

Abstract
Friend leukemia virus integration 1 (Fli-1) is an ETS transcription factor and a critical regulator of inflammatory mediators, including MCP-1, CCL5, IL-6, G-CSF, CXCL2, and caspase-1. GM-CSF is a regulator of granulocyte and macrophage lineage differentiation and a key player in the pathogenesis of inflammatory/autoimmune diseases. In this study, we demonstrated that Fli-1 regulates the expression of GM-CSF in both T cells and endothelial cells. The expression of GM-CSF was significantly reduced in T cells and endothelial cells when Fli-1 was reduced. We found that Fli-1 binds directly to the GM-CSF promoter using chromatin immunoprecipitation (ChIP) assay. Transient transfection assays indicated that Fli-1 drives transcription from the GM-CSF promoter in a dose-dependent manner and mutation of the Fli-1 DNA binding domain resulted in a significant loss of transcriptional activation. Mutation of a known phosphorylation site within the Fli-1 protein led to a significant increase in GM-CSF promoter activation. Thus, direct binding to the promoter and phosphorylation are two important mechanisms behind Fli-1 driven activation of the GM-CSF promoter. In addition, Fli-1 regulates GM-CSF expression in an additive manner with another transcription factor Sp1. Finally, we demonstrated that a low dose of a chemotherapeutic drug, camptothecin, inhibited expression of Fli-1 and reduced GM-CSF production in human T cells. These results demonstrate novel mechanisms for regulating the expression of GM-CSF and suggest that Fli-1 is a critical druggable regulator of inflammation and immunity.
Introduction
Granulocyte-macrophage colony-stimulating factor (GM-CSF), also known as colony-stimulating factor 2 (CSF-2), was initially identified as a cytokine that induces granulocyte and macrophage differentiation from bone marrow progenitors (1). GM-CSF maintains low basal levels under homeostatic conditions, but its levels can be elevated quickly when homeostasis is broken (2). Multiple studies have demonstrated that GM-CSF is rapidly produced in a variety of cell types, including endothelial and T cells during inflammatory or autoimmune reactions (2, 3).
Recent studies demonstrated that GM-CSF plays a critical role in chronic inflammation and autoimmunity by acting as a communicator and activator between tissue-invading lymphocytes and myeloid cells, which results in organ injury (3, 4). GM-CSF has been implicated in the pathogenesis of various inflammatory diseases, including graft-versus-host disease (GVHD) (5), autoimmune neuroinflammation (6, 7), sepsis (8), and leukemia (9). Targeting the link between lymphocytes and myeloid cells through GM-CSF is one of the hot topics for developing novel therapies for inflammatory and autoimmune diseases (3). Therefore, understanding the mechanisms of how GM-CSF expression is regulated will provide insight into the pathogenesis of diseases and lead to potential therapeutic targets.
It has been demonstrated that several transcription factors, including NF-κB, Sp1, AP-1, Runx1 and ETS family members ETS1 and ELF-1 regulate expression of GM-CSF (10–13). Peer et al. reported that lymphatic GM-CSF expression was affected by the E3 Ubiquitin Ligase Cbl-b in the murine experimental autoimmune encephalomyelitis (EAE) model (14). Transcriptomic analysis indicated that the transcription factor STAT5A/B directs GM-CSF signaling through the regulation of proliferation and cell survival genes (15). In addition, the transcription factor Runx1 (known as AML1) is essential for definitive hematopoiesis, and its protein binds to sites in the regulatory regions of several hematopoiesis-specific genes, including GM-CSF (16). Constitutively active phosphatase calcineurin (CN) displays synergistic activation of the GM-CSF promoter together with Runx1 or Runx2, and glycogen synthase kinase-3β-phosphorylated Ets1 is a target of AML1-recruited CN phosphatase at the GM-CSF promoter (16). Thomas et al. reported that NF-κB, AP-1 and ETS1 can transactivate the human GM-CSF promoter in T cells (17). Collectively, these studies indicate that ETS transcription factors may play a role in GM-CSF activation.
Friend leukemia virus integration 1 (Fli-1), an Ets family transcription factor, is involved in the pathogenesis and development of various diseases, including cancer, lupus, systemic sclerosis, and sepsis (18–24). Ets transcription factors traditionally bind to a short core consensus DNA binding motif GGAA/T through a winged helix-turn-helix domain (25). In previous studies, we have demonstrated that Fli-1 is a key regulator moderating the expression of several inflammatory mediators, including monocyte chemotactic protein 1 (MCP-1), chemokine ligand 5 (CCL5), interleukin 6 (IL-6), granulocyte colony-stimulating factor (G-CSF), chemokine ligand 2 (CXCL2), and IL-17A (26–32). Given the role of Fli-1 in autoimmune diseases (20, 33, 34) and that Ets transcription factors have been shown to modulate GM-CSF production in T cells (13, 17), the role of Fli-1 in promoting GM-CSF expression was investigated in this study. We report here that Fli-1 regulation of GM-CSF expression in T cells and endothelial cells by partial binding to the promoter, with phosphorylation of Fli-1, impacting its activation of the regulation of GM-CSF expression.
Materials and Methods
Cell cultures
The murine endothelial MS1 cells, embryonic fibroblast NIH3T3 cells, and Jurkat cells were purchased from the American Type Culture Collection (ATCC) and the first two cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Mediatech Inc., Manassas, VA) with 10% fetal bovine serum and 1% penicillin/streptomycin. The primary human umbilical vein endothelial cells (HUVECs) were purchased from ScienCell Research Laboratories (Carlsbad, CA) and maintained in endothelial cell medium (ScienCell Research Laboratories, Carlsbad, CA) according to the manufacturer’s instructions. The Jurkat cells were maintained in RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA) with 10% fetal bovine serum. All cells were maintained at 37°C with 5% CO2.
Cell transfection with siRNA and stimulation
MS1 cells were transfected with negative control or Fli-1 specific siRNA (Integrated DNA Technologies, Coralville, IA) for 24 hours and further stimulated with LPS (0.2 μg/ml; Sigma-Aldrich, St. Louis, MO) for another 24 hours. Lipofectamine 2000 (Invitrogen, Waltham, MA) and serum-free Opti-MEM (Thermo Fisher Scientific, Waltham, MA) were used for transfection. The cell supernatants were collected at different time points after stimulation (0, 6, and 24 hours), and GM-CSF cytokine production was measured by ELISA.
ELISA
GM-CSF protein concentrations in the supernatants collected from the MS1 cells and T cells, IL-2 production in the supernatants from the mouse and human T cells were measured by mouse or human GM-CSF or IL-2 ELISA kits (R&D Systems Inc., Minneapolis, MN) according to the manufacturer’s instructions.
Chromatin immunoprecipitation (ChIP) assay
The GM-CSF promoter was screened for putative Ets DNA binding sites (EBSs) through visual inspection and utilizing the MatInspector software tool (Genomatix, Ann Arbor, MI) (35). Four primer pairs were designed to cover the 14 putative EBSs identified and can be found in Table 1. ChIP assay was performed using an anti-Fli-1 rabbit polyclonal antibody and normal IgG control using EpiTect ChIP OneDay Kit (Qiagen, Germantown, MD) as described (27, 30). Briefly, chromatin was isolated from cross-linked HUVECs and Jurkat cells that were immunoprecipitated by antibodies specific for Fli-1 or normal rabbit IgG as a control. After immunoprecipitation, the DNA was purified and amplified by RT-PCR according to the manufacturer’s instructions (Qiagen, Germantown, MD). Fold change was calculated using the comparative Ct method 2− (ΔΔCT), where delta Ct was the difference between IgG and Fli-1 Ct values.
Table 1.
Primers used in the ChiP assay of the GM-CFS promoter1
| Primer Name | Forward Primer | Reverse Primer | Position from TSS2 | Amplicon Length |
|---|---|---|---|---|
| ChIP1 | 5′-GAAAACCCCCAAGCCT-3′ | 5′-CACTGGCAAAAGAGCTC-3′ | −622 to −459 | 163 |
| ChIP2 | 5′-TCAGGCCCATTCAGACTG-3′ | 5′-AGGCTTGGGGGTTTTC-3′ | −458 to −370 | 88 |
| ChIP3 | 5′-CTCACACTCAAGTCTCTC-3′ | 5′-CAGTCTGAATGGGCCTGA-3′ | −387 to −170 | 217 |
| ChIP4 | 5′-AGCTTGCTGAGAGTGG-3′ | 5′-GAGAGACTTGAGTGTGAG-3′ | −170 to −1 | 169 |
Primers are listed based on their distance from the TSS (transcription start site).
TSS: transcription start site.
Reporter and expression constructs
The full-length (655bp) human GM-CSF promoter cloned into the pXPG basic vector upstream of the Luciferase reporter gene was a gift from Peter Cockerill though Addgene (ID: 71250, Cambridge, MA) (36). The mouse Fli-1 gene was previously cloned into the pcDNA3.0 expression vector (Life Technologies, Rockville, MD) containing a 5′ Kozak sequence and FLAG tag (28). The following mutant Fli-1 proteins used in the study were provided by Dr. Maria Trojanowska at the Boston University: the human Fli-1 DNA binding domain mutant, cloned into the pSG5 expression vector, which contains a single amino acid mutation (tryptophan 321 to arginine) preventing Fli-1 from binding to DNA; the Fli-1 acetylation mutant, also in pSG5 (with lysine 380 replaced by arginine); and the Fli-1 phosphorylation mutant (with threonine 312 replaced with alanine) (26, 37, 38).
DNA transfection
For all the transient transfection studies, 24 hours prior to transfection, 4×105 NIH3T3 cells were seeded into six-well plates. The cells were transfected following the manufacturer’s instructions for the Fugene 6 transfection reagent (Promega, Madison, WI). The reporter constructs were transfected into the cells at a concentration of 2 μg (pXPG/basic, pXPG/GMCSF) for all experiments. All expression constructs (pcDNA3.0, pcDNA/Fl-i1, pSG5, pSG5/Fli-1, pSG5/Fli-1DNAbindMut, pSG5/PhosMut and pSG5/AceMut) were transfected into the cells at equimolar concentrations. The pcDNA/Fli-1 expression construct was transfected into the cells in increasing concentrations (0.025 μg, 0.05 μg, 0.1 μg, 0.2 μg, 0.25 μg, 0.5 μg, and 1 μg) for the dose-response study. In the binding domain mutant experiment, 0.75μg of the pSG/Fli-1 construct was used. In the phosphorylation/acetylation mutant experiment, 0.75 μg of the pcDNA/Fli-1, pSG/Fli-1, pSG/Fli-1Mut, pSG/Fli-1PhosMut, and pSG/Fli-1AceMut expression constructs were transfected into the cells. Based on dose-response curves for each construct, the following concentrations were used in the co-transfection experiments, 1 μg of NF-κB p50 and Sp1 and 0.1 μg of pcDNA/Fli-1, and NF-κB p65. Expression constructs containing the NF- κB family transcription factors p50 and p65, as well as Sp1 construct were generously donated from the Boss laboratory (Emory University School of Medicine, Atlanta, GA) (39). In all cases, empty expression vectors were used such that an equivalent amount of total DNA was transfected into the cells. A Renilla luciferase construct pRL/TK (Promega, Madison, WI), at a concentration of 200 ng, was used to control for transfection efficiency. The NIH3T3 cells were harvested 48 hours after transfection.
Luciferase reporter assay
The Dual-luciferase reporter assay system (Promega, Madison, WI) was utilized to determine the amount of luciferase activity for each experiment. Luminescence was measured on the Luminoskan Ascent Microplate Luminometer (Thermo Fisher Scientific, Waltham, MA). The Renilla luciferase construct was used to normalize activity for all transfections. The values reported are mean ± SEM for fold activation based on the activity compared to the empty pXPG/basic luciferase vector.
Western blotting
Cells were lysed in the Radioimmunoprecipitation assay (RIPA) buffer as previously described (33). After lysis, the supernatant was collected. Equal amounts of protein (20 μg-40 μg) were run on for 1.5 hours at 130V using 4–20% Criterion TGX Stain-Free Protein Gels (Bio-Rad, Hercules, CA) and electro-transferred to a PVDF membrane by Iblot2 transfer stacks (Invitrogen, Waltham, MA). Transferred proteins were probed with both a Fli-1 polyclonal antibody described previously (30) and an antibody to β-actin (Cell Signaling, Beverly, MA). The results were visualized using the Odyssey Imaging System (LI-COR, Lincoln, NE).
Mice with conditional deletion of Fli-1 and splenic T cell isolation
Mice with a Fli-1 conditional deletion (Fli-1flox/flox) were described previously (22, 40). A T-cell conditional deletion of Fli-1 exons 3 and 4 was mediated by a Cre/Lox system utilizing the CD4 promoter (41). Homozygous Fli-1 deletion in T cells was mediated via Fli-1flox/floxCD4Cre. Wild-type Fli-1 with CD4cre+ were used as controls (referred to as Fli-1+/+CD4cre). Spleens were harvested from Fli-1flox/floxCD4Cre and Fli-1+/+CD4Cre mice at the age of 8-10 weeks, minced, and dispersed into single cells in RPMI 1640 medium with 10% fetal bovine serum. Cells were filtered through a 40-μm pore-size cell strainer and washed with RPMI 1640 medium, and red blood cells (RBCs) were lysed using the mouse erythrocyte lysis kit (R&D Systems, Minneapolis, MN). CD4+ T cells were enriched by negative selection using a mouse CD4+ T cell isolation kit (Invitrogen, Waltham, MA) as instructed by the manufacturer. The cells were cultured at 1 million/mL of RPMI 1640 medium with recombinant mouse interleukin-2 (IL-2) (R&D Systems, Minneapolis, MN) at 20 IU/mL and CD28/CD3 beads (Mouse T-activator anti-CD3/CD28 Dynabeads, Gibco) that were added in a 1:1 ratio with the CD4+ T cells. The supernatants of T cells were collected after three days of stimulation, and GM-CSF cytokine production was measured by ELISA.
Human T cells isolation
Peripheral blood mononuclear cells from 5 healthy human volunteers were isolated using Ficoll-Paque (Miltenyi Biotec Inc., Auburn, CA), T cells were isolated with Dynabeads Untouched Human T cells kit (Thermo Fisher Scientific, Waltham, MA). Purified T cells were pretreated with 0.1 μM of camptothecin for 12 hours, then cultured at 1 million/mL of RPMI 1640 medium with recombinant human interleukin-2 (IL-2) (R&D Systems, Minneapolis, MN) at 20 IU/mL and CD28/CD3 beads (human T-activator anti-CD3/CD28 Dynabeads, Thermo Fisher Scientific) were added at a 1:1 ratio. The supernatants of T cells were collected after one and three days of stimulation, GM-CSF and IL-2 cytokine production was measured by ELISA.
Statistics
The data are expressed as mean ± SEM. GraphPad Prism 8 software (GraphPad Software, San Diego, CA) was used for statistical analysis. Two-group Student t-tests (paired or unpaired, as appropriate) were applied. Differences between the means of multiple groups were compared with one-way analysis of variance (ANOVA), followed by a Tukey multiple comparisons test. A value of p < 0.05 was considered statistically significant.
Results
Deletion of Fli-1 in T cells reveals decreased GM-CSF expression
To examine if Fli-1 regulates expression of GM-CSF in T cells, we isolated T cells from Fli-1+/+CD4cre and Fli-1flox/floxCD4cre mice. As shown in Fig. 1A, Fli-1 protein expression levels in T cells from the Fli-1flox/floxCD4cre mice were significantly reduced compared to Fli-1+/+CD4cre littermates by western blot analysis (Fig. 1A). T cells were stimulated with CD3/CD28 activation beads for three days, and the GM-CSF protein expression in supernatants was measured by ELISA. As shown in Fig. 1B, the concentration of GM-CSF from Fli-1 deficient T cells was significantly lower compared to the T cells from that of wild-type mice. Since it is known that expression of GM-CSF can be induced by IL-2-mediated signal transducer and activator of transcription 5 (STAT5) signaling (42), we measured IL-2 concentrations in the supernatants. As shown in Fig. 1C, the reduction of Fli-1 levels did not affect IL-2 expression. This data indicates that Fli-1 regulates GM-CSF expression in mouse splenic T cells.
Figure 1. Knockout of Fli-1 attenuates GM-CSF production in mouse splenic T cells.
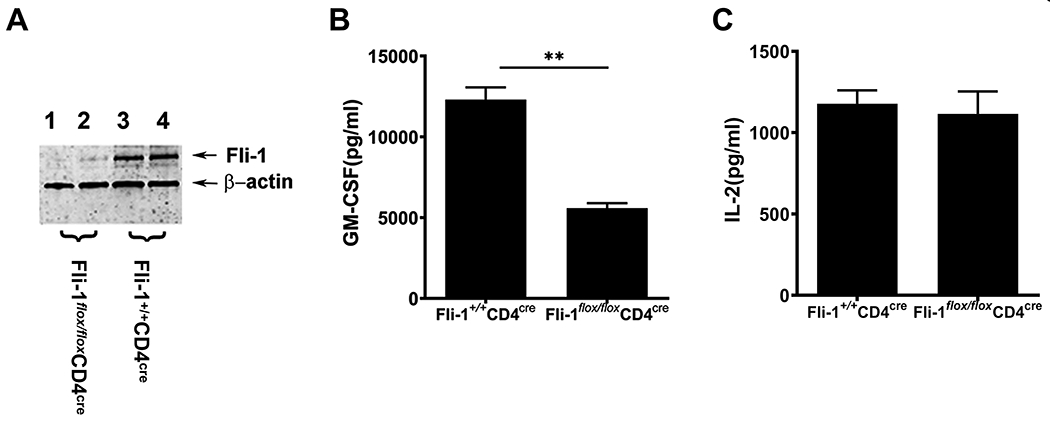
Splenic T cells were isolated from Fli-1+/+CD4cre and Fli-1flox/floxCD4Cre mice. (A) Decreased expression of Fli-1 protein in T cells from Fli-1flox/floxCD4Cre mice. T cells were isolated from Fli-1+/+CD4Cre and Fli-1flox/floxCD4Cre mice at the age of 8 weeks. Equal amounts of protein extracts from the T cells were loaded into each well and probed by an anti-Fli-1 antibody and β-actin. (B) Reduced GM-CSF production from Fli-1-deficient T cells. T cells were stimulated with CD3/CD28 for three days, and GM-CSF concentration in the supernatants was measured by ELISA (n=4 in each group). (C) Comparison of IL-2 levels between Fli-1-deficient T cells and normal T cells. IL-2 expression in supernatants was detected by ELISA. Data are presented as mean ± SEM from three independent experiments. **indicates p < 0.01.
Production of GM-CSF is reduced in endothelial cells upon selective inhibition of Fli-1
To study the effects of Fli-1 on GM-CSF production in endothelial cells, we inhibited Fli-1 expression in murine MS1 endothelial cells by transfecting Fli-1 specific siRNA. As shown in Fig. 2B, expression the Fli-1 protein was markedly decreased following the transfection of Fli-1 siRNA. The concentrations of GM-CSF from cells transfected with Fli-1 specific siRNA were significantly lower compared to the cells transfected with control siRNA at 6 and 24 hours following stimulation with LPS (Fig. 2A).
Figure 2. Knockdown of Fli-1 significantly decreased GM-CSF production in endothelial cells.
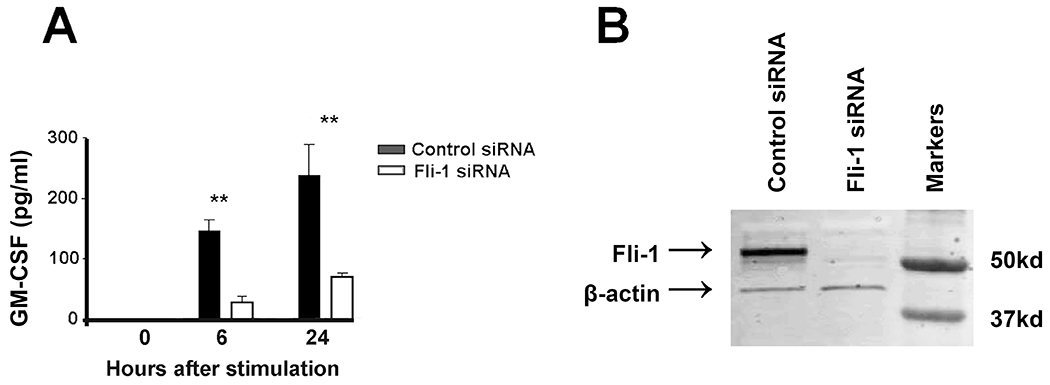
Murine MS1 cells were transfected with Fli-1 specific or negative control siRNA for 24 hours and further stimulated with LPS (0.2 μg/ml) for another 24 hours. (A) GM-CSF concentrations in murine MS1 cells were measured by ELISA at 0, 6, or 24 hours after stimulation. (B) Western blot results showing a specific knockdown of Fli-1 in murine MS1 cells. The data are presented as mean ± SEM. Data are representative of three independent experiments. ** indicates p < 0.01.
Fli-1 binds to the GM-CSF promoter
To determine if Fli-1 directly binds to the promoter of GM-CSF, we identified 14 putative Fli-1 binding sites within the human GM-CSF promoter and designed four pairs of primers to cover these sites (Fig. 3A and Table 1). We performed ChIP assay using these four pairs of primers with HUVECs and Jurkat cells. As shown in Fig. 3B, ChIP1, ChIP3, ChIP4 were significantly enriched for Fli-1 specific antibody compared to the IgG negative control in HUVECs. In Jurkat cells, Fli-1 binding was increased significantly, more than six-fold over the IgG control, for all four regions (ChIP1, ChIP2 ChIP3, and ChIP4). These results demonstrate that Fli-1 binds to the GM-CSF promoter in both HUVECs and Jurkat cells.
Figure 3. Fli-1 binds to the region of the GM-CSF promoter.
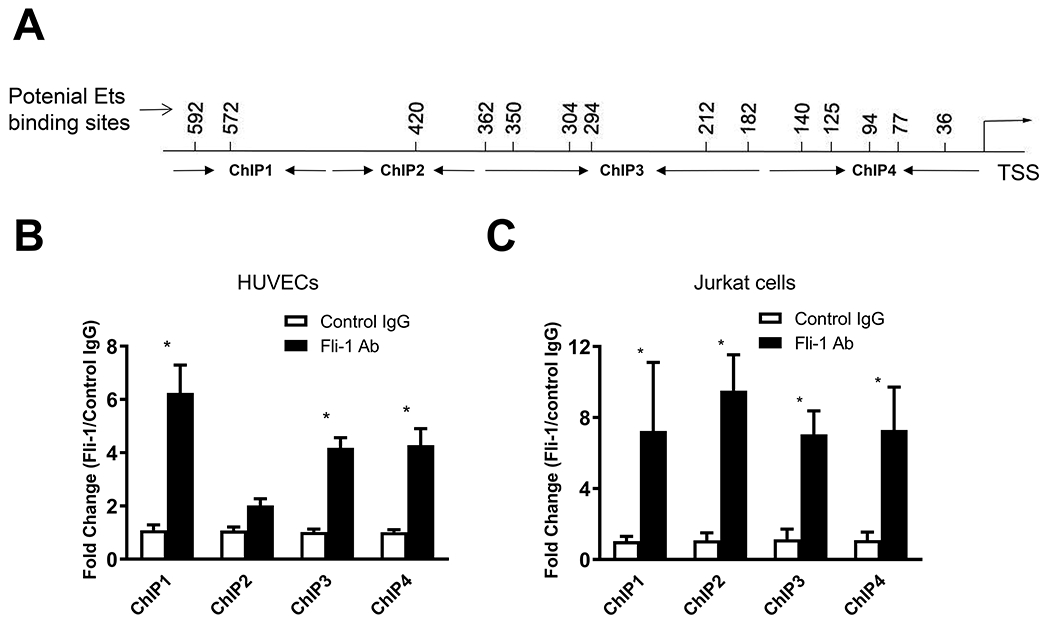
(A) Schematic diagram showing the location of the 14 putative Fli-1binding sites based on the MatInspector Software and visual inspection of the promoter. The location of the binding sites and four primers designed for ChIP, including distance from the transcription start site (TSS), can be found in the axis. ChIP analysis of Fli-1 binding to the GM-CSF promoter was performed in HUVECs (B) and Jurkat cells (C). The data are presented as mean ± SEM. Data are representative of four independent experiments. Fli-1 specific enrichment is compared to the IgG control. *p < 0.05.
Fli-1 drives transcriptional activation of the GM-CSF promoter in a dose-dependent manner
To investigate whether Fli-1 activates transcription from the GM-CSF promoter, transient transfection assays were performed in NIH3T3 cells. We have previously shown that NIH3T3 cells do not express detectable endogenous Fli-1 (26). The Fli-1 plasmid was transfected into NIH3T3 cells in increasing concentrations along with the full-length GM-CSF promoter/pXPG reporter construct. As shown in Fig. 4B, the Fli-1 transcription factor strongly induced activation from the GM-CSF promoter in a statistically significant manner when compared to the activation of the reporter construct. The results demonstrate that Fli-1 drives transcription from the GM-CSF promoter in a dose-dependent manner, with as little as 50 ng of Fli-1 needed to significantly activate transcription from the GM-CSF promoter. Fold activation begins to decrease when the Fli-1 plasmid concentration reaches 250 ng (Fig. 4B). We examined the Fli-1 protein expression after transfection by immunoblot; Fli-1 protein expression corresponds with increasing amounts of the Fli-1 plasmid transfected into the cells (Fig. 4A).
Figure 4. Fli-1 drives transcription from the human GM-CSF promoter.
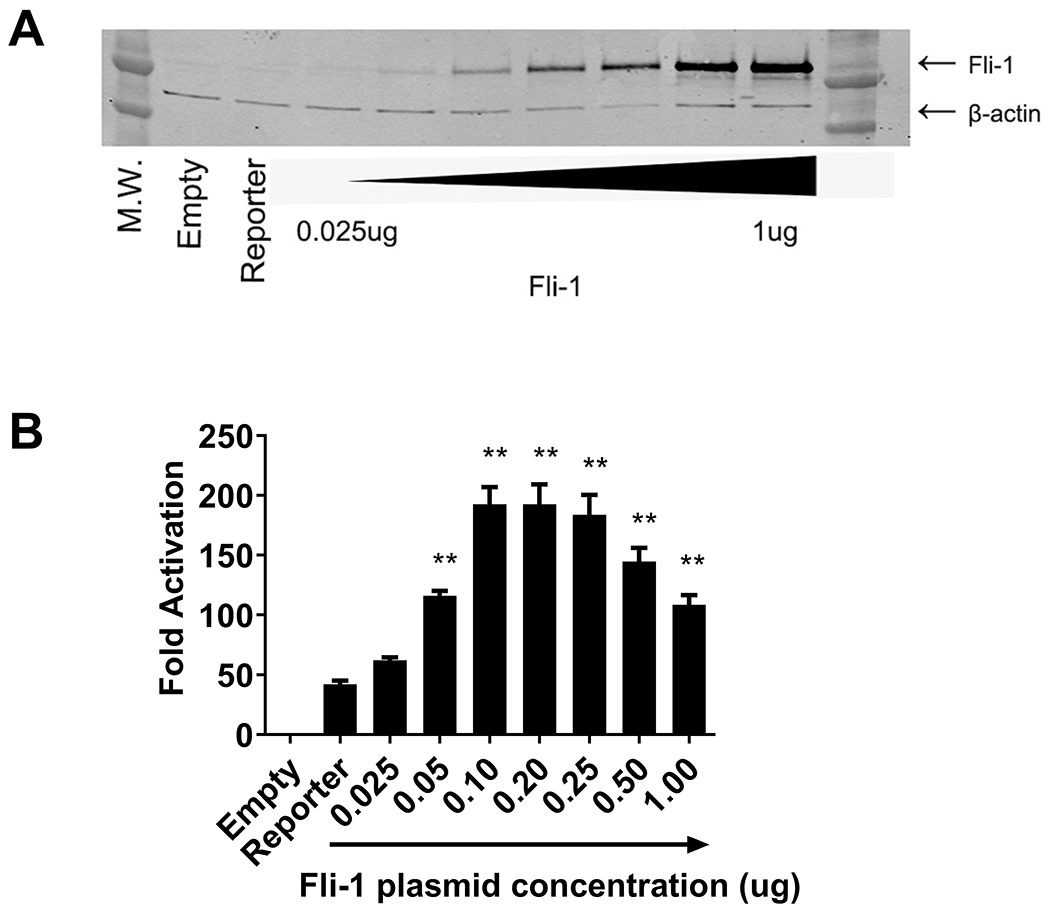
Transfections were carried out using increasing amounts of Fli-1 (from 0.025 ug to 1 ug) in NIH3T3 cells. (A) Fli-1 protein concentrations after transfection. (B) Graph depicting the results of the luciferase assay. Values shown are as fold activation over the empty vector control and are represented as mean ± SEM from three independent experiments (n=8). ** p < 0.01 compared to the activation of the reporter construct.
Fli-1 partially regulates activation of the GM-CSF promoter via direct binding
To further investigate whether Fli-1 mediated transcriptional activation from the GM-CSF promoter is through direct binding, wild-type Fli-1 construct and a Fli-1 DNA binding mutant construct containing a single amino acid mutation (tryptophan to arginine) at amino acid 321 that prevents Fli-1 from binding to the DNA were transfected into the NIH3T3 cells along with the GM-CSF promoter/pXPG reporter construct (43). As shown in Fig 5A, activation of the GM-CSF promoter by the Fli-1 DNA binding mutant was reduced by about 30% compared to the wild-type Fli-1 construct. Proteins for the wild-type Fli-1, and Fli-1 DNA binding mutant were expressed at similar levels as shown in the western blot (Fig. 5B). These results suggest that activation of the GM-CSF promoter by Fli-1 is partially through direct binding to the promoter and indicate that other indirect mechanisms may be involved in Fli-1 mediated activation.
Figure 5. Fli-1 partially regulates GM-CSF expression through direct binding to the GM-CSF promoter.
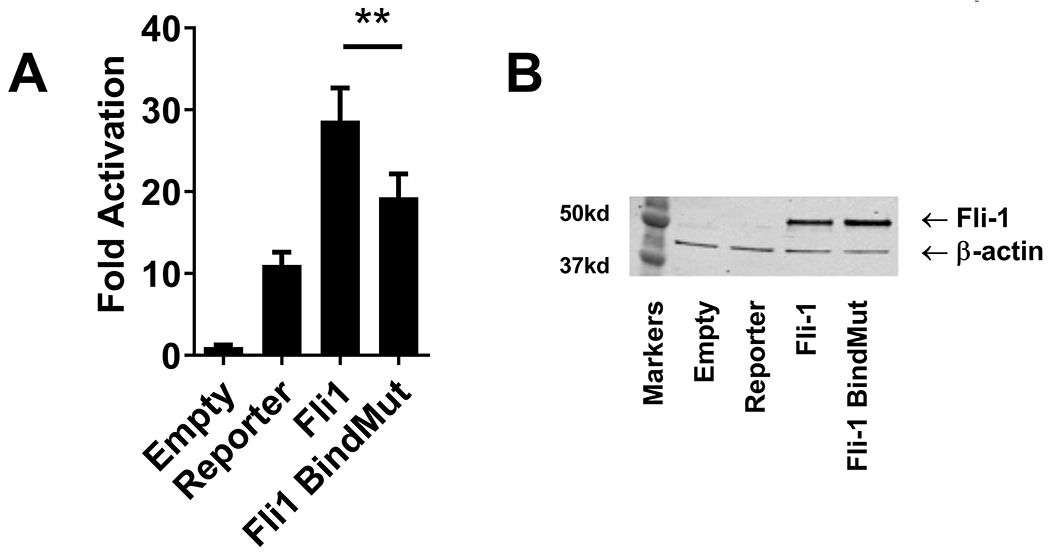
The wild-type Fli-1 construct and Fli-1 DNA binding mutant construct containing a single amino acid mutation (tryptophan to arginine) at amino acid 321 that prevents Fli-1 from binding to the DNA were transfected into the NIH3T3 cells along with the GM-CSF promoter/pXPG reporter construct. (A) Luciferase assay results. Values (GM-CSF promoter activation) shown are as fold activation over the empty vector control and are represented as mean ± SEM from three independent experiments (n=9). A two-tailed Student’s t-test with unequal variance was used to calculate significance, and ** indicate p < 0.01. (B) Fli-1 protein concentrations after transfection into the NIH3T3 cells were analyzed by western blot. Data are representative of three independent experiments.
Phosphorylation but not acetylation plays a role in Fli-1 driven activation of the GM-CSF promoter
Our previous study showed acetylation plays a key role in Fli-1 driven activation of the G-CSF promoter (26). Given that indirect mechanisms play a role in Fli-1 mediated regulation of the GM-CSF promoter, we next examined whether post-translational modification of Fli-1 affects the activation of the GM-CSF promoter. The wild-type Fli-1 construct, a Fli-1 phosphorylation mutant construct containing a mutation (threonine to alanine) at amino acid 312 which prevents phosphorylation of the Fli-1 protein, and a Fli-1 acetylation mutant construct containing a mutation (lysine to arginine) at amino acid 380 within the Fli-1 protein which prevents acetylation of the protein were transfected into the NIH3T3 cells along with the GM-CSF promoter/pXPG reporter construct. As shown in Fig. 6A, inhibiting phosphorylation of the amino acid 312 site resulted in a significant increase in Fli-1 driven activation of the GM-CSF promoter compared to the wild-type Fli-1; however, inhibiting acetylation of Fli-1 protein had a marginal effect on activation of the GM-CSF promoter. Proteins for the wild-type Fli-1, phosphorylation mutant, and acetylation mutant were expressed at similar levels as shown in the western blot (Fig. 6B). Therefore, phosphorylation of Fli-1 negatively impacts Fli-1 mediated activation of the GM-CSF promoter and acetylation does not.
Figure 6. Phosphorylation, not acetylation, plays a role in Fli-1-driven activation of the GM-CSF promoter.
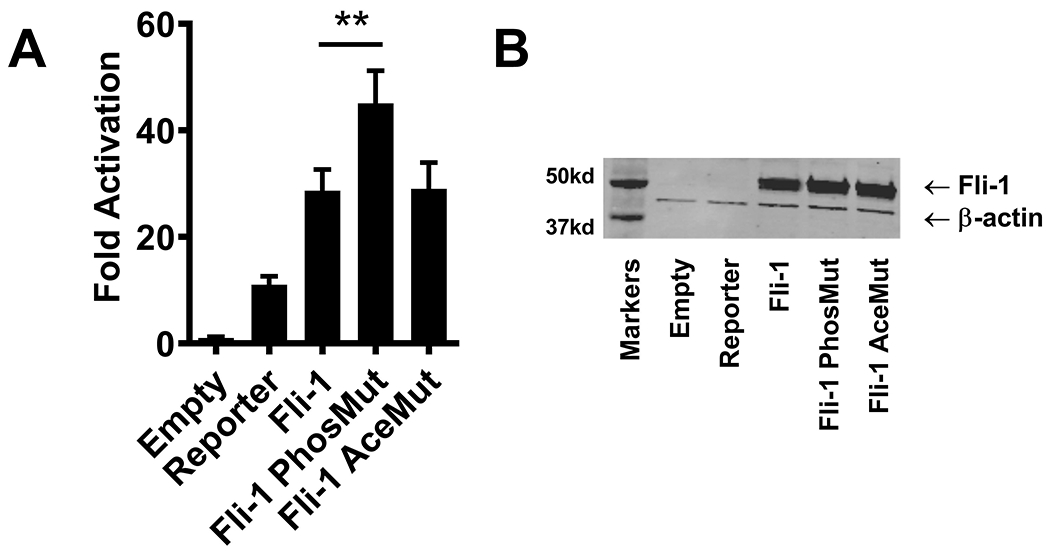
Wild-type Fli-1 construct, Fli-1 phosphorylation mutant construct containing a mutation (threonine to alanine) at amino acid 312, and Fli-1 acetylation mutant construct containing a mutation (lysine to arginine) at amino acid 380 were transfected into the NIH3T3 cells along with the GM-CSF promoter/pXPG reporter construct. (A) Graph depicting the results of the luciferase assay. Values shown are as fold activation over the empty vector control and are presented as mean ± SEM from three dependent experiments (n=9). A two-tailed Student’s t-test with unequal variance was used to calculate significance, and ** indicate p < 0.01. (B) Fli-1 protein concentrations after transfection into the NIH3T3 cells were analyzed by western blot. Data are representative of three independent experiments.
Fli-1 regulates the expression of GM-CSF in an additive manner with Sp1 but not NF-κB
We have previously shown that Fli-1 and NF-κB interact to enhance transcription from the MCP-1 promoter (27). Transcriptional factors Sp1 and NF-κB have been reported to regulate GM-CSF expression (7, 12, 44). To further understand the indirect mechanisms of Fli-1 mediated GM-CSF regulation, we next investigated if Fli-1 interacts with these transcription factors (Sp1 and NF-κB) in relation to the regulation of GM-CSF. Transfection of Sp1 and NF-κB p65 into the NIH3T3 cells along with the GM-CSF promoter led to a significant increase in promoter activation, however, NF-κB p50 significantly decreased GM-CSF activation (Fig. 7). When Fli-1 and Sp1 were transfected into the cells along with the GM-CSF promoter construct, a significant increase was observed in GM-CSF promoter activation compared to Fli-1 or Sp1 alone (Fig. 7); the effect of the two transcription factors together was additive, not synergistic. Abundant endogenous NF-κB expression was detected in NIH3T3 cells and the level of Sp1 was undetectable (Supplemental Fig. 1). Sp1 was not detected by co-immunoprecipitation with a specific anti-Fli-1 antibody in HUVECs (Supplemental Fig. 2). The Co-transfection of Fli-1 and NF-κB p65 did not increase the level of GM-CSF promoter activation (Fig. 7). While not conclusive, a significant decrease in GM-CSF activation by p50 appeared to be reduced by the addition of Fli-1 (Fig. 7). Thus, Fli-1 regulates GM-CSF independently with Sp1 but not NF-κB p65.
Figure 7. Fli-1 and Sp1 regulate the expression of GM-CSF in an additive manner.
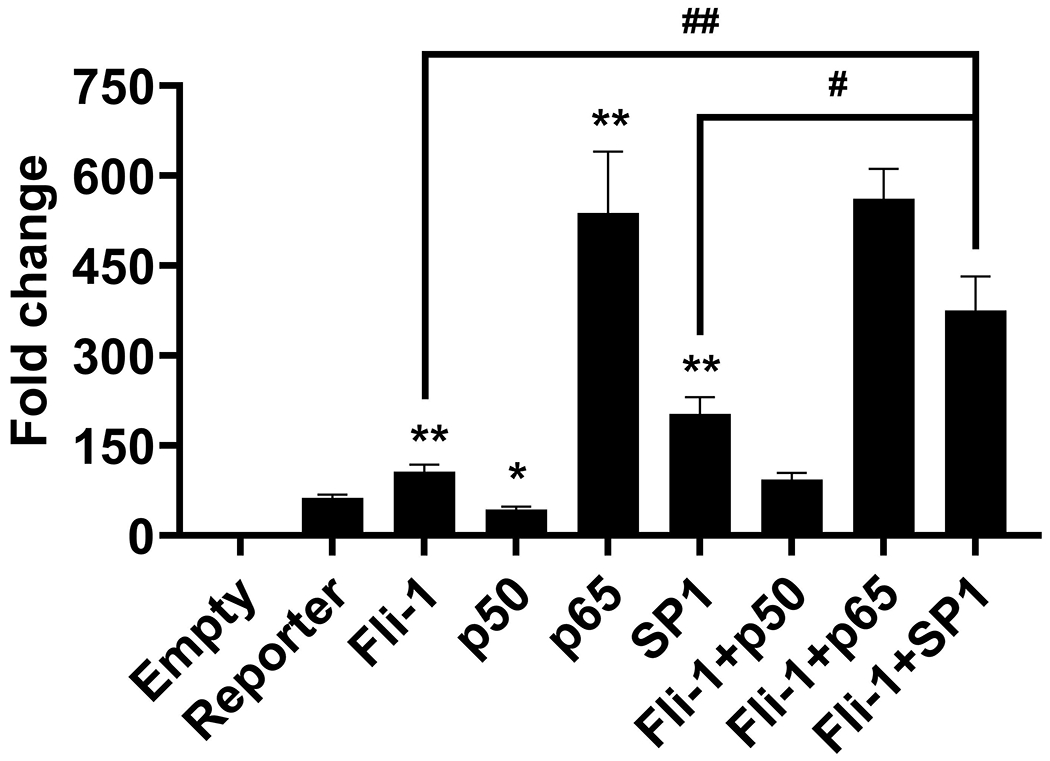
Fli-1 and the Sp1/ NF-κB transcription factors were co-transfected into NIH3T3 cells. Graph depicting the results of the luciferase assay after co-transfection of Fli-1 and Sp1, Fli-1 and NF-κB (p50 or p65) in NIH3T3 cells. Data presented are shown as fold activation over the empty vector control and are represented as mean ± SEM (n=10). *p < 0.05 and ** p<0.001 compared with reporter construct, #p < 0.05 and ## p<0.001compared with Fli-1+Sp1 group.
Expression of Fli-1 and production of GM-CSF in human T cells were decreased following treatment with camptothecin
It has been reported that the expression of Fli-1 was inhibited by camptothecin (CPT) (45). Next, we isolated human T cells from healthy volunteers. Purified T cells were treated with a control vehicle (0.1% DMSO) or 0.1μM of CPT for 12 hours, then stimulated with CD3/CD28 activation beads. As shown in Fig. 8A, Fli-1 protein levels were significantly decreased after camptothecin treatment (Fig. 8A). GM-CSF production was significantly lower on day 1 and 3 following stimulation of the cells treated with camptothecin compared with untreated cells (Fig. 8B). IL-2 levels were similar between the cells treated with camptothecin and controls.
Figure 8. Expression of Fli-1 and production of GM-CSF in human T cells were decreased following the treatment with camptothecin (CPT).
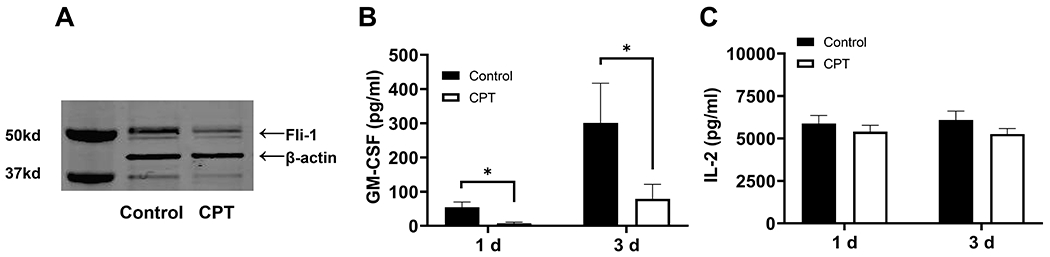
Human T cells were purified from 5 healthy volunteers and were treated with 0.1 μM of camptothecin or 0.1% DMSO (control) for 12 hours. T cells were stimulated with CD3/CD28 for three days. (A) Reduced expression of Fli-1 by camptothecin. Equal amounts of protein extracts from the human T cells were loaded into each well and probed by an anti-Fli-1 antibody and β-actin. Representative of 5 independent experiments. (B) Reduced GM-CSF in the supernatants from T cells treated with camptothecin. Concentrations of GM-CSF were measured by ELISA (n=5 in each group). (C) Comparison of IL-2 levels between human T cells treated with camptothecin and vehicle control. IL-2 expression in supernatants was detected by ELISA. Data are presented as mean ± SEM. A two-tailed Student’s t-test with unequal variance was used to calculate significance, and *indicates p < 0.05.
Discussion
We have demonstrated for the first time that expression of GM-CSF is regulated by the transcription factor Fli-1 in both T cells and endothelial cells. Results demonstrate that Fli-1 binds to several regions of the GM-CSF promoter in each cell type. Transient transfection assays demonstrated that Fli-1 drives transcription from the GM-CSF promoter in a dose-dependent manner. Fli-1 regulates the expression of GM-CSF by both direct and indirect means, and phosphorylation of Fli-1 negatively impacts activation and the regulation of GM-CSF expression.
Though GM-CSF was originally identified as a growth factor to promote differentiation of myeloid cells, recent evidence indicates that GM-CSF plays a critical role in local inflammation and autoimmune diseases, functioning as a communicator between tissue-invading lymphocytes and myeloid cells to stimulate and maintain inflammation (3). Hirota and colleagues have demonstrated that GM-CSF produced by Th17 cells enhanced chronic inflammation and that GM-CSF is an essential and crucial inflammatory mediator among Th17 cells, with synovial-resident innate lymphoid cells mediating inflammation and causing tissue damage in an autoimmune arthritis model (4). We previously demonstrated that reduced expression of Fli-1 resulted in significantly decreased inflammation in the kidney with diminished inflammatory cell infiltrations in two murine models of lupus, MRL/lpr mice and NZM2410 mice (30, 33, 34). We also found that reduced expression of Fli-1 led to significantly reduced amounts of Th17 cells in the kidney in MRL/lpr mice (31). Here, we establish that Fli-1 is a critical regulator in modulating the expression of GM-CSF. Tugues et al. demonstrated that GM-CSF produced by allogeneic T cells is the main player in promoting pathological damage and mediating lethal GVHD in the murine model, and elevated GM-CSF was similarly observed in human allogenic hematopoietic cell transplantation patients with severe GVHD (5). Significantly reduced organ damage and improved survival in mice were reported when the animals were given alloreactive Csf2−/− (GM-CSF-deficient) T cells in the study (5). We have also observed that mice that received T cells with reduced expression of Fli-1 had significantly reduced pathology in both acute GVHD or chronic GVHD (Schutt and Yu, manuscript in submission). Given the demonstrated effect of Fli-1 on GM-CSF and other inflammatory cytokine production, targeting Fli-1 may be an attractive strategy for treating GVHD without compromising the graft-versus-leukemia (GVL) effect. In this study, we have demonstrated that a low-dose of the chemotherapeutic drug camptothecin inhibited expression of Fli-1 and reduced the production of GM-CSF. Based on the effect of GM-CSF in GVHD and other diseases and the role of Fli-1 in regulating GM-CSF, future studies exploring the role of Fli-1 in these diseases such as experimental autoimmune encephalomyelitis are of interest.
Our previous studies have demonstrated that Fli-1 regulates the expression of several inflammatory cytokines and mediators, including MCP-1, CCL5, IL-6, G-CSF, CXCL2, and caspase-1 (26–30, 32, 46). An interesting observation from these findings is that Fli-1 regulates the expression of different cytokines through a variety of different mechanisms and our current results are consistent with this observation. A single amino acid mutation of the Fli-1 DNA binding domain led to a 30% reduction of activation from the GM-CSF promoter (Fig. 5A), suggesting Fli-1 regulates expression of GM-CSF through both direct and indirect binding. Whereas Fli-1 regulates expression of CCL5, G-CSF, and CXCL2 largely through direct binding (28, 47, 48), mutation of the Fli-1 DNA binding domain mostly abolished Fli-1 mediated activation of these promoters, Fli-1 regulates GM-CSF through indirect means as well, similar to MCP-1 (27). Our previous study showed that acetylation, not phosphorylation, plays a key role in Fli-1 driven activation of the G-CSF promoter (26). Conversely, no change in activation of the GM-CSF promoter was observed as a result of acetylation; however, mutation of the phosphorylation site at aa312 led to a significant increase in Fli-1 activation of the GM-CSF promoter (Fig. 6A). Thus, our data indicate that phosphorylation is an important mechanism behind Fli-1 driven activation of the GM-CSF promoter, again demonstrating that Fli-1 regulates each cytokine differently. Given the recognized role that Sp1, NF-κB p50, and p65 play in binding to the GM-CSF promoter, transcription factor complex assembly, and transactivation (49, 50), we examined if Fli-1 interacts with NF-κB and Sp-1 to synergistically enhance transcription from the GM-CSF promoter. Our results demonstrated that the co-transfection of Fli-1 and Sp1 led to an additive enhancement of transcription from the GM-CSF promoter, whereas there was no change in activation of the GM-CSF promoter when Fli-1 co-transfected with NF-κB p65 (Fig. 7). Results appear to suggest that Fli-1 may mitigate a significant reduction in GM-CSF activation by p50, when the two factors are transfected together (Fig. 7). Our previous data showed that Fli-1 interacts with p65 to synergistically enhance transcriptional activation of MCP-1, and the expression of CXCL2 is regulated by NF-κB and Fli-1 in an additive manner (27, 32). Collectively, our results indicate that Fli-1 regulates the expression of individual cytokines through a variety of different mechanisms.
In summary, we have identified that Fli-1 is a novel transcription factor regulating the expression of inflammatory chemokine GM-CSF, and could be targeted by a currently utilized therapeutic drug. Considering the emerging evidence that GM-CSF plays a critical role in inflammatory and autoimmune diseases, our findings provide new insights towards understanding the mechanisms of inflammation and disease development through GM-CSF and suggest that Fli-1 may represent a novel potential target for treating autoimmunity/inflammation-related diseases.
Supplementary Material
KEY POINTS.
Fli-1 regulates expression of GM-CSF by both direct and indirect means.
phosphorylation of Fli-1 negatively impacts the regulation of GM-CSF expression.
Fli-1 is a potential drug target.
Footnotes
The present study was supported in part by grants from the Lupus Research Alliance (519417), MUSC CCCR pilot award (X.Z.), and the National Institute of Health (1R01GM130653, H.F.) (R01 Al118305, X.Z.Y).
Abbreviations used in this manuscript:
CCL5, C-C Motif Chemokine Ligand 5; CPT, camptothecin; CXCL2, Chemokine (C-X-C motif) ligand 2; Fli-1, Friend leukemia virus integration 1; G-CSF, Granulocyte colony-stimulating factor; GM-CSF, Granulocyte-macrophage colony-stimulating factor; GVHD, graft-versus-host disease; HUVECS, human umbilical vein endothelial cells; IL-6, Interleukin 6; MCP-1, Monocyte chemoattractant protein-1.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Burgess AW, and Metcalf D. 1980. The nature and action of granulocyte-macrophage colony stimulating factors. Blood 56: 947–958. [PubMed] [Google Scholar]
- 2.Bhattacharya P, Thiruppathi M, Elshabrawy HA, Alharshawi K, Kumar P, and Prabhakar BS. 2015. GM-CSF: An immune modulatory cytokine that can suppress autoimmunity. Cytokine 75: 261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becher B, Tugues S, and Greter M. 2016. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 45: 963–973. [DOI] [PubMed] [Google Scholar]
- 4.Hirota K, Hashimoto M, Ito Y, Matsuura M, Ito H, Tanaka M, Watanabe H, Kondoh G, Tanaka A, Yasuda K, Kopf M, Potocnik AJ, Stockinger B, Sakaguchi N, and Sakaguchi S. 2018. Autoimmune Th17 Cells Induced Synovial Stromal and Innate Lymphoid Cell Secretion of the Cytokine GM-CSF to Initiate and Augment Autoimmune Arthritis. Immunity 48: 1220–1232. e1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tugues S, Amorim A, Spath S, Martin-Blondel G, Schreiner B, De Feo D, Lutz M, Guscetti F, Apostolova P, Haftmann C, Hasselblatt P, Núñez NG, Hottiger MO, van den Broek M, Manz MG, Zeiser R, and Becher B. 2018. Graft-versus-host disease, but not graft-versus-leukemia immunity, is mediated by GM-CSF-licensed myeloid cells. Science translational medicine 10. [DOI] [PubMed] [Google Scholar]
- 6.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, and Becher B. 2011. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nature immunology 12: 560–567. [DOI] [PubMed] [Google Scholar]
- 7.Yu J, Zhou X, Nakaya M, Jin W, Cheng X, and Sun SC. 2014. T cell-intrinsic function of the noncanonical NF-κB pathway in the regulation of GM-CSF expression and experimental autoimmune encephalomyelitis pathogenesis. Journal of immunology 193: 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mathias B, Szpila BE, Moore FA, Efron PA, and Moldawer LL. 2015. A Review of GM-CSF Therapy in Sepsis. Medicine 94: e2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchner T, Hiddemann W, Wormann B, Rottmann R, Maschmeyer G, Ludwig WD, Zuhlsdorf M, Buntkirchen K, Sander A, Aswald J, and et al. 1993. The role of GM-CSF in the treatment of acute myeloid leukemia. Leuk Lymphoma 11 Suppl 2: 21–24. [DOI] [PubMed] [Google Scholar]
- 10.Cockerill PN, Osborne CS, Bert AG, and Grotto RJ. 1996. Regulation of GM-CSF gene transcription by core-binding factor. Cell growth & differentiation : the molecular biology journal of the American Association for Cancer Research 7: 917–922. [PubMed] [Google Scholar]
- 11.Oakford PC, James SR, Qadi A, West AC, Ray SN, Bert AG, Cockerill PN, and Holloway AF. 2010. Transcriptional and epigenetic regulation of the GM-CSF promoter by RUNX1. Leuk Res 34: 1203–1213. [DOI] [PubMed] [Google Scholar]
- 12.Cakouros D, Cockerill PN, Bert AG, Mital R, Roberts DC, and Shannon MF. 2001. A NF-kappa B/Sp1 region is essential for chromatin remodeling and correct transcription of a human granulocyte-macrophage colony-stimulating factor transgene. Journal of immunology 167: 302–310. [DOI] [PubMed] [Google Scholar]
- 13.Wang CY, Bassuk AG, Boise LH, Thompson CB, Bravo R, and Leiden JM. 1994. Activation of the granulocyte-macrophage colony-stimulating factor promoter in T cells requires cooperative binding of Elf-1 and AP-1 transcription factors. Mol Cell Biol 14: 1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peer S, Cappellano G, Hermann-Kleiter N, Albrecht-Schgoer K, Hinterleitner R, Baier G, and Gruber T. 2018. Regulation of Lymphatic GM-CSF Expression by the E3 Ubiquitin Ligase Cbl-b. Frontiers in immunology 9: 2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura A, Rieger MA, Simone JM, Chen W, Wickre MC, Zhu BM, Hoppe PS, O’Shea JJ, Schroeder T, and Hennighausen L. 2009. The transcription factors STAT5A/B regulate GM-CSF-mediated granulopoiesis. Blood 114: 4721–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, Holm M, Xie XQ, Wolf-Watz M, and Grundstrom T. 2004. AML1/Runx1 recruits calcineurin to regulate granulocyte macrophage colony-stimulating factor by Ets1 activation. The Journal of biological chemistry 279: 29398–29408. [DOI] [PubMed] [Google Scholar]
- 17.Thomas RS, Tymms MJ, McKinlay LH, Shannon MF, Seth A, and Kola I. 1997. ETS1, NFkappaB and AP1 synergistically transactivate the human GM-CSF promoter. Oncogene 14: 2845–2855. [DOI] [PubMed] [Google Scholar]
- 18.Cui JW, Vecchiarelli-Federico LM, Li YJ, Wang GJ, and Ben-David Y. 2009. Continuous Fli-1 expression plays an essential role in the proliferation and survival of F-MuLV-induced erythroleukemia and human erythroleukemia. Leukemia 23: 1311–1319. [DOI] [PubMed] [Google Scholar]
- 19.Delattre O, Zucman J, Plougastel B, Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau G, and et al. 1992. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 359: 162–165. [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Eddy A, Teng YT, Fritzler M, Kluppel M, Melet F, and Bernstein A. 1995. An immunological renal disease in transgenic mice that overexpress Fli-1, a member of the ets family of transcription factor genes. Mol Cell Biol 15: 6961–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Noda S, Asano Y, Nishimura S, Taniguchi T, Fujiu K, Manabe I, Nakamura K, Yamashita T, Saigusa R, Akamata K, Takahashi T, Ichimura Y, Toyama T, Tsuruta D, Trojanowska M, Nagai R, and Sato S. 2014. Simultaneous downregulation of KLF5 and Fli1 is a key feature underlying systemic sclerosis. Nature communications 5: 5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li P, Zhou Y, Goodwin AJ, Cook JA, Halushka PV, Zhang XK, Wilson CL, Schnapp LM, Zingarelli B, and Fan H. 2018. Fli-1 Governs Pericyte Dysfunction in a Murine Model of Sepsis. J Infect Dis 218: 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sementchenko VI, and Watson DK. 2000. Ets target genes: past, present and future. Oncogene 19: 6533–6548. [DOI] [PubMed] [Google Scholar]
- 24.Karim FD, Urness LD, Thummel CS, Klemsz MJ, McKercher SR, Celada A, Van Beveren C, Maki RA, Gunther CV, Nye JA, and et al. 1990. The ETS-domain: a new DNA-binding motif that recognizes a purine-rich core DNA sequence. Genes & development 4: 1451–1453. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Luo H, Liu T, Zacksenhaus E, and Ben-David Y. 2015. The ets transcription factor Fli-1 in development, cancer and disease. Oncogene 34: 2022–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lennard Richard ML, Brandon D, Lou N, Sato S, Caldwell T, Nowling TK, Gilkeson G, and Zhang XK. 2016. Acetylation impacts Fli-1-driven regulation of granulocyte colony stimulating factor. European journal of immunology 46: 2322–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lennard Richard ML, Nowling TK, Brandon D, Watson DK, and Zhang XK. 2015. Fli-1 controls transcription from the MCP-1 gene promoter, which may provide a novel mechanism for chemokine and cytokine activation. Molecular immunology 63: 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lennard Richard ML, Sato S, Suzuki E, Williams S, Nowling TK, and Zhang XK. 2014. The Fli-1 Transcription Factor Regulates the Expression of CCL5/RANTES. Journal of immunology 193: 2661–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sato S, Lennard Richard M, Brandon D, Jones Buie JN, Oates JC, Gilkeson GS, and Zhang XK. 2014. A critical role of the transcription factor fli-1 in murine lupus development by regulation of interleukin-6 expression. Arthritis & rheumatology 66: 3436–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki E, Karam E, Williams S, Watson DK, Gilkeson G, and Zhang XK. 2012. Fli-1 transcription factor affects glomerulonephritis development by regulating expression of monocyte chemoattractant protein-1 in endothelial cells in the kidney. Clinical immunology 145: 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato S, Zhang XK, Temmoku J, Fujita Y, Matsuoka N, Yashiro-Furuya M, Asano T, Kobayashi H, Watanabe H, and Migita K. 2020. Ets Family Transcription Factor Fli-1 Promotes Leukocyte Recruitment and Production of IL-17A in the MRL/Lpr Mouse Model of Lupus Nephritis. Cells 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lou N, Lennard Richard ML, Yu J, Kindy M, and Zhang XK. 2017. The Fli-1 transcription factor is a critical regulator for controlling the expression of chemokine C-X-C motif ligand 2 (CXCL2). Molecular immunology 81: 59–66. [DOI] [PubMed] [Google Scholar]
- 33.Zhang XK, Gallant S, Molano I, Moussa OM, Ruiz P, Spyropoulos DD, Watson DK, and Gilkeson G. 2004. Decreased expression of the Ets family transcription factor Fli-1 markedly prolongs survival and significantly reduces renal disease in MRL/lpr mice. Journal of immunology 173: 6481–6489. [DOI] [PubMed] [Google Scholar]
- 34.Mathenia J, Reyes-Cortes E, Williams S, Molano I, Ruiz P, Watson DK, Gilkeson GS, and Zhang XK. 2010. Impact of Fli-1 transcription factor on autoantibody and lupus nephritis in NZM2410 mice. Clinical and experimental immunology 162: 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, and Werner T. 2005. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics (Oxford, England) 21: 2933–2942. [DOI] [PubMed] [Google Scholar]
- 36.Cockerill PN, Bert AG, Roberts D, and Vadas MA. 1999. The human granulocyte-macrophage colony-stimulating factor gene is autonomously regulated in vivo by an inducible tissue-specific enhancer. Proceedings of the National Academy of Sciences of the United States of America 96: 15097–15102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asano Y, Czuwara J, and Trojanowska M. 2007. Transforming growth factor-beta regulates DNA binding activity of transcription factor Fli1 by p300/CREB-binding protein-associated factor-dependent acetylation. The Journal of biological chemistry 282: 34672–34683. [DOI] [PubMed] [Google Scholar]
- 38.Asano Y, and Trojanowska M. 2009. Phosphorylation of Fli1 at threonine 312 by protein kinase C delta promotes its interaction with p300/CREB-binding protein-associated factor and subsequent acetylation in response to transforming growth factor beta. Molecular and cellular biology 29: 1882–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ping D, Boekhoudt G, Zhang F, Morris A, Philipsen S, Warren ST, and Boss JM. 2000. Sp1 binding is critical for promoter assembly and activation of the MCP-1 gene by tumor necrosis factor. The Journal of biological chemistry 275: 1708–1714. [DOI] [PubMed] [Google Scholar]
- 40.Asano Y, Stawski L, Hant F, Highland K, Silver R, Szalai G, Watson DK, and Trojanowska M. 2010. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. The American journal of pathology 176: 1983–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sawada S, Scarborough JD, Killeen N, and Littman DR. 1994. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell 77: 917–929. [DOI] [PubMed] [Google Scholar]
- 42.Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, Chang HD, Radbruch A, and Zielinski CE. 2014. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Science translational medicine 6: 241ra280. [DOI] [PubMed] [Google Scholar]
- 43.Czuwara-Ladykowska J, Shirasaki F, Jackers P, Watson DK, and Trojanowska M. 2001. Fli-1 inhibits collagen type I production in dermal fibroblasts via an Sp1-dependent pathway. The Journal of biological chemistry 276: 20839–20848. [DOI] [PubMed] [Google Scholar]
- 44.Yang TC, Chang PY, Kuo TL, and Lu SC. 2017. Electronegative L5-LDL induces the production of G-CSF and GM-CSF in human macrophages through LOX-1 involving NF-kappaB and ERK2 activation. Atherosclerosis 267: 1–9. [DOI] [PubMed] [Google Scholar]
- 45.Li YJ, Zhao X, Vecchiarelli-Federico LM, Li Y, Datti A, Cheng Y, and Ben-David Y. 2012. Drug-mediated inhibition of Fli-1 for the treatment of leukemia. Blood cancer journal 2: e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li P, Goodwin AJ, Cook JA, Halushka PV, Zhang XK, and Fan H. 2019. Fli-1 transcription factor regulates the expression of caspase-1 in lung pericytes. Molecular immunology 108: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lennard Richard ML, Brandon D, Ning Lou N, Sato S, Caldwell T, Nowling TK, Gilkeson G, and Zhang XK. 2016. Acetylation impacts Fli-1-driven regulation of granulocyte colony stimulating factor. European journal of immunology 46: 2322–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lou N, Lennard Richard ML, Yu J, Kindy M, and Zhang XK. 2016. The Fli-1 transcription factor is a critical regulator for controlling the expression of chemokine C-X-C motif ligand 2 (CXCL2). Molecular immunology 81: 59–66. [DOI] [PubMed] [Google Scholar]
- 49.Li T, Bai L, Li J, Igarashi S, and Ghishan FK. 2008. Sp1 is required for glucose-induced transcriptional regulation of mouse vesicular glutamate transporter 2 gene. Gastroenterology 134: 1994–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, Lakshmanan M, Raju A, Tenen DG, Cheng SY, Chuang KH, Li J, Prabhakar S, Li M, and Tergaonkar V. 2015. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol 17: 1327–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.