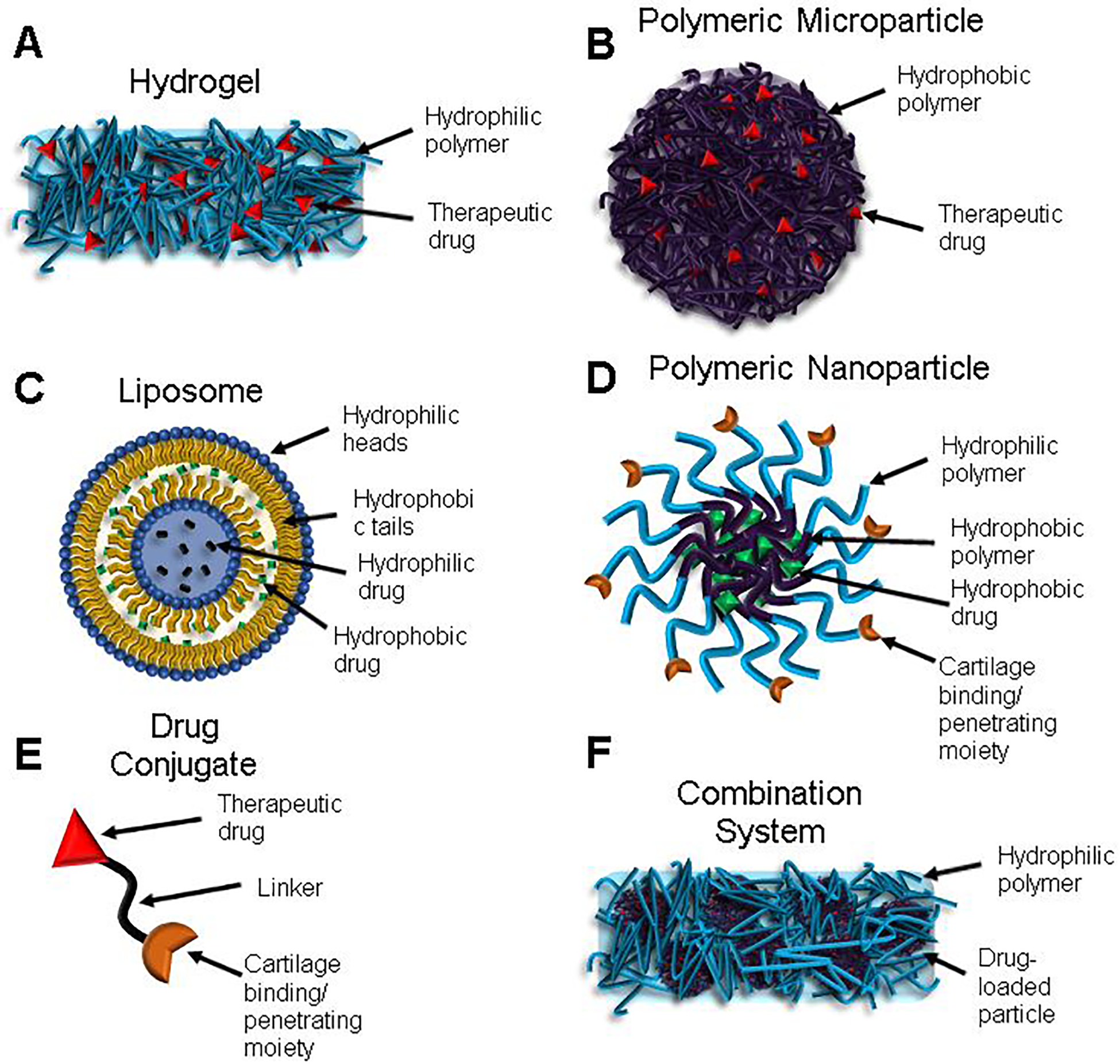
The preclinical data leading to clinical trials for Cingal has not been made available in peer-reviewed publications or FDA documents. Both Cingal and Monovisc are classified as medical devices as opposed to drugs, which generally reduces the level of preclinical data required for approval [45]. Additionally, the approval of Monovisc likely further reduces the preclinical testing requirement for Cingal, as Monovisc could be considered a predicate device by the FDA [43,46]. The first clinical trial for Cingal focused on efficacy of IA administration of Cingal (88 mg HA/18 mg TH), Monovisc (88 mg HA), or saline (placebo) (NCT01891396) [47] in 368 patients with knee OA. In the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscore, a questionnaire very commonly used to quantify OA disease progression in patients [48], Cingal provided a significant reduction compared to saline at all time points tested out to 26 weeks (−42.4 vs. −32.9 at week 26). Additionally, Cingal outperformed Monovisc at weeks 1 and 3, indicating more rapid pain relief with TH delivery (−34.6 vs. −29.6 at week 1) (Figure 4). This pattern was seen in other outcomes including evaluator Global Impression of Change score (GIC), patient GIC, WOMAC physical function subscore, and WOMAC stiffness subscore. GIC questionnaires are meant to capture patients’ wholistic change in health due to treatment [49,50]. The patients who received Cingal were later given a second injection to evaluate the safety of repeated treatments (NCT02381652) [51]; adverse events (AEs: arthralgia, injection site pain, swelling, and erythema) occurred in 4.3% of patients, a rate which was not significantly different than that for a single injection [52].
Figure 4.
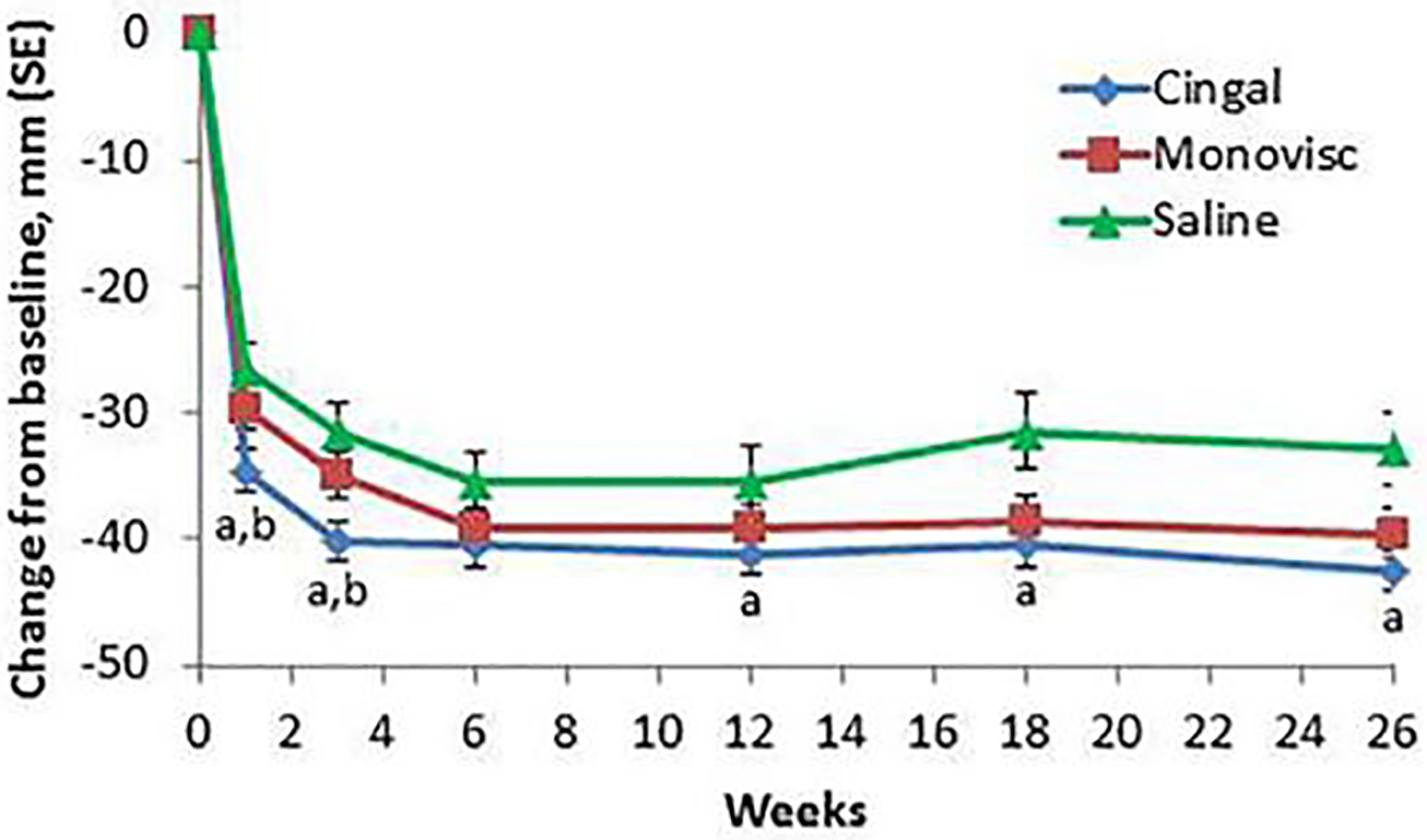
Mean changes from baseline for Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscore with different treatments (Cingal, Monovisc, or saline) over time. aP < 0.01 versus placebo; bP < 0.05 versus Monovisc. Reproduced under the terms of the Creative Commons Attribution-NonCommercial 4.0 License.[43] Copyright 2017, L. Hangody, R. Szody, P. Lukasik, W. Zgadzaj, E. Lénárt, E. Dokoupilova, D. Bichovsk, A. Berta, G. Vasarhelyi, A. Ficzere, G. Hangody, G. Stevens, and M. Szendroi.
Several other efficacy trials of Cingal have been completed or are ongoing. In 2017, a small trial in 60 patients with knee OA was conducted to compare the effect of Cingal and Monovisc on the WOMAC pain subscore (NCT03062787), for which results have not been made available [53]. A larger trial was conducted in 576 patients with knee OA comparing the effect of Cingal, Monovisc, or TH suspension (20 mg) on WOMAC pain subscore (NCT03191903) [54]. Anika published a press release stating that Cingal reduced pain in knee OA patients at 26 weeks but not in a manner that was a statistical improvement over free TH [55]. A follow-up study is ongoing to continue following these patients to 39 weeks to determine if Cingal outperforms free TH at this extended time point (NCT03390036) [56]. Finally, Anika has recently partnered with McMaster University to evaluate the reduction in patient-reported hip pain for Cingal in patients with hip OA up to 6 months post-injection (NCT04084704) [57].
Conclusions:
Cingal represents a combination of two well-known treatments for OA: IA corticosteroids and HA. The prolonged pain relief resulting from a single injection (at least 26 weeks) and relative simplicity of the product are beneficial for clinical success. Ideally, the viscosupplementation provided by HA and anti-inflammatory effects of TH will work in concert to improve pain relief for patients. The results to date suggest that the addition of TH could cause more rapid pain relief over HA alone, but that long-term benefits are similar. Additionally, current evidence suggests that Cingal and TH perform similarly, though it will be important to discover whether Cingal can extend the benefit of IA TH. Measurements of TH concentration over time in the joint would be critical to discovering if any additional benefit of HA+TH over either treatment alone is due to improved retention of TH or simply the combination of these two treatments.
2.1.2. SI-613
Seikagaku Co. is investigating an injectable HA formulation loaded with DF for IA joint therapy (SI-613). In this formulation, HA (600–1200 kDa) is tethered to DF via a 2-aminoethanol linker extended from glucuronic acid groups. The DF loading is 11.8% w/w [58,59].
In preclinical studies, SI-613 showed efficacy in rat and rabbit models of OA [58]. In a rat model of OA, SI-613 (0.5 mg) significantly improved pain score and weight-bearing rate 3 days post-injection compared to no treatment (~86% and ~120% improvements, respectively) and significantly outperformed a physical mixture of DF and HA (0.5 mg) and oral DF (2 mg/kg). SI-613 also significantly reduced pain score (~50% reduction) and synovial prostaglandin E2 levels (~70% reduction) compared to no treatment at days 1 and 2. These results were replicated in a rabbit model, as well: 5 mg SI-613 significantly reduced synovial prostaglandin E2 levels (~98% compared to phosphate buffered saline (PBS)) with no effect of oral DF or a physical mixture of HA and DF. SI-613 also reduced joint swelling compared to PBS. IA SI-613 exhibited an improved pharmacokinetic (PK) profile compared to oral DF or the mixture, with a much lower plasma maximum concentration (Cmax) of DF (1.343 vs. 621.0 and 125.9 ng ml−1, respectively) and a longer half-life (t1/2) (24 vs. 2.4 and 0.39 h, respectively).
Based on these preclinical data, a Phase 2 clinical trial was undertaken in 80 patients with knee OA focused on the improvement in the WOMAC pain subscore for SI-613 and placebo (NCT03209362) [60]. Results for this trial have not yet been made available. Notably, Seikagaku Co. has also been pursuing approval in Japan. In a Phase 3 study, patients received 3 injections of SI-613 each spaced 4 weeks apart and exhibited significant improvement in the WOMAC score compared to placebo [61]. Additionally, two other ongoing Phase 3 trials are investigating efficacy in shoulder, hip, elbow, and ankle, as well as the long-term safety of the product.
Conclusions:
SI-613 is based on a combination of HA and a NSAID, another accepted OA treatment. The IA strategy could avoid the known complications associated with oral NSAIDs, particularly in the GI system. The improved PK of the conjugated form over a physical mixture is encouraging; however, further extension of the t1/2 could improve the efficacy of this strategy. The idea that PK improvement is needed is supported by the dosing used in the Japanese trial, in which patients required 3 injections for extended pain relief.
2.1.3. Condrotide Plus
Researchers at the Italian Istituto Ortopedico Rizzoli are investigating a polynucleotide (PN)/HA injectable gel system for OA (Condrotide Plus). PNs are a mixture of purines, pyrimidines, deoxyribonucleotides, and deoxyribonucleosides which can be isolated from fish sperm or human placenta [62]. PNs have been shown to promote musculoskeletal tissue regeneration (mostly skin, but also bone, cartilage, and tendon) and reduce pain and inflammation in preclinical and clinical studies through activation of the adenosine A2A receptor [63]. Binding to the A2A receptor activates signal transduction via activation of the G protein leading to cyclic adenosine monophosphate (cAMP) signaling, which results in reduction in inflammatory cytokine reduction and increases cell proliferation [63,64]. Condrotide Plus consists of a mixture of PN isolated from fish sperm (molecular weight from 70–240 kDa) incorporated at 10 mg ml−1 combined with HA (800–1300 kDa) at 10 mg ml−1 (total gel concentration 20 mg ml−1) [62,65].
Similar to Cingal, preclinical data for Condrotide Plus is lacking due to its classification as a device. A single clinical trial of this product has been completed in 102 patients with knee OA, focused on efficacy of 40 mg PNHA (20 mg PN/20 mg HA) or 40 mg HA administered IA (NCT02417610) [62,66]. Patients received 3 injections each spaced 1 week apart. The main efficacy readout in this trial was the Knee Society Score (KSS), which is a scale originally developed to evaluate outcomes of total knee arthroplasty and is useful because it is specific to the knee [67]. At 2, 6, and 12 months after the third administration, patients receiving Condrotide Plus exhibited significantly higher KSS than patients receiving HA (mean score at 12 months of ~78 vs. ~73, respectively). They also demonstrated a significant reduction in the KSS pain subscore compared to HA at 2 and 12 months (~1.4 vs. ~1.8 at 12 months); however, there was no significant difference in the WOMAC pain subscore between treatments.
Conclusions:
Condrotide Plus is the only product currently being investigated which delivers a biologic with a purpose beyond pain relief. Viscosupplementation provided by HA is expected to relieve pain, while PNs could contribute to joint tissue regeneration. The regenerative quality of PNs could contribute to the longer-term efficacy seen in humans at 12 months, and a full year of efficacy could compensate for the initial need for 3 injections. Expanded studies investigating efficacy and PK in larger cohorts of patients will be informative, and preclinical investigation could provide more insight into the mechanism of action and stability of PNs+HA in diseased OA joints.
2.1.4. Terminated Development Programs
There are 2 examples of DDS products which were pursued for clinical treatment of OA, one of which is a hydrogel system. We included these systems because the problems encountered could be informative for researchers hoping to translate IA DDSs.
Hydros-TA
Carbylan Therapeutics developed an injectable, HA-based hydrogel system for TCA delivery (Hydros-TA). HA was functionalized with vinyl sulfone (HA-VS) and cross-linked with PEG dithiol (PEG-SH). The unloaded formulation was marketed as Hydros, while the TCA delivery formulation was marketed as Hydros-TA. This formulation delivered 10 mg TCA in a single injection [68,69]. In 2010, a clinical trial was conducted in 98 patients with knee OA focused on reduction of pain for 6 ml injections of either a commercial HA hydrogel (Synvisc-One, Anika Therapeutics), Hydros, or Hydros-TA (10 mg TCA) (NCT01134406) [69–71]. The changes in the WOMAC pain subscore for Synvisc-One, Hydros, and Hydros-TA at 26 weeks were 28.0, 30.5, and 34.4, respectively (no significant differences). Notably, at 2 weeks, the magnitude of the difference in pain score between Hydros and Hydros-TA was 12.4, indicating that corticosteroid-loaded HA provided faster onset of pain relief. These results led to a larger study in 510 patients with knee OA focused on the reduction in pain for Hydros, Hydros-TA, or TCA suspension (NCT02022930) [72]. Carbylan Therapeutics suspended production of Hydros-TA after this trial due to failure to meet the secondary endpoint (patients receiving free TCA improved at levels similar to the Hydros-TA cohort) [73]. Shortly after these results, Carbylan announced a merger with KalVista Pharmaceuticals Ltd., and KalVista has not pursued Hydros-TA or any other IA therapies for OA since the acquisition [74]. This example demonstrates a critical challenge in pursuing clinical development of new therapies. If results are not striking, the company’s landscape could shift in terms of relative emphasis on ongoing programs. Interestingly, Hydros-TA may have been more successful if a placebo-controlled trial had been undertaken; none of the clinically tested extended-release formulations discussed throughout this review have been shown to outperform their free drug counterpart.
2.2. Preclinical Hydrogel Studies
Multiple studies have investigated HA for delivery of small molecule therapeutics by simple mixing of the drug into the matrix, similar to Cingal. Kroin et al. investigated HA delivery of the corticosteroid TCA (5% HA, 100 μg TCA dose) compared with TCA suspension (Kenalog-40®, 40 μg dose) and HA alone [75]. Kenalog-40® significantly improved withdrawal force threshold 3 and 5 days after treatment compared to saline injection (~50% of pre-OA levels vs. ~17%). However, TCA loaded in the HA gels significantly improved withdrawal force threshold out to 28 days (~41% of pre-OA levels) before the effect waned, while unloaded HA had no effect. Zhang et al. also investigated HA hydrogels for corticosteroid delivery (dexamethasone, Dex) [76]. Four weeks after anterior cruciate ligament transection (ACLT), rats received a single IA injection of cross-linked HA (cHA), cHA-Dex (10 μg), or cHA-Dex (25 μg), and outcomes were evaluated at 8 weeks post-injection. High dose cHA-Dex significantly improved histological synovial inflammation by ~59% compared to saline and ~50% compared to cHA alone (Figure 5A), while both doses of cHA-Dex improved OA Research Society International (OARSI) histological score by ~50%. The high dose cHA-Dex treatment also significantly reduced osteophyte score compared to both saline (~73% improvement) and cHA alone (~50% improvement) (Figure 5B). Notably, low dose cHA-Dex significantly increased aggrecan mRNA expression in the tissue compared to saline and cHA, but this increase was not seen with high dose cHA-Dex (Figure 5C). These results demonstrate the delicate balance in dosing required for corticosteroids to achieve disease-modifying effects [15–17], with high concentrations adversely affecting ECM synthesis [22]. These two systems are very comparable to Cingal in that they use HA for delivery of corticosteroids. Both achieved therapeutic efficacy after a single injection and are relatively simple, indicating that they could follow the clinical development precedent set by Cingal.
Figure 5.
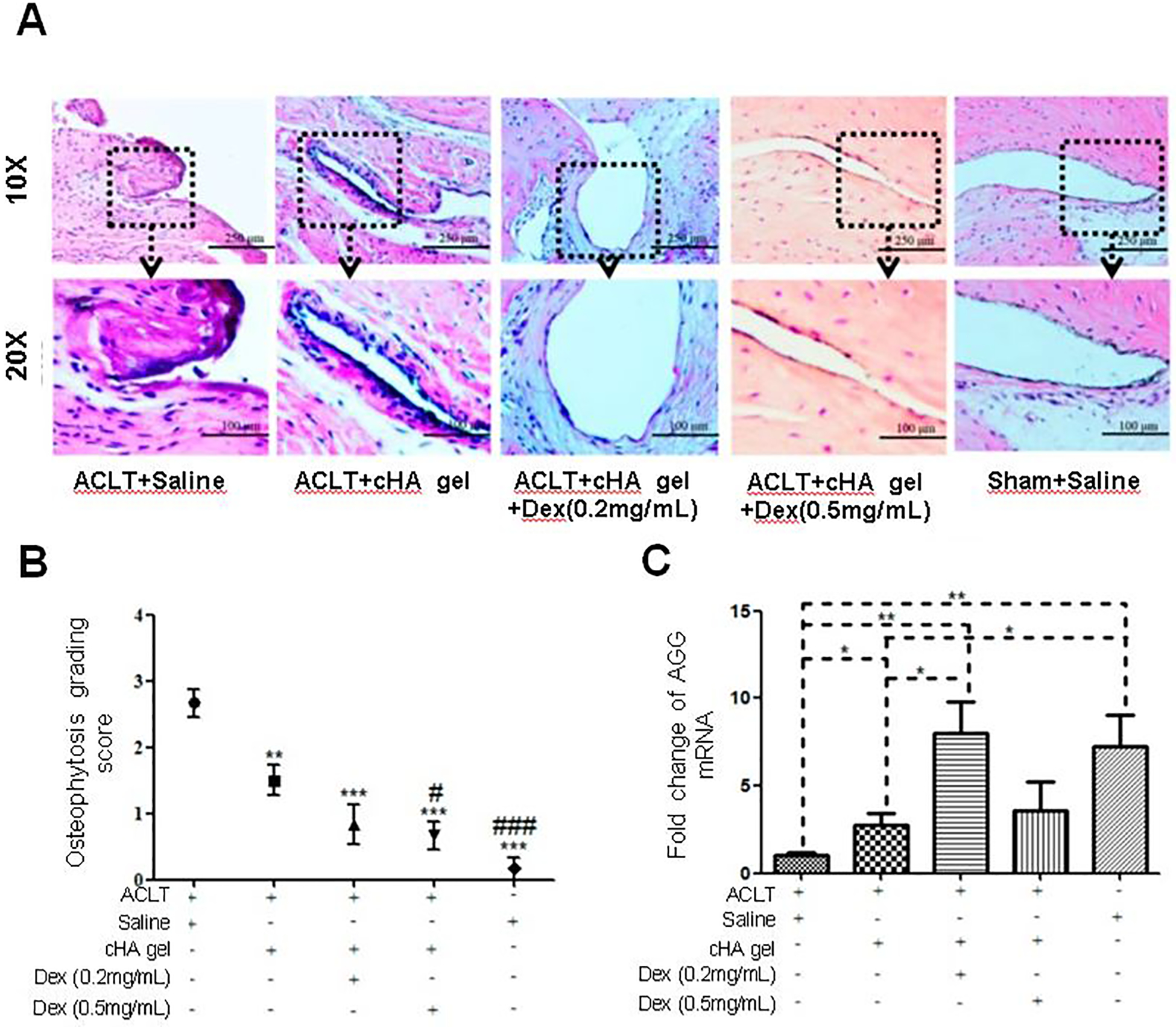
Investigation of intra-articular cross-linked hyaluronic acid (cHA) hydrogels for dexamethasone (Dex) delivery in rats with osteoarthritis (OA) induced via anterior cruciate ligament resection (ACLT). (A) Representative images of the synovial membrane after hematoxylin and eosin staining indicate decreased numbers of synovial lining cell layers and decreased infiltration of inflammatory cells in rat knees treated with the cHA gel + Dex. (B) cHA gel + Dex injections attenuated OA severity macroscopically, as shown by subjective grading of the severity of radiographic osteophytosis on a scale of 0 to 3: 0-normal, 1-mild, 2-moderate, 3-severe. Data were expressed as means ± standard deviation (SD). * used when compared with the ACLT + Saline group: designated as * p<0.05, ** p<0.01, and *** p<0.001. # used when compared with the ACLT + cHA gel group: designated as # p<0.05 and ### p<0.001. n=10. (C) Low dose cHA gel + Dex (0.2 mg/mL) injections increased mRNA levels of aggrecan (AGG) in comparison to the ACLT + Saline and cHA gel only groups (p<0.05). This was not the case for high dose cHA gel + Dex (0.5 mg/mL) injections. Data were expressed as means ± SD. * indicates a statistically significant difference: designated as * p<0.05, ** p<0.01, and *** p<0.001. n = 10. Adapted under the Creative Commons Attribution License CC BY 4.0.[76] Copyright 2016, Z. Zhang, X. Wei, J. Gao, Y. Zhao, Y. Zhao, L. Guo, C. Chen, Z. Duan, P. Li, and L. Wei.
Binding of small molecule drugs to HA, as with SI-613, could further extend drug residence in the joint. To this end, Mero et al. irreversibly conjugated salmon calcitonin (sCT) onto acetal-functionalized HA (HA-sCT) to promote joint retention [77]. CT is a 32-amino-acid peptide, which blocks bone resorption by binding CT receptors on osteoclasts. Oral sCT reduced subchondral bone damage and type II collagen degradation in a rat model of OA [78], but has exhibited disappointing results in clinical OA trials [79]. In Mero et al.’s study, IA injection of free sCT into the rat knee exhibited nearly immediate systemic exposure, causing up to a 50% reduction in plasma calcium concentrations at 5 hours post-injection, while HA-sCT exhibited a more sustained release profile (Cmax in serum ~5 ng ml−1 vs. ~15 ng ml−1 for sCT) and caused no hypocalcemia. In the rabbit ACLT OA model, Hyalastine (HA control), free sCT (400 IU), or HA-sCT (400 IU sCT) were injected IA 10, 17, and 24 days after surgery. sCT significantly improved macroscopic damage score compared to PBS in both the free and HA-sCT form (~2.5, ~1.5, and ~0.75, respectively), while only high-dose HA-sCT statistically outperformed Hyalastine (~0.75 vs. ~2.25). HA-sCT also significantly improved histological articular cartilage morphology score compared to PBS and Hyalastine. Notably, weekly injections were required to achieve these results, which is in contrast with the studies discussed above. This could be due to differences in the formulation of the HA (i.e. cross-linked vs. not) or the relative potency of the two drugs, with corticosteroids exhibiting longer-term effects compared to sCT. The dosing schedule is a barrier to the translation of this system, though the relative simplicity is an advantage, as SI-613 has already demonstrated the translational potential of HA-conjugated drugs.
As demonstrated by Condrotide Plus, HA can also be an effective delivery vehicle for biologics. In an interesting application, Lu et al. investigated a chemically cross-linked gelatin/HA/fucoidan (8% GLT, 1% HA, 1% FD) injectable hydrogel for the delivery of PRP in OA [80]. PRP is derived from whole blood and contains a concentrated cocktail of growth factors that has been shown to improve pain and function outcomes in clinical trials [81]. FD is a heparanoid polysaccharide which has been shown to interact with heparin-binding growth factors (like those found in PRP) and better retain them locally [82,83]. In vitro, GLT/HA gels loaded with PRP exhibited a significantly reduced burst release of platelet-derived growth factor (PDGF) compared to PRP alone. Inclusion of FD further slowed release, with ~30% less cumulative PDGF release at day 15 for GLT/HA/FD compared to GLT/HA alone. In a surgical rabbit model of OA, injection of GLT/HA/FD loaded with PRP reduced cartilage damage and rescued loss of proteoglycans by qualitative histological examination after 8 weeks. These results from a single injection are promising; however, more detailed efficacy studies would be useful to fully evaluate this system. Particularly, a comparison to PRP alone would be enlightening, as PRP is already clinically utilized for OA. Additionally, the relative complexity of this system could be a barrier to translation, especially due to regulation of multiple biological components. Despite this challenge, combining HA with potent biologics like PRP and PNs is exciting for the development of next-generation IA DMOAD therapies.
A limitation of HA as a drug delivery vehicle is its lack of an active physical or chemical crosslinking mechanism that occurs in situ to promote local retention. Thermogelling formulations have not yet reached the clinic; however, they have shown promise preclinically [84,85]. Hanafy et al. combined HA with the thermoresponsive copolymer Poloxamer 407 (PX) for IA delivery of diclofenac potassium (DK) [86]. Their formulation, termed Hyalomer, consisted of 18% PX (w/w), 0.25% HA (w/w), and 5 mg/ml DK and significantly slowed DK release in vitro. In a rat model of inflammatory OA, animals received no treatment or injections (twice per week) of Hyalomer (500 μg DK), unloaded Hyalomer (250 μg HA), HA only (250 μg), PX only (18 mg), or DK (2.5 mg kg−1, IA or oral). Two weeks after the first injection, Hyalomer-treated rats were the only group which demonstrated significantly less knee swelling and pain compared to no treatment. All treatments significantly reduced serum levels of cartilage oligomeric matrix protein (COMP, a marker of OA), IL-1β, and IL-6 compared to no treatment; Hyalomer represented the greatest reductions in these biomarkers (~60% reduction compared to no treatment). Finally, both Hyalomer and unloaded Hyalomer significantly reduced global joint damage scores compared to no treatment. This study represents an innovation to classically used HA hydrogels; however, the frequent injections indicate that the inclusion of PX did not improve HA retention, and further iteration will be needed to reach a clinically useful formulation.
Injectable hydrogels formulated from synthetic polymers (as opposed to HA) can be advantageous because they are more easily tuned for mechanical and degradation properties. No drug-loaded synthetic hydrogels are currently being clinically tested; however, synthetic products including Cartiva (implantable polyacrylamide) and Aquamid (injectable PVA) are being pursued. Cokelaere et al. investigated a synthetic thermoresponsive polymer for delivery of the NSAID celecoxib (CXB) [87]. CXB was loaded into a propyl-capped (poly(ε-caprolactone-co-lactide)-b-poly(ethylene glycol)-b-poly(ε-caprolactone-co-lactide) (PCLA-PEG-PCLA) hydrogel for IA injection in an equine model of synovitis. Horses received an injection of lipopolysaccharide (LPS) in the middle carpal (MC) joints followed by either CXB-loaded gels (~300 mg CXB) or PBS 2 hours after the initial LPS injection. Synovitis flares were induced with subsequent LPS injections at weeks 2 and 4. The peak synovial concentration of CXB was reached at 6h for the CXB-gel group (25.6±11.7 μg ml−1) and decreased to <0.1 μg ml−1 at day 14. CXB-gel significantly increased synovial glycosaminoglycan and collagen II synthesis compared to PBS; however, the effect was transient. Similarly, CXB-gel exhibited significantly lower levels of synovial prostaglandin E2 and white blood cell infiltration, but these anti-inflammatory effects only lasted ~4 hours. Notably, there was a significant increase in MMP activity 24 hours post-LPS treatment with CXB-gel compared to PBS, contradicting the encouraging ECM results and indicating the complexity of OA pathology in this model. These results demonstrate that an NSAID-loaded synthetic hydrogel could be a promising delivery system for OA, but further investigation is needed to fully understand the possible benefits for therapy. From a translational perspective, the demonstrated effectiveness in a large animal model is encouraging for the scaling up of this relatively simple system; however, more detailed efficacy studies should be carried out to fully understand the effects on OA pathology, particularly with comparison to oral or injected CXB. Additionally, the PK data demonstrate a burst release profile; slowing release and achieving a more sustained release profile would likely extend the beneficial effects seen at early time points.
Conclusions:
Hydrogels remain a promising avenue for OA therapy due to their ability to fill the narrow diseased joint space and potentially provide mechanical supplementation [35,36]. HA is a particularly attractive option because it is a native cartilage component and has regulatory precedent. Indeed, of the 6 local, sustained drug delivery technologies currently being clinically investigated for IA OA therapy, 3 are HA hydrogels (Figure 2C). However, the relatively fast clearance of HA from the joint limits its utility as a long-term drug delivery vehicle, and there is ample opportunity for innovation to improve PK of these systems. Synthetic polymeric hydrogels also expand the options for hydrogel-mediated drug delivery in the joint. In particular, synthetic hydrogels offer more facile routes to creating systems that are crosslinked to provide better physical properties and diffusional restraints for drug release or that are engineered with functionalities that provide affinity to the drug cargo in order to better sustained release. The more recent approvals of synthetic hydrogels (i.e. Cartiva) are encouraging for future products which combine both mechanical and pharmaceutical benefits.
3. Polymeric Microparticles
Polymeric MPs are formulated at ~1–100 μm, typically with a spherical morphology (Figure 3B), though other shapes are achievable [88,89]. They can be composed of natural materials such as GLT and chitosan or synthetic materials including PEG, poly(caprolactone) (PCL), poly(propylene sulfide) (PPS), poly(lactic-co-glycolic acid) (PLGA), or combinations of these and other polymers [4,90]. MPs (≤ ~10 μm) can be phagocytosed by superficial chondrocytes or synoviocytes [91], which can increase the potency and longevity of drug action. Larger MPs (10–100 μm) promote joint retention and increase drug residence time up to months [4], releasing the smaller, encapsulated free drug which can permeate throughout the tissue. By being engineered into a larger size range, larger MPs resist lymphatic and vascular drainage, a strategy which is clinically attractive due to its simplicity. However, large MPs with long residence times have been shown to promote a mild foreign body response in the joint [92], which indicates that the selection of polymeric material is critical. Additionally, their large size prevents MPs from penetrating the dense, ECM-rich cartilage matrix (pore size ~60–200 nm [93]). Therefore, MPs tend to form a long-term drug depot in the joint and could be best suited for drug delivery to the synovium or delivery of drugs or drug conjugates with cartilage-penetrating behavior (Section 5). Others have extensively reviewed polymeric particle systems for IA therapy of OA [4,90,94]; here, we highlight clinically and preclinically-tested polymer-based drug delivery MPs (Table 4).
Table 4:
Preclinical Microparticle Systems
| Polymer/ Drug/ Formulation | Model | Dosing Scheme (Dose) | Outcome | Ref. |
|---|---|---|---|---|
| PEA/ CXB/ MP | Rat ACLT/MMx model | Single injection (14.6 μg CXB) | PEA-CXB was detectable at 12 weeks but did not improve histological outcomes | [145] |
| PEA/ CXB/ MP | Rat ACLT/MMx model | Single injection (15, 115, 195 μg CXB) | PEA-CXB significantly improved bone outcomes independent of CXB dose; OARSI score was not improved | [146] |
| PEA/ TCA/ MP | Rat collagenase-induced model | Single injection (250 μg TCA) | PEA-TCA was detectable at 10 weeks and improved synovitis scores at 7 weeks. | [148] |
| PEA or PLGA/ TCA/ MP | Rat synovitis model | Single injection (62.5 μg TCA) | PEA-TCA significantly reduced joint swelling after second and third flares; PLGA-TCA did not; PEA-TCA significantly improved synovitis after the third flare | [149] |
| PLA-TGPS/ PH/ NPPs | Mouse DMM model | Injection on days 7 and 35 (2.5 mg/kg) | PH-NPPs significantly improved OARSI score and reduced serum cytokines at 9 weeks | [150] |
| PLA-TGPS/ KGN/ NPPs | Mouse DMM model | Injection on days 7 and 36 (2.25 mg/kg) | KGN-NPPs significantly improved OARSI score at day 63 | [152] |
Abbreviations: ACLT=anterior cruciate ligament transection; CXB=celecoxib; DMM=destabilization of the medial meniscus; KGN=kartogenin; MMx=medial meniscectomy; MP=microparticle; NPP=nanocrystal-polymer particle; OARSI=Osteoarthritis Research Society International; PEA=poly(ester amide); PH=PH-797804; PLA=poly(lactic acid); PLGA=poly(lactic-co-glycolic acid); TCA=triamcinolone acetonide; TGPS= D-α-tocopherol PEG 1000 succinate
3.1. Clinical Microparticle Products
3.1.1. FX006
FX006 (Zilretta™) is the first polymeric MP-based DDS to receive FDA approval for IA therapy of OA. FX006 comprises 75:25 PLGA (54 kDa) MPs loaded with TCA (Figure 6A). The formulation consists of 25% TCA and 75% PLGA by weight and results in MPs ranging in size from 20–100 μm. Solid TCA crystalline suspension is incorporated into the MPs using the solid-in-oil-in-water (S/O/W) emulsion technique: PLGA is dissolved in dichloromethane, solid TCA is added and sonicated, and the emulsion is formed in aqueous 0.3% PVA [95,96].
Figure 6.
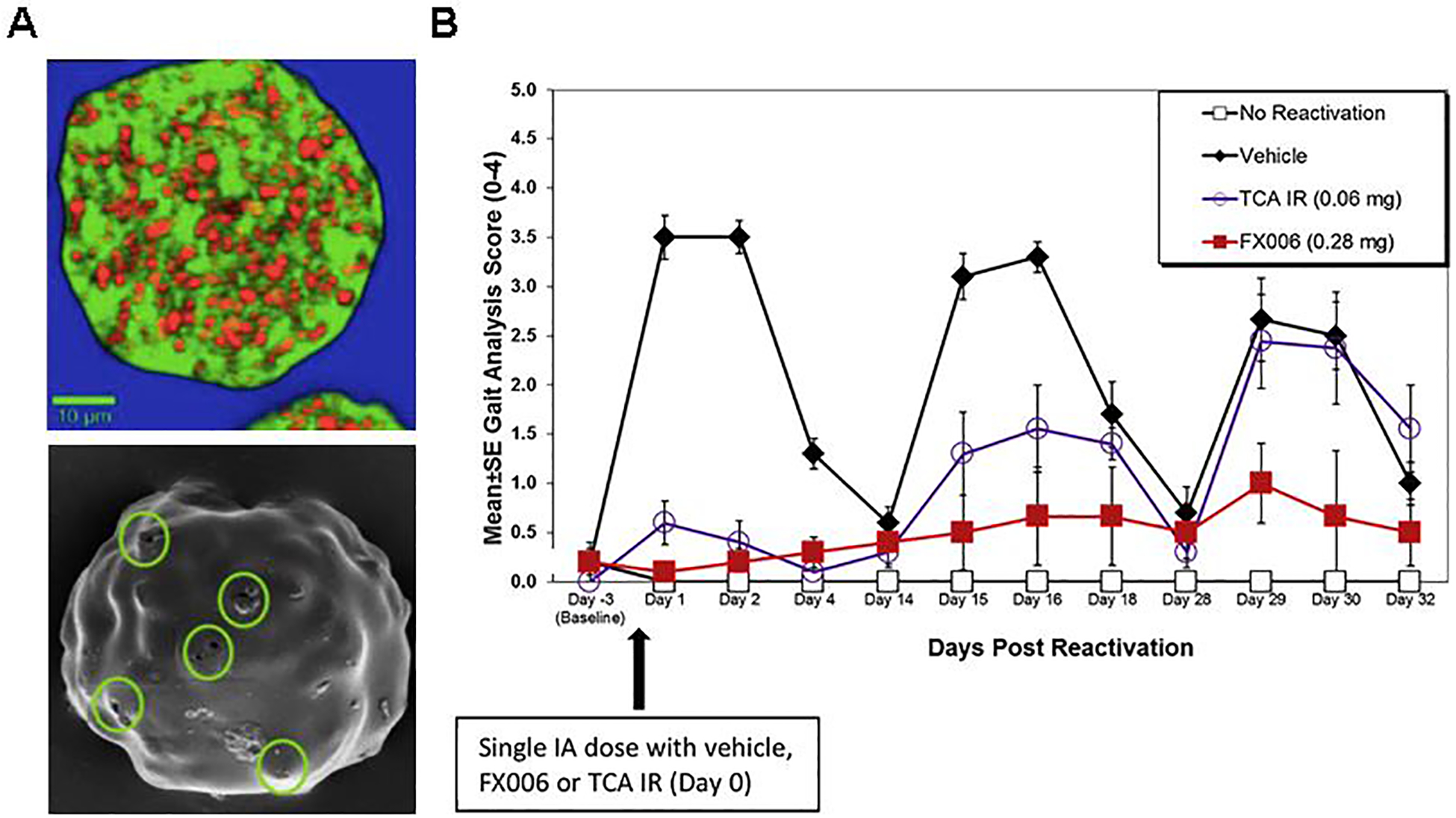
Pre-clinical investigation of FX006. (A) (Top) Raman microscope image of a cross-section of a FX006 microsphere. Small crystals of triamcinolone acetonide (TCA) (red) are embedded in a poly(lactic-co-glycolic acid) (PLGA) matrix (green). (Bottom) Scanning electron microscope image of a FX006 microsphere in the initial stages of release. The small channels, 500 nm in diameter, that appear on the smooth and largely intact surface of the microsphere are circled in green. (B) Gait analysis scores for control rats and groups treated with FX006 (0.28 mg) and TCA suspension (0.06 mg). Injection of FX006 (0.28 mg) on Day 0 resulted in significant sustained analgesic efficacy throughout the study period. Data were expressed as means ± standard error of the mean (SEM). (A) reproduced under the Creative Commons Attribution-Non Commercial-No Derivatives License 4.0 (CCBY-NC-ND).[120] Copyright 2018, P.G. Conaghan, D. J. Hunter, S. B. Cohen, V. B. Kraus, F. Berenbaum, J. R. Lieberman, D. G. Joness, A. I. Spitzer, D. S. Jevsevar, N. P. Katz, D. J. Burgess, J. Lufkin, J. R. Johnson, and N. Bodick. (B) Reproduced with permission.[99] Copyright 2014, Elsevier Ltd.
Preclinical Data
The preclinical data supporting the investigational new drug (IND) application for FX006 (IND #111325) can be broadly divided into safety, PK, and efficacy studies, with the majority of data focused on safety.
Preclinical Safety
Preclinical safety studies for FX006 were conducted in healthy rats and beagle dogs [92,97,98]. In both models, joints exposed to TCA (either as a suspension or as FX006) exhibited cartilage extracellular matrix loss compared to no treatment. Additionally, MP treatment (including blank PLGA control MPs) caused macrophage infiltration into the synovium, which appeared dependent on MP dose in beagle dogs. This finding indicates an unfavorable reaction inherent to the PLGA MPs, which could be attributable to foreign body response or direct damage to tissues from acidified degradation products causing macrophage recruitment. This is an important demonstration of a potential negative effect of the extended-release platform.
Beagle dogs were also used to investigate longer-term effects of FX006 and the effects of repeat dosing [92,97,98]. After a single injection, signs of foreign body response persisted out to 4 months but abated by month 6. Low levels of ECM damage were noted in TCA- and FX006-treated animals were evident out to 3 months but also reversed by month 6. The systemic findings for hypercortisolism (i.e. slight atrophy of adrenal gland) were similar between FX006 and TCA suspension for the same dose of TCA. This is notable because it allowed the study sponsors to pursue a regulatory bridge between FX006 and previously approved TCA suspension products; however, this finding creates uncertainty in the hypothesis that packaging TCA into PLGA microspheres will reduce systemic exposure and side effects. In the repeat-dose study in beagle dogs, 3 injections of FX006 worsened foreign body response as evidenced by macrophage infiltration, fibrosis, neovascularization, neutrophil infiltrates, and granulation tissue; these signs did not fully reverse 6 months after the last injection. High dose TCA suspension (18.75 mg ml−1) and FX006 (75 mg ml−1 MPs, 18.75 mg ml−1 TCA) caused corticosteroid-induced reduction in Safranin O staining out to 6 months. Overall, this study indicates that repeat dosing with FX006 should be approached cautiously.
Conclusions:
In preclinical studies, FX006 demonstrated acceptable safety outcomes. Local and systemic effects of TCA were generally similar between suspension and MP delivery. This finding allowed for regulatory similarity between FX006 and previously approved TCA suspension products; however, it casts doubt on the expected result that local, sustained release would reduce systemic side effects. Notably, MP presence in the joint caused mild foreign body response in a dose-dependent manner, which was not apparent for TCA suspension.
Preclinical Pharmacokinetics and Efficacy
Computational modeling was used to elucidate the PK parameters in rats and dogs [92]. Noncompartmental analysis showed a t1/2 in the rat joint of 451 h for FX006 vs. 107 h for TCA suspension (1.125 mg TCA). Additionally, Cmax of TCA in serum was reduced with FX006 (8.15 vs. 125 ng ml−1). Similar trends were predicted in dogs: t1/2 = 335 vs 33.2 h for 18.75 mg TCA; Cmax = 3.09 vs. 44.2 ng ml−1. Deeper compartmental analysis utilizing a 2-compartment structural model based on 2 first-order absorption processes revealed that the predicted mean residence time (MRT) in the joint for the early release phase was similar between FX006 and TCA suspension but that the MRT for the delayed release phase was significantly longer for FX006. Finally, by scaling the parameters studied for rats and dogs, a dose of 60 mg FX006 in humans was predicted to yield a TCA t1/2 of 539 h and plasma Cmax of 1.44 ng ml−1, which was expected to cause a clinically insignificant effect on endogenous cortisol production.
The major preclinical efficacy study for FX006 was performed in an inflammatory model of synovitis in rats, in which synovitis flares were induced on days 0, 14, and 28 [92,99,100]. FX006 at a dose of 0.28 mg significantly improved gait scores measured on a scale of 0–4 after each reactivation (gait score of ~1.0 at day 29) while TCA suspension did not provide a sustained therapeutic effect (gait score of ~2.5 at day 29) (Figure 6B). Composite histological scoring on a scale of 0–20 showed a statistically significant improvement in joint health for FX006 treated animals at day 32, after the third reactivation. Vehicle treatment resulted in a composite score of ~11, compared to ~7 for TCA suspension and ~1 for FX006. Additionally, FX006 had a plasma Cmax of TCA ~10x lower than for TCA suspension (1.68 ng ml−1 vs. 17.3 ng ml−1) and demonstrated no reduction of serum corticosterone levels.
Conclusions:
Computational PK modeling showed improved PK parameters consistent with a sustained-release profile for FX006, including longer t1/2, longer MRT in joint, and lower Cmax in plasma. In rats, FX006 provided sustained relief in an inflammatory synovitis model, outperforming TCA suspension. Experimental PK parameters in rats confirmed the predicted trend of reduced plasma Cmax for FX006 compared to TCA suspension. In sum, these results indicate that FX006 can extend TCA release, leading to improved outcomes in a preclinical model of OA.
Clinical Studies Leading to FDA Approval
Clinical data for FX006 can be broadly divided into PK studies and efficacy studies. Notably, all of the trials discussed in this section monitored safety in the form of AEs with a focus on treatment-emergent AEs (TEAEs). No life-threatening TEAEs occurred, and there was a very low incidence of serious TEAEs (<1%). The most common AEs experienced were injection site swelling, arthralgia, contusions, and headaches. A detailed analysis of TEAEs is provided in the FDA Clinical Review of FX006 [101].
Clinical Pharmacokinetics
Four clinical trials of FX006 focused on PK. The first occurred in 24 patients with knee OA and focused on PK and pharmacodynamics (PD) of a single injection of FX006 (10, 40, or 60 mg) or TCA suspension (40 mg) (NCT01487200) [102,103]. The primary PD outcome was change from baseline in the 24-hour weighted mean serum cortisol level; all groups significantly suppressed cortisol levels except 10 mg FX006. PK analysis revealed an approximately 30-fold decrease in the plasma TCA Cmax for 40 mg FX006 compared to 40 mg TCA suspension (1028 vs. 30,132 pg ml−1). Conversely, after 43 days, 40 mg FX006 exhibited a synovial fluid TCA concentration approximately 2000-fold higher than 40 mg TCA suspension (82,673 vs. 38.6 pg ml−1). A second PK study in 50 patients with knee OA focused on measuring synovial concentration of TCA after IA administration of FX006 (10 or 40 mg) or TCA suspension (40 mg) at longer time points (12–20 weeks post-treatment, NCT02003365) [103–105]. At 12 weeks, the mean synovial fluid concentration of TCA was 923.7 pg ml−1 when delivered as 40 mg FX006; all TCA suspension patients had levels below the limit of detection (50 pg ml−1). 40 mg FX006 maintained detectable TCA levels in the joint out to 20 weeks in some patients (mean 33.3 pg ml−1). Taken together, these studies supported the extended release profile of TCA in FX006 in humans.
The third and most critical FX006 clinical PK study was carried out in 81 patients with knee OA and focused on synovial fluid concentration of TCA after IA administration of 32 mg FX006 or 40 mg TCA suspension (NCT02637323) [106,107]. At 6 weeks, the mean TCA concentration in the joint was ~450-fold higher for FX006 than for TCA suspension (3590.0 vs. 7.7 pg ml−1). FX006 also maintained detectable TCA levels in the joint out to week 12 for 7 of 9 patients (mean = 290.6 pg ml−1) (Figure 7A). Additionally, the geometric mean (GM) of the plasma Cmax was approximately 10-fold lower (966.7 vs. 11,064.7 pg ml−1) and the t1/2 of TCA in the plasma was ~5x longer (347 vs. 72.5 hours) for FX006. This study was critical to approval of FX006 because it formed a regulatory bridge between the study drug and the already-approved suspension formulation of TCA (Kenalog-40®) by demonstrating that systemic exposure was reduced with FX006 [103].
Figure 7.
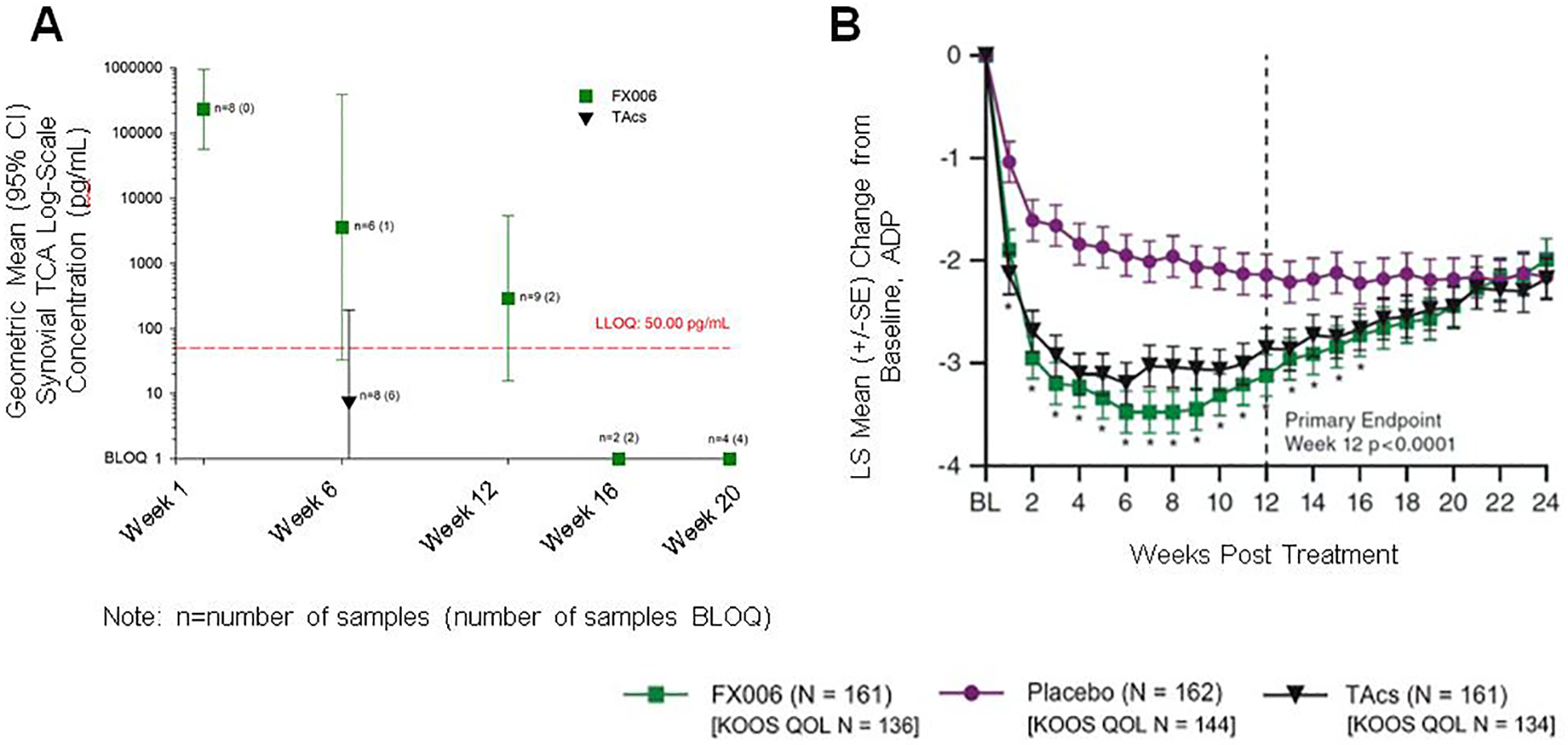
Clinical investigation of FX006 in patients with osteoarthritis (OA). (A) Synovial fluid triamcinolone acetonide (TCA) concentrations over time (log-linear scale) after a single intra-articular (IA) injection of FX006 or TCA crystalline suspension (TAcs). A scaler value of 1 was added to each observed concentration value in order to have a reasonable estimate of the geometric mean synovial fluid values. Error bars represent confidence intervals. LLOQ=lower limit of quantification; BLOQ=below limit of quantification. (B) Weekly records of the least squares mean (LSM) change from baseline (and standard error [SE]) for average daily pain (ADP)-intensity scores on a 0–10 numeric rating scale (n=484). (A) adapted with permission.[107] Copyright 2018, Elsevier Ltd. (B) reproduced under the Creative Commons Attribution-Non Commercial-No Derivatives License 4.0 (CCBY-NC-ND).[120] Copyright 2018, P.G. Conaghan, D. J. Hunter, S. B. Cohen, V. B. Kraus, F. Berenbaum, J. R. Lieberman, D. G. Joness, A. I. Spitzer, D. S. Jevsevar, N. P. Katz, D. J. Burgess, J. Lufkin, J. R. Johnson, and N. Bodick.
A related study investigating the PK benefits of extended-release TCA in FX006 was focused on patients with Type 2 diabetes mellitus (DM). Because these two diseases have common risk factors, particularly a sedentary lifestyle and obesity, they are common comorbidities, with approximately 30% of DM patients experiencing OA and approximately 14% of OA patients suffering from DM [108]. In DM patients, systemic exposure to corticosteroids from IA administration and subsequent efflux from the joint has been shown to cause an increase in blood glucose (BG), with one study finding ~23% of patients receiving IA TCA crystalline suspension experiencing elevated BG lasting 2.5–4 days [109]. Flexion investigated the effect of an IA injection of 32 mg FX006 or 40 mg TCA crystalline suspension on BG in 33 patients with knee OA and type 2 DM (NCT02762370) [110,111]. In these patients, mean average daily BG measured by continuous glucose monitoring-measured glucose (CGMG) increased from baseline by 8.2 mg/dl in FX006-treated patients and 37.1 mg/dl in TCA suspension-treated patients in days 1–3 after injection. The least squares mean (LSM) difference between these groups was 19.2 mg/dl and represented a statistically significant reduction. These results indicate that the extended-release profile of FX006 could have a specific benefit for the DM-afflicted patient population by reducing their risk for corticosteroid-induced hyperglycemia.
Conclusions:
The comprehensive clinical PK data around FX006 indicate that the extended-release formulation of TCA in FX006 reduces plasma Cmax and increases t1/2 in the joint relative to crystalline suspension of TCA. There were no indications that the extended release profile caused AEs at a rate higher than the classical suspension formulation. The trial in DM patients indicates that reducing systemic TCA exposure could be particularly beneficial to patients who have an established sensitivity or susceptibility to corticosteroid side effects.
Clinical Efficacy
Three efficacy studies contributed to FDA approval of FX006. The first occurred in 229 patients with knee OA, focused on magnitude and duration of pain relief for a single IA injection of FX006 (10, 40, or 60 mg) or TCA suspension (40 mg) (NCT01487161) [103,112]. Pain was measured using a patient-reported 11-point numeric rating scale (NRS, 0 = “no pain”; 10 = “pain as bad as you can imagine”). At weeks 8, 10, and 12, the change in the weekly mean of the average daily pain NRS was not significantly different for 10, 40, or 60 mg FX006 compared to 40 mg TCA suspension; however, all showed improvement (ranging from −3.2 to −4.3). Other outcomes studied (8 weeks post-injection) included the WOMAC score, responders according to the Outcome Measures in Rheumatoid Arthritis Clinical Trials/OARSI (OMERACT-OARSI) criteria, and the GIC score. The OMERACT-OARSI criteria represent a set of uniform outcome measures which define “responders” to a given treatment [113]. These secondary outcomes exhibited general improvement for all groups, with 10 and 40 mg FX006 statistically outperforming TCA suspension. A second Phase 2 trial in 306 patients with knee OA focused on magnitude and duration of pain relief for a single IA injection of FX006 (32 or 16 mg) or placebo (saline) (NCT02116972) [101,104,114,115]. The primary endpoint of the study was not met: at 12 weeks, the 32 mg FX006 group experienced a LSM change from baseline in the pain NRS of −3.08 compared to −2.50 for placebo (p=0.0821). However, there was a significant improvement for 32 mg FX006 over placebo at each of weeks 1–11 and week 13. Secondary analyses also demonstrated a significant improvement over saline in WOMAC pain, WOMAC physical function, and patient-reported GIC scores at weeks 4 and 8.
Flexion also attempted to conduct an efficacy trial in patients with PTOA as opposed to idiopathic OA (NCT02468583) [116]. The target population was patients 20–50 years of age with a confirmed diagnosis of PTOA of the knee. The intended enrollment was approximately 124 patients; however, the study was terminated due to low enrollment (6 patients). This case demonstrates the difficulty of studying PTOA in humans due to the fact that these patients are typically younger and can be relatively pain-free for years after their injury. The failure of this study raises an important conflict between clinical and preclinical OA studies: many animal models are surgically induced, modeling most closely PTOA, whereas a relative minority of human cases (~12% [117]) are post-traumatic. Kuyinu et al. recently reviewed animal models of OA and how they have been used to understand pathology of idiopathic and PTOA [118].
The pivotal efficacy trial leading to FDA approval focused on the magnitude and duration of pain relief for a single IA injection of 32 mg FX006, 40 mg TCA suspension, or saline (placebo) in 486 patients with knee OA (NCT02357459) [101,119,120]. At 12 weeks, FX006 outperformed placebo in improvement of pain NRS (−3.12 vs. −2.14) but did not perform significantly better than TCA suspension (−2.86) (Figure 7B). By week 24 of the study, the NRS improvements experienced by patients in the 3 groups were similar (~−2 for each). The trend in the area under the effect curve (AUE) was similar: FX006 significantly outperformed placebo (p<0.0001) but not TCA suspension (LSM = −247.3, −145.3, and −231.9, for FX006, placebo, and TCA suspension, respectively). FX006 also outperformed placebo at 12 weeks in WOMAC pain, stiffness, and function subscales.
Conclusions:
The above results were submitted to the FDA Division of Anesthesia, Analgesia, and Addiction Products (DAAAP) in NDA 208845 in December 2016 [104]. These studies demonstrated a clear therapeutic benefit over saline; however, the efficacy of FX006 was generally similar to TCA suspension. Based on its superiority over placebo, FX006 (Zilretta™) was approved for OA knee pain in 2017, making it the first microsphere-based DDS approved for IA treatment of OA.
Clinical Studies after FDA Approval
After receiving FDA approval for IA therapy of knee OA, Flexion began investigating expanded applications for FX006. A 2017 study in 208 patients with knee OA investigated the effect of repeat administration of FX006, with a focus on TEAEs and pain reduction for 32 mg FX006 administered twice to the same knee (NCT03046446) [121,122]. Patients received 1 injection on day 1 and received a repeat injection at week 12, 16, 20, or 24 depending on the return of pain. The overall incidence of TEAEs after both injections were similar (41.9 and 35.2% after the first and second injections, respectively), and the most common AE was arthralgia (10.6 and 19.0% during the first and second injection periods, respectively). Additionally, there were similar reductions of about −1.5 points in the WOMAC pain subscore after each injection, indicating similar efficacy for the repeat injections. These results indicate that FX006 could be appropriate for repeat dosing in the knee. In another exploration of dosing, a trial in 24 patients investigated FX006 therapy for bilateral OA, focused on systemic exposure to TCA after 2 IA injections of FX006 (32 mg per knee, total dose 64 mg) or TCA suspension (40 mg per knee, total dose 80 mg) (NCT03378076) [123–125]. The plasma Cmax of TCA was ~3-fold lower for FX006 compared to TCA suspension (2277.7 vs. 7394.7 pg ml−1). Systemic exposure measured as the area under the Cplasma-time curve was lower for FX006 than TCA suspension (911,096 vs. 1,253,714 h pg ml−1), while the MRT in the joint was longer with FX006 administration (324 vs 134 h). Notably, FX006 suppressed serum cortisol levels for a longer period than TCA suspension (29 vs. 8 days). Taken together, these results suggest that repeat injections of FX006 may be safer than bilateral injections, though both should be studied more extensively.
Another application that is being investigated is IA injections for hip and shoulder OA. Flexion initially planned a repeat dosing study in the hip similar to the above described in the knee; however, this study was terminated because there was a high incidence of incomplete study drug administration due to difficulties during injection (NCT03793010) [126]. Therefore, a small open-label, Phase 2 study was undertaken to verify successful administration of 32 mg FX006 into the acetabulofemoral joints in 16 patients with hip OA (NCT04065074) [127]. After administration issues were corrected, a trial was completed on 55 patients with OA of the shoulder or hip. This study focused on systemic TCA exposure after a single injection of 32 mg FX006 (12 shoulder, 15 hip) or 40 mg TCA suspension (13 shoulder, 15 hip) (NCT03382262) [128]. For the shoulder injections, serum TCA concentrations were similar over the 12-week study; however, there was ~4x less systemic TCA exposure with FX006 injection in the hip over days 0–3, after which the levels were comparable. Cmax values for shoulder injections were 1142.4 pg ml−1 for FX006 and 1689.6 pg ml−1 for TCA suspension. In the hip, Cmax values were 792.9 pg ml−1 for FX006 and 4672.6 pg ml−1 for TCA suspension. Notably, these values for free TCA were ~5–10x lower than those found for injection in the knee, while the FX006 Cmax values are similar for all 3 joints. These data indicate that the PK effects of FX006 are relatively consistent across injection sites, whereas TCA suspension might have more PK variability as a function of injection site.
There are currently 4 active trials for FX006. The first is planning to recruit 100 patients with OA of the knee (male and female, ≥40 years of age) to investigate reduction of synovial inflammation (measured by synovial volume) for a single injection of 32 mg FX006 (NCT03529942) [129]. The second is focused on improvement in physical performance measures with an expected recruitment of 70 patients (NCT03895840) [130]. The third has an intended enrollment of 35 patients with knee OA focused on improvement in pain, disability, physical performance, and physical activity levels (NCT04261049) [131]. Finally, an ongoing trial is investigating pain reduction in an anticipated cohort of 250 patients with either shoulder OA or shoulder adhesive capsulitis (NCT04160091) [132]. These active studies could further broaden the understanding of this formulation’s effect on various outcome measures associated with OA outside of pain relief and at sites other than the knee.
Conclusions:
Flexion has pioneered the clinical translation of MP corticosteroid delivery for OA. Their preclinical and clinical data show convincingly that the MP formulation prolongs TCA residence in the joint while reducing systemic exposure. However, it is still unclear if there is a measurable efficacy benefit following from this improved PK profile over TCA crystalline suspension. In light of our current knowledge regarding the adverse effect of corticosteroid exposure on ECM [22] and the preclinical data demonstrating reduction in ECM in FX006-exposed joints [92], it will be important to investigate long-term effects of FX006 injections on cartilage integrity. Another long-term outcome which will be interesting to study is whether FX006 has the ability to significantly delay time to total joint arthroplasty. Potentially the most promising application for FX006 is for patients who are uniquely susceptible to systemic corticosteroid off-target effects. The reduction in systemic exposure provided by the extended-release formulation could make corticosteroid treatment a viable option for these patients, expanding their opportunities for safe and effective pain relief.
3.1.2. EP-104IAR
Eupraxia Pharmaceuticals is developing a PVA-coated crystalline formulation of the corticosteroid fluticasone propionate (FP) for IA treatment of OA. The drug is first processed into crystals ~100 μm in diameter and then spray-coated with PVA, resulting in particles 60–150 μm in diameter. The particles are heat-treated to improve crystallinity of PVA. The final content of PVA is ~4.5% [133].
Preclinical data was collected in sheep and beagle dogs. In sheep, the particles were heat-treated at 130°C and injected at 0.25 mg kg−1. This formulation exhibited significant plasma FP concentrations (~2–10 ng ml−1) over 40 days, leading to the temperature at the heat-treatment step to be increased to 220°C [133]. This formulation was administered IA to beagle dogs (0.6 or 12 mg) [134]. Low-dose EP-104IAR did not exhibit detectable levels of FP in the plasma, while high-dose EP-104IAR demonstrated plasma concentrations of ~300 pg ml−1 3 days post-injection, which declined below detectable levels by day 7. High-dose EP-104IAR also maintained detectable synovial fluid levels of FP out to day 60 of the study (geometric mean 142.7 ng ml−1), a trend which was also reflected in cartilage levels of FP (geometric mean 53.9 ng ml−1). Chondrocyte vacuolation was identified histologically in both treatment groups at day 7, and this finding persisted at day 29 for high-dose EP-104IAR. Synovial intima vacuolation was also noted for this dose at day 7, which was not apparent at day 29. Vacuolation reversed by day 46; however, foreign material (likely PVA) was noted out to day 60. These data led to a Phase 1 trial in 32 patients with OA of the knee focused on PK and safety of a single IA injection of 15 mg EP-104IAR or placebo (NCT02609126) [135]. Though detailed results have not been reported for this trial, an abstract presented at the 2018 Pain Week conference stated that clinical PK data “support the non-clinical findings” and there were no safety concerns [136]. Eupraxia has initiated a Phase 2 trial with a planned recruitment of 238 patients with knee OA focused on PK, safety, and efficacy of EP-104IAR (25 mg) and placebo (NCT04120402) [137].
Conclusions:
The MP formulation of EP-104IAR provides a distinctly different strategy from FX006. Notably, the drug content of this formulation is much higher (~95%), which could reduce the foreign body response associated with MPs. Preclinical data did not investigate corticosteroid-induced proteoglycan loss, though cellular vacuolation is concerning. The results of the ongoing placebo-controlled trial will be informative for the future application of EP-104IAR.
3.1.3. Terminated Development Programs
FX005
Flexion Therapeutics partnered with AstraZeneca to investigate a PLGA MP formulation of a p38/MAPK inhibitor. The inhibitor, N-[5-[(cyclopropylamino)carbonyl]-2-methylphenyl]-3-fluoro-4-(pyridin-2-ylmethoxy)benzamide, was incorporated into PLGA MPs at a mass ratio of ~13%. In rats, the t1/2 of the free drug (15 ng) in the joint was 0.2 hours, while MP delivery (200 μg particles, ~26 ng drug) extended release out to 21 days (1 mM drug in synovial fluid). Serum levels remained low over this period (~10 to ~0.2 nM). In a therapeutic studyalso in rats, MP-mediated delivery of the inhibitor significantly improved weight bearing on the affected joint over 18 days, while the free drug had no effect [138]. A single clinical trial was undertaken in 140 patients with knee OA focused on safety, PK, and efficacy for FX005 (1, 10, or 45 mg) (NCT01291914) [139]. Flexion announced in a press release that FX005 improved joint pain and function compared to placebo at 4 weeks [140]. However, development was not continued, and Flexion terminated its partnership with AstraZeneca in 2017. The company did not expand on the reasons for the termination, but the approaching approval of FX006 could have contributed. The rights to FX005 reverted solely to AstraZeneca, which has not pursued development [141]. This case demonstrates the challenge of balancing clinical development of multiple drug candidates. Companies can halt development to focus on more advanced or potentially lucrative development programs. Business considerations like this will will impact the trajectory of IA DDS technologies for OA therapy, particularly in the pursuit of potential DMOADs, which could likely combine both new active agents and novel DDSs.
3.2. Preclinical Microparticle Systems
The clinical success of FX006 is encouraging for translation of other MP formulations for OA therapy. A polymer which has gained interest in recent years for IA MP-mediated drug delivery is poly(ester amide) (PEA). Because of the peptide bonds in the backbone of PEA, it is susceptible to protease degradation [142,143], particularly at elevated protease levels in the inflamed joint [144]. Janssen et al. investigated a PEA MP (10–100 μm) loaded with CXB (3.9%, w/w) formulated via an oil-in-water emulsion (Figure 8A) [145]. The authors demonstrated inflammation-triggered release in vitro by treating the particles with neutrophil lysates. In a rat OA model, PEA (375 μg, ± 14.6 μg CXB) was detectable in the joint tissue at 12 weeks (~17% remaining from initial injection) (Figure 8B); unfortunately, only PEA was measured, not CXB levels. Despite the impressive retention of PEA in the joint, CXB-loaded PEA MPs did not improve histological outcomes in joints compared with saline or unloaded MPs (Figure 8C). The same group performed a similar study focused on the dose of PEA-CXB MPs (~35 μm) [146]. OA rats received unloaded PEA MPs (1.75 mg) or PEA-CXB MPs (15, 115, or 195 μg CXB). The low-dose CXB MPs significantly increased weight bearing on the affected limb over the 15-week study, likely due to pain relief. Additionally, the low and high doses of CXB significantly improved histological synovial inflammation score (~36% and ~57% improvement, respectively); however, the total OARSI score, which focuses more on articular cartilage rather than the synovium [147], was not improved with any CXB doses. The 12-week retention of this system in the joint and its relative simplicity are encouraging for translation; however, the lack of dose response to CXB and lack of improvement in OARSI score indicates that a different therapeutic agent could be required to fully leverage the retention advantage toward achieving a DMOAD effect.
Figure 8.
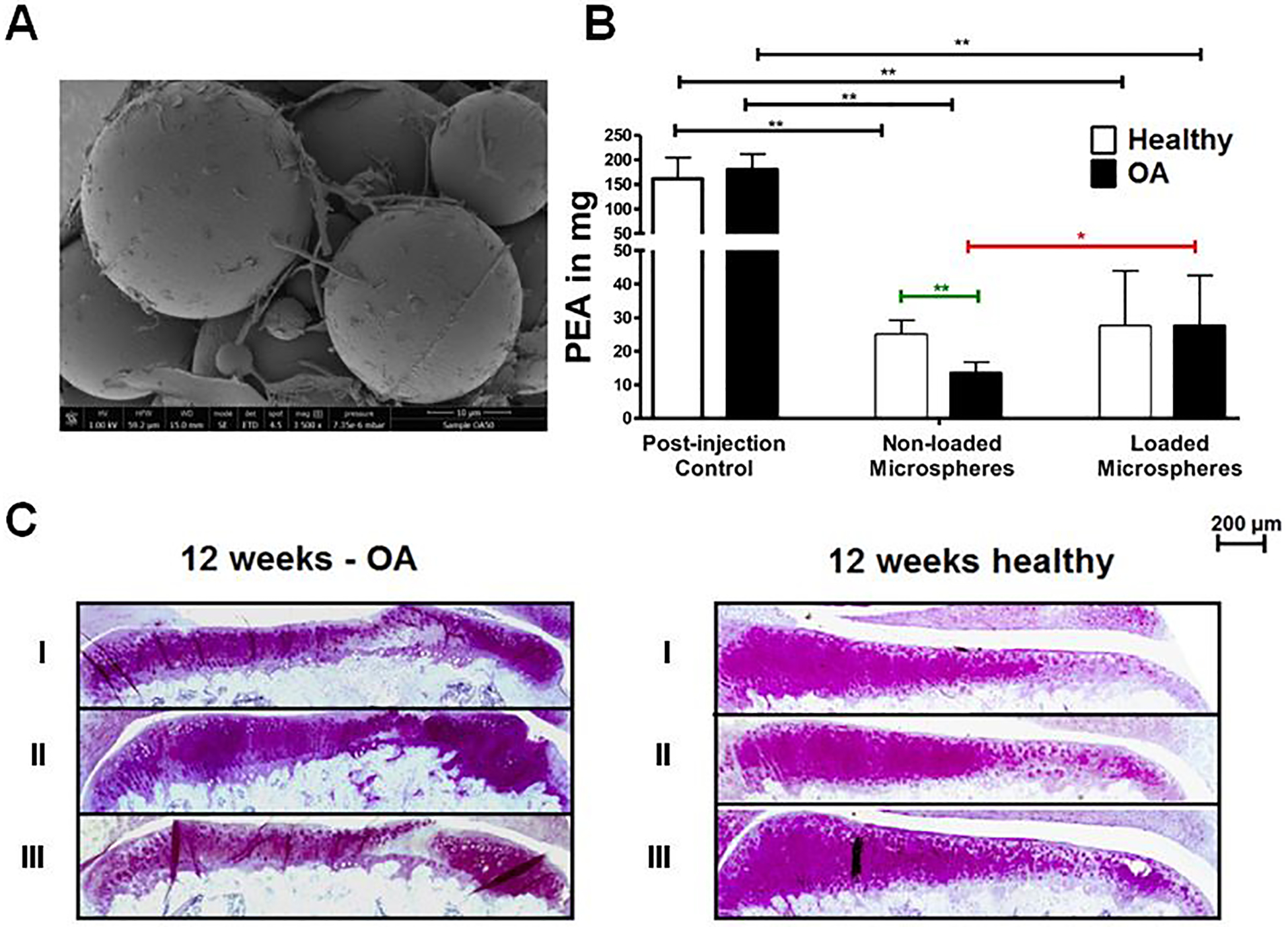
Poly(ester amide) (PEA) microparticle delivery of celecoxib (CXB) in rats with osteoarthritis (OA) induced via anterior cruciate ligament transection (ACLT)/partial medial meniscectomy (MMx) surgery. (A) Representative scanning electron microscope image of PEA microspheres. (B) In vivo degradation of both loaded and non-loaded PEA microspheres is shown by PEA levels in knees measured directly after injection (post-injection control) and twelve weeks after injection. Data are mean ± standard error of the mean (SEM), n = 6. * indicates a statistically significant difference: designated as * p<0.05 and ** p<0.005. (C) Representative images of histological sections of the medial tibial plateau for both OA-induced and healthy knees twelve weeks after treatment and thionin staining. Treatments were: 0.9% NaCl (I), non-loaded microspheres (II), and CXB-loaded microspheres (III). Reproduced with permission.[145] Copyright 2016, Elsevier Ltd.
Rudnik-Jansen et al. also investigated PEA MP-based sustained drug delivery in OA [148]. These MPs were 22 μm in mean diameter and loaded with 20% (w/w) of the corticosteroid TCA; this system is very similar to FX006 except for the different polymer composition. TCA suspension (250 μg) and PEA-TCA (1.25 mg, 250 μg TCA) were compared in a rat model of OA. Particles were detected up to 70 days post-injection, and peak serum levels of TCA were lower with PEA particles than TCA controls (12.5 vs. 50 μg ml−1). Neither TCA MPs nor bolus TCA had a significant effect on histologically assessed cartilage degradation, but TCA MPs significantly improved synovitis scores at 7 weeks (~66% improvement compared to unloaded particles). This group performed another study comparing PEA and PLGA MPs loaded with TCA [149]. The mean diameters and TCA loading were 24 vs. 39 μm and 28 vs. 23% (w/w) for PEA and PLGA, respectively. Rats underwent synovitis flares at days 0, 14, and 28 and received IA injection of empty PEA particles, TCA suspension, PLGA-TCA, or PEA-TCA (all 62.5 μg TCA). All TCA treatments significantly reduced joint swelling after the first flare (~85% reduction); however, only PEA-TCA significantly reduced swelling after the second and third flares (~50% and ~75% reduction). PLGA-TCA showed an intermediate reduction in joint swelling at these time points, but it was not significant. The histological synovitis score was significantly improved after the third flare with PEA-TCA compared to both empty PEA and TCA suspension (~64% improvement), but not PLGA-TCA. It is possible that PEA’s inherent sensitivity to inflammatory proteases could represent an improvement over PLGA; however, more comparative studies are needed to determine a benefit of possible disease-modified degradation over nonspecific hydrolysis. Disease-mediated drug release could represent an improvement over FX006 when retention is similar. Based on these studies, PEA could be a promising alternative to PLGA for MP therapy in OA. Because of their similarities, this system could follow a similar regulatory path as FX006; however, the clinical precedent for PEA lacks the extensive regulatory precedent of PLGA.
One attractive aspect of polymeric drug delivery is the opportunity to expand the classes of drugs which will be compatible and active in IA injection. Standard (non-MP) steroid formulations can reduce pain and inflammation, with sustained release formulations mostly providing benefits associated with less systemic side effects rather than actually improving OA outcomes. MPs as a platform can potentially be extended to load a wide variety of drug molecules. Maudens et al. sought this opportunity for delivery of PH-797804 (PH), a p38/mitogen-activated protein kinase (MAPK) inhibitor [150]. PH is an attractive candidate for OA due to the role of p38/MAPK signaling in inflammation, and an oral formulation has reached Phase 2 clinical testing (NCT01102660) [151]. PH or Dex were first recrystallized and stabilized with D-α-tocopherol PEG 1000 succinate (TGPS) to form nanocrystals. The drug nanocrystals were combined with poly(lactic acid) (PLA) and formulated into nanocrystal-polymer particles (NPPs) using a spray-drying formulation process (14.2 and 12.3 μm for PH and Dex, respectively) (Figure 9A). The authors expected that the nanocrystal formulation would improve drug incorporation into the MPs and slow release because the drug crystals form a slow-dissolving depot within the MP as opposed to dispersing throughout the polymer matrix. This nanocrystal strategy mimics the S/O/W process employed in the manufacturing of FX006, with relatively high drug loading of 25% (w/w). Control MPs were formulated with PLGA (75:25) and unmodified drug using a conventional emulsion-solvent extraction method with PH or Dex dissolved in the organic phase with PLGA (9.97 and 9.60 μm particles for PH and Dex, respectively). NPPs had approximately 10x higher drug loading than control MPs (~30 vs. ~3%, w/w) and significantly slower PH release in vitro over 90 days (~20% vs. ~80%) (Figure 9B). In the destabilization of the medial meniscus (DMM) model of PTOA, 2 injections of PH-NPPs (7 and 35 days after surgery, 2.5 mg kg−1 PH) significantly improved histological OARSI score and cartilage erosion (Figure 9C) and significantly reduced serum levels of IL-1β and tumor necrosis factor-α (TNF-α) compared to unloaded NPPs at day 63 (Figure 9D–E). There were numerical improvements in these metrics for PH-NPPs compared to Dex-NPPs, but these differences were not statistically significant. Notably, NPPs were still detectable in the joint at day 63. This group used a similar drug nanocrystal/spray drying formulation method to generate kartogenin (KGN)-PLA NPPs [152]. KGN is a CBFβ-RUNX1 pathway activator which is a potential DMOAD due to its ability to stimulate mesenchymal stem cell (MSC) chondrocyte differentiation [153]. Mice received 2 IA injections of unloaded NPPs, free KGN (1.1 μg kg−1 wk−1), or KGN-NPPs (2.25 mg kg−1 month−1 KGN). At 7 weeks after the first injection, ~25% of the combined KGN-NPP injections were retained in the knee joint. KGN-NPPs significantly reduced serum levels of VEGF compared to unloaded NPPs and significantly improved histological OARSI scores compared to unloaded NPPs and free KGN. The sustained-release capabilities of PLA MPs combined with the innovative nanocrystal drug format yields highly desirable drug loading with promise for long-lasting OA treatment. PH and KGN MP formulations show promise and warrant additional development toward clinical application as DMOADs. Notably, these studies demonstrated long-term retention of the polymeric MP, but drug levels in the joint were not directly assessed. Future work should measure drug retention in the joint directly and connect improved therapeutic outcomes with sustained release of the therapeutic.
Figure 9.

Investigation of PH797804 (PH)-loaded nanocrystal-polymer particles (NPPs) in the mouse destabilization of the medial meniscus (DMM) model of osteoarthritis (OA). (A) Schematic showing the structure of both conventional microparticles (MPs) and NPPs. (B) Characterization of MP and NPP cumulative drug release profiles based on an in vitro study over a three-month period. Data were expressed as means ± standard deviation (SD). n = 3. (C) Representative photomicrographs of mouse knees at 63 days with cartilage stained purple. Arrows indicate OA damage. Scale bar = 100 μm. Multiplex enzyme-linked immunosorbent assay (ELISA) analysis of (D) interleukin-1β (IL-1β) and (E) tumor necrosis factor-α (TNF-α) in mouse plasma after 63 days. Error bars represent standard deviation (SD). * indicates a statistically significant difference: designated as *p < 0.0332, **p < 0.0021, and ***p < 0.0002. Reproduced with permission.[150] Copyright 2018, Elsevier Ltd.
Conclusions:
The size-based retention of MPs can sustain local retention and reduce systemic exposure of drugs delivered IA. From the perspective of clinical translation, size-based retention allows for a relatively simple strategy for persistent drug release in the joint. Two of the 6 products investigated for OA therapy are MP formulations, including the only FDA-approved product (Figure 2C). The formulation of a microparticle depot is particularly suited for drugs which act in the synovium or outer cartilage layer, like corticosteroids. Future clinical work will benefit from further exploration of disease-responsive polymers such as PEA, improved formulation processes, and new drugs with the potential to be disease-modifying rather than purely palliative.
4. Liposomes
Liposomes are nano- to micro-sized phospholipid vesicles comprising a bilayer membrane envelope (Figure 3C) [154]. Drug loading can be achieved in the aqueous core (hydrophilic agents) or within the hydrocarbon membrane (hydrophobic agents) [154,155]. Liposomes can be formulated in a wide range of sizes (~20 nm to ~5 μm) depending on the composition and fabrication process [156]. Liposomes are attractive drug delivery vehicles because they can provide extended release of hydrophobic drugs, such as corticosteroids. Besides their capacity to act as slow-release drug depots, liposomes are of interest in the context of OA because they can also potentially provide lubrication to the joint [157]. For example, MM-II is a drug-free liposomal product currently in clinical trials for OA. The liposomes are formulated as multilamellar vesicles (MLVs, liposomes with concentric layers of lipids) from 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) at a molar ratio of 0.6:1.0 DMPC:DPPC [158,159]. In a clinical trial of 40 patients with knee OA, a single injection of MM-II exhibited a larger reduction in pain out to 90 days post-injection compared to commercial HA (Durolane™). The WOMAC pain subscore a questionnaire very commonly used to quantify OA disease progression in patients [48], showed an improvement of ~−38 vs. ~+28 for MM-II vs. HA at day 7 and ~−42 vs. ~−27 at day 90, indicating both a faster onset of pain relief and a larger magnitude of relief [160]. The sustained efficacy of MM-II out to 90 days is impressive for long-term pain relief and is presumed to be due to lubrication of the joint. Improved pain relief over HA indicates that joint lubrication via liposomes could be an alternative strategy to viscosupplementation; however, these products should be compared in larger cohorts of patients. Others have reviewed liposomal drug formulations for OA [90]; here, we highlight the clinical product TLC599 and preclinical systems for OA drug delivery (Table 5).
Table 5:
Preclinical Nanoparticle, Conjugate, and Combination Systems
| Polymer/ Drug/ Formulation | Model | Dosing Scheme (Dose) | Outcome | Ref. |
|---|---|---|---|---|
| LNP/ siIhh/ Liposome | Rat ACLT model | Five injections every 2 weeks for 10 weeks (2.5 and 5 nM LNP) | LNP-siIhh improved histological morphology and decreased MMP13 staining | [171] |
| Collagomer/ DF/ Liposome | Rat MIA model | Single injection (1 mg/kg DF) | Collagomer-DF reduced inflammation volume similar to free DF and unloaded collagomer | [175] |
| Polyurethane/ KGN/ NP | Rat ACLT model | Injections at weeks 0, 3, 6, and 9 (100 μM KGN) | PN-KGN significantly improved OARSI score and qualitatively improved collagen II staining | [178] |
| PLGA-GA/ indomethacin/ NP | Rat AIA model | Injections at weeks 2 and 3 (500 μg SANS3/8-GA2) | SANS3/8-GA2 reduced knee swelling and synovial TNF-α | [179] |
| Chitosan-HA/ Curcuminoid/ NP | Rat ACLT/MCLT model | Four injections once per week (0.38 mg cucrcuminoid) | HA-CNP significantly reduced MMP levels and preserved histological cartilage integrity | [180] |
| Chitosan-MoS2/ Dex/ Nanosheet | Mouse papain-induced model | Injections every 3 days for 28 days (1 mg/kg MCD) | MCD decreased degradation enzymes and improved histological score and functional outcomes | [181] |
| DMAEMA-co-BMA-co-PEG/ siMMP13/ NP | Mouse mechanical loading model | Six injections once per week (0.5 mg/kg siMMP13) | mAb-CII targeted NPs significantly improved joint retention over non-targeted NPs and demonstrated MMP13 silencing and improved joint outcomes compared to scrambled siRNA. | [177] |
| Biotinylated PEG-Avidin/ Dex/ conjugate | Rabbit ACLT model | One injection (0.5 mg Dex) | Avidin-Dex significantly rescued joint space thickness and improved bone outcomes but reduced Safranin-O staining at 3 weeks. | [192] |
| PAMAM/ IGF-1/ conjugate | Rat ACLT/MMx model | Single injection (6 μM IGF-1) | PAMAM-IGF-1 reduced cartilage degradation and osteophyte volume at 4 weeks | [190] |
| HAMA-chitosan/ Cordycepin/ hydrogel-MP hybrid | Mouse ACLT model | Injections once per week (5 mg/kg cordycepin) | CM-cordycepin +HAMA significantly improved OARSI score, decreased proteinase levels, increased ECM proteins at 8 weeks | [193] |
| GelMA-chitosan/ SIN/ hydrogel-MP hybrid | Mouse ACLT model | Injections once per week (5 mg/kg SIN) | CM-SIN+HAMA significantly improved OARSI score, decreased proteinase levels, increased ECM proteins at 8 weeks | [195] |
| HA-PEG/ KGN/ hydrogel-NP hybrid | Rat ACLT/MMx model | Injection on weeks 7 and 10 (50 mg) | HA-PEG-KGN reduced OARSI score and increased ECM staining at 8 weeks after first injection | [196] |
| PEG-4MAL-PLGA/ Dex/ hydrogel-NP hybrid | Mouse mechanical loading model | Single injection (1.8 μg Dex) | PEG-4MAL hydrogels ± PLGA-Dex NPs improved OARSI score and reduced osteophyte size at 2 weeks | [197] |
| HA-mixed lipids/ CXB/ hydrogel-liposome hybrid | Rabbit ACLT/ MCLT/ PCLT/MMx model | Single injection (0.15 mg CXB) | HA-CXB liposomes reduced global joint damage scores at 2 weeks | [198] |
Abbreviations: ACLT=anterior cruciate ligament transection; AIA=antigen-induced arthritis; BMA=butyl methacrylate; CM=chitosan microparticle; CNP=curcuminoid nanoparticle; CXB=celecoxib; Dex=dexamethasone; DF=Diclofenac; DMAEMA=2-(dimethylamino)ethyl methacrylate; ECM=extracellular matrix; GA=glucosamine; GelMA=gelatin methacrylate; HA=hyaluronic acid; HAMA=hyaluronic acid methacrylate; IGF-1=insulin-like growth factor-1; KGN=kartogenin; LNP=lipid nanoparticle; mAb-CII=monoclonal antibody against collagen II; MCD=molybdenum disulfide-chitosan nanosheet loaded with dexamethasone; MCLT=medial cruciate ligament transection; MIA=monoiodoacetate; MMP13=matrix metalloproteinase-13; MoS2=molybdenum disulfide; MMx=medial meniscectomy; MP=microparticle; NP=nanoparticle; OARSI=Osteoarthritis Research Society International; PAMAM=poly(amidoamine); PCLT=posterior cruciate ligament transection; PEG=poly(ethylene glycol); PEG-4MAL=4-arm maleimide-functionalized polyethylene glycol; PLGA=poly(lactic-co-glycolic acid); PN=polyurethane nanoparticle; SANS=self-assembling nanosystem; siIhh=small interfering RNA against Indian Hedgehog; siMMP13=small interfering RNA against matrix metalloproteinase-13; SIN=sinomenium; siRNA=small interfering RNA; TNF-α=tumor necrosis factor-α.
4.1. Clinical Liposomal Products
4.1.1. TLC599
Taiwan Liposome Co. (TLC) is developing a liposomal formulation of Dex (TLC599) for IA treatment of OA. The liposome is composed of 67.5% 1,2- dioleoyl-sn-glycero-3- phosphatidylcholine (DOPC), 7.5% l,2-dioleoyl-sn-glycero-3-phosphoglycerol (DOPG), and 25% cholesterol. Dex-sodium phosphate (DSP) is incorporated at 12 mg per 100 μmol of phospholipid, and the final particles are ~130 nm in diameter [161].
Preclinical Studies
To investigate the safety and PK of TLC599, multiple dose-finding studies were conducted in rabbits and beagle dogs. In one study, healthy dogs and rabbits received an injection of TLC599 (single dose 12 mg per knee, dogs; single or repeat dose at day 31, 1.2 mg per knee, rabbits) [162]. In all cases, there was no difference in toluidine blue staining in the cartilage at any time point assessed compared to initial staining levels. A second study compared safety of TLC599 (12 mg per knee, Dex dose equivalent to 60 mg TCA), free TCA (2.1 and 18.75 mg per knee), and extended-release TCA (ER-TCA, 2.1 and 18.75 mg per knee). The composition of ER-TCA was not clear from this study. At 30 days, TLC599 demonstrated comparable levels of proteoglycans in the joint as saline injections. However, high-dose TCA and both low- and high-dose ER-TCA formulations caused significant proteoglycan loss, indicating that TLC599 could cause less chondrotoxicity than ER-TCA. To investigate PK, beagle dogs received a bilateral injection of 18 mg per knee, and synovial fluid was collected over time [163]. DSP was detected at ~100 ng ml−1 out to 120 days post-injection, indicating sustained release of the drug (Figure 10A). A complementary study investigated the effect of multiple bilateral injections of 18 mg per knee TLC599 (once every 13 weeks for a total of 4 injections). The serum Cmax of DSP was similar after the first and fourth doses (450 and 522 ng ml−1 respectively), indicating similar systemic drug exposure for consecutive injections with 13 weeks of spacing. The authors did not report any local effects on cartilage after this multiple-dose regimen. Notably, their PK data demonstrates extended release of DSP, but they do not report proteoglycan loss as demonstrated with ER-TCA.
Figure 10.

Preclinical and clinical investigation of intra-articular (IA) TLC599 for osteoarthritis (OA) therapy. (A) Mean concentration of dexamethasone phosphate (DSP) in canine synovial fluid. (B) Least-squares (LS) mean change from baseline (and standard error [SE]) in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) pain subscore on a 0–4 numeric rating scale. (A) reproduced with permission.[163] Copyright 2019, BMJ Publishing Group Ltd. (B) adapted with permission.[168] Copyright 2019, Elsevier Ltd.
Early Clinical Studies
The preclinical data outlined above led to 2 clinical trials. The first focused on safety of 2 doses of IA TLC599 (6 and 12 mg DSP) in 40 patients with knee OA (NCT02803307) [164]. TLC presented these results at the 20th Asia Pacific League of Associations for Rheumatology Congress in 2018, which indicated that both doses had minimal effect on serum cortisol levels and significantly reduced patient-reported pain and WOMAC scores [165,166]. These data led to a trial in 77 patients with knee OA focused on efficacy of IA injections of saline compared to TLC599 (12 or 18 mg DSP) (NCT03005873) [167]. 12 mg TLC599 significantly reduced WOMAC pain subscore vs. placebo at all time points (0–24 weeks), with LSM change of −0.95 compared to −0.6 for TLC599 and placebo, respectively, at 24 weeks (Figure 10B). 52% of patients treated with 12 mg TLC599 experienced >30% pain relief over 24 weeks, compared to 22% of patients treated with saline [168]. Two trials of TLC599 are currently active. The first is recruiting patients with OA of the knee (target 90 patients) for a study focused on PK for a single injection of TLC599 (6 or 12 mg) or free DSP (4 mg) (NCT03754049) [169]. The second is focused on efficacy of TLC599, free DSP (4 mg), or saline (placebo) in patients with knee OA (target enrollment of 500) (NCT04123561) [170].
Conclusions:
The early results for TLC599 are encouraging for OA pain reduction. Better maintenance of proteoglycan levels for TLC599 compared to ER-TCA in preclinical trials indicates that this formulation could reduce ECM loss compared to ER-TCA. It will be interesting to understand if this extended-release formulation outperforms free Dex in ongoing clinical trials, as this would represent an advantage over polymeric MP-based drug developed to date. The performance of the drug-free liposome MM-II is also a consideration for the future application of this product, as it will be important to understand if the addition of Dex or other drugs provide additional benefit to this liposomal OA therapy.
4.2. Preclinical Liposomal Studies
Similar to hydrogels and polymeric MPs, preclinical liposome studies have explored a wider range of drugs for OA than previous or ongoing clinical work. For example, Wang et al. investigated a solid lipid NP (LNP) for Indian Hedgehog (Ihh) small interfering RNA (siRNA) for IA therapy of OA (Figure 11A) [171]. Ihh is normally involved in chondrocyte differentiation, maturation, and proliferation but has been implicated in the progression of OA [172,173]. The LNP was composed of 23% Dlin-KC2-DMA (a commercial cationic lipid for siRNA packaging), 28% DPPC, 8% cholesterol, and 41% C16 ceramide-mPEG2000 (w/w) and encapsulated ~0.2–0.4 nmol siRNA (~67 nm in diameter). IA injections in vivo in healthy rats demonstrated fluorescence throughout the cartilage with LNP delivery, with no signal using free siRNA (Figure 11B), and achieved ~80% Ihh silencing 24 hours post-injection. In a therapeutic study, LNPs (2.5 or 5 nM siRNA) or free siRNA (2.5 nM) were injected every 2 weeks for 10 weeks. Both LNP doses significantly increased histological staining of type II collagen while decreasing MMP13, type X collagen, and Runx2 compared to free siRNA. Histological cartilage damage score for 2.5 and 5 nM LNP-Ihh was significantly reduced by ~57 and ~82%, respectively, compared to free siIhh (Figure 11C). This study demonstrates the advantage of utilizing a drug carrier to improve efficacy, particularly for an intracellular-acting biologic like siRNA. LNPs improved cartilage penetration and likely facilitated cell uptake of siIhh in the joint tissue; however, achieving longer-term retention of the system is an opportunity for improvement when considering clinical translation to avoid injections every 2 weeks. This intensive dosing schedule is in contrast with the relatively long-term action of both MM-II and TLC599 after a single injection. Additionally, the relative complexity of this system compared to TLC599 would be an additional barrier to regulatory approval, though the recent approval of a siRNA liposome for hereditary transthyretin-mediated amyloidosis is encouraging [174].
Figure 11.
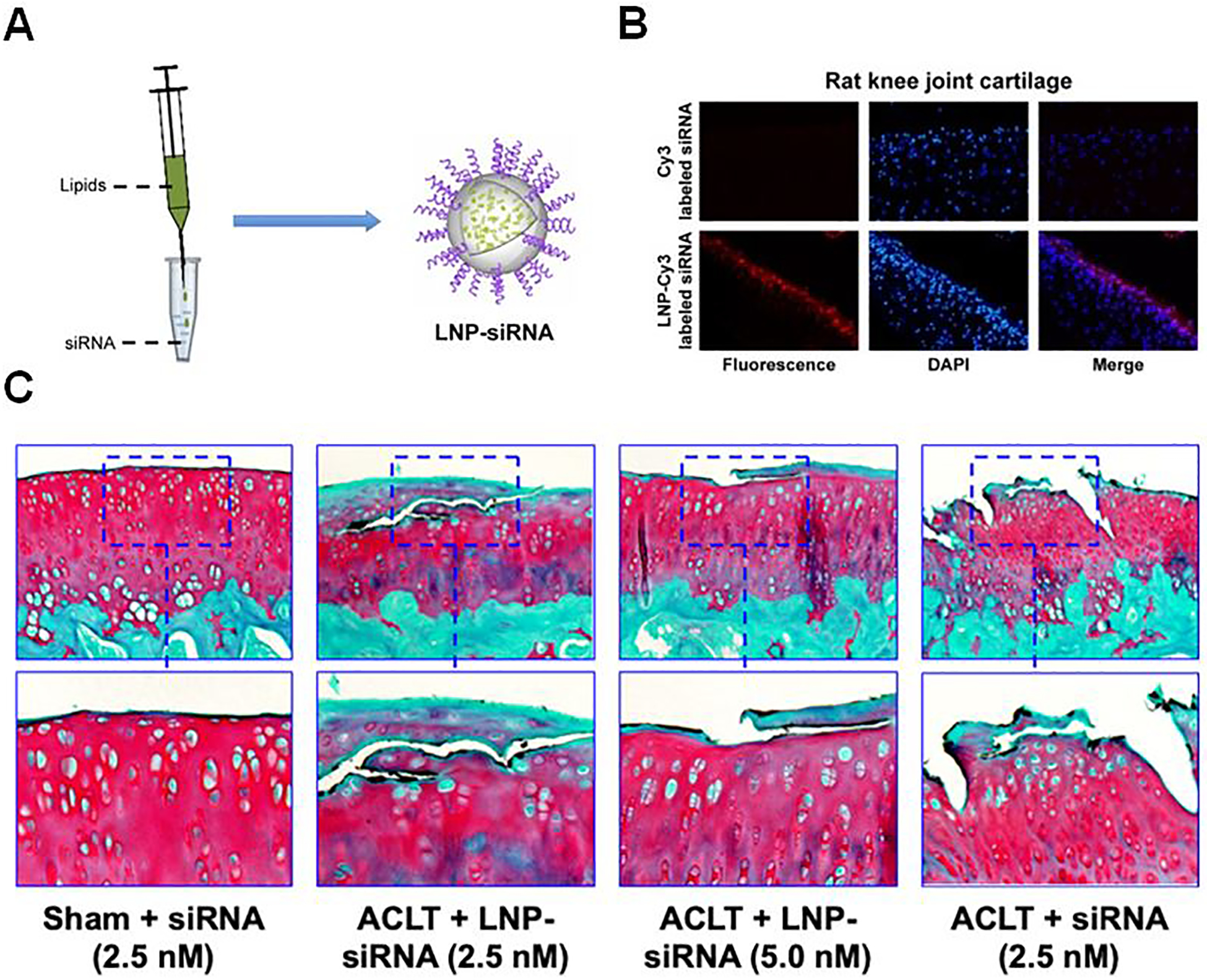
Lipid nanoparticles (LNPs) loaded with small interfering RNA against Indian Hedgehog (siIhh) for osteoarthritis (OA) therapy in a rat anterior cruciate ligament transection (ACLT) model. (A) Schematic of LNP production: produced by mixing lipid solution and siRNA solution drop by drop under strong vortex followed by ultracentrifugation to remove ethanol. (B) Using fluorescence microscopy (10x magnification), fluorescent signals were detected in the LNP-Cy3-labeled siRNA treated rat knee cartilage while not in the free Cy3-labeled siRNA treated rat knee cartilage. (C) Representative images of articular cartilage specimens stained with Safranin O from rats treated with LNP-siIhh showed less surface damage compared with the untreated controls. Reproduced with permission.[171] Copyright 2014, Dove Medical Press Limited.
Another recently explored strategy is to combine phospholipids with other biologic macromolecules such as extracellular matrix proteins. Elron-Gross et al. formulated a collagen type I/dipalmitoyl phosphatidylethanolamine (DPPE) liposome system (collagomer) loaded with the NSAID DF [175]. Collagen and DPPE were combined at a ratio of 1:5 (w:w) and chemically cross-linked, and DF was loaded by re-hydrating lyophilized collagomers in an aqueous solution of the drug (encapsulation efficiency ~85%). The resulting particles were ~5 μm in diameter, ~1 order of magnitude larger than TLC599. Rats with OA received IA injection of DF-collagomer (1 mg kg−1 DF), free DF (1 mg kg−1), unloaded collagomers, or no treatment. A final group received oral gavage of DF (1 mg kg−1). At day 18, DF-collagomer reduced inflammation volume by ~75% compared to initial levels, which was significantly better than no treatment (~50%) and DF by oral gavage (~38%). The loaded collagomer did not significantly outperform unloaded collagomers (~45%) or IA DF (~45%), mainly due to very high variability in these groups. These results show that both unloaded collagomers and DF reduce inflammation, but there is not strong evidence for added benefit from a combination. The murky evidence for improved efficacy with a DDS compared to the free drug, particularly for corticosteroids and NSAIDs, is not exclusive to this example and was a consistent trend for Cingal, FX006, and multiple other preclinical systems.
Conclusions:
The promising clinical results from the unloaded liposomal product MM-II and the regulatory approval of liposomes in other diseases are both encouraging signs for clinical translation of drug-loaded liposomes. It will be interesting to discover if the addition of classical agents like NSAIDs and corticosteroids (as in TLC599) will provide added benefit over liposomes alone. The potential for lubrication effects of liposomes to provide even added if not synergistic benefit over their drug retention function provides a potential competitive advantage relative to most polymeric MPs. Potentially most exciting is the possibility for liposomes to expand the repertoire of therapies which can be delivered IA, by increasing retention, facilitating cellular uptake, and allowing co-delivery of lipophilic and hydrophilic drugs.
5. Polymeric Nanoparticles
Polymeric NPs fall in a size range of ~10–900 nm (Figure 2D). These systems are wide-ranging in terms of the accessible compositions and types of applications that are pursued for delivery of small molecule drugs, proteins, and nucleic acids [90]. Various retention strategies can be employed to delay joint clearance for these smaller particles. For example, the particle surface can be decorated with targeting moieties to promote binding to cells or other tissue components to improve retention [176,177], or small positively-charged vehicles can penetrate and adhere to the negatively-charged cartilage matrix [94,176]. Additionally, chondrocytes and synoviocytes can take up NPs, similar to small MPs. However, there are contradicting design considerations, because these smaller particles are very susceptible to swift lymphatic clearance, particularly if cartilage penetration or tissue binding is weak or inefficient [4]. NP strategies for OA have been reviewed by others [90]; here, we specifically highlight studies which have demonstrated a therapeutic benefit in animal models of OA (Table 5).
Though there are no clinically investigated polymeric NPs for IA OA therapy, there are many attractive aspects of NPs which are being investigated preclinically. For example, hydrophobic agents can be packaged into small micellar structures for improved PK profiles and tissue penetration. Utilizing this strategy, Fan et al. formulated a polyurethane-KGN NP [178]. PEG was combined with hexamethylene diisocyanate (HDI) which formed an amphiphilic polyurethane with pendant amino groups. The resulting polymer was either left unmodified (polyurethane NP, PN) or was conjugated with KGN (PN-KGN). The resulting nanospheres were approximately 25 nm in diameter. In vitro, PN-KGN released ~20% of loaded KGN over 4 weeks. In a rat OA model, animals received IA injections of saline, PN (118 μg), KGN (100 μM), or PN-KGN (118 μg NP, 100 μM KGN) at weeks 0, 3, 6, and 9. 12 weeks after surgery and the first injection, PN-KGN significantly improved histological OARSI score over free KGN (~65% improvement over PBS vs. ~26% improvement) and qualitatively increased collagen II staining. The improvement over free KGN indicates an advantage with NP delivery; however, the frequent dosing schedule could be a barrier to translation of this system.
Others have combined synthetic polymers with biomolecules to leverage the tunability of synthetics and the inherent biologically functional biologics in a single nanomedicine. Kamel et al. formulated a self-assembling nanosystem (SANS) from PLGA (50:50) loaded with the NSAID indomethacin via a thin film hydration method using 2 high molecular weight surfactants (Poloxamer 407 and Tetronic 90R4) [179]. The SANS were surface modified with collagen type I, GLT, or glucosamine, causing an increase in particle size (85.7 nm unmodified, 174–920 nm modified). These biomolecules were expected to improve biocompatibility and potentially stimulate regenerative effects. Notably, their optimized SANS formulation (SANS3/8) released ~60% of the loaded indomethacin within 4 hours in vitro. The authors compared IA treatment with free indomethacin, SANS3/8, a formulation lacking PLGA (SANS3/0), and SANS3/8 modified with glucosamine (SANS3/8-GA2) in a rat antigen-induced arthritis (AIA) model. All treatments were 500 μg total (drug+NP) and were administered 2 and 3 weeks after the initial immunization. At week 4 (1 week after the second treatment) SANS3/8-GA2 outperformed the other formulations in joint swelling (~−0.1 mm compared to ~1.0 mm for no treatment) and synovial fluid TNF-α levels (~65% reduction compared to no treatment). Qualitative histopathology showed reduction in synovial thickening, infiltrating cells, and cartilage damage with SANS3/8-GA2 compared to no treatment. The improvement of glucosamine-modified SANS over SANS alone demonstrates potential benefits for combining synthetic and biologic components for OA drug delivery. However, the system should be iterated to improve retention and reduce dosing frequency, with investigation at later time points to demonstrate translational potential.
NPs formulated purely from biologic polymers have also demonstrated efficacy in OA. For example, Wang et al. investigated a combination chitosan/HA NP system for IA therapy of OA. Chitosan (10–40 kDa) was co-dissolved with curcuminoid (an antioxidant and anti-inflammatory drug) and ionically crosslinked to form NPs ~42 nm in diameter [180]. N-hydroxysuccinimide (NHS)-HA was added to formulate HA-chitosan NPs (HA-CNP) ~165 nm in diameter loaded with ~38% (w/w) curcuminoid. In chondrocytes treated with IL-1β and TNF-α, HA-CNP significantly reduced both mRNA and protein expression of MMP1 and MMP13 and reduced chondrocyte apoptosis compared to no treatment. In a rat model of OA, animals received IA injections of saline, HA (1 mg), or HA-CNP (1 mg NPs, 0.38 mg curcuminoid) once per week for 4 weeks. Both HA and HA-CNP significantly reduced MMP1 and MMP13 mRNA and protein expression in the cartilage while increasing collagen II mRNA and protein expression, with a larger magnitude effect from HA-CNP. Qualitative histological assessment showed that HA-CNP preserved the cartilage integrity and cellularity compared to the saline group. The lack of difference between HA and HA-CNP indicates that the mechanical benefit of HA could be dominant over the anti-inflammatory effects of curcuminoid; unloaded HA NP and free curcuminoid controls would have facilitated the deconvolution of these variables.
Biologic polymers can also be used to modify inorganic materials for OA therapy. Zhao et al. investigated a chitosan-coated molybdenum disulfide (MoS2) nanosheet system loaded with Dex (MCD) for photothermal on-demand Dex delivery in the joint (Figure 12A) [181]. The nanosheets were ~70 nm across and 2–3 nm thick. Because of its photothermal properties, MCD released Dex in an intensity and time-dependent manner when stimulated with near-infrared (NIR) light. In healthy mice, MCD (1 mg kg−1) was retained in the knee for 48 hours (Figure 12B). In a therapeutic study, IA treatments of saline, Dex (1 mg kg−1) or MCD (1 mg kg−1) were administered, and the joints were irradiated every hour for 8 hours. This treatment scheme was repeated every 3 days for 28 days. MCD treatment significantly reduced histological staining and serum levels of TNF-α, IL-1β, and IL-8 compared to saline and free Dex. MCD also significantly increased aggrecan levels while decreasing MMP13 expression, ADAMTS5 expression (Figure 12C), and OARSI histological scores, all compared to both saline and free Dex. Free Dex demonstrated an intermediate effect on these metrics. Finally, MCD treatment improved functional outcomes including walking distance and velocity relative to saline and the free drug. These results support previous findings that controlled doses of corticosteroids can demonstrate disease-modifying effects. Reliable, on-demand corticosteroid release in the joint by photothermal stimulation is a unique strategy for pain management, though upscaling this to the larger joints of patients may prove problematic. Also, the relatively short retention time in the joint and intensive dosing schedule are concerns for clinical translation of this system.
Figure 12.

Molybdenum disulfide (MoS2) nanosheet delivery of dexamethasone (Dex) in a mouse model of papain-induced osteoarthritis (OA). (A) Schematic showing the synthesis scheme for the drug-loaded nanosystem and its drug release mechanism via response to near-infrared (NIR) light in vitro (I). Mechanism of the drug-loaded nanosystem in vivo after intra-articular (IA) injection and NIR light exposure for the treatment of OA (II). (B) Retention time of MoS2-chitosan-Dex (MCD) is shown by representative photoacoustic images of mouse knee joints at 0, 24, and 48 h timepoints after IA injection of MCD at 1 mg kg-1. (C) Representative images of mouse joint tissue after 28 days of treatment indicates the therapeutic effect of MCD based on cartilage reconstruction and immunofluorescence staining of several proteins that are associated with OA pathology: matrix metalloproteinase 13 (MMP13), a disintigrin and metalloproteinase with thrombospondin motifs 5 (ADAMTS5), Aggrecan. The proteins are fluorescently labeled in green and nuclei in blue. The NIR radiation power density is 0.4 W cm-2. Scale bar is 50 μm. Reproduced with permission.[181] Copyright 2019, American Chemical Society.
Conjugation of cartilage matrix-binding ligands to NPs is one option to potentially improve retention and reduce injection frequency. Bedingfield et al. pursued this strategy for siRNA delivery against MMP13 loaded in polymeric micelles (~100 nm) with anti-collagen II antibody (mAb-CII) conjugated to the corona [177]. siMMP13 was complexed in a 2-(dimethylamino)ethyl methacrylate (DMAEMA)-co-butyl methacrylate (BMA) hydrophobic core with a hydrophilic PEG corona. mAb-CII conjugation significantly improved retention at 72 h compared to bare siRNA and non-targeted controls and exhibited ~95% MMP13 silencing in vivo at this time point. In a load-induced model of PTOA, mice received injections of siMMP13-NPs once per week for 6 weeks (0.5 mg kg−1 siRNA). Treatment with targeted siMMP13-NPs qualitatively reduced MMP13 histological staining in the cartilage and synovium compared to a scrambled control sequence. OARSI cartilage score, joint MMP activity, and joint damage measured by collagen II binding were reduced for siMMP13-NPs compared to scrambled sequence by ~75%, ~40%, and ~70%, respectively. These results demonstrate the potential advantage of employing a cartilage-binding moiety for IA therapies, though therapeutic outcomes were not compared between targeted and non-targeted NPs. Additionally, the once per week dosing schedule indicates that cartilage binding may require iteration to further improve retention and reduce injection frequency. This study represents a striking demonstration of NPs to expand efficacy of biologic drugs like siRNA, where the free drug was poorly retained.
Conclusions:
Because NP therapy in OA can be limited by the relatively small size and short retention time of the DDS, the studies reviewed in this section all required multiple injections [178–181]. The most promising formulations will likely utilize cartilage binding or another strategy to promote retention; an advantage of NPs is that the synthetic strategies are better established for surface functionalization for binding to extracellular matrix to promote joint retention as in the Bedingfield study [176,177]. Also, NP chemistries are well-established for alternate drug loading mechanisms, including encapsulation of hydrophilic drugs (polymersomes) and direct drug conjugation onto NP constituents through reversible covalent linkages. However, these strategies are still being investigated for OA.
6. Cartilage-penetrating Drug Conjugates
Cartilage consists of densely packed ECM including collagen fibrils, aggrecan proteoglycans, and other extracellular macromolecules (Figure 13A). The aggrecan aggregates contain a high density of negatively charged glycosaminoglycans; therefore, transport into cartilage is limited by both size and charge [182,183]. Direct conjugation of therapeutics to tissue-penetrating agents has been pursued to overcome these barriers (Figure 2E). This strategy is attractive due to the reduced complexity of the system and the smaller size of the therapeutic relative to MP or NP formulations. It has been found that substances with a hydrodynamic diameter <15 nm have the ability to permeate deep into the cartilage matrix [184], while larger particles are size limited. A great deal of progress has been made investigating cartilage penetration by conjugation to a heparan-binding domain [185], cationic peptides [186,187], positively-charged proteins such as avidin [16,184,188] and modified green fluorescent protein (GFP) [189], and positively-charged polymers [190]. These strategies have been reviewed in detail recently [182,191]; here, we highlight several studies which demonstrated therapeutic efficacy in animal models of OA (Table 5).
Figure 13.
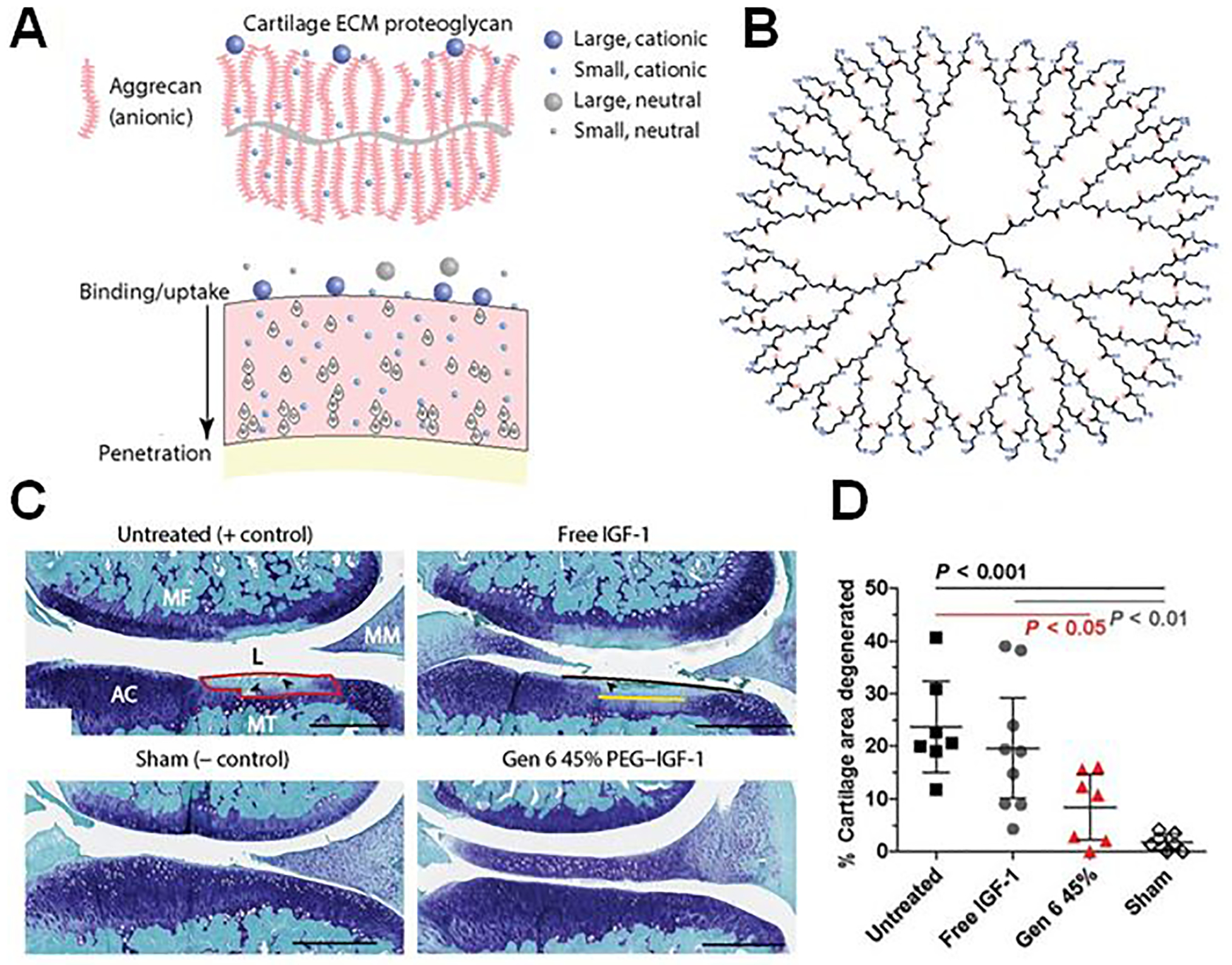
Poly(amidoamine) (PAMAM) dendrimer delivery of insulin-like growth factor-1 (IGF-1) in the rat anterior cruciate ligament (ACLT)/ partial medial meniscectomy (MMx) model of osteoarthritis (OA). (A) Schematic of the mechanism of retention and penetration of nanocarriers based on size and charge within the joint after intra-articular injection. Cationic particles can bind to the anionic aggrecan-dense extracellular matrix (ECM) of cartilage. (B) Chemical structure and design of PAMAM dendrimers formulated as cartilage-penetrating nanocarriers. (C) Representative images of the joint four weeks after surgery stained with toluidine blue/fast green indicate the dendrimer-IGF-1 conjugate (denoted Gen 6 45% PEG-IGF-1) reduces cartilage degeneration. Area of degeneration, total widths of degeneration, and significant widths of degeneration are outlined in red, black, and yellow, respectively, and matrix loss is shown as black arrowheads. MF, medial femur; MT, medial tibia; MM, medial meniscus; AC, articular cartilage; L, lesion. Scale bars, 500 μm. (D) The area of degenerated cartilage tissue of the medial tibia was quantified for each rat as a percentage of total cartilage area in the section. Reproduced with permission.[190] Copyright 2018, American Association for the Advancement of Science.
To facilitate penetration into the densely negative cartilage matrix, Bajpayee et al. investigated a biotinylated PEG-Dex conjugated to positively charged avidin (Av-Dex) [192]. In a rabbit model of OA, 0.5 mg Dex in the Av-Dex form significantly reduced the change in joint space thickness at 3 weeks (approximately −18% for saline, −16% for free Dex, and −7% for Av-Dex). Av-Dex significantly reduced osteophyte severity score (1.6±0.3 vs. 0.1±0.2 for free and Av-Dex, respectively) and preserved trabecular bone volume/total volume (BV/TV) ratio. Free Dex-treated knees exhibited a ~5-fold increase in both IL-1β and MMP1 gene expression compared to sham controls; these increases were not seen with Av-Dex treatment. However, histological Safranin-O staining demonstrated a significant worsening of joint score in the Av-Dex group which was not seen in free Dex or saline. Because avidin-only controls exhibited an ~10% reduction in GAG concentration, the loss of Safranin-O staining could be due to a combination of avidin and extended exposure to Dex in the Av-Dex group. This result indicates that corticosteroids, which have been documented to reduce cartilage proteoglycans [22], may not be appropriate for long-term, intra-cartilage drug delivery. However, utilization of avidin as a cartilage-penetrating agent for drug conjugation represents a strategy with translational potential due to its relative simplicity and effectiveness, and it would be interesting to test this strategy for other drugs that may have chondroprotective or chondrogenic properties.
Geiger et al. also took advantage of a positively-charged carrier to facilitate penetration and retention into the densely negative cartilage matrix (Figure 13A) [190]. In this study, poly(amidoamine) (PAMAM) dendrimers were loaded with insulin-like growth factor-1 (IGF-1) (Figure 13B), forming nanocarriers 4 and 7 nm in diameter depending on the molecular weight of PAMAM used. PAMAM was PEGylated with NHS methyl-PEG and NHS-maleimide PEG, and IGF-1-SH was conjugated to the maleimide-PEG groups. In vitro, PAMAM-IGF-1 and free IGF-1 demonstrated statistically similar increases in GAG synthesis and percent of cells in S phase of proliferation. Using fluorescently labeled IGF-1, the t1/2 in the joint was found to be 0.41 and 4.21 days for free IGF-1 and 58 kDa PAMAM-IGF-1, respectively. In a rat model of OA, PAMAM-mediated delivery significantly reduced the percent of cartilage area with degenerative changes over those with no treatment (~25% vs. ~9%) but was not significantly better than free IGF-1 (~20%) (Figure 13C–D). Similarly, dendrimer-IGF-1 significantly reduced osteophyte volume compared to no treatment (~0.2 vs. ~1.1 mm3), but not compared to free IGF-1. The discrepancy between improved IGF-1 t1/2 (~10-fold increase with dendrimer packaging) and the therapeutic benefit may reflect the potency of the drug, such that its effect is not improved by a t1/2 which is still relatively short. The simplicity and efficacy of this system make it attractive for clinical translation; however, in its current form it is difficult to justify utilizing dendrimer delivery with no significant improvement over the free drug.
Conclusions:
Cartilage penetration is a facet of OA drug delivery which has been missing from clinical strategies thus far. Drug depot strategies like microspheres and hydrogels largely reside in the synovium and joint cavity, respectively, following injection. Presumably, release from these depots provides drug bioavailability throughout the whole joint, including the cartilage, to some degree, but may not efficiently deliver bioactive drug concentrations to the deeper chondrocytes. Modification with penetrating agents, typically carrying a positive charge, is a promising strategy to realize small, tissue-penetrating drug conjugates for IA therapy of OA.
7. Hybrid Systems
Hybrid systems are attractive because they can leverage benefits of multiple classes of delivery systems and biomaterials simultaneously. For example, hydrogels often provide mechanical supplementation but are not effective at sustaining long-term delivery relative to particulate systems, and vice versa. Therefore, encapsulation of polymeric particles into hydrogels provides cushioning and viscosupplementation from the bulk hydrogel and also enables extended-release from the particle component (Figure 2F). Another possible hybrid implementation is the use of polymeric MPs as depots for smaller, cartilage-penetrating NPs or conjugates. Though behavior of such systems can be difficult to predict since they exhibit complex drug release kinetics due to combined diffusional barriers, the combined benefits of multiple DDSs can be an overall advantage for IA therapy. Another limitation of hybrid systems is that their formulation process tends to be more complex and possibly less reproducible relative to single component systems. Though drug-loaded hydrogels, liposomes, and polymeric particles have been tested in clinical trials, manufacturing and regulatory complexity may be part of the reason that hybrid systems have not yet been tested in patients. Here, we highlight promising combination systems that have been shown to have efficacy in animal models of OA (Table 5).
HA is an attractive candidate for polymeric particle delivery for the reasons discussed above (FDA approved, easily modifiable, biocompatible). To leverage these advantages, Xia et al. prepared a combination HA hydrogel/chitosan MP platform for IA treatment of OA [193]. HA was modified with methacrylate (HAMA) groups for UV crosslinking. Chitosan microspheres (CM, ~100 μm) were loaded with cordycepin (~1.9% w/w) and incorporated into HAMA gels. Cordycepin is a cellular autophagy promoter, and could have therapeutic potential due to evidence that dysregulation of autophagy in chondrocytes contributes to OA [194]. Notably, there was no difference in the release rate between CM’s with and without HAMA, indicating the gel did not contribute to slowing drug release. In a mouse model of OA, mice received IA injection of CM+HAMA, CM-cordycepin alone (5 mg kg−1 cordycepin), or CM-cordycepin loaded in HAMA hydrogels (5 mg kg−1 cordycepin) once per week over the course of the study. At 4 and 8 weeks, CM-cordycepin+HAMA statistically outperformed all other groups in histological OARSI score (8 weeks: ~65, ~54, and ~40% improvement compared to PBS, CM+HAMA, and CM-cordycepin, respectively). Additionally, at both 4 and 8 weeks, the histological levels of MMP13 and ADAMTS5 were significantly lower while levels of collagen II and aggrecan were significantly higher compared to all other groups. Finally, the combination system significantly increased histological levels of microtubule-associated protein 1A/1B light chain 3 (LC3), indicating that increased autophagy could be contributing to improved outcomes. This group pursued a similar system for delivery of the autophagy promoter sinomenium (SIN) [195]. In this study, SIN-loaded CMs (~100 μm, 0.004% SIN w/w) were loaded into a photo-crosslinkable gelatin methacrylate (GelMA) hydrogel. OA mice received IA injections of GelMA (10 μl), SIN (5 mg kg−1), CM-SIN (5 mg kg−1 SIN), or CM-SIN+GelMA (5 mg kg−1 SIN) once per week throughout the study. Once again, the hybrid system significantly outperformed all other groups in histological OARSI score, reduction of ADAMTS5 and MMP13 (statistically similar to CM-SIN), and increase in collagen II and aggrecan levels. These and other results are encouraging for autophagy promoters as potential new DMOADs; however, the frequency of injections (weekly) in these studies warrant further investigation of extended-release formulations.
In another exploration of hydrogel-mediated particle delivery, Kang et al. loaded KGN-PEG micelles in HA hydrogels [196]. Because NPs often lack the ability to resist lymphatic clearance that larger MPs exhibit, NPs could benefit from hydrogel delivery more than MPs. In this study, PEGylated KGN formed micelles ~340 nm which were then covalently bound to HA modified with ethylenediamine (EDA) via carboxy-modified end groups on the PEG chains. Scanning electron microscopy (SEM) of the system revealed that HA binding formed an outer layer on the micelles, increasing hydrodynamic diameter to ~425 nm by dynamic light scattering (DLS). HA packaging significantly slowed KGN release in vitro, with ~70% drug release from NPs alone compared to ~30% release for NPs+HA over 5 days. The authors attribute this effect to the increased diffusional barrier presented by HA. In a rat model of OA, rats received 2 injections spaced 3 weeks apart: PBS, free KGN (100 μM), HA hydrogels (50 mg), or HA/KGN-NP hydrogels (50 mg). Eight weeks after the first injection, histological OARSI and Mankin scores (a scale indicating cartilage damage, with 14 indicating the most severe) were significantly lower in the HA/PEG-KGN group compared to all other groups, exhibiting ~80% improvement compared to PBS and ~50–70% improvement compared to HA. Qualitative immunohistochemical examination also showed increased Col2 and aggrecan staining compared to PBS. These results are promising, though it would have been informative to compare the combination system to KGN-PEG micelles or HA-KGN hydrogels alone to probe the true benefit of both micelle packaging of KGN and HA-mediated delivery in vivo. Notably, there appears to be a significant PK benefit of chemical conjugation between NPs and the HA hydrogel as opposed to physical mixing of the two components based on the in vitro release data.
One option to improve gel residence in the joint is to utilize synthetic polymers which are more easily modified than natural materials like HA or GLT. To this end, Holyoak et al. pursued a 4-armed maleimide-functionalized PEG (PEG-4MAL) hydrogel-based “mechanical pillow” system loaded with PLGA-Dex NPs (~203 nm) (Figure 14A) [197]. The PEG-4MAL hydrogels was cross-linked with 50% non-degradable dithiothreitol (DTT) and 50% MMP-degradable peptide crosslinker (GCRDVPMSMRGGDRCG, VDM). This formulation was resistant to mechanical load-induced release but susceptible to collagenase-induced release for on-demand drug delivery in the joint. OA mice received IA injection of bolus Dex (10 μg), PLGA-Dex NPs (1.8 μg Dex), PEG-4MAL hydrogel+PLGA-Dex NPs (1.8 μg Dex), or PEG-4MAL hydrogel alone. Two weeks after injection, both NP-loaded hydrogels and hydrogels alone significantly reduced osteophyte size compared to non-hydrogel-treated groups (~160 mm vs. ~25 mm) (Figure 14B). Similarly, both hydrogel-treated groups significantly improved histological OARSI score by ~50% compared to the other 3 groups (Figure 14C). These results suggest that the cushioning provided by the hydrogel could be more beneficial than extended Dex release. It will be worthwhile to extend this into future studies to investigate if Dex is more beneficial at time points later than 2 weeks.
Figure 14.
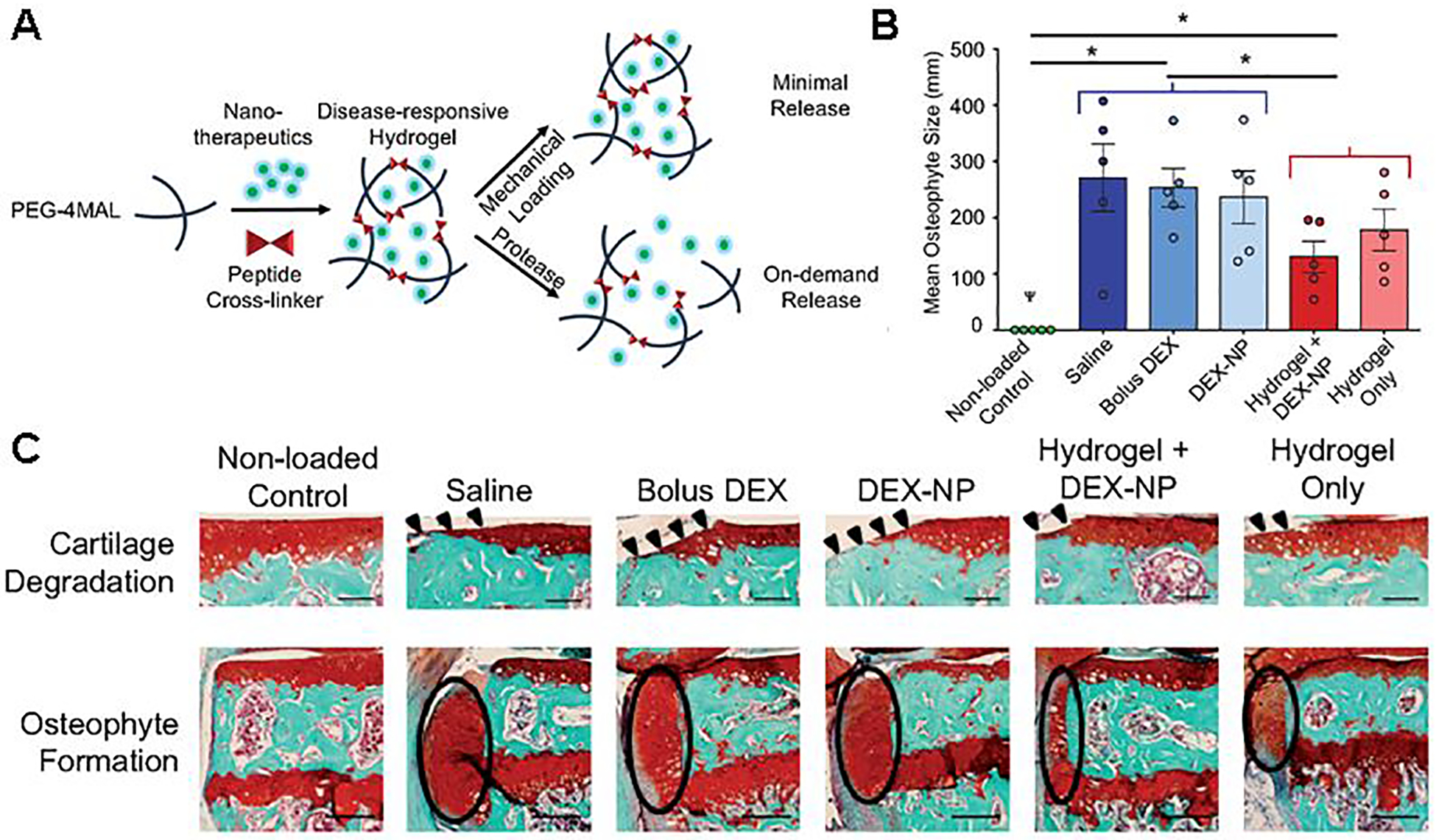
Poly(ethylene glycol)-4-maleimide (PEG-4MAL) “mechanical pillows” loaded with poly(lactic-co-glycolic acid) (PLGA)-dexamethasone (Dex) nanoparticles (NPs) in a load-induced model of osteoarthritis (OA). (A) Schematic showing the formation and delivery of PEG-4MAL hydrogels with NPs and their method of therapeutic retention while maintaining viscoelastic properties under daily mechanical loading. The drug is released under protease-rich conditions. (B) In OA-induced mice, these “mechanical pillows” attenuated in vivo osteophyte formation, as shown by the mean medial-lateral width of the osteophytes from three representative sections in the joint (posterior, middle, and anterior). n = 5 mice/group. φp < 0.05 for loading; and *p < 0.05 for hydrogel vs. no hydrogel nested by loading. (C) This, along with attenuation of cartilage damage, is shown by representative images of the joint with Safranin O and fast green-staining. Cartilage erosion is indicated with arrowheads and osteophyte formation with ellipses. Cartilage scale bars = 100 μm. Osteophyte scale bars = 200 μm. Reproduced under the Creative Commons CC BY License.[197] Copyright 2019, D. T. Holyoak, T. A. Wheeler, M. C. H. van der Muelen, and A. Singh.
Liposomes, like polymeric particles, can also benefit from hydrogel delivery for extended release. Dong et al. pursued this strategy by loading CXB-liposomes into HA hydrogels for OA therapy [198]. The liposomes were ~5 μm in diameter with a CXB encapsulation efficiency of 99%. In vitro, HA gels reduced burst release compared to liposomes alone; however, both systems released ~90% of loaded CXB at 48 hours. In a rabbit model of OA, animals received IA injections of saline, CXB liposomes (0.15 mg CXB), HA (3 mg), or CXB liposomes in HA (0.15 mg CXB, 3 mg HA). The hybrid liposome-HA group significantly improved weight bearing on the affected limb compared to saline at 24 and 48 hours post-injection; CXB liposomes alone were only effective at 24 hours, indicating a benefit from extended release from the hydrogel. Additionally, 2 weeks post-injection, global histological joint scores were significantly improved for the hybrid system compared to saline; the other treatments did not reach statistical significance. These results indicate that hydrogel encapsulation could be an effective strategy for prolonging liposomal drug release, though longer time points should be investigated to confirm this idea in a more translationally relevant context.
Conclusions:
Combination systems are still in the early phases of development for IA OA therapy. The potential for leveraging a combination of both slow release from MPs and viscosupplementation from hydrogels is exciting for the OA drug delivery field. Alternatively, encapsulation of cartilage-penetrating conjugates in slow release MPs or hydrogels could further extend the efficacy of these agents. However, a great deal of work is needed to determine the most advantageous combinations for hybrid strategies. As development programs progress for hydrogels, liposomes, NPs, MPs, and conjugates, it is anticipated that more work on optimal integration of these primary components with secondary mechanobiological devices will follow.
8. Conclusions and Future Outlook
As a localized, progressive, chronic, and currently irreversible disease, OA is an ideal candidate for local sustained release of therapeutic agents. An optimal system would provide long-lasting, therapeutic concentrations of the drug in the joint space with minimal systemic exposure. One major challenge for OA therapy is the fact that most patients are already afflicted with late-stage disease, have lost a large percentage of healthy tissue, and can at best hope for pain relief. In these cases, IA therapy with a corticosteroid or NSAID can provide short-term alleviation. However, the future of OA therapy should forge beyond pain relief, focusing on restoration of joint function through methods including new drug development, earlier diagnosis, and advanced bioengineering tissue replacement/reparative approaches. These advances will emerge from a wide array of disciplines and collaborative expertise, will manifest differently across patients, and will undoubtedly include IA therapies.
The preclinical setting provides broad freedom to investigate innovative combinations of drug delivery formulations and therapeutics. One of the exciting aspects of advanced drug delivery formulations is the possibility to expand the repertoire of therapeutic agents that can be applied to disease, whether by improving PK and bioavailability, enhancing action, facilitating cartilage penetration, reducing off-target toxicities, or converting previously ineffective drugs into viable treatment options. Some of the preclinical studies reviewed here pursued this possibility for exciting small-molecule drugs like MSC stimulators and autophagy promoters as well as biologics like siRNA and growth factors. While the free forms of these drugs were largely ineffective, their controlled release formulations showed promise as potential DMOADs.
In contrast, all clinical formulations (and many preclinical) except PNHA were limited to corticosteroid or NSAID delivery. The heavy precedent for these therapies in clinical practice, particularly corticosteroids for IA treatment, gives them an easier regulatory pathway for achieving translation in the context of DDSs. Preclinical and clinical data has established that synovial drug release kinetics can be slowed, prolonged for months, with DDS technologies, particularly for MP formulations. However, the expected clinical benefit of more favorable PK behavior is rarely realized. FX006 and Cingal failed to relieve pain more effectively than their drug suspension counterparts, and a similar result for Hydros-TA led to termination of development. This lack of improvement over unencapsulated corticosteroids for these DDS products could be due to the mechanism of action of these corticosteroids and their potency in pain relief. Patient variability and the placebo effect of IA injections also confound interpretation of efficacy data for these systems [26]. Notably, preclinical investigations allowed for more differentiation between DDS and free drug with histological and molecular investigation, as opposed to just pain relief. Future clinical evaluation of DDS DMOAD candidates would benefit from outcomes outside pain scores, such as new biomarkers or advanced imaging modalities which provide insight into the progression of disease. Additionally, troubling preclinical evidence has shown adverse effects of prolonging corticosteroid residence in the joint, particularly local loss of proteoglycans.
The FDA approval of FX006 represents a milestone in the field of advanced DDS IA therapy for OA. However, current evidence is weak that extending residence time of corticosteroids and NSAIDs will improve therapy significantly. Though a discussion of new therapeutics for OA is outside the scope of this review, it is clear that true disease-modifying agents are needed. Novel formulations will likely play an integral role in achieving this goal by maximizing drug activity, safety, and patient compliance. Complementary investigation into pathological mechanisms, new drug targets, therapeutic design, and DDS design will pave the way to the next generation of IA therapies for OA patients.
Acknowledgements
This work has been supported in part by the National Institutes of Health, National Institute of Biomedical Imaging and Bioengineering (training Grant No. T32-EB021937), the National Science Foundation (GRFP DGE-1937963), the Department of Defense (DOD CDMRP 0R130302), and the Veterans’ Administration (I01 BX004151, BLRD VA Merit Award).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Karen A. Hasty, Department of Orthopedic Surgery and Biomedical Engineering, University of Tennessee Health Science Center, 1211 Union Ave. Suite 520, Memphis, TN 38104, United States Department of Veterans’ Affairs, Memphis VA Medical Center, 1030 Jefferson Ave., Memphis, TN 38104, United States.
Leslie J. Crofford, Department of Medicine, Division of Rheumatology and Immunology, Vanderbilt University Medical Center, 1161 21st Ave. S., Nashville, TN 37232, United States
Craig L. Duvall, Department of Biomedical Engineering, Vanderbilt University, 5824 Stevenson Center, Nashville, TN 37232, United States
9. References
- [1].Martel-Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl AJ, Pelletier J-P, Nat. Rev. Dis. Primers 2016, 2, 16072. [DOI] [PubMed] [Google Scholar]
- [2].Pereira D, Ramos E, Branco J, Acta Méd. Port 2015, 1. [DOI] [PubMed] [Google Scholar]
- [3].Rosenthal AK, Curr. Opin. Rheumatol 2011, 23, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Maudens P, Jordan O, Allémann E, Drug Discovery Today 2018, 23, 1761. [DOI] [PubMed] [Google Scholar]
- [5].Ondrésik M, Azevedo Maia FR, da Silva Morais A, Gertrudes AC, Dias Bacelar AH, Correia C, Gonçalves C, Radhouani H, Amandi Sousa R, Oliveira JM, Reis RL, Biotechnol. Bioeng 2017, 114, 717. [DOI] [PubMed] [Google Scholar]
- [6].Altman RD, Barthel HR, Drugs 2011, 71, 1259. [DOI] [PubMed] [Google Scholar]
- [7].Argoff CE, Mayo Clin. Proc 2013, 88, 195. [DOI] [PubMed] [Google Scholar]
- [8].Miceli-Richard C, Le Bars M, Schmidely N, Dougados M, Ann. Rheum. Dis 2004, 63, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lanas A, Tornero J, Zamorano JL, Ann. Rheum. Dis 2010, 69, 1453. [DOI] [PubMed] [Google Scholar]
- [10].Asadullah K, Schäcke H, Cato ACB, Trends Immunol. 2002, 23, 120. [DOI] [PubMed] [Google Scholar]
- [11].Vanden Berghe W, Vermeulen L, De Wilde G, De Bosscher K, Boone E, Haegeman G, Biochem. Pharmacol 2000, 60, 1185. [DOI] [PubMed] [Google Scholar]
- [12].Grewe M, Gausling R, Gyufko K, Hoffmann R, Decker K, J. Hepatol 1994, 20, 811. [DOI] [PubMed] [Google Scholar]
- [13].Bendrups A, Hilton A, Meager A, Hamilton JA, Rheumatol. Int 1992, 12, 217. [DOI] [PubMed] [Google Scholar]
- [14].Lukiw WJ, Pelaez RP, Martinez J, Bazan NG, Proc. Natl. Acad. Sci 1998, 95, 3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lu YCS, Evans CH, Grodzinsky AJ, Arthritis Res. Ther 2011, 13, R142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bajpayee AG, Quadir MA, Hammond PT, Grodzinsky AJ, Osteoarthritis Cartilage 2016, 24, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Huebner KD, Shrive NG, Frank CB, J. Orthop. Res 2014, 32, 566. [DOI] [PubMed] [Google Scholar]
- [18].Sinusas K, Am. Fam. Physician 2012, 85, 49. [PubMed] [Google Scholar]
- [19].MacMahon PJ, Eustace SJ, Kavanagh EC, Radiology 2009, 252, 647. [DOI] [PubMed] [Google Scholar]
- [20].Wright J, Cowper J, Page DT, Knight C, Clin. Exp. Rheumatol 1983, 1, 137. [PubMed] [Google Scholar]
- [21].Bettencourt RB, Linder MM, Primary Care 2010, 37, 691. [DOI] [PubMed] [Google Scholar]
- [22].McAlindon TE, LaValley MP, Harvey WF, Price LL, Driban JB, Zhang M, Ward RJ, JAMA, J. Am. Med. Assoc 2017, 317, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lo G, LaValley M, McAlindon T, Felson D, JAMA, J. Am. Med. Assoc 2003, 290, 3115. [DOI] [PubMed] [Google Scholar]
- [24].Bannuru RR, Brodie CR, Sullivan MC, McAlindon TE, Cartilage 2016, 7, 322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Levy DM, Petersen KA, Scalley Vaught M, Christian DR, Cole BJ, Arthroscopy: J. Arthroscopic Rel. Surg 2018, 34, 1730. [DOI] [PubMed] [Google Scholar]
- [26].Jones IA, Togashi R, Wilson ML, Heckmann N, Vangsness CT, Nat. Rev. Rheumatol 2019, 15, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Askari A, Gholami T, NaghiZadeh MM, Farjam M, Kouhpayeh SA, Shahabfard Z, SpringerPlus 2016, 5, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Katz JN, Best Prac. & Res. Clin. Rheumatol 2006, 20, 145. [DOI] [PubMed] [Google Scholar]
- [29].Ong KL, Runa M, Lau E, Altman RD, ClinicoEcon. Outcomes Res 2019, 11, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Furman BD, Mangiapani DS, Zeitler E, Bailey KN, Horne PH, Huebner JL, Kraus VB, Guilak F, Olson SA, Arthritis Res. Ther 2014, 16, R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jimmy B, Jose J, Oman. Med. J 2011, 26, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Davies PSE, Graham SM, MacFarlane RJ, Leonidou A, Mantalaris A, Tsiridis E, Expert Opin. Invest. Drugs 2013, 22, 423. [DOI] [PubMed] [Google Scholar]
- [33].Qvist P, Bay-Jensen AC, Christiansen C, Dam EB, Pastoureau P, Karsdal MA, Pharmacol. Res 2008, 58, 1. [DOI] [PubMed] [Google Scholar]
- [34].Hoffman A, Adv. Drug Delivery Reviews 2012, 64, 18. [Google Scholar]
- [35].Pascual-Garrido C, Rodriguez-Fontan F, Aisenbrey EA, Payne KA, Chahla J, Goodrich LR, Bryant SJ, J. Orthop. Res 2018, 36, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].He Z, Wang B, Hu C, Zhao J, Colloids Surf., B 2017, 154, 33. [DOI] [PubMed] [Google Scholar]
- [37].Vilela C, Correia C, Oliveira J, Sousa R, Espregueira-Mendes J, Reis R, ACS Biomater. Sci. Eng 2015, 1, 726. [DOI] [PubMed] [Google Scholar]
- [38].Bannuru R, Natov N, Obadan I, Price L, Schmid C, McAlindon T, Arthritis Rheum.-Arthritis Care Res 2009, 61, 1704. [DOI] [PubMed] [Google Scholar]
- [39].Bliddal H, Evaluation of the Safety of Intraarticular Aquamid Reconstruction Injection for Knee Osteoarthritis in Humans, https://clinicaltrials.gov/ct2/show/NCT03060421?term=aquamid&draw=2&rank=1, accessed: February, 2020.
- [40].Sahin S, Tuncel S, Salimi K, Bilgic E, Korkusuz P, Korkusuz F, Chun H, Park K, Kim C, Khang G, Novel Biomater. Regener. Med 2018, 1077, 183. [DOI] [PubMed] [Google Scholar]
- [41].Sadozai KK, Gooding TB, Bui K, Sherwood CH (Anika Therapeutics Inc.), EP2656833B1, 2018.
- [42].Dernek B, Duymus TM, Koseoglu PK, Aydin T, Kesiktas FN, Aksoy C, Mutlu S, J. Phys. Ther. Sci 2016, 28, 3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Hangody L, Szody R, Lukasik P, Zgadzaj W, Lénárt E, Dokoupilova E, Bichovsk D, Berta A, Vasarhelyi G, Ficzere A, Hangody G, Stevens G, Szendroi M, Cartilage 2017, 9, 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Anika Therapeutics, Inc., Cingal, https://www.anikatherapeutics.com/products/orthobiologics/cingal/, accessed: March, 2020.
- [45].Van Norman GA, JACC: Basic to Transl. Sci 2016, 1, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Van Norman GA, JACC: Basic to Transl. Sci 2016, 1, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Anika Therapeutics, Inc., A Randomized, Double-Blind, Placebo Controlled, Multi-Center Study of a Single Injection Cross-Linked Sodium Hyaluronate Combined With Triamcinolone Hexacetonide (Cingal) to Provide Symptomatic Relief of Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT01891396, accessed: February, 2020.
- [48].Collins NJ, Misra D, Felson DT, Crossley KM, Roos EM, Arthritis Care Res. (Hoboken) 2011, 63, S208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hurst H, Bolton J, J. Manipulative Physiol. Ther 2004, 27, 26. [DOI] [PubMed] [Google Scholar]
- [50].Schneider LS, Olin JT, Int. Psychogeriatr 1996, 8, 277. [DOI] [PubMed] [Google Scholar]
- [51].Anika Therapeutics, Inc., Cingal 13–02: An Open-Label, Follow-On Study to Cingal 13–01 to Evaluate the Safety of a Repeat Injection of Cross-Linked Sodium Hyaluronate Combined With Triamcinolone Hexacetonide (Cingal ®) to Provide Symptomatic Relief of Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT02381652, accessed: February, 2020.
- [52].Anika Therapeutics, Inc., Cingal.com, Clinical Evidence, https://www.cingal.com/for-physicians/clinical-evidence/, accessed: February, 2020.
- [53].Anika Therapeutics, Inc., Effect of Cingal ® Compared to Monovisc ® in Patients with Osteoarthritis of the Knee (EEFFEK Study), https://clinicaltrials.gov/ct2/show/NCT03062787, accessed: February 10, 2020.
- [54].Anika Therapeutics, Inc., Randomized, Double-Blind, Active Comparator Controlled, Multi-Center Study of a Single Injection Cross-Linked Sodium Hyaluronate Combined With Triamcinolone Hexacetonide (Cingal ™) to Provide Symptomatic Relief of Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT03191903, accessed: February, 2020.
- [55].Anika Therapeutics, Inc., Anika Therapeutics Announces Top-Line Results from CINGAL 16–02 Clinical Trial in Knee Osteoarthritis, https://ir.anikatherapeutics.com/news-releases/news-release-details/anika-therapeutics-announces-top-line-results-cingal-16-02, accessed: February 2020.
- [56].Anika Therapeutics, Inc., Extension Study to Cingal 16–02: Trial Extension to 39 Week Follow Up in the Randomized, Double-Blind, Active Comparator Controlled, Multi-Center Study of a Single Injection Cross-Linked Sodium Hyaluronate Combined With Triamcinolone Hexacetonide (Cingal ®) to Provide Symptomatic Relief of Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT03390036, accessed: February, 2020.
- [57].McMaster University, A Prospective Evaluation of the Cingal Injection for Hip Osteoarthritis (ECHO Study), https://clinicaltrials.gov/ct2/show/NCT04084704, accessed: February, 2020.
- [58].Yoshioka K, Kisukeda T, Zuinen R, Yasuda Y, Miyamoto K, BMC Musculoskelet. Disord 2018, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kisukeda T, Onaya J, Yoshioka K, BMC Musculoskelet. Disord 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Seikagaku Co., A Multicenter, Randomized, Double-blind, Placebo-controlled Study to Evaluate the Efficacy and Safety of Repeated Intra-Articular Injection of SI-613 in Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT03209362?term=si-613&draw=2&rank=1, accessed: February, 2020.
- [61].Seikagaku Co., Ono Pharmaceuticals Co., Ono and Seikagaku Announce the Topline Results from a Phase III Confirmatory Study of ONO-5704/SI-613 in Patients with Knee Osteoarthritis in Japan, https://www.seikagaku.co.jp/en/news/news-7734112843911996096/main/0/link/20190219-e.pdf, accessed: March, 2020.
- [62].Dallari D, Sabbioni G, Del Piccolo N, Carubbi C, Veronesi F, Torricelli P, Fini M, Clin. J. Sport Med 2020, 30. [DOI] [PubMed] [Google Scholar]
- [63].Veronesi F, Dallari D, Sabbioni G, Carubbi C, Martini L, Fini M, J. Cell. Physiol 2017, 232, 2299. [DOI] [PubMed] [Google Scholar]
- [64].Altavilla D, Squadrito F, Polito F, Irrera N, Calò M, Lo Cascio P, Galeano M, La Cava L, Minutoli L, Marini H, Bitto A, Surgery 2011, 149, 253. [DOI] [PubMed] [Google Scholar]
- [65].Cattarini Mastelli L, Cattarini Mastelli G, US9220734B2, 2015.
- [66].Istituto Ortopedico Rizzoli, Comparative Assessment of Viscosupplementation With Polynucleotides and Hyaluronic Acid to Viscosupplementation With Hyaluronic Acid in the Treatment of Osteoarthritis of the Knee. Randomized, Double-blind, Controlled Study, https://clinicaltrials.gov/ct2/show/NCT02417610, accessed: February, 2020.
- [67].Scuderi GR, Bourne RB, Noble PC, Benjamin JB, Lonner JH, Scott WN, Clin. Orthop. Relat. Res 2012, 470, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Daniloff GY, Gravett DM, He P (Carbylan Therapeutics, Inc.), AU2012245517A1, 2013. [Google Scholar]
- [69].Petrella R, Emans P, Alleyne J, Dellaert F, Gill D, Maroney M, BMC Musculoskelet. Disord 2015, 16, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Carbylan Therapeutics, Inc., A Prospective, Multi-center, Randomized, Double-blind Feasibility Study to Evaluate the Safety and Performance of Hydros Joint Therapy and Hydros-TA Joint Therapy for Management of Pain Associated With Osteoarthritis in the Knee, https://clinicaltrials.gov/ct2/show/NCT01134406, accessed: February, 2020.
- [71].Petrella R, Eamans P, Alleyne J, Maroney M, Osteoarthritis Cartilage 2012, 20, S172. [Google Scholar]
- [72].Carbylan Therapeutics, Inc., A Multi-center, Randomized, Double-blind Study to Evaluate the Safety and Efficacy of Hydros and Hydros-TA Joint Therapies for Management of Pain Associated With Osteoarthritis in the Knee, https://clinicaltrials.gov/ct2/show/NCT02022930, accessed: February, 2020.
- [73].Keown A, Bay Area’s Carbylan to Suspend Clinical Development of Hydros-TA and Slash Jobs to Conserve Cash, https://www.biospace.com/article/bay-area-s-carbylan-to-suspend-clinical-development-of-hydros-ta-and-slash-jobs-to-conserve-cash-/, accessed: February, 2020.
- [74].Renzi D, Crockett A, Carbylan Therapeutics, Inc. and KalVista Pharmaceuticals Ltd. Enter into Share Purchase Agreement, http://www.globenewswire.com/news-release/2016/06/15/848731/0/en/Carbylan-Therapeutics-Inc-and-KalVista-Pharmaceuticals-Ltd-Enter-into-Share-Purchase-Agreement.html, accessed: February 2020.
- [75].Kroin JS, Kc R, Li X, Hamilton JL, Das V, van Wijnen AJ, Dall OM, Shelly DA, Kenworth T, Im H-J, Gene 2016, 591, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Zhang Z, Wei X, Gao J, Zhao Y, Zhao Y, Guo L, Chen C, Duan Z, Li P, Wei L, Int. J. Mol. Sci 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Mero A, Campisi M, Favero M, Barbera C, Secchieri C, Dayer JM, Goldring MB, Goldring SR, Pasut G, J. Control. Release 2014, 187, 30. [DOI] [PubMed] [Google Scholar]
- [78].Nielsen RH, Bay-Jensen AC, Byrjalsen I, Karsdal MA, Osteoarthritis Cartilage 2011, 19, 466. [DOI] [PubMed] [Google Scholar]
- [79].Karsdal MA, Byrjalsen I, Alexandersen P, Bihlet A, Andersen JR, Riis BJ, Bay-Jensen AC, Christiansen C, Osteoarthritis Cartilage 2015, 23, 532. [DOI] [PubMed] [Google Scholar]
- [80].Lu H, Chang W, Tsai M, Chen C, Chen W, Mi F, Mar. Drugs 2019, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Meheux CJ, McCulloch PC, Lintner DM, Varner KE, Harris JD, Arthroscopy 2016, 32, 495. [DOI] [PubMed] [Google Scholar]
- [82].Hamidi S, Letourneur D, Aid-Launais R, Di Stefano A, Vainchenker W, Norol F, Le Visage C, Tissue Eng., Part A 2013, 20, 1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Purnama A, Aid-Launais R, Haddad O, Maire M, Mantovani D, Letourneur D, Hlawaty H, Le Visage C, Drug Delivery Transl. Res 2015, 5, 187. [DOI] [PubMed] [Google Scholar]
- [84].Petit A, Sandker M, Müller B, Meyboom R, van Midwoud P, Bruin P, Redout EM, Versluijs-Helder M, van der Lest CHA, Buwalda SJ, de Leede LGJ, Vermonden T, Kok RJ, Weinans H, Hennink WE, Biomater. 2014, 35, 7919. [DOI] [PubMed] [Google Scholar]
- [85].Wang SJ, Qin JZ, Zhang TE, Xia C, Front. Chem 2019, 7, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hanafy AS, El-Ganainy SO, Int. J. Pharm 2020, 573, 118859. [DOI] [PubMed] [Google Scholar]
- [87].Cokelaere SM, Plomp SGM, de Boef E, de Leeuw M, Bool S, van de Lest CHA, van Weeren PR, Korthagen NM, Eur. J. Pharm. Biopharm 2018, 128, 327. [DOI] [PubMed] [Google Scholar]
- [88].Bhaskar S, Pollock KM, Yoshida M, Lahann J, Small 2010, 6, 404. [DOI] [PubMed] [Google Scholar]
- [89].He Y, Park K, Mol. Pharmaceutics 2016, 13, 2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Kavanaugh TE, Werfel TA, Cho H, Hasty KA, Duvall CL, Drug Delivery Transl. Res 2016, 6, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Butoescu N, Seemayer CA, Foti M, Jordan O, Doelker E, Biomater. 2009, 30, 1772. [DOI] [PubMed] [Google Scholar]
- [92].Ahn M, Chang JH, Mellon RD, Hertz S, Compton K, Pharmacology/Toxicology NDA Review and Evaluation, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208845Orig1s000PharmRedt.pdf, accessed: January, 2020.
- [93].Hall BK, Newman SA, Cartilage Molecular Aspects. 1991.
- [94].Periyasamy PC, Leijten JCH, Dijkstra PJ, Karparien M, Post JN, J. Nanomater 2012, 2012, 1. [Google Scholar]
- [95].Bodick N, Lufkin J, Willwerth C, Kumar A, Bolognese J, Schoonmaker C, Ballal R, Hunter D, Clayman M, JBJS 2015, 97, 877. [DOI] [PubMed] [Google Scholar]
- [96].Bodick N, Blanks RC, Kumar A, Clayman MD, Moran M (Flexion Therapeutics, Inc.), 2017.
- [97].Kumar A, Walz A, Garlic D, Spainhour C, Bodick N, Osteoarthritis Cartilage 2014, 22, S463. [Google Scholar]
- [98].Williamson T, Walz A, Garlick D, Lightfoot-Dunn R, Schafer K, Kelley S, Bodick N, Osteoarthritis Cartilage 2017, 25, S431. [Google Scholar]
- [99].Kumar A, Bendele A, Blanks R, Bodick N, Osteoarthritis Cartilage 2015, 23, 151. [DOI] [PubMed] [Google Scholar]
- [100].Kumar A, Bendele A, Blanks R, Bodick N, Osteoarthritis Cartilage 2012, 20, S289. [DOI] [PubMed] [Google Scholar]
- [101].Horn P, Clinial Review, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208845Orig1s000MedR.pdf, accessed: January 2020.
- [102].Flexion Therapeutics, Inc., A Double-Blind, Randomized, Parallel Group, Active Comparator Study to Evaluate the Safety, Pharmacokinetics, and Pharmacodynamic Effects (HPA Axis) of FX006 in Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT01487200, accessed: January, 2020.
- [103].Qiu W, Clinical Pharmacology and Biopharmaceutics Review(s), https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208845Orig1s000ClinPharmR.pdf, accessed: January 2020.
- [104].Hertz S, Summary Review, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/208845Orig1s000SumRedt.pdf, accessed: January 2020.
- [105].Flexion Therapeutics, Inc., An Open-label, Single Administration Study to Characterize the Local Duration of Exposure of Triamcinolone Acetonide From FX006 in Patients With Osteoarthritis (OA) of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT02003365, accessed: January, 2020.
- [106].Flexion Therapeutics, Inc., An Open-label, Single Administration Study to Characterize the Systemic Pharmacokinetics and Local Extent and Duration of Exposure of Triamcinolone Acetonide from FX006 in Patients with Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT02637323, accessed: January, 2020.
- [107].Kraus V, Conaghan P, Aazami H, Mehra P, Kivitz A, Lufkin J, Hauben J, Johnson J, Bodick N, Osteoarthritis Cartilage 2018, 26, 34. [DOI] [PubMed] [Google Scholar]
- [108].Louati K, Vidal C, Berenbaum F, Sellam J, RMD Open 2015, 1, e000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Habib GS, Miari W, JCR: J. Clin. Rheumatol 2011, 17, 302. [DOI] [PubMed] [Google Scholar]
- [110].Russell SJ, Sala R, Conaghan PG, Habib G, Vo Q, Manning R, Kivitz A, Davis Y, Lufkin J, Johnson JR, Kelley S, Bodick N, Rheumatol. 2018, 57, 2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Flexion Therapeutics, Inc., A Double-Blind, Randomized, Parallel Group Comparison of the Effects of FX006 and TCA IR (Triamcinolone Acetonide Suspension) on Blood Glucose in Patients With Osteoarthritis of the Knee and Type 2 Diabetes Mellitus, https://clinicaltrials.gov/ct2/show/NCT02762370?term=fx006&cond=diabetes&draw=2&rank=1, accessed: March, 2020.
- [112].Flexion Therapeutics, Inc., A Double-Blind, Randomized, Parallel Group, Dose-Ranging Study Comparing FX006 to Commercially Available Triamcinolone Acetonide Injectable Suspension in Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT01487161, accessed: January, 2020.
- [113].Pham T, van der Heijde D, Altman RD, Anderson JJ, Bellamy N, Hochberg M, Simon L, Strand V, Woodworth T, Dougados M, Osteoarthritis Cartilage 2004, 12, 389. [DOI] [PubMed] [Google Scholar]
- [114].Flexion Therapeutics, Inc., A Double-Blind, Randomized, Parallel Group, Dose-Ranging Study to Assess the Safety and Efficacy of FX006 for the Treatment of Pain in Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT02116972, accessed: January, 2020.
- [115].Conaghan P, Cohen S, Berenbaum F, Lufkin J, Johnson J, Bodick N, Arthritis Rheumatol. 2018, 70, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Flexion Therapeutics, Inc., A Double-Blind, Randomized, Parallel Group, Proof of Concept Study Comparing FX006 to Kenalog-40 in Patients with Post-Traumatic Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT02468583, accessed: January, 2020.
- [117].Thomas AC, Hubbard-Turner T, Wikstrom EA, Palmieri-Smith RM, J. Athletic Training 2017, 52, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Kuyinu EL, Narayanan G, Nair LS, Laurencin CT, J. Orthop. Surg. Res 2016, 11, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Flexion Therapeutics, Inc., A Double-Blind, Randomized, Single-Dose Study to Assess the Safety and Efficacy of FX006 for the Treatment of Pain in Patients with Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT02357459, accessed: January, 2020.
- [120].Conaghan P, Hunter D, Cohen S, Kraus V, Berenbaum F, Lieberman J, Jones D, Spitzer A, Jevsevar D, Katz N, Burgess D, Lufkin J, Johnson J, Bodick N, F.−.-P. Inves, F.−.-P. Inves, J.Bone Jt. Surg., Am. Vol 2018, 100, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Spitzer AI, Richmond JC, Kraus VB, Gomoll A, Jones DG, Huffman KM, Peterfy C, Cinar A, Lufkin J, Kelley SD, Rheumatol. and Therapy 2019, 6, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Flexion Therapeutics, Inc., An Open-label Study to Assess the Safety of Repeat Administration of FX006 to Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT03046446?term=NCT03046446&draw=2&rank=1, accessed: March 2020.
- [123].Flexion Therapeutics, Inc., A Randomized, Open-label, Parallel Group Study in Patients With Bilateral Knee Osteoarthritis Comparing the Systemic Exposure of Triamcinolone Acetonide Following Administration Into Both Knees of Either Extended-release FX006 or Immediate-release TAcs (Triamcinolone Acetonide Suspension), https://clinicaltrials.gov/ct2/show/study/NCT03378076, accessed: January, 2020.
- [124].Kivitz A, Kwong L, Shlotzhauer T, Lufkin J, Curto T, Kelley S, Arthritis & Rheumatol. 2018, 70. [Google Scholar]
- [125].Kivitz A, Kwong L, Shlotzhauer T, Lufkin J, Cinar A, Kelley S, Ther. Adv. Musculoskelet. Dis 2019, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Flexion Therapeutics, Inc., A Randomized, Double-blind, Placebo-controlled, Parallel-group Study to Evaluate the Efficacy and Safety of FX006 in Patients with Hip Osteoarthritis, https://clinicaltrials.gov/ct2/show/NCT03793010?term=NCT03793010&draw=2&rank=1, accessed: March, 2020.
- [127].Flexion Therapeutics, Inc., A Single-arm, Open-label Study to Evaluate a Procedure for Intra-articular (IA) Injection of FX006 in Patients With Osteoarthritis (OA) of the Hip, https://clinicaltrials.gov/ct2/show/NCT04065074?term=NCT04065074&draw=2&rank=1, accessed: March, 2020.
- [128].Flexion Therapeutics, Inc., A Randomized, Open-label Study Comparing the Systemic Exposure to Triamcinolone Acetonide Following a Single Intra-articular Dose of Extended-release FX006 or Immediate-release TAcs (Triamcinolone Acetonide Suspension) in Patients With Osteoarthritis of the Shoulder (Glenohumeral Joint) or Hip, https://clinicaltrials.gov/ct2/show/study/NCT03382262, accessed: February, 2020.
- [129].Flexion Therapeutics, Inc., An Open-Label Study to Evaluate the Effect of the Administration of FX006 on Synovial Inflammation in Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/study/NCT03529942, accessed: February, 2020.
- [130].Flexion Therapeutics, Inc., The Effect of Intra-articular Bilateral Knee Injections of Zilretta on Osteoarthritis Research Society International (OARSI) Recommended Physical Performance Measures in Adults With Knee Osteoarthritis, https://clinicaltrials.gov/ct2/show/NCT03895840, accessed: February, 2020.
- [131].Flexion Therapeutics, Inc., Effect of ZILRETTA Injection on Neuromuscular Function, Gait Biomechanics and Physical Performance, https://clinicaltrials.gov/ct2/show/NCT04261049?term=NCT04261049&draw=2&rank=1, accessed: March, 2020.
- [132].Flexion Therapeutics, Inc., A Phase 2, Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate the Efficacy and Safety of FX006 in Patients with Gelohumeral Osteoarthritis or Shoulder Adhesive Capsulitis (RANGE), https://clinicaltrials.gov/ct2/show/NCT04160091?term=NCT04160091&draw=2&rank=1, accessed: March, 2020.
- [133].Helliwell JA, Malone AM, Smith TJ, Baum MM (Eupraxia Pharmaceuticals USA LLC), US20180071223A1, 2018.
- [134].Getgood A, Dhollander A, Malone A, Price J, Helliwell J, Cartilage 2019, 10, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Eupraxia Pharmaceuticals, A Randomized, Double-Blind, Placebo-Controlled, Phase I Trial Evaluating the Pharmacokinetics, Safety and Preliminary Efficacy of EP-104IAR (Long-Acting Fluticasone Propionate) in Patients with Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT02609126?term=EP-104IAR&draw=2&rank=2, accessed: February, 2020.
- [136].Postgrad. Med 2018, 130, 1.29190175 [Google Scholar]
- [137].Eupraxia Pharmaceuticals, A Phase II, Randomized, Double-Blind, Vehicle-Controlled Study Evaluating the Safety, Efficacy, and Pharmacokinetics of Single Dose EP-104IAR for 24 Weeks in Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT04120402?term=EP-104IAR&draw=2&rank=1, accessed: February, 2020.
- [138].Bateman NF, Macfaul PA, Nash IA (Astrazeneca Ltd.), WO2009125226A1, 2008.
- [139].Flexion Therapeutics, Inc., A Double-Blind, Randomized, Placebo-Controlled, Two-Phase Study (a Single Ascending Dose Phase Followed by a Proof of Concept Phase) to Assess the Safety, Efficacy and Pharmacokinetics of FX005 for the Treatment of Pain in Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT01291914?term=fx005&draw=2&rank=1, accessed: February, 2020.
- [140].Weintraub A, Flexion Overhaul Bears Fruit With Upbeat Data From Arthritis Trial, https://xconomy.com/boston/2012/05/30/flexion-overhaul-bears-fruit-with-upbeat-data-from-arthritis-trial/2/, accessed: February 2020.
- [141].Keown A, Massachussetts’ Flexion Terminates R&D Deal With AstraZeneca PLC, https://www.biospace.com/article/massachusetts-flexion-terminates-r-and-d-deal-with-astrazeneca-plc-/, accessed: February 2020.
- [142].Botines E, Franco L, Puiggali J, J. Appl. Polym. Sci 2006, 102, 5545. [Google Scholar]
- [143].Murase S, Lv L, Kaltbeitzel A, Landfester K, del Valle L, Katsarava R, Puiggali J, Crespy D, RSC Adv. 2015, 5, 55006. [Google Scholar]
- [144].Nakano S, Ikata T, Kinoshita I, Kanematsu J, Yasuoka S, Clin. Exp. Rheumatol 1999, 17, 161. [PubMed] [Google Scholar]
- [145].Janssen M, Timur UT, Woike N, Welting TJM, Draaisma G, Gijbels M, van Rhijn LW, Mihov G, Thies J, Emans PJ, J. Control. Release 2016, 244, 30. [DOI] [PubMed] [Google Scholar]
- [146].Tellegen AR, Rudnik-Jansen I, Pouran B, de Visser HM, Weinans HH, Thomas RE, Kik MJL, Grinwis GCM, Thies JC, Woike N, Mihov G, Emans PJ, Meij BP, Creemers LB, Tryfonidou MA, Drug Delivery 2018, 25, 1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Glasson SS, Chambers MG, Van Den Berg WB, Little CB, Osteoarthritis Cartilage 2010, 18, S17. [DOI] [PubMed] [Google Scholar]
- [148].Rudnik-Jansen I, Colen S, Berard J, Plomp S, Que I, van Rijen M, Woike N, Egas A, van Osch G, van Maarseveen E, Messier K, Chan A, Thies J, Creemers L, J. Control. Release 2017, 253, 64. [DOI] [PubMed] [Google Scholar]
- [149].Rudnik-Jansen I, Schrijver K, Woike N, Tellegen A, Versteeg S, Emans P, Mihov G, Thies J, Eijkelkamp N, Tryfonidou M, Creemers L, Drug Delivery 2019, 26, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Maudens P, Seemayer CA, Pfefferlé F, Jordan O, Allémann E, J. Control. Release 2018, 276, 102. [DOI] [PubMed] [Google Scholar]
- [151].Pfizer A Randomized, Double-Blind, Double-Dummy, Placebo And Active Controlled Two Period Four Treatment Cross-Over Clinical Trial To Examine The Pain Relief Of PH-797804 Alone Or With Naproxen In Subjects With Flare-Enriched Osteoarthritis Of The Knee, https://clinicaltrials.gov/ct2/show/NCT01102660, accessed: February, 2020.
- [152].Maudens P, Seemayer CA, Thauvin C, Gabay C, Jordan O, Allémann E, Small 2018, 14, 1703108. [DOI] [PubMed] [Google Scholar]
- [153].Johnson K, Zhu S, Tremblay M, Payette J, Wang J, Bouchez L, Meeusen S, Althage A, Cho C, Wu X, Schultz P, Science 2012, 336, 717. [DOI] [PubMed] [Google Scholar]
- [154].Torchilin V, Nat. Rev. Drug Disc 2005, 4, 145. [DOI] [PubMed] [Google Scholar]
- [155].Lee Y, Thompson D, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Mozafari M, Cell. Mol. Biol. Lett 2005, 10, 711. [PubMed] [Google Scholar]
- [157].Sivan S, Schroeder A, Verberne G, Merkher Y, Diminsky D, Priev A, Maroudas A, Halperin G, Nitzan D, Etsion I, Barenholz Y, Langmuir 2010, 26, 1107. [DOI] [PubMed] [Google Scholar]
- [158].Verberne G, Schroeder A, Halperin G, Barenholz Y, Etsion I, Wear 2010, 268, 1037. [DOI] [PubMed] [Google Scholar]
- [159].Barenholz Y, Dolev Y, Turgeman K, Sarfati G, Hershkovitz MA (Moebius Medical Ltd.), WO2019038763A1, 2017.
- [160].Kandel L, Dolev Y, Rivkind G, Liebergall M, Mattan Y, Barenholz Y, Shimonov R, Chevalier X, Osteoarthritis Cartilage 2014, 22, S193. [Google Scholar]
- [161].Shih SF, Chang PC, Wu MJ (Taiwan Liposome Co., Ltd.), W02020014118A1, 2018.
- [162].Yu W, Wu M, Chang P, Shih S, Osteoarthritis Cartilage 2019, 27, S161. [Google Scholar]
- [163].Wu TJ, Yu W, Wu M, Chang P, Shih S, Ann. Rheum. Dis 2019, 78, 523. [Google Scholar]
- [164].Taiwan Liposome Co., A Randomized, Open-label, Parallel, Phase I/II Single-Dose Administration trial of TLC599 in Subjects with Osteoarthritis (OA) of the Knee, https://clinicaltrials.gov/ct2/show/NCT02803307?term=tlc599&draw=1&rank=2, accessed: February, 2020.
- [165].Taiwan Liposome Co., Taiwan Liposome Company to Present Full Data from TLC599 Phase I/II Study in Knee Osteoarthritis at 20th Asia Pacific League of Associations for Rheumatology Conference., http://ir.tlcbio.com/static-files/d7da6f0a-6535-454f-ad7b-2b61e01f46f9, accessed: February, 2020. [Google Scholar]
- [166].Lai CC, Chiang CC, Lee CS, Chang CC, Lin HY, Chen TL, Shih SF, presented at 20th Asia Pacific League of Assoc. for Rheumatol. Congress, Kaohsiung, Taiwan, September, 2018. [Google Scholar]
- [167].Taiwan Liposome Co., A Phase IIa, Randomized, Double blinded, Placebo controlled, Dose finding Study for Single dose Administration of TLC599 in Patients with Osteoarthritis (OA) of Knee, https://clinicaltrials.gov/ct2/show/study/NCT03005873?term=tlc599&draw=1&rank=3, accessed: February, 2020.
- [168].Hunter D, Chang C, Wei J, Lin H, Brown C, Shih S, Osteoarthritis Cartilage 2019, 27, S87. [Google Scholar]
- [169].Taiwan Liposome Co., A Phase 2, Open-label, Pharmacokinetic Study of a Single Intra-articular Administration of TLC599 in Subjects With Mild to Moderate Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT03754049?term=tlc599&draw=1&rank=1, accessed: February, 2020.
- [170].Taiwan Liposome Co., A Phase 3, Randomized, Double-blind, Placebo- and Active-controlled Study to Evaluate the Efficacy and Safety of TLC599 in Patients With Osteoarthritis of the Knee, https://clinicaltrials.gov/ct2/show/NCT04123561?term=tlc599&draw=1&rank=4, accessed: February, 2020.
- [171].Wang S, Wei X, Sun X, Chen C, Zhou J, Zhang G, Wu H, Guo B, Wei L, Int. J. Nanomed 2018, 13, 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Wei F, Zhou J, Wei X, Zhang J, Fleming B, Terek R, Pei M, Chen Q, Liu T, Wei L, Osteoarthritis Cartilage 2012, 20, 755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Zhang C, Wei X, Chen C, Cao K, Li Y, Jiao Q, Ding J, Zhou J, Fleming B, Chen Q, Shang X, Wei L, Int. J. Mol. Sci 2014, 15, 7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Akinc A, Maier MA, Manoharan M, Fitzgerald K, Jayaraman M, Barros S, Ansell S, Du X, Hope MJ, Madden TD, Mui BL, Semple SC, Tam YK, Ciufolini M, Witzigmann D, Kulkarni JA, van der Meel R, Cullis PR, Nat. Nanotechnol 2019, 14, 1084. [DOI] [PubMed] [Google Scholar]
- [175].Elron-Gross I, Glucksam Y, Biton IE, Margalit R, J. Control. Release 2009, 135, 65. [DOI] [PubMed] [Google Scholar]
- [176].Rothenfluh DA, Bermudez H, O’Neil CP, Hubbell JA, Nat. Mater 2008, 7, 248. [DOI] [PubMed] [Google Scholar]
- [177].Bedingfield SK, Yu F, Liu DD, Jackson MA, Himmel LE, Cho H, Colazo JM, Crofford LJ, Hasty KA, Duvall CL, bioRxiv 2020, 2020.01.30.925321. [DOI] [PMC free article] [PubMed]
- [178].Fan W, Li J, Yuan L, Chen J, Wang Z, Wang Y, Guo C, Mo X, Yan Z, Drug Delivery 2018, 25, 1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Kamel R, Salama AH, Mahmoud AA, Int. J. Pharm 2016, 515, 657. [DOI] [PubMed] [Google Scholar]
- [180].Wang J, Wang X, Cao Y, Huang T, Song D-X, Tao H-R, Int. J. Mol. Med 2018, 42, 2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Zhao Y, Wei C, Chen X, Liu J, Yu Q, Liu Y, Liu J, ACS Appl. Mater. Interfaces 2019, 11, 11587. [DOI] [PubMed] [Google Scholar]
- [182].Bajpayee AG, Grodzinsky AJ, Nat. Rev. Rheumatol 2017, 13, 183. [DOI] [PubMed] [Google Scholar]
- [183].Evans CH, Kraus VB, Setton LA, Nat. Rev. Rheumatol 2014, 10, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Bajpayee AG, Wong CR, Bawendi MG, Frank EH, Grodzinsky AJ, Biomater. 2014, 35, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Miller RE, Grodzinsky AJ, Cummings K, Plaas AHK, Cole AA, Lee RT, Patwari P, Arthritis Rheum. 2010, 62, 3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Vedadghavami A, Wagner EK, Mehta S, He T, Zhang C, Bajpayee AG, Acta Biomater. 2019, 93, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Cook Sangar ML, Girard EJ, Hopping G, Yin C, Pakiam F, Brusniak M-Y, Nguyen E, Ruff R, Gewe MM, Byrnes-Blake K, Nairn NW, Miller DM, Mehlin C, Strand AD, Mhyre AJ, Correnti CE, Strong RK, Simon JA, Olson JM, Sci. Transl. Med 2020, 12, eaay1041. [DOI] [PubMed] [Google Scholar]
- [188].He T, Zhang C, Vedadghavami A, Mehta S, Clark HA, Porter RM, Bajpayee AG, J. Control. Release 2020, 318, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Krishnan Y, Rees HA, Rossitto CP, Kim S-E, Hung H-HK, Frank EH, Olsen BD, Liu DR, Hammond PT, Grodzinsky AJ, Biomater. 2018, 183, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Geiger BC, Wang S, Padera RF, Grodzinsky AJ, Hammond PT, Sci. Transl. Med 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Vedadghavami A, Zhang C, Bajpayee AG, Nano Today 2020, 34, 100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Bajpayee AG, De la Vega RE, Scheu M, Varady NH, Yannatos IA, Brown LA, Krishnan Y, Fitzsimons TJ, Bhattacharya P, Frank EH, Grodzinsky AJ, Porter RM, Eur. Cells Mater 2017, 34, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Xia C, Chen P, Mei S, Ning L, Lei C, Wang J, Zhang J, Ma J, Fan S, Oncotarget 2017, 8, 2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Carames B, Taniguchi N, Otsuki S, Blanco F, Lotz M, Arthritis Rheum. 2010, 62, 791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Chen P, Xia C, Mei S, Wang J, Shan Z, Lin X, Fan S, Biomater. 2016, 81, 1. [DOI] [PubMed] [Google Scholar]
- [196].Kang M-L, Jeong S-Y, Im G-I, Tissue Eng., Part A 2017, 23, 630. [DOI] [PubMed] [Google Scholar]
- [197].Holyoak D, Wheeler T, van der Meulen M, Singh A, Regener. Biomater 2019, 6, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Dong J, Jiang D, Wang Z, Wu G, Miao L, Huang L, Int. J. Pharm 2013, 441, 285. [DOI] [PubMed] [Google Scholar]