

Abstract
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disorder that leads to neuronal death in the brain and spinal cord. Over the last decades, evidence has emerged regarding the functional diversity of astrocytes, microglia, and T cells in the central nervous system (CNS), and the role of neuroinflammation in ALS. In this review, we summarize current knowledge regarding neuroinflammation in ALS, both at the level of specific molecular pathways and potential cellular pathways as well as outline questions about the immune mechanisms involved in ALS pathogenesis.
Keywords: ALS, neuroinflammation, autoimmunity, glial cells
Introduction
Amyotrophic lateral sclerosis (ALS) is a multifactorial neurodegenerative disorder characterized by progressive degeneration of upper and lower motor neurons, which leads to muscle weakness, paralysis, and eventually death, usually within 2–5 years after diagnosis (1). About 95% of cases are sporadic (SALS; i.e., occurring randomly without a gene family history), while about 5% are familial ALS (FALS), suggesting a potential genetic basis (2). While more than 25 genes have been associated with ALS, alterations in few genes occur most frequently in both SALS and FALS (3–6). These are the Cu/Zn superoxide dismutase (SOD1) gene (encoding the SOD1 dismutase), TAR DNA binding protein 43 (TARDPB, encoding TDP-43), and chromosome 9 open reading frame 72 (C9ORF72, encoding C9orf72). As discussed below, each of these alterations could modulate immune reactivity in ALS progression (7).
Several studies suggest that changes in immune responses can contribute to the pathogenesis of ALS in humans and mice (8). Neurodegeneration in ALS is associated the aggregation of misfolded proteins, which leads to inclusion bodies formation in the central nervous system (CNS) and may promote an inflammatory response by resident and/or infiltrating immune cells (9).
Three cell types have in particular attracted attention for roles in neuroinflammatory processes potentially associated with ALS: resident astrocytes, microglial cells, and CNS-infiltrating T cells. The goal of this review is to discuss findings related to the current knowledge of immune-mediated alterations and disorders in ALS disease, and to identify knowledge gaps and areas for further investigation.
Mutant SOD1 and alterations of the inflammasome complex in ALS
Mutations in the SOD1 gene account for 12–23.5% of FALS and ~7.3% of apparent SALS, on the basis of multiple large population studies in Caucasians. There is no information about specific variants worldwide (10). SOD1 dismutase is a ubiquitous antioxidant enzyme, which accounts for 1-2% of the total cytoplasmic protein in the CNS. The role of SOD1 is to catalyze the dismutation of superoxide radicals to molecular oxygen or hydrogen peroxide neutralizing dangerous free superoxide radical (11). To date, more than 185 ALS-associated SOD1 variants have been described (12). Notably, the SOD1 variant (G93A) has been strongly associated with rapid progression and shortened lifespan (13). Although the SOD1G93A not cause loss of CuZn-superoxide dismutase activity, it confers a toxic gain-of-function linked to the propensity to promote mutant SOD1 (mSOD1) misfolding and aggregation (12). The generation of human-derived antibody targeting SOD1G93A corroborates the idea that mutations may induce some degree of misfolding which leads to neo-epitopes exposure and triggering immune responses (14). Moreover, it has been described that wild-type (WT) and misfolded SOD1 protein are secreted by a broad range of cells (15). Once secreted, mSOD1 protein may act as a damage-associated molecular pattern (DAMP), trigger inflammasome activation, and mediate neurotoxicity in ALS (16). Transgenic mice carrying a mutant SOD 1G93A were developed as a mouse model for ALS disease (17).
The NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome is a multiprotein complex that assembles in response to pathogen-associated molecular patterns (PAMPs) or DAMPs, to induce the secretion of the proinflammatory cytokines IL-1β and IL-18 (18). SOD1G93A mice exhibit a higher expression of NLRP3 in spinal cord astrocytes in pre- and early-symptomatic periods and increased IL-1β levels, implicating inflammasome activation in ALS pathogenesis (16). During the pre-symptomatic stages, astrocytes from SOD1G93A mice exhibit increased expression of IFN-γ responsive genes, leading to neuronal death through IFN-γ release (19). Spinal cord astrocytes from ALS patients also show higher expression of NLRP3 with increased IL-18 and active caspase-1 further suggesting a possible role for the inflammasome complex (16).
Studies have suggested that the number of mSOD1 gene copies has a significant impact on disease severity (20). Indeed, compared to SOD1G93A mice, the SOD1G37R mice displayed slowed disease onset and progression due to low expression of mSOD1 (14). Furthermore, genetic excision of mSOD1 in astrocytes led to delayed microglial activation and slower disease progression in SOD1G37R mice, demonstrating that its overexpression in astrocyte alter their crosstalk with microglia (21). TGF-β1 upregulation in astrocytes from ALS patients and SOD1G93A mice accelerated disease progression, while excision of SOD1G93A decreased TGF-β1 levels and slowed disease progression (22). Levels of TGF-β1 were higher in the serum, plasma, and cerebrospinal fluid (CSF) of ALS patients and correlated with clinical outcomes (23). Furthermore, administration of the TGF-β1 inhibitor SB-431542, extended survival in SOD1G93A mice (22).
Finally, the P2X7 receptor mediate NLRP3 inflammasome activation in response to extracellular ATP. SOD1G93A spinal cord astrocytes are prone to P2X7 activation and show increased proliferation compared to WT astrocytes. Moreover, blockade of ATP signaling in SOD1G93A astrocytes prevents motor neuron death by inhibiting neurotoxic astrocyte proliferation (24). Taken together, these studies suggest a role for alterations of the inflammasome complex and SOD1 in ALS.
Toll-like receptors (TLRs) and ALS
TLRs are expressed in neurons, astrocytes, and microglia (25) and recognize PAMPs, such as lipopolysaccharides (LPS), and DAMPs, released during tissue damage (26). Several TLRs, including TLR4 and TLR2, have been implicated in ALS pathogenesis (27).
Microglial cells transfected with SOD1G93A responded to TLR2 stimulation with a higher oxidative stress marker response and released larger amounts of TNF-α than controls expressing WT SOD1 (28). The TLR4 agonist LPS increased reactive oxygen species (ROS) and nitric oxide (NO) production, activated NF-κB inducing death of motor neurons (29).
TLR4 antagonists prevent motor neuron death in spinal cord cultures from SOD1G93A mice through inhibition of IL-1β release by LPS-stimulated microglia (30), treatment with recombinant IL-1 receptor antagonist extended the lifespan of SOD1G93A mice (31) and TLR4 is upregulated in the lumbar spinal cord of SOD1G93A mice and ALS patients (32). In conclusion, signaling through different TLRs may contribute to a chronic inflammatory state in ALS.
The interplay between TDP-43 alterations and innate immunity
Aberrant C-terminal fragments of TDP-43 protein are ubiquitinated, hyperphosphorylated, and accumulate as cellular inclusions in neurons and glia (33). Extracellular TDP-43 accumulation also occurs, perhaps as a consequence of cell death caused by intracellular cytotoxicity of TDP-43 inclusion bodies. ALS patients showed higher levels of TDP-43 in CSF than age-matched healthy controls (34). Furthermore, TDP-43 protein has been found in secreted exosomes from primary neurons, but not from astrocytes and microglia, which may contribute to both propagation and cellular clearance of TDP-43 in ALS brain (35).
In microglial cultures, mutant TDP-43 interacts with the TLR4 coreceptor CD14 triggering NF-κB and NLRP3 inflammasome activation, promoting TNF-α release, and leading to motor neuron death (36). Since inflammatory stimuli such as LPS or TNF-α induce TDP-43 aggregation in the cytoplasm of microglia, secreted TNF-α may exacerbate TDP-43 proteinopathy (37). Internalization of TDP-43 aggregates by microglia, promotes dynamic changes such as ubiquitination, proteolytic cleavage, translocation of s TDP-43 from the nucleus to the cytoplasm and triggers NLRP3, caspase activation leading to the release of IL-1β and IL-18 (38).
The role of astrocytes in ALS
Astrocytes are immune-competent cells and the most abundant glial cells in the CNS. They play an essential role in the blood-brain barrier maintenance and permeability, control immune cell trafficking, and promote both suppressive and pro-inflammatory responses (39). Astrocytes are found in postmortem brain tissue, in the subcortical white matter and spinal cord of both SALS and FALS patients, suggesting that astrocytes respond to CNS injury (40,41). Relatedly, astrocytes derived from both ALS patients and animal models induce death in vitro of motor neurons that display low expression of the major histocompatibility complex class I (MHC-I) molecule. In contrast, sustained MHC-I expression prevents astrocyte-mediated toxicity, thus increasing functional performance and motor neuron survival (42).
Furthermore, cortical astrocytes isolated from SOD1G93A at an early disease stage exhibit a neuroprotective profile with decreased cell proliferation, low expression of NF-κB and micro(miR)-146a, and dampening pro-inflammatory pathways, while of NF-κB and micro(miR)-146a are upregulated at the symptomatic stage (43).
The neuronal Ephrin Type-B Receptor 1 (EphB1) is associated with increased levels of phosphorylated STAT3, and pre-treatment with EphB1 small interfering RNA (siRNA) prevented this effect. Moreover, SOD1 G93A astrocytes from both mice and humans do not display STAT3 phosphorylation following EphB1 stimulation and this neuroprotective response is impaired in ALS (44).
Astrocytes can produce a wide array of cytokines in response to neuronal activity through a process known as astrogliosis (45). Recently, it was demonstrated that lysophosphatidylcholine (LPC), a lipid associated with neurodegeneration, acts as a trigger of NLRP3 inflammasome activation in astrocytes. NLRP3-deficient mice showed decreased levels of the LPC receptor and reduced astrogliosis (46), indicating that the NLRP3 inflammasome is involved in the astrocytic component of neuroinflammation.
The role of microglia in ALS disease
Microglia are CNS-resident cells that play an important role in immune surveillance of the brain and spinal cord. While astrocytes originate from neuroepithelial progenitors, microglia are derived from yolk sac macrophage progenitors that migrate into the brain early during embryonic development. Under physiological conditions, microglia exist in a resting state. When stimulated, these cells adopt either a pro-inflammatory (M1-like) or the anti-inflammatory (M2-like) state, depending on the disease stage and surrounding inflammatory milieu (47). M1-like microglia shows phagocytic activity, and produce pro-inflammatory mediators, including, ROS and NO, IL-1β, IL-6, TNF-α, and IFN-γ. In contrast, M2-like microglia are associated with the production of anti-inflammatory cytokines such as IL-4, IL-10 and IL-13 (9).
Visualization of microglial activation using Positron Emission Tomography (PET) reveals microglia activation in the motor cortex and precentral gyrus of ALS patients, which correlates with disease severity (48).
Several reports have demonstrated that mSOD1 microglia may contribute to the progression of ALS. Overexpression has shown that SOD1G93A increased expression of RAGE, a pro-inflammatory pattern-recognition receptor, and released exosomes enriched in HMGB1 and SOD1 which may act as a DAMP and interact with RAGE to promote a pro-inflammatory response (49,50). Indeed, HMGB1 blocking using anti-HMGB1 antibody reduces neurotoxicity in SOD1G93A mice at the pre-onset stage (51). Moreover, SOD1G93A mice lacking RAGE had slower disease progression and extended lifespan. This effect was associated with reduced numbers of microglia and astrocytes, and decreased expression of TNF-α, C1q, and IL-1β in the spinal cord (52). Remarkably, secretion of TNF-α, C1q and IL-1α mediated astrocyte activation by microglia, and knockout of these factors dampened reactive astrogliosis (53). Thus, the HMGB1/RAGE axis emerged as a potential therapeutic target for ALS.
Selective excision of SOD1G37R from microglia and macrophages extends survival of transgenic mice (54). Restoration of microglial cells after bone marrow (BM) transplantation in mSOD1/PU.1−/− mice, which lack endogenous microglia at birth, slows disease progression and extends survival (55).
Animal models suggest a shift in microglia subsets throughout the disease course of ALS. At disease onset, SOD1G93A microglia showed increased levels of M2 markers, such as Ym1, CD163, and BDNF and lower levels of M1 markers, compared to end-stage disease (56). Moreover, following LPS stimulation, SOD1G93A microglia was associated with increased IL-10 production and lower levels of TLR2 expression at the pre-symptomatic stage (57).
Zhao et al. 2020 demonstrated that M2-like cells derived from iPSCs (induced pluripotent stem cells) of ALS patients suppressed the production of IL-6 and TNF-α by M1-like cells, and inhibited effector T cell proliferation in vitro. These M2-like cells converted effector T cells into regulatory T cells (Tregs) with sustained FoxP3 expression and were capable of rescuing the immunomodulatory capacity of Tregs from ALS patients (58). Overall, these studies point towards the heterogeneity of microglial functions that display either neuroprotective and neurotoxicity properties throughout the course of disease progression and shed some light on its therapeutic potential.
C9orf72-mediated immune response in ALS
Expansion of a GGGGCC (G4C2) hexanucleotide repeat in the intronic region of the C9orf72 gene is the most common genetic cause of ALS and frontotemporal dementia (FTD), accounting for up to 40% of FALS cases and 10% of SALS (59). While healthy individuals have between 2-23 G4C2 repeats, ALS, and/or FTD, patients may have hundreds to thousands (60). To date, three mechanisms have been proposed to underlie how C9orf72 mutations contribute to ALS pathogenesis, which includes: [1] decreased expression of C9orf72 transcripts and loss-of-function of the C9orf72 protein, also known as C9ORF72 haploinsufficiency; [2] RNA-mediated toxicity due to accumulation of the repeat-containing transcripts that form RNA foci in the nucleus; [3] toxicity mediated by increased dipeptide repeat protein synthesis through a process termed Repeat-associated non-AUG (RAN) translation (61).
The C9orf72 gene expression is higher in microglia and other myeloid cells than in neurons suggesting a potential role in innate immune responses and/or other microglia functions. C9orf72−/− deficient mice exhibit progressive splenomegaly and lymphadenopathy, develop mild age-related neuroinflammation, and show upregulation of pro-inflammatory-related genes as well as increased levels of IL-6 and IL-1β (62,63) Similarly, ALS patients with C9orf72 repeat expansions showed an association between IL-1β and lifespan (64). Moreover, the loss of C9orf72 leads to increased autophagic activity and promotes lysosomal trafficking defects in microglia, which can disrupt the clearance of aberrant proteins (63,65). Furthermore, post-mortem human brain samples from ALS patients carrying C9orf72 mutations have shown increased microglial activation with a positive correlation between CD86 and Iba1, markers of microglial activation, and disease severity (66).
Recent studies indicate that C9orf72 mutation is associated with autoimmunity. C9orf72−/− mice exhibit high titers of autoantibodies to antinuclear antibodies (ANA), anti-double-stranded DNA (dsDNA), and anti-rheumatoid factor (RF) compared to WT mice and C9orf72+/− mice (67). In contrast, C9ORF72 knockout mice’s repopulation with bone marrow from WT mice promotes immune tolerance (68). Taken together, these findings may explain why ALS and FTD patients carrying the C9ORF72 mutation seem to be more susceptible to autoimmune disorders.
The role of T cells in ALS
The data reviewed above suggest that astrocytes and microglia alterations in ALS are mediated by genes and proteins associated with cell metabolism, immune activation and innate immunity. The data also highlight alterations in key proteins, such as TDP-43, prone to aggregation and likely to be internalized and processed by phagocytic cells. A logical hypothesis is that these events might trigger an adaptive immune response mediated by T cells.
The concept that ALS is associated with autoimmune features was first introduced by Appel et al. more than 30 years ago (69) and several studies reported infiltration of T cells in the brain and spinal cord, and abnormalities in the T cell subsets in peripheral blood from ALS patients and murine disease models (70–73). When T cell recruitment is blocked in SOD1G93A mice, either through CCR2 (CD192) inhibition or CD4+ T cell depletion, mice have shorter lifespans and increased expression of pro-inflammatory genes (74). In addition, SOD1RAG2−/− mice, an animal model lacking mature lymphocytes, showed increased microglial activation at early but not in later stages compared to SOD1RAG2+/+ mice (75). Taken together, these data suggest a potential interaction between T cells and microglia.
The infiltrating cytotoxic CD8+ T cells (CTL) have been reported in the CNS of both ALS patients, and SOD1G93A mice (75,76). Consistently, the ablation of MHC-I in microglia and lack of CD8+ T infiltration in the spinal cord of SOD1G93A mice is associated with motor neuron survival and extended lifespan. Recently, Coque et al. demonstrated that SOD1G93A CD8+ T cells recognize MHC-I restricted self-peptides presented by motor neurons, leading to neuronal death mediated through the FasL/Granzyme B pathway. Activated SOD1 CD8+ T cells produced IFN-γ, which induced the expression of MHC-I on motor neurons. Notably, CD8+ T cells infiltrated in CNS of SOD1G93A mice displayed an oligoclonal TCRVβ repertoire compared to peripheral CD8+ T cells, and a specific clonotype (mTRBV15) was found within lymph nodes and CNS of SOD1G93A mice (77). Altogether, these data suggest that these cells may contribute to motor neuron death in an MHC-I-dependent manner.
Regulatory T cells (Tregs) and ALS
CD4+CD25hiFoxP3+ Tregs are essential for the maintenance of self-tolerance and immune homeostasis in humans and mice. Indeed, Tregs can inhibit pro-inflammatory T cells and activated microglia in vitro and in vivo (78). IL-4+ Tregs from spinal cords of SOD1G93A mice were increased at early disease stages and were associated with M2-like microglia. Treg function decreased in the rapid progression stage, with loss of FoxP3 expression (74). Consistent with these observations, ALS patients showed decreased numbers of FoxP3+ Tregs in peripheral blood at the rapid progression stage, as well as low levels of TGF-β and IL-4. Treg numbers inversely correlated with disease progression and severity (79–81).
Tregs from ALS patients were impaired in suppressive function, but after in vitro stimulation recovered their suppressive capacity, suggesting a potential therapeutic strategy (82). Alsuliman et al. developed an approach to isolate autologous Tregs from ALS patients to expand them ex vivo in the presence of IL-2 and rapamycin for clinical use. A phase I trial demonstrated a potential benefit of autologous Treg infusions, and this correlated with a potential slowing of disease progression (83,84). A randomized, double-blind phase II clinical trial is underway to evaluate safety and efficacy (MIROCALS: Modifying Immune Response and Outcomes in ALS).
Antigen specific CD4+ T cells and ALS
Since the CNS is considered an immune-privileged site, it is assumed that reactive CD4+ T cell response to CNS-derived antigens (self or non-self) are primed in the periphery. As summarized in Figure 1, antigen-presenting cells (APCs), such as dendritic cells (DC), may capture aggregated CNS-derived antigens and display them through MHC-II for recognition by CD4+ T cells in the draining lymph nodes. Once primed, CD4+ T cells may differentiate into distinct Th lineages after recognizing their cognate antigen in the CNS (85). Microglia are the predominant cell type expressing MHC-II in the CNS (86) and may act as APCs and modulate the reactivity of incoming T cells. Under steady-state conditions, microglia express low levels of MHC-II, but expression is increased upon activation (87).
Figure 1. CD4+ T cell reactivity in ALS disease.
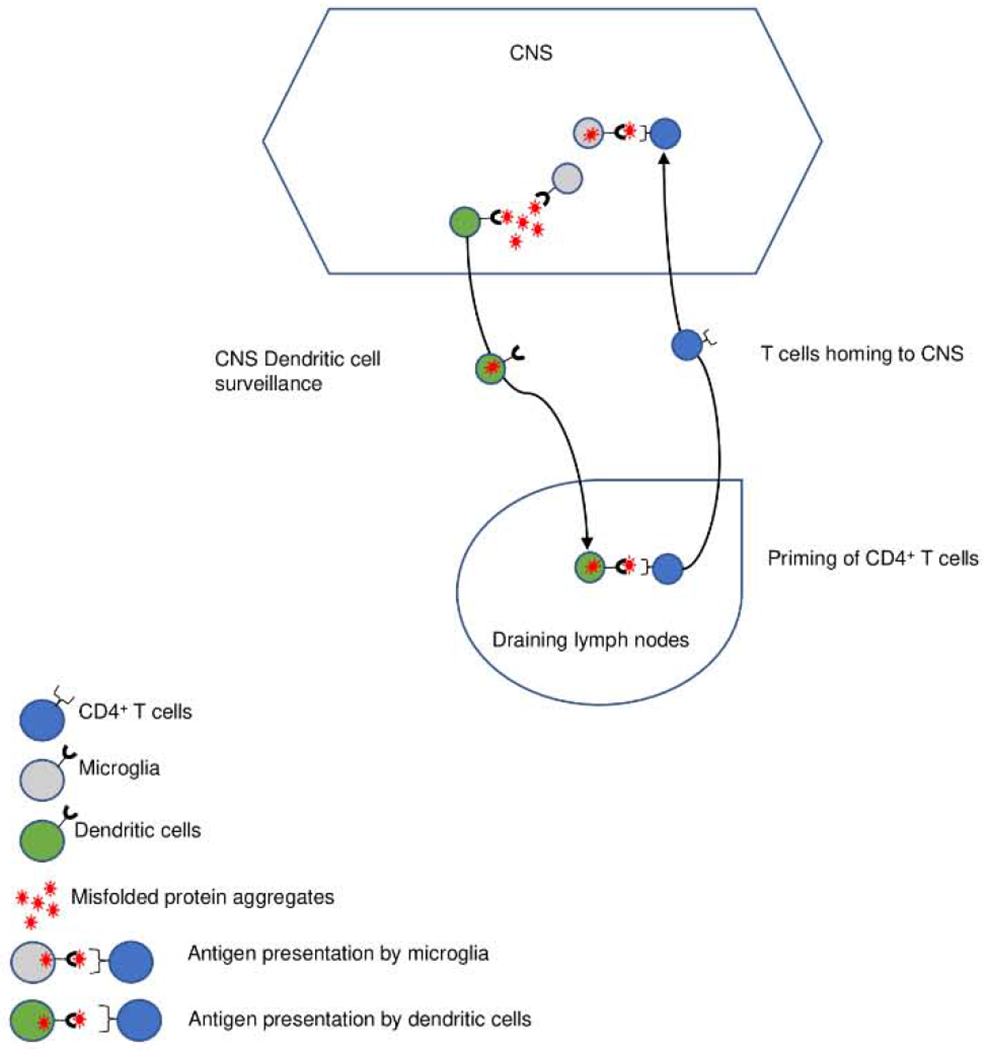
Misfolded protein aggregates are taken up by dendritic cells and reach the draining lymph nodes, where proteins can be presented as self-peptides through MHC class II for recognition by CD4+ T cells. After priming, antigen-specific T cells migrate to the CNS. Upon activation, microglia upregulate MHC class II and act as antigen-presenting cells to restimulate CNS-infiltrating T cells, promoting neuroinflammation.
Misfolded proteins are targeted for degradation by the ubiquitin/proteasome system and the resulting peptide fragments associate MHC-II molecules and are presented to T cells (88). Indeed, several studies have shown that CD4+ T cells may recognize neurodegeneration-associated proteins, thus suggesting autoimmune features of neurodegenerative disorders (89–92). Monsonego et al. identified CD4+ T cells specific for amyloid β (Aβ) in Alzheimer’s disease (AD) patients. These Aβ-reactive T cells exhibited different cytokine profiles, including Th1 (associated with IFN-γ production) and Th2 (associated with IL-5 and IL-13 production) (89). Recently, we demonstrated that the CD4+ T cell reactivity to epitopes derived from α-synuclein (α-syn) in Parkinson’s disease (PD) patients (90). In a follow up study, α-syn T cell reactivity was high even before the onset of motor symptoms and PD diagnosis, suggesting a pivotal role of α-syn-specific CD4+ T cells at the preclinical PD stage (91). These studies suggest a role for T cells in neurodegenerative diseases and that aggregated neuronal proteins can be engulfed, processed and presented by APCs.
Increased levels of TCRBV2 gene transcripts were observed in the CSF of ALS patients suggesting an antigen-mediated T cell expansion (93). Consistent with this observation, microglia upregulate MHC-II expression in the CSF, which may reflect increased antigen presentation (75). As described above, Tregs are decreased in the rapid progression stage of ALS. However, Tregs deficiency would not be expected to cause damage in itself, which would rather be likely resulting from a no longer regulated antigen-specific T cell response.
Thus, taken together, we hypothesize that TDP-43 and SOD1 proteins, which forms aggregates in ALS patients, may be recognized by CD4+ T cells and trigger an autoimmune response in the CNS. In this scenario, it would be of interest to identify the neuronal antigens that drive specific T cell responses, and characterize the phenotype of responding cells. This would enable studies of how epitope-specific T cells may modulate responses and influence different stages of ALS.
CONCLUSIONS
Several studies have suggested that neuroinflammation is involved in ALS disease progression. The molecular pathways implicated include the SOD1 and NLRP3 inflammasome complex, leading to release of large amounts of IL-1β and IL-18; and in turn motor neuron death. Moreover, recent evidence supports the interplay between TLR signaling, TDP-43 proteinopathy and motor neuron death. At the cellular level, both astrocytes and microglia are implicated. Microglial are identified as potential APCs engulfing aggregated proteins, degrading and presenting processed peptides bound to MHC-II for immune recognition by T cells. While a potential role for Tregs in ALS has been reported, autoimmune damage is likely mediated by effector T cells causing neuronal damage. While the antigens associated with effector CD4+ and CD8+ T cell reactivity to CNS-derived antigens in other neurodegenerative diseases have been described, no information is currently available about the targets of neuro-antigen specific T cells in ALS. The identification of these targets, would allow to study the contribution of effector T cells to ALS pathogenesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURES
The authors declare no competing interests
REFERENCES
- 1.Mehta P, Kaye W, Raymond J, Punjani R, Larson T, Cohen J. Prevalence of Amyotrophic Lateral Sclerosis. Morb Mortal Wkly Rep. 2018;67(46):1285–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sabatelli M, Conte A, Zollino M. Clinical and genetic heterogeneity of amyotrophic lateral sclerosis. Clin Genet. 2013;83(5):408–16. [DOI] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. [DOI] [PubMed] [Google Scholar]
- 4.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science (80-). 2008;319(5870):1668–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron [Internet]. 2011;72(2):245–56. Available from: 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nguyen HP, Van Broeckhoven C, van der Zee J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet [Internet]. 2018;34(6):404–23. Available from: 10.1016/j.tig.2018.03.001 [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Wang F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front Immunol. 2017;8(AUG):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beers DR, Zhao W, Liao B, Kano O, Wang J, Huang A, et al. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav Immun [Internet]. 2011;25(5):1025–35. Available from: 10.1016/j.bbi.2010.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hooten KG, Beers DR, Zhao W, Appel SH. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics. 2015;12(2):364–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei Q, Zhou Q, Chen Y, Ou R, Cao B, Xu Y, et al. Analysis of SOD1 mutations in a Chinese population with amyotrophic lateral sclerosis: A case-control study and literature review. Sci Rep. 2017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bunton-Stasyshyn RKA, Saccon RA, Fratta P, Fisher EMC. SOD1 Function and Its Implications for Amyotrophic Lateral Sclerosis Pathology: New and Renascent Themes. Neuroscientist. 2015;21(5):519–29. [DOI] [PubMed] [Google Scholar]
- 12.Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front Neurosci. 2019;13(December):1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamashita S, Ando Y. Genotype-phenotype relationship in hereditary amyotrophic lateral sclerosis. Translational Neurodegeneration. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maier M, Welt T, Wirth F, Montrasio F, Preisig D, McAfoose J, et al. A human-derived antibody targets misfolded SOD1 and ameliorates motor symptoms in mouse models of amyotrophic lateral sclerosis. Sci Transl Med. 2018; [DOI] [PubMed] [Google Scholar]
- 15.Mondola P, Damiano S, Sasso A, Santillo M. The Cu, Zn superoxide dismutase: Not only a dismutase enzyme. Front Physiol. 2016;7(NOV):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, et al. NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia. 2015;63(12):2260–73. [DOI] [PubMed] [Google Scholar]
- 17.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science (80-). 1994;264(5166): 1772–5. [DOI] [PubMed] [Google Scholar]
- 18.Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis [Internet], 2019; 10(2). Available from: 10.1038/s41419-019-1413-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aebischer J, Cassina P, Otsmane B, Moumen A, Seilhean D, Meininger V, et al. IFNγ triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ [Internet], 2011; 18(5):754–68. Available from: 10.1038/cdd.2010.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majchrzak M, Drela K, Andrzejewska A, Rogujski P, Figurska S, Fiedorowicz M, et al. SOD1/Rag2 Mice with Low Copy Number of SOD1 Gene as a New Long-Living Immunodeficient Model of ALS. Sci Rep [Internet], 2019;9(1): 1–13. Available from: 10.1038/s41598-018-37235-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Endo F, Komine O, Fujimori-Tonou N, Katsuno M, Jin S, Watanabe S, et al. Astrocyte-Derived TGF-β1 Accelerates Disease Progression in ALS Mice by Interfering with the Neuroprotective Functions of Microglia and T Cells. Cell Rep [Internet], 2015;11(4):592–604. Available from: 10.1016/j.celrep.2015.03.053 [DOI] [PubMed] [Google Scholar]
- 23.Itzecka J, Stelmasiak Z, Dobosz B. Transforming growth factor-beta 1 (TGF-beta 1) in patients with amyotrophic lateral sclerosis. Cytokine. 2002;20(5):239–43. [DOI] [PubMed] [Google Scholar]
- 24.Gandelman M, Peluffo H, Beckman JS, Cassina P, Barbeito L. Extracellular ATP and the P2X7receptor in astrocyte-mediated motor neuron death: Implications for amyotrophic lateral sclerosis. J Neuroinflammation. 2010;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rietdijk CD, Wezel RJA Van, Garssen J, Kraneveld AD. Neuronal toll-like receptors and neuro-immunity in Parkinson’s disease, Alzheimer’s disease and stroke. 2016;27–37. [Google Scholar]
- 26.Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol Mech Dis. 2020;15(1):493–518. [DOI] [PubMed] [Google Scholar]
- 27.Letiembre M, Liu Y, Walter S, Hao W, Pfander T, Wrede A, et al. Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiol Aging. 2009;30(5):759–68. [DOI] [PubMed] [Google Scholar]
- 28.Liu Y, Hao W, Dawson A, Liu S, Fassbender K. Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J Biol Chem. 2009;284(6):3691–9. [DOI] [PubMed] [Google Scholar]
- 29.Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circulation Research. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Paola M, Sestito SE, Mariani A, Memo C, Fanelli R, Freschi M, et al. Synthetic and natural small molecule TLR4 antagonists inhibit motoneuron death in cultures from ALS mouse model. Pharmacol Res [Internet], 2016;103:180–7. Available from: 10.1016/j.phrs.2015.11.020 [DOI] [PubMed] [Google Scholar]
- 31.Meissner F, Molawi K, Zychlinsky A. Mutant superoxide dismutase 1-induced IL-1β accelerates ALS pathogenesis. Proc Natl Acad Sci U S A. 2010; 107(29): 13046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Casula M, Iyer AM, Spliet WGM, Anink JJ, Steentjes K, Sta M, et al. Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience [Internet], 2011;179:233–43. Available from: 10.1016/j.neuroscience.2011.02.001 [DOI] [PubMed] [Google Scholar]
- 33.Prasad A, Bharathi V, Sivalingam V, Girdhar A, Patel BK. Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis. Front Mol Neurosci. 2019; 12(February): 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yl Noto, Shibuya K, Sato Y, Kanai K, Misawa S, Sawai S, et al. Elevated CSF TDP-43 levels in amyotrophic lateral sclerosis: Specificity, sensitivity, and a possible prognostic value. Amyotroph Lateral Scler. 2011. ;12(2): 140–3. [DOI] [PubMed] [Google Scholar]
- 35.Iguchi Y, Eid L, Parent M, Soucy G, Bareil C, Riku Y, et al. Exosome secretion is a key pathway for clearance of pathological TDP-43. Brain. 2016;139(12):3187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao W, Beers DR, Bell S, Wang J, Wen S, Baloh RH, et al. TDP-43 activates microglia through NF-κB and NLRP3 inflammasome. Exp Neurol [Internet], 2015;273:24–35. Available from: 10.1016/j.expneurol.2015.07.019 [DOI] [PubMed] [Google Scholar]
- 37.Correia AS, Patel P, Dutta K, Julien JP. Inflammation induces TDP-43 mislocalization and aggregation. PLoS One. 2015; 10(10): 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leal-Lasarte MM, Franco JM, Labrador-Garrido A, Pozo D, Roodveldt C. Extracellular TDP-43 aggregates target MAPK/MAK/MRK overlapping kinase (MOK) and trigger caspase-3/IL-18 signaling in microglia. FASEB J. 2017;31(7):2797–816. [DOI] [PubMed] [Google Scholar]
- 39.Colombo E, Farina C. Astrocytes: Key Regulators of Neuroinflammation. Trends Immunol. 2016;37(9):608–20. [DOI] [PubMed] [Google Scholar]
- 40.Stephenson DT, Stephenson DT, Wright S. Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. Vol. 50, Journal of Neuropathology and Experimental Neurology. 1991. p. 263–77. [DOI] [PubMed] [Google Scholar]
- 41.Schiffer D, Cordera S, Cavalla P, Migheli A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J Neurol Sci. 1996;139(SUPPL.):27–33. [DOI] [PubMed] [Google Scholar]
- 42.Song SW, Miranda CJ, Braun L, Meyer K, Frakes AE, Ferraiuolo L, et al. Major histocompatibility complex class I molecules protect motor neurons from astrocyte-induced toxicity in amyotrophic lateral sclerosis. Nat Med. 2016;22(4):397–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomes C, Cunha C, Nascimento F, Ribeiro JA, Vaz AR, Brites D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and miR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model. Mol Neurobiol. 2019;56(3):2137–58. [DOI] [PubMed] [Google Scholar]
- 44.Tyzack GE, Hall CE, Sibley CR, Cymes T, Forostyak S, Carlino G, et al. A neuroprotective astrocyte state is induced by neuronal signal EphB1 but fails in ALS models. Nat Commun [Internet]. 2017;8(1). Available from: 10.1038/s41467-017-01283-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Voet S, Srinivasan S, Lamkanfi M, Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med. 2019;11(6):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JPY. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214(5):1351–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wolf SA, Boddeke HWGM, Kettenmann H. Microglia in Physiology and Disease. Annu Rev Physiol. 2017;79(1):619–43. [DOI] [PubMed] [Google Scholar]
- 48.Turner MR, Cagnin A, Turkheimer FE, Miller CCJ, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15(3):601–9. [DOI] [PubMed] [Google Scholar]
- 49.Vaz AR, Pinto S, Ezequiel C, Cunha C, Carvalho LA, Moreira R, et al. Phenotypic effects of wild-type and mutant SOD1 expression in n9 murine microglia at steady state, inflammatory and immunomodulatory conditions. Front Cell Neurosci. 2019;13(April):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ray R, Juranek JK, Rai V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci Biobehav Rev [Internet]. 2016;62:48–55. Available from: 10.1016/j.neubiorev.2015.12.006 [DOI] [PubMed] [Google Scholar]
- 51.Lee JD, Liu N, Levin SC, Ottosson L, Andersson U, Harris HE, et al. Therapeutic blockade of HMGB1 reduces early motor deficits, but not survival in the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Neuroinflammation. 2019;16(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JD, McDonald TS, Fung JNT, Woodruff TM. Absence of Receptor for Advanced Glycation End Product (RAGE) Reduces Inflammation and Extends Survival in the hSOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol Neurobiol. 2020;57(10):4143–55. [DOI] [PubMed] [Google Scholar]
- 53.Guttenplan KA, Weigel MK, Adler DI, Couthouis J, Liddelow SA, Gitler AD, et al. Knockout of reactive astrocyte activating factors slows disease progression in an ALS mouse model. Nat Commun [Internet]. 2020;11(1):1–9. Available from: 10.1038/s41467-020-17514-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science (80-). 2006;312(5778):1389–92. [DOI] [PubMed] [Google Scholar]
- 55.Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103(43):16021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol [Internet]. 2012;237(1):147–52. Available from: 10.1016/j.expneurol.2012.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gravel M, Beland LC, Soucy G, Abdelhamid E, Rahimian R, Gravel C, et al. II-10 controls early microglial phenotypes and disease onset in ALS caused by misfolded superoxide dismutase 1. J Neurosci. 2016;36(3):1031–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhao W, Beers DR, Thonhoff JR, Thome AD, Faridar A, Wang J, et al. Immunosuppressive Functions of M2 Macrophages Derived from iPSCs of Patients with ALS and Healthy Controls. Vol. 23, iScience. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lall D, Baloh RH. Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. J Clin Invest. 2017;127(9):3250–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang Q, Jiao B, Shen L. The Development of C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Disorders. Frontiers in Genetics. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AKMG, et al. C9orf72 is required for proper macrophage and microglial function in mice. Science (80-). 2016;351(6279):1324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olesen MN, Wuolikainen A, Nilsson AC, Wirenfeldt M, Forsberg K, Madsen JS, et al. Inflammatory profiles relate to survival in subtypes of amyotrophic lateral sclerosis. Neurol Neuroimmunol neuroinflammation. 2020;7(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, et al. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun [Internet]. 2016;4(1):51. Available from: 10.1186/s40478-016-0324-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brettschneider J, Toledo JB, van Deerlin VM, Elman L, McCluskey L, Lee VMY, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS One. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Appel SH, Stockton Appel V, Stewart SS, Kerman RH. Amyotrophic Lateral Sclerosis: Associated Clinical Disorders and Immunological Evaluations. Arch Neurol. 1986;43(3):234–8. [DOI] [PubMed] [Google Scholar]
- 70.TROOST D, van den OORD JJ, JONG JMBV DE. Immunohistochemical characterization of the inflammatory infiltrate in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 1990;16(5):401–10. [DOI] [PubMed] [Google Scholar]
- 71.Engelhardt JI, Tajti J, Appel SH. Lymphocytic Infiltrates in the Spinal Cord in Amyotrophic Lateral Sclerosis. Arch Neurol. 1993;50(1):30–6. [DOI] [PubMed] [Google Scholar]
- 72.Graves MC, Fiala M, Dinglasan LAV, Liu NQ, Sayre J, Chiappelli F, et al. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and t cells. Amyotroph Lateral Scler Other Mot Neuron Disord. 2004;5(4):213–9. [DOI] [PubMed] [Google Scholar]
- 73.Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M, et al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J Neuroimmunol [Internet]. 2009;210(1–2):73–9. Available from: 10.1016/j.jneuroim.2009.02.012 [DOI] [PubMed] [Google Scholar]
- 74.Beers DR, Henkel JS, Zhao W, Wang J, Appel SH. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A. 2008;105(40):15558–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiu IM, Chen A, Zheng Y, Kosaras B, Tsiftsoglou SA, Vartanian TK, et al. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci U S A. 2008;105(46):17913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sta M, Sylva-Steenland RMR, Casula M, Jong JMBV, Troost D, Aronica E, et al. Innate and adaptive immunity in amyotrophic lateral sclerosis: Evidence of complement activation. Neurobiol Dis. 2011; [DOI] [PubMed] [Google Scholar]
- 77.Coque E, Salsac C, Espinosa-Carrasco G, Varga B, Degauque N, Cadoux M, et al. Cytotoxic CD8 + T lymphocytes expressing ALS-causing SOD1 mutant selectively trigger death of spinal motoneurons. Proc Natl Acad Sci U S A. 2019;116(6):2312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lucca LE, Dominguez-Villar M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat Rev Immunol [Internet]. 2020; Available from: 10.1038/s41577-020-0296-3 [DOI] [PubMed]
- 79.Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134(5):1293–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5(1):64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sheean RK, McKay FC, Cretney E, Bye CR, Perera ND, Tomas D, et al. Association of regulatory T-Cell Expansion with progression of amyotrophic lateral sclerosis a study of humans and a transgenic mouse model. JAMA Neurol. 2018;75(6):681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beers DR. Tregs in ALS are Dysfunctional and Predict Progression Rate and Severity. JCI Insight. 2017;2(5):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alsuliman A, Appel SH, Beers DR, Basar R, Shaim H, Kaur I, et al. A robust, good manufacturing practice–compliant, clinical-scale procedure to generate regulatory T cells from patients with amyotrophic lateral sclerosis for adoptive cell therapy. Cytotherapy [Internet]. 2016;18(10):1312–24. Available from: 10.1016/j.jcyt.201606012 [DOI] [PubMed] [Google Scholar]
- 84.Thonhoff JR, Beers DR, Zhao W, Pleitez M, Simpson EP, Berry JD, et al. Expanded autologous regulatory T-lymphocyte infusions in ALS A phase I, first-in human study. Neurol Neuroimmunol NeuroInflammation. 2018;5(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Korn T, Kallies A. T cell responses in the central nervous system. Nat Rev Immunol. 2017;17(3):179–94. [DOI] [PubMed] [Google Scholar]
- 86.Hayes GM, Woodroofe MN, Cuzner ML. Microglia are the major cell type expressing MHC class II in human white matter. J Neurol Sci. 1987;80(1):25–37. [DOI] [PubMed] [Google Scholar]
- 87.Abellanas MA, Zamarbide M, Basurco L, Luquin E, Garcia-Granero M, Clavero P, et al. Midbrain microglia mediate a specific immunosuppressive response under inflammatory conditions. J Neuroinflammation. 2019;16(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arase N, Arase H. Cellular misfolded proteins rescued from degradation by MHC class II molecules are possible targets for autoimmune diseases. J Biochem. 2015; 158(5):367–72. [DOI] [PubMed] [Google Scholar]
- 89.Monsonego A, Zota V, Karni A, Krieger J I, Bar-or A, Bitan G, et al. Increased T cell reactivity to amyloid β protein in older humans. J Clin Invest. 2003; 112(3):415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature [Internet], 2017;546(7660):656–61. Available from: 10.1038/nature22815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lindestam Arlehamn CS, Dhanwani R, Pham J, Kuan R, Frazier A, Rezende Dutra J, et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun. 2020; 11(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lindestam Arlehamn CS, Pham J, Alcalay RN, Frazier A, Shorr E, Carpenter C, et al. Widespread Tau-Specific CD4 T Cell Reactivity in the General Population. J Immunol. 2019;203(1):84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Panzara MA, Gussoni E, Begovich AB, Murray RS, Zang YQ, Appel SH, et al. T cell receptor BV gene rearrangements in the spinal cords and cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Neurobiol Dis. 1999;6(5):392–405. [DOI] [PubMed] [Google Scholar]