

Abstract
Modular strategies to fabricate gels with tailorable chemical functionalities are relevant to applications spanning from biomedicine to analytical chemistry. Here, the properties of clickable poly(acrylamide-co-propargyl acrylate) (pAPA) hydrogels are modified via sequential in-gel copper-catalyzed azide-alkyne cycloaddition (CuAAC) reactions. Under optimized conditions, each in-gel CuAAC reaction proceeds with rate constants of ~0.003 s−1, ensuring uniform modifications for gels < 200 μm thick. Using the modular functionalization approach and a cleavable disulfide linker, pAPA gels were modified with benzophenone and acrylate groups. Benzophenone groups allow gel functionalization with unmodified proteins using photoactivation. Acrylate groups enabled copolymer grafting onto the gels. To release the functionalized unit, pAPA gels were treated with disulfide reducing agents, which triggered ~50 % release of immobilized protein and grafted copolymers. The molecular mass of grafted copolymers (~6.2 kDa) was estimated by monitoring the release process, expanding the tools available to characterize copolymers grafted onto hydrogels. Investigation of the efficiency of in-gel CuAAC reactions revealed limitations of the sequential modification approach, as well as guidelines to convert a pAPA gel with a single functional group into a gel with three distinct functionalities. Taken together, we see this modular framework to engineer multifunctional hydrogels as benefiting applications of hydrogels in drug delivery, tissue engineering, and separation science.
Keywords: hydrogels, stimuli-responsive materials, click chemistry, drug delivery, tissue engineering
Graphical Abstract
A modular framework to prototype multifunctional hydrogels for applications in drug delivery, tissue engineering, and separation science. Starting with clickable copolymer gels of acrylamide and propargyl acrylate (pAPA gels), new chemical functionalities were imparted via sequential in-gel click reactions. Using this flexible hydrogel engineering strategy, pAPA gels were reversibly functionalized with unmodified proteins and graft copolymers.
1. Introduction
Multifunctional hydrogels underpin advances in drug delivery,[1a] mechanobiology,[1b] and analytical chemistry,[1c] among many other areas. Polyacrylamide (pAAm) gels, in particular, are ubiquitous in separation science and biomedicine[2]. Though pAAm gels are chemically inert under mild conditions,[3] modified pAAm gels have broad utility in demanding applications. A prevailing strategy to confer new mechanical, chemical, and biological properties to pAAm gels is copolymerization with other monomers. Such pAAm copolymer gels have bolstered emerging applications in separation science,[1c] tissue engineering,[2b] bioassays,[2c] and other fields. Since functional comonomers are often produced to fulfill the needs of a specific application, the resulting pAAm copolymer gels have a narrow range of functionality.
In combination with copolymerization, the specificity and efficiency of click chemistry[4] offers an alternative approach to tailor the properties of pAAm. Soon after the inception of click chemistry,[4] many groups leveraged click reactions in polymer, hydrogel, and surface science.[5] Yet, despite the ubiquity of copper-catalyzed azide-alkyne cycloaddition (CuAAC) click reactions, examples of heterogeneous, in-gel CuAAC reactions are limited.[6] The success of combinatorial medicinal chemistry approaches, which are also based on click chemistry,[7] has fueled the growing availability of CuAAC building blocks, presenting an opportunity to engineer hydrogels in a modular fashion. As is the focus of this work, modular hydrogel engineering can accelerate the development of hydrogels with diverse biological and chemical properties without the need for de novo monomer synthesis.
Transiently modifying the biological properties of materials with immobilized proteins is important for development of biosensors[8a] and scaffolds for guided cellular behavior,[8b] among others. However, many reversible immobilization strategies rely on upstream chemical modifications to the target polypeptide,[8] and thus are not compatible with samples available only in sparing quantities. Furthermore, release methods relying on hydrogel degradation[9] are compatible with unmodified protein samples, but only when structural integrity of the gel is not critical. For compatibility with sparingly available samples such as the protein contents of individual cells,[1c] our group introduced benzophenone-modified pAAm (BPMA-pAAm) copolymer gels.[10] Yet, the robust C-C bond linking proteins to the BPMA-pAAm gel matrix can withstand many chemical and thermal treatments.[1c, 11] As a result, an alternative strategy is needed to achieve transient modification of pAAm hydrogels with unmodified protein samples, while preserving the structural integrity of the hydrogel.
Similarly, grafting copolymers to or from hydrogel materials can confer new or enhanced physical and chemical properties to the gels.[12] Grafting onto pAAm gels is relatively rare in comparison to the converse modification.[13] A possible reason is that free radical polymerization of acrylamide/bis-acrylamide to produce pAAm gels proceeds with high efficiency,[14] limiting availability of unsaturated acrylate groups for direct graft copolymerization. To address this challenge, some groups have leveraged the differential reactivity of secondary and tertiary free radicals to fabricate hydrogels with unsaturated acrylate groups.[15] Savina and colleagues circumvented the lack of unsaturated acrylate groups with a method to graft copolymers from the readily available amide groups on pAAm.[3] The primary drawback of either approach is that copolymers grafted from non-cleavable groups available on the hydrogel backbone yield permanent modifications, demanding a different approach to reversibly modify pAAm hydrogels with graft copolymers.
Here, we introduce a modular strategy to engineer the biological and chemical properties of pAAm gels based on copolymerization of AAm with a click-reactive comonomer, and sequential, in-gel CuAAC reactions. We present a generalizable framework to fabricate clickable gels comprised of AAm and propargyl acrylate comonomers (pAPA gels), and optimize in-gel CuAAC reactions to convert clickable groups on pAPA gels to new chemical functionalities. Furthermore, we leverage the modularity and versatility of our approach to impart new biological and chemical properties to pAPA gels. Namely, we functionalize pAPA gels with a cleavable linker followed by benzophenone or acrylate capping groups; the respective capping groups underlie reversible functionalization of pAPA gels with protein and graft copolymers. We evaluate the protein immobilization performance of benzophenone-functionalized pAPA gels relative to a published non-reversible analogous material,[10] and scrutinize the properties of copolymers grafted onto acrylate-functionalized pAPA gels. Importantly, we investigate the efficiency of in-gel CuAAC reactions, elucidating limitations of sequential in-gel CuAAC reactions as well as opportunities to predictably convert the single functional group in pAPA gels into multiple chemical functions on the same gel.
2. Results and Discussion
2.1. A modular approach to engineer chemical properties of pAPA gels.
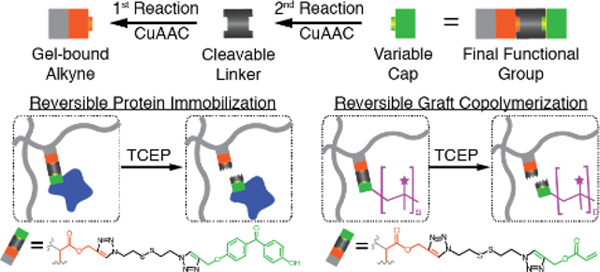
We devised a three-stage, modular approach to engineer the chemical functionality of pAAm copolymer gels. Our strategy comprises fabrication of clickable hydrogels, an initial in-gel CuAAC reaction with a bifunctional linker group, and a second in-gel CuAAC reaction with a capping group (Figure 1A). The properties of the variable linker and capping groups dictate the final chemical function of the modified clickable gel (Figure 1A). The clickable gels serve as reactive scaffolds, accelerating material prototyping and functionality testing with commercially available click chemistry building blocks. As the sequential modification approach is the most generalizable, we developed a framework to optimize functionalization of clickable gels through sequential in-gel CuAAC reactions.
Figure 1.

A modular strategy to confer new chemical properties to clickable hydrogels via sequential CuAAC reactions. (A) Schematic of sequential CuAAC reactions to introduce cleavable chemical functions to clickable poly(acrylamide-co-propargyl acrylate) (pAPA) gels. To emphasize the modularity of the sequential in-gel CuAAC reaction approach, we depict reacting species as LEGO®-like blocks where protruding ends represent pendant alkynes and indented ends represent azides. In a model system, we functionalized pAPA gels with cleavable Rhodamine110 groups through an initial in-gel CuAAC reaction with the bifunctional N3-SS-N3 linker followed by a second reaction with a Rhodamine110-alkyne capping group. (B) Representative photograph of pAPA gel regions independently exposed to Rhodamine110-N3 shows retention of the chromophore in the gel due to the in-gel CuAAC reaction. Scale bar = 6.5 mm (C) Representative UV-Vis absorbance spectra of pAPA gel regions exposed to 500 μM solutions of Rhodamine110-N3 for increasing amounts of time. (D) Reaction progress curve for the first in-gel CuAAC reaction between alkyne groups on the pAPA gels and 500 μM solutions of Rhodamine110-N3. (E) Reaction progress curve for the second in-gel CuAAC reaction, where we first exposed pAPA gels to 500 μM solutions of the N3-SS-N3 linker for 90 min followed by a second reaction with 250 μM solutions of Rhodamine110-alkyne. All error bars represent one standard deviation of the mean of 5 independent measurements per condition.
Unless noted in the text, our experimental system comprised thin hydrogel layers (~ 40 μm thick) bonded covalently to a conventional microscopy glass slide. The glass backing facilitated spectroscopic and optical measurements to evaluate retention and release of materials in the hydrogels. Furthermore, we utilized a gasket assembly to create isolated ~ 42 mm2 hydrogel regions, allowing us to selectively introduce or wash out reagents in the independent hydrogel regions via diffusion.[17] All gels in this study had a final formulation of 8 %T (0.08 g mL−1 total acrylamide per mL) with 3.3 % cross-linker (0.033 g of bis-acrylamide per g of total acrylamide), and if appropriate, complemented with comonomers (e.g., propargyl acrylate) to a final acrylamide to comonomer mol:mol ratio of 377:1 (3 mM).
In the first stage of our approach, we aimed to fabricate clickable pAAm copolymer gels. Inspired in the work of Ciftci and colleagues,[6] we copolymerized acrylamide/bis-acrylamide and propargyl acrylate, yielding cross-linked poly(acrylamide-co-propargyl acrylate) (pAPA) gels (Figure 1A). Although the alkyne group in propargyl acrylate may participate in free radical polymerization, the reactivity of alkynes towards free radicals is ~100-fold lower than that of alkenes.[6] Therefore, we expected pAPA gels to exhibit pendant, click-reactive, alkyne groups. To validate availability of clickable alkyne groups in pAPA gels, we reacted pAPA gels with Rhodamine110-azide (Rhodamine110-N3) under CuAAC reaction conditions. Retention of the dye after washing excess reagents from the gels (Figure 1B and Figure S1) confirmed reactivity towards azides. Next, we sought to optimize each in-gel CuAAC reaction to maximize conversion of alkyne groups in pAPA gels to the new chemical functions.
2.2. Optimization of in-gel CuAAC reactions.
First, we aimed to establish adequate reaction times for each in-gel CuAAC reaction. In the first reaction, we functionalize pAPA gels with the N3-SS-N3 linker to introduce a cleavable link to the hydrogel matrix through the reducible disulfide bond, as well as azide functional groups for a subsequent CuAAC reaction with alkyne-functionalized capping groups (Figure 1A). In the second reaction, we react the now azide-functionalized pAPA gels with a variable alkyne-functionalized capping group to introduce the final chemical function. To reversibly functionalize pAPA gels with protein or graft copolymers, we utilized benzophenone-alkyne or propargyl acrylate capping groups, respectively. To maximize the rate of each in-gel CuAAC reaction, we selected the highest concentrations of N3-SS-N3 and benzophenone-alkyne (the least soluble capping group) that produced solutions free of visible precipitates (see Supporting Information). Unless otherwise noted, we utilize 500 μM and 250 μM concentrations for the first and second in-gel CuAAC reactions, respectively. For purposes of in-gel CuAAC reaction optimization, we substituted the cleavable N3-SS-N3 linker with Rhodamine110-N3 (Figure 1B – 1D) and the capping groups with Rhodamine110-alkyne (Figure 1E). Since the identity of the substituent groups has little effect on the efficiency of CuAAC,[4b,16] the fluorescent proxies (i.e., Rhodamine110-N3 and Rhodamine110-alkyne) were not only representative of the linker and capping groups, but also provided a simple readout to track the progress of each reaction.
To determine adequate reaction times, we reacted pAPA gels with the corresponding fluorescent proxies and measured accumulation of Rhodamine110 in the gel as a function of time (Figure 1B – 1D). Notably, we observed fluorescence quenching upon accumulation of Rhodamine110 in the gel (Figure S2), which confounded quantitation. In contrast, UV-Vis absorbance at 504 nm increased monotonically with increasing Rhodamine110 accumulation in the pAPA gels (Figure 1B, 1C). The first reaction reached equilibrium after 30 min of exposure to the 500 μM Rhodamine110-N3 solution (Figure 1D). We followed the same approach to optimize the second in-gel CuAAC reaction. First, however, we modified pAPA gels with the N3-SS-N3 linker for 90 min to assure reaction equilibration, yielding gel-bound azide groups. Thereafter, we exposed the now azide-functionalized pAPA gels with Rhodamine110-alkyne and measured accumulation of the dye in pAPA gels as a function of reaction time (Figure 1E). The second in-gel CuAAC reaction also reached equilibrium after 30 min of exposure to the Rhodamine110-alkyne solution. Consequently, we selected a conservative 90 min reaction time for each in-gel CuAAC modification, ensuring maximal conversion of the gel-bound groups to the final chemical function in each reaction. Following the second in-gel CuAAC reaction with Rhodamine110 alkyne, pAPA gels exhibited cleavable Rhodamine110 groups (Figure 1A). To assess reversible functionalization of pAPA gels with Rhodamine110, we treated the gels with a solution of tris(2-carboxyethyl)phosphine (TCEP) for 30 min, triggering cleavage of the disulfide bond in the N3-SS-N3 linker. TCEP treatment triggered 99.1 ± 10.0% release of the Rhodamine110 capping groups from pAPA gels (Figure S3), validating operation of the release function.
Spatial uniformity can be critical for biological applications, where non-uniform distribution of physical and biological cues[8b] may drive differential responses. Mass transport limitations may produce spatial gradients of the in-gel concentrations of soluble reactants, and thus non-uniform modifications to the gel. We utilized the dimensionless Damköhler number (Da) to determine evaluate the presence and severity of mass transport limitations in our system. Da is defined as the ratio of the characteristic reaction rate to the mass transport rate. In our experimental set up, each in-gel CuAAC reaction occurred in fluidically isolated ~42 mm2 gel areas,[17] where the gel volume is submerged under the soluble reagent solution present in ~50-fold volumetric excess. Therefore, we expect the concentration of the soluble reagent to remain approximately constant, allowing reaction modeling as a pseudo-first order reaction:
| (1) |
where kobs is the characteristic reaction rate, t is time, Ao, At, and A∞ are the measured 504 nm absorbances prior to the reaction, at time t, and at equilibrium, respectively. To extract kobs,[18] we substituted corresponding values for Ao, At, and A∞ (Figure 1D – 1E) and fit a linear model. As kobs (2.7 × 10−3 s−1) was highest for the second reaction (i.e., azide-functionalized pAPA gel with soluble Rhodamine110-alkyne), we utilize this value as a conservative estimate for dimensionless analysis. To calculate Da, we take the ratio of kobs to the inverse of the characteristic diffusion time of small molecules in a 40 μm thick hydrogel with a conservative diffusivity estimate of 10−6 cm2 s−1 (), which yields Da of 0.04. As Da << 1, we conclude in-gel CuAAC reactions in our system proceed in a reaction limited regime, and thus negligibly affected by mass transport limitations. Dimensionless analysis can be extended to anticipate the effects of geometric changes or changes to the transport properties, such as size-exclusion from the gel.[19] For example, under the same reaction conditions, we expect mass transport limitations to become non-negligible as the characteristic diffusion length approaches 200 μm, when Da ~ 1. Similarly, size-exclusion effects may arise from changes to the hydrogel formulation (e.g., higher cross-linker density) or changes to the soluble species introduced (e.g., clickable macromolecules). Importantly, kobs values in our system are comparable to those reported for uncatalyzed azide-alkyne cycloadditions on a alkyne-functionalized surface, but an order of magnitude slower than CuAAC reactions on the same system.[18] We attribute the lower CuAAC reaction rates in our system to the lack of convective transport inside the gel matrix, which replenishes depleted reagents at surfaces. Importantly, fluorescent proxies used for in-gel CuAAC reaction optimization may be combined with confocal microscopy to directly evaluate the spatial reaction uniformity, complementing surface (e.g., FTIR[20a]) and bulk (e.g., magic spinning angle NMR[20b]) chemical analysis tools.
2.3. Reversible functionalization of pAPA gels with unmodified protein.
Next, we sought to develop a reversible protein immobilization strategy compatible with unmodified (e.g., native) proteins. Benzophenone reacts readily with unmodified proteins upon UV irradiation, which produces C-C bonds between benzophenone and the unmodified protein backbone through hydrogen abstraction and free radical recombination.[21] UV actuation of benzophenone, however, poses a constraint on the release mechanism as common photoactivated release methods also require UV illumination.[8b,22] Chemically cleavable linkers, such as N3-SS-N3, can decouple the protein immobilization and release processes. Taken together, we aimed to introduce reversible, photoactivated immobilization of unmodified proteins in pAPA gels through sequential in-gel CuAAC reactions: first with the cleavable N3-SS-N3 linker and second with a benzophenone-alkyne capping group (Figure 2A). Overall, we expected benzophenone-functionalized pAPA gels to retain protein upon UV exposure, and release the captured protein in response to disulfide reducing agents (Figure 2B).
Figure 2.

Functionalization of pAPA gels with cleavable benzophenone capping groups confers reversible protein immobilization. (A) Schematic of sequential CuAAC reactions with the chemically cleavable N3-SS-N3 linker first and benzophenone capping groups second. (B) UV irradiation triggers benzophenone to react covalently with unmodified proteins; disulfide reducing agents (e.g., TCEP) trigger release of immobilized protein from the gel. (C) We introduced fluorescently labeled protein into gels of the indicated type, exposed to UV light, and measured protein fluorescence signal remaining in the gel after an overnight wash (post-immobilization, blue). To evaluate release of immobilized protein, we washed the gels in a 10 mM solution of TCEP for 30 min at room temperature and measured protein fluorescence signal remaining in the gels (post-release, gray). Representative fluorescence micrographs show benzophenone-modified pAPA gels after protein immobilization and after triggering release (scale bars: 1000 μm). (D) Time-resolved protein fluorescence measurements of the release process in a solution of 10 mM solution of TCEP (TCEP(+), circle) relative to a negative control pAPA gel placed in borate buffer (TCEP(−), triangle). We normalized protein fluorescence measurements to the initial reading (t = 0 s), and fit an exponential decay function (Fit, black line) to the normalized data. Representative fluorescence micrographs show temporal progression of the release process (scale bars: 500 μm). All error bars represent one standard deviation of the mean of 6 and 4 independent measurements per condition for panels C and D, respectively.
First, we investigated benzophenone-mediated protein immobilization in correspondingly modified pAPA gels. We incubated benzophenone-functionalized pAPA gels with solutions of fluorescently-labelled trypsin inhibitor (21 kDa), exposed the gel and protein solution to UV light, and measured retention of protein fluorescence in the gels after washing unbound protein (Figure 2C). To control for non-specific protein retention in the hydrogels due to phenomena such as entropic trapping,[23] we performed the same procedure on pAAm gels (i.e., no benzophenone). Similarly, we benchmarked protein immobilization performance in benzophenone-functionalized pAPA gels against a published non-cleavable benzophenone-methacrylamide and AAm copolymer gel (BPMA-pAAm).[10] Relative to pAAm negative control gels, benzophenone-modified pAPA gels retained 9.0-fold more protein (p < 0.05 ANOVA with Tukey’s Test). Similarly, the BPMA-pAAm positive control gels retained 141.3-fold more protein than pAAm gels (p < 0.05, ANOVA with Tukey’s Test). As immobilization conditions (e.g., protein preparation, illumination source, time and power) remained the same for all gel types, the 15.7-fold difference in protein retention between benzophenone-modified pAPA gels and BPMA-pAAm gels suggests a difference in the concentration of benzophenone groups between the two gel types (Figure S4). We investigate the root cause of this discrepancy in the last section of this manuscript. Nevertheless, functionalization of pAPA gels with benzophenone capping groups introduced the desired UV-activated protein immobilization functionality, and thus we scrutinized the release function next.
We hypothesized that cleavage of the N3-SS-N3 linker would trigger release of immobilized protein only in benzophenone-functionalized pAPA gels. In BPMA-pAAM control gels, the immobilized protein is covalently linked to the benzophenone (BPMA) group copolymerized in pAAm gel. By contrast, the disulfide bond on the N3-SS-N3 linker group is subject to cleavage upon treatment with reducing agents (Figure 2B). To assess chemically triggered release of protein immobilized in benzophenone-containing gels, we incubated the gels in a TCEP solution for 30 min at room temperature, and measured protein fluorescence after washing away unbound protein (Figure 2C). Consistent with the chemical properties of and BPMA-pAAm and benzophenone-functionalized pAPA gels, TCEP treatment produced a significant 59.8 ± 29.4% loss (p < 0.05, t-test) of protein fluorescence signal only in the benzophenone-functionalized pAPA gels (Figure 2C). As is relevant for applications in drug delivery and tissue engineering,[24] biological reducing agents (i.e., cysteine, glutathione) also triggered comparable levels of protein release (Figure S5).
Reducing agents cleave disulfide bonds with characteristic times of < 20 s at room temperature;[25] therefore, we hypothesized that protein diffusion would dictate the rate of release from pAPA gels. To evaluate this hypothesis, we measured the change in protein fluorescence signal as it washed away from pAPA gels submerged in a TCEP solution (Figure 2D). At each time point, we normalized the protein fluorescence signal to the protein signal immediately after application of the TCEP treatment (t = 0 s) and modeled diffusive transport out of the gel with an exponential decay function () (Figure 2D). τ is the characteristic diffusion time (), and thus indicates whether transport out of the gel is consistent with expected diffusion timescales. Accounting only for the effect of pore size on protein diffusivity, we expected τprotein of 1520 s out of a 250 μm thick, 8 %T pAPA gel for trypsin inhibitor (~21 kDa).[26] Notably, we required thicker gels (250 μm vs 40 μm) to measure protein release kinetics due to the dynamic range of our imaging system. Empirically, we measured τprotein of 1390.8 s for immobilized trypsin inhibitor (Figure 2D). Over the course of the release experiment, photobleaching accounted for 7.5 ± 3 % of signal loss, indicating that cleavage of the N3-SS-N3 linker drives protein signal loss from the pAPA gels. As the measured and theoretical τprotein values are comparable (within 9 %), we conclude that protein diffusivity limits the rate of release from benzophenone-functionalized pAPA gels. Moreover, large deviations from the theoretical τprotein could point to changes in gel hydrophobicity, charge, or average porosity.[27] The results indicate that modification of pAPA gels via sequential in-gel CuAAC reactions has negligible effects on the underlying hydrogel properties. Furthermore, we observed gradual elution of immobilized protein from pAPA gels over the course of a 12-day wash (Figure S6). Therefore, as is relevant for controlled release applications,[24] release of protein immobilized in benzophenone-functionalized pAPA gels follows two independent regimes: burst release upon cleavage of the N3-SS-N3 linker, and slow release during extended wash conditions.
Incomplete protein release is not necessarily unexpected. Near complete release of protein from o-nitrobenzyl-modified PEG hydrogels was only achieved consistently at high UV illumination powers.[8b] Similarly, the release efficiency of nucleic acids from psoralen-modified pAAm gels ranged from ~64 – 82 % with dependencies on molecular mass and UV illumination time.[28a] Critically, our results compare favorably with chemically activated release from pAAm hydrogels functionalized with N-succinimidyl ester groups through a disulfide linker.[28b] Under comparable release conditions, reducing agents triggered > 90 % release of small ligands but release of whole proteins ranged from 49 – 62 %.[28b] Incomplete release of protein immobilized in benzophenone-functionalized pAPA gels may also arise from UV activated cleavage of the N3-SS-N3 linker.[29] Thiyl radicals formed under UV illumination can provide permanent anchoring sites. As discussed, next, we also observed incomplete release of copolymers grafted onto pAPA gels, suggesting that a UV activated mechanism for incomplete release may be dominant.
2.4. Reversible functionalization of pAPA gels with graft copolymers.
Next, we aimed to transiently alter the chemical properties of pAPA gels via reversible grafting of copolymers. To enable this capability, we sought to introduce cleavable acrylate groups to pAPA gels independently from the gel fabrication process. To achieve this goal, we modified pAPA gels with the cleavable N3-SS-N3 linker first and propargyl acrylate as the capping group via sequential CuAAC reactions (Figure 3A). We hypothesized that acrylate capping groups on pAPA gels would participate in free radical polymerization,[30] offering grafting sites for polymer chains growing in the gel (Figure 3B). In contrast to benzophenone-mediated protein immobilization (Figure 2B), the grafting-from copolymerization process is not dependent on UV illumination, but rather on the presence of a free radical initiator. Overall, in the presence of a monomer and a photoinitiator, we expect incorporation of graft copolymers onto acrylate-functionalized pAPA gels. Similarly, we expect disulfide reducing agents to trigger release of the graft copolymers from the gel (Figure 3B).
Figure 3.

Functionalization of pAPA gels with cleavable acrylate capping groups confers reversible functionalization with graft copolymers. (A) Schematic of sequential CuAAC reactions with the cleavable N3-SS-N3 linker first and the acrylate capping groups second. (B) Under UV-initiated free radical polymerization conditions, acrylate groups are incorporated into polymer chains growing in the gel. Treating acrylate-functionalized pAPA gels with disulfide reducing agents (e.g., TCEP) triggers release of grafted copolymers from the gel. (C) We introduced methacrylated RhodamineB (RhoB) and a photoinitiator into gels of the indicated type, exposed to UV light, and measured RhoB fluorescence signal remaining in the gel after an overnight wash (post-graft, magenta). To evaluate release, we washed the gels in a 10 mM solution of TCEP for 30 min at room temperature and measured RhoB fluorescence signal remaining in the gels (post-release, gray). Representative fluorescence micrographs show pAPA gels before and after triggering release of grafted RhoB copolymers (scale bars: 1000 μm). (D) Time-resolved RhoB fluorescence measurements during release from acrylate-functionalized pAPA gels placed in a solution of 10 mM solution of TCEP (TCEP(+), circle) relative to a negative control pAPA gel placed in borate buffer (TCEP(−), triangle). We normalized RhoB fluorescence measurements to the initial reading (t = 0 s) and fit an exponential decay function (Fit, black line) to the normalized release data. Representative fluorescence micrographs show temporal progression of the release process (scale bars: 500 μm). All error bars represent one standard deviation of the mean of 6 and 8 independent measurements per condition for panels C and D, respectively.
First, we evaluated copolymer grafting from acrylate capping groups in correspondingly functionalized pAPA gels. We incubated acrylate-functionalized pAPA gels with solutions of methacrylated Rhodamine B (RhoB, the soluble monomer) and a photoinitiator, exposed the gel and monomer solution to UV light, and measured retention of RhoB fluorescence signal in the gels after an overnight wash (Figure 3C). Simultaneously, we tested pAAm gels to control for polymer entrapment in the hydrogel[31] as well as grafting from sites other than acrylate groups. Acrylate-functionalized pAPA gels retained > 10-fold more RhoB fluorescence signal than pAAm hydrogels. Moreover, we observed minimal adsorption[32] of unpolymerized RhoB monomer on the control pAPA and pAAm gels incubated with monomer solutions not containing photoinitiator (Figure S7), indicating that the photoinitiated reaction drives RhoB retention in pAPA gels. As expected, a 30 min treatment with a TCEP solution produced 68.0 ± 4.6 % reduction in RhoB fluorescence signal only in the acrylate-functionalized pAPA gels (p < 0.05, t-test, Figure 3C). Therefore, we conclude that unsaturated acrylate groups drive retention of RhoB signal in acrylate-functionalized pAPA gels, and cleavage of the N3-SS-N3 linker triggers loss of RhoB fluorescence signal from pAPA gels. To assess whether the RhoB fluorescence signal observed in acrylate-functionalized pAPA gels stemmed from a polymeric form of RhoB, we examine the kinetics of the release process next.
As discussed above for protein, the in-gel diffusivity (Dgel) of the solute limits the rate of release from pAPA gels. Therefore, we hypothesized that polymeric forms of RhoB would diffuse out of the gel with characteristic diffusion times in excess of the estimated τ of the unpolymerized RhoB monomer (τRhoB). Assuming a conservative diffusivity value of 10−6 cm2 s−1,[33] we estimated τRhoB of ~9 s out of a 40 μm thick pAPA gel. To measure τ of RhoB species in the acrylate-functionalized pAPA gels, we measured the change in RhoB fluorescence signal as it washed away from pAPA gels submerged in a TCEP solution (Figure 3D). At each time point, we normalized the RhoB fluorescence signal to the RhoB signal immediately after application of the TCEP treatment (t = 0 s). To estimate τ, we fit an exponential decay function to the normalized data (Figure 3D). Consistent with our hypothesis, the measured τ (253.4 s) exceeded τRhoB (τ for unpolymerized RhoB) by 28-fold. Therefore, we attribute the fluorescence RhoB signal in acrylate-functionalized pAPA gels to a polymerized form of RhoB. Importantly, photobleaching of RhoB accounted for 0.9 ± 1.4 % of signal loss during the first 30 mins of the time course experiment (Figure 3D), confirming that release of grafted RhoB copolymers dominates loss of RhoB fluorescence signal from pAPA gels.
The measured τg-RhoB (τ of grafted RhoB) offered an alternative method to estimate the molecular mass of RhoB copolymer grafted onto pAPA gels based on the diffusivity of the released graft copolymer. The solution diffusivity of the graft copolymer Dsol is related to Dgel by the empirical model proposed by Park et al. for diffusion of macromolecules in hydrogels[19,26b]:
| (2) |
where Φ is the polymer volume fraction of the gel matrix. Φ can be expressed as ,[26c] where T is the concentration of monomer in the gel precursor (0.08 g mL−1 in this report). We arrived at this expression for Φ based on the linear relationship betweenΦ and T presented previously by Baselga and colleagues.[26c] Rearranging the diffusion equation to solve for Dgel (3.16 × 10−8 cm2 s−1) and substituting into the model we obtain Dsol of 5.97 × 10−7 cm2 s-1. Interpolating from Dsol values reported for linear PEG polymers,[36a] we estimate the grafted RhoB copolymer released from pAPA gels had an average molecular mass of ~6.2 kDa. Estimating molecular mass relative to the properties of standards of known molecular weight is analogous to molecular mass determination with accepted methods such as GPC. Moreover, collecting graft copolymers released from pAPA gels for GPC[36b] analysis could provide insight into the distribution of molecular mass, as well as analysis of non-fluorescent graft copolymers. Taken together, on-demand release from a gel enables molecular mass analysis of grafted copolymers, expanding the suite of tools available to characterize copolymers grafted onto hydrogels.
Overall, we demonstrate that functionalization of pAPA gels with unsaturated acrylate groups through a chemically cleavable linker produces hydrogels that can be reversibly functionalized with graft copolymers. More importantly, functionalizing hydrogels with unsaturated acrylates enables thiol-ene and Michael addition reactions,[9b,37] expanding the range of transient chemical functions that may be introduced to pAPA gels via sequential in-gel CuAAC reactions. Next, we discuss the limitations and opportunities of imparting new properties to pAPA gels through sequential in-gel CuAAC reactions.
2.5. Evaluating the efficiency of in-gel CuAAC reactions.
We observed lower protein immobilization performance of benzophenone-functionalized pAPA gels relative to BPMA-pAAm gels, suggesting a difference in the availability of benzophenone groups between the two materials. To achieve equivalent benzophenone concentrations between the two types of materials we made two assumptions: comparable availability of alkyne groups in pAPA gels as that of benzophenone groups in BPMA-pAAm gels and high conversion efficiency of alkyne groups in pAPA gels to benzophenone groups through the sequential CuAAC reactions. We deemed the first assumption as accurate due to equivalent comonomer concentrations (3 mM propargyl acrylate or BPMA) in the gel precursor formulations and the reactivity of the corresponding comonomer type towards acrylamide (Figure S8).[38] Consequently, we turned our attention to the second assumption and assessed the efficiency of sequential in-gel CuAAC reactions.
The overall efficiency in our system is the product of the efficiencies of each sequential in-gel CuAAC reaction. Specifically, for benzophenone-functionalized pAPA gels, the efficiency of the first reaction is the fraction of gel-bound alkyne groups converted to azide groups after modification with the bifunctional N3-SS-N3 linker (Figure 2A). Similarly, the efficiency of the second reaction is the fraction of the gel-bound azide groups converted to benzophenone groups during the second CuAAC reaction with the benzophenone-alkyne capping group (Figure 2A). However, the homobifunctional N3-SS-N3 linker can react with two neighboring gel-bound alkyne groups to produce unintended chemical cross-links. Gel-bound alkyne groups consumed in cross-linking reactions are not converted to gel-bound azides, reducing overall efficiency of the system. Therefore, we hypothesized that cross-linking reactions during the first in-gel CuAAC reaction with the N3-SS-N3 linker artificially limited the benzophenone groups available in benzophenone-functionalized pAPA gels. N3-SS-N3 mediated cross-links can increase the cross-linking density of the gel. Yet, based on the gel formulation, N3-SS-N3 mediated cross-links can only yield < 0.1 % change in the overall gel cross-linking density, which is beyond the resolution of mechanical characterization methods.[39] Alternatively, we hypothesized that we could detect formation of N3-SS-N3 mediated cross-links as increased consumption of gel-bound alkyne groups. To test this hypothesis, we sought to develop a method to measure and predict consumption of gel-bound alkynes on pAPA gels.
During CuAAC reactions, multiple intermediates form reversibly prior to the irreversible formation of the final triazole product.[40] As the intermediates can exist in equilibrium, the final concentration of the triazole product must increase in proportion to the equilibrium concentration of the intermediates. In turn, the latter depends on the concentration of the soluble azide reagent. Therefore, we predicted that at low concentrations of soluble azides, only a fraction of gel-bound alkynes would be consumed in the in-gel CuAAC reaction, leaving unreacted groups available for another reaction. To evaluate this hypothesis, we reacted pAPA gels with solutions of azide-functionalized Cy5 (Cy5-N3) and measured retention of the dye in the gel after washing unbound species. Exposing pAPA gels to a non-saturating concentration (125 μM) of Cy5-N3 produced an initial change in the UV-Vis absorption at 654 nm of pAPA gels (ΔAbs 1st Exposure, Figure 4A) proportional to the consumption of gel-bound alkynes. As unreacted gel-bound alkyne groups remain available for subsequent CuAAC reactions, exposing the same pAPA gel to a fresh 500 μM Cy5-N3 solution produced an additional increase in the 654 nm absorbance (ΔAbs 2nd Exposure, Figure 4A) proportional to the quantity of available (unreacted) alkynes in the gel. To establish reaction equilibration times at non-saturating (< 500 μM) concentrations of Cy5-N3, we reacted pAPA gels with 125 and 500 μM solutions of Cy5-N3 and measured accumulation of Cy5 in the gel as a function of time (Figure 4B). CuAAC reactions between pAPA gels and Cy5-N3 solutions at 125 and 500 μM reached equilibrium within 30 min. Therefore, we selected a conservative 90 min reaction time for all remaining in-gel CuAAC reactions. Next, we aimed to develop a predictive model to estimate consumption and availability of gel-bound alkyne groups on pAPA gels.
Figure 4.

N3-SS-N3 mediated cross-links detected as increased consumption of gel-bound alkyne groups on pAPA gels. (A) Representative UV-Vis absorbance spectrum of a pAPA gel region after an in-gel CuAAC reaction with a 125 μM (grey) solution of Cy5-N3, producing an initial change in the 654 nm absorbance of the gel (ΔAbs 1st Exposure). A second in-gel CuAAC reaction with a 500 μM solution of Cy5-N3 (black) produced an additional increase in 654 nm absorbance (ΔAbs 2nd Exposure) proportional to the availability of gel-bound alkyne groups remaining after the first reaction. Corresponding schematics show partial and full consumption of gel-bound alkynes after each reaction. (B) Reaction progress curves for in-gel CuAAC reactions with 125 μM (black circle) and 500 μM (open triangle) solutions of Cy5-N3. (C) Retention of Cy5 dye in pAPA gels reacted with various concentrations of Cy5-N3 measured as ΔAbs 1st Exposure (black circles). Relative to ΔAbs 1st Exposure, ΔAbs 2nd Exposure (open triangle) is the net increase in Cy5 retention after a second in-gel CuAAC reaction at 500 μM Cy5-N3. (D-i) ΔAbs 1st Exposure of a control in-gel CuAAC reaction between pAPA gels and a 500 μM Cy5-N3 solution. (D-ii) ΔAbs 1st Exposure of a control in-gel CuAAC reaction between pAPA gels and a 125 μM Cy5-N3 solution. (D-iii) ΔAbs 2nd Exposure of pAPA gels initially reacted with a 62.5 μM solution of the bifunctional N3-SS-N3 linker followed by a second in-gel CuAAC reaction with a 500 μM solution of Cy5-N3. Corresponding schematics depict complete (D-i) and partial (D-ii) consumption of gel-bound alkynes. We detected N3-SS-N3 mediated cross-links (D-iii, left) as reduced availability of gel-bound alkynes for a second reaction with 500 μM Cy5-N3 relative to the 125 μM Cy5-N3 condition. All error bars represent one standard deviation of the mean of 4 independent measurements per condition, except for the 125 μM data on panel B, which contains 3 independent measurements.
We required a model to predict consumption and availability of gel-bound alkynes to estimate the relative increase in gel-bound alkyne consumption due to N3-SS-N3 cross-links. To determine a suitable concentration regime for prediction, we exposed pAPA gels to increasing concentrations of Cy5-N3 and measured retention of dye in the gel via UV-Vis absorbance (ΔAbs 1st Exposure, Figure 4C). We observed a monotonic increase in retention of dye as function of azide concentration. Furthermore, exposing the same pAPA gels to fresh 500 μM solutions of Cy5-N3 produced an additional change in 654 nm absorbance (ΔAbs 2nd Exposure), which was inversely proportional to ΔAbs 1st Exposure. These results provided insight in two important ways. First, the negligible magnitude of ΔAbs 2nd Exposure for gels initially treated with 500 μM solutions of Cy5-N3 indicates that concentrations ≥ 500 μM yield nearly complete conversion of alkyne groups on pAPA gels (Figure 4C). For this reason, we refer to soluble azide concentrations ≥ 500 μM as saturating. Second, consistent with the stoichiometry of CuAAC reactions, the consumption of alkyne groups on pAPA gels, measured as ΔAbs 1st Exposure, followed a linear trend proportional the concentration of Cy5-N3 at concentrations < 200 μM (Figure 4C). The first power proportionality breaks as the concentration of soluble azides approaches the saturating limit likely due to depletion of the reactants. Based on this rationale, we normalized the ΔAbs 1st Exposure data to the 500 μM condition and fit a linear model up to the 175 μM Cy5-N3 condition (y = 0.0040x + 0.0042, R2 = 0.95, Figure S9). Next, we utilize the model to scrutinize N3-SS-N3 mediated cross-links.
Relative to an equivalent reaction with the monofunctional Cy5-N3, we hypothesized that reactions with N3-SS-N3 would consume a larger fraction of gel-bound alkynes due to cross-linking. Importantly, our predictive linear model is relative to the saturating concentration condition (500 μM Cy5-N3). Therefore, all percent values presented below are relative to control reactions conducted at 500 μM Cy5-N3. The model predicted that exposing the pAPA gels to a 125 μM azide solution would result in 50.7 ± 5.9 % consumption of gel-bound alkyne groups. Experimentally, ΔAbs 1st Exposure in the gel regions exposed to 125 μM Cy5-N3 reached 56.7 ± 5.3 % of that observed in the 500 μM Cy5-N3 control gels, validating the model (Figure 4D–i – 4D–ii). By extension, in the absence of possible cross-linking reactions we expect ΔAbs 2nd Exposure to reach 43.3 ± 5.3 % of ΔAbs 1st Exposure for the 500 μM control condition. To study formation of N3-SS-N3 mediated cross-links, we measured ΔAbs 2nd Exposure on pAPA gels after an initial exposure to a 62.5 μM solution of the bifunctional N3-SS-N3 (Figure 4D–iii). Note that in order to reach an equivalent total azide concentration of 125 μM, the concentration of the bifunctional N3-SS-N3 linker must be reduced by half. After exposing the same gels to a fresh 500 μM solution of Cy5-N3, ΔAbs 2nd Exposure reached 19.2 ± 2.3 % of the control 500 μM Cy5-N3 condition (Figure 4D–iii). Consequently, we conclude that the first reaction with the bifunctional N3-SS-N3 linker consumed 24.1 ± 5.8 % more gel-bound alkyne groups than what would be expected with the monofunctional Cy5-N3. The increased consumption of gel-bound alkyne groups indicates that the N3-SS-N3 linker participates in cross-linking reactions between neighboring gel-bound alkynes.
Despite evidence of N3-SS-N3 mediated cross-links, the sequential in-gel CuAAC reaction approach still produced pAPA gels with the desired functionalities (i.e., protein immobilization and copolymer grafting). In this report, we detected the deleterious effects of N3-SS-N3 mediated cross-links as lower protein immobilization performance of benzophenone-modified pAPA gels relative to that the BPMA-pAAm controls. However, at higher alkyne contents, N3-SS-N3 mediated cross-links may yield appreciable changes to the mechanical properties and porous microstructure of the gel.[39] Overall, we conclude that the sequential in-gel CuAAC reaction approach is best suited to rapidly prototype and test functionality of modified pAPA gels. Optimal performance, however, may require upstream preparation of the final functional group to introduce the final chemical function to pAPA gels in a single in-gel CuAAC reaction. Alternatively, heterobifunctional linkers may circumvent cross-link formation[41] while preserving compatibility with the sequential modification approach.
Importantly, the results of the root cause investigation above point to an unexpected advantage of functionalizing pAPA gels with non-saturating concentrations of soluble azides. Namely, since consumption of gel-bound alkynes can be predictably tuned through adjustments to the concentration of the soluble azide reagent, we can fabricate pAPA gels with multiple combinations of chemical functions through sequential in-gel CuAAC reactions. To demonstrate such capability, we combined the reversible grafting-from copolymerization functionality with an irreversible modification with Cy5-N3 (Figure S10), effectively converting the single alkyne functional group in pAPA gels into three distinct chemical functionalities.
3. Conclusion
To accelerate development of multifunctional hydrogels through prototyping, we devised a versatile, modular strategy to engineer the chemical properties of pAAm copolymer gels. Our generalizable approach employs sequential, in-gel CuAAC reactions to impart new chemical functionalities to clickable copolymer hydrogels comprised of acrylamide/bis-acrylamide and propargyl acrylate (pAPA gels). Under optimized conditions, each in-gel CuAAC reaction proceeded in the reaction-limited regime (Da << 1) ensuring uniform reaction rates throughout the gel slabs < 200 μm in characteristic diffusion length. Leveraging this modular approach, we functionalized pAPA gels with benzophenone and acrylate functional groups through a cleavable linker. Benzophenone-functionalized pAPA gels retained > 9-fold more unmodified protein than unmodified pAAm hydrogels. Similarly, acrylate groups on correspondingly modified pAPA gels provided grafting sites for polymer chains growing in the pAPA gel matrix. We achieved on-demand release of ~50 % of immobilized protein and graft copolymers upon treating the gels with solutions of disulfide reducing agents with diffusion-limited rates of release. In-depth characterization of the efficiency of in-gel CuAAC reactions elucidated limitations of the sequential pAPA gel modification approach, as well as guidelines to predictably control the efficiency of in-gel CuAAC reactions. The latter allowed us to convert the single alkyne functional group in pAPA gels into three different chemical functionalities. Taken together, we present a straightforward framework to engineer the biological and chemical properties of hydrogel materials with broad applicability in biomedicine and analytical chemistry.
4. Experimental Section
Only essential experimental details of pAPA gel fabrication and in-gel CuAAC reactions are provided here; refer to the Supplementary Information document for complete experimental details.
pAPA Gel Fabrication:
8 %T, 3.3 %C acrylamide/bis-acrylamide gel precursor solutions were complemented with 3 mM propargyl acrylate and chemically polymerized with 0.08 % ammonium persulfate (w/v) and 0.08 % N,N,N′,N′-tetramethylethylenediamine (v/v). Gels were prepared between microfabricated SU-8 molds (~40 μm or ~250 μm high features) and methacrylate-functionalized microscopy glass slides, resulting in gels grafted onto the glass surface.
In-gel CuAAC Reactions:
CuAAC reaction solutions were prepared with the indicated concentrations of soluble azide or alkyne reagents. Each solution was prepared with 125 μM copper (II) sulfate (CuSO4), 625 μM Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), and 2500 μM sodium ascorbate (1:5:20 ratio) in 1X phosphate buffered saline (PBS). Critically, CuSO4 and THPTA should be mixed prior to mixing with PBS to prevent formation of precipitates. After preparation, the CuAAC reaction solution was delivered over the gel for the designated period of time (usually 90 min) on a table top shaker followed by a wash in fresh PBS with multiple buffer exchanges.
Supplementary Material
Acknowledgements
The authors are grateful for insightful discussions with Drs. J. Vlassakis, Y. Zhang, and other Herr lab members, as well as Profs. Z. Gartner (UCSF) and M. Landry (UC Berkeley). This work was supported by National Institutes of Health NIH training grant (T32GM008155), National Cancer Institute Innovative Molecular Analysis Technologies grant (R21CA193679, A.E.H.), National Cancer Institute R01CA203018 (A.E.H.), Ford Foundation Pre-Doctoral Fellowship (H.D.N.), UC Berkeley College of Engineering Fellowship (H.D.N.), NSF Graduate Research Fellowship Program (DGE 1106400, S.J.), and NIH Stem Cell Engineering Training Program (T32 GM098218, S.J.).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Hector D. Neira, Department of Bioengineering, University of California Berkeley Berkeley, CA 94720 (USA) The UC Berkeley/UCSF Graduate Program in Bioengineering Berkeley, CA 94720 (USA).
Shaheen Jeeawoody, Department of Bioengineering, University of California Berkeley Berkeley, CA 94720 (USA) The UC Berkeley/UCSF Graduate Program in Bioengineering Berkeley, CA 94720 (USA).
Amy E. Herr, Department of Bioengineering, University of California Berkeley Berkeley, CA 94720 (USA) The UC Berkeley/UCSF Graduate Program in Bioengineering Berkeley, CA 94720 (USA).
References
- [1].a) Gupta MK, Martin JR, Dollinger BR, Hattaway ME, Duvall CL, Adv. Funct. Mater. 2017, 27, 1704107; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kloxin AM, Tibbitt MW, Kasko AM, Fairbairn JA, Anseth KS, Adv. Mater. 2010, 22, 61; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Duncombe TA, Kang CC, Maity S, Ward TM, Pegram MD, Murthy N, Herr AE, Adv. Mater. 2016, 28, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Issaq HJ, Veenstra TD, Biotechniques 2008, 44, 697; [DOI] [PubMed] [Google Scholar]; b) Han L, Xu J, Lu X, Gan D, Wang Z, Wang K, Zhang H, Yuan H, Weng J, J. Mater. Chem. B 2017, 5, 731; [DOI] [PubMed] [Google Scholar]; c) Frey MT, Wang YL, Soft Matter 2009, 5, 1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Savina IN, Mattiasson B, Galaev IY, Polymer, 2005, 46, 9596. [Google Scholar]
- [4].a) Tornøe CW, Christensen C, Meldal M, J. Org. Chem. 2002, 67, 3057; [DOI] [PubMed] [Google Scholar]; b) Rostovtsev V, Green L, Fokin V, Sharpless K, Angew. Chem. Int. Ed. 2002, 41, 2596; Angew. Chemie 2002, 114, 2708; [DOI] [PubMed] [Google Scholar]; c) Kolb HC, Finn MG, Sharpless KB, Angew. Chem. Int. Ed. 2001, 40, 2004; Angew. Chemie 2001, 113, 2056. [DOI] [PubMed] [Google Scholar]
- [5].a) Koyama Y, Yonekawa M, Takata T, Chem. Lett. 2008, 37, 918; [Google Scholar]; b) Malkoch M, Vestberg R, Malkoch N, Vestberg R, Gupta N, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsburye K, Hawker CJ, Chem. Commun. 2006, 26, 2774; [DOI] [PubMed] [Google Scholar]; c) Krouit M, Bras J, Belgacem MN, Eur. Polym. J. 2008, 44, 4074. [Google Scholar]
- [6].Ciftci M, Kahveci MU, Yagci Y, Allonas X, Ley C, Tar H, Chem. Commun. 2012, 48, 10252. [DOI] [PubMed] [Google Scholar]
- [7].Wang X, Huang B, Liu X, Zhan P, Drug Discov. Today 2016, 21, 118. [DOI] [PubMed] [Google Scholar]
- [8].a) Ananth A, Genua M, Aissaoui N, Díaz L, Eisele NB, Frey S, Dekker C, Richter RP, Görlich D, Small 2018, 14, 1703357; [DOI] [PubMed] [Google Scholar]; b) DeForest CA, Tirrell DA, Nat. Mater. 2015, 14, 523; [DOI] [PubMed] [Google Scholar]; c) Luo Y, Shoichet MS, Biomacromol 2004, 5, 2315. [DOI] [PubMed] [Google Scholar]
- [9].a) Kloxin AM, Kasko AM, Salinas CN, Anseth KS, Science 2009, 324, 59; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) DeForest CA, Anseth KS, Nat. Chem. 2011, 3, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hughes AJ, Lin RKC, Peehl DM, Herr AE, Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Gopal A, Herr AE, Sci. Rep. 2019, 9, 15389; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sinkala E, Sollier-Christen E, Renier C, Rosàs-Canyelles E, Che J, Heirich K, Duncombe TA, Vlassakis J, Yamauchi KA, Huang H, Jeffrey SS, Herr AE, Nat. Commun. 2017, 8, 14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Nasef MM, Güven O, Prog. Polym. Sci. 2012, 37, 1597; [Google Scholar]; b) Minko S, in Polymer Surfaces and Interfaces (Ed: Stamm M), Springer, Berlin, Heidelberg, Germany: 2008, Ch. 11, 215. [Google Scholar]
- [13].a) Biswal DR, Singh RP, Carbohydr. Polym. 2004, 57, 379; [Google Scholar]; b) Patil SB, Inamdar SZ, Reddy KR, Raghu AV, Soni SK, Kulkarni RV, J. Microbiol. Methods 2019, 159, 200. [DOI] [PubMed] [Google Scholar]
- [14].a) Tobita H, Hamielec AE, Polym. Int. 1993, 30, 177; [Google Scholar]; b) Caglio S, Righetti PG, Electrophoresis 1993, 14, 554. [DOI] [PubMed] [Google Scholar]
- [15].Beria L, Gevrek TN, Erdog A, Sanyal R, Pasini D, Sanyal A, Biomater. Sci. 2014, 2, 67. [DOI] [PubMed] [Google Scholar]
- [16].Kislukhin AA, Hong VP, Breitenkamp KE, Finn MG, Bioconjug. Chem. 2013, 24, 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Neira HD, Herr AE, Anal. Chem. 2017, 89, 10311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Orski SV, Sheppard GR, Arumugam S, Arnold RM, Popik VV, Locklin J, Langmuir 2012, 28, 14693. [DOI] [PubMed] [Google Scholar]
- [19].Tong J, Anderson JL, Biophys. J. 1996, 70, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Murtezi E, Ciftci M, Yagci Y, Polym. Int. 2015, 64, 588; [Google Scholar]; b) Shapiro YE, Prog. Polym. Sci. 2011, 36, 1184. [Google Scholar]
- [21].Dormán G, Nakamura H, Pulsipher A, Prestwich GD, Chem. Rev. 2016, 116, 15284. [DOI] [PubMed] [Google Scholar]
- [22].Leriche G, Chisholm L, Wagner A, Bioorganic Med. Chem. 2012, 20, 571. [DOI] [PubMed] [Google Scholar]
- [23].Liu L, Li P, Asher SA, Nature 1999, 397, 141. [DOI] [PubMed] [Google Scholar]
- [24].a) Zhu Y, Shi J, Shen W, Dong X, Feng J, Ruan M, Li Y, Angew. Chemie - Int. Ed. 2005, 44, 5083; Angew. Chemie 2005, 117, 5213; [DOI] [PubMed] [Google Scholar]; b) Giri S, Trewyn BG, Stellmaker MP, Lin VSY, Angew. Chemie - Int. Ed. 2005, 44, 5038; Angew. Chemie 2005, 117, 5166 [DOI] [PubMed] [Google Scholar]
- [25].Liang J, Fernández JM, J. Am. Chem. Soc. 2011, 133, 3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].a) Erickson HP, Biol. Proced. Online 2009, 11, 32; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Park IH, Johnson CS, Gabriel DA, Macromolecules 1990, 23, 1548; [Google Scholar]; c) Baselga J, Hernandez-Fuentes I, Masegosa RM, Llorente MA, Polym J 1989, 21, 467. [Google Scholar]
- [27].Amsden B, Macromolecules 1998, 31, 8382 [Google Scholar]
- [28].a) Zhang Y, Chan PPY, Herr AE, Angew. Chemie - Int. Ed. 2018, 57, 2357; Angew. Chemie 2018, 130, 2381; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schnaar RL, Langer BG, Brandley BK, Anal. Biochem. 1985, 151, 268. [DOI] [PubMed] [Google Scholar]
- [29].a) Fiore GL, Rowan SJ, Weder C, Chem. Soc. Rev. 2013, 42, 7278; [DOI] [PubMed] [Google Scholar]; b) Du X, Li J, Welle A, Li L, Feng W, Levkin PA, Adv. Mater. 2015, 27, 4997. [DOI] [PubMed] [Google Scholar]
- [30].Kirby BJ, Wheeler AR, Zare RN, Fruetel JA, Shepodd TJ, Lab Chip 2003, 3, 5. [DOI] [PubMed] [Google Scholar]
- [31].Tsukeshiba H, Huang M, Na YH, Kurokawa T, Kuwabara R, Tanaka Y, Furukawa H, Osada Y, Gong JP, J. Phys. Chem. B 2005, 109, 16304. [DOI] [PubMed] [Google Scholar]
- [32].Sasaki H, Onoe H, Osaki T, Kawano R, Takeuchi S, Sens. Actuators B Chem. 2010, 150, 478. [Google Scholar]
- [33].White ML, Dorion GH, J. Polym. Sci. 1961, 55, 731. [Google Scholar]
- [34].Michalek L, Barner L, Barner-Kowollik C, Adv. Mater. 2018, 30, 1706321. [DOI] [PubMed] [Google Scholar]
- [35].a) Wolski K, Gruszkiewicz A, Wytrwal-Sarna M, Bernasik A, Zapotoczny S, Polym. Chem. 2017, 8, 6250; [Google Scholar]; b) Wang W, Wang W, Li H, Lu X, Chen J, Kang NG, Zhang Q, Ind. Eng. Chem. Res. 2015, 54, 1292; [Google Scholar]; c) Kostruba A, Stetsyshyn Y, Mayevska S, Yakovlev M, Vankevych P, Nastishin Y, Kravets V, Soft Matter 2018, 14, 1016. [DOI] [PubMed] [Google Scholar]
- [36].a) Waggoner RA, Blum FD, Lang JC, Waggoner RA, Macromolecules 1995, 28, 2658; [Google Scholar]; b) Nagase K, Kobayashi J, Kikuchi A, Akiyama Y, Kanazawa H, Okano T, ACS Appl. Mater. Interfaces 2012, 4, 1998. [DOI] [PubMed] [Google Scholar]
- [37].a) Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN, Chem. Mater. 2014, 26, 724; [Google Scholar]; b) Wu D, Liu Y, He C, Chung T, Goh S, Macromolecules 2004, 37, 6763. [Google Scholar]
- [38].a) Dainton FS, Sisley WD, Trans. Faraday Soc. 1963, 59, 1385; [Google Scholar]; b) Kim WS, Lee SH, Kang IK, Park NK, J. Control. Release 1989, 9, 281. [Google Scholar]
- [39].Denisin AK, Pruitt BL, ACS Appl. Mater. Interfaces 2016, 8, 21893. [DOI] [PubMed] [Google Scholar]
- [40].Berg R, Straub BF, Beilstein J. Org. Chem. 2013, 9, 2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Arumugam S, Popik VV, J. Org. Chem. 2014, 79, 2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.