

Abstract
Loss of the neuromuscular junction (NMJ) is an early and critical hallmark in all forms of ALS. The study design was to develop a functional NMJ disease model by integrating motoneurons (MNs) differentiated from multiple ALS-patients’ induced pluripotent stem cells (iPSCs) and primary human muscle into a chambered system. NMJ functionality was tested by recording myotube contractions while stimulating MNs by field electrodes and a set of clinically relevant parameters were defined to characterize the NMJ function. Three ALS lines were analyzed, 2 with SOD1 mutations and 1 with a FUS mutation. The ALS-MNs reproduced pathological phenotypes, including increased axonal varicosities, reduced axonal branching and elongation and increased excitability. These MNs formed functional NMJs with wild type muscle, but with significant deficits in NMJ quantity, fidelity and fatigue index. Furthermore, treatment with the Deana protocol was found to correct the NMJ deficits in all the ALS mutant lines tested. Quantitative analysis also revealed the variations inherent in each mutant lines. This functional NMJ system provides a platform for the study of both fALS and sALS and has the capability of being adapted into subtype-specific or patient-specific models for ALS etiological investigation and patient stratification for drug testing.
Keywords: neuromuscular junction, ALS, human-based, functional model, patient-specific
Graphical Abstract
1. Introduction
ALS is an adult-onset, fatal neurodegenerative disease characterized by progressive muscle weakening, loss of muscle control, degeneration of motoneurons (MNs) and eventual muscle paralysis. The symptoms usually begin with muscle spasticity and weakness in limbs or bulbar locations and progress for 2~5 years before the patients die due to failure of the respiratory system. Over 6000 people in the U.S. are diagnosed with ALS each year and currently there is no cure despite decades of study. To date only two FDA-approved drugs are available, but their methods of action only reduces disease progression by a few months. The paucity of approved therapies, wide epidemiology, and poor preclinical models require the development of new methodologies to enable better treatments to not only extend life expectancy but ultimately discover a cure for the disease.
One major obstacle inherent to treating ALS is the heterogeneity of this disease, for which causative variables have been postulated to be from genetic causes, varied cellular and molecular mechanisms, participation of different cell types and clinical manifestation.[1] Depending on whether there is a clear genetic marker or family history, ALS can be grouped into familial ALS (fALS) which accounts for only 5 ~10% of its incidence and the remaining 90% has been labelled as sporadic ALS (sALS). More than 20 ALS genes have been identified in fALS, and the number is still growing, while no clear genetic association has been identified for those sALS cases. Based on extensive studies, mostly from fALS, it is evident that each gene contributes to the eventual deterioration of MNs through different cellular molecular mechanisms; protein aggregation in the SOD1 mutation, RNA foci and protein nucleocytoplasm mislocalization in FUS and TDP 43 mutations, and toxicity of hexanucleotide-related RNA and the dipeptide repeat protein in the C9ORF72orf72 mutation.[2] Additionally, multiple cell types have been found to be active players in driving MN deterioration and death, such as astrocytes and microglia, each through a different mechanism.[3] Clinical manifestations of ALS exist in a continuum, from the disease onset age and site, involvement of upper and lower MNs, to progression rate and survival time.[4] Therefore, the multi-faceted heterogeneity nature of ALS makes it difficult to develop a model or find a therapy that can be applicable to all ALS cases. However, one common pathology is shared by all ALS patients, which is the impaired and progressive loss of NMJs and ultimately MN death.[5] NMJ function represents an initial common vector for all ALS subtypes, both fALS and sALS, and any therapies that can protect or restore NMJ integrity are potentially beneficial for global ALS treatment. Accordingly, a human functional NMJ model would be of great value for overall ALS pathological studies and drug development.
Accumulative evidence suggests that the loss of MN-muscle connections represents an early critical stage of this disease.[6] A major hypothesis in ALS research is distal axonopathy that induces pathological changes that occur at the NMJ at very early stages of ALS prior to MN degeneration and clinical phenotype manifestations.[7] Early pathological events that occur at the NMJ may ultimately be responsible for the early NMJ disruption rather than MN dysfunction and death.[8] Denervation of muscles by MNs is an early and important characteristic of ALS progression in both mouse models and ALS patients,[9] as shown by EMG (electromyography) in early clinical diagnosis of the disease. Studies from ALS mice and patients carrying SOD1 mutations indicated that muscle denervation occurred even before the activation of astrocytes or microglia, and significantly before nerve withdrawal and MN death.[9a] Therapeutic rescue of MNs without concomitant NMJ protection provides minimal improvements in overall outcomes such as life span.[10] Clinically, ALS patients commonly demonstrate reduced sustainability of NMJ mediated voluntary muscle tone or increased fatigability, which is defined as an inability to sustain a predicable maximal force during voluntary contraction, even in muscles that don’t show clear weakness.[11] Although this progressive muscle weakness has multiple sources from central to peripheral, the deterioration of NMJ function is an important contributor.[12] A phenotypic model that could reproduce these clinical NMJ’s deficits to enable the analysis using a human-based in vitro NMJ system could define meaningful parameters that represents clinical phenotypes while being employed for in vitro evaluation of potential therapeutics.
Numerous biological model systems have been developed for ALS, with the SOD1 mutant animal models and human stem cell-derived cellular models as the most popular. The majority of studies using animals have been performed in a model that overexpressed human SOD1 mutant genes,[13] which did not produce the mutant protein at pathophysiological levels. These models have provided invaluable insights into the understanding of the pathology and cellular mechanisms in ALS. However, not all the clinical pathologies are recapitulated[14] and there has been an overwhelming failure of translation from animal pre-clinical models to impactful clinical therapies, highlighting the issue of species gap.[15] Correspondently, more human-based systems have been employed. The actual diseased tissue itself clearly holds significant value in elucidating pathogenic mechanisms but typically represents late stages of ALS and does not represent early pathogenic processes. In addition, the in vivo environment may easily compensate for the initial cellular/molecular perturbations before any functional deficits are manifested. Human iPSC technology has increasingly been utilized in disease modeling[9] as these systems can not only better capture early stages of disease progression, but also makes it possible to develop patient-specific models because iPSC generated MNs retain the patients’ full genetic information. This is particularly important for ALS considering the heterogeneity of the disease, especially for the large number of sporadic cases in which the genetic information is mostly undefined. A functional NMJ system for ALS utilizing patient-derived iPSCs would establish a more relevant model for understanding the different disease phenotypes as well as for therapeutic screening in a high-content system.
Several functional NMJ systems have been developed with animal cells,[16] in animal-human hybrids,[17] and human-based NMJ systems.[18] However, these systems are limited functionally due to utilization of dual-patch electrophysiology, pharmacological excitation or lack of physical segregation of the MNs and muscle. Recent integration with optogenetics[19] and Bio-MEMs technology[20] has enabled these in vitro NMJs to be analyzed in a more controlled, non-invasive way and at a higher resolution. However, none of these systems were fabricated for isolation of the NMJs from the motoneuron cell bodies to allow cell specific interrogation of the therapeutics and selective stimulation of motoneuron and muscle. To address the need for a sensitive, human-based functional NMJ model with segregated readouts, a high-content phenotypic NMJ system was developed by integrating the biological system with a BioMEMs construct which allows repetitive temporal interrogation of NMJ function and relevant drug testing in a separated platform.[21] In the current study, ALS patient iPSC-derived MNs were combined with this Bio-MEMs engineered NMJ system to develop a defined functional neuromuscular system to study NMJ function of different ALS phenotypes and their chronological changes. The function of the established NMJs were characterized and clinically relevant phenotypes were identified from these ALS-NMJs and correspondent parameters have been defined for this ALS-NMJ model system. Moreover, a holistic ALS treatment, the Deanna Protocol (DP), exhibited positive effects in rescuing each of these functional mutant phenotypes. This study has established a human-based high content functional ALS-NMJ system and a set of clinically relevant parameters in NMJ number, fidelity and fatigue index that recapitulates and translates to measured clinical ALS neuromuscular parameters. This model can be easily adapted to also examine sALS patients’ cells, for both pathological investigation and therapeutic testing.
2. Results
A study design was developed to determine if different ALS mutations all had deficits in NMJ function and whether a therapeutic treatment could reverse this function irregardless of mutation-specific mechanisms.
2.1. Differentiation of ALS-MNs from patient-derived iPSCs
In order to develop patient specific NMJ models, functional MNs were differentiated from iPSCs from ALS patients and healthy controls based on protocols described in[22] with modifications detailed in the Materials and Methods (Figure 1. I). The differentiated neurons were positive for the neuronal markers βIII-Tubulin and MAP2, and to traditional MN markers HB9 [23], Islet1 [24] and SMI32 [25] based on immunocytochemistry (Figure 1.II A–D). Functional MNs were successfully differentiated from all tested ALS-iPSC lines by utilizing this protocol. Based on immunocytochemistry characterization and flow cytometry quantification, over 90% of the neurons were MNs. Patch clamp analysis was performed to evaluate the electrophysiological function of the MNs from multiple differentiation batches for each iPSC line and all demonstrated electrophysiological competency (Figure 1.IIE). To determine if the MNs could be excited by the neurotransmitter glutamate as in in vivo, a small aliquot of 30 μl glutamate (50 mM, final 500 μM) was applied to the MNs during GAP-free recording and intensive action potentials were elicited (Figure 1IIF), confirming their capability to respond to glutamate, which is a prerequisite for their circuit function in vivo.
Figure 1.
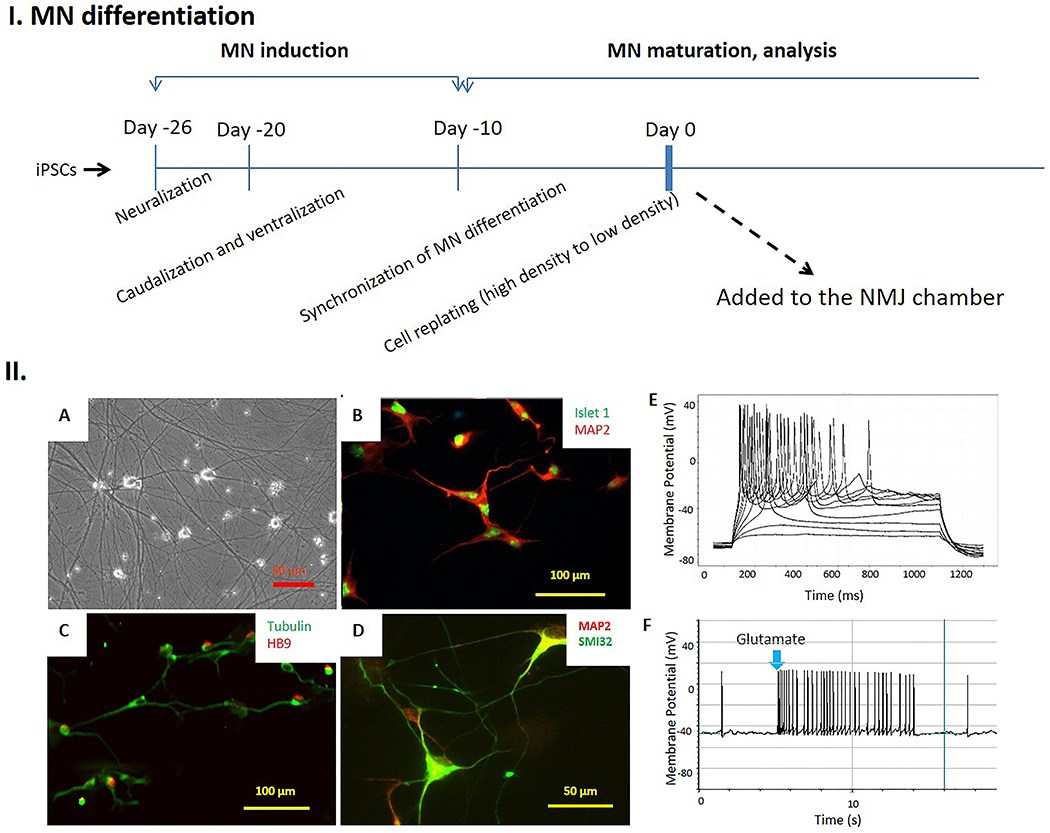
Differentiation of functional motoneurons from human iPSCs. I. Scheme illustrating the major differentiation stages of the protocol for generating motoneurons from iPSCs. II. Immunocytochemical and electrophysiological characterization of hMNs differentiated from hiPSCs. A). Phase micrographs of the iPSC-derived hMNs. B-D). The neurons expressed motoneuron markers Islet1 (B), HB9 (C) and SMI32 (D) as well as the pan-neuronal markers MAP2 or β III Tubulin. E). Patch-clamp recordings from the differentiated neurons at Day 27 demonstrated repetitive firing during current clamp, indicating their excitability. F). Application of glutamate (500 µM) to the patched neurons elicited active firings, confirming their responsiveness to Glutamate.
2.2. Characterization of ALS-hMNs demonstrated ALS pathology
To generate a representative sample of human MNs that harbor ALS mutations, 3 iPSC lines derived from ALS patients were selected because previous data indicated they had defined genetic and clinical features related to ALS. The ALS genes of these iPSC lines were examined and the ALS-relevant mutations identified as: SOD1 L144P, SOD1 D90A, and FUS G522A (Table S1). The ALS disease onset ages were relatively early ranging from 35 to 48, with the onset locations all from lower MNs (limbs). EMG recordings from the patients all indicated acute denervation at lumbosacral/lower limbs, cervical/upper limbs or thoracic sites. The two patients with SOD1 mutations were free of dementia and other neurodegenerative diseases, while Alzheimer’s disease was identified in the patient with the FUS mutation. In the literature, identification of the SOD1 D90A mutation has been reported multiple times and is identified as being both recessive and dominant depending on the genetic background.[26] The reports on SOD1 L144P and FUS G522A mutations are not as common but available.[27] The derived iPSC lines were well characterized by Coriell for their genetic integrity and pluripotency. MNs were differentiated from these iPSC lines according to the procedures described above.
To determine whether the MNs derived from ALS patients’ iPSCs (ALS-MNs) retained ALS-relevant deficits, these MNs were characterized to compare to previous literature reports for viability, axonal growth rate, axonal/dendritic branching, the presence of axonal varicosities and electrophysiological function as a control for our studies. With the assays employed in this study, as indicated in Figure 2A, the ALS MNs with SOD1 mutations displayed reduced viability while those with FUS did not. Impaired axonal growth and branching have been reported in ALS-MNs.[20d, 28] Significant difficulty of axonal regeneration after replating has been observed in some lines of ALS-MNs under phase microscopy, therefore, morphology of MNs after replating were examined by immunocytochemistry and followed for 2 days. As in Fig 2B–E, MNs from all the ALS lines demonstrated significant reduction in both axonal branching and total axonal/dendritic length per neuron, and the reductions in the MNs of both SOD1 lines are more severe than that in FUS MNs. Accordingly, the growth rate, which can be retrieved by comparing the change from Day 1 to Day 2, is also lower in SOD1 MNs compared to FUS MNs. Increased axonal varicosities have also been reported in several neurodegenerative diseases and has been considered a sign cells are under intracellular or environmental stress.[29] Multiple studies, including one from our group utilizing MNs from SOD1 L144P,[30] indicated that an increase in varicosities is a representative phenotype for ALS MNs.[31] MNs from the other 2 ALS lines in this study also demonstrated a significant increase in axonal varicosities (Figure 2 F and G), although the increase in FUS MNs was not as severe as those of SOD1 MNs. The varicosity phenotype is proposed to be linked to deficits in axonal transportation[30, 32] and may contribute to defects in NMJ function. In addition, since hyper- and hypo-excitability phenotypes have been frequently reported for ALS MNs,[33] the electrophysiological properties of these differentiated ALS-MNs were analyzed by patch clamp. The ratio of delayed rectifier potassium current over sodium current (IK+dr/INa+) was quantified weekly during weeks 2 to 4 and compared between mutants and healthy controls. As in Figure 2 H and I, the ratio did not indicate significant differences between healthy controls and mutants in weeks 2 & 3, but dropped significantly in week 4 in SOD1 mutant MNs and slightly in FUS MNs, suggesting a hyperexcitability phenotype in these MNs.[33b, 34] The phenotypic analysis of ALS-MNs indicated that they all demonstrated some facets of the ALS phenotypes reported previously. The type and severity of the phenotype varied depending on the specific mutation and possibly other unidentified factors.
Figure 2.
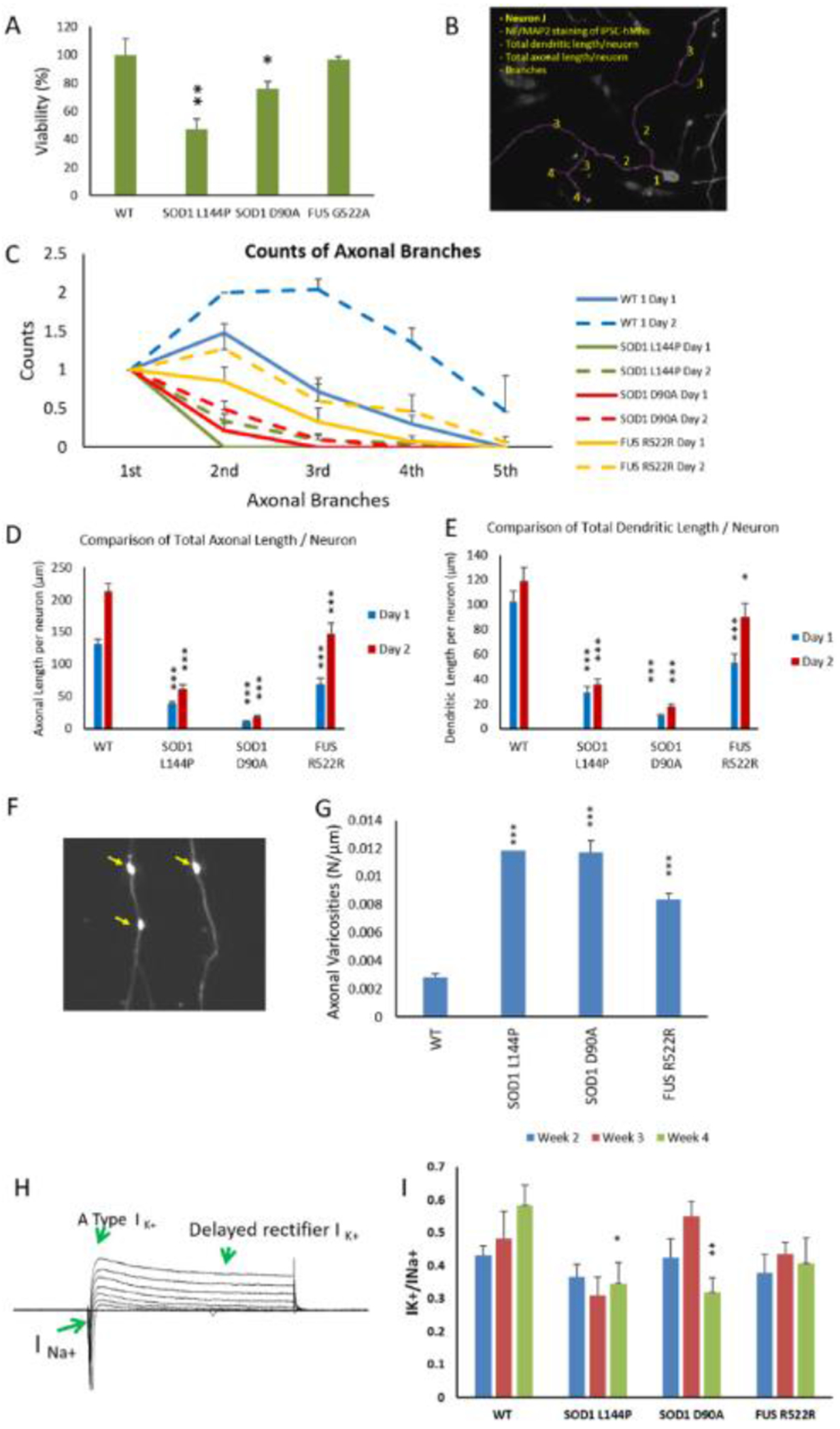
Phenotypic analysis of ALS-MNs. MNs from 3 ALS-iPSC lines were analyzed and compared to WT. A) Viability of MNs derived from different iPSC lines. Cultures from the same batch of platings were fixed at D1 and D7 separately and immunostained with neuronal markers Neurofilament and MAP2. The number of MNs were imaged at 20X from the same area size of the coverslips, quantified and normalized to Day 1 within the same plating batch to eliminate any batch-related variation. All data from the mutant lines were then normalized to WT to eliminate cell death caused by any experimental procedure as a determinant variable. At least two coverslips were analyzed for each time point in each batch and all the data were normalized to the average of 3 coverslips from day 1 within the batch. For each genetic group, at least 2 batches of analysis were quantified. B-E) Morphological analysis of ALS-MNs. IPSC MNs from DIV1 and DIV2 were immunostained for neurofilament (NF) and MAP2 to visualize the axonal and dendritic morphology, respectively. The images were analyzed in Neuron J for quantification of branching and measurement of process length (an illustration of the morphological analysis procedure is displayed in (B)). ALS mutant MNs demonstrated reduced counts of axonal branches per neuron (C), total axonal length per neuron (D) and total dendritic length per neuron (E) on Day 1 and Day 2 of the cultures. All the comparisons were between mutant and WT on the same day for each ALS line. F-G) Quantification of axonal varicosities in each ALS-MN line. MNs on D17 were immunostained with SMI32 and axonal varicosities per neuron were counted in image J and normalized to axonal length (μm) (F). A significant increase of axonal varicosities was observed in all ALS-MN groups (G). H-I) Patch clamp analysis of ALS-MNs. (H) is a sample set of current recordings under voltage clamp. (I) Quantifications of the Na+ and K+ currents revealed a significant reduction of the K+(DR) /Na+ current ratio on week 4 in all three ALS-MN lines compared to the WT-MN line, suggesting a hyper-excitability phenotype. Data represent mean ± SEM. Asterisks indicate that the condition is significantly different than the WT at the same time point (Dunnett’s test): * p<0.1, ** p<0.01, *** P<0.001. For B-G, n≥ 30 neurons from at least 3 batches of culture were analyzed. For H-I, n≥5~17 neurons from at least 2 batches were analyzed.
2.3. A human-based NMJ system for functional ALS mutant characterization
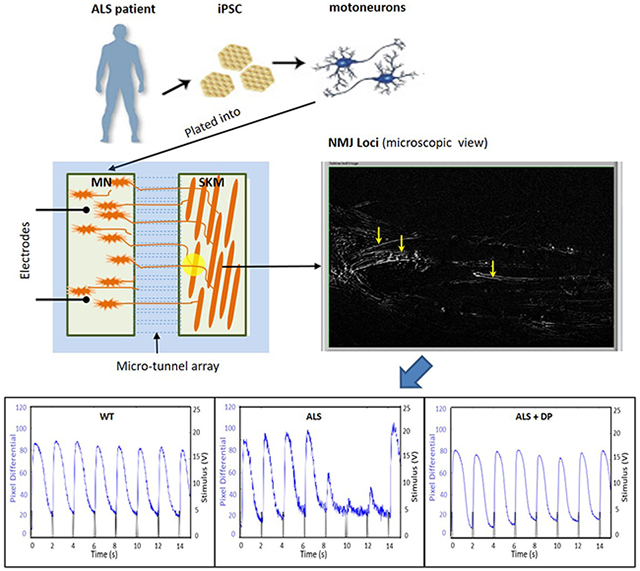
Previously, we established the first functional human NMJ system that allows for isolated treatment of MNs and muscle, and demonstrated the capacity of generating dose response curves to perform toxicity testing and therapeutic evaluations.[21a] This high content NMJ system was adapted for ALS investigation by integrating patient-derived MNs into the MN chambers and NMJs were established with primary muscle differentiated from satellite cells/myoblasts from a healthy subject. The cell culture scheme for establishing the ALS-NMJ system is shown in Figure 3 A–B. The experimental time points for the NMJ system were counted from the day SKM differentiation was initiated unless otherwise specified. Myoblasts were plated into the muscle chamber first, and four days after the initiation of myotube differentiation, differentiated MNs were cultured into the MN-side of the chamber. This schedule was utilized because myotubes have already started to form at about Day 4 in culture. Approximately 2-3 days later the number of axons that has transversed through the tunnels is then sufficent to effectively innervate the myotubes. The SKM culture promoted axon progression into the microtunnels to establish NMJ formation (Figure 3C). Except for these axonal connections, the two chambers are isolated both electronically and chemically, allowing independent stimulation and treatment of each chamber.[21a] To examine the functional connection between MNs and SKMs through NMJs, MNs were excited by field electrical stimulation, while the contraction of correspondent myofibers was monitored by differential phase contrast microscopy (supplement videos 1 A and B). The phase images in Figure 3C demonstrated the morphology of MNs in the MN chamber and axonal distribution in the muscle chamber. Immunocytochemistry of the SKM chamber revealed the physical association of axonal terminals (neurofilament) and myofibers (myosin heavy chain) (Figure 3D) and close apposition of presynaptic terminals visualized by the synaptophysin marker and post-synaptic receptors visualized by BTX-488 (Figure 3E), suggesting synaptic formation.
Figure 3.
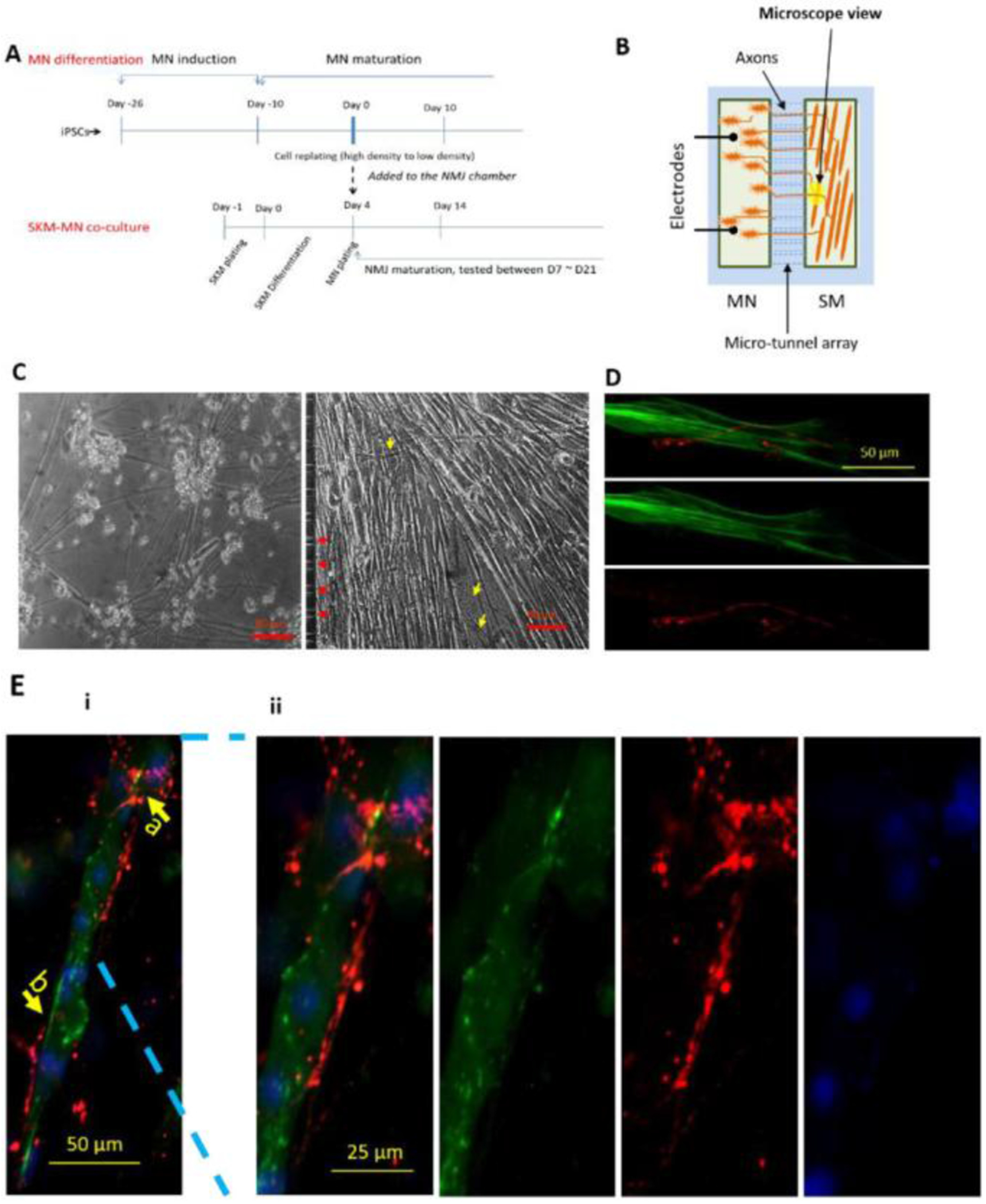
Illustration of the NMJ system. IPSC-MNs were co-cultured separately with WT-SKM where the MN chamber and SKM chamber were connected through micro-tunnels that were axonal permissible, but chemical and electrical impermeable. A) The cell plating scheme for the NMJ cultures inside the micro-chambers. B) Diagram of NMJ chamber system illustrating the MN chamber and SKM chamber are connected via microtunnels, through which MNs sent axons to reach the muscle. MNs were stimulated by field electrodes and induced myofiber contractions were captured by pixel differentials through a phase contrast microscope connected to a video camera. C) Sample phase images of cells in the NMJ system demonstrating MNs in the MN chamber and myofibers in the muscle chamber. Red arrows indicated the tunnel openings for axonal exits and yellow arrows point to some axons in the muscle chamber. D and E) Immunocytochemistry analysis of NMJs in the chambers. D) An image from the muscle chamber indicating an axonal terminal (red, stained with neurofilament) branched at the terminal and wrapped around the myotube which was visualized with the marker myosin heavy chain (MHC, green). Ei) Co-immunostaining with Synaptophysin (red) and Bungarotoxin-488 (green) indicating potential synaptic sites. Arrow b points to a location where the presyanptic terminal is in close alignment with a post-synaptic receptor, while arrow a indicates a close apposition of the two. ii) An enlarged view of i) highlighting the detailed morphology at location a.
2.4. Functional NMJ evaluation of ALS mutants
According to the study design, this NMJ platform was developed to uncover deficits in function similar to clinical measurements to reveal mutant specific ALS pathologies without cell death. Initially, the number of functional NMJs formed per chamber system for each mutant was characterized and compared to the healthy controls. Single field electrical stimulations (2V) were applied to the MN chamber while the resulting contracting myotubes within the muscle chamber were identified by pixel subtraction (indirect stimulation). Video 1A is a sample recording of myotube contractions under indirect stimulation while video 1B depicts the direct stimulation of all myotubes in the muscle chamber (Supplementary materials). The total number of contracting myotubes in response to electrical stimulation of the MNs was quantified and compared between ALS-NMJs and the healthy controls. Functional NMJs were detected as early as Day 7 of SKM differentiation (or Day 3 after MN plating). The NMJ number generally peaked between Days 12~17 and gradually decayed afterwards, partially due to the detachment of myotubes caused by extensive contractions. Considering the difference in MN axonal growth rate between mutants and the healthy controls (Figure 2), consequently resulting in differences in NMJ number at early culture days, the total NMJ numbers per chamber were analyzed on D14 and D17.
All the mutant lines demonstrated a reduction in NMJ number as demonstrated in Figure 4. The difference in SOD1 D90A was not statistically significant on D17 but was on D14 (P<0.1). SOD1 L144P demonstrated a reduction of NMJ number on D14 (P<0.001) and D17 (P<0.1), while FUS G522A demonstrated a reduction on both D14 and D17 but the change was not significant compared to the control. A decrease in NMJ number at an early stage of testing may imply a difficulty in NMJ regeneration while those at later testing days suggests a defect in NMJ maintenance. Both SOD1 NMJs demonstrated more significant reduction at D14 than D17, suggesting a difficulty in NMJ formation which supports the slower axonal regeneration phenotype (Figure 2D). Accordingly, the mild NMJ reduction phenotype in FUS mutant supports the mild MN phenotype as in Figure 2. Additional comparisons of D14 vs D17 in Figure 4 suggests an increase in NMJ numbers from D14 to D17 for the SOD1 L144P mutation that could reveal generation of additiional NMJs over time, while the decreases shown in SOD1 D90A and FUS R522A could reflect pathologic degeneration of NMJs. However, a quantitative comparison between D14 vs D17 indicated no significant difference except in the SOD1 D90A mutation. In vivo, muscle denervation normally induces a compensating mechanism, nerve terminal sprouting and re-innervation, which has been observed in multiple conditions such as spinal cord injury and ALS.[12, 35] The active denervation and re-innervation events occurring at ALS muscle[35b] suggests this plays an important role in the terminal muscle denervation, MN death and disease progression.[12, 36] The results from this ALS-NMJ system suggests that a difficulty in NMJ regeneration (as in D14) is likely the primary deficit contributing to the ultimate muscle denervation and paralysis which is typical in clinical ALS.
Figure 4.
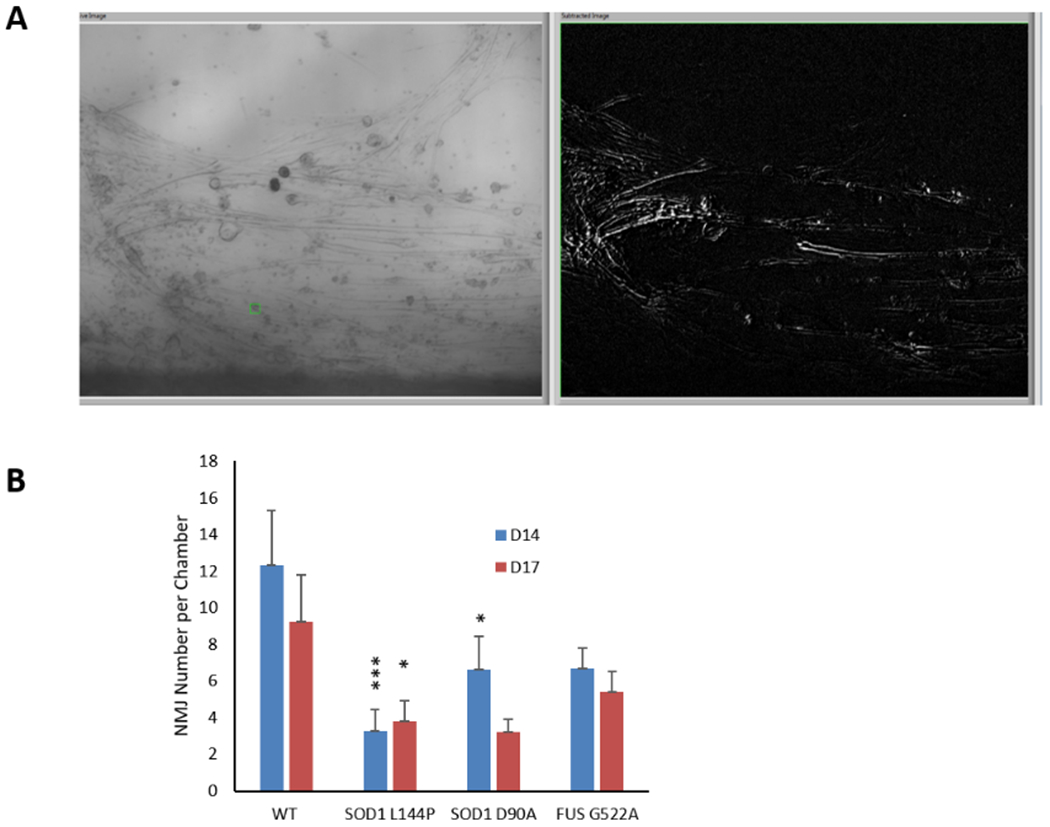
The number of functional NMJs per chamber was quantified by field-electrical stimulation of MNs in the MN chamber while recording induced myofiber contractions in the SKM chamber. A) A screen shot of an NMJ recording indicating multiple contracting myofibers upon electrical stimulation in the MN chamber. B) The number of functional NMJs per chamber were quantified and reduction of NMJ numbers was observed in all 3 mutant lines of MNs compared to WT (Dunnett’s test). Data represent Mean ± SEM. N≥ 20. Asterisks indicate that the condition is significantly different than the WT at the same time point (Dunnett’s test): *, P<0.1; **, P<0.01; ***, P<0.001.
NMJ fidelity was the function most closely correlated with clinical measures and so was investigated in detail. This feature of NMJ function is the ability to reliably transmit electrical activity from a MN to muscle to induce muscle contraction at increasing stimulation frequencies. The transmission reliability, defined as NMJ fidelity, is the percentage of MN-induced muscle contractions over the total number of MN stimulations (equation 1) over a set duration (15 sec in this case) of MN stimulation. When NMJ transmission is impaired, NMJ fidelity can be compromised, resulting in failure of muscle contraction upon MN stimulation or “skips”, which can be considered a correlate to limb spasticity. To quantify NMJ fidelity, an innervated myotube demonstrating prominent contractions under repetitive indirect stimulations was identified for fidelity testing. Stimulation pulses to MNs at increasing frequencies (0.33 Hz, 0.5 Hz, 1 Hz and 2 Hz; each for 15 sec) were applied and correspondent myotube contractions were recorded. Video 2 in the Supplemental Materials is presented as an example. During this stimulation series, healthy controls normally responded reliably under all frequencies, while ALS-NMJs demonstrated variations in severity of defects from “skips” to complete failure (Figure 5A). All of the tested mutant NMJs responded well to low stimulation frequency (0.33 Hz) but demonstrated increased number of skips as the frequency was increased. As quantified in Figure 5B, ALS-NMJs from all mutant lines demonstrated a decrease in NMJ fidelity compared to WT-NMJs at correspondent frequencies and testing days (* P < 0.1, ** P < 0.01, *** P < 0.001), among which the decrease in FUS mutant at D14 is the most prominent. In vivo, increased MN stimulation frequency usually induces tetanus in muscle contraction due to temporal summation effects and insufficient time for the muscle to relax. This temporal summation effect, or tetanus, was also observed in the NMJ system (Fig 5A). While the tetanus response was well maintained in healthy control-NMJs during stimulation, it was frequently interrupted or unattainable in ALS-NMJs for all the mutant lines tested, which can be correlated clinically with sudden muscle weakness. Muscle contractions under direct stimulations were recorded as a control. A tetanic muscle response relates to muscle tone, for which higher input frequency induces high muscle tone, or isometric strength.[37] The NMJ failure at high frequencies caused tetanus failure (Figure 5A), which is reminiscent of the reduction or loss of muscle tone, a commonly reported symptom in ALS patients[38] and may relate to the frequent tripping and falling of patients at early stage of the disease.
Figure 5.
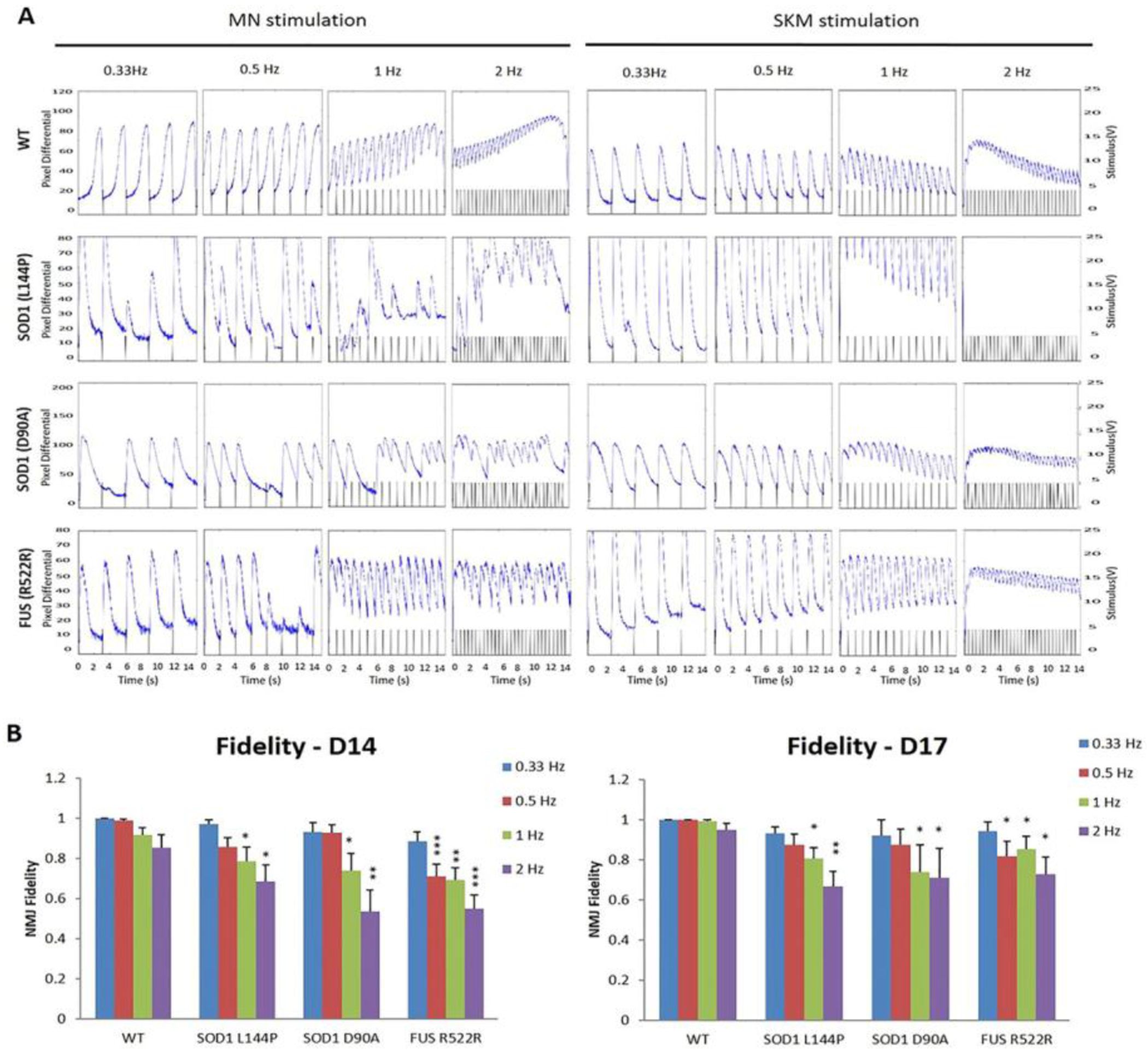
NMJ fidelity was quantified as the percentage of successful induction of muscle contractions induced by motoneuron stimulation under four testing frequencies (0.3, 0.5, 1, and 2 Hz) at Days 14 & 17 in ALS-NMJ systems compared to WT controls. A) Sample traces of myofiber contractions under motoneuron stimulation and muscle direct stimulation, respectively, at four different frequencies from 3 ALS-NMJ systems and the WT controls. B) Quantification of NMJ fidelity compared between ALS mutant and WT groups (Dunnett’s test). Data represent Mean ± SEM. N≥10. Asterisks indicate that the condition is significantly different than the WT at the same time point (Dunnett’s test): * p<0.1, ** p<0.01, *** p<0.001.
Muscle fatigue is also one of the hallmarks for ALS diagnosis and clinical progression.[39] Clinically, it is measured by quantifying the reduction of maximal voluntary contraction fatigue (MVCF) over a certain period of time during which the subject is asked to voluntarily contract the muscle in a specific protocol.[40] To quantify the reduction of tetanic amplitude during the 15 sec stimulation duration, the fatigue index, a clinical parameter used for measuring muscle fatigue, was adapted to measure fatigue in this in vitro NMJ system (equation 2, Figure 6A). Quantification of traces demonstrating full or partial tetanus revealed an increased fatigue index in all the ALS-NMJs on at least one testing day (* P < 0.1, ** P < 0.01, *** P < 0.001 depending on the mutant line, testing frequency and testing Day) (Figure 6B).
Figure 6.
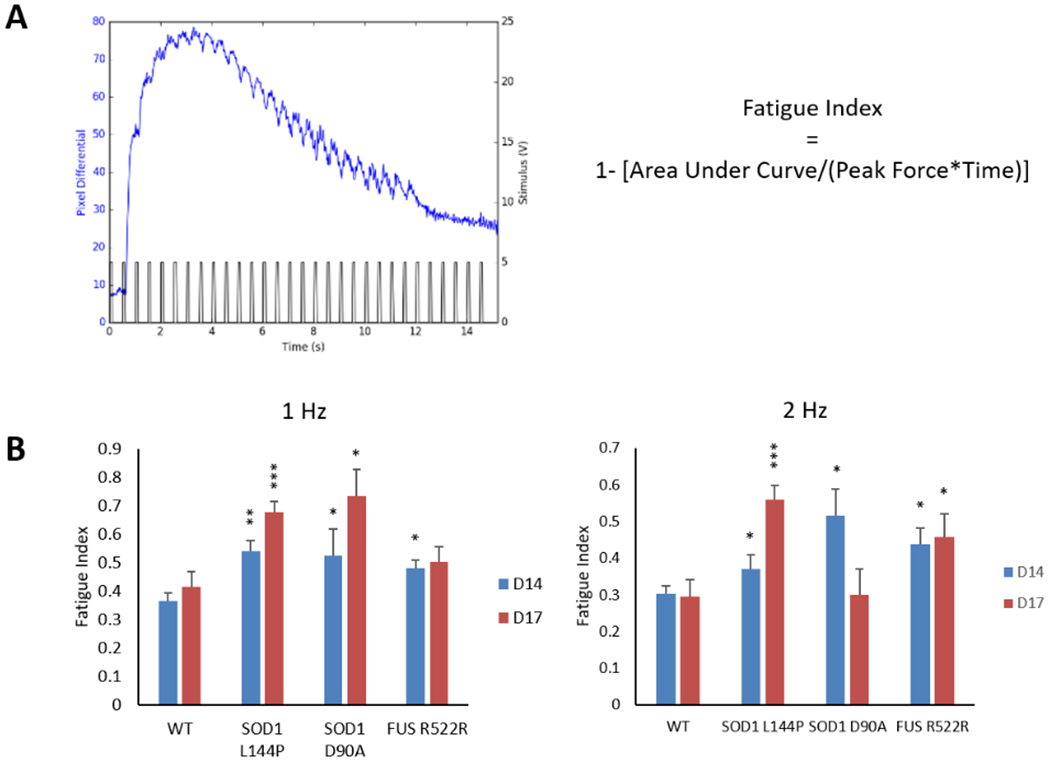
Determination of fatigue Index in the ALS-NMJs. A) A representative myofiber contraction trace under 2 Hz stimulation from the MN chamber. The formula for calculating NMJ fatigue is the same as the physiological measurement of in vivo fatigue. B) Fatigue Index in ALS-NMJs were significantly higher than those in WT-NMJs under both 1 Hz and 2 Hz stimulations. Quantification of each ALS mutant was compared to WT for correspondent stimulation frequencies and culture days (Dunnett’s test). Data represent Mean ± SEM. N≥3. Asterisks indicate that the condition is significantly different than the WT at the same time point (Dunnett’s test): *, P<0.1; **, P<0.01; ***, P<0.001.
2.5. Therapeutic treatment of ALS-NMJ mutants utilizing the Deana Protocol
The mutant ALS-NMJs demonstrated significant functional deficits which mirrored clinical observations. Two FDA approved drugs for ALS, Riluzole and Edaravone, target only limited cellular mechanisms, mainly anti-glutamatergic toxicity and reducing oxidative stress respectively, showing limited efficacy in some specific subgroups of patients within a particular treatment window.[41] Another therapeutic that has been reported effective in ALS patients and an animal model, is the Deana Protocol (DP), a regimen of nutritional supplement proposed as a treatment for ALS.[30, 42] DP contains supplements targeting a spectrum of cellular mechanisms, especially in facilitating cell metabolism and reducing oxidative stress, cellular mechanisms shared by almost all ALS subtypes and as such the DP potentially has a wide target population. The DP was optimized in a previous study where it was shown to be effective in correcting the ALS-related axonal phenotype in one of the ALS-MN lines included in this study, SOD1 L144P, as well as the axonal varicosities induced by glutamate excitotoxicity.[30] To evaluate the DP over a statistically relevant subset of ALS phenotypes as indicated in the study design, it was tested for reversal of ALS-NMJ deficits.
DP treatment to the MN-side of the chamber was initiated 3 days post MN plating and replenished during subsequent MN feeding every other day. The function of treated NMJs was analyzed on D14 and D17 as described above. The number of NMJs per chamber was significantly improved by DP treatment for all the mutant lines tested, with more prominent increases in ALS-NMJs and slight increase in the healthy controls (P<0.1 at D14) (Figure 7A). In Figure 7B, the DP revealed improvements in NMJ fidelity in all the mutant lines tested. While the rescue effect was more pronounced for both SOD1 lines on both testing days, the rescue for FUS line was only observed on Day 14. This may suggest that deficits in NMJ fidelity in FUS on the two days are caused by different mechanisms. Similarly, after DP treatment, the fatigue index was reduced significantly in the ALS-NMJs of all the lines tested on D14 and/or D17 compared to untreated controls, and the fatigue index after rescue reached a level insignificantly different from untreated healthy controls (Figure 7C). The MN and NMJ phenotypes of all the ALS lines and their response to DP treatment are summarized in Table 1.
Figure 7.
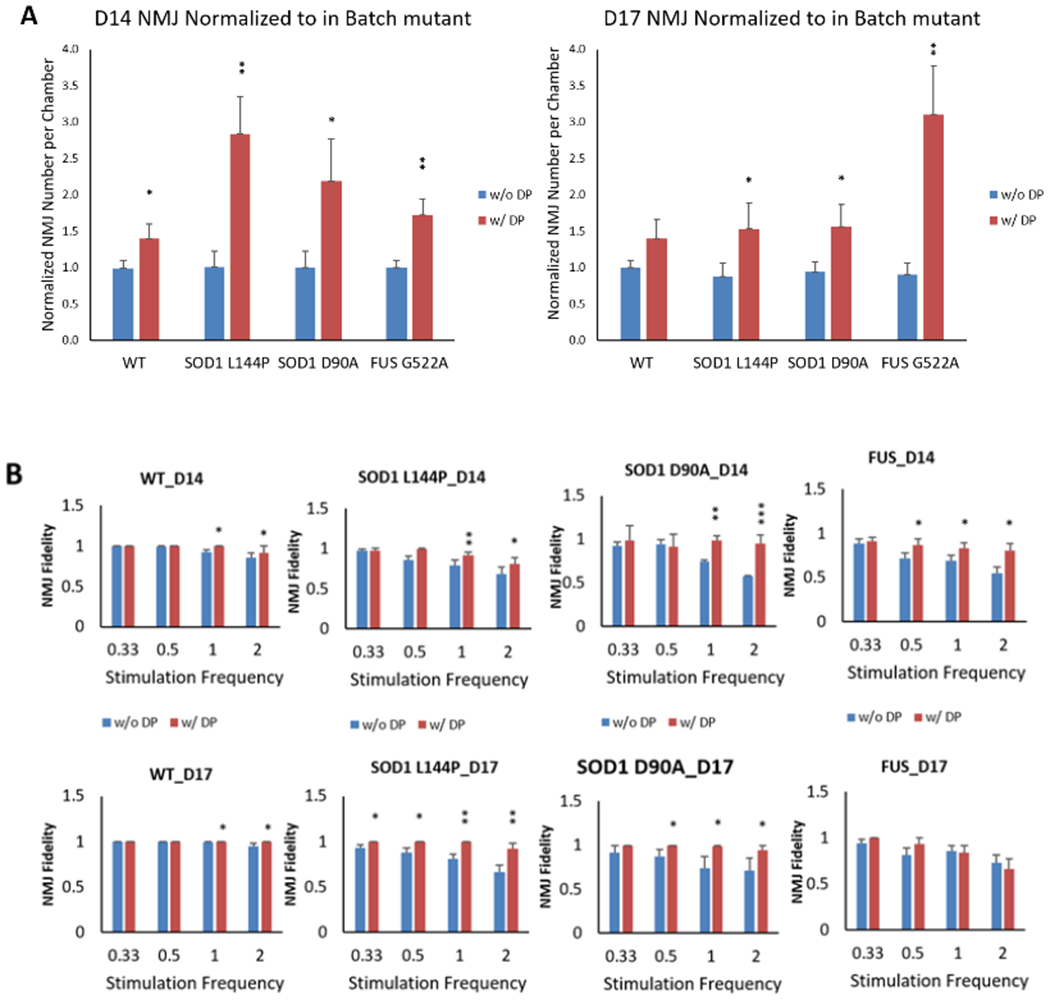
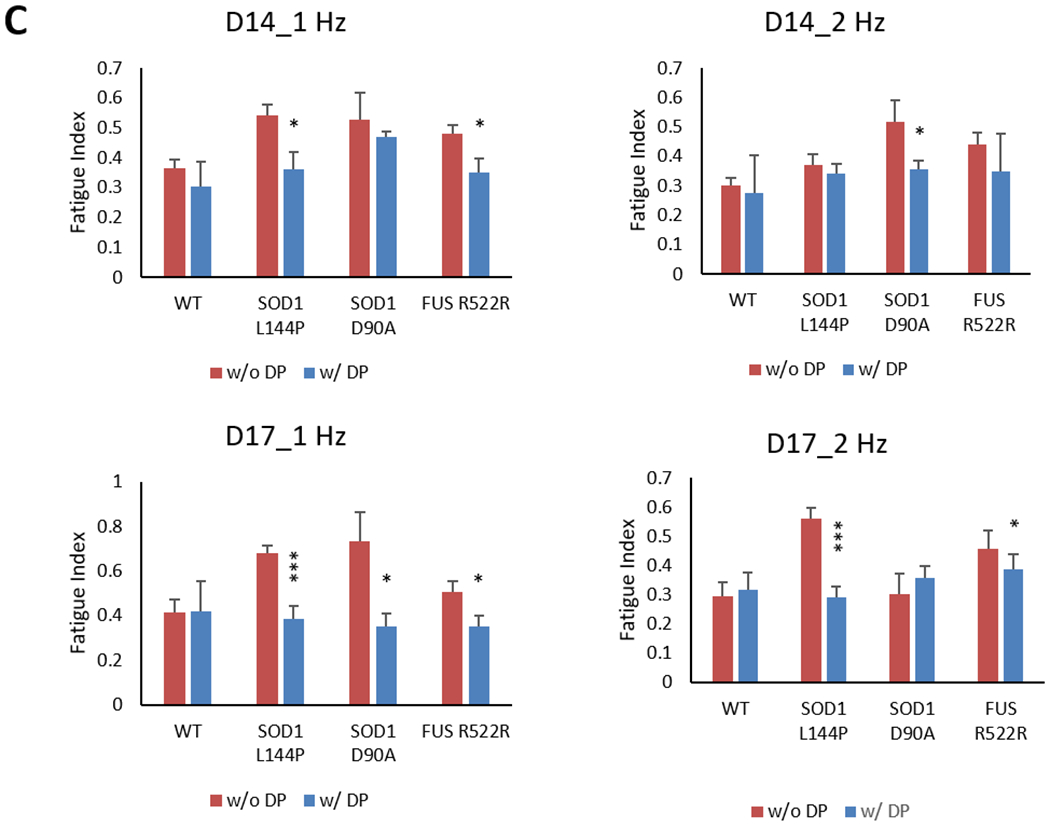
ALS-NMJ functional phenotypic deficits were improved by the DP drug treatment. A) The NMJ numbers per chamber normalized to in-batch control without DP for each genetic line. NMJ number was increased in all the ALS-NMJ conditions after DP treatment, while FUS-NMJs showed more improvement at D17 than SOD1-NMJ mutant at D14. N≥9. B) NMJ fidelity was improved by DP treatment for all the ALS mutants at D17, but only for SOD1 mutants at D14. N≥5. C) NMJ fatigue Index was corrected by DP treatment for all the mutants. Quantifications were compared between with and without DP treatment for each mutant (Student’s t-test, two-tailed). Data represent Mean ± SEM. N≥3. For all analysis, asterisks indicate that the condition is significantly different than the WT at the same time point (Dunnett’s test): *, P<0.1; **, P<0.01; ***, P<0.001.
Table 1.
Summary of the NMJ phenotype analysis for the three ALS lines and their response to DP treatment.
| MN phenotype | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ALS Lines | Viability | Axonal Branches | Axonal Length/Neuron | Dendritic Length/Neuron | Axonal varicosity | IK+ dr/INa+ | ||||||||
| Day 1 | Day 2 | Day 1 | Day 2 | Day 1 | Day 2 | Wk 2 | Wk 3 | Wk 4 | ||||||
| SOD1 L144P | ** | *** | *** | *** | *** | *** | *** | *** | * | |||||
| SOD1 D90A | * | ** | *** | *** | *** | *** | *** | *** | ** | |||||
| FUS G522A | * | ** | *** | *** | *** | * | *** | |||||||
| NMJ phenotype | ||||||||||||||
| ALS Lines | NMJ Number | NMJ Fidelity | Fatigue Index | |||||||||||
| D14 | D17 | D14 | D17 | D 14 | D17 | |||||||||
| 0.33 Hz | 0.5 Hz | 1 Hz | 2 Hz | 0.33 Hz | 0.5 Hz | 1 Hz | 2 Hz | 1 Hz | 2 Hz | 1 Hz | 2 Hz | |||
| SOD1 L144P | *** | * | * | * | ** | *** | * | *** | *** | |||||
| SOD1 D90A | * | * | * | ** | * | * | * | * | * | |||||
| FUS G522A | *** | ** | ** | * | * | * | * | ** | * | |||||
| Treatment effect by DP | ||||||||||||||
| ALS Lines | NMJ Number | NMJ Fidelity | Fatigue Index | |||||||||||
| D14 | D17 | D14 | D17 | Day 14 | D17 | |||||||||
| 0.33 Hz | 0.5 Hz | 1 Hz | 2 Hz | 0.33 Hz | 0.5 Hz | 1 Hz | 2 Hz | 1 Hz | 2 Hz | 1 Hz | 2 Hz | |||
| SOD1 L144P | ** | * | ** | * | * | * | ** | ** | * | *** | *** | |||
| SOD1 D90A | * | * | ** | *** | * | * | * | * | * | |||||
| FUS G522A | ** | ** | * | * | * | * | * | * | ||||||
P < 0.1,
P < 0.01,
P < 0.001
3. Discussion
This study has established a functional human-based ALS-NMJ model by integrating ALS patient-derived iPSC-MNs and primary human skeletal muscle into a compartmentalized chamber and NMJ function was analyzed for clinically-relevant phenotypes. The MNs derived from ALS patients (2 SOD1, 1 FUS) demonstrated previously observed pathological phenotypes, including increased axonal varicosities, reduced axonal branching and growth rate and increased excitability to validate our differentiation methodology. When introduced into the NMJ chamber system, these ALS-MNs formed functional NMJs with healthy muscle. However, the ALS-NMJs demonstrated significant, distinct functional deficits including a reduced number of functional NMJs, reduced NMJ fidelity and increased fatigue index. These findings correspond to clinical symptoms observed in ALS patients such as muscle weakness, fatigue and wasting. Thus, the results from evaluation of potential therapeutics in this in vitro model could be used to predict clinical outcomes for a common interaction. To assess this concept, this functional ALS-NMJ system was further employed for therapeutic evaluation. Treatment of ALS-MNs with the Deanna Protocol was found to correct the NMJ phenotypes in all the ALS mutant lines tested, confirming this NMJ model could be used as an efficient functional system for evaluating drug efficacy. The combination of the high content NMJ system with the ALS patient-derived MNs enables not only in-depth investigation of the NMJ pathology, but also screening for patient-specific therapies.
Clinically, early signs and symptoms of ALS include muscle weakness (or reduced muscle tone) and increased muscle fatigue resulting in difficulty in walking and other normal daily activities. To analyze the NMJ deficits in detail, several functional parameters were defined to characterize NMJ function in vitro in an effort to represent clinical motor phenotypes. The first parameter, NMJ number, reflects the capability of NMJ formation and NMJ maintenance. The summation of these two mechanisms determines the extent of muscle innervation. In this study, two different time points – D14 and D17 were tested, as the most significant NMJ formation efficacy was between these two time points. Many factors can affect the NMJ total during the NMJ development stage including MN number (MN viability), axonal growth rate, axonal branching and the activity level of the MNs. The second parameter, NMJ fidelity, characterizes how reliable NMJ transmission is upon repeated MN stimulation at increasing frequencies. Low stimulation frequencies (0.33 and 0.5 Hz) normally results in individual contractions of myotubes with each stimulation pulse detected as single contraction peaks, however, higher stimulation frequencies (1 and 2 Hz) generally induces a fused or unfused tetanic response. Consistent NMJ fidelity depends on proper function of a series of mechanisms including reliable excitation of MNs upon each stimulation, efficient transportation of neurotransmitters to the synaptic terminal, reliable terminal depolarization, sufficient neurotransmitter release upon each excitation, and binding/activation of acetylcholine receptors to cause myotube contraction. Since patch clamp analysis indicated normal excitability of ALS-MNs before and during weeks 2~3 which corresponds to D17~24 of the NMJ systems, the fidelity defects in these mutant ALS-NMJs was unlikely to be attributable to defects in MN excitation. However, previously it had been shown that the axonal varicosity phenotype in ALS-MNs suggests compromised axonal transportation,[29–30] which could cause insufficient neurotransmitter supply and quick exhaustion of functional mitochondria storage at synaptic terminals. This hypothesis is supported by the observation of faster decay of NMJ fidelity under higher frequency stimulations during fidelity testing. The number of NMJs and functional fidelity of each NMJ relates directly to muscle tone, which relies on the number of recruited motor units, size of motor units and the fidelity of each NMJ. The third parameter, NMJ fatigue index, reflects the decline of myotube tension over time under tetanic conditions at high stimulation frequency (some 1Hz and almost all the 2 Hz recordings). Normally, myotube tension under tetanus contraction is elevated due to the summation effect, caused by incremental Ca2+ accumulation inside the sarcolemma under high frequency stimulation. However due to fatigue of NMJ function caused by decline of MN stimuli, failure of the MN-muscle synaptic apparatus and muscle exhaustion, tension will decline over time, which is identified as fatigue. In this system, only MNs harboring the ALS mutations were utilized while the muscle was from normal primary muscle, so the increased fatigue in the ALS-NMJs most likely originated from deficits in the MNs. This parameter was previously only used clinically as a result of muscle fatigue. However in the clinic, in addition to the fatigue from an individual NMJ, the alterations of motor unit size under disease condition and the reduction of recruited motor unit number over time are also important contributors to the decline of muscle tension over a period of maximum voluntary contraction.[43] This difference between in vitro and in vivo systems can be related by comparing the NMJ formation and phenotype maintenance using NMJ number analysis. Overall, the combination of these three parameters presents a comprehensive picture of the functional defects of individual NMJs and the overall number of functional NMJs, which together generate a useful reflection of ALS mutant phenotypes: muscle tone and muscle fatigue.
More than one human based functional NMJ system has been reported by utilizing human stem cells as the source.[18a, 19, 20d, 21a] The approaches to achieve non-invasive and MN-specific stimulations include selective chemical stimulation such as N-Methyl-D-aspartate,[18a] optogenetic technology,[19] or electrical stimulation in compartmentalized systems.[21a] Electrical stimulation is preferred since the other approaches either lack temporal control or require genetic labeling of MNs. In addition, compartmentalized chambers are electrically and chemically insolated by the barrier, therefore providing an avenue for chamber-selective treatment which is important for mechanistic investigations and therapeutic testing. Similarly, to achieve non-invasive monitoring of muscle excitation/contraction, multiple approaches have been utilized including Ca2+ imaging, differential phase contrast imaging and pillar deflection, in which the latter two are preferred since they directly measure contraction. Our NMJ system combines electrical stimulation of MNs and phase differential monitoring of muscle contraction in a compartmentalized chamber platform. Recently, a microphysiological 3D NMJ model, formed between human iPSC-derived muscle bundles on pillars and iPSC-derived optogenetic motoneuron spheroids, was applied to the study of ALS by including ALS-MN spheroids derived from a single sALS patient.[20d] This study reported reduced MN fascicles and neurite elongation rate, decreased muscle contraction force as well as increased skips of muscle contraction under MN stimulation. Our study included 3 different mutations, 2 SOD1 mutations and 1 FUS, to establish a general mechanistic model focused on the NMJ defects. Furthermore, to extend the NMJ model to the next level for the study of ALS and drug testing, a set of functional parameters were defined to recapitulate and quantitatively analyze the clinical correlation: NMJ number, fidelity and fatigue index. Application of these parameters to these ALS-NMJ mutants uncovered clinically relevant defects that could be used to predict in vivo results.
The three ALS mutant lines used in this study all demonstrated significant deficits in both MN phenotype and NMJ function with statistically significant variations between the three mutations. MN phenotypic analysis indicated that the two SOD1 mutations demonstrated more extensive deficits compared to the FUS mutation, i.e. MN viability, MN morphology, and axonal varicosity formation. SOD1 L144P demonstrated the most severe deficits consistently for all the MN phenotypic parameters. Deficits in SOD1 D90A were relatively milder for most of the parameters analyzed, and the FUS G55A mutant was the mildest phenotype. However, this tendency was only partially preserved in the NMJ functional analysis. SOD1 L144P still demonstrated more severe deficits in all the NMJ parameters analyzed than the two other mutants, except for NMJ fidelity at D14, in which FUS G522A presented the most severe phenotype. SOD1 L144P had more severe deficits at fatigue index while FUS G522A was more significant for NMJ fidelity. This relative rise in disease phenotype severity in the FUS mutant when utilizing a more functional platform (NMJ vs. MN alone) implies more prevalent deficits were revealed after interaction with the muscle. This severe NMJ fidelity phenotype is in line with the clinical situation of this patient, early disease onset at 35 years old and with the comorbidity of AD and heart disease. In addition, ALS and AD are both neurodegenerative diseases and they share some common physiological or pathological pathways. For example, soluble Amyloid β precursor protein (sAPP), FUS, TDP-43 and SOD1 all share the same axonal transportation mechanisms [44]. So the comorbidity of AD and ALS in the patient harboring the FUS R522A mutation may reciprocately aggravate each disease and caused the early onset of ALS in this patient, although the MN phenotype alone is not as severe as the two other ALS lines. As in Table 1, each mutant displayed a unique phenotype “signature”. Correspondently, DP treatment caused different levels of effectiveness and parameter preference in the different ALS mutants. Considering the etiology, the low MN viability and the slow axonal growth can cause the reduction and delay of NMJ formation respectively. This could be the case for SOD1 L144P, which showed the most severe phenotype in MN viability, axonal length and D14 NMJ number. Its severely impaired NMJ fatigue index and reduced D17 fidelity could be a result of axonal transport problems as indicated by the significant increase in axonal varicosities. Interestingly, FUS G522A, while displaying a relative mild MN phenotype from morphological analysis and in D14 NMJ number formation, demonstrated the most severe reduction in D14 fidelity. Overall, these data suggest special concerns are necessary when dealing with different ALS cases, even those having mutations in the same ALS genes, and caution is needed when translating data between different ALS models, i.e. from a MN-only model to the functional NMJ model. These observations again emphasize the necessity for developing patient-specific disease models or the establishment of a common functional target for different ALS mutations.
ALS generally presents with progressive degeneration of both upper and lower MN phenotypes. Hence, a dearth of research has focused on the cellular and molecular mechanisms that initiate and subsequently drive MN death. For example, the protein aggregation of SOD1 mutation, RNA foci in FUS mutation, the nucleocytoplasmic transport defects in the mutations of C9ORF72, FUS and TDP43 have been thoroughly evaluated.[45] Multiple ALS models based on these ALS MNs have been developed. These studies have dramatically advanced our knowledge concerning ALS etiology, and revealed the complexity and heterogeneity of this disease. However, studies based on a gene-specific MN phenotypic marker may only reveal the etiology relevant to the particular gene group, and the drugs validated based on a particular MN marker may only be effective for a small subset of the patient population. This ALS-NMJ model is applicable to the pathological study for all types of ALS, both sporadic and familial. In addition, deficits in NMJ function occur before MN death and are an earlier pathological marker common to all ALS cases. The patch clamp data indicated that the MN excitability phenotype was not exhibited until 4 weeks after plating, but the NMJ phenotype was able to be detected in less than two weeks, suggesting this phenotypic NMJ system is more sensitive than a phenotypic MN-only system.
Despite of the heterogeneity of ALS pathological mechanism and NMJ phenotype, different ALS mutants all displayed certain improvements in NMJ function after treatment with the Deana Protocol. This could be due to the fact that the DP treatment contains five components targeting a spectrum of cellular mechanisms that are known to be altered in ALS pathology including counteracting oxidative metabolism, mitochondria protection, glutamate detoxification and energy production.[42, 46] Its effect has been confirmed in correcting the axonal varicosity phenotype in human MNs harboring SOD1 mutations and those induced by glutamate neurotoxicity.[30] Anecdotal reports of ALS patients utilizing the DP supplement cocktail suggests the effectiveness of the treatment as indicated by an improvement in motor function.[42] While the specific cellular mechanisms that have been targeted and corrected in each tested mutant by DP can be further dissected in future studies, the preliminary testing of this drug in our ALS-NMJ model underscores its wide spectrum of effectiveness in ALS treatment.
While being a good model to study the autologous effect of MNs, this functional ALS-NMJ system also provides a platform for dissecting the non-autologous mechanisms in ALS pathology. The other cellular component of the NMJ, muscle, also plays an important role in axonal retraction and muscle denervation.[47] Future integration of ALS mutant derived muscle into this system alone or together with ALS-MNs would allow for the evaluation of the role of muscle in ALS pathology. Moreover, the capacity of isolating the drug treatment for MNs and/or muscle in this compartmentalized system would allow for identification of cell targets and detailed mechanistic observations. Additionally, astrocytes, microglia and Schwann cells are all found to be important components in the etiology of ALS disease and are also potential therapeutic targets,[3b, 48] and their roles can also be investigated after being introduced into the current bipartite system. Therefore, this model not only constitutes an important tool for de-convoluting NMJ pathology in ALS and drug testing, but also provides a broader platform for investigating the role of each interacting cell types and ultimately establishing the complete cell ontology image for ALS in a common functional model.
4. Conclusion
Overall, this study established a phenotypic ALS-NMJ system by utilizing iPSC-MNs derived from ALS patients, and by defining a set of parameters to uncover clinically-relevant NMJ deficits. The utilization of three ALS mutant lines in the study design revealed significant NMJ deficits in all the mutant lines. However, the NMJ phenotype demonstrated some variations in severity and parameter selection, as well as in response to drug treatment, highlighting the need for common indications in patient-specific models. This functional ALS-NMJ platform can be patient-specific and applicable to all ALS cases for etiological study and drug testing.
5. Experimental Section
Study design
This study was aimed at investigating pathology of NMJs caused by iPSC derived ALS-MN relevant mutations, as well as the effects of DP treatment on NMJs that exhibit ALS-disease pathology. To investigate ALS - related NMJ dysfunctions as well as DP as a generalized treatment for ALS-related NMJ dysfunction, ALS-MNs were developed from disparate genetic backgrounds: one line with a FUS mutation, and two lines with SOD1 mutations. The same protocols and time frames were followed for the differentiation, cryopreservation, thawing/plating and analysis of the motoneurons from all the iPSC lines, as well as the plating, culture and functional testing of corresponent ALS-NMJ systems. The NMJs produced from these ALS-MNs all showed similar dysfunction, enabling the investigation of the DP treatment on ALS-related NMJ dysfunction independent of mutation-specific mechanisms. Of particular note, this study is specifically not designed to investigate the effects of specific genetic mutations (eg FUS or SOD1) on NMJ dysfunction nor their mechanisms but only to establish an ALS functional deficit phenotype. Characterized diseased motoneurons were differentiated from ALS patient iPSCs and cultured in the BioMEMs NMJ system with skeletal muscle differentiated from primary satellite cells/myoblasts isolated from biopsies of normal subjects. After innervation, functional measurements of NMJ number, fidelity and fatigue index were obtained to establish a signature of each ALS mutant compared to the NMJs using MNs from healthy controls. Each experiment was repeated at least 5 times. Outliers were only excluded if data from direct stimulation of the healthy primary skeletal muscle (SKM) revealed abnormal and/or unavoidable deficits, such as SKM detachment or muscle spontaneous contractions.
The number of systems tested was determined statistically so as to detect a difference in NMJ fidelity of at least 30%, with a type I error rate (α) of 0.05 and type II error rate (β) of 0.2 using a Dunnetts’s multiple-comparisons test to the control condition.
Microfabrication, assembly and coating of the BioMEMs NMJ co-culture system
Polydimethylsilxane (PDMS) chambers were designed, casted and cut as previously described.[21a] The PDMS NMJ chambers were incubated for 24 hours in 70% isopropanol to remove any unpolymerized monomers that could be toxic to the cells. The chambers were dipped in 100% ethanol and dried under sterile conditions for at least 2-hours prior to assembly. Glass coverslips (22 mm x 22 mm) were cleaned by plasma treatment for 2-minutes under oxygen pressure of 750 mTorr, sterilized with 70% ethanol and air-dried under sterile conditions before assembly. The sterile PDMS chambers adhered to the clean coverslips by applying gentle pressure around the edges and gently tapping on the tunnels. The SKM-side and MN-side of the chamber were coated with rat tail collagen I (ThermoFisher A1048301; 60 μg/mL) and laminin (ThermoFisher 23017015; 3 μg/mL), respectively. The collagen was removed after a 2-hour incubation period and rinsed twice with 1X phosphate buffer solution (PBS). The muscle proliferation medium (Adult Growth Medium, AGM) was added to the muscle-side before storing the systems at 4°C overnight. The laminin remained on the MN-side. After 24 hours, the laminin solution on the MN-side was removed and replaced with human motoneuron media (HMN). The NMJ systems were stored at 37°C and 5% CO2 1-hour before cell culture plating for equilibration.
Plating of MNs and skeletal muscle (SKM) plating in the BioMEMs NMJ system
Primary human skeletal muscle myoblasts were obtained from Lonza, Allendale, NJ, USA, and were passaged once in Adult Growth Medium (AGM)[49] and cryopreserved. These cells were seeded at a density of 500 cells/mm2 in the BioMEMs NMJ system in AGM. Upon 80% confluence near the tunnels, the myoblasts were switched to a serum-free differentiation medium (NBActiv4; Brain Bits). The differentiated MNs were plated in the BioMEMs NMJ system at a density of 1500 cells/mm2 five days after myoblasts plating (Figure 3A). The day after plating, MNs were thoroughly rinsed with fresh human motoneuron medium (hMN) to remove any detached/dead cells and to support optimal environment for axonal progression through the microtunnels. The systems were fed every two days by a half media change with NBActiv4 for the SKM side and MN medium for MN side up to D17, counted from the day of muscle differentiation.
Differentiation of human motoneurons from iPSCs
Human iPSCs were obtained from the Coriell Institute, Camden, NJ, from healthy subjects (line ND41865) as well as from ALS patients with the following mutations: SOD1 (D90A; ND35660), SOD1 (L144P; line ND39032) and FUS (G522A; ND39034). The differentiation protocol for motoneurons from iPSCs was based on the one described by Qu et al.[22] with modifications for replacing Component C with LDN 193189 (0.1 mM) and SB431542 (6 mM). Each iPSC line received was considered passage 0 and passaged up to P10 utilizing the established protocols recommended by the NIH. Cells at passages P6–10 were used for motoneuron induction.
Patch clamp analysis of iPSC-MNs
The electrophysiological properties of iPSC-derived MNs were investigated using whole-cell patch-clamp recording techniques as previously detailed.[50] Briefly, cultured neurons maintained on glass coverslips were placed in the recording chamber of a Zeiss Axioscope 2FS Plus upright microscope. In culture, the motoneurons were visually distinguished from non-neuronal cells using an infrared differential interference contrast (DIC) video-microscope. Borosilicate glass patch pipettes (BF 150-86-10; Sutter Instrument Company), with a resistance of 6 to 10 MΩ, were made using a Sutter P97 pipette puller (Sutter Instrument Company). Both current-clamp and voltage-clamp recordings were taken using a Multiclamp 700A amplifier (Axon instruments). The pipette (intracellular) solution contained 1 mM EGTA, 140 mM K-gluconate, 2 mM MgCl2, 2 mM Na2ATP and 10 mM HEPES (pH 7.2). The motoneuron medium was supplemented with 10 mM HEPES (pH 7.2) and was used as the extracellular solution for all patch experiments.
Following the formation of a giga-Ω seal and membrane puncture, the cell capacitance was compensated. Signals were filtered at 3 kHz and sampled at 20 kHz using a Digidata 1322A interface (Axon Instruments). Data recording and analysis were performed using the pClamp8 software (Axon Instruments). Membrane potentials were corrected by subtraction of a 15 mV tip potential, which was calculated using Axon’s pClamp8 program. Depolarization-evoked inward and outward currents were examined in voltage-clamp mode. Depolarization-evoked action potentials were examined in current-clamp mode and induced using 1 second depolarizing current injections from a −70 mV holding potential. Spontaneous firing and glutamate-induced firing were recorded from GAP-free mode.
Deanna Protocol (DP) formulation and treatment
Based on the DP treatment results from our previous study,[30] the DP formulation was composed of the following components: L-Arginine (310 μM), α-Ketoglutarate (310 μM), γ-Aminobutyric acid (0.8 μM), 5-Hydroxytryptophan (0.8 μM), and Glutamate oxaloacetate transaminase (7100 mU/L). The DP components were diluted in hMN medium and introduced to the MN-side of the BioMEMs NMJ system on D7. During feeding (every two days), the MN-side would be dosed with the DP medium. Functional testing would be conducted at 7 and 10 days after initial dosing with DP.
Preparation and functional testing of the co-culture in the Bio-MEMS NMJ system
A stimulation apparatus consisting of chlorinated silver wires connected to a pulse stimulator (A-M Systems, model 2100) was used to electrically stimulate either the muscle (direct) or MNs (indirect) by submerging Ag-Cl wires on the corresponding side of the NMJ chamber. A video camera recording pixel subtraction of the muscle contraction was used to obtain all functional data. The videos were recorded using a Hamamatsu digital camera (model C8484-05G) with a high-speed acquisition of 50 frames per second and LabView software for recording, pulse stimulation control and data analysis. Electrical pulses (2V) were applied at 0.33 Hz, 0.5 Hz, 1 Hz, and 2 Hz for a set duration of 15 seconds. During testing, systems were maintained in the corresponding medium with or without DP treatment at 37°C on a temperature-controlled heating stage.
Data analysis of NMJ recording
The video data was analyzed in Python using the OpenCV library. Briefly, the pixel values of the first frame of the video was subtracted from the pixel values of all subsequent frames, quantifying the degrees to which subsequent frames differ from the first. This difference was used to identify the contraction of the cells, as the movement of the cells generated a pixel differential, which increased as the cell contracted and decreased as the cell relaxed. This pixel differential was co-plotted with the stimulation pulse, allowing the identification of any correlation between the electrical stimulation and cell response to stimuli. Skipping during the duration of each stimulation frequency was also recorded. The number of synchronized contractions (Ns) at a given pulse was divided by the number of stimulation pulses within a set duration (Nτ) which gives the NMJ fidelity:
| equation. 1 |
To measure the fatigue index of NMJ, the muscle contraction traces demonstrating complete and partial tetanus at 1 Hz and 2 Hz were analyzed. Both the area under curve and the peak amplitude of each trace were measured using a customized Python script. The NMJ fatigue index[40] is defined as:
| equation. 2 |
Immunofluorescence and microscopy
Cells were fixed with 4% paraformaldehyde for 15 minutes at room temperature, rinsed twice with 1X PBS, and then blocked and permeabilized for 1-hour at room temperature in the blocking buffer (2.5% donkey serum, 1% BSA) plus 0.1% Triton-X. Primary antibodies against NFH (EMD Millipore; 1:1000), SMI32 (Calbiochem; 1:100), βIII Tubulin (Sigma; 1:1000), Islet-1 (Developmental Studies Hybridoma Bank (DSHB), 2 μg/ml), HB9 (DSHB, 2 μg/ml) and MAP2 (Millipore; 1:1000), Myosin Heavy Chain (DSHB, A4_1025, 2 μg/ml), Synaptophysin (abcam, 1:100) were diluted in blocking buffer and incubated overnight at 4°C. Samples were then incubated with species-specific secondary antibodies (Invitrogen; 1:250) for 2-hours at room temperature. Bungarotoxin-488 (BTX-488) (ThermoFisher, 1:250) was added during the secondary antibody incubation step. For visualization of nuclei in myotubes and MNs, DAPI (300 nM) was used. ProLong® Gold Antifade Mountant (P36930) was used to mount coverslips onto the slides. For staining the chambers, the PDMS component was not removed to avoid disturbance of the axons in the tunnels and innervation of myotubes. Fluorescence Imaging was performed using UltraView™ spinning disk confocal microscope (PerkinElmer). Volocity software was used to process Z-stack projections of scanned images.
Statistical analysis
For each mutant, at least five separate culture chambers spanning two or three separate culture batches were used for testing. In each chamber, a single myotube was selected for recording. This myotube was selected based on the reliability of a synchronized contraction under MN-side bulk field stimulation. Once selected, the myotube would be subjected to the testing regimen. Any data pre-processing such as normalization has been specified in corresponding text or Figure legends. Data are represented as mean ± standard error of the mean, and values were compared across conditions using one- and two-way ANOVA followed by Dunnett’s test for comparison to the control condition, incorporating data transformations when appropriate to improve homoscedasticity and/or normality, or student’s T-test (2-tail, unequal variance) for DP treatment experiments for comparison to the untreated condition. Excel was used for statistical analysis.
Supplementary Material
Acknowledgements
X. Guo and V. Smith contributed equally to this work. We would like to thank Drs. Brian Wainger and Steve Lambert for their helpful discussion.
Funding: Research supported by Department of Defense grant number AL130166 and the National Institutes of Health grant number R01-NS050452.
Competing interests: The authors confirm that competing financial interests exist but there has been no financial support for this research that could have influenced its outcome. However, JJH has ownership interest and is Chief Scientist and member of the Board of Directors in a company that may benefit financially as a result of the outcomes of the research or work reported in this publication.
Contributor Information
Xiufang Guo, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Virginia Smith, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Max Jackson, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
My Tran, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Michael Thomas, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Aakash Patel, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Eric Lorusso, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Siddharth Nimbalkar, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Yunqing Cai, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA.
Christopher W. McAleer, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA
Ying Wang, Department of Biomedical Engineering, 305 Weill Hall, Cornell University, Ithaca, NY, 14853, USA.
Christopher J. Long, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA
James J. Hickman, NanoScience Technology Center, University of Central Florida, 12424 Research Parkway, Suite 400, Orlando, FL 32826, USA
References
- [1].a) Ticozzi N, and Silani V, in Neurodegenerative Diseases: Clinical Aspects, Molecular Genetics and Biomarkers, Vol ; [Google Scholar]; b) Beghi E, Mennini T, Bendotti C, Bigini P, Logroscino G, Chio A, Hardiman O, Mitchell D, Swingler R, Traynor BJ, and Al-Chalabi A, Curr. Med. Chem. 2007, 14, 3185–200. [DOI] [PubMed] [Google Scholar]
- [2].a) Rohrer JD, Isaacs AM, Mizielinska S, Mead S, Lashley T, Wray S, Sidle K, Fratta P, Orrell RW, Hardy J, Holton J, Revesz T, Rossor MN, and Warren JD, The Lancet Neurology 2015, 14, 291–301; [DOI] [PubMed] [Google Scholar]; b) Taylor JP, Brown RH, and Cleveland DW, Nature 2016, 539, 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Serio A, and Patani R, Stem Cells 2018, 36, 293–303; [DOI] [PubMed] [Google Scholar]; b) Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, D. H., Takahashi R, Misawa H, and Cleveland DW, Nat. Neurosci. 2008, 11, 251–253; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Frakes Ashley E., Ferraiuolo L, Haidet-Phillips Amanda M., Schmelzer L, Braun L, Miranda Carlos J., Ladner Katherine J., Bevan Adam K., Foust Kevin D., Godbout Jonathan P., Popovich Phillip G., Guttridge Denis C., and Kaspar Brian K., Neuron 81, 1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sabatelli M, Conte A, and Zollino M, Clin. Genet. 2013, 83, 408–416. [DOI] [PubMed] [Google Scholar]
- [5].Campanari M-L, García-Ayllón M-S, Ciura S, Sáez-Valero J, and Kabashi E, Front. Mol. Neurosci. 2016, 9, 160–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cappello V, and Francolini M, Int. J. Mol. Sci. 2017, 18, 2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].So E, Mitchell JC, Memmi C, Chennell G, Vizcay-Barrena G, Allison L, Shaw CE, and Vance C, Hum. Mol. Genet. 2018, 27, 463–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Murray LM, Talbot K, and Gillingwater TH, Neuropathol. Appl. Neurobiol. 2010, 36, 133–56; [DOI] [PubMed] [Google Scholar]; b) Thomson SR, Wishart TM, Patani R, Chandran S, and Gillingwater TH, J. Anat. 2012, 220, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, and Glass JD, Exp. Neurol. 2004, 185, 232–240; [DOI] [PubMed] [Google Scholar]; b) Clark JA, Southam KA, Blizzard CA, King AE, and Dickson TC, J. Chem. Neuroanat. 2016, 76, 35–47. [DOI] [PubMed] [Google Scholar]
- [10].a) Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, and Oppenheim RW, The Journal of Neuroscience 2006, 26, 8774–8786; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rouaux C, Panteleeva I, René F. d. r., Gonzalez de Aguilar J-L, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier A-L, and Loeffler J-P, The Journal of Neuroscience 2007, 27, 5535–5545; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sironi F, Vallarola A, Violatto MB, Talamini L, Freschi M, De Gioia R, Capelli C, Agostini A, Moscatelli D, Tortarolo M, Bigini P, Introna M, and Bendotti C, Stem Cell Research 2017, 25, 166–178. [DOI] [PubMed] [Google Scholar]
- [11].a) Sanjak M, Brinkmann J, Belden DS, Roelke K, Waclawik A, Neville HE, Ringel SP, Murphy JR, and Brooks BR, J. Neurol. Sci. 2001, 191, 55–59; [DOI] [PubMed] [Google Scholar]; b) Sharma KR, Kent-Braun JA, Majumdar S, Huang Y, Mynhier M, Weiner MW, and Miller RG, Neurology 1995, 45, 733–40. [DOI] [PubMed] [Google Scholar]
- [12].Thomas CK, and Zijdewind I, Muscle Nerve 2006, 33, 21–41. [DOI] [PubMed] [Google Scholar]
- [13].a) Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, and Eggan K, Nat. Neurosci. 2007, 10, 608–614; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, and Przedborski S, Nat. Neurosci. 2007, 10, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, and Lee VM-Y, Science 2006, 314, 130–133. [DOI] [PubMed] [Google Scholar]
- [15].a) Mohs RC, and Greig NH, Alzheimers Dement (N Y) 2017, 3, 651–657; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mak IW, Evaniew N, and Ghert M, Am J Transl Res 2014, 6, 114–118. [PMC free article] [PubMed] [Google Scholar]
- [16].Fischbach GD, Dev. Biol. 1972, 28, 407–429. [DOI] [PubMed] [Google Scholar]
- [17].a) Li X-J, Du Z-W, Zarnowska ED, Pankratz M, Hansen LO, Pearce RA, and Zhang S-C, Nat. Biotechnol. 2005, 23, 215; [DOI] [PubMed] [Google Scholar]; b) Guo X, Das M, Rumsey J, Gonzalez M, Stancescu M, and Hickman J, Tissue engineering. Part C, Methods 2010, 16, 1347–55; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Umbach JA, Adams KL, Gundersen CB, and Novitch BG, PLoS One 2012, 7, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Puttonen K, Ruponen M, Naumenko N, Hovatta OH, Tavi P, and Koistinaho J, Front. Cell. Neurosci. 2015, 9, ; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Demestre M, Orth M, Föhr KJ, Achberger K, Ludolph AC, Liebau S, and Boeckers TM, Stem Cell Research 2015, 15, 328–336. [DOI] [PubMed] [Google Scholar]
- [19].Steinbeck Julius A., Jaiswal Manoj K., Calder Elizabeth L., Kishinevsky S, Weishaupt A, Toyka Klaus V., Goldstein Peter A., and Studer L, Cell Stem Cell 2016, 18, 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].a) Ionescu A, Zahavi EE, Gradus T, Ben-Yaakov K, and Perlson E, Eur. J. Cell Biol. 2016, 95, 69–88; [DOI] [PubMed] [Google Scholar]; b) Southam KA, King AE, Blizzard CA, McCormack GH, and Dickson TC, J. Neurosci. Methods 2013, 218, 164–169; [DOI] [PubMed] [Google Scholar]; c) Smith AST, Long CJ, Pirozzi K, and Hickman JJ, Technology 2013, 1, 37–48; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Osaki T, Uzel SGM, and Kamm RD, Science Advances 2018, 4, eaat5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Santhanam N, Kumanchik L, Guo X, Sommerhage F, Cai Y, Jackson M, Martin C, Saad G, McAleer CW, Wang Y, Lavado A, Long CJ, and Hickman JJ, Biomaterials 2018, 166, 64–78; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zahavi EE, Ionescu A, Gluska S, Gradus T, Ben-Yaakov K, and Perlson E, J. Cell Sci. 2015, 128, 1241–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Qu Q, Li D, Louis KR, Li X, Yang H, Sun Q, Crandall SR, Tsang S, Zhou J, Cox CL, Cheng J, and Wang F, Nat Commun 2014, 5, 3449. [DOI] [PubMed] [Google Scholar]
- [23].a) Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, and Sockanathan S, Neuron 1999, 23, 659–674; [DOI] [PubMed] [Google Scholar]; b) Sances S, Bruijn LI, Chandran S, Eggan K, Ho R, Klim JR, Livesey MR, Lowry E, Macklis JD, Rushton D, Sadegh C, Sareen D, Wichterle H, Zhang S-C, and Svendsen CN, Nat. Neurosci. 2016, 19, 542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].a) Ericson J, Thor S, Edlund T, Jessell T, and Yamada T, Science 1992, 256, 1555–1560; [DOI] [PubMed] [Google Scholar]; b) Qu Q, Li D, Louis KR, Li X, Yang H, Sun Q, Crandall SR, Tsang S, Zhou J, Cox CL, Cheng J, and Wang F, Nature Communications 2014, 5, 3449. [DOI] [PubMed] [Google Scholar]
- [25].a) Kawada J, Kaneda S, Kirihara T, Maroof A, Levi T, Eggan K, Fujii T, and Ikeuchi Y, Stem Cell Reports 2017, 9, 1441–1449; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Carriedo SG, Yin HZ, and Weiss JH, J. Neurosci. 1996, 16, 4069–4079; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tsang YM, Chiong F, Kuznetsov D, Kasarskis E, and Geula C, Brain Res. 2000, 861, 45–58; [DOI] [PubMed] [Google Scholar]; d) Grunseich C, Zukosky K, Kats IR, Ghosh L, Harmison GG, Bott LC, Rinaldi C, Chen K.-l., Chen G, Boehm M, and Fischbeck KH, Neurobiol. Dis. 2014, 70, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kunst CB, Am. J. Hum. Genet. 2004, 75, 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Huey ED, Ferrari R, Moreno JH, Jensen C, Morris CM, Potocnik F, Kalaria RN, Tierney M, Wassermann EM, Hardy J, Grafman J, and Momeni P, Neurobiol. Aging 2012, 33, 1016.e9–1016.e1.016E17; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Balasubramanian U, Cohen D, Zhang P-W, Mosmiller E, Sattler R, Maragakis NJ, and Rothstein JD, PLoS One 2015, 10, e0118266–e0118266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Körner S, Böselt S, Wichmann K, Thau-Habermann N, Zapf A, Knippenberg S, Dengler R, and Petri S, J. Neuropathol. Exp. Neurol. 2016, 75, 326–333. [DOI] [PubMed] [Google Scholar]
- [29].Coleman M, Nat. Rev. Neurosci. 2005, 6, 889–898. [DOI] [PubMed] [Google Scholar]
- [30].Lavado A, Guo X, Smith AS, Akanda N, Martin C, Cai Y, Elbrecht D, Tran M, Bryant J-P, Colon A, Long CJ, Lambert S, Morgan D, and Hickman JJ, Int J Pharm Pharm Res 2017, 11, 348–374. [PMC free article] [PubMed] [Google Scholar]
- [31].a) Chen H, Qian K, Du Z, Cao J, Petersen A, Liu H, Blackbourn Lisle W., Huang C-L, Errigo A, Yin Y, Lu J, Ayala M, and Zhang S-C, Cell Stem Cell 2014, 14, 796–809; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Morrison BM, Shu IW, Wilcox AL, Gordon JW, and Morrison JH, Exp. Neurol. 2000, 165, 207–220. [DOI] [PubMed] [Google Scholar]
- [32].Williamson TL, and Cleveland DW, Nat. Neurosci. 1999, 2, 50–56. [DOI] [PubMed] [Google Scholar]
- [33].a) Naujock M, Stanslowsky N, Bufler S, Naumann M, Reinhardt P, Sterneckert J, Kefalakes E, Kassebaum C, Bursch F, Lojewski X, Storch A, Frickenhaus M, Boeckers TM, Putz S, Demestre M, Liebau S, Klingenstein M, Ludolph AC, Dengler R, Kim K-S, Hermann A, Wegner F, and Petri S, Stem Cells 2016, 34, 1563–1575; [DOI] [PubMed] [Google Scholar]; b) Wainger Brian J., Kiskinis E, Mellin C, Wiskow O, Han Steve S. W., Sandoe J, Perez Numa P., Williams Luis A., Lee S, Boulting G, Berry James D., Brown Robert H., Cudkowicz Merit E., Bean Bruce P., Eggan K, and Woolf Clifford J., Cell Reports 2014, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gunthorpe MJ, Large CH, and Sankar R, Epilepsia 2012, 53, 412–424. [DOI] [PubMed] [Google Scholar]
- [35].a) McComas AJ, Skeletal muscle: form and function, Champaign, IL: Human Kinetics, (1996); [Google Scholar]; b) Jensen L, #xf8, rgensen LH, Bech RD, Frandsen U, Schr, #xf8, and der HD, BioMed Research International 2016, 2016, 12. [Google Scholar]
- [36].a) McComas AJ, Sica REP, Campbell MJ, and Upton ARM, Journal of Neurology, Neurosurgery, and Psychiatry 1971, 34, 453–460; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wohlfart G, Neurology 1957, 7, 124–124. [DOI] [PubMed] [Google Scholar]
- [37].Dreibati B, Lavet C, Pinti A, and Poumarat G, Ann. Phys. Rehabil. Med. 2010, 53, 266–277. [DOI] [PubMed] [Google Scholar]
- [38].Brooks BR, Miller RG, Swash M, and Munsat TL, Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000, 1, 293–9. [DOI] [PubMed] [Google Scholar]
- [39].a) Sanjak M, Konopacki R, Capasso R, Roelke KA, Peper SM, Houdek AM, Waclawik A, and Brooks BR, Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2004, 5, 26–32; [DOI] [PubMed] [Google Scholar]; b) Gibbons CJ, Thornton EW, and Young CA, Front. Psychol. 2013, 4, 788–788; [DOI] [PMC free article] [PubMed] [Google Scholar]; c)C. National Clinical Guideline, in Motor Neurone Disease: Assessment and Management, Vol [Google Scholar]
- [40].Lou J-S, Phys. Med. Rehabil. Clin. N. Am. 2005, 16, 1063–1079. [DOI] [PubMed] [Google Scholar]
- [41].a) Zoccolella S, Beghi E, Palagano G, Fraddosio A, Guerra V, Samarelli V, Lepore V, Simone IL, Lamberti P, Serlenga L, and Logroscino G, Eur. J. Neurol. 2007, 14, 262–268; [DOI] [PubMed] [Google Scholar]; b) Fang T, Al Khleifat A, Meurgey J-H, Jones A, Leigh PN, Bensimon G, and Al-Chalabi A, Lancet Neurol. 2018, ; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cruz MP, Pharmacy and Therapeutics 2018, 43, 25–28.29290672 [Google Scholar]
- [42].Group TA, Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration 2013, 14, 319–323. [DOI] [PubMed] [Google Scholar]
- [43].a) Piotrkiewicz M, and Hausmanowa-Petrusewicz I, Front. Aging Neurosci. 2013, 5, 7-7; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Martineau É, Di Polo A, Vande Velde C, and Robitaille R, eLife 2018, 7, e41973; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Henderson RD, and McCombe PA, Neurotherapeutics 2017, 14, 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Muresan V, and Ladescu Muresan Z, Neurodegener. Dis. 2016, 16, 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].a) Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao F-B, and Taylor JP, Nature 2015, 525, 129; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul Iii JW, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, and Gitler AD, Nat. Neurosci. 2015, 18, 1226; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chou C-C, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin-Asp PG, Chen YH, Duong DM, Seyfried NT, Powers MA, Kukar T, Hales CM, Gearing M, Cairns NJ, Boylan KB, Dickson DW, Rademakers R, Zhang Y-J, Petrucelli L, Sattler R, Zarnescu DC, Glass JD, and Rossoll W, Nat. Neurosci. 2018, 21, 228–239; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Aulas A, and Vande Velde C, Front. Cell. Neurosci. 2015, 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].a) Ari C, Poff AM, Held HE, Landon CS, Goldhagen CR, Mavromates N, and D’Agostino DP, PLoS One 2014, 9, e103526; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Muyderman H, and Chen T, Br. J. Pharmacol. 2014, 171, 2191–2205; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cozzolino M, and Carrì MT, Prog. Neurobiol. 2012, 97, 54–66. [DOI] [PubMed] [Google Scholar]
- [47].Maimon R, and Perlson E, Neural Regeneration Research 2019, 14, 969–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Madill M, McDonagh K, Ma J, Vajda A, McLoughlin P, O’Brien T, Hardiman O, and Shen S, Mol. Brain 2017, 10, 22; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Haidet-Phillips AM, Hester ME, Miranda CJ, Meyer K, Braun L, Frakes A, Song S, Likhite S, Murtha MJ, Foust KD, Rao M, Eagle A, Kammesheidt A, Christensen A, Mendell JR, Burghes AH, and Kaspar BK, Nat. Biotechnol. 2011, 29, 824–8; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, Chintawar S, Schnell C, Antel JP, Allen ND, Cader MZ, Wade-Martins R, James WS, and Cowley SA, Stem Cell Reports 8, 1727–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].McAleer CW, Rumsey JW, Stancescu M, and Hickman JJ, Biotechnol. Progr. 2015, 31, 997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Guo X, Johe K, Molnar P, Davis H, and Hickman J, Journal of Tissue Engineering Regenerative Medicine 2010, 4, 181–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.