

SUMMARY
Adverse environmental conditions reduce crop productivity and often increase a load of unfolded or misfolded proteins in the endoplasmic reticulum (ER). This potentially lethal condition, known as ER stress, is buffered by the unfolded protein response (UPR), a set of signaling pathways designed to either recover ER functionality or ignite programmed cell death. Despite the biological significance of the UPR to the life of the organism, the regulatory transcriptional landscape underpinning ER stress management is largely unmapped, especially in crops. To fill this significant knowledge gap, we performed a large-scale systems-level analysis of protein-DNA interaction (PDI) network in maize (Zea mays). Using 23 promoter fragments of six UPR marker genes in a high-throughput enhanced yeast one-hybrid (eY1H) assay, we identified a highly interconnected network of 262 transcription factors (TFs) associated with significant biological traits and 831 PDIs underlying the UPR. We established a temporal hierarchy of TF binding to gene promoters within the same family as well as across different families of TFs. Cistrome analysis revealed the dynamic activities of a variety of cis-regulatory elements (CREs) in ER stress-responsive gene promoters. By integrating the cistrome results into a TF network analysis, we mapped a subnetwork of TFs associated with a CRE that may contribute to the UPR management. Finally, we validated the role of a predicted network hub gene using the Arabidopsis system. The PDIs, TF networks and CREs identified in our work are foundational resources for understanding transcription regulatory mechanisms in the stress responses and crop improvement.
Keywords: Transcription factors, UPR, ER stress, gene regulation, yeast one-hybrid, TF network, protein-DNA interaction, cistrome
INTRODUCTION
Growth and agronomic performance of maize, one of the most important crops in the world, are threatened by pathogen attack (Oerke, 2006, Mueller et al., 2016) and extreme weather (Wuebbles et al., 2017, Webber et al., 2018). A recent report showed that 9-10% of cereal production has been lost to drought and extreme heat (Lesk et al., 2016). Abiotic and biotic stress can increase the demand of protein folding beyond the biosynthetic capacity of the ER (Bao and Howell, 2017), causing a potentially lethal condition called ER stress. To maintain the ER homeostasis, plants initiate the UPR, a set of highly conserved surveillance pathways designed to clear the ER of misfolded proteins. Insufficient UPR ignites programmed cell death. The actuation and termination of UPR are achieved predominantly by gene expression reprogramming in association with cellular and metabolic changes during ER stress. Therefore, a better understanding of the gene regulation in the UPR is necessary to maintain, and possibly boost, crop productivity, which has to be doubled to feed the 9 billion world population estimated by 2050 (Godfray et al., 2010, Ray et al., 2013). However, little is known about how the UPR target genes are transcriptionally modulated during ER stress especially in crops such as maize.
In plants, the UPR has mainly been studied in the model species Arabidopsis thaliana and is coordinated by two branches: an ER membrane-associated basic leucine zipper (bZIP) TF [bZIP28 in plants; ACTIVATING TRANSCRIPTION FACTOR 6 (ATF6) in mammals] and the protein kinase and ribonuclease, inositol-requiring enzyme 1 (IRE1), and its splicing target [bZIP60 in plants; X-box binding protein 1 (XBP1) in metazoans; HAC1 in yeast] (Walter and Ron, 2011, Howell, 2013). Upon ER stress, bZIP28 (via proteolysis in Golgi) and bZIP60 (via IRE1-mediated unconventional splicing) are translocated into the nucleus where they control the expression of target genes, acting as master regulators. Orthologous genes encoding these core components have been identified or characterized in crops such as maize (Li et al., 2012, Srivastava et al., 2018, Kanodia et al., 2020) and rice (Oono et al., 2010, Hayashi et al., 2012, Lu et al., 2012, Qian et al., 2018, Wang et al., 2018b). Growing evidence, however, suggests that gene regulation of the UPR is more complex than previously thought. First, the transcriptional targets of bZIP28 and bZIP60 are only partially overlapping (Ruberti et al., 2018). Second, these TFs undergo unique transcriptional regulation during ER stress in addition to the posttranscriptional and/or posttranslational regulation (Iwata et al., 2008, Srivastava et al., 2018). Third, the transcriptional activity of bZIP28 and bZIP60 is modulated via an interaction with other transcriptional regulators and epigenetic components on their binding sites (Liu and Howell, 2010, Song et al., 2015, Nawkar et al., 2017, Lai et al., 2018). Finally, UPR biomarker genes, which are highly conserved in eukaryotes, show unique expression signature during a time-course of ER stress (Travers et al., 2000, Martínez and Chrispeels, 2003, Srivastava et al., 2018). Therefore, different yet unknown TFs acting with bZIP28 and bZIP60 are likely involved in the regulation of UPR genes in a time-specific manner. However, despite the biological significance of the UPR, there is no comprehensive interactive map between UPR TFs and their targets in plants.
To fill this knowledge gap, we carried out a TF network analysis built on hundreds of PDIs obtained from eY1H screen, which, like any Y1H screen, can lead to the identification of transcriptional regulators bound to DNA sequences, irrespective of their activation or repression roles (Brady et al., 2011, Gaudinier et al., 2011). A total of 23 promoter fragments of six UPR biomarker genes whose expression is sequentially modulated during ER stress in maize was implemented for the eY1H screen. Our work identified TFs associated with a variety of biological processes and physically bound to the promoters of UPR genes in a highly interconnected network, potentially linking the UPR to significant biological processes. Our network model allowed us to develop testable hypotheses on the temporal hierarchy of gene regulation in the UPR, identify key regulatory TFs and reveal temporal activities of CREs responsive to ER stress. We also validated our predictive pipeline using Arabidopsis as an experimental platform. Together, our analyses provide the resources to map the complexity of the dynamics of gene expression in the maize UPR with potential translational relevance to other organisms for conserved stress-responsive pathways.
RESULTS
Identification of PDIs underlying the dynamics of UPR gene expression
The bulk transcriptional changes in the UPR have been established in Arabidopsis (Martínez and Chrispeels, 2003), maize (Srivastava et al., 2018, Kanodia et al., 2020) and metazoan species (Travers et al., 2000, Han et al., 2013), but the identity and dynamics of the gene-regulatory hubs underlying the progression of the ER stress responses are yet unmapped in any of these species. To fill this gap and identify the transcriptional regulators upstream of the UPR in maize, we sought to identify the TFs binding to the promoters of genes representing distinct phases of ER stress responses. We hypothesized that the promoters of temporally expressed UPR target genes would be targeted by multi-functional TF s in specific gene-regulatory modules linking the UPR to diverse biological processes for restoration of cell homeostasis (early phase of the UPR) or ignition of cell death (late phase of the UPR in unresolved ER stress conditions). To test this, we analyzed an RNA-sequencing (RNA-seq) dataset that we acquired during a time-course of ER stress in maize roots exposed to the ER stress inducer tunicamycin (Tm) (Srivastava et al., 2018). We selected six genes, hereafter named UPR target genes: Binding protein2 (ZmBiP2, Zm00001d014993), ZmbZIP60 (Zm00001d046718), Bax inhibitor1 (ZmBi1, Zm00001d015091), Peroxidase1 (ZmPRX1, Zm00001d022279), Cystein protease1 (ZmCP1, Zm00001d007049) and Secretion-associated Ras-related 1 (ZmSAR1, Zm00001d049068). We selected these genes because they showed unique expression signatures during the time-course of ER stress and are associated directly with significant cellular and metabolic processes required for ER homeostasis (Figure 1a). As such, these genes would allow us to dissect the architecture of the upstream transcriptional network underlying the UPR. For example, ZmPRX1, one of the peroxidase family genes critical for oxidative stress response (Cosio and Dunand, 2009, Wang et al., 2015b), was rapidly down-regulated at the early stage of ER stress when pro-survival events are prevalent (Srivastava et al., 2018). This phase was then followed by only partially distinct temporal surges in the expression of ZmBiP2 (the most known UPR biomarker gene encoding an ER chaperone protein), ZmSAR1 (encoding small GTPase associated with membrane trafficking), ZmBi1 (critical for cell death) and ZmbZIP60 (UPR regulator) (Nakańo and Muramatsu, 1989, Cebulski et al., 2011, Walter and Ron, 2011). At the late stage of ER stress, when pro-death processes are activated (Srivastava et al., 2018), ZmCP1, a closest homolog of an Arabidopsis executioner protease associated with programmed cell death (Zhang et al., 2014b), showed a high level of transcriptional induction. These features make ZmCP1 an effective transcriptional marker in the late stage of ER stress in our analysis, despite the lack of signal peptide and ER retention signal, which are present in the Arabidopsis homolog (Solomon et al., 1999, Zhang et al., 2014a).
Figure 1.
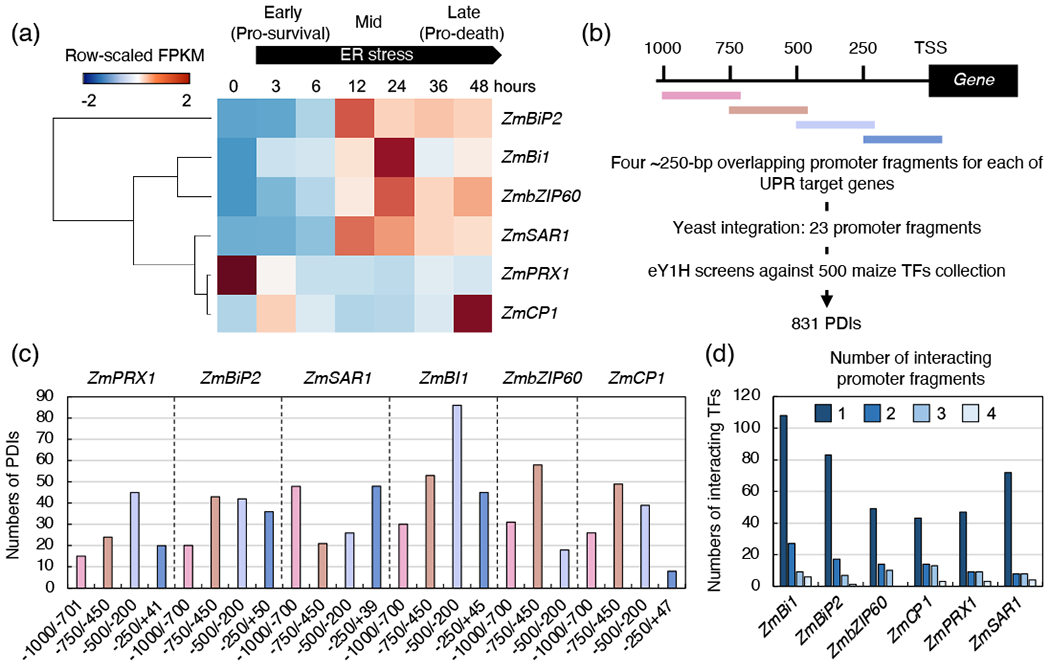
eY1H screen of UPR target genes against a collection of 500 maize TF. (a) Expression patterns of UPR target genes during a time-course of ER stress in maize. Fold change of a fragments per kb exon model per million mapped fragments (FPKM) to 0 h was log2-transformed at each time point. Genes were hierarchically clustered. (b) Scheme of the screened promoter fragment baits and workflow of the eY1H screen. (c) PDIs obtained from the eY1H screen. The exact coordinate of each promoter fragment is shown on the X-axis. The full list of PDIs is available in Table S1. (d) The majority of PDIs for each gene is fragment specific.
To identify the TFs binding to the promoters of the UPR genes, we performed an eY1H screen, which is an established semi-automated platform that allows to confidently identify TFs binding to promoter sequences as baits (Gaudinier et al., 2011, Reece-Hoyes et al., 2011, Sparks et al., 2013, Koryachko et al., 2015, Taylor-Teeples et al., 2015, Gaudinier et al., 2018, Ikeuchi et al., 2018, Li et al., 2018, Smit et al., 2020). To perform the eY1H screen for the promoters of the selected UPR genes against a collection of 500 maize TFs (Burdo et al., 2014), we took a strategic approach in which the 1-kb long promoters were split into four ~250-bp long partially overlapping fragments (Figure 1b and Table S1). We chose 1-kb promoter regions upstream of the transcriptional start site because it generally has the highest binding frequency or chromatin accessibility in many plant species (Maher et al., 2018). Also this approach has been successfully used in a previous Y1H screen in maize (Yang et al., 2017). Using partially overlapping promoter regions is an advantageous approach over using an intact 1-kb long promoter fragment because it increases assay sensitivity and decreases false-positives in PDIs (Pruneda-Paz et al., 2014). Furthermore, the approach provides topological information for each PDI within the 1-kb long promoter and ultimately allows mapping the spatial dynamics of PDIs within the gene promoters at a fine resolution. Except for a fragment of the ZmbZIP60 promoter (−263/+23-bp), which failed to be transformed into yeast, all promoter fragments from the six genes were successfully integrated in the yeast genome. We identified a total of 831 PDIs across 23 promoter fragments and 262 binding TFs (Figure 1c and Table S2). These results point to a combinatorial action of multiple TFs to lead the expression dynamics of the UPR target genes, in line with our initial hypothesis. The number of PDIs varied across different promoter fragments of each UPR target gene. For example, ZmBi1 and ZmPRX1 showed the highest number of PDIs with the −500/−200-bp fragment while ZmBiP2, ZmbZIP60 and ZmCP1 did so with the −750/−450-bp fragment. ZmSAR1 displayed the highest number of PDIs with the −1000/−700-bp and −250/+39-bp fragments. Interestingly, the majority of PDIs resulted to be fragment-specific as an average of 70% of TFs was found to interact with a single promoter fragment of the UPR target genes (Figure 1d), possibly driven by a sequence-specific binding. In our screen results, we also found that ZmbZIP17a (Zm00001d007042) and ZmbZIP17b (Zm00001d038189), both predicted to be homologs of the Arabidopsis bZIP17 and/or bZIP28 (Srivastava et al., 2018, Zhu et al., 2019), and ZmbZIP60 TFs bound to the promoter fragments of ZmBiP2. Such TF-DNA interactions have extensively been characterized in Arabidopsis (Zhang et al., 2017) and metazoans (Fink et al., 2018), suggesting a conserved transcriptional regulation for this chaperone gene and potentially other UPR biomarkers in maize. Although our data do not include the binding of ZmbZIP17a to the promoter of ZmbZIP60, which was previously identified by chromatin immunoprecipitation followed by deep-sequencing (ChIP-seq) (Srivastava et al., 2018), we identified binding of ZmbZIP17b, the close paralog of ZmbZIP17a, to the promoter of ZmbZIP60, supporting a transcriptional regulation between these UPR modulators. The results also support the strength of our approach. Furthermore, in addition to the expected interactions, our screen identified novel putative PDIs on the UPR genes. To validate our approach, we compared PDIs in our dataset with a publicly available pre-release ChIP-seq dataset of 110 non-redundant TFs analyzed in maize leaves (http://www.epigenome.cuhk.edu.hk/C3C4.html). Among the 110 TFs in the database, 25 TFs were included in our PDIs, among which 16 TFs had genomic coordinates of peaks available. Despite the distinct biological (i.e., unicellular vs. multicellular) and technical (i.e., in vitro vs. in vivo) setups of the eY1H and ChIP-Seq analyses, the limited number of TFs represented in the ChIP database and the fact that the occurrence of TF binding to promoters depends largely on growth and experimental conditions, we found that among a total of 48 PDIs associated with the 16 TFs, 13 PDIs (27%) were confirmed by the ChIP-seq dataset (Table S2). Therefore, despite the unavoidable false negatives and false positives in network approaches like our own, our dataset includes PDIs that are conserved cross-species and validated by other experimental methods.
Mapping the TF network underlying the dynamics of gene expression in the UPR
We next sought to delineate the hierarchy of the TFs for the transcriptional regulation in the UPR. To do so, we aimed to map a TF network by integrating the 831 PDIs obtained from our eY1H screen with the transcriptomic profiles during the course of ER stress (Figure 2a). We named the resulting landscape “TF network underlying the UPR” (TNU). The TNU comprises 126 TFs that bind the promoter of a single UPR target gene and 136 TFs that bind the promoters of multiple UPR target genes, including 15 TFs that bind the promoters of all six UPR target genes. A large number of genes encoding the TFs were differentially expressed during the ER stress in a temporal manner, revealing a transcriptional signal cascade in the TNU in ER stress. Intriguingly, not all TF genes were differentially expressed during ER stress, supporting the possibility that posttranscriptional or posttranslational modifications could be critical to the transcriptional activity of these TFs. Interestingly, the exclusive and non-exclusive binding of TFs to each target gene coincided with the initiation of differential expression of UPR target genes (Figure 1a and 2b and Figure S1). The promoter fragments of ZmBi1, whose increasing expression peaked at 24 h of ER stress, were bound by a total of 149 TFs including 41 TFs that bound the promoter in an exclusive manner. However, the promoter fragments of the early silenced ZmPRX1 and of the late induced ZmCP1 were bound by only 68 TFs (17 exclusively) and 73 TFs (11 exclusively), respectively. These data support a regulatory complexity led by multiple TFs on UPR target gene promoters, which likely depends on the specific gene function during the course of ER stress. In line with this, genes, whose expression is altered in the mid-phase of adaptive ER stress (i.e., ZmBi1) when multiple biological pathways might interplay (Srivastava et al., 2018), could require a higher level of the regulatory inputs than early or late phase genes, as reflected by the larger node size of differentially expressed TFs converging onto ZmBi1 (Figure 2a).
Figure 2.
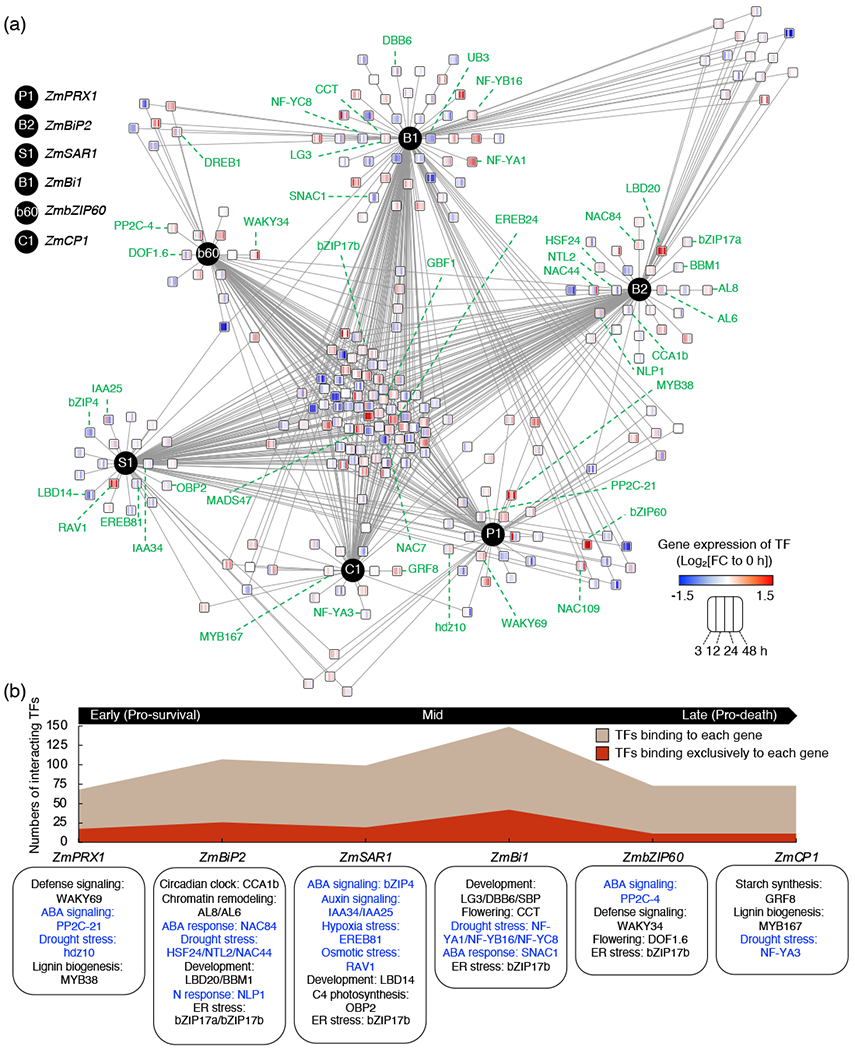
A TF network underlying the UPR. (a) The TF network underlying the UPR (TNU) constructed by integrating PDIs obtained from our eY1H screen with the transcriptome profile (Srivastava et al., 2018). UPR target genes are indicated by black circles. Square nodes represent TFs. Gene expression change at 3, 12, 24 and 48 h, relative to 0 h, is visualized in the heatmap in each node. Due to the limited space, the abbreviation Zm for each gene was removed. TFs that have been either functionally characterized or annotated are indicated by green dot lines. (b) Temporal dynamics of the TNU. At X-axis, bait genes are placed where their expression begins to be altered during the adaptive ER stress (Figure 1a). Biological processes of TFs that bound exclusively to each bait gene are shown in the boxed text at the bottom. The complete set of biological processes and references is provided in Table 1. Biological processes associated with abiotic stress are indicated in blue font.
Next, we used publicly available data to annotate the biological processes associated with each regulatory TF module. Although only 45 maize TFs have been either functionally characterized or analyzed in vivo thus far (Table 1), the TNU represents an array of diverse biological pathways, other than the UPR, among which the major category comprises abiotic stress responses (28 TFs associated with abiotic stress among 45 TFs annotated: 62%). Therefore, not only the TNU confirms the expected association of UPR with abiotic stress in other plant species (Zhang et al., 2008, Deng et al., 2011) and biotic stress (Pajerowska-Mukhtar et al., 2012, Nagashima et al., 2014, Lai et al., 2018), it also reveals the potential existence of yet uncharacterized molecular links of the UPR with other biological traits (e.g., meristem development and secondary metabolism) via TF binding to promoters of genes involved in such processes. This is exemplified by a maize homolog of the CIRCADIAN CLOCK ASSOCIATED1 (ZmCCA1b), which was found to be exclusively bound to the promoter fragments of ZmBiP2 (an early-mid ER stress phase induced gene) in our dataset. Therefore, we investigated whether the binding of ZmCCA1b to the promoter of ZmBiP2 is specific to a CRE by scanning the sequences of the promoter fragments (Figure S2a). ZmCCA1b bound to −1000/−700, −750/−450 and −500/200-bp ZmBiP2 promoter fragments all of which have one of the ZmCCA1b binding motifs (A/C/GAAATA; evening-like element) (Ko et al., 2016). The evidence that ZmCCA1b did not bind to the −250/+50-bp fragment, which has no evening-like element, emphasizes the sequence-specific binding of the TF and further validates our eY1H screen results. Then, to link the PDIs to gene expression, we checked whether expression of ZmBiP2 is under diurnal oscillation, potentially through ZmCCA1b binding, using the DIURNAL database (Mockler et al., 2007). In a circadian condition (constant light) following entrainment on a diurnal condition (long-day), the expression of ZmBiP2 increased from zeitgeber time 0 (ZT0 = dawn), peaked at ZT4 and lowered at dusk (Figure S2b). As the diurnal expression pattern of ZmCCA1b highly overlaps with the one of ZmBiP2 (Hayes et al., 2010, Ko et al., 2016), these findings indicate that ZmCCA1b may be a positive regulator of ZmBiP2 in a circadian clock manner. We also found an evening-like element on promoter fragments of other bait genes, but their diurnal expression was not detected in the DIURNAL database. In addition to the circadian clock pathway, an indication of a link between cell wall lignin biogenesis and the UPR is provided in the TNU by the exclusive physical binding of the lignin master regulators MYB DNA-binding domain protein38 (ZmMYB38) (Barrière et al., 2015) and ZmMYB167 (Bhatia et al., 2019) to ZmPRX1 and ZmCP1 respectively (Figure 2b). Because ER stress negatively affects plant growth (Chen and Brandizzi, 2012, Ruberti et al., 2018), these results are consistent with the possibility that specific early- and late-phase modulated genes in the UPR are responsible for modifications of the cell wall during the ER stress. Taken together, mapping the TNU revealed a complex regulatory framework in which TFs associated with important biological processes bind redundantly or specifically to UPR target genes that are dynamically expressed in response to ER stress in maize.
Table 1.
A full list of TFs that are shown in Figure 2a. These TFs have been functionally characterized or analyzed in published literatures (see References). The entire dataset of 831 PDIs is provided in Table S2.
| Gene ID | Gene Name | Biological process | Target gene | Reference |
|---|---|---|---|---|
| Zm00001d046828 | ZmPP2C-21 | ABA signaling | ZmPRX1 | (Wang et al., 2014) |
| Zm00001d032024 | ZmMYB38 | Lignin biosynthesis | ZmPRX1 | (Barrière et al., 2015) |
| Zm00001d043569 | ZmWAKY69 | Defense signaling | ZmPRX1 | (Agostini et al., 2019) |
| Zm00001d005951 | Zmhdz10 | Drought response | ZmPRX1 | (Zhao et al., 2014) |
| Zm00001d049543 | ZmCCA1b | Circadian clock | ZmBiP2 | (Hayes et al., 2010, Ko et al., 2016) |
| Zm00001d007042 | ZmbZIP17a | ER stress | ZmBiP2 | (Srivastava et al., 2018) |
| Zm00001d016861 | ZmAL8 | Chromatin remodeling | ZmBiP2 | (Wang et al., 2015a) |
| Zm00001d032923 | ZmHSF24 | Drought stress | ZmBiP2 | (Du et al., 2017b) |
| Zm00001d039506 | ZmNAC84 | ABA response | ZmBiP2 | (Zhu et al., 2016b) |
| Zm00001d040004 | ZmLBD20 | Development | ZmBiP2 | (Zhang et al., 2014c) |
| Zm00001d050816 | ZmAL6 | Chromatin remodeling | ZmBiP2 | (Wang et al., 2015a) |
| Zm00001d027510 | ZmNLP1 | Nitrogen response | ZmBiP2 | (Ge et al., 2018) |
| Zm00001d042492 | ZmBBM1 | Development | ZmBiP2 | (Salvo et al., 2014) |
| Zm00001d049913 | ZmNTL2 | Drought stress | ZmBiP2 | (Xue et al., 2013) |
| Zm00001d028999 | ZmNAC44 | Drought stress | ZmBiP2 | (Ding et al., 2014) |
| Zm00001d018178 | ZmbZIP4 | ABA response | ZmSAR1 | (Ma et al., 2018) |
| Zm00001d049141 | ZmIAA34 | Auxin signaling | ZmSAR1 | (Ludwig et al., 2013) |
| Zm00001d006739 | ZmLBD14 | Development | ZmSAR1 | (Zhang et al., 2014c) |
| Zm00001d018973 | ZmIAA25 | Auxin signaling | ZmSAR1 | (Pautler et al., 2015) |
| Zm00001d043782 | ZmRAV1 | Osmotic stress | ZmSAR1 | (Min et al., 2014) |
| Zm00001d030727 | ZmOBP2 | C4 photosynthesis | ZmSAR1 | (Skirycz et al., 2006) |
| Zm00001d035512 | ZmEREB81 | Hypoxia stress | ZmSAR1 | (Du et al., 2014) |
| Zm00001d017147 | ZmDBB6 | Development | ZmBi1 | (Li et al., 2017) |
| Zm00001d040611 | ZmLG3 | Development | ZmBi1 | (Mantilla-Perez and Salas Fernandez, 2017) |
| Zm00001d022099 | ZmNF-YB16 | Drought stress | ZmBi1 | (Wang et al., 2018a) |
| Zm00001d027874 | ZmNF-YA1 | Drought stress | ZmBi1 | (Luan et al., 2015) |
| Zm00001d036648 | ZmNF-YC8 | Drought stress | ZmBi1 | (Song et al., 2017) |
| Zm00001d034601 | ZmSNAC1 | ABA response | ZmBi1 | (Yue et al., 2015) |
| Zm00001d048369 | ZmCCT | Flowering | ZmBi1 | (Jin et al., 2018) |
| Zm00001d052890 | ZmUB3 | Development | ZmBi1 | (Chuck et al., 2014) |
| Zm00001d009939 | ZmWAKY34 | Defense signaling | ZmbZIP60 | (Kebede et al., 2018) |
| Zm00001d026628 | ZmDOF1.6 | Flowering | ZmbZIP60 | (Song et al., 2017) |
| Zm00001d012962 | ZmPP2C-4 | ABA signaling | ZmbZIP60 | (Wang et al., 2014) |
| Zm00001d002429 | ZmGRF8 | Starch synthesis | ZmCP1 | (Zhang et al., 2019) |
| Zm00001d032032 | ZmMYB167 | Cell wall lignin | ZmCP1 | (Bhatia et al., 2019) |
| Zm00001d006835 | ZmNF-YA3 | Drought stress | ZmCP1 | (Su et al., 2018) |
| Zm00001d027957 | ZmMADS47 | Kernel storage | ZmBiP2/ZmSAR1 | (Qiao et al., 2016) |
| Zm00001d042609 | ZmNAC109 | Drought stress | ZmSAR1ZmBi1 | (Wang et al., 2019) |
| Zm00001d046718 | ZmbZIP60 | ER stress | ZmSAR1/ZmBi1 | (Srivastava et al., 2018) |
| Zm00001d053859 | ZmEREB98 | Osmotic stress | ZmbZIP60/ZmCP1 | (Yu et al., 2018) |
| Zm00001d032295 | ZmDREBl | Cold stress | ZmBi1/ZmbZIP60 | (Zheng et al., 2006, Avila et al., 2018) |
| Zm00001d038189 | ZmbZIP17b | ER stress |
ZmBi1/ZmBiP2/ZmS AR1/ZmbZIP60 |
(Srivastava et al., 2018) |
| Zm00001d039065 | ZmGBF1 | Hypoxia stress |
ZmBi1/ZmBiP2/ZmS AR1/ZmbZIP60/Zm CP1/ZmPRX1 |
(De Vetten and Ferl, 1995) |
| Zm00001d041472 | ZmNAC7 | Growth and fitness |
ZmBi1/ZmBiP2/ZmS AR1/ZmbZIP60/Zm CP1/ZmPRX1 |
(Zhang et al., 2019) |
| Zm00001d002025 | ZmEREB24 | Hypoxia stress |
ZmBi1/ZmBiP2/ZmS AR1/ZmbZIP60/Zm CP1/ZmPRX1 |
(Du et al., 2017a) |
Prediction of protein-protein interactions on the promoters of the UPR genes
Because TFs generally behave as a protein complex on the gene regulatory elements (Lemon and Tjian, 2000), we tested whether the TFs binding to the promoters of UPR target genes could interact with other TFs. Among the 36 TF families included in the 831 PDIs, the five most represented families were the basic leucine zipper domain (bZIP), APETALA2/ethylene responsive element binding factor (AP2/EREBP), NAM, ATAF and CUC (NAC), MYB (Figure 3a). To test whether these TF families exhibited a binding preference for promoter fragments of the UPR target genes, we measured the interaction frequency of the TFs of each of the five families with the four (or three of ZmbZIP60) promoter fragments of each UPR target gene. Each TF family showed different enrichment for the interactions in specific promoter fragments (Figure 3b). For example, bZIP TFs were bound to the −500/−200-bp fragment of ZmBi1 with the highest enrichment (43.2%) but for ZmBiP2 the highest enrichment (45.7%) was found at the −250/+50-bp fragment. Furthermore, 45% of the interactions of ERF TFs with ZmBAX1 promoters occurred at the −750/−450-bp fragment although the highest binding enrichment (40% and 42.8%) was found at the −500/−200-bp fragment of ZmCP1 and ZmPRX1, respectively. Next, because the binding preference of a TF family can depend on other TF families (Lemon and Tjian, 2000, Zhu et al., 2018), we aimed to quantify the TF-DNA binding frequency correlation of the TF families identified in our eY1H screen. To do so, we calculated the Spearman rank correlation of the TF-DNA binding frequency over promoter fragments of each gene across the TF families (Figure S3). We found that the binding preference of WAKY TFs was highly correlated with that of bZIP, MYB and NAC TFs on the promoters of ZmBi1, ZmBiP2 and ZmbZIP60, respectively (Spearman rank correlation = 1.0). In addition to these strong correlations, multiple TF families showed a high correlation with other families (Spearman rank correlation ≥ 0.8). These results indicate that, when binding to the UPR target genes, TFs may interact with TFs of the same family or different families during ER stress, consistent with the earlier findings (Liu and Howell, 2010, Meng et al., 2017, Nawkar et al., 2017, Lai et al., 2018).
Figure 3. Interactions between TFs on promoters of UPR genes.
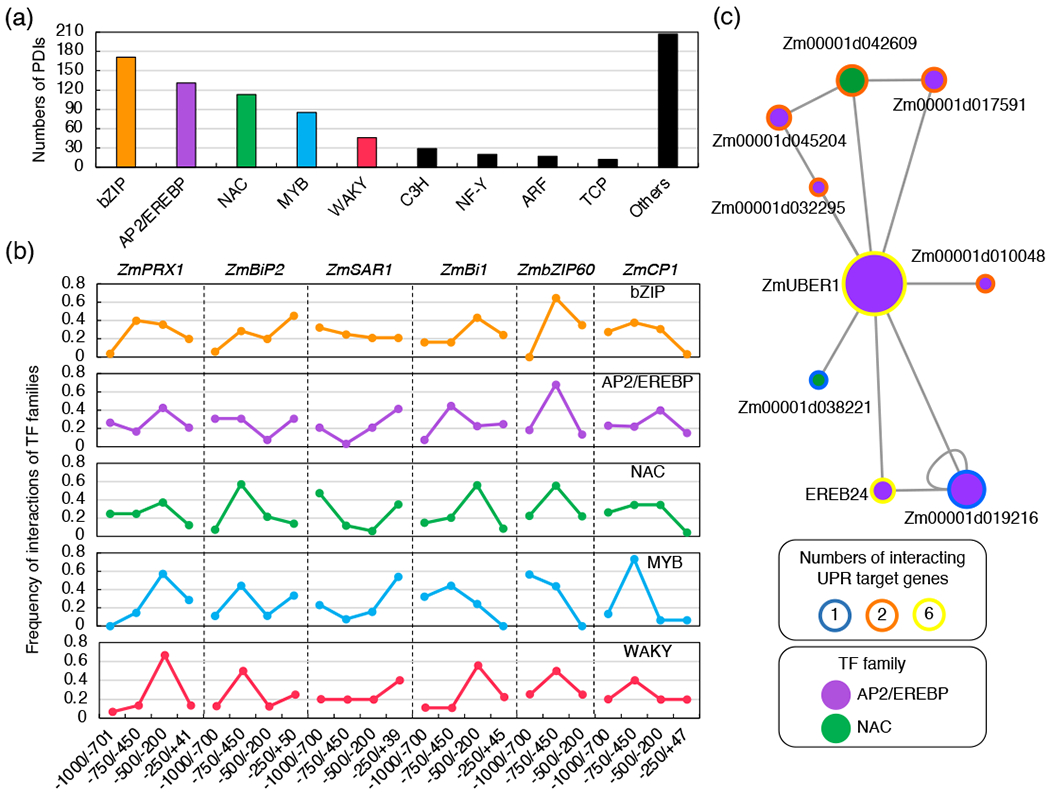
(a) Numbers of PDIs associated with TF families. The full list of PDIs with the TF family membership is available in Table S2. (b) Frequency distribution for the binding of multiple TF family members to each of promoter fragments used in the eY1H screens. TF families of which the number of PDIs was greater than 45 are shown. (c) TF interaction networks of ERF and NAC TFs that bound to promoters of the UPR genes. The size of the nodes is proportional to the number of interactions. The color of nodes indicates the number of interactions of each TF with UPR target genes.
Next, to identify TFs that function as interacting hubs in the TNU, we investigated potential interactions across the TFs using the protein-protein interaction database for maize (PPIM) (http://comp-sysbio.org/ppim/), which consists of more than 2 million protein-protein interactions (PPIs) accurately predicted or experimentally determined (Zhu et al., 2016a). To effectively integrate the PPIM into our eY1H dataset, we identified interactions of the TFs that both bound each of the UPR target genes in our eY1H screen, and then consolidated them. This generated a total of 20 non-redundant PPIs, all of which were inferred as regulatory PPI in maize leaves (Yu et al., 2015, Zhu et al., 2016a) (Table S3). Among the TFs that were bound to at least one UPR target gene, we identified an ERF TF (Zm00001d043205), hereafter named as UPR BINDING ERF 1 (ZmUBER1), which was found to interact with six ERF TFs as well as two NAC TFs (Figure 3c). Importantly, in our eY1H screen ZmUBER1 was found to bind the promoters of all six UPR target genes (Figure 3c and Table S2). Thus, our data indicate that ZmUBER1 is at the core of a significant hub and possibly another UPR master regulator during the progression of ER stress responses. The results also support that the promoter binding activity of ZmUBER1 for UPR target gene expression is likely modulated, or at least coordinated, via an interaction with different TFs.
Identification of de novo CREs in the TF network underlying the UPR
In the UPR, the regulation of gene expression is primarily dependent on the activity of CREs that are responsive to ER stress (Yoshida et al., 1998, Oh et al., 2003, Liu and Howell, 2010, Hayashi et al., 2013). These CREs are highly conserved in eukaryotes and include the ER stress response element-I (ERSE-I, 5’-CCAAT-N10-CACG-3’), ERSE-II (5’-ATTGG-N2-CACG-3’), ERSE-III (5’-TCATCG-3’), UPRE-I (5’-TGACGTGR-3’) and UPRE-II (5’-GATGACGCGTAC-3’). The temporal activity of these elements during ER stress progression is not well defined, especially in plants. To fill this knowledge gap, we used the AME software (Buske et al., 2010) and measured enrichment of the canonical CREs and their variants (i.e., with a mismatch on the 3’) along 1-kb in the promoters of the differentially expressed genes (DEGs) during ER stress (Figure 4A). As a result, we found a discrete enrichment distribution of the CREs along the progression of ER stress. For example, while the enrichment of UPRE-I was high on promoters of DEGs at 3 h, it gradually declined as ER stress progressed until 48 h when it increased again. After showing slight, yet significant, enrichment on the promoters of DEGs at 6, 12 and 24 h, ERSE-I was highly enriched on promoters of DEGs at 48 h, supporting a late involvement of this CRE in the later phases of the UPR. The other CREs, such ERSE-II (Kokame et al., 2001), UPRE-II (Hayashi et al., 2013) and UPRE-III (Sun et al., 2013), which are known to be active in the UPR in non-maize species showed lower enrichment compared to ERSE-I, UPRE-I and their variants (Figure 4a). These results indicate that the majority of the canonical CREs and their variants are differentially enriched in the promoters of DEGs in a time-dependent fashion during ER stress. The data also underscore that the maize UPR may rely primarily on a subset of CREs and possibly unique or at least ones not previously reported. To test this, we performed de novo motif analysis on the 1-kb promoters of the DEGs during ER stress progression using the MEME software (Bailey and Elkan, 1994) (Figure 4b). We identified five de novo motifs that were significantly enriched in the promoters of DEGs, but not in the promoters of randomly selected genes. Among the motifs, the TCGGC motif was the most strongly enriched in all time-points analyzed except for the 3 h time point, suggesting a role in gene activity regulation after the early stages of the UPR. By comparing with the Arabidopsis cistrome database built on DNA Affinity Purification and sequencing (DAP-seq) (O’Malley et al., 2016) with the motif comparison tool, TOMTOM (Gupta et al., 2007), we established that the TCGGC motif statistically matches with the binding site of the Arabidopsis AP2/EREBP TF (P-value = 0.0024), encoded by the AT1G1210 locus (Figure 4c). These observations support that the TCGGC motif may mediate transcriptional regulation in the UPR by recruiting AP2/EREBP TFs to the promoters of ER stress-responsive genes. Interestingly and consistent with this hypothesis, among the UPR target genes, ZmBiP2, ZmbZIP60 and ZmSAR1 have a TCGGC motif in their promoters (Figure 4c) and were bound by 20 AP2/EREBP TFs in our eY1H analyses (Table S1). As a result, the significant representation of the TCGGC motif generates a subnetwork of the TNU (Figure 4d). Zm00001d002025, an AP2/EREBP TF, was found to bind all promoter fragments containing the TCGGC motif (Table S2), suggesting that it could act as regulatory-hub TF in regulating UPR responsive genes via the TCGGC motif. Interestingly also, ZmUBER1, which is at the core of a highly connected hub in the TNU (Figure 3c), also belongs to the TCGGC subnetwork, binding to three promoter fragments (Figure 4d). Taken together, our cistrome analysis revealed the dynamic activities of canonical CREs, as well as potential CREs active in ER stress responses as well as a subnetwork of the TNU that is likely mediated by the TCGGC.
Figure 4.
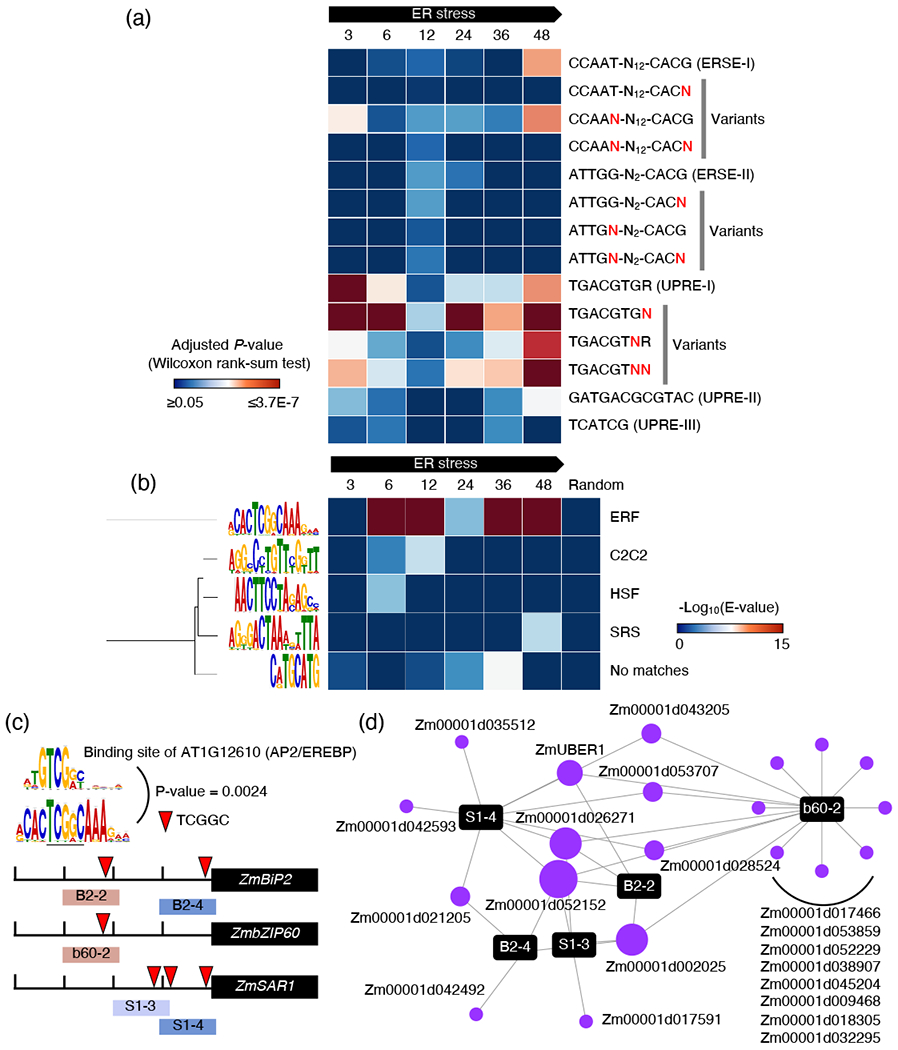
A TF network mediated by novel CRE underlying gene expression in the UPR. (a) Enrichments of canonical ER stress-responsive CRE with or without mismatch on 1-kb promoters of DEGs in maize (n = 1232 for 3 h, 2157 for 6 h, 351 for 12 h, 992 for 24 h, 1231 for 36 h and 2023 for 48 h). The analysis was carried out using AME (Buske et al., 2010) (Wilcoxon rank-sum test, Adjusted P-value by Bonferroni correction). N marked by red color indicates a random nucleotide. (b) Enrichments of de novo motifs on promoters of DEGs in maize during the adaptive ER stress. De novo motif analysis was performed using MEME (Bailey and Elkan, 1994). Motifs are displayed on the left of each row. The best match to a transcription factor family for the motif was identified from the Arabidopsis cistrome database (O’Malley et al., 2016) using TOMTOM (Gupta et al., 2007) and displayed to the right of each row. The median number of DEGs across time points is 280. We randomly selected 280 protein-coding genes in the maize genome and included the de novo motif analysis as a control. (c) Location of the highly enriched de novo motif on promoters of ZmBiP2, ZmbZIP60 and ZmSAR1. (d) A network of AP2/EREBP TFs on UPR promoters containing the de novo motif. The size of the nodes is proportional to the number of PDIs.
The Arabidopsis homolog of ZmUBER1 has an important role in the UPR
Our PPI network analysis (Figure 3c) and cistrome-based subnetwork analysis (Figure 4d) pinpointed ZmUBER1 as a potential regulatory hub in the TNU. To investigate the relevance of ZmUBER1 in the UPR and experimentally validate our genomic pipeline, we tested the role of its sequence homolog in A. thaliana in which UPR regulators have extensively been investigated so far (Howell, 2013, Pastor-Cantizano et al., 2020). AtERF9 was found to be the closest homolog of ZmUBER1 (E-value = 2e-22 and identity = 81%, BLASTP) in the Arabidopsis genome (TAIR10). Furthermore, the APETALA2 (AP2) domain is highly conserved in AtERF9 and ZmUBER1 (Figure S4). The core binding site (CGGC) of AtERF9 identified by the DAP-seq analysis (O’Malley et al., 2016) is present on the promoters of known UPR biomarker genes (e.g., AtBiP3, AtbZIP28 and AtbZIP60), suggesting that AtERF9 could bind to these promoters via the CRE (Figure 5a). Indeed, AtERF9 binding to the promoter of AtbZIP60 was also identified in an earlier eY1H screening (Sparks et al., 2016). Thus, we investigated the expression of the UPR biomarker genes in WT and an AtERF9 loss-of-function T-DNA insertion mutant (erf9) in response to ER stress (Figure 5b). While the transcriptional induction of AtBiP3, AtbZIP28 and AtERdj3B in Tm-treated samples relative to one in the corresponding mock samples was not changed in erf9 compared to WT, the abundance of spliced AtbZIP60 transcript was significantly enhanced in erf9. Importantly also, erf9 showed less sensitivity to ER stress compared to WT when seeds were directly germinated on growth media containing the ER stress inducer tunicamycin (Tm), a treatment that causes chronic/unresolved ER stress (Martínez and Chrispeels, 2003, Chen and Brandizzi, 2012, Deng et al., 2013) (Figure 5d). Our data not only validate the functional importance of ZmUBER1 in the TNU and the genomic pipeline, but also suggest that its regulatory role is at least partially conserved in Arabidopsis.
Figure 5.
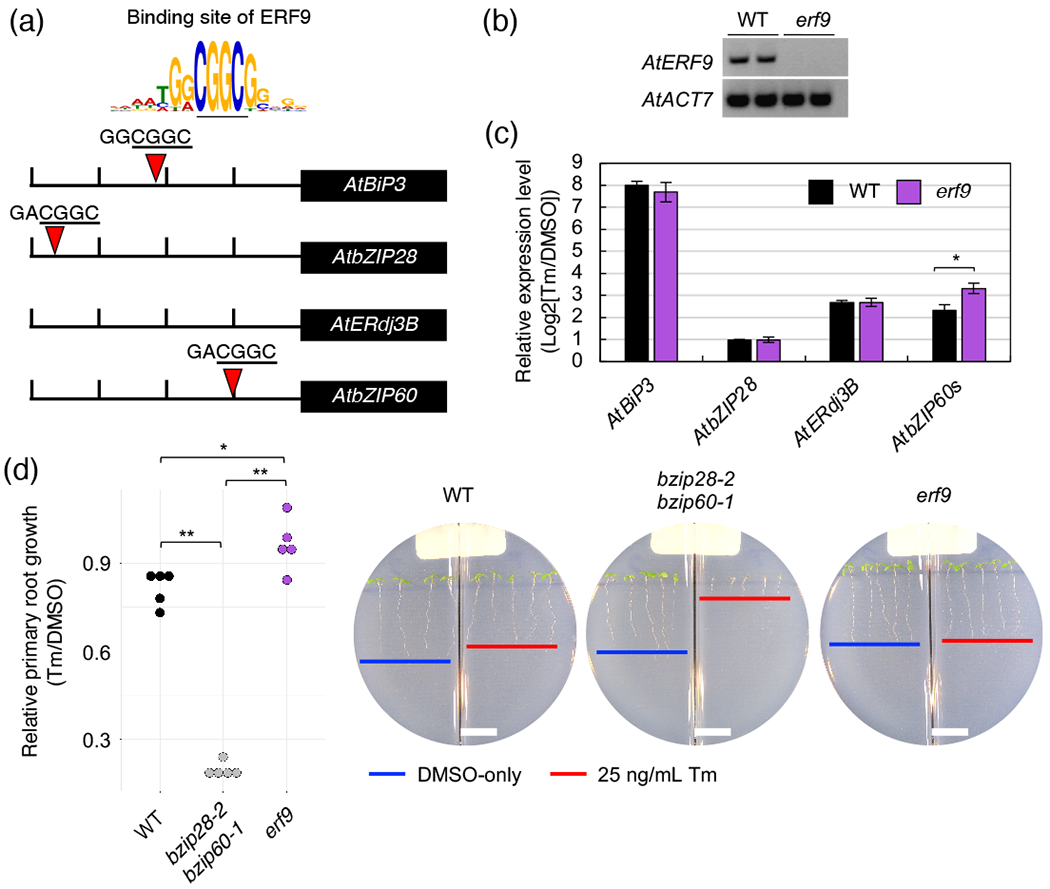
The knock-out of Arabidopsis ZmUBER1 homolog gene reduces the sensitivity of root growth to chronic ER stress. (a) Location of AtERF9 binding motif on the promoters of UPR genes. The core element is underlined. (b) RT-PCR showing transcript level of AtERF9 in WT and erf9 T-DNA mutant. AtACT7 was used as a loading control. (c) qRT-PCR showing the relative expression levels of UPR genes normalized to the mock control (DMSO-only) at 24 h of Tm treatment. Means ± SEM; three biological replicates. *P < 0.05, Student’s t-test. (d) Relative growth rate of the primary root of WT, erf9 and bzip28-2 bzip60-1 in chronic ER stress. bzip28-2 bzip60-1, which is known to be lethal in chronic ER stress (Deng et al., 2013), was used as a negative control in the experiment. Seeds were directly germinated and grown on the growth media in split plates of which one half contained Tm (25 ng/mL) while the other half contained only DMSO (corresponding mock control). The relative growth rate was measured seven days after growth. Means ± SEM; five biological replicates (n = 4-5 per replicate, with five seedlings for replicate). *P < 0.05, **P < 0.001 (Student’s t-test). Representative pictures are shown. Scale bar (black) = 1.3 cm.
DISCUSSION
The present study provides valuable resources and insights into the transcription regulatory program underlying the temporal dynamics of gene expression in the maize UPR. Using a systems-level approach based on eY1H screen assay and TF network mapping, we identified 831 PDIs between TFs associated with an array of biological processes and promoters of UPR target genes. The identified PDIs were partially validated by other experimental approaches and allowed to build a comprehensive TF network in the maize UPR. In the network, the positional bias of binding preference of TF families to specific fragments within 1-kb promoters not only emphasizes the potential interactions across TFs but also pinpoints a regulatory interacting hub based on the PPIM (Zhu et al., 2016a). Our cistrome analysis revealed a putative CRE, the TCGGC motif, which is highly enriched in the promoters of the DEGs during ER stress and that maps into a subnetwork of the TNU where multiple TFs converge into regulatory hubs. Finally, a functional analysis validated the relevance of the predicted regulatory hub TF, ZmUBER1, in the TNU using Arabidopsis as an experimental platform. Overall, the results provide interesting insights and generate testable hypotheses into the gene architecture potentially operating in the UPR in maize, as well as an eY1H resource that can be used as a springboard for future DNA-TF interaction studies in stress biology analyses.
Network analyses reveal a significant convergence of TF-mediated regulation of UPR gene responses and biological processes during ER stress responses
ER homeostasis is constantly challenged during development and in response to environmental cues (Pastor-Cantizano et al., 2020). Therefore, the UPR is inevitably associated with a variety of physiological processes and stresses, but the underlying mechanisms are still largely unknown. In this work, we demonstrate that multi-functional TFs are physically bound to the promoters of representative UPR target genes, potentially wiring the UPR to a variety of biological pathways involved in ER stress responses. It is noteworthy that a majority of TFs in the TNU are associated with abiotic stress, which is in line with the general view that ER stress is triggered directly or indirectly by abiotic stress (Liu et al., 2007, Moreno et al., 2012, Zhang et al., 2017). Furthermore, the UPR has been reported to be associated with vegetative and reproductive development (Iwata et al., 2008, Deng et al., 2013). Accordingly, our dataset identifies PDIs for potential association of the UPR with such pathways. The evidence that the clock oscillator TF, ZmCCA1b, binds to the promoter fragments of the UPR gene ZmBiP2, in which the CRE recognized by ZmCCA1b is abundantly represented, is a good example of such interactions between the UPR and physiological processes. The open question is how the ZmBiP2 promoter, but not others, is exclusively bound by ZmCCA1b. One possible explanation is that ZmBiP2, as a highly conserved molecular chaperone, may have a specific role to anticipate and adapt to daily rhythmic changes in the protein folding demand. This hypothesis is supported by a recent study in mammalian cells that the peak in the diurnally oscillated expression of BiP is narrowly followed by a surge in collagen synthesis at night, likely to prevent protein misfolding and ER stress (Pickard et al., 2019). Also, ER stress responses have been shown to be influenced by the circadian clock in mammalian cells (Maillo et al., 2017, Bu et al., 2018, Pickard et al., 2019). Additional experiments are needed to establish how the oscillation of ZmBiP2 expression is associated with protein homeostasis in maize. Nonetheless, collectively, these results and the evidence provided in our work that the promoter of individual UPR target genes can be bound by different TFs associated with a variety of biological processes support the hypothesis that a temporally-executed combinatorial binding of a gene promoter by multiple TFs is necessary to actuate ER stress responses in coordination with other biological processes. Our dataset represents a substantial data resource to test this hypothesis in the future.
A cistrome analysis delineates the dynamics of gene regulation in the UPR
Because the transcriptional regulation in the UPR has to be orchestrated by sequence-specific bindings of TFs, there have been significant efforts to identify CREs responsible for the gene expression changes in ER stress. Multiple highly conserved CREs that are responsive to ER stress have been reported (Yoshida et al., 1998, Iwata et al., 2008, Liu and Howell, 2010). In our work, not only we confirmed the conserved enrichment of some of these canonical CREs (ERSE-I, UPRE-I and their variants) in maize ER stress responses, but also established a dynamic temporal change of the enrichment levels during the time-course of ER stress. Our data support a complex nature of the TF-promoter binding landscape in the UPR well beyond the canonical CREs. Indeed, we identified a previously non-reported CRE (TCGGC motif) that was strongly enriched in promoters of DEGs from 3 h to 48 h of ER stress in our analysis. Although the role of the TCGGC motif as a CRE in the UPR has yet to be established experimentally, its enrichment in DEGs and the finding of a TCGGC motif-mediated network module, which consists of AP2/EREBP TFs and their UPR targets, strongly supports the hypothesis that TCGGC may be another key CRE in the UPR of maize. Because the AP2/EREBP is a large plant-specific TF family that is responsive to a broad array of stresses (Riechmann and Meyerowitz, 1998), the TCGGC motif and its binding-TFs could have evolved specifically in plants which, as a sessile organism, must maintain the ER homeostasis against a variety of unfavorable environmental conditions. Although the TCGGC motif has not been previously reported as an ER stress responsive-CRE in Arabidopsis, the core element (CGGC), which is recognized by a large number of AP2/EREBP TFs (O’Malley et al., 2016), is located in the promoters of the selected UPR biomarker genes in Arabidopsis. It is possible that the DNA sequence context surrounding the motif may differ across plant species. A maize-specific cistrome database, such as the one recently reported for AUXIN RESPONSE FACTORS TF family (Galli et al., 2018), would help to increase the accuracy for the motif-to-TF mapping. Nonetheless, the CRE analyses, along with the PDI and protein homology analyses, allowed us to identify the Arabidopsis homolog of ZmUBER1 for functional characterization. Our data indicated that the loss of the Arabidopsis homolog AtERF9 lead to an increase of transcript of spliced AtbZIP60 and enhanced resistance to chronic ER stress. These results pose that in ER stress conditions AtERF9 functions as a repressor of the adaptive UPR and ER stress defense mechanisms underpinning growth. Interestingly, in conditions of ER stress the erf9 mutant is viable, and despite the increase in spliced AtbZIP60 abundance verified in this mutant, the expression of AtBiP3, a target of bZIP60 (Iwata et al., 2008) was unaffected. These results are consistent with the possibility that the transcriptional regulation exerted by AtERF9 on the promoters of AtbZIP60 that contain the AtERF9 CRE depends on the presence of other transcription regulators acting in combination with AtERF9. In the alternative, possibly because of the functional redundancy of AtERF9 with other UPR regulators, the loss of AtERF9 may induce spliced AtbZIP60 transcripts to levels that are insufficient to trigger downstream responses.
Taken together, by integrating multiple molecular measurements (PDIs, transcriptome, PPIs, and cistrome) into a network context, our systems-level approach reveals a multilayered picture of the TF binding-mediated gene regulation in the UPR that significantly advances the understanding of the transcriptional landscape of ER stress. Therefore, the information presented in this work represents a rich resource for the research community to further elucidate the molecular basis of UPR at an experimental level and improve the agronomic performance of maize.
METHODS
Promoter cloning, yeast transformation and eY1H.
The genomic DNA of maize B73 was extracted from mature leaf of 2-weeks old plant using the DNeasy Plant Mini Kit (Qiagen, Valencia, CA, USA). Promoter fragments as described in Figure 1b and Table S1, were amplified from the maize B73 genomic DNA using Phusion High-Fidelity DNA Polymerase (New England BioLabs, Beverly, MA, USA). Promoter fragments were recombined either into pDONR P4-P1R using BP clonase II or pENTR 5’-TOPO (Life Technologies, Grand Island, NY, USA). The entry clones were fully sequenced, and then recombined into both pMW2 and pMW3 using LR clonase II (Life Technologies, Grand Island, NY, USA). The resulting pMW2 and pMW3 constructs were sequence-confirmed, and then transformed into the yeast strain YM4271, as previously described (Deplancke et al., 2004). See Table S1 for details of primer sequence, coordinates and cloning methods. The bZIP60-4 fragment failed to be transformed into the yeast genome despite multiple attempts. Thus, a total of 23 promoter fragments were used for the screening. Yeast colonies were screened for autoactivation and construct presence. Promoter strains were mated against a collection of 500 maize TF strains (Burdo et al., 2014) using a Singer Instruments ROTOR HDA robot platform in the Yeast One Hybrid Services Core at the Genome Center at the University of California Davis, as previously described (Gaudinier et al., 2011, Reece-Hoyes et al., 2011, Gaudinier et al., 2018). Each TF was screened twice in two technical replicates.
RNA-seq analysis.
The maize ER stress expression dataset (Srivastava et al., 2018) was downloaded from Gene Expression Omnibus (GSE111250). This expression dataset consists of control and the time-course of ER stress conditions in three biological replicates. The quality of raw reads was evaluated using FastQC (version 0.11.5). Reads were cleaned for quality and adapters with Cutadapt (version 1.8.1) (Martin, 2011) using a minimum base quality of 20 retaining reads with a minimum length of 30 nucleotides after trimming. Quality-filtered reads were aligned to the maize genome (B73 RefGen_v4) using Bowtie (version 2.2.4) (Langmead and Salzberg, 2012) and TopHat (version 2.0.14) (Kim et al., 2013) with a 10 bp minimum intron length and 15,000 bp maximum intron length. Fragments per kilobase exon model per million mapped reads (FPKM) were calculated using B73 RefGen_v4 gene model annotation with Cufflinks (version 1.3.0) (Trapnell et al., 2010). Per-gene read counts were measured using HTSeq (version 0.6.1p1) (Anders et al., 2015) in the union mode with a minimum mapping quality of 20 with stranded=reverse counting. Differential gene expression analysis was performed in each sample relative to the mock control using DESeq2 (version 1.16.1) (Love et al., 2014) within R (version 3.4.0). Genes of which the total count is < 100 were not included in the analysis. DEGs were obtained based on adjusted P-value < 0.01 and absolute log2-transformed fold change > 2. The heatmap of expression fold changes was produced using R package ggplots.
Validation of PDIs using the pre-release ChIP-seq dataset.
The ChIP-seq database consists of binding peaks of 110 TFs, among which 25 TFs participated in our PDIs. Peak coordinates of 16 TFs were available for downloading in bed format (http://www.epigenome.cuhk.edu.hk/C3C4.html. Downloaded peaks were merged and sorted using custom scripts. Genomic coordinates of promoter fragments of UPR target genes were identified by BLASTN searches against the B73 genome (maizesequence.org), saved in bed format and intersected with the ChIP-seq peaks using BEDTools (version 2.28.0) (Quinlan and Hall, 2010).
Visualization of networks.
All networks were visualized using Cytoscape (version 3.7.2) (Shannon et al., 2003). For the TNU (Figure 2a), a text file was compiled in which each row corresponded to one interaction and each interaction between source and target had four values of log2-transformed expression fold-change of source TF gene (3, 12, 24 and 48 h). Source files of all network construction are provided in Network S1 for Figure 2a, Network S2 for Figure 3c and Network S3 for Figure 4d.
Identification of cistrome.
The cistrome analysis was performed using tools of MEME suite (version 5.0.5) (http://meme-suite.org) with default parameters and modifications indicated below. Random model letter frequencies were calculated based on GC content in the maize genome (https://www.ncbi.nlm.nih.gov/genome/12). 1-kb upstream sequence of the transcription start site (TSS), hereafter called 1-kb promoter, in the B73 RefGen_v4 annotation was used. Enrichment of canonical cis-elements and their variants on promoters of ER stress-responsive genes was measured using AME (Buske et al., 2010). The same number of protein-coding genes with one of DEGs was randomly selected at each time-point. The 1-kb promoter sequences of randomly selected genes were used for input control. De novo motif discovery in the 1-kb promoters of ER stress-responsive genes was performed using MEME (Bailey and Elkan, 1994). To eliminate any de novo motifs not specific to ER stress-responsive genes, 280 protein-coding genes (a median number of DEGs across time-points) were randomly selected and served as a control for the analysis. Similarity of enriched motifs with the Plant Cistrome Database (O’Malley et al., 2016) was assessed using TOMTOM (Gupta et al., 2007). The heatmap of enrichment was produced using R package ggplots. Spearman rank correlation of binding frequency of TF families was calculated in R and visualized in heatmaps using R package corrplot.
Plant genotypes and growth
A. thaliana Columbia-0 (Col) ecotype was used as the WT control. The knock-out T-DNA mutant of AtERF9 (SALK_043407) was used for gene expression analyses and chronic ER stress assays in which the bzip28-2 (SALK_132285) bzip60-1 (SALK_050203) double mutant (Deng et al., 2013) was used as a negative control. T-DNA mutants were genotyped before use. Surface-sterilized seeds were plated on half-strength Linsmaier Skoog (LS) medium (Caisson Labs, Ontario, Canada) supplemented with 1% sucrose (Sigma-Aldrich, St. Louis, MO, USA), and 1.2% Agar (Acumedia, Lansing, MI, USA). After stratification in the dark at 4 °C for two days, plates were transferred to a growth chamber with 80 μmol m−2 s−1 under 16 h light:8 h dark with temperature 22 °C. For adaptive ER stress, seven-day-old seedlings were transferred to half-strength LS liquid buffer containing either 0.5 μg/mL Tm (Sigma-Aldrich, St. Louis, MO, USA) or DMSO alone as mock, and treated for 24 hours. After the treatment, whole seedlings were collected in three biological replicates (n = 12 per replicate), and immediately frozen in the liquid nitrogen. For chronic ER stress assay, seeds were plated on half-strength LS medium (Caisson Labs, Ontario, Canada) supplemented with 1% sucrose (Sigma-Aldrich, St. Louis, MO, USA), 1.2% Agar (Acumedia, Lansing, MI, USA) and 25 ng/mL Tm (Sigma-Aldrich, St. Louis, MO, USA) or DMSO alone as mock in five biological replicates (n = 4-5 per replicate, with five seedlings for replicate). Plates were photo-scanned seven days after growth. The relative length of primary roots was analyzed using ImageJ software (https://imagej.nih.gov).
RNA extraction, RT-PCR and qRT-PCR analyses
The frozen samples described above were ground to a fine powder in liquid nitrogen using a Retch MM400 Mixer Mill with zirconium oxide balls. Total RNA was extracted using the NucleoSpin RNA Plant kit (MACHEREY-NAGEL, Düren, Germany) according to the manufacturer’s instruction. cDNA was synthesized from 1 μg of DNaseI-treated total RNA using iScript cDNA Synthesis Kit (BIO-RAD, Hercules, CA, USA) according to the manufacturer’s instruction. For qRT-PCR, Fast SYBR Green Master Mix (Applied Biosystems, Foster City, CA, USA) was used in the presence of gene-specific primers and template cDNAs in an ABI7500 (Applied Biosystems, Foster City, CA, USA). The list of primers used in qRT-PCR is provided in Table S4. For RT-PCR, GoTaq DNA polymerase (Promega, Madison, WI, USA) was used in the presence of gene-specific primers and template cDNAs.
Data Availability Statement
The full result of eY1H assay including gene accession numbers is available in Table S2. Source files for network construction are provided in Network S1–3.
Supplementary Material
Figure S1. eY1H screens for UPR target gene promoters in maize. Six-way Venn diagram showing TFs bindings uniquely or redundantly to the UPR target genes. The total number of TFs binding to each gene is provided in parenthesis. A full list of TFs is provided in Table S2.
Figure S2. ZmCCA1b binds specifically to ZmBiP2 promoter fragments containing the evening-like element, generating a rhythmic expression of ZmBiP2. (a), Schemes of the 1-kb promoter of UPR target genes. Promoter fragments displayed with solid blue bar indicate possession of interaction with ZmCCA1b while the promoter fragment displayed by the dashed bar indicates no interaction. (b), Rhythmic diurnal oscillation of ZmBiP2 expression under constant light condition following entraining in the long-day condition obtained from the DIURNAL database (Mockler et al., 2007). The modeled pattern was also obtained from the DIURNAL for better visualization. White and dashed black bars on X-axis indicate the subjected day and night, respectively.
Figure S3. Correlation of TF binding preference to promoter fragments of each UPR target gene. Heatmaps of Spearman rank correlation calculated using the binding frequency to promoter fragments (Figure 3b). The colors represent the correlation value from inverse correlation (blue) and perfect correlation (red). (a), ZmPRX1. (b), ZmBiP2. (c), ZmSAR1. (d), ZmBi1. (e), ZmbZIP60. (f), ZmCP1.
Figure S4. Protein sequence alignment of ZmUBER1 and AtERF9. The highly conserved AP2 domain is highlighted with an orange box. The analysis was performed using the Clustal Omega program (Sievers et al., 2011).
Table S1. Creation of entry clones in our eY1H screen. For BP cloning, attB4 (GGGGACAACTTTGTATAGAAAAGTTG) and attB1R (GGGGACTGCTTTTTTGTACAAACTTG) sites are attached at 5’ of the forward and reverse primers, respectively.
Table S2. PDIs obtained from our eY1H screen. Each row indicates a PDI.
Table S3. Experimentally determined protein-protein interactions among TFs that bound to UPR target genes.
Table S4. List of primers used for genotyping and gene expression analyses.
Network S1. Network source file of Figure 2a.
Network S2. Network source file of Figure 3c.
Network S3. Network source file of Figure 4d.
Acknowledgements
This study was supported primarily by the National Science Foundation Plant Genome Research Program award (IOS 1444339) with contributing support from by the Great Lakes Bioenergy Research Center, U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research (Award DE-SC0018409), the National Institutes of Health (GM136637), Chemical Sciences, Geoscience and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy (award number DE-FG02-91ER20021) and MSU AgBioResearch (MICL02598). We thank Dr. Stephen H. Howell (Iowa State University) for helpful discussion. We also thank the Genome Center and Proteomics Core, University of California, Davis for the eY1H screen.
Footnotes
Conflict of Interest Statement
The authors declare no conflicts of interest.
REFERENCES
- Agostini RB, Postigo A, Rius SP, Rech GE, Campos-Bermudez VA and Vargas WA (2019) Long-Lasting Primed State in Maize Plants: Salicylic Acid and Steroid Signaling Pathways as Key Players in the Early Activation of Immune Responses in Silks. Mol Plant Microbe Interact, 32, 95–106. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT and Huber W (2015) HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila LM, Obeidat W, Earl H, Niu X, Hargreaves W and Lukens L (2018) Shared and genetically distinct Zea mays transcriptome responses to ongoing and past low temperature exposure. BMC genomics, 19, 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL and Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol, 2, 28–36. [PubMed] [Google Scholar]
- Bao Y and Howell SH (2017) The Unfolded Protein Response Supports Plant Development and Defense as well as Responses to Abiotic Stress. Front Plant Sci, 8, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrière Y, Courtial A, Soler M and Grima-Pettenati J (2015) Toward the identification of genes underlying maize QTLs for lignin content, focusing on colocalizations with lignin biosynthetic genes and their regulatory MYB and NAC transcription factors. Molecular breeding, 35, 87. [Google Scholar]
- Bhatia R, Dalton S, Roberts LA, Moron-Garcia OM, Iacono R, Kosik O, Gallagher JA and Bosch M (2019) Modified expression of ZmMYB167 in Brachypodium distachyon and Zea mays leads to increased cell wall lignin and phenolic content. Scientific Reports, 9, 8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady SM, Zhang L, Megraw M, Martinez NJ, Jiang E, Yi CS, Liu W, Zeng A, Taylor-Teeples M, Kim D, Ahnert S, Ohler U, Ware D, Walhout AJ and Benfey PN (2011) A stele-enriched gene regulatory network in the Arabidopsis root. Mol Syst Biol, 7, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu Y, Yoshida A, Chitnis N, Altman BJ, Tameire F, Oran A, Gennaro V, Armeson KE, McMahon SB, Wertheim GB, Dang CV, Ruggero D, Koumenis C, Fuchs SY and Diehl JA (2018) A PERK-miR-211 axis suppresses circadian regulators and protein synthesis to promote cancer cell survival. Nat Cell Biol, 20, 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdo B, Gray J, Goetting-Minesky MP, Wittler B, Hunt M, Li T, Velliquette D, Thomas J, Gentzel I, dos Santos Brito M, Mejía-Guerra MK, Connolly LN, Qaisi D, Li W, Casas MI, Doseff AI and Grotewold E (2014) The Maize TFome--development of a transcription factor open reading frame collection for functional genomics. Plant J, 80, 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske FA, Bodén M, Bauer DC and Bailey TL (2010) Assigning roles to DNA regulatory motifs using comparative genomics. Bioinformatics, 26, 860–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebulski J, Malouin J, Pinches N, Cascio V and Austriaco N (2011) Yeast Bax inhibitor, Bxi1p, is an ER-localized protein that links the unfolded protein response and programmed cell death in Saccharomyces cerevisiae. PLoS One, 6, e20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y and Brandizzi F (2012) AtIRE1A/AtIRE1B and AGB1 independently control two essential unfolded protein response pathways in Arabidopsis. Plant J, 69, 266–277. [DOI] [PubMed] [Google Scholar]
- Chuck GS, Brown PJ, Meeley R and Hake S (2014) Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation. Proceedings of the National Academy of Sciences, 111, 18775–18780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosio C and Dunand C (2009) Specific functions of individual class III peroxidase genes. J Exp Bot, 60, 391–408. [DOI] [PubMed] [Google Scholar]
- De Vetten NC and Ferl RJ (1995) Characterization of a maize G-box binding factor that is induced by hypoxia. The Plant Journal, 7, 589–601. [DOI] [PubMed] [Google Scholar]
- Deng Y, Humbert S, Liu JX, Srivastava R, Rothstein SJ and Howell SH (2011) Heat induces the splicing by IRE1 of a mRNA encoding a transcription factor involved in the unfolded protein response in Arabidopsis. Proc Natl Acad Sci U S A, 108, 7247–7252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Srivastava R and Howell SH (2013) Protein kinase and ribonuclease domains of IRE1 confer stress tolerance, vegetative growth, and reproductive development in Arabidopsis. Proc Natl Acad Sci U S A, 110, 19633–19638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplancke B, Dupuy D, Vidal M and Walhout AJ (2004) A gateway-compatible yeast one-hybrid system. Genome Res, 14, 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Virlouvet L, Liu N, Riethoven JJ, Fromm M and Avramova Z (2014) Dehydration stress memory genes of Zea mays; comparison with Arabidopsis thaliana. BMC Plant Biol, 14, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Huang M, Zhang Z and Cheng S (2014) Genome-wide analysis of the AP2/ERF gene family in maize waterlogging stress response. Euphytica, 198, 115–126. [Google Scholar]
- Du H, Zhu J, Su H, Huang M, Wang H, Ding S, Zhang B, Luo A, Wei S and Tian X (2017a) Bulked segregant RNA-Seq reveals differential expression and SNPs of candidate genes associated with Waterlogging tolerance in maize. Frontiers in plant science, 8, 1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Wang G, Ji J, Shi L, Guan C and Jin C (2017b) Comparative transcriptome analysis of transcription factors in different maize varieties under salt stress conditions. Plant growth regulation, 81, 183–195. [Google Scholar]
- Fink EE, Moparthy S, Bagati A, Bianchi-Smiraglia A, Lipchick BC, Wolff DW, Roll MV, Wang J, Liu S, Bakin AV, Kandel ES, Lee AH and Nikiforov MA (2018) XBP1-KLF9 Axis Acts as a Molecular Rheostat to Control the Transition from Adaptive to Cytotoxic Unfolded Protein Response. Cell Rep, 25, 212–223.e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli M, Khakhar A, Lu Z, Chen Z, Sen S, Joshi T, Nemhauser JL, Schmitz RJ and Gallavotti A (2018) The DNA binding landscape of the maize AUXIN RESPONSE FACTOR family. Nat Commun, 9, 4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudinier A, Rodriguez-Medina J, Zhang L, Olson A, Liseron-Monfils C, Bågman AM, Foret J, Abbitt S, Tang M, Li B, Runcie DE, Kliebenstein DJ, Shen B, Frank MJ, Ware D and Brady SM (2018) Transcriptional regulation of nitrogen-associated metabolism and growth. Nature, 563, 259–264. [DOI] [PubMed] [Google Scholar]
- Gaudinier A, Zhang L, Reece-Hoyes JS, Taylor-Teeples M, Pu L, Liu Z, Breton G, Pruneda-Paz JL, Kim D, Kay SA, Walhout AJ, Ware D and Brady SM (2011) Enhanced Y1H assays for Arabidopsis. Nat Methods, 8, 1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge M, Liu Y, Jiang L, Wang Y, Lv Y, Zhou L, Liang S, Bao H and Zhao H (2018) Genome-wide analysis of maize NLP transcription factor family revealed the roles in nitrogen response. Plant growth regulation, 84, 95–105. [Google Scholar]
- Godfray HC, Beddington JR, Crute IR, Haddad L, Lawrence D, Muir JF, Pretty J, Robinson S, Thomas SM and Toulmin C (2010) Food security: the challenge of feeding 9 billion people. Science, 327, 812–818. [DOI] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL and Noble WS (2007) Quantifying similarity between motifs. Genome Biol, 8, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, Kilberg MS, Sartor MA and Kaufman RJ (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol, 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, Takahashi H, Wakasa Y, Kawakatsu T and Takaiwa F (2013) Identification of a cis-element that mediates multiple pathways of the endoplasmic reticulum stress response in rice. Plant J, 74, 248–257. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Wakasa Y, Takahashi H, Kawakatsu T and Takaiwa F (2012) Signal transduction by IRE1-mediated splicing of bZIP50 and other stress sensors in the endoplasmic reticulum stress response of rice. The Plant Journal, 69, 946–956. [DOI] [PubMed] [Google Scholar]
- Hayes KR, Beatty M, Meng X, Simmons CR, Habben JE and Danilevskaya ON (2010) Maize global transcriptomics reveals pervasive leaf diurnal rhythms but rhythms in developing ears are largely limited to the core oscillator. PLoS One, 5, e12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell SH (2013) Endoplasmic reticulum stress responses in plants. Annu Rev Plant Biol, 64, 477–499. [DOI] [PubMed] [Google Scholar]
- Ikeuchi M, Shibata M, Rymen B, Iwase A, Bågman A-M, Watt L, Coleman D, Favero DS, Takahashi T and Ahnert SE (2018) A gene regulatory network for cellular reprogramming in plant regeneration. Plant and Cell Physiology, 59, 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata Y, Fedoroff NV and Koizumi N (2008) Arabidopsis bZIP60 is a proteolysis-activated transcription factor involved in the endoplasmic reticulum stress response. Plant Cell, 20, 3107–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Liu X, Jia W, Liu H, Li W, Peng Y, Du Y, Wang Y, Yin Y and Zhang X (2018) ZmCOL3, a CCT gene represses flowering in maize by interfering with the circadian clock and activating expression of ZmCCT. Journal of integrative plant biology, 60, 465–480. [DOI] [PubMed] [Google Scholar]
- Kanodia P, Vijayapalani P, Srivastava R, Bi R, Liu P, Miller WA and Howell SH (2020) Control of translation during the unfolded protein response in maize seedlings: Life without PERKs. Plant Direct, 4, e00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebede AZ, Johnston A, Schneiderman D, Bosnich W and Harris LJ (2018) Transcriptome profiling of two maize inbreds with distinct responses to Gibberella ear rot disease to identify candidate resistance genes. BMC genomics, 19, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R and Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol, 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko DK, Rohozinski D, Song Q, Taylor SH, Juenger TE, Harmon FG and Chen ZJ (2016) Temporal Shift of Circadian-Mediated Gene Expression and Carbon Fixation Contributes to Biomass Heterosis in Maize Hybrids. PLoS Genet, 12, e1006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokame K, Kato H and Miyata T (2001) Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J Biol Chem, 276, 9199–9205. [DOI] [PubMed] [Google Scholar]
- Koryachko A, Matthiadis A, Muhammad D, Foret J, Brady SM, Ducoste JJ, Tuck J, Long TA and Williams C (2015) Clustering and Differential Alignment Algorithm: Identification of Early Stage Regulators in the Arabidopsis thaliana Iron Deficiency Response. PLoS One, 10, e0136591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YS, Renna L, Yarema J, Ruberti C, He SY and Brandizzi F (2018) Salicylic acid-independent role of NPR1 is required for protection from proteotoxic stress in the plant endoplasmic reticulum. Proc Natl Acad Sci U S A, 115, E5203–E5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B and Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon B and Tjian R (2000) Orchestrated response: a symphony of transcription factors for gene control. Genes Dev, 14, 2551–2569. [DOI] [PubMed] [Google Scholar]
- Lesk C, Rowhani P and Ramankutty N (2016) Influence of extreme weather disasters on global crop production. Nature, 529, 84–87. [DOI] [PubMed] [Google Scholar]
- Li B, Tang M, Nelson A, Caligagan H, Zhou X, Clark-Wiest C, Ngo R, Brady SM and Kliebenstein DJ (2018) Network-Guided Discovery of Extensive Epistasis between Transcription Factors Involved in Aliphatic Glucosinolate Biosynthesis. Plant Cell, 30, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Wang J, Sun Q, Li W, Yu Y, Zhao M and Meng Z (2017) Expression analysis of genes encoding double B-box zinc finger proteins in maize. Functional & integrative genomics, 17, 653–666. [DOI] [PubMed] [Google Scholar]
- Li Y, Humbert S and Howell SH (2012) ZmbZIP60 mRNA is spliced in maize in response to ER stress. BMC Res Notes, 5, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX and Howell SH (2010) bZIP28 and NF-Y transcription factors are activated by ER stress and assemble into a transcriptional complex to regulate stress response genes in Arabidopsis. Plant Cell, 22, 782–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Srivastava R, Che P and Howell SH (2007) Salt stress responses in Arabidopsis utilize a signal transduction pathway related to endoplasmic reticulum stress signaling. The Plant Journal, 51, 897–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W and Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SJ, Yang ZT, Sun L, Song ZT and Liu JX (2012) Conservation of IRE1-regulated bZIP74 mRNA unconventional splicing in rice (Oryza sativa L.) involved in ER stress responses. Mol Plant, 5, 504–514. [DOI] [PubMed] [Google Scholar]
- Luan M, Xu M, Lu Y, Zhang L, Fan Y and Wang L (2015) Expression of zma-miR169 miRNAs and their target ZmNF-YA genes in response to abiotic stress in maize leaves. Gene, 555, 178–185. [DOI] [PubMed] [Google Scholar]
- Ludwig Y, Zhang Y and Hochholdinger F (2013) The maize (Zea mays L.) AUXIN/INDOLE-3-ACETIC ACID gene family: phylogeny, synteny, and unique root-type and tissue-specific expression patterns during development. PLoS One, 8, e78859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Liu C, Li Z, Ran Q, Xie G, Wang B, Fang S, Chu J and Zhang J (2018) ZmbZIP4 Contributes to Stress Resistance in Maize by Regulating ABA Synthesis and Root Development. Plant Physiol, 178, 753–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher KA, Bajic M, Kajala K, Reynoso M, Pauluzzi G, West DA, Zumstein K, Woodhouse M, Bubb K, Dorrity MW, Queitsch C, Bailey-Serres J, Sinha N, Brady SM and Deal RB (2018) Profiling of Accessible Chromatin Regions across Multiple Plant Species and Cell Types Reveals Common Gene Regulatory Principles and New Control Modules. Plant Cell, 30, 15–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillo C, Martín J, Sebastián D, Hernández-Alvarez M, García-Rocha M, Reina O, Zorzano A, Fernandez M and Méndez R (2017) Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat Cell Biol, 19, 94–105. [DOI] [PubMed] [Google Scholar]
- Mantilla-Perez MB and Salas Fernandez MG (2017) Differential manipulation of leaf angle throughout the canopy: current status and prospects. Journal of Experimental Botany, 68, 5699–5717. [DOI] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal, 17, 10–12. [Google Scholar]
- Martínez IM and Chrispeels MJ (2003) Genomic analysis of the unfolded protein response in Arabidopsis shows its connection to important cellular processes. Plant Cell, 15, 561–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Ruberti C, Gong Z and Brandizzi F (2017) CPR5 modulates salicylic acid and the unfolded protein response to manage tradeoffs between plant growth and stress responses. Plant J, 89, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min H, Zheng J and Wang J (2014) Maize ZmRAV1 contributes to salt and osmotic stress tolerance in transgenic arabidopsis. Journal of Plant Biology, 57, 28–42. [Google Scholar]
- Mockler TC, Michael TP, Priest HD, Shen R, Sullivan CM, Givan SA, McEntee C, Kay SA and Chory J (2007) The DIURNAL project: DIURNAL and circadian expression profiling, model-based pattern matching, and promoter analysis. Cold Spring Harb Symp Quant Biol, 72, 353–363. [DOI] [PubMed] [Google Scholar]
- Moreno AA, Mukhtar MS, Blanco F, Boatwright JL, Moreno I, Jordan MR, Chen Y, Brandizzi F, Dong X and Orellana A (2012) IRE1/bZIP60-mediated unfolded protein response plays distinct roles in plant immunity and abiotic stress responses. PLoS One, 7, e31944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller DS, Wise KA, Sisson AJ, Allen TW, Bergstrom GC, Bosley DB, Bradley CA, Broders KD, Byamukama E and Chilvers MI (2016) Corn yield loss estimates due to diseases in the United States and Ontario, Canada from 2012 to 2015. Plant health progress, 17, 211–222. [Google Scholar]
- Nagashima Y, Iwata Y, Ashida M, Mishiba K and Koizumi N (2014) Exogenous salicylic acid activates two signaling arms of the unfolded protein response in Arabidopsis. Plant Cell Physiol, 55, 1772–1778. [DOI] [PubMed] [Google Scholar]
- Nakańo A and Muramatsu M (1989) A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol, 109, 2677–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawkar GM, Kang CH, Maibam P, Park JH, Jung YJ, Chae HB, Chi YH, Jung IJ, Kim WY, Yun DJ and Lee SY (2017) HY5, a positive regulator of light signaling, negatively controls the unfolded protein response in. Proc Natl Acad Sci U S A, 114, 2084–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley RC, Huang SC, Song L, Lewsey MG, Bartlett A, Nery JR, Galli M, Gallavotti A and Ecker JR (2016) Cistrome and Epicistrome Features Shape the Regulatory DNA Landscape. Cell, 165, 1280–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oerke E-C (2006) Crop losses to pests. The Journal of Agricultural Science, 144, 31–43. [Google Scholar]
- Oh DH, Kwon CS, Sano H, Chung WI and Koizumi N (2003) Conservation between animals and plants of the cis-acting element involved in the unfolded protein response. Biochem Biophys Res Commun, 301, 225–230. [DOI] [PubMed] [Google Scholar]
- Oono Y, Wakasa Y, Hirose S, Yang L, Sakuta C and Takaiwa F (2010) Analysis of ER stress in developing rice endosperm accumulating beta-amyloid peptide. Plant Biotechnol J, 8, 691–718. [DOI] [PubMed] [Google Scholar]
- Pajerowska-Mukhtar KM, Wang W, Tada Y, Oka N, Tucker CL, Fonseca JP and Dong X (2012) The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-defense transition. Curr Biol, 22, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor-Cantizano N, Ko DK, Angelos E, Pu Y and Brandizzi F (2020) Functional Diversification of ER Stress Responses in Arabidopsis. Trends Biochem Sci, 45, 123–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautler M, Eveland AL, LaRue T, Yang F, Weeks R, Lunde C, Je BI, Meeley R, Komatsu M, Vollbrecht E, Sakai H and Jackson D (2015) FASCIATED EAR4 encodes a bZIP transcription factor that regulates shoot meristem size in maize. Plant Cell, 27, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard A, Chang J, Alachkar N, Calverley B, Garva R, Arvan P, Meng QJ and Kadler KE (2019) Preservation of circadian rhythms by the protein folding chaperone, BiP. FASEB J, 33, 7479–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda-Paz JL, Breton G, Nagel DH, Kang SE, Bonaldi K, Doherty CJ, Ravelo S, Galli M, Ecker JR and Kay SA (2014) A genome-scale resource for the functional characterization of Arabidopsis transcription factors. Cell Rep, 8, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian D, Chen G, Tian L and Qu LQ (2018) OsDER1 Is an ER-Associated Protein Degradation Factor That Responds to ER Stress. Plant Physiol, 178, 402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Z, Qi W, Wang Q, Feng Y.n., Yang Q, Zhang N, Wang S, Tang Y and Song R (2016) ZmMADS47 regulates zein gene transcription through interaction with Opaque2. PLoS genetics, 12, e1005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR and Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray DK, Mueller ND, West PC and Foley JA (2013) Yield Trends Are Insufficient to Double Global Crop Production by 2050. PLoS One, 8, e66428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Diallo A, Lajoie B, Kent A, Shrestha S, Kadreppa S, Pesyna C, Dekker J, Myers CL and Walhout AJ (2011) Enhanced yeast one-hybrid assays for high-throughput gene-centered regulatory network mapping. Nat Methods, 8, 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann JL and Meyerowitz EM (1998) The AP2/EREBP family of plant transcription factors. Biol Chem, 379, 633–646. [DOI] [PubMed] [Google Scholar]
- Ruberti C, Lai Y and Brandizzi F (2018) Recovery from temporary endoplasmic reticulum stress in plants relies on the tissue-specific and largely independent roles of bZIP28 and bZIP60, as well as an antagonizing function of BAX-Inhibitor 1 upon the pro-adaptive signaling mediated by bZIP28. Plant J, 93, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvo SA, Hirsch CN, Buell CR, Kaeppler SM and Kaeppler HF (2014) Whole transcriptome profiling of maize during early somatic embryogenesis reveals altered expression of stress factors and embryogenesis-related genes. PLoS One, 9, e111407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD and Higgins DG (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skirycz A, Reichelt M, Burow M, Birkemeyer C, Rolcik J, Kopka J, Zanor MI, Gershenzon J, Strnad M and Szopa J (2006) DOF transcription factor AtDof1. 1 (OBP2) is part of a regulatory network controlling glucosinolate biosynthesis in Arabidopsis. The Plant Journal, 47, 10–24. [DOI] [PubMed] [Google Scholar]
- Smit ME, McGregor SR, Sun H, Gough C, Bågman A-M, Soyars CL, Kroon JT, Gaudinier A, Williams CJ and Yang X (2020) A PXY-mediated transcriptional network integrates signaling mechanisms to control vascular development in Arabidopsis. The Plant Cell, 32, 319–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon M, Belenghi B, Delledonne M, Menachem E and Levine A (1999) The involvement of cysteine proteases and protease inhibitor genes in the regulation of programmed cell death in plants. The Plant Cell, 11, 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K, Kim HC, Shin S, Kim K-H, Moon J-C, Kim JY and Lee B-M (2017) Transcriptome analysis of flowering time genes under drought stress in maize leaves. Frontiers in plant science, 8, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song ZT, Sun L, Lu SJ, Tian Y, Ding Y and Liu JX (2015) Transcription factor interaction with COMPASS-like complex regulates histone H3K4 trimethylation for specific gene expression in plants. Proc Natl Acad Sci U S A, 112, 2900–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks E, Wachsman G and Benfey PN (2013) Spatiotemporal signalling in plant development. Nat Rev Genet, 14, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks EE, Drapek C, Gaudinier A, Li S, Ansariola M, Shen N, Hennacy JH, Zhang J, Turco G, Petricka JJ, Foret J, Hartemink AJ, Gordân R, Megraw M, Brady SM and Benfey PN (2016) Establishment of Expression in the SHORTROOT-SCARECROW Transcriptional Cascade through Opposing Activities of Both Activators and Repressors. Dev Cell, 39, 585–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava R, Li Z, Russo G, Tang J, Bi R, Muppirala U, Chudalayandi S, Severin A, He M, Vaitkevicius SI, Lawrence-Dill CJ, Liu P, Stapleton AE, Bassham DC, Brandizzi F and Howell SH (2018) Response to Persistent ER Stress in Plants: A Multiphasic Process That Transitions Cells from Prosurvival Activities to Cell Death. Plant Cell, 30, 1220–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Cao Y, Ku L, Yao W, Cao Y, Ren Z, Dou D, Wang H, Ren Z and Liu H (2018) Dual functions of ZmNF-YA3 in photoperiod-dependent flowering and abiotic stress responses in maize. Journal of experimental botany, 69, 5177–5189. [DOI] [PubMed] [Google Scholar]
- Sun L, Yang ZT, Song ZT, Wang MJ, Lu SJ and Liu JX (2013) The plant-specific transcription factor gene NAC103 is induced by bZIP60 through a new cis-regulatory element to modulate the unfolded protein response in Arabidopsis. Plant J, 76, 274–286. [DOI] [PubMed] [Google Scholar]
- Taylor-Teeples M, Lin L, de Lucas M, Turco G, Toal TW, Gaudinier A, Young NF, Trabucco GM, Veling MT, Lamothe R, Handakumbura PP, Xiong G, Wang C, Corwin J, Tsoukalas A, Zhang L, Ware D, Pauly M, Kliebenstein DJ, Dehesh K, Tagkopoulos I, Breton G, Pruneda-Paz JL, Ahnert SE, Kay SA, Hazen SP and Brady SM (2015) An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature, 517, 571–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ and Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol, 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS and Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell, 101, 249–258. [DOI] [PubMed] [Google Scholar]
- Walter P and Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science, 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang B, Li Z, Ran Q, Li P, Peng Z and Zhang J (2018a) ZmNF-YB16 overexpression improves drought resistance and yield by enhancing photosynthesis and the antioxidant capacity of maize plants. Frontiers in plant science, 9, 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Liu C, Zhang D, He C, Zhang J and Li Z (2019) Effects of maize organ-specific drought stress response on yields from transcriptome analysis. BMC plant biology, 19, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Liu J, Wang Y, Zhao Y, Jiang H and Cheng B (2015a) Systematic Analysis of the Maize PHD-Finger Gene Family Reveals a Subfamily Involved in Abiotic Stress Response. Int J Mol Sci, 16, 23517–23544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QL, Sun AZ, Chen ST, Chen LS and Guo FQ (2018b) SPL6 represses signalling outputs of ER stress in control of panicle cell death in rice. Nat Plants, 4, 280–288. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang Q, Zhao Y, Han G and Zhu S (2015b) Systematic analysis of maize class III peroxidase gene family reveals a conserved subfamily involved in abiotic stress response. Gene, 566, 95–108. [DOI] [PubMed] [Google Scholar]
- Wang YG, Yu HQ, Zhang YY, Lai CX, She YH, Li WC and Fu FL (2014) Interaction between abscisic acid receptor PYL3 and protein phosphatase type 2C in response to ABA signaling in maize. Gene, 549, 179–185. [DOI] [PubMed] [Google Scholar]
- Webber H, Ewert F, Olesen JE, Müller C, Fronzek S, Ruane AC, Bourgault M, Martre P, Ababaei B, Bindi M, Ferrise R, Finger R, Fodor N, Gabaldón-Leal C, Gaiser T, Jabloun M, Kersebaum KC, Lizaso JI, Lorite IJ, Manceau L, Moriondo M, Nendel C, Rodríguez A, Ruiz-Ramos M, Semenov MA, Siebert S, Stella T, Stratonovitch P, Trombi G and Wallach D (2018) Diverging importance of drought stress for maize and winter wheat in Europe. Nat Commun, 9, 4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuebbles DJ, Fahey DW and Hibbard KA (2017) Climate science special report: fourth national climate assessment, volume I. [Google Scholar]
- Xue Y, Warburton ML, Sawkins M, Zhang X, Setter T, Xu Y, Grudloyma P, Gethi J, Ribaut J-M and Li W (2013) Genome-wide association analysis for nine agronomic traits in maize under well-watered and water-stressed conditions. Theoretical and applied genetics, 126, 2587–2596. [DOI] [PubMed] [Google Scholar]
- Yang F, Li W, Jiang N, Yu H, Morohashi K, Ouma WZ, Morales-Mantilla DE, Gomez-Cano FA, Mukundi E, Prada-Salcedo LD, Velazquez RA, Valentin J, Mejía-Guerra MK, Gray J, Doseff AI and Grotewold E (2017) A Maize Gene Regulatory Network for Phenolic Metabolism. Mol Plant, 10, 498–515. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Haze K, Yanagi H, Yura T and Mori K (1998) Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem, 273, 33741–33749. [DOI] [PubMed] [Google Scholar]
- Yu CP, Chen SC, Chang YM, Liu WY, Lin HH, Lin JJ, Chen HJ, Lu YJ, Wu YH, Lu MY, Lu CH, Shih AC, Ku MS, Shiu SH, Wu SH and Li WH (2015) Transcriptome dynamics of developing maize leaves and genomewide prediction of cis elements and their cognate transcription factors. Proc Natl Acad Sci U S A, 112, E2477–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Meng X, Liu Y, Li N, Zhang A, Wang T-J, Jiang L, Pang J, Zhao X and Qi X (2018) The chromatin remodeler ZmCHB101 impacts expression of osmotic stress-responsive genes in maize. Plant molecular biology, 97, 451–465. [DOI] [PubMed] [Google Scholar]
- Yue R, Lu C, Sun T, Peng T, Han X, Qi J, Yan S and Tie S (2015) Identification and expression profiling analysis of calmodulin-binding transcription activator genes in maize (Zea mays L.) under abiotic and biotic stresses. Frontiers in plant science, 6, 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Liu D, Lv X, Wang Y, Xun Z, Liu Z, Li F and Lu H (2014a) The cysteine protease CEP1, a key executor involved in tapetal programmed cell death, regulates pollen development in Arabidopsis. The Plant Cell, 26, 2939–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Liu D, Lv X, Wang Y, Xun Z, Liu Z, Li F and Lu H (2014b) The cysteine protease CEP1, a key executor involved in tapetal programmed cell death, regulates pollen development in Arabidopsis. Plant Cell, 26, 2939–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ohyama K, Boudet J, Chen Z, Yang J, Zhang M, Muranaka T, Maurel C, Zhu JK and Gong Z (2008) Dolichol biosynthesis and its effects on the unfolded protein response and abiotic stress resistance in Arabidopsis. Plant Cell, 20, 1879–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SS, Yang H, Ding L, Song ZT, Ma H, Chang F and Liu JX (2017) Tissue-Specific Transcriptomics Reveals an Important Role of the Unfolded Protein Response in Maintaining Fertility upon Heat Stress in Arabidopsis. Plant Cell, 29, 1007–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Xie S, Han J, Zhou Y, Liu C, Zhou Z, Wang F, Cheng Z, Zhang J and Hu Y (2019) Integrated transcriptome, small RNA, and degradome analysis reveals the complex network regulating starch biosynthesis in maize. BMC genomics, 20, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y-M, Zhang S-Z and Zheng C-C (2014c) Genomewide analysis of LATERAL ORGAN BOUNDARIES Domain gene family in Zea mays. Journal of genetics, 93, 79–91. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Ma Q, Jin X, Peng X, Liu J, Deng L, Yan H, Sheng L, Jiang H and Cheng B (2014) A novel maize homeodomain-leucine zipper (HD-Zip) I gene, Zmhdz10, positively regulates drought and salt tolerance in both rice and Arabidopsis. Plant Cell Physiol, 55, 1142–1156. [DOI] [PubMed] [Google Scholar]
- Zheng J, Zhao J, Zhang J, Fu J, Gou M, Dong Z, Hou W, Huang Q and Wang G (2006) Comparative expression profiles of maize genes from a water stress-specific cDNA macroarray in response to high-salinity, cold or abscisic acid. Plant science, 170, 1125–1132. [Google Scholar]
- Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, Cramer P and Taipale J (2018) The interaction landscape between transcription factors and the nucleosome. Nature, 562, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Wu A, Xu XJ, Xiao PP, Lu L, Liu J, Cao Y, Chen L, Wu J and Zhao XM (2016a) PPIM: A Protein-Protein Interaction Database for Maize. Plant Physiol, 170, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Wang G, Li C, Li Q, Gao Y, Chen F and Xia G (2019) Maize Sep15-like functions in endoplasmic reticulum and reactive oxygen species homeostasis to promote salt and osmotic stress resistance. Plant Cell Environ, 42, 1486–1502. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Yan J, Liu W, Liu L, Sheng Y, Sun Y, Li Y, Scheller HV, Jiang M, Hou X, Ni L and Zhang A (2016b) Phosphorylation of a NAC Transcription Factor by a Calcium/Calmodulin-Dependent Protein Kinase Regulates Abscisic Acid-Induced Antioxidant Defense in Maize. Plant Physiol, 171, 1651–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. eY1H screens for UPR target gene promoters in maize. Six-way Venn diagram showing TFs bindings uniquely or redundantly to the UPR target genes. The total number of TFs binding to each gene is provided in parenthesis. A full list of TFs is provided in Table S2.
Figure S2. ZmCCA1b binds specifically to ZmBiP2 promoter fragments containing the evening-like element, generating a rhythmic expression of ZmBiP2. (a), Schemes of the 1-kb promoter of UPR target genes. Promoter fragments displayed with solid blue bar indicate possession of interaction with ZmCCA1b while the promoter fragment displayed by the dashed bar indicates no interaction. (b), Rhythmic diurnal oscillation of ZmBiP2 expression under constant light condition following entraining in the long-day condition obtained from the DIURNAL database (Mockler et al., 2007). The modeled pattern was also obtained from the DIURNAL for better visualization. White and dashed black bars on X-axis indicate the subjected day and night, respectively.
Figure S3. Correlation of TF binding preference to promoter fragments of each UPR target gene. Heatmaps of Spearman rank correlation calculated using the binding frequency to promoter fragments (Figure 3b). The colors represent the correlation value from inverse correlation (blue) and perfect correlation (red). (a), ZmPRX1. (b), ZmBiP2. (c), ZmSAR1. (d), ZmBi1. (e), ZmbZIP60. (f), ZmCP1.
Figure S4. Protein sequence alignment of ZmUBER1 and AtERF9. The highly conserved AP2 domain is highlighted with an orange box. The analysis was performed using the Clustal Omega program (Sievers et al., 2011).
Table S1. Creation of entry clones in our eY1H screen. For BP cloning, attB4 (GGGGACAACTTTGTATAGAAAAGTTG) and attB1R (GGGGACTGCTTTTTTGTACAAACTTG) sites are attached at 5’ of the forward and reverse primers, respectively.
Table S2. PDIs obtained from our eY1H screen. Each row indicates a PDI.
Table S3. Experimentally determined protein-protein interactions among TFs that bound to UPR target genes.
Table S4. List of primers used for genotyping and gene expression analyses.
Network S1. Network source file of Figure 2a.
Network S2. Network source file of Figure 3c.
Network S3. Network source file of Figure 4d.
Data Availability Statement
The full result of eY1H assay including gene accession numbers is available in Table S2. Source files for network construction are provided in Network S1–3.