

Abstract
Emerging evidence is demonstrating the extent of T-cell infiltration within the tumor microenvironment has favorable prognostic and therapeutic implications. Hence, immunotherapeutic strategies that augment the T-cell signature of tumors hold promising therapeutic potential. Recently, immunotherapy based on intratumoral injection of mannan-BAM, toll-like receptor ligands and anti-CD40 antibody (MBTA) demonstrated promising potential to modulate the immune phenotype of injected tumors. The strategy promotes the phagocytosis of tumor cells to facilitate the recognition of tumor antigens and induce a tumor-specific adaptive immune response. Using a syngeneic colon carcinoma model, we demonstrate MBTA’s potential to augment CD8+ T-cell tumor infiltrate when administered intratumorally or subcutaneously as part of a whole tumor cell vaccine. Both immunotherapeutic strategies proved effective at controlling tumor growth, prolonged survival and induced immunological memory against the parental cell line. Collectively, our investigation demonstrates MBTA’s potential to trigger a potent anti-tumor immune response.
Keywords: immunogenic tumor cell vaccine, adjuvant, TLR, irradiation
Table of Contents
Numerous investigations are demonstrating the extent of T-cell inflammation within the tumor microenvironment may predict tumor responsiveness to immunotherapy. Hence, immunotherapeutic strategies that augment the T-cell signature of tumors hold promising therapeutic potential. In this study, vaccination with irradiated tumor cells mixed with amphiphilic phagocytic agonists and immunostimulatory adjuvants effectively trigger a potent anti-tumor immune response.
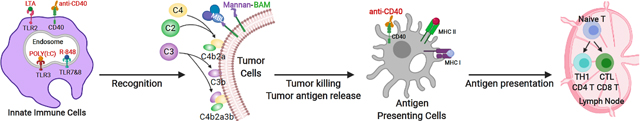
1. Introduction
There is growing recognition that the effectiveness of cancer immunotherapies depends on the presence of a pre-existing immune response and in the ability to harness its full potential.[1–3] This understanding has served as the impetus for designing new cancer classifications based on the type, density and location of immune cells within the tumor, a concept known as the “immune contexture” of tumors.[3,4] Within this context, the extent of T-cell infiltration within the tumor microenvironment and their functional characteristics takes center stage. Tumors with more T-cell inflamed and activated phenotypes are associated with better response to immunotherapeutic treatments and improved survival.[3–7] As such, immunotherapeutic strategies that augment the T-cell infiltration of tumors and induce more favorable immunologic contexture signatures hold promising therapeutic potential.
Recently, a promising immunotherapeutic strategy consisting of a combination of mannan, a polysaccharide derived from Saccharomyces cerevisiae, toll-like receptor (TLR) ligands and agonistic anti-CD40-monoclonal antibody (abbreviated as MBTA) demonstrated potent antitumor responses in several murine cancer models when injected intratumorally (in situ).[8,9] Mechanistically, mannan serves as a phagocytosis-stimulating ligand when conjugated to Biocompatible Anchor for Cell Membrane (BAM).[10,11] The linkage of mannan to BAM (Mannan-BAM) facilitates anchoring of mannan to cell membranes via BAM’s hydrophobic oleyl group and subsequently exploits mannan recognition by pattern recognition receptors, leading to complement activation and opsonophagocytosis of tumor cells.[11, 12]
To bolster mannan-BAM’s inductive effect on innate immune cells, three TLR ligands (lipoteichoic acid (LTA), polyinosinic-polycytidylic acid (poly(I:C)) and resiquimod (R-848)) and immunostimulatory anti-CD40-mAb were incorporated as adjuvants.[8,9,11,13–15] LTA from Bacillus subtilis activates TLR2 mediated inflammatory pathways known to increase TNFα secretion.[16, 17] Polyinosinic-polycytidylic acid (poly(I:C)), a synthetic analog of viral dsRNA, activates TLR3 mediated signaling previously demonstrated to activate antigen-presenting cells (APCs), modulate the phenotype of tumor associated macrophages to more immunosupportive phenotypes, and induce tumor cell apoptosis.[17, 18] Resiquimod (R-848), an imidazoquinolinamine and synthetic analog of viral ssRNA activates TLR7/8 pathways in humans (TLR7 pathway in mice), resulting in activation of innate immune cells and the induction of Th1 cell-mediated immunity.[17,19, 20] Lastly, agonistic anti-CD40-mAb is introduced to mimic the natural ligand of CD40 on T helper cells and APCs, including dendritic cells (DC), B cells, and monocytes. CD40 ligation with anti-CD40-mAb results in APC activation and induction of adaptive immunity.[13,21–23]
Prior MBTA studies demonstrated striking efficacy in regressing primary tumors, injected with MBTA.[8,9] However, the efficacy and characterization of the immune response generated against distal, untreated tumors was not explored. Additionally, previous experimental mouse models of MBTA have relied on the in-situ injection of MBTA to generate an antitumor immune response.[8,9] Such therapeutic delivery strategy has demonstrated efficacy in improving tumor growth control and overall mouse survival. However, a significant limitation of the in-situ injection strategy is its reliance on an anatomically accessible tumor to inject MBTA. Given that primary tumors are commonly surgically removed in the clinical setting or may be located in poorly accessible anatomical locations, we sought to develop an alternative form of therapeutic delivery that does not depend on the intratumoral injection of MBTA. Considering the amphiphilic biochemical properties of mannan-BAM, and previous pre-clinical[24–26] and clinical[27–29] studies demonstrating promising antitumor effects of irradiated whole tumor cell vaccines, we assessed if an irradiated whole tumor cell vaccine, pulsed with MBTA, would generate a similar therapeutic effect.
In the present study, we demonstrated that MBTA injection can induce an adaptive immune response to a distally untreated tumor using a murine colon carcinoma model. We also showed that MBTA could be delivered in combination with whole tumor cells to create an effective vaccine against established tumors.
2. Results
2.1. MBTA elicits rejection of primary and metastatic (distal) CT26 tumors
To test the hypothesis that local intratumoral injection of MBTA could generate an adaptive immune response against distal tumors, we used CT26, a murine colon carcinoma cell line (Figure. 1). First, we established a subcutaneous tumor model by inoculating CT26 tumor cells (5.5 × 105) in the right flank, creating a representative primary tumor, while a distal representative metastatic tumor was established by inoculating CT26 tumor cells (2.5 × 105) in the left flank at the same time (Figure. 2A and Figure S1, Supporting Information). After 10 days, mice bearing left flank tumors (average left flank tumor volume - 31.3 mm3) were randomized into two treatment arms: normal saline (control) and in-situ injection of MBTA into right flank tumors. Treatments were administered every day for 3 days then repeated weekly for a total of 4 weeks (Figure. 2B). Tumor growth was assessed twice a week until survival end point.
Figure 1: MBTA Immunotherapy.

The linkage of mannan to biocompatible anchor for cell membrane (BAM) facilitates the anchoring of mannan to the lipid bilayer of tumor cells via BAM’s hydrophobic oleyl group. Intratumoral injection of mannan-BAM exploits mannan recognition by pattern recognition receptors (MBL), leading to complement activation and opsonophagocytosis of tumor cells. To bolster mannan-BAM’s inductive effect on innate immune cells, three TLR ligands (lipoteichoic acid (LTA), polyinosinic-polycytidylic acid (poly(I:C)) and resiquimod (R-848)) and immunostimulatory anti-CD40-mAb are incorporated as immunological adjuvants. Activation of innate immune cells with TLR agonists generates chemokines and inflammatory cytokines that promote APC maturation and tumor antigen processing. CD40, a tumor necrosis factor receptor, is expressed on T helper cells and APC’s, including dendritic cells (DC), B cells (not shown), and monocytes. CD40 ligation with anti-CD40-mAb results in APC activation and induction of adaptive immunity.
Figure 2: In-situ MBTA injection at representative primary tumors suppresses distant tumor growth via a T-cell dependent immune response.

A) BALB/c female mice were injected into right flank tumors (in-situ) with either normal saline (control) or with MBTA. Representative pictures were taken 10 days after the start of treatment. Additional representative pictures are presented in Figure. S1 (Supporting Information); B) Treatment Schema: Mice were inoculated with CT26 cells subcutaneously over the right and left flanks, respectively. After 10 days, mice were randomized into two treatment groups: in-situ injection of either normal saline (control) or MBTA. Treatments were injected into right flank tumors three consecutive days per week for four weeks (12 dosages). Experiment was performed twice; C & D) Tumor growth curves: Tumor volumes over time of in-situ saline (black; n=7) and MBTA treated (pink; n=7) mice; E) Cumulative survival of mice over time. ***p = 0.0002 by Log-rank (Mantel-Cox) test; F & G) Tumor growth curves: Tumor volumes over time of saline (black; n=9), CD8+ Depleted (turquoise; n=5), CD4+ Depleted (purple; n=5) and non-T-cell depleted (pink; n=9) in-situ MBTA treated mice. H) Cumulative survival of mice over time by Log-rank (Mantel-Cox) test with Tukey-Kramer post hoc test. Saline vs. MBTA Treatment (***p = 0.0002); saline vs. CD4+ depletion (p > 0.05); saline vs. CD8+ depletion (p > 0.05).
Four days after the start of treatment, a Mann-Whitney U test revealed a statistically significant difference in both right and left flank tumor volumes between in-situ MBTA injected mice versus saline treated mice (median tumor volumes ± SEM of in-situ MBTA vs saline treated mice: right flank treated tumors - 74.4 ± 8.8 vs 150.2 ± 28.1 mm3, p = 0.0041; left flank distant tumors - 39.7 ± 5.3 vs 111.0 ± 11.5 mm3, p = 0.0006) (Figure. 2C & D, Figure S2 and Table S3A & B, Supporting Information). Additionally, in-situ MBTA injected mice demonstrated a significantly improved survival when compared to saline treated mice (median survival ± SEM of in-situ MBTA vs saline treated mice: 67 ± 11.7 vs. 18 ± 1.2 days, p = 0.0002 by Log-Rank test) (Figure. 2E). Of note, 2/7 (28.6%) MBTA treated mice achieved complete regression (CR) for the duration of the study. These results suggest that in-situ injection of MBTA into right flank tumors suppressed distal tumor growths.
2.2. Effect of MBTA on distal tumors is dependent on adaptive immunity
CT26 tumor-bearing mice were subjected to CD4+ or CD8+ T-cell ablation to assess if the observed antitumor effect is a T-cell mediated process. Depletion antibodies were administered both prior to and during treatment. T-cell depletion was confirmed via flow cytometry 16 days after initiating treatment (Figure. S4, Supporting Information). CD4+ and CD8+ T-cell depleted mice were subjected to the same in-situ MBTA treatment schedule as above.
Ten days after the start of treatment, a Kruskal-Wallis test revealed a statistically significant difference in both right and left flank tumor volumes (p = 0.0019 and 0.0057, respectively, Table S5A and B) corresponding to the following four treatment groups: non-T-cell depleted in-situ MBTA treated mice, CD4+ depleted in-situ MBTA treated mice, CD8+ depleted in-situ MBTA treated mice, and saline treated mice (Figure. 2F & G). A post hoc analysis of median tumor volumes on treatment day 10 revealed that there was no significant difference in right flank treated tumor volumes between CD4+ depleted in-situ MBTA treated mice when compared to the right flank treated tumor volumes of non-T-cell depleted in-situ MBTA treated mice (Figure. 2F and Table S5A, Supporting Information) (median tumor volumes ± SEM: CD4+ depleted vs non-T-cell depleted in-situ MBTA treated mice – 199.6 ± 71.3 vs 136.9 ± 19.8 mm3, Bonferroni adjusted p > 0.05). In contrast, right flank tumor volumes of both CD8+ depleted in-situ MBTA treated mice and saline treated control mice were significantly larger when compared to right flank tumors of non-T-cell depleted in-situ MBTA treated mice (median tumor volumes ± SEM: CD8+ depleted vs non-T-Cell depleted in-situ MBTA treated mice – 222.4 ± 31.9 vs 136.9 ± 19.8 mm3, Bonferroni adjusted p = 0.0420; saline treated vs non-T-cell depleted in-situ MBTA treated mice – 595.4 ± 133.6 vs 136.9 ± 19.8 mm3, Bonferroni adjusted p = 0.0111). Collectively, right flank tumor growth data suggests that CD8+ T-cells significantly affect tumor growth control of right flank tumors.
A post hoc analysis of median tumor volumes on treatment day 10 also revealed that there was no significant difference in left flank distant tumor volumes between CD4+ or CD8+ depleted in-situ MBTA treated mice when compared to the left flank distant tumor volumes of saline treated control mice (Figure. 2G and Table S5B, Supporting Information) (median tumor volumes ± SEM: CD4+ depleted in-situ MBTA treated vs saline treated mice – 448.7 ± 291.1 vs 607.4 ± 121.1 mm3, Bonferroni adjusted p > 0.05; CD8+ depleted in-situ MBTA treated vs saline treated mice - 582.5 ± 75.6 vs 607.4 ± 121.1 mm3, Bonferroni adjusted p > 0.05). In contrast, left flank distant tumor volumes of non-T-cell depleted in-situ MBTA treated mice were significantly smaller when compared to the left flank distant tumor volumes of saline treated control mice (median tumor volumes ± SEM: non-T-cell depleted in-situ MBTA treated vs saline treated mice – 158.7 ± 72.5 vs 607.4 ± 121.1 mm3, Bonferroni adjusted p = 0.0074). Left flank tumor growth data suggests that CD4+ and CD8+ T-cells significantly affect tumor growth control of distal left flank tumors.
In addition to differences in tumor growth, T-cell depletion studies also revealed differences in survival between the aforementioned 4 treatment groups (Log-Rank test p = 0.0003). Both CD4+ and CD8+ depleted in-situ MBTA treated mice demonstrated no significant improvement in survival when compared to saline treated control mice (median survival ± SEM of CD4+ depleted in-situ MBTA vs saline treated mice: 21 ± 3.2 vs 17 ± 0.9 days, Tukey-Kramer adjusted p > 0.05; CD8+ depleted in-situ MBTA vs saline treated mice: 17 ± 0.6 vs 17 ± 0.9 days, Tukey-Kramer adjusted p > 0.05) (Figure. 2H). In contrast, non-T-cell depleted in-situ MBTA treated mice demonstrated a significant improvement in survival when compared to saline treated control mice (median survival ± SEM of non-T-cell depleted in-situ MBTA treated mice vs saline treated mice: 28 ± 2.4 vs 17 ± 0.9, Tukey-Kramer adjusted p = 0.0002). Collectively, CD4+ and CD8+ T-cell depletion data suggests MBTA’s therapeutic efficacy at distal, left flank tumors is dependent on T-cells.
When compared to the median survival of in-situ MBTA injected mice in Section 2.1, mice in the non-T-cell depleted in-situ MBTA injected treatment arm demonstrated a decreased median survival time (median survival ± SEM of in-situ MBTA injected mice in Section 2.1 vs non-T-cell depleted in-situ MBTA injected mice: 67 ± 11.7 vs 28 ± 2.4 days). The difference in survival time can be attributed to the experimental design of each experiment. Animals corresponding to Results Section 2.1 were subcutaneously inoculated with 2.5 × 105 CT26 tumor cells in the left flank to establish a distant (representative metastatic) tumor, while animals in this experiment were inoculated with 5.0 × 105 CT26 tumor cells. At the start of treatment, animals in this section had developed significantly larger left flank distal tumors compared to those in Section 2.1 and therefore reached the study end point substantially faster. Nevertheless, despite the aforementioned differences in the number of CT26 cells used to establish left flank tumors, both experiments independently confirmed that in-situ treatment with MBTA into right flank tumors significantly reduced the tumor growth of distal left flank tumors and significantly improved the median survival of MBTA treated mice when compared to saline treated control mice.
2.3. MBTA stimulates the innate and adaptive immune systems to elicit rejection of primary and metastatic CT26 tumors
Immunophenotyping (I.P.) of tumors were performed to further assess the immune profile of the tumor microenvironment using the subcutaneous tumor model. Both right and left flank tumors were harvested 10 days and 16 days after the start of MBTA treatment. The timing of the I.P. analyses in this study deserves specific explanation. To elucidate temporal changes in immune cell populations, I.P. analyses were completed at two distinct time points (Figure. 3A). Specifically, the first timepoint was carried out 10 days after first treatment and within 6 hours following in-situ injection of MBTA. Day 10 was chosen to coincide with the final injection of the 2nd set of treatments and would provide insight into the acute changes of immune cell populations immediately following treatment. The second time point was carried out 6 days later, on day 16, to assess interim changes in immune cell composition.
Figure 3: Enhanced innate immunity against CT26 tumors after MBTA treatment.

A) Timing of tumor immunophenotyping (I.P.) experiments: I.P. experiments were completed on days 10 and 16 from the start of treatment to assess for immune cell populations at right and left flank tumors. Notably, I.P. Day 10 was completed 6 hours after in-situ injection of MBTA or saline (control) at the right flank tumors, whereas I.P. Day 16 was completed 6 days later to facilitate assessment of interim changes in immune cell populations; B & C) Percentage of CD45+ cells in total live tumor dissociated cells. I.P. Day 10 and I.P. 16 analyses demonstrate MBTA treated mice (pink) had significantly higher immune cells within both right and left flank tumors than saline-treated control mice (black). ** p < 0.01 by Mann-Whitney U test; D & E) Percentage of innate immune cells in total live tumor dissociated cells. Innate immune cells found within right and left flank tumors corresponding to I.P. Day 10 and 16 analyses are shown. Data is shown as individual data plots with median (line). *p < 0.05, ** p < 0.01 by Mann-Whitney U test. Example gating strategy for each cell population is demonstrated in Figure. S11 (Supporting Information). A total of 20 mice were used to complete immunophenotyping (I.P.) experiments. These were allocated to the following treatment groups: saline I.P. Day 10 (n=5); MBTA treatment I.P. Day 10 (n=5); saline I.P. Day 16 (n=5); MBTA treatment I.P. Day 16 (n=5).
Results from both I.P. (Day 10) and I.P. (Day 16) analyses revealed that in-situ injection of MBTA into right flank tumors significantly increased the infiltration of immune cells (CD45+ cells) into both right and left flank tumors of MBTA treated mice compared to saline treated (control) mice (Figure. 3B & C). Assessment of the innate leukocyte subpopulations in right flank tumors on I.P. (Day 10) revealed a striking dendritic (CD45.2+CD11c+MHCII+) and neutrophilic (CD45.2+ CD11c-CD11b+ Ly6G+) inflammatory response in MBTA treated mice versus control mice (Figure. 3D). Six days later, results from I.P. (Day 16) analysis of right flank tumors revealed a significant decrease in neutrophils and a significant increase in antigen presenting cells (APCs), including dendritic cells and MHC class II positive monocytes (CD45.2+ CD11c-CD11b+Ly6G-Ly6C high MHC II+) (Figure. 3D and Figure. S6A). Additionally, assessments of the innate immune cell populations at distal, non-treated left flank tumors similarly revealed an increased, though not significant neutrophilic immune response on I.P. (Day 10), followed by a significant increase in APCs (dendritic cells and MHC class II positive monocytes) on I.P. (Day 16) in tumors of MBTA treated mice versus control (Figure. 3E and Figure. S6B). These results suggest MBTA therapy facilitates neutrophil and APC trafficking to tumors, thereby promoting phagocytosis of tumor cells and processing for the further development of tumor-specific adaptive immune responses.
Although no significant changes were appreciated in the overall quantity of macrophages (CD45.2+CD11c-CD11b+ Ly6G- Ly6C-/low) (Figure. 3D & E), an assessment of the CD206 positive alternatively activated macrophage (AAM) population, also known as M2-macrophages (CD45.2+ CD11c-CD11b+ Ly6G- Ly6C-/low CD206+), demonstrated that the percentage of AAM was significantly decreased in primary (MBTA treated) tumors on both I.P. (Day 10) and I.P. (Day 16) analyses (Figure. S6C, Supporting Information). Similarly, distal tumors from the MBTA treatment arm also revealed a significant decrease in the percentage of AAM on I.P. (Day 10) (Figure. S6D, Supporting Information). This finding is consistent with previous investigations demonstrating that TLR agonist and anti-CD40 antibody can skew the polarization of tumor associated macrophages (TAM) towards an M1-like phenotype.30
Results from I.P. (Day 10) and I.P. (Day 16) analyses also revealed an overall increase in adaptive immune cells found at both right and left flank tumors of MBTA treated mice compared to saline treated mice (Figure. 4A & B). At right flank treated tumors, I.P. (Day 16) analysis revealed that MBTA treated mice demonstrated a significant increase in CD8+ T-cells (CD45.2+TCRβ+CD4-CD8+) and B-cells (CD45.2+TCRβ-CD19+) (Figure. 4A). In contrast, at left flank distant tumors, results from I.P. (Day 10) and I.P. (Day 16) analyses revealed that both CD8+ T-cell and B cell populations were significantly increased in tumors extracted from MBTA treated mice compared to saline treated mice (Figure. 4B). These results suggest that in-situ injection of MBTA at right flank tumors augments the overall quantity of adaptive immune cells to distal, non-treated left flank tumors. Additionally, these results underscore MBTA’s therapeutic potential to modulate the T-cell inflammatory response at representative metastatic lesions.
Figure 4: Enhanced adaptive immunity against CT26 tumors after MBTA treatment.

A & B) Percentage of adaptive immune cells in total live tumor dissociated cells. Adaptive immune cells found within right and left flank tumors corresponding to I.P. Day 10 and 16 analyses are shown; C) Percentage of IFNγ or TNFα positive CD4+ or CD8+ T-cells per total CD4+ or CD8+ T-cells, respectively. Harvested left flank CD4+ and CD8+ T-cells corresponding to Day 10 and 16 analyses were stimulated for 5 h with PMA/Ionomycin ex-vivo and intracellular IFNγ and TNFα was determined by flow cytometry; D & E) Percentage of AH-1-specific CD8+ T-cells extracted from whole blood and left flank tumors, respectively. Saline-treated control mice (black); in-situ MBTA treated mice (pink); Data is shown as individual plots with median. *p < 0.05, ** p < 0.01 by Mann-Whitney U test. Example gating strategy for each cell population is demonstrated in Figure. S11–13 (Supporting Information). A total of 20 mice were used to complete immunophenotyping (I.P.) experiments. These were allocated to the following treatment groups: Saline I.P. Day 10 (n=5); MBTA Treatment I.P. Day 10 (n=5); Saline I.P. Day 16 (n=5); MBTA Treatment I.P. Day 16 (n=5); For blood tetramer experiments, 5 mice were in the saline treatment arm and 6 mice were in the MBTA treatment arm. For tumor tetramer experiments, 4 mice were in the saline treatment arm and 5 mice were in the MBTA treatment arm.
Cytokine secretion analyses of CD4+ (CD45.2+TCRβ+CD4+CD8-) and CD8+ T-cell populations extracted from left flank distant tumors were also completed to assess the functional effect of in-situ injection of MBTA on T-cells. Upon extraction, T-cells were stimulated with PMA/Ionomycin in vitro and assessed for intracellular expression of IFNγ, TNFα and Granzyme B. T-cells harvested from MBTA treated mice on I.P. (Day 10) and (Day 16) demonstrated higher production of IFNγ and TNFα (Figure. 4C). Notably, CD8+ T-cells demonstrated a significant increase in IFNγ on I.P. (Day 10) and a significant increase in TNFα during the six-day interval between I.P. (Day 10) and (Day 16). We also noticed increased Granzyme B positive CD8+ T cells, however, the increase did not reach statistical significance (Figure. S6E, Supporting Information). These results reveal that MBTA treatment significantly augmented the quantity and activation of CD8+ T-cells within left flank distant tumors, suggesting their major role in MBTA-induced tumor growth inhibition.
To assess if MBTA therapy generated higher quantities of CD8+ T-cells (CTLs) against the immunodominant epitope of CT26, gp70423–431 (AH-1)[31], AH-1 loaded H-2Ld tetramer was used. We asked if in-situ MBTA treated mice had a higher quantity of AH-1/H-2Ld-specific CD8+ T-cells relative to saline treated (control) mice. Peripheral blood samples and left flank tumors of in-situ MBTA treated and control mice were collected 11 days after the start of treatment for analysis. In-situ MBTA treated mice had a significantly higher percentage of AH-1/ H-2Ld-specific CD8+ T-cells in whole blood relative to control mice (p = 0.02 by Mann-Whitney U test, Figure. 4D). In-situ MBTA treated mice also demonstrated a trend of increase in the percentage of AH-1/H-2Ld-specific CD8+ T-cells within left flank tumors relative to control mice (p = 0.06 by Mann-Whitney U test, Figure. 4E). These results suggest that in-situ MBTA injections increase the AH-1-specific CD8+ T-cell clones circulating in blood and in representative metastatic left flank tumors within 11 days from the start of MBTA treatment.
Taken together, in-situ injection of MBTA resulted in significant changes in the immunophenotypes of representative primary and metastatic tumors. At representative primary right flank tumors, MBTA treatment generated an innate inflammatory response that was first characterized by a strong neutrophilic and dendritic cell predominance and later followed by a significant increase in APC populations (MHC II+ monocytes and dendritic cells). At representative metastatic left flank tumors, MBTA treatment strengthened the adaptive immune response by increasing the overall supply of CD8+ T-cells and B-cells infiltrating left flank tumors. T-cells extracted from left flank tumors were not only higher in quantity, but also demonstrated higher expressions of IFNγ and TNFα cytokines, validating their enhanced tumoricidal activity. Collectively, these findings are consistent with the observation that in-situ MBTA treatment elicits tumor rejection of representative metastatic tumors.
2.4. Irradiated whole tumor cell-MBTA vaccine generates potent antitumor immunity
Aforementioned MBTA experiments have relied on the presence of a pre-established tumor at the right flank for in-situ injection of MBTA. This therapeutic method has indeed shown efficacy in slowing tumor growth, prolonging survival and achieving complete regression in a subset of treated animals. However, recognizing that primary tumors are commonly debulked surgically in the clinical setting or may be nestled in poorly accessible anatomical locations, we sought to develop an alternative form of therapeutic delivery that does not depend on the presence of a pre-established primary tumor to render therapeutic efficacy at distal, non-treated tumors.
Given mannan-BAM’s efficacy in anchoring into tumor cell membrane, we assessed if subcutaneous vaccinations consisting of sublethally irradiated autologous whole tumor cells (Figure S7, Supporting Information) pulsed with MBTA in-vitro would render a similar therapeutic effect as the in-situ MBTA injection strategy (Figure. 5A). To address this question, we inoculated mice with CT26 cells (2.5 × 105) at the left flank. After 11 days, mice bearing tumors (average tumor volume – 42.4 mm3) were randomized into 3 treatment arms – normal saline (control), radiated CT26 cells (rCT26) or rCT26-MBTA vaccine. In accordance with the previously described therapeutic schedule, mice received a total of 12 vaccines according to their respective treatment group (Figure. 5B). Vaccines were delivered subcutaneously into the right flank and tumor growth was assessed twice a week until survival end point.
Figure 5: rCT26-MBTA vaccines generate a potent antitumor immune response in CT26-bearing mice.

A) Development of rCT26-MBTA vaccine: CT26 cells are expanded in vitro (I) and subsequently aliquoted into 1×106 million cells per vaccine dose (II). CT26 cells are sublethally irradiated using a 137Cs MARK I model irradiator to induce tumor cell apoptosis and prevent engraftment of tumor cells at the vaccine site (III). Irradiated tumor cells (III) are subsequently incubated with MBTA (IV) for an hour, facilitating the in-vitro integration of mannan-BAM into tumor cell membranes (V). The therapeutic mixture consisting of irradiated CT26 tumor cells and MBTA is subcutaneously injected into CT26 tumor bearing animals. B) Treatment Schema: For the rCT26-MBTA vaccine experiments, female BALB/c mice were inoculated with CT26 cells subcutaneously at the left flank. After 11 days, mice were randomized into three treatment groups: normal saline (control; n=7), irradiated CT26 cells (rCT26; n=7) or rCT26 pulsed with MBTA vaccines (rCT26-MBTA; n=8). Treatments were injected subcutaneously into the right flank three consecutive times per week for four weeks (12 dosages). Experiment was performed twice; C) Tumor growth curves: saline (black), rCT26 (orange) and rCT26-MBTA (pink) vaccinated mice; D) Cumulative survival of mice over time by Log-rank (Mantel-Cox) test with Tukey-Kramer post hoc test. Saline vs. rCT26-MBTA Treatment (**p = 0.0071); rCT26 vs. rCT26-MBTA Treatment (**p = 0.0034); Saline vs. rCT26 Treatment (p = 0.9846); E & F) Percentage of AH-1-specific CD8+ T-cells extracted from whole blood (E) and flank tumors (F), respectively. normal saline (control, black); rCT26 (orange); rCT26-MBTA (pink). Data is shown as individual plots with median. *p < 0.05, ** p < 0.01 by Kruskal-Wallis test. For blood tetramer experiments, 5 mice were in the saline treatment arm, 5 mice were in the rCT26 treatment arm, and 4 mice were in the rCT26-MBTA treatment arm. For tumor tetramer experiments, 3 mice were in the saline treatment arm, 5 mice were in the rCT26 treatment arm, and 4 mice were in the rCT26-MBTA treatment arm.
After 6 days of treatment, a Kruskal-Wallis test revealed a statistically significant difference in tumor volumes corresponding to the saline treated control, rCT26 and rCT26-MBTA treatment arms (p = 0.0108) (Figure. 5C, Table S8A). A post hoc analysis revealed that rCT26-MBTA vaccinated mice demonstrated significantly smaller median tumor volumes compared to rCT26 or saline vaccinated mice after 6 and 15 days of treatment, respectively (Day 6: median tumor volumes ± SEM of rCT26-MBTA vs rCT26 vaccinated mice – 47.7 ± 19.0 vs 247.1 ± 37.3 mm3, Bonferroni adjusted p = 0.0279; Day 15: median tumor volumes of rCT26-MBTA vs saline vaccinated mice – 67.6 ± 137.7 vs 971.2 ± 320.3 mm3, Bonferroni adjusted p = 0.0420). Of note, no significant difference in tumor volumes were appreciated between rCT26 and saline treatment arms (Bonferroni adjusted p > 0.05).
In addition to improved tumor growth control, mice in the rCT26-MBTA vaccine treatment group also demonstrated a significant increase in survival when compared to saline or rCT26 vaccinated mice (median survival ± SEM of rCT26-MBTA vs saline treated mice: 44 ± 9.2 vs 20 ± 1.9 days, Tukey-Kramer adjusted p = 0.0071; median survival of rCT26-MBTA vs rCT26 vaccinated mice: 44 ± 9.2 vs 20 ± 2.1 days, Tukey-Kramer adjusted p = 0.0034) (Figure. 5D). In contrast, mice in the rCT26 vaccine treatment group demonstrated no significant increase in survival when compared to saline treated mice (median survival ± SEM of rCT26 vs saline treated mice: 20 ± 2.1 vs 20 ± 1.9 days, Tukey-Kramer adjusted p > 0.05). Of note, 1/8 (12.5%) of rCT26-MBTA vaccine treated mice achieved complete remission (CR) for the duration of the study. (Figure. 5D). These results suggest that treatment with a total of 12 rCT26-MBTA vaccines can successfully generate a potent antitumor immune response.
We also evaluated the quantities of AH-1/H-2Ld-specific CD8+ T-cells found in peripheral blood samples and within distal, non-treated flank tumors. Peripheral blood samples were collected 10 days after the start of treatment and after receiving a total of 2 vaccinations. Tumors were harvested 22 days after the start of treatment and after receiving a total of 4 vaccinations which is in accordance with the known timeline for T-cell activation after initial antigen stimulation.[32] rCT26-MBTA vaccine treated mice demonstrated a significantly higher percentage of AH-1/H-2Ld-specific CD8+ T-cells in whole blood relative to saline treated (control) mice (p = 0.0018 by Kruskal-Wallis test, Figure. 5E).
Tumor infiltrating lymphocytes were isolated and analyzed by flow cytometry to assess the presence of AH-1/H-2Ld-specific CD8+ T-cells in tumors. rCT26-MBTA vaccine treated mice similarly had a significantly higher percentage of AH-1/H-2Ld-specific CD8+ T-cells within tumors relative to control (p = 0.0169 by Kruskal-Wallis test, Figure. 5F). Although not statistically significant, rCT26 vaccine treated mice demonstrated a trend of increase in the percentage of AH-1/H-2Ld-specific CD8+ T-cells found in whole blood and within tumors when compared to saline treated mice (p = 0.1642 and 0.6469, respectively, by Kruskal-Wallis test). Collectively, AH-1-MHC tetramer results suggest that a significantly higher quantity of AH-1-specific CD8+ T-cell clones were found in whole blood and within tumors of rCT26-MBTA vaccine treated mice when compared to saline treated control mice, underscoring its potential to generate CD8+ T-cell responses against CT26-specific antigens.
To assess for the general tolerability of MBTA treatments, we compared body weight changes associated with both in-situ MBTA and rCT26-MBTA vaccine treatments. In-situ MBTA treated mice demonstrated a significant acute drop in mean body weight 3 days after the start of treatment when compared to pre-treatment levels (Figure. S9A, Supporting Information). The loss in body weight was recuperated 7 days after the start of treatment. In contrast, mice treated with rCT26-MBTA vaccines demonstrated no significant changes in body weight (Figure. S9B, Supporting Information). Of note, none of the in-situ MBTA or rCT26-MBTA vaccinated mice died unexpectedly in our investigations, suggesting that treatment with MBTA was overall well-tolerated.
2.5. MBTA therapy induces antigen-specific long-term memory
A series of tumor re-challenge experiments were performed to assess if treatment with MBTA induced immunological memory against CT26 tumors. All mice that achieved complete regression (CR, n = 4) of CT26 tumors were challenged after a minimum of 50 days had passed from their last day of treatment. Naïve BALB/c (Control) and MBTA treated mice were inoculated with CT26 cells in the left flank, opposite from the site of initial treatment. All control mice demonstrated development of tumors and reached corresponding end points. In contrast, all CR mice displayed no evidence of tumor growth, confirming the induction of an effective immunological memory against CT26 tumor antigens (Figure. 6A & B and Table S8B, Supporting Information).
Figure 6: MBTA treatment induces immunological memory against CT26 cells.

A) Tumor growth curves: Mice cured of CT26 colon carcinoma were re-challenged subcutaneously with 4.5×105 CT26 cells after 50 days from last treatment day. CT26 Naïve (black, n=4) and MBTA Pre-treated (pink, n=4) mice; B) Cumulative survival of re-challenged mice over time. Naïve vs MBTA pre-treated: **p = 0.0069 by Log-rank (Mantel-Cox) test; C) Representative bioluminescence imaging of intracranially re-challenged mice. Mice cured of CT26 colon carcinoma were also re-challenged intracranially with 1×104 CT26-Luc cells in the right frontal lobe (1 mm rostral of the bregma, 2 mm right of midline and 2 mm deep from the skull surface). MBTA Pre-treated (left, n=4) and Naïve (right, n=4); D) Cumulative survival of intracranially re-challenged mice over time; Naïve vs MBTA Pre-treated mice: *p = 0.0100 by Log-rank (Mantel-Cox) test.
To assess if immunological memory extended beyond peripheral circulation and could engage CT26 tumors within the central nervous system (CNS), we challenged the same MBTA treated mice that achieved CR intracranially. CT26-Luc cells with stable luciferase expression were used in this experiment to facilitate monitoring of intracranial tumor growth. MBTA treated and control mice were inoculated with CT26-Luc cells in the frontal lobe. All control mice developed intracranial tumors and reached corresponding end points while none of the previously treated mice demonstrated evidence of intracranial tumor formation (Figure. 6C & D). Taken together, re-challenge experiments revealed that MBTA treated mice developed immunological memory against CT26 cells that is not only durable, but also sufficiently robust to prevent the relapse of parental tumors in both the periphery and intracranially.
3. Discussion
We investigated the immunotherapeutic potential of MBTA to modulate an antitumor immune response against representative primary and metastatic tumors of the CT26 colon carcinoma cell line. The immunotherapeutic strategy leverages the use of phagocytosis stimulating ligands and immunostimulatory adjuvants for directing an immune response against tumor-specific antigens (TSA). We find that MBTA delivered as an in-situ tumor injection or as part of a whole tumor cell vaccine generates a potent tumor-specific adaptive immune response capable of controlling tumor growth and inducing tumor regression in a subset of representative metastatic tumors. Notably, both forms of MBTA delivery achieve long-lasting immunological memory against the CT26 colon carcinoma cell line that is effective at clearing reappearing CT26-associated antigens in immunized mice.
Previous MBTA investigations demonstrated that in-situ injection of MBTA induced potent innate immune responses against vaccinated tumors.[8,9] More recently, in a pheochromocytoma mouse model, Caisova et al. demonstrated that in-situ injection of MBTA resulted in lower tumor burden of metastatic organ lesions when compared to PBS-treated control mice and significantly prolonged survival.[8] However, a limitation of the study included the lack of assessment of the immune cells infiltrating metastatic organ lesions. Hence, the efficacy and the characterization of the immune response at distant untreated tumors remained to be investigated and was a point of focus in this study.
Our experimental results demonstrated that in-situ injection of MBTA culminated in the induction of a systemic immune response capable of delaying tumor growth and inducing complete tumor regression in a subset of untreated representative metastatic tumors. Moreover, through immunophenotyping (I.P.) analyses of both in-situ MBTA treated tumors and distant untreated tumors, we make several unique observations pertaining to MBTA’s antitumor effect.
First, in-situ injected tumors demonstrate a significant increase in the percentage of infiltrating neutrophils and dendritic cells when compared to saline treated controls. In a second set of I.P. analyses completed 6 days after in-situ MBTA treatment, we found dendritic cells and monocytes became the predominant immune cell population within in-situ vaccinated tumors. Considering that tumors commonly display genomic instability and develop TSA unique to patients,[33,34] the increased trafficking of APC’s to MBTA treated tumors provides a favorable immunological condition for APC processing of TSA and for the subsequent development of tumor-specific adaptive immune responses.
Assessment of the adaptive immune cell populations within in-situ injected and distant untreated tumors provides a second set of observations relating to MBTA’s potential to stimulate adaptive immune responses against representative metastatic tumors. Notably, MBTA treated mice demonstrated a significant increase in CD8+ T-cells and B cells within representative metastatic tumors. Moreover, further investigation into the functional states of the T-cells extracted from representative metastatic tumors revealed that CD8+ T-cells from MBTA treated mice were more cytotoxic as indicated by TNFα and IFNγ expressions. Collectively, these findings validate MBTA’s potential to modulate the immune phenotype of tumors, which may be an important consideration for predicting tumor response to immune check point inhibitors and other immunotherapeutic strategies.[34–36]
The aforementioned experimental results demonstrate the efficacy of in-situ injection of MBTA in generating a systemic antitumor immune response. However, a key limitation in the translational applicability of in-situ treatment with MBTA lies in its reliance on a pre-established tumor to generate its antitumor effect. Considering the wide anatomic variability of tumors encountered in the clinical setting and the fact that many primary tumors are surgically excised, we designed a new therapeutic delivery for MBTA that circumvented the need for in-situ delivery of MBTA. Specifically, we are the first to report that a vaccine composed of irradiated autologous tumor cells pulsed with MBTA (rCT26-MBTA vaccine) induces a potent tumor-specific adaptive immune response.
The design of the rCT26-MBTA vaccine provides several advantages over the in-situ delivery of MBTA. First, similar to the in-situ injection strategy, the rCT26-MBTA vaccine leverages the acquired amphiphilic properties of mannan when linked to BAM. Notably, because colon carcinoma cells are pulsed with MBTA in vitro, the interactions between mannan-BAM and tumor cell membranes are more specific, reducing the potential for off-target interactions between mannan-BAM and non-tumorous cells. Second, it is well established that the tumor microenvironment plays an important role in promoting tumor progression, including precluding effective processing of TSA by APCs.[37–39] In this context, the rCT26-MBTA vaccine provides a more favorable immunological setting for APCs to recognize, process and present tumor-specific antigens to effectors of the adaptive immune system. Lastly, although MBTA-pulsed tumor cells are initially irradiated to prevent tumor outgrowth, tumor cell irradiation itself provides an additional pro-inflammatory component targeted by dendritic cells and promote immunogenic cell death.[40, 41]
Collectively, our investigation into immunotherapy with MBTA underscores its potential to render a potent antitumor immune response against both primary and representative metastatic tumors. The observed antitumor effect can be generated via in-situ injection of MBTA or through a subcutaneous vaccination consisting of irradiated whole tumor cells pulsed with MBTA. Immunotherapy with MBTA addresses two important challenges in cancer immunology, namely, the effective recognition of tumor specific neoantigens and the induction of more favorable T-cell signatures within the tumor microenvironment. Given the current therapeutic limitations for addressing solid tumors and metastatic disease, we see great translational potential for adapting MBTA immunotherapy into the clinical setting. Specifically, we envision MBTA immunotherapy playing a distinct role in addressing primary tumors located in anatomically sensitive or difficult to reach locations and secondly as part of a whole tumor cell vaccine regimen intended to reinforce T-cell migration to distant metastatic lesions.
4. Experimental Section
Experimental Design:
Our first objective was to characterize the abscopal therapeutic effect resulting from intratumoral (in-situ) injection of MBTA. To advance our investigation, we simultaneously implanted syngeneic CT26 tumors at two separate anatomical locations on mice. Tumors located on the right flank served as the site for MBTA injection, while tumors on the left flank remained untreated. Both tumor sites were routinely monitored and measured to assess for tumor growth. Innate and adaptive immune cell populations were analyzed by flow cytometry. Cytotoxicity activities of adaptive immune cells were assessed by cytokine (IFNγ, TNFα and Granzyme B) staining and AH-1-tetramer staining. Mice that achieved complete regression of CT26 tumors were re-challenged with the parental cell line to confirm the development of immunological memory.
Our second objective was to assess if a subcutaneous vaccine composed of irradiated CT26 cells pulsed with MBTA (rCT26-MBTA vaccine) was effective in controlling tumor growth at distant tumors. To advance this investigation, we implanted syngeneic CT26 tumors over the left flank of mice and subcutaneously delivered the rCT26-MBTA vaccine into the right flank. Tumors were routinely monitored and measured to assess for tumor growth. Additionally, each mouse treated with the rCT26-MBTA vaccine was monitored to confirm no tumor development in the right flank. The cytotoxicity activities of CD8+ T-cells in the tumors was assessed by AH1-tetramer staining. Mice that achieved complete regression of CT26 tumors were similarly re-challenged with the parental cell line to confirm the development of immunological memory.
Drugs and Mannan-BAM Synthesis:
Mannan and polyinosinic–polycytidylic acid sodium salt were obtained from Sigma-Aldrich (St. Louis, Mo). Lipoteichoic acid was obtained from Sigma-Aldrich (St. Louis, Mo) and InvivoGen (San Diego, CA). Resiquimod was obtained from Tocris Bioscience (Minneapolis MN). Anti-mouse CD40 (clone: FGK4.5/GFK45) was obtained from BioXCell (West Lebanon, NH). Biocompatible anchor for cell membrane (BAM) was obtained from NOF America Corporation (White Plains, NY).
Mannan-BAM synthesis was performed as previously reported.[9,11,14] Aminated mannan was prepared by reductive amination.[42] Mannan solution in an environment of ammonium acetate (300 mg ml−1) was reduced by 0.2 M sodium cyanoborohydride at pH 7.5 and 50 °C for five days. Solution was further dialyzed using MWCO 3500 dialysis tubing (Serva, Heidelberg, Germany) against PBS at 4 °C overnight. Binding of BAM on amino group of mannan was performed at pH 7.3 according to Kato et al.[10] During one hour at room temperature N-hydroxysuccinimide (NHS) group of BAM reacted with amino group of mannan. Solutions obtained after dialysis as above was stored frozen at −20 0C until use.
Cell-Lines:
CT26 colon carcinoma cells were obtained from ATCC. Tumor cells were cultured in complete medium (RPMI 1640, Gibco) containing 10% (vol/ vol) FBS (Gibco), 100 U ml−1 penicillin, 100 μg ml−1 streptomycin (Gibco). We verified that none of the cell lines used in this study were found in the Register of Misidentified Cell Lines maintained by the International Cell Line Authentication Committee (http://iclac.org/databases/cross-contaminations/). The cell lines were tested and shown to be negative for mycoplasma contamination using PCR amplification. CT26-Luc cells were generated by stable transduction of luciferase-containing lentivirus (EF1a-ffLuc2-eGFP) into naïve CT26 cells.
Syngeneic Tumor Models:
Mice were maintained, and experiments were conducted with the approval of the NCI Animal Use and Care Committees. For CT26 tumors, female BALB/c (6–8-week-old) were purchased from Charles River Laboratory. CT26 cells (2.5 – 5.5 × 105) suspended in 100–200 μL PBS were subcutaneously injected into right or left flanks. Tumor volume was measured twice per week using a caliper and tumor volume was calculated according to the formula: Volume (mm3) = L×W2 /2, where L is the length and W is the width of the tumor (in millimeters).
For the T-cells depletion studies, mice in the CD8+ and CD4+ depletion groups were injected with 250 μg of CD8-depleting antibodies (clone 53–6.7; BioXcell) and CD4-depleting antibodies (clone GK1.5; BioXcell), respectively. Injections were given −2 day, −1 day, and on the day of therapy initiation (day −2, −1, 0) then weekly thereafter.
Sample sizes of animal studies were determined empirically based on previous pilot experiments. Pre-established starting tumor size used as inclusion criteria for randomization was determined by pilot experiments. Investigators performed randomization manually and were not blinded from the group allocation during outcome measurement. Survival end point for all animal studies were defined according to the following criteria: (1) tumor volume exceeding 2000 mm3, (2) tumor diameter exceeding 2 cm, (3) severe non-healing skin necrosis over the tumor.
For tumor re-challenge studies, mice that achieved complete regression of CT26 tumors (n=4) and naïve female BALB/c mice were inoculated with 4.5 × 105 CT26 cells suspended in 100 μl PBS into left flank. For intracranial tumor challenge, both CR (n=4) and naïve female BALB/c mice were inoculated with CT26-Luc cells. Briefly, using a Hamilton syringe (Hamilton Company), 1.0 × 104 CT26-Luc cells in 2 μL HBSS containing 5 ug/mL DNase I were stereotactically injected through an entry site at a point 1 mm rostral of the bregma, 2 mm right of midline, and 2 mm deep from the skull surface in the right frontal lobe, as previously reported[43]. Surgical anesthesia was obtained via vaporized isoflurane.
In-situ MBTA Injections:
For in-situ MBTA injections, 50 μl of the therapeutic mixture consisting of 0.5 mg R-848 (HCl form), 0.5 mg poly(I:C), 0.5 mg LTA, and 20 μg anti-CD40 per ml of 0.2 mM mannan-BAM in PBS was injected intratumorally.
Irradiated whole tumor cell-MBTA vaccine:
For rCT26-MBTA vaccines, 100 μL of the following therapeutic mixture were injected subcutaneously into the right flanks of rCT26-MBTA treated mice: a) 1 × 106 irradiated CT26 cells suspended in 50 μl PBS and b) 50 μl of the therapeutic mixture consisting of 0.5 mg R-848 (HCl form), 0.5 mg poly(I:C), 0.5 mg LTA, and 20 μg anti-CD40 per ml of 0.2 mM mannan-BAM in PBS. The irradiated whole tumor cell (CT26)-MBTA vaccine (rCT26-MBTA vaccine) was generated by first irradiating tumor cells (1 × 106 tumor cells suspended in 50 μl PBS, dose per mouse) with 50 Gy in a 137Cs MARK I model irradiator (JL Shepherd & Associates, San Fernando, CA). CT26 tumor cells were initially irradiated to induce tumor cell apoptosis and prevent tumor outgrowth when used as a component in the rCT26-MBTA vaccine (Figure. S7, Supporting Information). Irradiated cells were then pulsed (incubated) with MBTA (50 μl, dose per mouse) for 1 hour and subsequently injected subcutaneously to the right flank of treated animals according to the specified therapeutic schedule.
Mice receiving the rCT26 vaccine were injected with 1 × 106 irradiated tumor cells suspended in 100 μl PBS (dose per mouse).
Immunophenotyping (I.P) of tumors with flow cytometry:
CT26 tumors were established by injecting 5.0 × 105 CT26 cells to the right and left flanks. Two independent sets of I.P experiments were completed after 10 and 16 days from the start of treatment. Each I.P experiment consisted of control (n = 5) and MBTA treated mice (n = 5). After mice were sacrificed, both right and left flank tumors were excised and subjected to mechanical disruption using a GentleMACS Dissociator (Miltenyi Biotec) in the presence of enzymatic digestion using Tumor Dissociation Kit (Miltenyi Biotec).
Antibodies for flow cytometry staining are listed in Figure. S10, supporting information. Gating strategy used for I.P analyses of tumors was performed as previously reported (Figure. S11–13).[44] Specific immune cell populations were defined as follows: Dendritic cells (CD45.2+CD11c+MHCII+); Macrophages (CD45.2+CD11c-CD11b+Ly6G- Ly6C-/low); Monocytes (CD45.2+CD11c-CD11b+Ly6G-Ly6C high); MHC class II+ Monocytes (CD45.2+CD11c-CD11b+Ly6G-Ly6C high MHCII+); Neutrophils (CD45.2+CD11c-CD11b+Ly6G+); CD4+ T-cells (CD45.2+TCRβ+ CD4+CD8-); CD8+ T-cell (CD45.2+TCRβ+CD4-CD8+); B cells (CD45.2+TCRβ-CD19+).
Intracellular cytokine staining and flow cytometry – Suspensions containing T-cells were stained with a fixable live/dead stain (Invitrogen) in PBS followed by surface antibody staining in FACS buffer (PBS with 0.5% BSA and 0.1% sodium azide). For intracellular staining, cells were first stimulated with Cell Stimulation Cocktail (eBioscience) containing PMA/Ionomycin and protein transport inhibitor for five hours prior to undergoing staining. Next, cells were stained for surface molecules following fixation and permeabilization (eBioscience), and then stained with cytokine antibodies. Stained cells were analyzed by flow cytometry (LSRII; BD Bioscience). Data analysis was performed using FlowJo software (TreeStar).
Statistical Analysis:
Tables S3, S5, and S8 demonstrate tumor growth data for all time points up to the point that all animals within each group were still alive. For each experiment, exact nonparametric methods, Kruskal-Wallis (for levels of treatment factor >2) or Mann-Whitney U test (for levels of treatment factor =2), were performed to evaluate treatment effect on tumor volume at a single timepoint. For the experiment with factor level greater than two, Mann-Whitney U test was applied to multiple comparisons and the exact p-values are adjusted by Bonferroni method. For the T-cell depletion study, right and left sides were accounted in the adjustment. The overall survival was defined as the time (days) from the first day of treatment to survival end point. Animals that survived more than 100 days were censored. Survival curves were drawn using Kaplan-Meier method and were compared using log-rank test. The Tukey-Kramer method was used for pairwise multiple comparisons.
The statistical analyses were performed using SAS version 9.4 and GraphPad Prism. The reported p-values were adjusted for multiple comparison within each experiment, but not adjusted for multiple experiments. P values < 0.05 were regarded as statistically significant.
Supplementary Material
Acknowledgements
We thank the NIH Tetramer Facility (Emory University, Atlanta, GA) for providing the AH-1 tetramer used in this study. We also thank the members of the NCI CCR Flow Cytometry Core for expertise with flow cytometry.
Funding: The research was supported by the Intramural Research Program of the National Institute of Neurological Diseases and Stroke, the National Cancer Institute and the National Institute of Child Health and Human Development of the National Institutes of Health. Author R.M. was partly supported by the NIH Medical Research Scholars Program, a public–private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH. For a complete list of donors, please visit http://fnih.org/work/education-training-0/medical-research-scholars-program.
Footnotes
Data and materials availability: The authors declare that all the other data supporting the findings of this study are available within the article and its Supplementalry information files and from the corresponding author upon reasonable request.
References
- 1.Gajewski TF, Schreiber H, Fu Y-X. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi: 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349 [DOI] [PubMed] [Google Scholar]
- 3.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. doi: 10.1038/s41573-018-0007-y [DOI] [PubMed] [Google Scholar]
- 4.Galon J, Angell HK, Bedognetti D, Marincola FM. The Continuum of Cancer Immunosurveillance: Prognostic, Predictive, and Mechanistic Signatures. Immunity. 2013;39(1):11–26. doi: 10.1016/j.immuni.2013.07.008 [DOI] [PubMed] [Google Scholar]
- 5.Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. doi: 10.1038/nrclinonc.2017.101 [DOI] [PubMed] [Google Scholar]
- 6.Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front Immunol. 2018;9(JAN):14. doi: 10.3389/fimmu.2018.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galon J, Costes A, Sanchez-Cabo F, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (80- ). 2006;313(5795):1960–1964. doi: 10.1126/science.1129139 [DOI] [PubMed] [Google Scholar]
- 8.Caisova V, Li L, Gupta G, et al. The Significant Reduction or Complete Eradication of Subcutaneous and Metastatic Lesions in a Pheochromocytoma Mouse Model after Immunotherapy Using Mannan-BAM, TLR Ligands, and Anti-CD40. Cancers (Basel). 2019;11(5):654. doi: 10.3390/cancers11050654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caisová V, Uher O, Nedbalová P, et al. Effective cancer immunotherapy based on combination of TLR agonists with stimulation of phagocytosis. Int Immunopharmacol. 2018;59:86–96. doi: 10.1016/J.INTIMP.2018.03.038 [DOI] [PubMed] [Google Scholar]
- 10.Kato K, Itoh C, Yasukouchi T, Nagamune T. Rapid Protein Anchoring into the Membranes of Mammalian Cells Using Oleyl Chain and Poly(ethylene glycol) Derivatives. Biotechnol Prog. 2004;20(3):897–904. doi: 10.1021/bp0342093 [DOI] [PubMed] [Google Scholar]
- 11.Janotová T, Jalovecká M, Auerová M, et al. The use of anchored agonists of phagocytic receptors for cancer immunotherapy: B16-F10 murine melanoma model. Gabriele L, ed. PLoS One. 2014;9(1):e85222. doi: 10.1371/journal.pone.0085222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Figueiredo RT, Carneiro LAM, Bozza MT. Fungal surface and innate immune recognition of filamentous fungi. Front Microbiol. 2011;2:248. doi: 10.3389/fmicb.2011.00248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hassan SB, Sørensen JF, Olsen BN, Pedersen AE. Anti-CD40-mediated cancer immunotherapy: an update of recent and ongoing clinical trials. Immunopharmacol Immunotoxicol. 2014;36(2):96–104. doi: 10.3109/08923973.2014.890626 [DOI] [PubMed] [Google Scholar]
- 14.Caisová V, Vieru A, Kumžáková Z, et al. Innate immunity based cancer immunotherapy: B16-F10 murine melanoma model. BMC Cancer. 2016;16(1):940. doi: 10.1186/s12885-016-2982-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urban-Wojciuk Z, Khan MM, Oyler BL, et al. The role of tlrs in anti-cancer immunity and tumor rejection. Front Immunol. 2019;10(OCT):2388. doi: 10.3389/fimmu.2019.02388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seo HS, Michalek SM, Nahm MH. Lipoteichoic acid is important in innate immune responses to gram-positive bacteria. Infect Immun. 2008;76(1):206–213. doi: 10.1128/IAI.01140-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinhagen F, Kinjo T, Bode C, Klinman DM. TLR-based immune adjuvants. Vaccine. 2011;29(17):3341–3355. doi: 10.1016/j.vaccine.2010.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi F, Pretto S, Tagliabue E, Balsari A, Sfondrini L. Exploiting poly(I:C) to induce cancer cell apoptosis. Cancer Biol Ther. 2017;18(10):747–756. doi: 10.1080/15384047.2017.1373220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rook AH, Gelfand JC, Wysocka M, et al. Topical resiquimod can induce disease regression and enhance T-cell effector functions in cutaneous T-cell lymphoma. Blood. 2015;126(12):1452–1461. doi: 10.1182/blood-2015-02-630335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.WU J, HUANG D, TYRING S. Resiquimod: a new immune response modifier with potential as a vaccine adjuvant for Th1 immune responses. Antiviral Res. 2004;64(2):79–83. doi: 10.1016/j.antiviral.2004.07.002 [DOI] [PubMed] [Google Scholar]
- 21.Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–1043. doi: 10.1158/1078-0432.CCR-12-2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreau M, Yasmin-Karim S, Kunjachan S, et al. Priming the abscopal effect using multifunctional smart radiotherapy biomaterials loaded with immunoadjuvants. Front Oncol. 2018;8(MAR). doi: 10.3389/fonc.2018.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fransen MF, Sluijter M, Morreau H, Arens R, Melief CJM. Local activation of CD8 T cells and systemic tumor eradication without toxicity via slow release and local delivery of agonistic CD40 antibody. Clin Cancer Res. 2011;17(8):2270–2280. doi: 10.1158/1078-0432.CCR-10-2888 [DOI] [PubMed] [Google Scholar]
- 24.Hunn MK, Farrand KJ, Broadley KWR, et al. Vaccination with irradiated tumor cells pulsed with an adjuvant that stimulates NKT cells is an effective treatment for glioma. Clin Cancer Res. 2012;18(23):6446–6459. doi: 10.1158/1078-0432.CCR-12-0704 [DOI] [PubMed] [Google Scholar]
- 25.Curry WT, Gorrepati R, Piesche M, et al. Cancer Therapy: Clinical Vaccination with Irradiated Autologous Tumor Cells Mixed with Irradiated GM-K562 Cells Stimulates Antitumor Immunity and T Lymphocyte Activation in Patients with Recurrent Malignant Glioma. 2016. doi: 10.1158/1078-0432.CCR-15-2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian H, Shi G, Yang G, et al. Cellular immunotherapy using irradiated lung cancer cell vaccine co-expressing GM-CSF and IL-18 can induce significant antitumor effects. BMC Cancer. 2014;14(1):48. doi: 10.1186/1471-2407-14-48 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Koster BD, Santegoets SJAM, Harting J, et al. Autologous tumor cell vaccination combined with systemic CpG-B and IFN-α promotes immune activation and induces clinical responses in patients with metastatic renal cell carcinoma: a phase II trial. Cancer Immunol Immunother. 2019;68(6):1025–1035. doi: 10.1007/s00262-019-02320-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uyl-de Groot CA, Vermorken JB, Hanna MG, et al. Immunotherapy with autologous tumor cell-BCG vaccine in patients with colon cancer: a prospective study of medical and economic benefits. Vaccine. 2005;23(17–18):2379–2387. doi: 10.1016/J.VACCINE.2005.01.015 [DOI] [PubMed] [Google Scholar]
- 29.Steiner HH, Bonsanto MM, Beckhove P, et al. Antitumor vaccination of patients with glioblastoma multiforme: A pilot study to assess feasibility, safety, and clinical benefits. J Clin Oncol. 2004;22(21):4272–4281. doi: 10.1200/JCO.2004.09.038 [DOI] [PubMed] [Google Scholar]
- 30.Van Dalen FJ, Van Stevendaal MHME, Fennemann, Verdoes M, Ilina O. Molecular repolarisation of tumour-associated macrophages. Molecules. 2019;24(1). doi: 10.3390/molecules24010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang AYC, Guldent PH, Woods AS, et al. The Immunodominant Major Histocompatibility Complex Class I-Restricted Antigen of a Murine Colon Tumor Derives from an Endogenous Retroviral Gene Product (Murine Tumor Antigens/Cytotoxic T Lymphocytes/Endogenous Murine Leukemia Virus/Tandem Mass Spectrome. Vol 93.; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: Naïve to memory and everything in between. Am J Physiol - Adv Physiol Educ. 2013;37(4):273–283. doi: 10.1152/advan.00066.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonaventura P, Shekarian T, Alcazer V, et al. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol. 2019;10:168. doi: 10.3389/fimmu.2019.00168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):165. doi: 10.1038/s12276-018-0191-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275. doi: 10.1038/NRC.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ganesh K, Stadler ZK, Cercek A, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16(6):361–375. doi: 10.1038/s41575-019-0126-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moynihan KD, Irvine DJ. Roles for innate immunity in combination immunotherapies. Cancer Res. 2017;77(19):5215–5221. doi: 10.1158/0008-5472.CAN-17-1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ahonen CL, Engle X, Martinez DG, et al. In situ Stimulation of CD40 and Toll-like Receptor 3 Transforms Ovarian Cancer-Infiltrating Dendritic Cells from Immunosuppressive to Immunostimulatory Cells. Cancer Res. 2009;69(18):7329–7337. doi: 10.1158/0008-5472.can-09-0835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bandola-Simon J, Roche PA. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol Immunol. April 2018. doi: 10.1016/J.MOLIMM.2018.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zitvogel L, Kepp O, Kroemer G. Decoding Cell Death Signals in Inflammation and Immunity. Cell. 2010;140(6):798–804. doi: 10.1016/j.cell.2010.02.015 [DOI] [PubMed] [Google Scholar]
- 41.Scheffer SR, Nave H, Korangy F, et al. Apoptotic, but not necrotic, tumor cell vaccines induce a potent immune response in vivo. Int J Cancer. 2003;103(2):205–211. doi: 10.1002/ijc.10777 [DOI] [PubMed] [Google Scholar]
- 42.Torosantucci A, Bromuro C, Chiani P, et al. A novel glyco-conjugate vaccine against fungal pathogens. J Exp Med. 2005;202(5):597–606. doi: 10.1084/jem.20050749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toda M, Iizuka Y, Kawase T, Uyemura K, Kawakami Y. Immuno-viral therapy of brain tumors by combination of viral therapy with cancer vaccination using a replication-conditional HSV. Cancer Gene Ther. 2002;9(4):356–364. doi: 10.1038/sj.cgt.7700446 [DOI] [PubMed] [Google Scholar]
- 44.Unsworth A, Anderson R, Haynes N, Britt K. OMIP-032 : Two Multi-Color Immunophenotyping Panels for Assessing the Innate and Adaptive Immune Cells in the Mouse Mammary Gland. 2016:527–530. doi: 10.1002/cyto.a.22867 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.