

Abstract
Live biotherapeutic products (LBPs) are an emerging therapeutic modality that are clinically investigated for treating pathogenic infections and inflammatory diseases. A major class of LBPs are feces derived microbial consortiums which require numerous process development steps (e.g. separation, purification, blending) to facilitate LBP formulation into oral dosage forms. A subset of these LBPs circumvent the need for continuous fecal processing by batch culture for individual strains of microbes that are rationally defined and combined in the final LBP formulation. Separately, delivery formulations (e.g. polymer encapsulation) are being developed for LBPs to improve storage and intestinal engraftment; however, formulation requires additional manufacturing processes distinct from fecal processing or batch culture. Here, a streamlined approach termed batch culture formulation (BCF) is developed to combine the individual batch culture and formulation processes into a single-step process. Based on a previously described polymeric film formulation that encapsulates LBPs, BCF is shown to reduce the number of required processes to formulate LBP-films without altering LBP phenotype, function, or storage profiles compared to the standard LBP-film formulation approach. Additionally, it is demonstrated that BCF facilitates scaled-fabrication from the milligram to gram scale with predictable loading, highlighting the potential that BCF has for clinical translation.
Keywords: microbe, drug delivery, process development, scale-up, polymeric film
Graphical Abstract
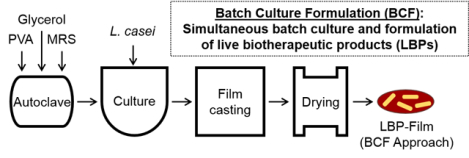
Live biotherapeutic products (LBPs) are an emerging therapeutic class for prevention or treatment of diseases. Here, we describe a new approach that combines batch culture and formulation to reduce the number of processing steps towards formulating LBPs into oral dosage forms. This approach is scalable from miligram to gram scale, highlighting its translational potential.
1. Introduction
The role of the microbiome in regulating health and disease has motivated efforts to develop therapeutic approaches that modulate the microbiome.[1] An emerging therapeutic modality that relies on microbiome modulation for the treatment or prevention of pathogenic infections and inflammatory diseases are live biotherapeutic products (LBPs).[2] A major class of LBPs are feces-derived microbial consortiums,[2a] which are predominately inspired by the success of fecal microbiota transplants.[3] Much like fecal microbiota transplants (FMT), these LBPs require human donor feces; as such, there is a constant need for fecal donations to provide the source material for these LBPs.[4] Recently, LBPs have achieved several milestones in clinical trials for treatment of Clostridium difficile infections, highlighting their therapeutic potential.[2a, 4–5] Unlike small molecule drugs or biologics, LBPs are living organisms that sustainably interact with the existing microbiome through colonization to shift dysbiosis towards non-diseased, protective, states. Resulting from these differences with traditional therapeutic modalities, LBPs require different manufacturing, formulation, and delivery considerations than small molecules and biologics do.[2b, 5b, 6]
LBPs that are derived from human donor feces must undergo multiple processing steps (e.g. filtering, biomass separation, pathogen neutralization, LBP isolation) prior to administration;[5a, 7] when these LBP processing steps are applied to unique microbiota compositions of each fecal donor, variations in the composition of LBPs are possible.[4, 8] A distinct subset of LBPs that can potentially standardize LBP composition are being developed as defined microbial consortiums of specific microbes that are individually cultured and specifically combined in LBP formulations.[9] These next-generation LBPs can potentially eliminate the repeated isolation and purification processes needed to manufacture LBPs from feces by introducing batch culture approaches. Separately, delivery formulations (e.g. polymer encapsulation, surface modifications) are being developed for LBPs to improve storage and intestinal engraftment;[10] however, formulation requires additional and specific manufacturing processes (e.g. excipient sterilization, excipient blending) distinct from fecal processing or batch culture. [11] As such, there exists opportunity to create new process development strategies that optimize the formulation of next-generation LBPs.
Motivated by next-generation LBPs that are either batch cultured or formulated for enhanced storage or delivery, we developed a new approach termed batch culture formulation (BCF) that introduces formulation components during the batch culture process. Previously, we developed a poly(vinyl alcohol) (PVA)-based film formulation that encapsulates and enables long-term storage, mucoadhesion, and controlled release of a model LBP,[12] Lactobacillus casei ATCC 393 (L. casei ATCC 393).[13] Briefly, fabrication of LBP-films involves independent processes including preparation and sterilization of both LBP growth media and formulation components, LBP batch culture, biomass separation, mixing of LBP and formulation components after batch culture, solution casting of the LBP/formulation mixture, and solvent evaporation to facilitate film formation (Figure 1A). Here, we apply our BCF approach towards the fabrication of LBP-films by batch culturing LBPs in the presence of formulation components (Figure 1B). This BCF approach decreases the number of processing steps while maintaining similar formulation performance as compared to LBP films that are formulated after batch culture. We then demonstrate that BCF can be used to scale-up LBP-film fabrication three orders of magnitude while providing predictable intra- and inter-batch loading of LBPs.
Figure 1.

Process development for LBP-film fabrication. (A) Standard formulation approach for LBP-films and (B) batch culture formulation (BCF) approach for LBP-films.
2. Results
2.1. Rationale for batch culture formulation (BCF)
Previously, we reported an approach (Figure 1A) to formulate PVA-based films for LBP encapsulation (LBP-films) that enabled long-term storage and versatile delivery functionalities. [12] Similar to other encapsulation methods for LBPs,[10d, 14] multiple processes in series were required to prepare and batch culture LBPs, prepare formulation components, and subsequently encapsulate LBPs (Figure 1A). To decrease process burden and facilitate formulation scale-up, we envisioned a BCF approach for LBPs (Figure 1B). In BCF, LBPs are inoculated into DeMan-Rogosa-Sharpe (MRS) growth media that already contains the sterilized polymer encapsulant, PVA, and the excipients, glycerol and MRS. Upon reaching a desired optical density (OD), BCF LBPs can be directly solvent cast on substrates for evaporation-based LBP-film formation. By using BCF, process burden could be reduced by eliminating the following processes: separate sterilization of MRS media and formulation components, solubilization of formulation components, centrifugation to separate LBP biomass, and mixing of LBPs, excipients, and polymer encapsulants.
2.2. Optimization of drying conditions
Since thin films are a widely investigated oral dosage form used for drug delivery, approaches to accelerate drying after solvent casting to improve film-fabrication speed while maintaining drug stability have been broadly explored.[15] Our standard LBP-film fabrication process, as previously reported,[12] requires a 2 day drying step at 4°C to facilitate LBP-film formation. Towards improving our overall LBP-film fabrication process and potentially decreasing drying time, we first investigated the role that environmental conditions have on film formation during drying. We hypothesized that higher drying temperatures and lower moisture would decrease film drying time by accelerating evaporation. As such, we examined two drying conditions at room temperature and reduced humidity, a vacuum desiccator and an anaerobic chamber. In the desiccator, LBP-films formed after 2 weeks, or 12 days longer than our standard drying process at 4°C; additionally, viability was not detectable after complete drying (Figure 2). In the anaerobic chamber, LBP-films dried after 2 days and were subsequently transferred to 4°C for storage. However, LBP viability in these films significantly increased at day 2, as compared to the initial loading at day 0 (Figure 2). This dynamic loading and unpredictability in the LBP-films would be unsuitable for clinical applications, since dosing must be consistent and controlled. These findings indicated that neither of these room temperature drying conditions facilitated fabrication of LBP films with viability consistent to their initial loading in the aqueous formulation. In contrast to these two conditions at room temperature, LBP viability remained constant over 2 weeks when LBP-films were dried at 4°C (Figure 2), in agreement with our previously published results.[12] Importantly, LBP-films dried at 4°C formed after 2 days of drying, indicating that higher temperature under the conditions evaluated here did not accelerate drying time. As such, the subsequent studies were performed using the 4°C condition for film drying to provide consistent and predictable LBP loading in LBP-films.
Figure 2.
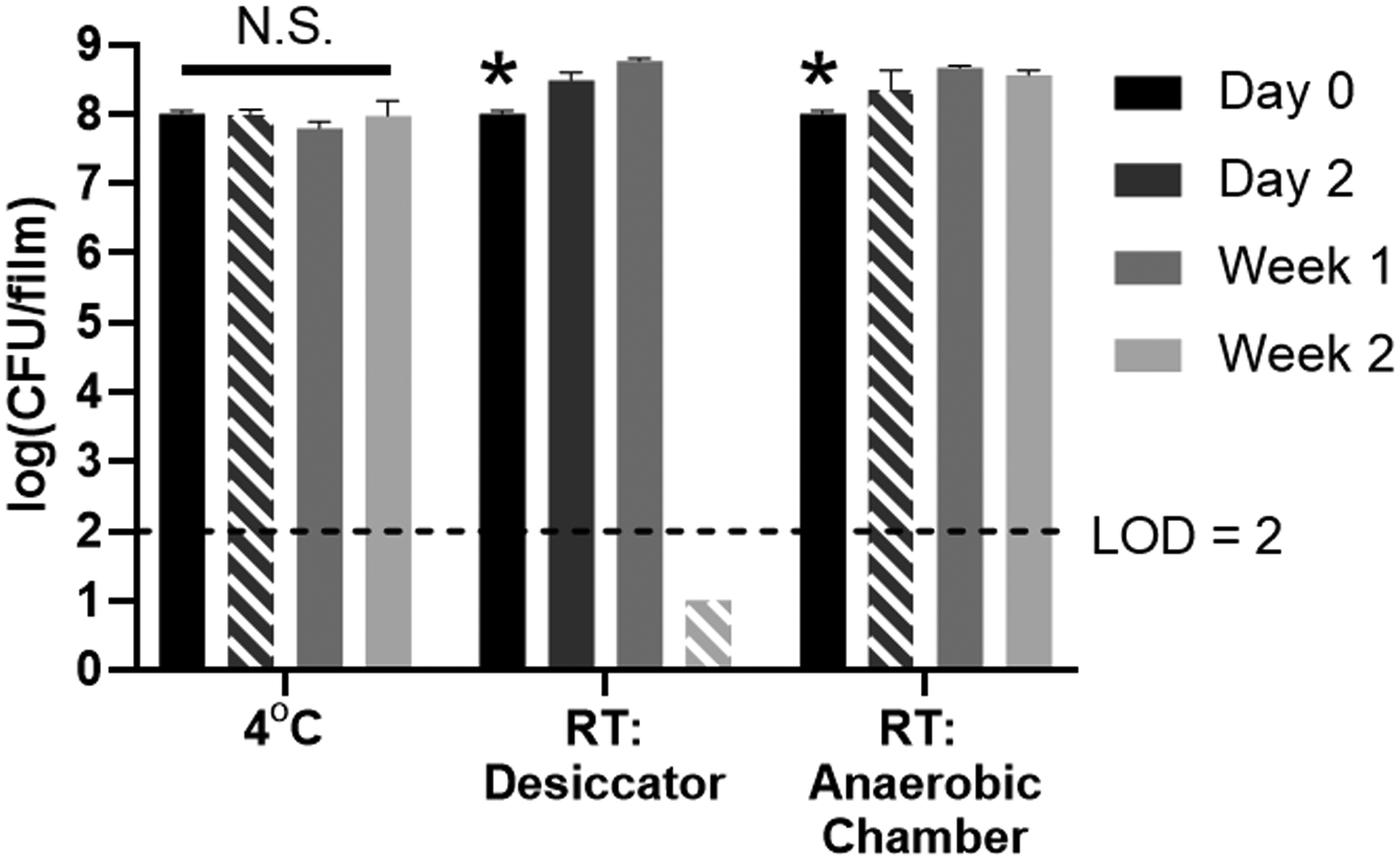
Viability of L. casei ATCC 393 during drying at various environmental conditions (4°C, room temperature in a vacuum dessicator, or room temperature in an anaerobic chamber) and after storage. Upon complete drying, LBP-films were placed at 4°C for storage. Hatched bars indicate the timepoint at which LBP-films were dry for each environmental condition. Each error bar represents standard deviation (n = 3). In each drying condition, statistical analysis was conducted using one-way ANOVA followed by post hoc Dunnett’s test for pairwise comparison (statistical significance defined at p < 0.05). *: significantly different from each of the other groups. N.S.: not significant. LOD = Limit of detection. RT = Room temperature.
2.3. Evaluating LBP physiological function during BCF
The BCF approach is dependent on the growth of LBPs in the presence of polymer encapsulants and storage excipients. Previous work has demonstrated that polymers in the aqueous environment interact with LBP surfaces and influence their behavior.[12, 16] However, this has yet to be studied or evaluated in the context of formulation and when including formulation components during batch culture. As such, we evaluated if the PVA (polymer encapsulant) and glycerol (excipient) affects the growth rate or metabolic activity of the L. casei ATCC 393 during BCF. As compared to standard growth in MRS, additional presence of PVA and glycerol did not affect the growth of L. casei ATCC 393 at concentrations required for film formulation (Figure 3A). Likewise, we demonstrated that the secretion rates of L-lactate, a known metabolite of L. casei ATCC 393,[17] using the BCF approach (with PVA and glycerol present) were comparable to standard growth conditions over a 24 hour period (Figure 3B). These findings confirmed the feasibility of culturing LBPs in the PVA and glycerol-containing media without altering key LBP physiological functions such as growth and metabolite secretion.
Figure 3.
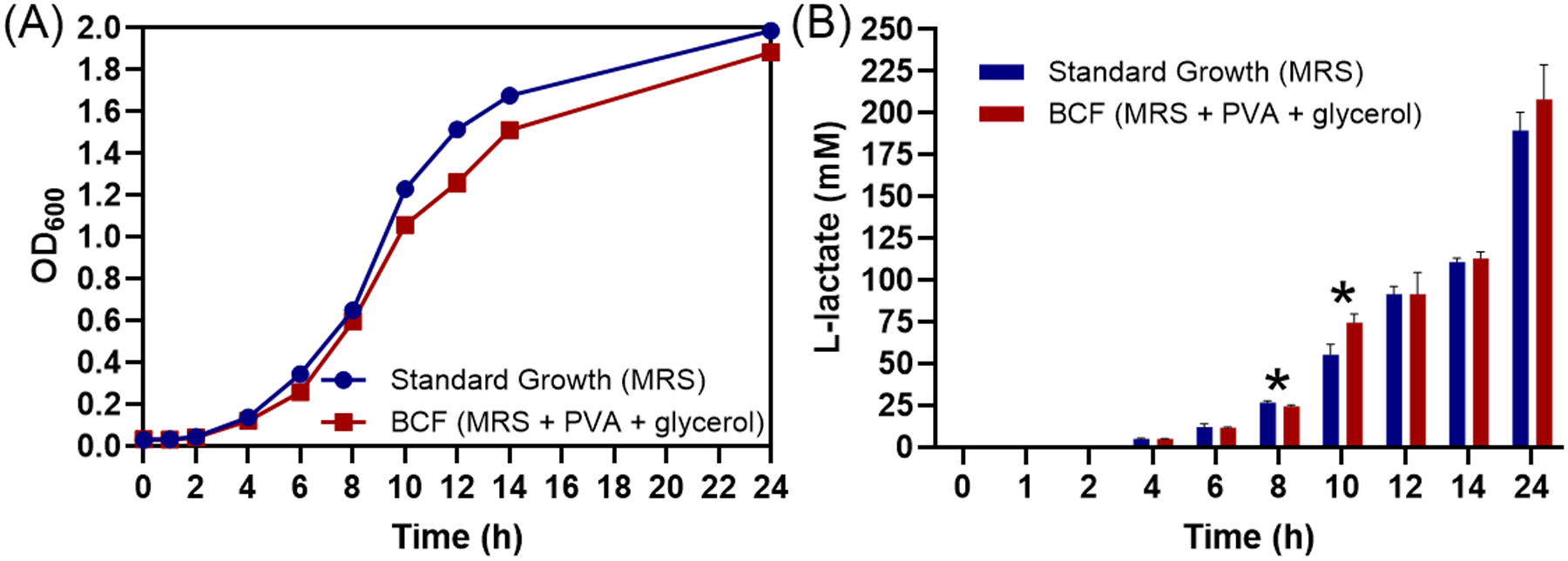
Effects of additional PVA and glycerol on growth and metabolite secretion of L. casei ATCC 393 during BCF. Standard growth media consisted of MRS. BCF conditions contained MRS with addition of 3% wt vol−1 PVA and 1.26% wt vol−1 glycerol. Comparison of (A) growth curves and (B) cumulative L-lactate secretion over 24 hours at 37°C for standard (blue) and BCF (red) growth conditions. Each error bar represents standard deviation (n = 3). * Denotes statistical difference (P < 0.05) using two-sided Student’s t-test between standard growth and BCF groups at each time point.
2.4. Evaluating the storage potential of LBPs using BCF
Since LBP-films using the BCF approach would have: (1) additional LBP metabolites, resulting from eliminating the biomass separation (centrifugation) process, and (2) fewer protectants (e.g. glucose, amino acids), due to their consumption from MRS during batch culture, we directly compared the storage of LBP-films that were formulated using the standard (Figure 1A) and the BCF (Figure 1B) approaches. As expected, and in agreement with our previous work,[12] the viability of LBPs encapsulated utilizing the standard LBP-film fabrication approach remained at the initial CFU loading (Figure 4, blue bars) over 6 weeks. Similarly, viability of LBPs encapsulated using BCF also maintained the initial CFU loading over a 6 week period (Figure 4, red bars). Importantly, the differences in viability at each timepoint were statistically insignificant when comparing the standard approach to the BCF approach, indicating that the BCF approach can provide the same level of storage benefits as compared to standard formulation approach at 6 weeks.
Figure 4.
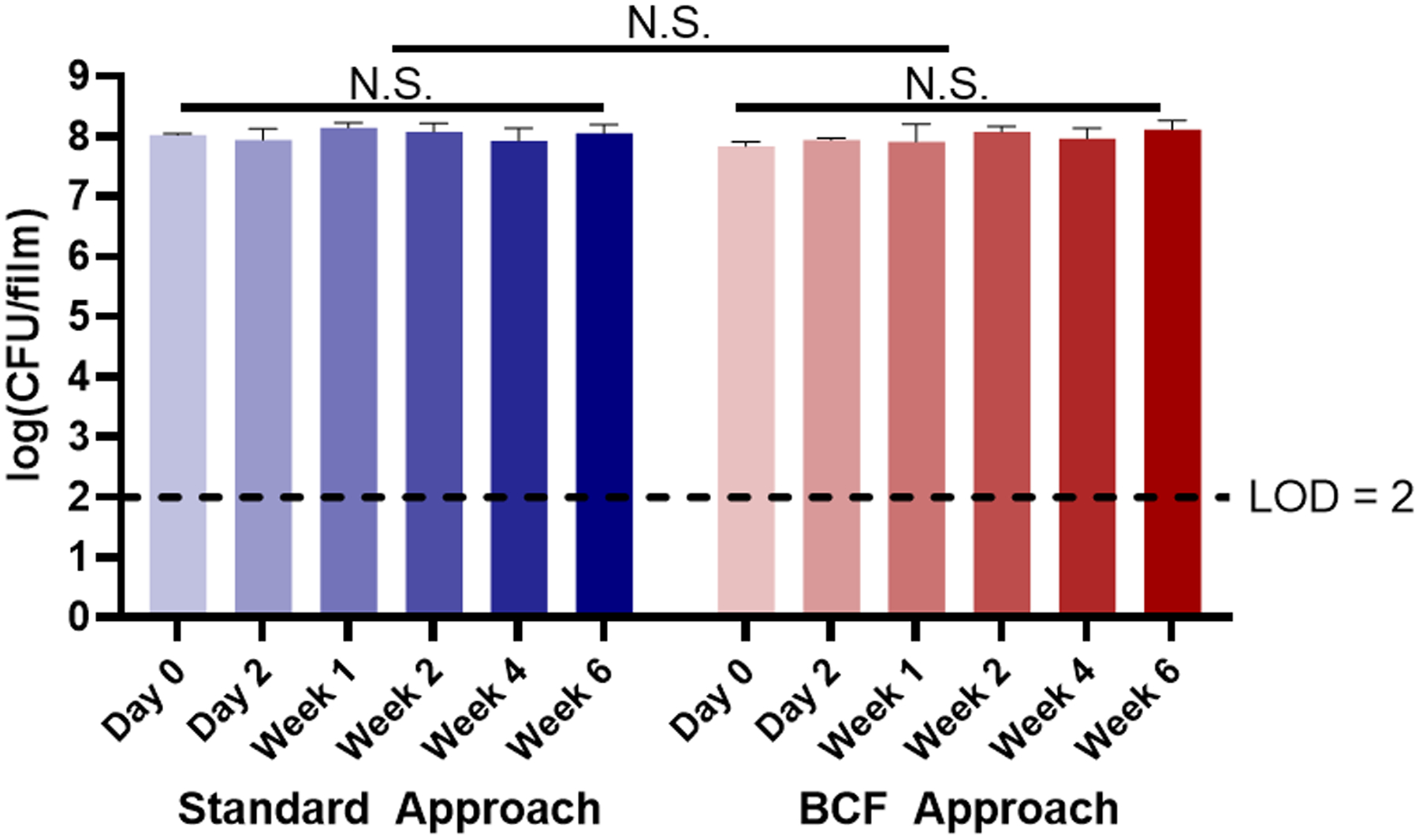
Storage profiles of L. casei ATCC 393 in LBP-films fabricated with standard formulation and BCF approaches. LBP-films were dried at 4°C and remained refrigerated for the duration of the study. Each error bar represents standard deviation (n = 3). Statistical analysis was conducted using two-way ANOVA (statistical significance defined at p < 0.05). N.S.: not significant. LOD = Limit of detection.
2.5. Scaled fabrication of LBP-films using BCF
We evaluated the suitability of the BCF approach in transitioning rapidly to larger processes for scale-up purposes. Up until this point, all LBP-films were fabricated in 24-well plates at the milligram scale. Based on our previous work, this is sufficient for fabrication of LBP-films that can meet clinically-relevant dosing (up to 1010 CFU/capsule);[12] however, a larger-scale process that can fabricate LBP-films at the same dose would result in more total doses per batch. As such, we evaluated the BCF approach at larger scale using a borosilicate glass substrate (Figure 5Ai) with a surface area of over 600-fold higher than that of a single well in a 24-well plate. As previously performed in 24-well plates, we cast the LBP-film mixture on the glass substrate (Figure 5Aii), allowed it to dry for 2 days at 4°C, and then sectioned the scaled LBP-film into 36 representative films (Figure 5Aiii). We used this approach to make three independent larger-scale batches of LBP-films to evaluate consistency of our BCF scale-up approach. Within these 3 batches, the thickness of dried and sectioned LBP-films were measured and ranged from 0.03 to 0.07 mm (Figure 5B). The total mass of dried and sectioned LBP-films ranged from 96.8 to 178.5 milligrams (Figure 5C); the total mass (including non-sampled sections) of the complete LBP-film ranged from 7.9 to 8.4 grams. To determine the consistency of LBP loading within and across batches, we quantified CFU per mass unit (CFU/mg). Three batches produced dried and sectioned LBP-films with loading between 5.4 and 7.0 log(CFU/mg) (Figure 5D). Analysis revealed statistically significant differences in thickness, mass, and loading density of sampled films across the three independent batches (Figure 5B–D). Detailed process design formulation parameters for each batch are presented in Table 1. We then compared the loading density of scaled LBP-films to LBP-films fabricated in the 24-well plate. A single LBP-film in a 24-well plate represents ~0.37% of the mass of the completely dried LBP-film fabricated at the gram scale. When comparing the loading density of dried and sectioned LBP-films with 24-well plate LBP-films, no statistical differences in loading density (CFU/mg) were observed (Figure 5E), indicating the consistency of loading density for each of the three individual batches.
Figure 5.

Scale-up of L. casei ATCC 393 LBP-films fabricated via BCF approach from the milligram to gram scale. (A) Representative images of scaled LBP-film fabrication: (i) glass substrate, (ii) top-down and side views with rhodamine-dyed LBP-film solution, (iii) top-down and side views of 6×6 sectioned dried LBP-film, and (iv) remaining LBP-film after 6×6 section sampling. LBP-film parameters for 3 independent batches (batch 1: grey, batch 2: yellow, batch 3: blue) of sampled sections from scaled-LBP-films: (B) thickness, (C) mass, and (D) bacterial loading density. (E) Comparison of bacterial loading density between control LBP-films (fabricated in 24-well plates, six biological replicates for each batch) and scaled-LBP-films (fabricated via BCF, thirty-six biological replicates for each batch) from three matching (control and BCF) independent batches. Individual data points (batch 1: grey, batch 2: yellow, batch 3: blue) represent means of individual batches for scaled (circles) and 24-well plate control (triangles) LBP-films. In (B-D), each error bar represents standard deviation of batches (n = 36). Statistical analysis was conducted using one-way ANOVA (statistical significance defined at p < 0.05). *: significant. In (E), each error bar represents standard deviation of batches (n = 3). Statistical analysis was conducted on means from three batches using two-sided paired Student’s t-test (statistical significance defined at p < 0.05). N.S.: not significant.
Table 1.
Formulation parameters for each scaled-batch of BCF LBP-film and the associated controls.
| Approach | Sample | Formulation Parameters | Batch 1 | Batch 2 | Batch 3 |
|---|---|---|---|---|---|
| Scaled LBP formulation | Whole film | Casting mass [g]a) | 81.9 | 81.9 | 82.6 |
| Casting loading [log(CFU)]b) | 10.5 | 10.5 | 10.5 | ||
| Total loading after film drying [log(CFU)] | 10.5 | 10.3 | 10.3 | ||
| Total mass after film drying [g] | 7.9 | 8.1 | 8.4 | ||
| Loading density after film drying [log(CFU/mg)]c) | 6.6 | 6.4 | 6.3 | ||
| Sampled films | Total mass [g] {% of whole film} | 4.42 {55.8} | 4.49 {54.9} | 4.82 {57.3} | |
| Total loading [log(CFU)] {% of whole film} | 10.2 {61.2} | 10.0 {54.0} | 9.9 {41.6} | ||
| Average mass [mg] (±SDd)) | 122.9 (16.9) | 124.7 (12.8) | 134.0 (19.3) | ||
| Average thickness [mm]e) (±SD) | 0.05 (0.01) | 0.04 (0.01) | 0.04 (0.01) | ||
| Average loading [log(CFU)] (±SD) | 8.7 (0.2) | 8.4 (0.2) | 8.3 (0.3) | ||
| Average loading density [log(CFU/mg)] (±SD) | 6.6 (0.2) | 6.3 (0.2) | 6.1 (0.3) | ||
| Standard LBP formulation | 24-well plate | Casting volume [mL]f) | 0.3 | 0.3 | 0.3 |
| Casting loading [log(CFU)] | 8.0 | 8.1 | 8.1 | ||
| Average mass [mg] (±SD) | 28.9 (0.4) | 29.3 (0.6) | 32.4 (2.5) | ||
| Average thickness [mm] (±SD) | 0.01 (0) | 0.01 (0) | 0.01 (0) | ||
| Average loading [log(CFU)](±SD) | 8.1 (0.1) | 8.0 (0.1) | 7.8 (0.2) | ||
| Average loading density [log(CFU/mg)] (±SD) | 6.7 (0.1) | 6.6 (0.1) | 6.3 (0.2) |
g = grams;
log(CFU) = log(colony forming units);
log(CFU/mg) = log(CFU/milligrams);
SD = standard deviation;
mm = millimeters;
mL = milliliters.
2.6. BCF is compatible with standard LBP storage methods
The BCF approach (Figure 1B) is a continuous process that facilitates preparation of LBPs from batch culture to capsule-compatible oral dosage form. To explore possible alternatives in the BCF process that could create opportunities for personalized or on-demand modifications to the LBP-films, we investigated the potential of storing the aqueous film solution prior to solvent casting and LBP-film formation. As such, we aliquoted and stored the fresh aqueous BCF pre-film solutions consisting of the LBP, polymer encapsulant, and storage excipients. We then thawed these BCF stocks for evaluation at various timepoints for viability and formulation into LBP-films. Viability of L. casei ATCC 393 and LBP-film performance post storage was benchmarked to fresh, non-stored, LBP-films using BCF. First, we applied Fourier-transform infrared (FTIR) spectroscopy to compare the chemical features of the films since PVA is known to undergo changes upon freeze-thaw cycles.[18] LBP-films fabricated through the standard and BCF approaches exhibited similar spectra (Figure 6A). Notably, no apparent peak shifts or appearances were observed for any group, indicating that storing BCF aqueous solution at −80°C for 30 days did not alter the interactions amongst the polymer, excipients, or LBPs. Additionally, these results provide supporting evidence that LBP-films fabricated using the BCF approach are chemically indistinguishable from standard LBP-films. Next, we evaluated LBP-film properties by confirming that BCF LBP-films, formulated from pre-film solution stored at −80°C for 30 days, remained compatible with standard oral dosage forms; specifically, we demonstrate insertion of LBP-films into a 00-sized human oral capsule (Figure 6B). Finally, we compared the viability of L. casei ATCC 393 in the pre- and post-storage aqueous culture and the corresponding films upon drying. No significant differences of viability were identified in either the aqueous pre-film solutions or the final dried LBP-films (Figure 6C).
Figure 6.
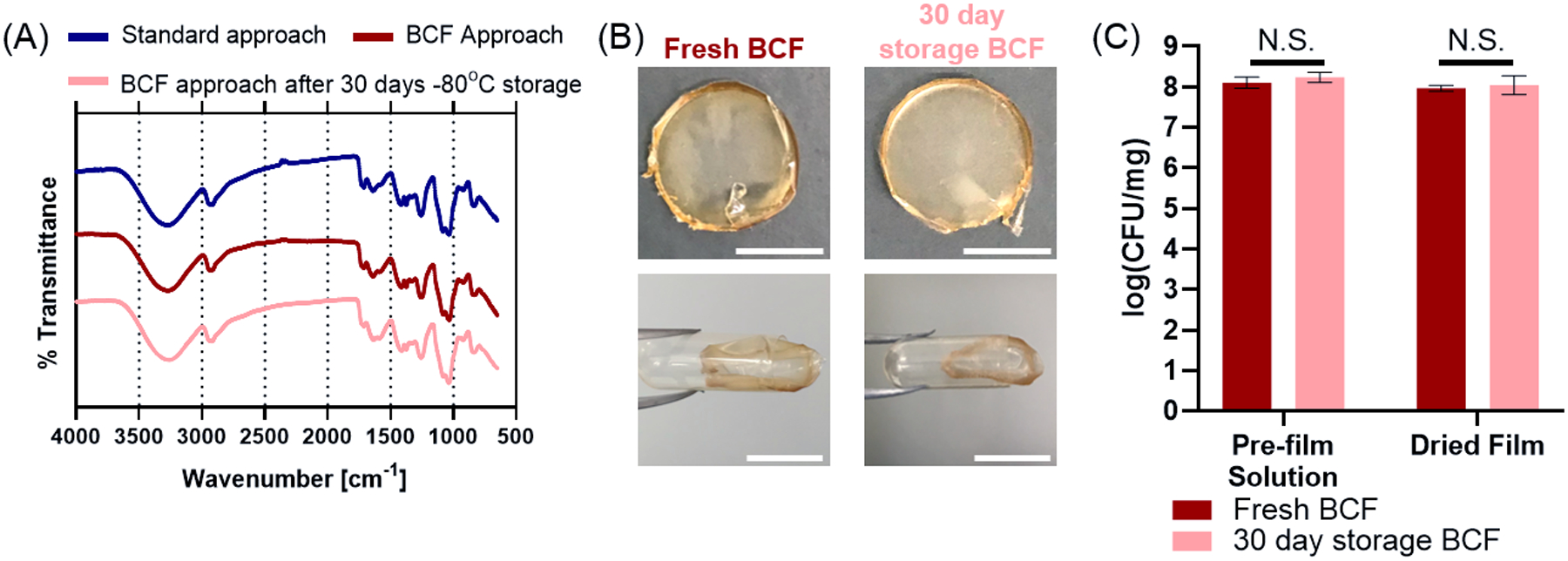
Storage characterization and performance of BCF LBP-films. (A) FTIR spectra of LBP-films fabricated with the standard approach, BCF approach, and BCF approach stored as pre-film solution for 30 days at −80°C prior to LBP-film formation. (B) Representative images of oral capsule-compatible LBP-films cast from fresh BCF (control) or pre-film BCF solution stored at −80°C for 30 days. Scale bar = 1 cm. (C) LBP viability of pre-film solution and after drying LBP-film for fresh BCF (control) and −80°C stored BCF. Each error bar represents standard deviation (n = 3). Statistical analysis was conducted using two-sided Student’s t-test (statistical significance defined at p < 0.05). N.S.: not significant.
3. Discussion
Here, we described a streamlined and scalable process, BCF, to synthesize delivery systems for LBPs during their batch culture. L. casei ATCC 393, a model LBP, was inoculated into a sterilized solution to facilitate batch culture in the presence of polymer encapsulants and storage excipients. The pre-film solution, directly from batch culture, was solvent cast on a glass substrate and, upon air-drying, formed LBP-films consisting of the LBP, polymer encapsulants, and excipients in the pre-film batch cultured solution. As compared to standard formulation after batch culture (Figure 1A), the BCF approach removes high-burden processing steps (Figure 1B) such as separate sterilization/solubilization of growth media and formulation components, centrifugation to separate LBP biomass, and mixing of LBPs, polymer encapsulants, and excipients. As such, BCF substantially reduces complexity for manufacturing LBP delivery systems.
Through BCF, L. casei ATCC 393 maintained similar growth and metabolic activities in the aqueous solution as compared to standard batch culture (Figure 3). While our previous study suggested interactions between PVA and cell walls,[12] these results indicate that the interactions are not sufficient to hinder nutrient metabolism and cell division. These findings also indicate that other LBP components do not interfere with the growth or metabolism of LBPs during BCF. Storage profiles in LBP-films using BCF were non-inferior to standard formulation approach up to 6 weeks at 4°C (Figure 4). These results suggest that the unconsumed protectants remaining in MRS (e.g. glucose, amino acids) during BCF are sufficient to confer the same storage benefits as compared to the standard LBP-film process that adds fresh MRS (without consumed protectants) after batch culture. Considering the high cost of MRS and other complex and defined culture mediums, the BCF approach can potentially be used to provide the same formulation benefits (e.g. enhanced storage) while decreasing the total amount of raw materials. Combined with a reduction in the total number of processes, BCF may be a more cost-effective approach as compared to standard formulation approaches that occur after batch culture. The non-inferior storage profile also indicates that the presence of LBP metabolites in final films, such as L-lactate, did not cause viability loss over the study period, supporting the feasibility of removing centrifugation steps. The success of BCF with L. casei ATCC 393 implicates that BCF is potentially suitable for any bacteria with known media that can: (i) support bacterial growth and (ii) provide storage benefits after film drying. In particular, as MRS allows growth of many lactic-acid secreting bacterial genera, including Lactobacilli,[19] Bacilli,[20] and Bifidobacteria,[21] it is possible that BCF can be applied to bacteria under these genera. Additionally, under the circumstances where the media does not provide desired storage benefits, other storage excipients can be introduced to the culture before film casting to enable centrifugation-free formulation processes.
After demonstrating that the BCF approach could produce non-inferior LBP-films, we focused our efforts on the scale-up of LBP-films using BCF, since solvent casting has proven to be a scalable process in formulating several commercially available pharmaceutics.[15] Indeed, LBP-films using the BCF approach were produced at the gram-scale (Figure 5); while inconsistencies across batches existed (Figure 5B, 5C, 5D), appropriate low-volume controls that determine loading density (CFU/mg of film) can be used to accurately determine and predict loading of the final LBP-film (Figure 5E). Broadly, these results highlight that intra-batch loading density of LBP-films can be accurately estimated using smaller-scale films (~0.37% of the total LBP-film mass); in practice, this approach could be used to determine CFU/mg for LBP-films for accurate dosing. Importantly, suboptimal LBP doses have been associated with reduced engraftment and efficacy in clinical trials,[7b] and as such, confirming that the BCF approach can be scaled and used to formulate LBP-films with predictable doses highlights the robustness and utility of the BCF approach. To our knowledge, efforts on scaled-fabrication of LBPs formulations are scarcely reported in the literature, and as such, our work highlights important considerations when transitioning fabrication of LBP formulations from the milligram to gram scale.
The aqueous formulation from BCF withstood −80°C storage for 30 days without significant viability loss, and maintained properties required for compatibility with standard oral dosage forms (Figure 6). These results indicate that the BCF approach is compatible with clinically-relevant storage conditions and can be stored prior to formation of LBP-films. This could facilitate fabrication of on-demand tunable LBP-films, directly from storage, by enabling modifications to the film solution such as: (i) inclusion of additional LBPs, prebiotics, or excipients after batch culture and storage, (ii) modifications to the final dosage form such as film geometry or loading per dose (via casting larger or smaller volumes of the stored pre-film solution), or (iii) a retainable frozen stock for quality control analysis.
Recently, the FDA issued safety alerts after identification of an FMT-associated death in 2019[22] and emergent concerns have arisen for SARS-Cov-2-borne risks in donor feces.[23] LBPs grown via batch culture, rather than directly sourced from fecal donations, have potential benefits for manufacturing processes, given that reliance on donor feces will always present risks of pathogens or viruses. As such, the BCF approach, which enables facile and scalable fabrication of LBP-films from batch culture, may prove to be highly valuable toward accelerating clinical application of LBP oral dosage forms. Future in vivo studies will evaluate how BCF LBP-films influence LBP survival, distribution, and colonization along the gastrointestinal tract in healthy and diseased lab rodents, compared to non-encapsulated LBPs and LBP-films fabricated through standard, non-BCF approaches. These insights from in vivo evaluation will in turn dictate directions to optimize BCF for specific indications. Toward clinical translation, the compatibility of this platform with more clinically relevant LBPs must be assessed as many clinically promising LBPs consist of microbial consortiums rather than single strains,[9] and involve obligate anaerobes with oxygen sensitivity.[24] Larger manufacturing scale-up with detailed characterization of film components and batch-to-batch consistency are still needed to confirm the potential of LBP-films to satisfy regulatory guidelines and future clinical investigation.[25]
4. Conclusion
In summary, we describe a new strategy, BCF, to fabricate LBP- formulations that incorporates formulation components in LBP batch culture. This BCF approach reduces the number of processing steps and maintains similar formulation performance as compared to LBP films that are formulated after batch culture in a standard process. We also demonstrate that BCF can be used to scale-up LBP-film fabrication three orders of magnitude while providing predictable intra- and inter-batch loading of LBPs. In addition, BCF is compatible with clinically-relevant storage conditions, enabling personalized and on-demand LBP-film fabrication. Collectively, this new BCF approach can potentially be applied to next-generation LBPs that require both batch culture/fermentation and formulation.
5. Methods
Materials:
Poly(vinyl alcohol) (PVA, 87%-90% hydrolyzed, 30–70 kDa) was purchased from Sigma-Aldrich (Missouri, USA). Glycerol was purchased from Fisher Scientific (Massachusetts, USA). DeMan-Rogosa-Sharpe (MRS) broth and Lactobacillus casei ATCC 393 were purchased from Thermo Scientific (California, USA). MRS agar was purchased from Becton, Dickinson and Company. All the water used in this paper was sterilized Milli-Q water.
Bacterial Growth and Film Fabrication:
LBP-films from the standard approach were fabricated as described previously.[12] Briefly, L. casei ATCC 393 was inoculated into autoclaved MRS broth (5.2% wt vol−1) and grew overnight statically at 37°C in sealed conical tubes or glass containers. The overnight culture was diluted down with MRS broth to OD600 < 0.3 and harvested when OD600 reached 0.50 ± 0.05. Then the culture was centrifuged at 4000 rpm for 10 minutes at room temperature and the pellet was washed once in sterile water. OD600 values were measured by GENESYS 30 visible spectrophotometer (Thermo Scientific, California, USA) with bacteria-free media subtracted. Polymer solutions were prepared by autoclaving MRS broth and then adding PVA (3% wt vol−1) and glycerol (1.26% wt vol−1) in the autoclaved MRS broth at 80°C until complete dissolution. After polymer solutions were cooled down to room temperature, L. casei ATCC 393 (108 CFU mL−1) was added and stirred until homogenization. Finally, the prefabricated film solution (300 μL) was cast into individual wells of 24-well plates and dried under one of the following conditions as indicated in the main text: (i) air-dry under normal atmosphere at 4°C for 2 days; (ii) air-dry in the anaerobic chamber at room temperature for 2 days or (iii) vacuum-dry in a desiccator at room temperature for 2 weeks. In the BCF approach, polymer solutions were prepared by autoclaving (121°C, 20 min) MRS (5.2% wt vol−1), glycerol (0.63% wt vol−1), and PVA in water (3% wt vol−1). When cooled down, glycerol stock of L. casei ATCC393 (containing 31.5% wt vol −1 glycerol) was inoculated into the autoclaved polymer solutions at 1:50 volume ratio to ensure the final glycerol concentration at 1.26% wt vol−1. Then bacteria were cultured statically at 37°C and the culture (300 μL) was directly cast into individual wells of 24-well plates for film drying at 4°C for 2 days when OD600 reached 0.4 – 0.8. For scaled LBP-film fabrication, the culture (approximately 82 g) was cast onto a borosilicate glass sheet (30.5 × 30.5 cm2) and dried as described above. Rhodamine B (0.03 mg/mL) was dissolved in the culture to visualize the scaling-up process. Film thickness was measured by a digital caliper (World Precision Instruments, Florida, USA) with limit of detection at 0.01 mm.
L-lactate Measurement:
At indicated time points, 1 mL of culture was sampled and centrifuged at 4000 rpm for 10 min. L-lactate in the supernatant was quantified with an EnzyChrom™ Lactate Assay Kit (BioAssay Systems, California, USA) according to the instruction manual.
Quantification of Viability of Bacteria for Storage Study:
Films were dissolved in sterile water under shaking for 10–20 minutes. Dissolved films solutions were serially diluted and drop-plated (10 μL) on MRS agar, incubated at 37°C for 48–60 hours, and enumerated for colony forming units (CFUs). Prefabricated aqueous formulation was used as control (day 0).
Stability Assessment of Prefabricated Aqueous Formulation:
Aqueous formulation was prepared through the BCF approach, aliquoted and frozen at −80°C. At indicated time points, one aliquot was completely thawed in a water bath at room temperature and then cast into 24-well plates as described above. Bacterial viability in freeze-thaw treated formulation and the corresponding films were quantified as described above. FTIR measurement was conducted on a Bruker Hyperion microscope with a single bounce Ge ATR attachment. A resolution of 4 cm−1 was used, and both background and sample scans were acquired with 64 scans.
Statistical Analysis:
All CFU data were presented after log transformation unless otherwise noted. Experiments were conducted in triplicate unless otherwise noted. The data were presented as mean ± SD. Significant differences were assessed with parametric two-sided Student’s t-test, one-way ANOVA or two-way ANOVA as indicated in relevant figure legends. α = 0.05. Dunnett’s test was chosen for post hoc comparison. P- value less than 0.05 was considered significantly different. All statistical analysis was conducted with Prism (version 8.4.3, Graphpad Software, LLC).
Acknowledgement
Research reported in this publication was supported by the National Institute Of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health under Award Number R21DK123583 and the National Center For Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR002489. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. FTIR measurement was assisted by Dr. Carrie L. Donley at the Chapel Hill Analytical and Nanofabrication Laboratory, CHANL, a member of the North Carolina Research Triangle Nanotechnology Network, RTNN, which is supported by the National Science Foundation, Grant ECCS-1542015, as part of the National Nanotechnology Coordinated Infrastructure, NNCI.
Footnotes
Conflict of interest
K.Q. and A.C.A. are inventors on a patent application filed by The University of North Carolina at Chapel Hill on aspects of the work presented here.
References
- [1].a) Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI, Nature 2007, 449, 804; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cho I, Blaser MJ, Nature Reviews Genetics 2012, 13, 260; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Young VB, Clin Microbiol Infect 2016, 22, 905;27619640 [Google Scholar]; d) Schmidt TSB, Raes J, Bork P, Cell 2018, 172, 1198. [DOI] [PubMed] [Google Scholar]
- [2].a) Vargason AM, Anselmo AC, Bioeng Transl Med 2018, 3, 124; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Oka A, Sartor RB, Dig Dis Sci 2020, 65, 757; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) O’Toole PW, Marchesi JR, Hill C, Nature microbiology 2017, 2, 1. [DOI] [PubMed] [Google Scholar]
- [3].a) Smits LP, Bouter KE, de Vos WM, Borody TJ, Nieuwdorp M, Gastroenterology 2013, 145, 946; [DOI] [PubMed] [Google Scholar]; b) Borody TJ, Khoruts A, Nature reviews Gastroenterology & hepatology 2012, 9, 88. [DOI] [PubMed] [Google Scholar]
- [4].Khoruts A, Hoffmann DE, Palumbo FB, J Law Med Ethics 2019, 47, 482. [DOI] [PubMed] [Google Scholar]
- [5].a) Khanna S, Pardi DS, Kelly CR, Kraft CS, Dhere T, Henn MR, Lombardo MJ, Vulic M, Ohsumi T, Winkler, Pindar, McGovern, Pomerantz RJ, Aunins JG, Cook DN, Hohmann, J Infect Dis 2016, 214, 173; [DOI] [PubMed] [Google Scholar]; b) Garber K, Nat Rev Drug Discov 2020, 19, 655. [DOI] [PubMed] [Google Scholar]
- [6].a) Markey KA, van den Brink MR, Peled JU, Cell Host & Microbe 2020, 27, 169; [DOI] [PubMed] [Google Scholar]; b) Carlson PE Jr, Cell Host & Microbe 2020, 27, 173. [DOI] [PubMed] [Google Scholar]
- [7].a) Lagier JC, Dubourg G, Million M, Cadoret F, Bilen M, Fenollar F, Levasseur A, Rolain JM, Fournier PE, Raoult D, Nat Rev Microbiol 2018, 16, 540; [DOI] [PubMed] [Google Scholar]; b) McGovern BH, Ford CB, Henn MR, Pardi DS, Khanna S, Hohmann EL, O’Brien EJ, Desjardins CA, Bernardo P, Wortman JR, Lombardo MJ, Litcofsky KD, Winkler JA, McChalicher CWJ, Li SS, Tomlinson AD, Nandakumar M, Cook DN, Pomerantz RJ, Aunins JG, Trucksis, Clin Infect Dis 2020, DOI: 10.1093/cid/ciaa387. [DOI] [Google Scholar]
- [8].Franks AH, Harmsen HJ, Raangs GC, Jansen GJ, Schut F, Welling GW, Appl Environ Microbiol 1998, 64, 3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, Shiota A, Takeshita K, Yasuma-Mitobe K, Riethmacher D, Kaisho T, Norman JM, Mucida D, Suematsu M, Yaguchi T, Bucci V, Inoue T, Kawakami Y, Olle B, Roberts B, Hattori M, Xavier RJ, Atarashi K, Honda K, Nature 2019, 565, 600; [DOI] [PubMed] [Google Scholar]; b) Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K, Nature 2013, 500, 232; [DOI] [PubMed] [Google Scholar]; c) Wortman J, Lachey, Lombardo M, Cambridge, MA: Seres Therapeutics 2016; [Google Scholar]; d) Ford C, Litcofsky K, McGovern B, Pardi D, Nathan R, Hansen V, Brennan R, Pullman J, Bernardo P, Tomlinson A, presented at Open Forum Infectious Diseases 2019. [Google Scholar]
- [10].a) Jimenez M, Langer R, Traverso G, J Exp Med 2019, 216, 1005; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang X, Cao Z, Zhang M, Meng L, Ming Z, Liu J, Sci Adv 2020, 6, eabb1952; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vargason AM, Santhosh S, Anselmo AC, Small 2020, 16, e2001705; [DOI] [PubMed] [Google Scholar]; d) Anselmo AC, McHugh KJ, Webster J, Langer R, Jaklenec A, Adv Mater 2016, 28, 9486; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Li S, Jiang W, Zheng C, Shao D, Liu Y, Huang S, Han J, Ding J, Tao Y, Li M, J Control Release 2020, DOI: 10.1016/j.jconrel.2020.09.011; [DOI] [PubMed] [Google Scholar]; f) Zheng D-W, Pan P, Chen K-W, Fan J-X, Li C-X, Cheng H, Zhang X-Z, Nat Biomed Eng 2020, 1. [DOI] [PubMed] [Google Scholar]
- [11].Rokka S, Rantamäki P, European Food Research and Technology 2010, 231, 1. [Google Scholar]
- [12].Qiu K, Young I, Woodburn BM, Huang Y, Anselmo AC, Adv Healthc Mater 2020, 9, e1901643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Eun CS, Kim YS, Han DS, Choi JH, Lee AR, Park YK, APMIS 2011, 119, 49; [DOI] [PubMed] [Google Scholar]; b) Tien MT, Girardin SE, Regnault B, Bourhis L. Le, Dillies MA, Coppee JY, Bourdet-Sicard R, Sansonetti PJ, Pedron T, J Immunol 2006, 176, 1228; [DOI] [PubMed] [Google Scholar]; c) Llopis M, Antolin M, Carol M, Borruel N, Casellas, Martinez, E., Guarner F, Malagelada JR, Inflamm Bowel Dis 2009, 15, 275. [DOI] [PubMed] [Google Scholar]
- [14].a) Jonas AM, Glinel K, Behrens A, Anselmo AC, Langer RS, Jaklenec A, ACS Appl Mater Interfaces 2018, 10, 16250; [DOI] [PubMed] [Google Scholar]; b) Zhang H, Yang C, Zhou W, Luan, Li W, Deng Q, Dong, Tang H, Huang F, ACS Sustainable Chemistry & Engineering 2018, 6, 13924; [Google Scholar]; c) Yao M, Wu J, Li B, Xiao H, McClements DJ, Li L, Food hydrocolloids 2017, 72, 228. [Google Scholar]
- [15].Karki S, Kim H, Na S-J, Shin D, Jo K, Lee J, Asian Journal of Pharmaceutical Sciences 2016, 11, 559. [Google Scholar]
- [16].a) Châtellier X, Bottero J-Y, Petit J. Le, Langmuir 2001, 17, 2791; [Google Scholar]; b) Helander IM, Nurmiaho-Lassila EL, Ahvenainen R, Rhoades J, Roller S, Int J Food Microbiol 2001, 71, 235; [DOI] [PubMed] [Google Scholar]; c) Grimoud J, Durand H, Courtin C, Monsan P, Ouarne F, Theodorou V, Roques C, Anaerobe 2010, 16, 493. [DOI] [PubMed] [Google Scholar]
- [17].Ding S, Tan T, Process Biochemistry 2006, 41, 1451. [Google Scholar]
- [18].Hassan CM, Peppas NA, Macromolecules 2000, 33, 2472. [Google Scholar]
- [19].De Man J, Rogosa d., Sharpe ME, Journal of applied Bacteriology 1960, 23, 130. [Google Scholar]
- [20].Nikiforova OA, Klykov S, Volski A, Dicks LM, Chikindas ML, Annals of Microbiology 2016, 66, 661. [Google Scholar]
- [21].Vinderola C, Reinheimer J, International Dairy Journal 1999, 9, 497. [Google Scholar]
- [22].Carlson PE, Cell Host & Microbe 2020, 27, 173. [DOI] [PubMed] [Google Scholar]
- [23].Green CA, Quraishi MN, Shabir S, Sharma N, Hansen R, Gaya DR, Hart AL, Loman NJ, Iqbal TH, The Lancet Gastroenterology & Hepatology 2020, 5, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Khan MT, van Dijl JM, Harmsen HJ, PLoS One 2014, 9, e96097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Cordaillat-Simmons M, Rouanet A, Pot B, Experimental & Molecular Medicine 2020, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]