

Abstract
Background
Down syndrome (DS) is characterized by a genome-wide profile of differential DNA methylation that is skewed towards hypermethylation in most tissues, including brain, and includes pan-tissue differential methylation. The molecular mechanisms involve the overexpression of genes related to DNA methylation on chromosome 21. Here, we stably overexpressed the chromosome 21 gene DNA methyltransferase 3L (DNMT3L) in the human SH-SY5Y neuroblastoma cell line and assayed DNA methylation at over 26 million CpGs by whole genome bisulfite sequencing (WGBS) at three different developmental phases (undifferentiated, differentiating, and differentiated).
Results
DNMT3L overexpression resulted in global CpG and CpG island hypermethylation as well as thousands of differentially methylated regions (DMRs). The DNMT3L DMRs were skewed towards hypermethylation and mapped to genes involved in neurodevelopment, cellular signaling, and gene regulation. Consensus DNMT3L DMRs showed that cell lines clustered by genotype and then differentiation phase, demonstrating sets of common genes affected across neuronal differentiation. The hypermethylated DNMT3L DMRs from all pairwise comparisons were enriched for regions of bivalent chromatin marked by H3K4me3 as well as differentially methylated sites from previous DS studies of diverse tissues. In contrast, the hypomethylated DNMT3L DMRs from all pairwise comparisons displayed a tissue-specific profile enriched for regions of heterochromatin marked by H3K9me3 during embryonic development.
Conclusions
Taken together, these results support a mechanism whereby regions of bivalent chromatin that lose H3K4me3 during neuronal differentiation are targeted by excess DNMT3L and become hypermethylated. Overall, these findings demonstrate that DNMT3L overexpression during neurodevelopment recreates a facet of the genome-wide DS DNA methylation signature by targeting known genes and gene clusters that display pan-tissue differential methylation in DS.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13072-021-00387-7.
Keywords: DNMT3L, Down syndrome, Whole genome bisulfite sequencing, Bivalent chromatin, Epigenomics, PiggyBac transgenesis
Background
DS is the leading genetic cause of intellectual disability and results from trisomy 21 [1]. However, genes outside of chromosome 21 are also altered in DS and differences in gene expression and DNA methylation are observed across the entire genome. Most DS tissues exhibit differentially methylated sites that tend to be hypermethylated when compared to typically developing controls [2]. Furthermore, some genes show pan-tissue differential methylation, which is also skewed heavily towards hypermethylation. Mechanistically, there are a number of genes located on chromosome 21 that belong to pathways related to DNA methylation and have the potential to result in the hypermethylation pattern observed in DS [2]. Functional experimentation into the cause of hypermethylation has demonstrated a key role of the chromosome 21 encoded DNA methyltransferase DNMT3L at select genes [3]. While DNMT3L is catalytically inactive, it is a regulatory factor that binds to and stimulates the de novo methyltransferases DNMT3A and DNMT3B [4–6].
Structurally, DNMT3L and DNMT3A form elongated heterotetramers through their C-terminal domains (DNMT3L–DNMT3A–DNMT3A–DNMT3L), and this complex multimerizes on DNA to form nucleoprotein filaments that spread DNA methylation over larger regions, such as CpG islands [7–9]. Members of the de novo methyltransferase family (DNMT3A,B,L) contain an ATRX–DNMT3–DNMT3L (ADD) domain that binds to the unmodified histone H3 tail (H3K4me0), to localize DNA methylation to previously unmethylated regions, and this binding is inhibited by methylation at lysine 4 of H3 (H3K4me3) [10–12].
In mouse embryonic stem cells (mESCs), DNMT3L has been reported to be either a positive or negative regulator of DNA methylation depending on genomic context [13]; however, another report found it to only function as a positive regulator [14]. This difference appears to be due to the mESCs cell lines used as well as the methodologies used to assay DNA methylation, since the studies examined different regions of the genome at different levels of resolution. Experiments using transient DNMT3L overexpression in mESCs and somatic cells have demonstrated a role for DNMT3L in assembling a repressive chromatin modifying complex that silences retroviral sequences through de novo DNA methylation and methylation-independent mechanisms [15]. Finally, DNMT3L is also required for the establishment of parental imprinting patterns in developing gametes [16, 17].
While DNMT3L is a prime candidate for the genome-wide DS DNA hypermethylation profiles, the genome-wide effect of DNMT3L overexpression has not yet been characterized. Previously, we profiled post-mortem DS brain and compared to matched controls using WGBS. We observed a genome-wide wide impact on DNA methylation profiles, where ~ 75% of the differentially methylated regions (DMRs) were hypermethylated and mapped to genes involved in one-carbon metabolism, membrane transport, and neurotransmission. Given these observations, we sought to characterize the effect of DNMT3L overexpression on different phases of neuronal development by assaying differentiating SH-SY5Y cells.
SH-SY5Y cells are a human neuronal cell line that have been subcloned three times from the SK-N-SH cell line, which was derived from metastatic cells of a 4-year-old female with neuroblastoma. Unlike many other cancer lines, the SH-SY5Y cell line has a stable karyotype with a dominant ploidy of 2, which includes trisomy 1q and 7, gains on 2p and 17q, and losses on 14q and 22q [18–20]. Similar to other cancer cell lines, SH-SY5Y cells have a relatively hypomethylated genome when compared to neurons from human post-mortem brain samples [21, 22]. SH-SY5Y cells display a mixed morphology that is primarily neuroblast-like with short processes; however, there is also a smaller proportion of epithelial-like cells, which likely reflects the cell lines multipotency and an origin from the neural crest. SH-SY5Y cells can be consistently differentiated from a neuroblast-like state into a homogenous population of mature dopaminergic neurons in 18 days [23]. The differentiation protocol is a three-phased reprogramming approach, which involves gradual serum deprivation and the addition of retinoic acid during the first two phases and then a transition to an extracellular matrix where the cells are given a neuronal media that lacks serum. The neuron-like cells become dependent on neurotrophic factors, such as BDNF, for their survival and the epithelial-like cells, which require serum, do not survive. Notably, during this differentiation process, serial splitting via a brief incubation with low strength trypsin allows for the selection of the less adherent neuron-like cells (for the next phase or harvest) and ultimately results in a morphologically pure culture of mature dopaminergic neurons in the final phase. Thus, SH-SY5Y cells serve as an ideal model system to investigate the methylome-wide effects of DNMT3L overexpression on undifferentiated, differentiating, and differentiated human neurons.
Here, we interrogated the contribution of DNMT3L, a chromosome 21 encoded gene, in DS-associated genome-wide changes in DNA methylation. Stable overexpression of DNMT3L in human SH-SY5Y cells provided a model system to test the effects of this known methylation regulator on genome-wide DNA methylation across neuronal differentiation. We found that DNMT3L overexpression recreated a facet of the genome-wide DNA profile observed in DS tissues, including brain. Interestingly, chromatin state analyses revealed that the DNMT3L-induced hypermethylated DMRs were enriched within bivalent domains, whereas the hypomethylated DMRs were enriched within heterochromatin. These results indicate that an increase in DNMT3L copy number during neuronal differentiation leads to chromatin-specific effects on DNA methylation.
Results
Stable DNMT3L Overexpression in SH-SY5Y Cells
SH-SY5Y cells were transfected with either myc-tagged human DNMT3L [4] and enhanced GFP (eGFP) via a piggyBac transposase [24] construct or a matched control construct without DNMT3L (Fig. 1a). eGFP-positive cells were selected by flow cytometry and then cultured again to enrich for a cell line with stable DNMT3L overexpression. For the experimental investigations, three developmental phases were assayed: 1) undifferentiated cells (growth), 2) the first phase of differentiation (phase 1), and 3) the final phase of differentiation (phase 3) that represents the pure and mature neuronal culture (Fig. 1b). When compared to the growth phase, phase 1 neurons displayed a relatively elongated cell body with an increased number of longer neurites with neuronal growth cones that were starting to form synapses with their neighboring cells. Notably, phase 1 neurons are the most commonly assayed form of differentiated SH-SY5Y cells in the literature. Phase 2 is a relatively brief intermediate phase for the final selection that occurs in phase 3 and, thus, was not examined. After being plated as a monolayer, phase 3 neurons migrated to form large spheres of highly interconnected neurons, which were similar to neurospheres; however, they are were attached to an extracellular matrix and produced a three-dimensional network with a larger-scale organization that consists of long tracts of bundled processes. Notably, the eGFP-only control cells displayed visibly higher fluorescence levels than those with DNMT3L + eGFP, a result that was observed in all cell lines. Endogenous DNMT3L expression was confirmed by Western blot in all transfected cell lines and compared to a non-transfected cell line (WT). As expected, DNMT3L expression was much higher in the transgenic DNMT3L cell lines and only low basal levels were detected in the WT and eGFP-only control cell lines (Fig. 1c).
Fig. 1.

Stable overexpression of human DNMT3L in SH-SY5Y cells at different phases of neural differentiation. a PiggyBac vector for stable integration of human DNMT3L (hDNMT3L), which was co-expressed with eGFP. Upon translation the self-cleaving peptide (P2A) sequence between the polypeptide resulted in separate proteins. The vector was flanked by inverted terminal repeats (ITRs) that were recognized by the PiggyBac transposon. b Representative live cell images the DNMT3L + eGFP and eGFP cell lines during the different phases assayed in either the GFP or phase contrast channels. c Western blot of DNMT3L and GAPDH in wild-type SH-SY5Y cells (WT) and the transgenic cell lines
Stable DNMT3L overexpression results in global and CpG island hypermethylation
WGBS libraries were constructed using DNA isolated from all cell lines and sequenced to > 10 × coverage, resulting in > 26 million assayed CpGs. There was a significant (p = 0.016) increase in global CpG methylation (Fig. 2a) and a significant (p = 0.004) increase in global CpG island methylation in cells with DNMT3L overexpression (3L) when compared to vector-only (eGFP) controls. Cell developmental phase, herein referred to as “phase”, also had a significant (p = 0.004) effect on global CpG methylation and a significant (p = 0.030) effect on global CpG island methylation. In the growth phase, DNMT3L cells showed 69.70% global methylation and 34.11% CpG island methylation, while the eGFP cells showed 69.43 and 33.82%, respectively. In phase 1 of differentiation, DNMT3L cells showed 70.38% global methylation and 34.39% CpG island methylation, while eGFP cells showed 69.93 and 33.89%, respectively. In phase 3 of differentiation, the DNMT3L cells showed 69.68% global methylation and 33.95% CpG island methylation, while eGFP cells showed 69.33 and 33.52%, respectively. Notably, both global CpG methylation and global CpG island methylation levels were the highest in phase 1 and the lowest in phase 3. Principal component analysis (PCA) revealed that the cells clustered together by phase for both the 20 Kb window and single CpG approaches (Fig. 2b), but clustered by genotype for the CpG island approach (Fig. 2c).
Fig. 2.
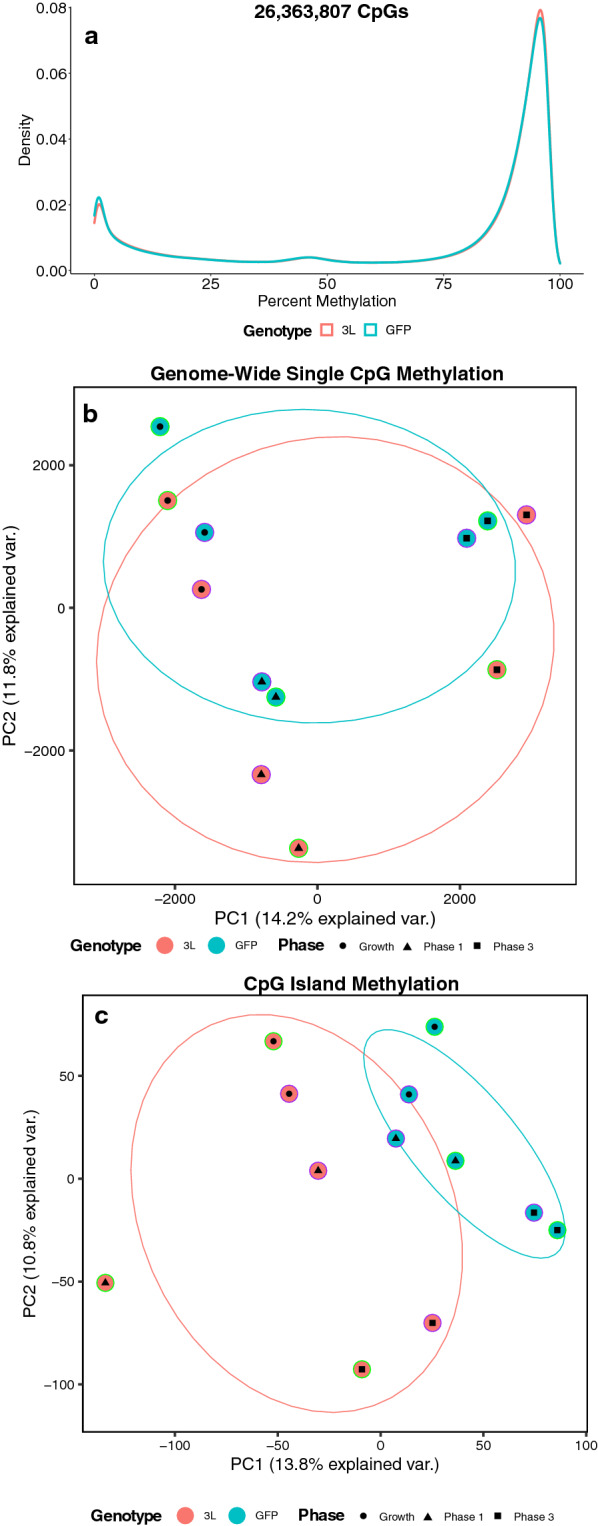
Global methylation profiles of DNMT3L overexpression. a Density plots of the mean of smoothed individual CpG methylation values for each cell line at the different phases. b Principal component analysis (PCA) of smoothed individual global CpG methylation values. c PCA of smoothed individual CpG island methylation values. For each PCA, the color of the outermost shape represents the cell line, where green represents the first batch of cell lines and purple represents the second. The ellipses represent the 68% confidence interval, which represents 1 standard deviation from the mean for data with a normal distribution
DNMT3L DMRs map to genes related to neurodevelopment, cellular signaling, and gene regulation
To examine the specific genetic loci altered by DNMT3L overexpression during neuronal differentiation, differentially methylated regions (DMRs) were identified using pairwise genotype comparisons of all three phases. The replicate cell lines were utilized as biological replicates and batch was directly adjusted for in the analyses. The significant (permutation p < 0.05) DMRs from each pairwise phase comparison were skewed towards hypermethylation (Additional File 1: Supplementary Table 1). In the growth phase, 62% of the 6746 DMRs were hypermethylated, as well as 71% of the 5954 DMRs in phase 1, and 68% of the 6389 DMRs in phase 3 (Fig. 3). DMRs were mapped to genes and gene ontology (GO) terms were slimmed to identify the least dispensable significant (p < 0.05) terms. Across all phases, when examining the effect of DNMT3L overexpression, these slimmed GO terms represented different neurodevelopmental processes, including synapse, signaling, and biological adhesion. However, there were also phase-specific GO terms, including the glucuronidation hierarchy of GO terms in the phase 1 comparison. While represented by xenobiotic glucuronidation, the glucuronidation hierarchy appears to represent retinoic acid, which is added to the media in phase 1, since the glucuronidation of retinoic acid is involved in neurodevelopment [25].
Fig. 3.

Differentially methylated region (DMR) profiles and slimmed gene ontology (GO) enrichments for DNMT3L overexpression at the different phases of neural differentiation. The heatmaps represent hierarchal clustering of Z-scores, which represent the number of standard deviations from the mean of the non-adjusted individual smoothed methylation value for each DMR. The bar plots represent the least dispensable significant enrichments (p < 0.05)
To further test the functional relevance of DNA methylation changes resulting from DNMT3L overexpression, we tested the DMRs for enrichments within genomic regions annotated as CpG islands, CpG shores, CpG shelves, or open sea, as well as for enrichments within promoter and gene body annotations. Both the hypermethylated and hypomethylated DMRs from all pairwise phase comparisons were significantly (q < 0.05) enriched within CpG islands (Fig. 4a) and significantly de-enriched within intergenic regions (Fig. 4b). Furthermore, the hypermethylated DMRs from all pairwise phase comparisons were significantly (q < 0.05) enriched within promoters and regions of the gene body; while, the hypomethylated DMRs from all pairwise phase comparisons were significantly (q < 0.05) enriched within introns.
Fig. 4.

Annotation enrichments for DMRs from the growth, phase 1, and phase 3 comparisons. a CpG and b genic annotation enrichments for hypermethylated and hypomethylated DMRs. * = q < 0.05
To understand the similarities and differences between the impact of DNMT3L overexpression on each developmental phase, DMRs from all pairwise comparisons were compared for overlap by both genomic coordinate and gene association. Overlaps of the DMRs by gene symbol included 962 in common to all three phases (Fig. 5a), even though only 12 overlapped by genomic coordinate in all three phases. The DMRs from all phase comparisons were then merged by sequence overlap to produce a consensus DMR profile, and hierarchal clustering analysis revealed that the cells clustered primarily by genotype and then developmental phase, and not by cell line of origin (Fig. 5b). The consensus DMR profile highlighted not only the overall skew towards DMR hypermethylation, but also clusters of DMRs that were specific to differentiation phase. A meta p-value of analysis of the GO terms from all pairwise phase comparisons revealed that the least dispensable significant (p < 0.05) terms were largely similar to the individual comparisons and primarily represented neurodevelopment (Fig. 5c).
Fig. 5.
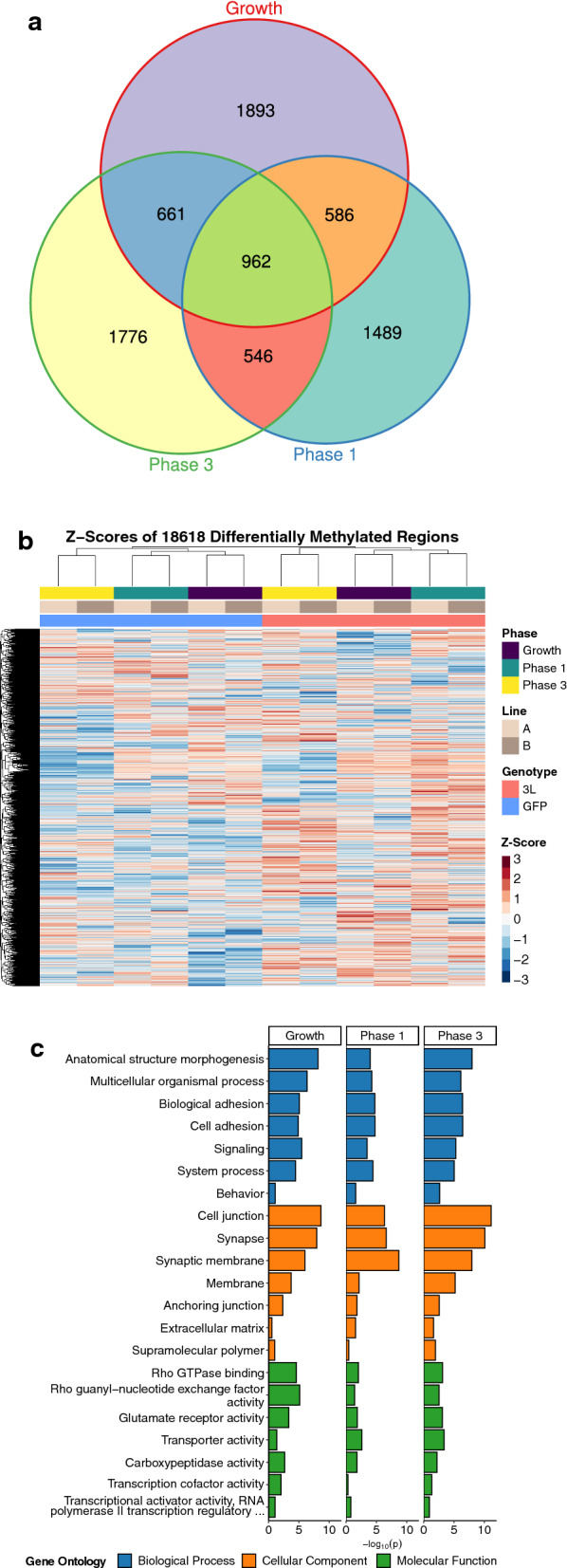
Consensus DMR profiles for DNMT3L overexpression across all phases of neural differentiation. a Euler diagram of gene symbol overlaps for the genotype comparison at each phase of neural differentiation. b Heatmap of hierarchal clustering of Z-scores for the consensus DMRs that are derived from merging the DMRs from each phase comparison by sequence overlap. c Bar plot of comparison specific p-values from the meta-analysis of the least dispensable significant slimmed GO enrichments (pmeta < 0.05)
The hypermethylated DMRs are enriched for bivalent chromatin and DS cross-tissue regions
To further investigate the functional relevance between the hypermethylated and hypomethylated DNMT3L DMRs, enrichment analyses within chromatin state maps from the reference epigenomes were performed. The most significant (q < 0.05) chromHMM chromatin state enrichments overall were for the hypermethylated DMRs in regions of bivalent chromatin (Fig. 6a) marked by H3K4me3 (Fig. 6b) in stem cells. This effect was most pronounced in the differentiated cells. In contrast, the hypomethylated DMRs were most significantly (q < 0.05) enriched within heterochromatic regions (Fig. 6a) marked by H3K9me3 (Fig. 6b) in ESCs.
Fig. 6.

Reference epigenome enrichment analyses for the hypermethylated and hypomethylated DMRs from the genotype comparisons at each phase of neural differentiation. a Summary heatmap of top q-values for the chromHMM core 15-state enrichment analyses for the brain, embryonic stem cell derivatives (ES-derivatives), embryonic stem cells (ESC), induced pluripotent stem cells (iPSC), and neurospheres (Neurosph) categories. b Summary heatmap of q-values for Roadmap epigenomics 127 reference epigenomes 5 core histone modification enrichment analyses for the Brain, ESC, and iPSC categories. All enrichments are relative to background regions. * = q < 0.05
To test the hypothesis that the DNMT3L DMRs represent a facet of the differentially methylated genes observed in DS, cross-tissue and pan-tissue comparisons with differentially methylated DS sites across diverse tissues were performed [26–35]. These results revealed a significant (q < 0.05) enrichment of the DNMT3L DMRs within DS differentially methylated sites identified from multiple tissues (Fig. 7a). This enrichment was predominantly for the hypermethylated DNMT3L DMRs from all pairwise phase comparisons. The only significant (q < 0.05) enrichment for the hypomethylated DNMT3L DMRs was from the growth comparison and within fetal frontal cortex CpGs. To further understand the DS differentially methylated genes most impacted by DNMT3L, genes with DNMT3L-associated differential methylation in all three phases of neural differentiation were filtered to include those also differentially methylated in at least 5 DS methylome datasets, and also investigated for overlap with embryonic bivalent chromatin marks from a previous study [36] (Fig. 7b). In this analysis, the common pan-tissue genes mapping to the DNMT3L-specific DMRs were generally from perinatal or neuronal DS datasets and most also mapped to bivalent chromatin. Notably, in the DNMT3L overexpression cells lines, one of the pan-tissue DMR sites identified maps to the promoter of TBX1 (Fig. 7c). TBX1 is a transcription factor involved in embryonic development and a key gene in 22q11.2 deletion syndrome (DiGeorge syndrome). DiGeorge syndrome shares phenotypic similarities with DS, which include congenital facial and cardiac abnormalities as well as intellectual disability [37]. Additionally, TBX1 is regulated by retinoic acid, which is a key component of the SH-SY5Y differentiation protocol utilized [23].
Fig. 7.
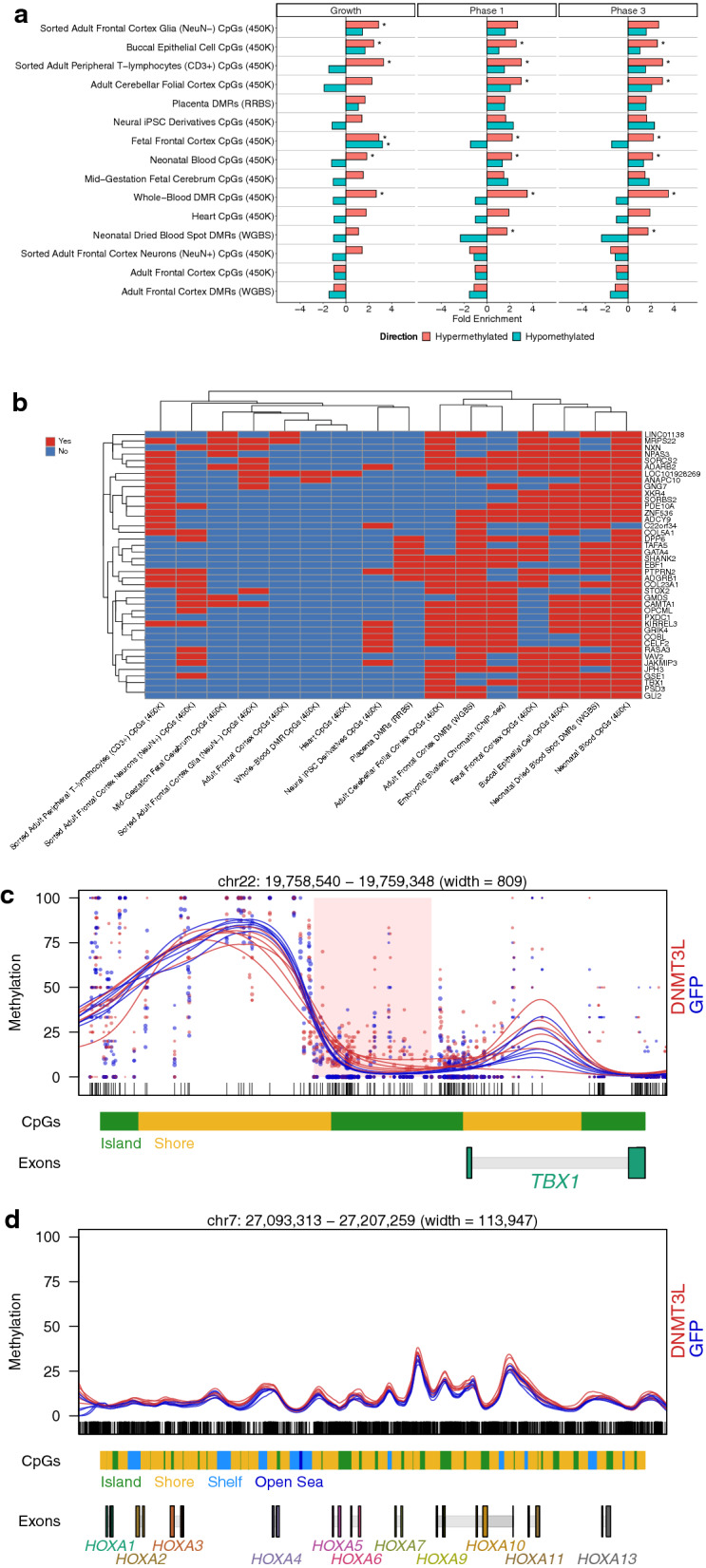
Enrichments of previously reported Down syndrome differentially methylated loci among the DNMT3L overexpression DMRs at each phase of neural differentiation. a Bar plots of enrichments of Down syndrome differentially methylated loci among the DNMT3L DMRs, shown for each of the available Down syndrome methylation datasets. The enrichments are relative to background regions. * = q < 0.05. b Heatmap (hierarchal clustering) of pan-tissue Down syndrome differentially methylated genes (DMRs in at least 5 datasets from previous studies), color coded for concordance or lack of concordance with the differential methylation produced by DNMT3L overexpression in SH-SY5Y neuroblastoma cells. Intergenic mappings were excluded. c Plot of the DNMT3L overexpression DMRs within the TBX1 promoter region. Samples at each phase of neural differentiation have been color coded by genotype. The dots represent the methylation value of a single CpG site and their size represents coverage, while the lines represent an estimate of the individual methylation value for a sample. d Plot of hypermethylation of the HOXA cluster in DNMT3L-overexpressing cells
Also present amongst the DNMT3L differentially methylated genes were subsets of the 25 previously known pan-tissue and multi-tissue DS genes [2], which were all hypermethylated. This observation is in line with the previous observation that 24 out of the 25 pan-tissue and multi-tissue DS genes are hypermethylated. The growth comparison contained DMRs mapping to ZNF837, RYR1, BCL9L, VPS37B, and RUNX1. The phase 1 comparison contained a DMR mapping to TEX14. The phase 3 comparison contained DMRs mapping to BCL9L, RFPL2, RYR1, ADAMTS10, MZF1, and RUNX1.
The DNMT3L DMRs also mapped to two clusters of genes previously known to be altered in DS and important in neurodevelopment, specifically the protocadherin and HOX genes clusters [2]. The clustered protocadherin locus is located on chromosome 5 and involved in the establishment of single-cell neuronal identity [38]. Notably, the protocadherin gamma cluster contains one of the previously known 25 pan-tissue DS genes (PCDHGA2) [2], and differential methylation within the cluster has been observed in diverse DS tissues, which include fetal brain, adult brain, neonatal blood, T-lymphocytes, buccal epithelial cells, and placenta [29–31, 33–35]. The protocadherin gamma cluster contained two hypermethylated DNMT3L DMRs and one hypomethylated DMR in the phase 1 comparison. The protocadherin beta cluster contained a hypermethylated DNMT3L DMR in the growth comparison and a different hypermethylated DMR in the phase 3 comparison. The clustered HOX genes, which are involved in early developmental patterning, contained DNMT3L DMRs in all phase comparisons. The HOXA gene cluster also contains one of the previously known 25 pan-tissue genes (HOXA4) [2], and large-scale hypermethylation of the cluster was apparent in DNMT3L-overexpressing cells at all phases of differentiation (Fig. 7d). Differential methylation of the HOX clusters has been observed in a number of tissues, which includes adult brain, fetal brain, neonatal blood, whole-blood, T-lymphocytes, placenta, buccal epithelial cells, and neural induced pluripotent stem cell (iPSC) derivatives [26, 27, 30–35]. DNMT3L DMRs mapping to HOXB7, HOXC4, HOXC5, HOXD10, and HOXD12 were present in the growth comparison, while those mapping to HOXD-AS2, HOTAIR (“HOX transcript antisense RNA” for the HOXC cluster), HOXC5, HOXC6, HOXC12, and HOXC13, and HOXD12 were present in the phase 1 comparison, and those mapping to HOTTIP (“HOXA distal transcript antisense RNA”), HOXC-AS2, HOXC-AS3, HOXC12, and HOTTAIR were present in the phase 3 comparison. The presence of DNMT3L DMRs in the HOX gene clusters is also notable given that retinoic acid is known to regulate their expression and chromatin state in mESCs [39].
While the DNMT3L DMRs overlapped with many of the 106 imprinted genes in the human genome, these results were not significantly higher than expected in enrichment analyses. The growth DMRs mapped to 18 imprinted genes: SNRPN, ZNF597, ZFAT, NTM, LIN28B, GRB10, MIMT1, KCNQ1, ADTRP, ANO1, OSBPL5, FAM50B, ATP10A, SNORD116, MIR296, GLIS3, PPP1R9A, and KCNK9. The phase 1 DMRs mapped to 19 imprinted genes: ATP10A, MKRN3, PHLDA2, RB1, MESTIT1, IGF2, DGCR6L, ZFAT, NTM, MIR296, MAGI2, ZIM2, SNORD116, KCNK9, ZDBF2, NAA60, KCNQ1, GNAS, and KCNQ1OT1. The phase 3 DMRs mapped to 22 imprinted genes: OSBPL5, TP73, MKRN3, WT1, KCNK9, NAA60, PPP1R9A, ATP10A, SNRPN, PLAGL1, GRB10, SNORD116, SLC22A3, MEG8, MEG3, ZIM2, MIMT1, GNAS, UBE3A, NDN, IGF2, and ADTRP. This lack of enrichment is consistent with the literature, as although DNMT3L is required for the establishment of imprinting, it is not required for its maintenance [14].
Finally, we examined the effect of lot-to-lot variation on DNMT3L overexpression in cell culture. This involved generating an additional set of replicate SH-SY5Y cell lines from a more recent lot of cells than the one used for the first two sets of transfection experiments. While this additional set of cell lines clustered differently from the other two and showed a stronger hypermethylation profile in DNMT3L-overexpressing cells compared to control cells, a replication analysis that included all three transfection experiments still reproduced the main the findings (Additional File 1: Supplementary Table 2 and Additional File 2: Supplementary Note and Supplementary Figures S1–S7). Together, these results demonstrate the reproducibility of the main findings of this study, which is that DNMT3L overexpression results in hypermethylation of genes differentially methylated in DS and regions of bivalent chromatin.
Discussion
The results of this study confirmed previous observations and provided novel findings that are relevant to understanding the role of DNMT3L in establishing DNA methylation profiles during development and in DS. DNMT3L overexpression in undifferentiated, differentiating, and differentiated SH-SY5Y neurons resulted in a global increase in CpG and CpG island methylation. The DMRs mapped to genes involved in neurodevelopment, cellular signaling and gene regulation. Both DNMT3L genotype and cell differentiation phase were inter-related in the consensus DMR clustering, demonstrating that different regions of the same genes are affected during development. Furthermore, these results demonstrate that DNMT3L overexpression results in differential methylation of cross-tissue and pan-tissue sites within genes identified in 15 previous DS datasets from diverse tissues, with a majority of the pan-tissue genes mapping to bivalent chromatin. However, our results also highlight a directional dichotomy in the DNA methylation changes associated with DNMT3L overexpression. The hypermethylated DNMT3L DMRs showed a cross-tissue DS profile, in contrast to the hypomethylated DMRs that showed a distinct profile related to early neurodevelopment. There were also distinct differences in the chromatin state enrichments for the hypermethylated and hypomethylated DMRs.
The hypermethylated DMRs were enriched within regions of bivalent chromatin marked by H3K4me3. Bivalent chromatin consists of regions marked with both activating H3K4me3 and repressive H3K27me3 and serves to regulate key developmental genes in ESCs by repressing them during pluripotency and poising them for rapid activation upon removal of H3K27me3 during differentiation [40, 41]. Notably, while bivalent promoters in ESCs are unmethylated or hypomethylated, upon differentiation some bivalent promoters lose H3K4me3 and gain DNA methylation [14, 42, 43]. Disruption of bivalent chromatin has been previously implicated in DS [29, 35, 44]. Since DNMT3L recognizes unmethylated H3K4 [10], a possible mechanism is that regions of bivalent chromatin lose H3K4me3 as they differentiate and become hypermethylated by the excess DNMT3L that recognizes the H3K4me0. This hypothesis is consistent with the observation that phases 1 and 3 DNMT3L DMRs show much stronger enrichments within regions marked by H3K4me3 when compared to the growth phase. This proposed mechanism is also consistent with the growth phase still showing enrichment within regions marked by H3K4me3, as the cells are already committed to the neural crest lineage. Additionally, while bivalent chromatin is generally hypomethylated in normal cells, in cancer cells, bivalent chromatin becomes hypermethylated and is associated with increased developmental gene expression (45, 46). In neural progenitors, many promoters with the H3K27me3 modification gain DNA methylation during differentiation [47].
In comparison to the hypermethylated DMRs, the hypomethylated DNMT3L DMRs in our study were enriched within regions of heterochromatin marked by H3K9me3. Notably, the observation of DNMT3L causing hypomethylation has also been previously established. Knockdown of DNMT3L in mESCs resulted in 14,107 regions showing a decrease in methylation and 5,724 showing a gain in methylation [13]. This ratio is comparable to our observations and the regions identified significantly (p = 0.0002) overlap with the genomic coordinates for the consensus DNMT3L DMRs. Mechanistically, DNMT3L is known to either release or sequester DNMT3A from heterochromatin, which has been hypothesized to enable preferential targeting of euchromatin [48]. Taken together, we hypothesize that in our study, the release or sequestering of DNMT3A from heterochromatin by excess DNMT3L resulted in the preferential targeting of bivalent chromatin within regions that recently lost H3K4me3 as the cells differentiated.
Conclusions
While DNMT3L overexpression during neuronal differentiation recapitulates an important facet of the epigenetic signature of DS, it is not the only mechanism. The other methylation differences observed in DS brain are likely related to age, medication exposure, and an increase in copy number of the other genes encoding proteins related to DNA methylation and one-carbon metabolism that are also located on chromosome 21 [2]. For example, we observed hypermethylation of the chromosome 21 RUNX1 locus in both the growth phase and phase 3 cells. Previously, we observed hypermethylation of RUNX1 as well as genome-wide hypomethylation of RUNX1-binding sites in neonatal DS when compared to non-DS control blood [35]. However, we did not observe hypomethylation of RUNX1-binding sites in DNMT3L-overexpressing cells, likely because RUNX1 was diploid in SH-SY5Y cells. The results of our study also demonstrate reproducibility in gene ontologies and chromatin states impacted by DNMT3L overexpression, despite variation inherent to individual cell lines. Overall, our findings suggest that DNMT3L overexpression during neuronal differentiation recreates a facet of the aberrant Down syndrome DNA methylation signature by targeting specific chromatin states that regulate genes important for neurodevelopment.
Methods
Plasmids and cloning
DNMT3L was cloned into an expression plasmid by Gibson assembly. First, human wild-type DNMT3L cDNA (IMAGE clone 1541874) was PCR amplified from pD3LMyc [4]. PCR amplification primers were design with the complementary overhangs necessary for Gibson assembly using NEBuilder (New England Biolabs). The sequence for the forward primer was: TGTCTCATCATTTTGGCAAAATGGAGCAGAAGCTGATCTCAGAGGAGGAC. The sequence of the reverse primer was: TCACCGCATGTTAGCAGACTTCCTCTGCCCTCGCCACCTCCGCTGCCGCCTAAAGAGGAAGTGAGTTCTGTTGAAAAATACTTG. The reverse primer was designed to include a sequence coding for a self-cleaving P2A peptide inframe of DNMT3L. A PiggyBac-compatible expression plasmid, PbCAG-eGFP, was a gift from Joseph Loturco (Addgene plasmid # 40973) [24]. PbCAG-eGFP was linearized with EcoRI. Both the linearized plasmid and the DNMT3L PCR amplicon were run on an agarose gel and purified in a silica column (Qiagen). Purified fragments were mixed at 1:3 molar ratio and assembled with NEBuilder HiFi DNA Assembly Mater Mix (New England Biolabs) according to the manufacturer’s instructions. Assembled products were then introduced into NEB Turbo E. coli competent bacteria by chemical transformation, plated on agar plates supplemented with ampicillin, and grown at 37 °C according to the manufacturer’s instructions. Colonies were then grown in LB and the plasmid was purified in spin silica columns according to manufacturer’s instructions. Resulting pb-DNMT3L-P2A-eGFP plasmids were confirmed by Sanger sequencing before proceeding to cell culture transfection experiments.
Cell culture and transfection
Low passage SH-SY5Y cells in the undifferentiated growth phase were transfected with the plasmids via Lipofectamine 3000 (Invitrogen) according to the manufacturer’s instructions. After transfection, the cells were grown to a previously established timepoint when transient expression was shown to dissipate, so that only cells with stable integration could be sorted for GFP via flow cytometry. After flow cytometry, the population of > 95% GFP-positive cells were re-cultured, expanded for ~ 5 passages, and then differentiated according to Shipley et al. [23] except that the newer B-27 plus neuronal cell culture system (Gibco) was used and the retinoic acid was dissolved in DMSO. DNA was isolated from flash frozen cell pellets using the QIAamp DNA Micro kit (Qiagen) according to the manufacturer’s instructions.
Western Blot
Protein was prepared by homogenizing cells in RIPA buffer (Alfa Aesar) and quantitated by Bicinchoninic Acid (Thermo Fisher) following the manufacturers' instructions. Ten micrograms of protein per sample were loaded on Bis–Tris gel (Thermo Fisher Scientific) and transferred to a PVDF membrane (Bio-Rad) according to the manufacturers' instructions. Immunoblotting was visualized on a LiCor Odyssey instrument. Antibodies for immunoblotting were: DNMT3L (Santa Cruz Biotechnology, Catalog # sc-393603) and GAPDH (Advanced ImmunoChemicals Inc., Catalog # 2-RGM2). Molecular weights were determined using known weight markers and reported weights.
WGBS
WGBS library preparation was performed using the post-bisulfite adaptor tagging (PBAT) method with the terminal deoxyribonucleotidyl transferase-assisted adenylate connector-mediated single-stranded-DNA ligation technique [49, 50] via the Accel-NGS Methyl-Seq DNA Library Kit (Swift Biosciences) with the Methyl-Seq Combinatorial Dual Indexing Kit (Swift Biosciences) according to the manufacturer’s instructions. The library pool was sequenced across 2 lanes on an Illumina NovaSeq 6000 S4 flow cell for 150 bp paired end reads to generate ~ 150 million unique read-pairs (~ 10X coverage) of the genome per sample.
Bioinformatic analyses
Trimming of adapters and methylation bias, screening for contaminating genomes, alignments to hg38, deduplication, calculation of coverage and insert size metrics, extraction of CpG methylation values, generation of genome-wide cytosine reports (CpG count matrix), and examination of quality control metrics were performed using CpG_Me (https://github.com/ben-laufer/CpG_Me) [35, 51–53]. DMR calling as well as most downstream analyses were performed using DMRichR, which utilizes the dmrseq and bsseq algorithms (https://github.com/ben-laufer/DMRichR) [35, 54, 55]. Linear mixed-effects models of the average methylation level were utilized for global and CpG island methylation level analyses, where genotype (DNMT3L or eGFP) and cellular differentiation phase were direct effects, while cell line of origin was a random effect. Transcription factor motif enrichment testing using HOMER [56], DS cross-tissue enrichment testing via GAT [57], as well as enrichment testing for histone post-translational modifications (5 marks, 127 epigenomes) and chromatin states (15-state chromHMM model) [58, 59] through LOLA [60], were all performed as previously described [35].
Supplementary Information
Additional file 1: Table 1. Testable background regions and significant DMRs for DNMT3L overexpression in A) growth phase, B) phase 1, and C) phase 3. Table 2. Testable background regions and significant DMRs for DNMT3L overexpression in all 3 cell line replicates for A) growth phase, B) phase 1, and C) phase 3 (XLSX 78248 KB)
Additional file 2: Supplementary note. Figures S1–S7. Additional replicate cell line set analysis (PDF 10503 KB)
Acknowledgements
Not applicable.
Abbreviations
- DS
Down syndrome
- DNMT3L
DNA methyltransferase 3L
- DMRs
Differentially methylated regions
- WGBS
Whole genome bisulfite sequencing
- ADD
ATRX–DNMT3–DNMT3L
- mESCs
Mouse embryonic stem cells
- eGFP
Enhanced green fluorescent protein
- PCA
Principal component analysis
- iPSC
Induced pluripotent stem cell
Authors’ contributions
BIL, JAG, and JML designed the study. JML acquired funding and supervised the project. JAG designed, cloned, and validated the constructs. JAG and BIL performed the transfections. BIL performed the cell culture differentiation and microscopy. JAG and JMJ performed the western blots. BIL performed the bioinformatic analyses. BIL interpreted the results and wrote the manuscript with intellectual contributions and edits from JAG and JML. All authors reviewed and approved the final manuscript.
Funding
This work was supported by a National Institutes of Health (NIH) grant [3R01ES015359-10S1] to JML and I. Hertz-Picciotto, a Canadian Institutes of Health Research (CIHR) postdoctoral fellowship [MFE-146824] to BIL, a CIHR Banting postdoctoral fellowship [BPF-162684] to BIL, a Jane Coffin Childs Memorial Fund for Medical Research fellowship to JAG, a NIH T32 Fellowship to JAG [5T32CA108459], and the UC Davis Intellectual and Developmental Disabilities Research Center (IDDRC) [P50HD103526]. The flow cytometry was carried out by the UC Davis Flow Cytometry Shared Resource Laboratory and was supported by the UC Davis Comprehensive Cancer Center Support Grant (CCSG) awarded by the National Cancer Institute [NCI P30CA093373]. The library preparation and sequencing was carried out by the DNA Technologies and Expression Analysis Cores at the UC Davis Genome Center and was supported by a NIH Shared Instrumentation Grant [1S10OD010786-01].
Availability of Data and Materials
The datasets generated and/or analyzed during the current study are available in the NCBI Gene Expression Omnibus (GEO) repository, through accession number: GSE168276.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Benjamin I. Laufer and J. Antonio Gomez contributed equally to the manuscript
References
- 1.Presson AP, Partyka G, Jensen KM, Devine OJ, Rasmussen SA, McCabe LL, McCabe ERB. Current estimate of down syndrome population prevalence in the United States. J Pediatr. 2013;163:1163–1168. doi: 10.1016/j.jpeds.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Do C, Xing Z, Yu YE, Tycko B. Trans-acting epigenetic effects of chromosomal aneuploidies: lessons from Down syndrome and mouse models. Epigenomics. 2017;9:189–207. doi: 10.2217/epi-2016-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu J, Mccarter M, Lian G, Esposito G, Capoccia E, Delli-Bovi LC, Hecht J, Sheen V. Global hypermethylation in fetal cortex of Down syndrome due to DNMT3L overexpression. Hum Mol Genet. 2016;25:1714–1727. doi: 10.1093/hmg/ddw043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chedin F, Lieber MR, Hsieh C-L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci. 2002;99:16916–16921. doi: 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Z-X, Mann JR, Hsieh C-L, Riggs AD, Chédin F. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J Cell Biochem. 2005;95:902–917. doi: 10.1002/jcb.20447. [DOI] [PubMed] [Google Scholar]
- 6.Jeltsch A, Jurkowska RZ. Allosteric control of mammalian DNA methyltransferases—a new regulatory paradigm. Nucleic Acids Res. 2016;44:8556–8575. doi: 10.1093/nar/gkw723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jurkowska RZ, Anspach N, Urbanke C, Jia D, Reinhardt R, Nellen W, Cheng X, Jeltsch A. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res. 2008;36:6656–6663. doi: 10.1093/nar/gkn747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajavelu A, Jurkowska RZ, Fritz J, Jeltsch A. Function and disruption of DNA Methyltransferase 3a cooperative DNA binding and nucleoprotein filament formation. Nucleic Acids Res. 2012;40:569–580. doi: 10.1093/nar/gkr753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stepper P, Kungulovski G, Jurkowska RZ, Chandra T, Krueger F, Reinhardt R, Reik W, Jeltsch A, Jurkowski TP. Efficient targeted DNA methylation with chimeric dCas9-Dnmt3a-Dnmt3L methyltransferase. Nucleic Acids Res. 2017;45:1703–1713. doi: 10.1093/nar/gkw1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ooi SKT, Qiu C, Bernstein E, Li K, Jia D, Yang Z, Erdjument-Bromage H, Tempst P, Lin S-P, Allis CD, et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature. 2007;448:714–717. doi: 10.1038/nature05987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otani J, Nankumo T, Arita K, Inamoto S, Ariyoshi M, Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009;10:1235–1241. doi: 10.1038/embor.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Jurkowska R, Soeroes S, Rajavelu A, Dhayalan A, Bock I, Rathert P, Brandt O, Reinhardt R, Fischle W, et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010;38:4246–4253. doi: 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F, Matarese F, Stunnenberg HG, Oliviero S. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell. 2013;155:121. doi: 10.1016/j.cell.2013.08.056. [DOI] [PubMed] [Google Scholar]
- 14.Veland N, Lu Y, Hardikar S, Gaddis S, Zeng Y, Liu B, Estecio MR, Takata Y, Lin K, Tomida MW, et al. DNMT3L facilitates DNA methylation partly by maintaining DNMT3A stability in mouse embryonic stem cells. Nucleic Acids Res. 2019;47:152–167. doi: 10.1093/nar/gky947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kao T-H, Liao H-F, Wolf D, Tai K-Y, Chuang C-Y, Lee H-S, Kuo H-C, Hata K, Zhang X, Cheng X, et al. Ectopic DNMT3L triggers assembly of a repressive complex for retroviral silencing in somatic cells. J Virol. 2014;88:10680–10695. doi: 10.1128/JVI.01176-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bourc’his, D., Xu, G. L., Lin, C. S., Bollman, B. and Bestor, T. H. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536–2539. doi: 10.1126/science.1065848. [DOI] [PubMed] [Google Scholar]
- 17.Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129:1983–1993. doi: 10.1242/dev.129.8.1983. [DOI] [PubMed] [Google Scholar]
- 18.Do JH, Kim IS, Park T-K, Choi D-K. Genome-wide examination of chromosomal aberrations in neuroblastoma SH-SY5Y cells by array-based comparative genomic hybridization. Mol Cells. 2007;24:105–112. [PubMed] [Google Scholar]
- 19.Yusuf M, Leung K, Morris KJ, Volpi EV. Comprehensive cytogenomic profile of the in vitro neuronal model SH-SY5Y. Neurogenetics. 2013;14:63–70. doi: 10.1007/s10048-012-0350-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krishna A, Biryukov M, Trefois C, Antony PMA, Hussong R, Lin J, Heinäniemi M, Glusman G, Köglsberger S, Boyd O, et al. Systems genomics evaluation of the SH-SY5Y neuroblastoma cell line as a model for Parkinson’s disease. BMC Genomics. 2014;15:1154. doi: 10.1186/1471-2164-15-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroeder DI, Lott P, Korf I, LaSalle JM. Large-scale methylation domains mark a functional subset of neuronally expressed genes. Genome Res. 2011;21:1583–1591. doi: 10.1101/gr.119131.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunaway K, Goorha S, Matelski L, Urraca N, Lein PJ, Korf I, Reiter LT, LaSalle JM. Dental pulp stem cells model early life and imprinted DNA methylation patterns. Stem Cells. 2017;35:981–988. doi: 10.1002/stem.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shipley MM, Mangold CA, Szpara ML. Differentiation of the SH-SY5Y human neuroblastoma cell line. J Vis Exp. 2016 doi: 10.3791/53193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen F, LoTurco J. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J Neurosci Methods. 2012;207:172–180. doi: 10.1016/j.jneumeth.2012.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reay WR, Cairns MJ. The role of the retinoids in schizophrenia: genomic and clinical perspectives. Mol Psychiatry. 2020;25:706–718. doi: 10.1038/s41380-019-0566-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henneman P, Bouman A, Mul A, Knegt L, Van Der Kevie-Kersemaekers A-MM, Zwaveling-Soonawala N, Meijers-Heijboer HEJJ, Paul van Trotsenburg AS, Mannens MM, van Trotsenburg ASP, et al. Widespread domain-like perturbations of DNA methylation in whole blood of Down syndrome neonates. PLoS ONE. 2018;13:e0194938. doi: 10.1371/journal.pone.0194938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bacalini MG, Gentilini D, Boattini A, Giampieri E, Pirazzini C, Giuliani C, Fontanesi E, Scurti M, Remondini D, Capri M, et al. Identification of a DNA methylation signature in blood cells from persons with down syndrome. Aging (Albany, NY) 2015;7:82–96. doi: 10.18632/aging.100715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cejas RB, Wang J, Hageman-Blair R, Liu S, Blanco JG. Comparative genome-wide DNA methylation analysis in myocardial tissue from donors with andwithout Down syndrome. Gene. 2021;764:145099. doi: 10.1016/j.gene.2020.145099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mendioroz M, Do C, Jiang X, Liu C, Darbary HK, Lang CF, Lin J, Thomas A, Abu-Amero S, Stanier P, et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 2015;16:263. doi: 10.1186/s13059-015-0827-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones MJ, Farré P, McEwen LM, MacIsaac JL, Watt K, Neumann SM, Emberly E, Cynader MS, Virji-Babul N, Kobor MS. Distinct DNA methylation patterns of cognitive impairment and trisomy 21 in down syndrome. BMC Med Genomics. 2013;6:58. doi: 10.1186/1755-8794-6-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin S, Lee YK, Lim YC, Zheng Z, Lin XM, Ng DPY, Holbrook JD, Law HY, Kwek KYC, Yeo GSH, et al. Global DNA hypermethylation in down syndrome placenta. PLoS Genet. 2013;9:e1003515. doi: 10.1371/journal.pgen.1003515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laan L, Klar J, Sobol M, Hoeber J, Shahsavani M, Kele M, Fatima A, Zakaria M, Annerén G, Falk A, et al. DNA methylation changes in down syndrome derived neural iPSCs uncover co-dysregulation of ZNF and HOX3 families of transcription factors. Clin Epigenetics. 2020;12:9. doi: 10.1186/s13148-019-0803-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.El Hajj N, Dittrich M, Böck J, Kraus TFJ, Nanda I, Müller T, Seidmann L, Tralau T, Galetzka D, Schneider E, et al. Epigenetic dysregulation in the developing Down syndrome cortex. Epigenetics. 2016;11:563–578. doi: 10.1080/15592294.2016.1192736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laufer BI, Hwang H, Vogel Ciernia A, Mordaunt CE, LaSalle JM. Whole genome bisulfite sequencing of Down syndrome brain reveals regional DNA hypermethylation and novel disorder insights. Epigenetics. 2019;14:672–684. doi: 10.1080/15592294.2019.1609867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laufer BI, Hwang H, Jianu JM, Mordaunt CE, Korf IF, Hertz-Picciotto I, LaSalle JM. Low-pass whole genome bisulfite sequencing of neonatal dried blood spots identifies a role for RUNX1 in down syndrome DNA methylation profiles. Hum Mol Genet. 2020;11(8):563. doi: 10.1093/hmg/ddaa218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Court F, Arnaud P. An annotated list of bivalent chromatin regions in human ES cells: a new tool for cancer epigenetic research. Oncotarget. 2017;8:4110–4124. doi: 10.18632/oncotarget.13746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Du Q, de la Morena MT, van Oers NSC. (2020) The genetics and epigenetics of 22q11.2 deletion syndrome. Front Genet. 2020;10:1365. doi: 10.3389/fgene.2019.01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen WV, Maniatis T. Clustered protocadherins. Development. 2013;140:3297–3302. doi: 10.1242/dev.090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Kumar B, Parrish ME, Slaughter BD, Unruh JR, Gogol M, Seidel C, Paulson A, Li H, Gaudenz K, Peak A, et al. Analysis of dynamic changes in retinoid-induced transcription and epigenetic profiles of murine Hox clusters in ES cells. Genome Res. 2015;25:1229–1243. doi: 10.1101/gr.184978.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 41.Vastenhouw NL, Schier AF. Bivalent histone modifications in early embryogenesis. Curr Opin Cell Biol. 2012;24:374–386. doi: 10.1016/j.ceb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galonska C, Charlton J, Mattei AL, Donaghey J, Clement K, Gu H, Mohammad AW, Stamenova EK, Cacchiarelli D, Klages S, et al. Genome-wide tracking of dCas9-methyltransferase footprints. Nat Commun. 2018 doi: 10.1038/s41467-017-02708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lane AA, Chapuy B, Lin CY, Tivey T, Li H, Townsend EC, Van Bodegom D, Day TA, Wu SC, Liu H, et al. Triplication of a 21q22 region contributes to B cell transformation through HMGN1 overexpression and loss of histone H3 Lys27 trimethylation. Nat Genet. 2014;46:618–623. doi: 10.1038/ng.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernhart SH, Kretzmer H, Holdt LM, Jühling F, Ammerpohl O, Bergmann AK, Northoff BH, Doose G, Siebert R, Stadler PF, et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci Rep. 2016;6:1–18. doi: 10.1038/srep37393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunican DS, Mjoseng HK, Duthie L, Flyamer IM, Bickmore WA, Meehan RR. Bivalent promoter hypermethylation in cancer is linked to the H327me3/H3K4me3 ratio in embryonic stem cells. BMC Biol. 2020;18:25. doi: 10.1186/s12915-020-0752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohn F, Weber M, Rebhan M, Roloff TC, Richter J, Stadler MB, Bibel M, Schübeler D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008;30:755–766. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 48.Jurkowska RZ, Rajavelu A, Anspach N, Urbanke C, Jankevicius G, Ragozin S, Nellen W, Jeltsch A. Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: Heterochromatic localization and role of Dnmt3L. J Biol Chem. 2011;286:24200–24207. doi: 10.1074/jbc.M111.254987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miura F, Enomoto Y, Dairiki R, Ito T. Amplification-free whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2012;40:e136. doi: 10.1093/nar/gks454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miura F, Shibata Y, Miura M, Sangatsuda Y, Hisano O, Araki H, Ito T. Highly efficient single-stranded DNA ligation technique improves low-input whole-genome bisulfite sequencing by post-bisulfite adaptor tagging. Nucleic Acids Res. 2019;47:e85. doi: 10.1093/nar/gkz435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wingett SW, Andrews S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research. 2018;7:1338. doi: 10.12688/f1000research.15931.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–1572. doi: 10.1093/bioinformatics/btr167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Korthauer K, Chakraborty S, Benjamini Y, Irizarry RA. Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics. 2018;20:367–383. doi: 10.1093/biostatistics/kxy007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansen KD, Langmead B, Irizarry RA. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 2012;13:R83. doi: 10.1186/gb-2012-13-10-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heger A, Webber C, Goodson M, Ponting CP, Lunter G. GAT: a simulation framework for testing the association of genomic intervals. Bioinformatics. 2013;29:2046–2048. doi: 10.1093/bioinformatics/btt343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ernst J, Kellis M. ChromHMM: Automating chromatin-state discovery and characterization. ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods. 2012;2012(9):215–216. doi: 10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J et al. Integrative analysis of111 reference human epigenomes. Nature 2015; 518: 317–329. [DOI] [PMC free article] [PubMed]
- 60.Sheffield NC, Bock C. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2015;32:587–589. doi: 10.1093/bioinformatics/btv612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table 1. Testable background regions and significant DMRs for DNMT3L overexpression in A) growth phase, B) phase 1, and C) phase 3. Table 2. Testable background regions and significant DMRs for DNMT3L overexpression in all 3 cell line replicates for A) growth phase, B) phase 1, and C) phase 3 (XLSX 78248 KB)
Additional file 2: Supplementary note. Figures S1–S7. Additional replicate cell line set analysis (PDF 10503 KB)
Data Availability Statement
The datasets generated and/or analyzed during the current study are available in the NCBI Gene Expression Omnibus (GEO) repository, through accession number: GSE168276.