

Abstract
An intrinsic property of the heart is an ability to rapidly and coordinately adjust flux through metabolic pathways in response to physiologic stimuli (termed metabolic flexibility). Cardiac metabolism also fluctuates across the 24‐hours day, in association with diurnal sleep‐wake and fasting‐feeding cycles. Although loss of metabolic flexibility has been proposed to play a causal role in the pathogenesis of cardiac disease, it is currently unknown whether day‐night variations in cardiac metabolism are altered during disease states. Here, we tested the hypothesis that diet‐induced obesity disrupts cardiac “diurnal metabolic flexibility”, which is normalized by time‐of‐day‐restricted feeding. Chronic high fat feeding (20‐wk)‐induced obesity in mice, abolished diurnal rhythms in whole body metabolic flexibility, and increased markers of adverse cardiac remodeling (hypertrophy, fibrosis, and steatosis). RNAseq analysis revealed that 24‐hours rhythms in the cardiac transcriptome were dramatically altered during obesity; only 22% of rhythmic transcripts in control hearts were unaffected by obesity. However, day‐night differences in cardiac substrate oxidation were essentially identical in control and high fat fed mice. In contrast, day‐night differences in both cardiac triglyceride synthesis and lipidome were abolished during obesity. Next, a subset of obese mice (induced by 18‐wks ad libitum high fat feeding) were allowed access to the high fat diet only during the 12‐hours dark (active) phase, for a 2‐wk period. Dark phase restricted feeding partially restored whole body metabolic flexibility, as well as day‐night differences in cardiac triglyceride synthesis and lipidome. Moreover, this intervention partially reversed adverse cardiac remodeling in obese mice. Collectively, these studies reveal diurnal metabolic inflexibility of the heart during obesity specifically for nonoxidative lipid metabolism (but not for substrate oxidation), and that restricting food intake to the active period partially reverses obesity‐induced cardiac lipid metabolism abnormalities and adverse remodeling of the heart.
Keywords: chronobiology, gene expression, heart, metabolism, obesity
Abbreviations
- abhd5
abhydrolase domain containing 5
- accα
acetyl‐CoA carboxylase α
- acot1
acyl‐coA thioesterase 1
- agpat3
1‐acylglycerol‐3‐phosphate O‐acyltransferase 3
- Akt
protein kinase B
- ATGL
adipose triglyceride lipase
- bmal1
brain and muscle ARNT‐like 1
- BVW/TL
biventricular weight to tibia length ratio
- cd36
fatty acid translocase
- CE
cholesteryl ester
- Cer
ceramide
- CGI58
comparative gene identification 58
- CHREBP
Carbohydrate response element binding protein
- CLAMS
comprehensive laboratory animal monitoring system
- clock
circadian locomotor output cycles kaput
- cry2
cryptochrome 2
- CVD
cardiovascular disease
- DG
diglyceride
- fabp4
fatty acid binding protein 4
- GLUT1
glucose transporter 1
- GLUT4
glucose transporter 4
- HFpEF
heart failure with preserved ejection fraction
- lcad
long chain acyl‐CoA dehydrogenase
- LC‐MS
liquid chromatography‐mass spectrometry
- lipe
hormone sensitive lipase
- LXRα
liver X receptor α
- mcd
malonyl‐CoA decarboxylase
- mlxipl
MLX interacting protein like
- nampt
nicotinamide phosphoribosyltransferase
- NEFA
non‐esterified fatty acid
- npas2
neuronal PAS domain protein 2
- nr1d1
nuclear receptor family 1, member D1
- nr1d2
nuclear receptor family 1, member D2
- nr1h3
nuclear receptor family 1, member H3
- PC
phosphatidylcholine
- pdk4
pyruvate dehydrogenase kinase 4
- PE(O)
ether‐linked phosphatidylethanolamine
- PE
phosphatidylethanolamine
- per2
period 2
- per3
period 3
- PI
phosphatidylinositol
- pnpla2
patatin like phospholipase domain containing 2
- RER
respiratory exchange ratio
- sgpl1
sphingosine‐1‐phosphate lyase 1
- slc2a1
solute carrier family 2, member 1
- slc2a4
solute carrier family 2, member 4
- SM
sphingomyelin
- srebf1
sterol regulatory element binding transcription factor 1
- SREBP1
sterol regulatory element binding protein 1
- TG
triglyceride
- ZT
zeitgeber time
1. INTRODUCTION
The prevalence of obesity has risen steadily over the last few decades. 1 Obesity increases risk of multiple comorbidities, such as type 2 diabetes mellitus and cardiovascular diseases (CVD). 2 CVD is the leading cause of death globally. 3 In the US alone, approximately 2,300 individuals die from CVD daily. 4 Obesity associated CVD comprise hypertension, atherosclerosis, and heart failure. 5 , 6 , 7 The latter includes obesity cardiomyopathy, often characterized as heart failure with preserved ejection fraction (HFpEF). 8 Putative mechanisms contributing toward the pathogenesis of obesity cardiomyopathy include diminished coronary reserve, chronic low‐grade inflammation, increased fibrosis coupled with myocardial stiffening, excessive oxidative stress, and numerous metabolic derangements (such as lipotoxicity, mitochondrial dysfunction, impaired energetics, and metabolic inflexibility). 9 , 10
Cardiac metabolism and contractile function are inextricably interlinked. For example, during acute periods of increased workload (eg, physical activity), elevated energetic demand is countered by increased rates of cardiac glucose utilization. 11 , 12 Conversely, during acute periods of increased fatty acid availability (eg, following a high fat meal, or during short‐term fasting), cardiac fatty acid utilization is augmented, thereby preventing accumulation of excess lipid species in the heart (ie, lipotoxicity) and preserving cardiac function. 13 , 14 This ability of the heart to acutely shift metabolic fluxes in response to a physiologic perturbation is termed metabolic flexibility. 15 However, chronic alterations in workload (as might occur during hypertension) and/or substrate availability (eg, during ischemic heart disease, diabetes mellitus, obesity) often cause more permanent changes in cardiac metabolism, leading to metabolic inflexibility. 10 , 14 , 16 It has been postulated that metabolic inflexibility plays a causal role in the development of contractile dysfunction during disease states. 15
Cardiac metabolism displays dramatic fluctuations over the course of the day, in association with diurnal sleep/wake and fasting/feeding cycles. 17 Upon awakening, both blood pressure and heart rate rise steeply, in parallel with increased mental and physical activity. 18 At this time, the heart increases glucose utilization, presumably to balance energetic demand with ATP generation. 19 Toward the end of the active period, nutrient stores (both glycogen and triglyceride) are replenished in the heart. 19 , 20 The sleep period is considered a time of growth and repair, during which cardiac cellular constituent turnover increases, likely ensuring optimal cardiomyocyte function in preparation of the upcoming awake period. 17 , 21 Temporal partitioning of cardiac metabolic processes appears to be governed not only by behavioral cycles over the course of the day (and associated neurohumoral fluctuations), but are also orchestrated by intrinsic mechanisms (ie, cell autonomous circadian clocks). 17 Indeed, genetic disruption of the murine cardiomyocyte circadian clock abolished 24‐hours rhythms in cardiac metabolism (in the absence of perturbed sleep/wake and fasting/feeding cycles), as well as development of severe cardiomyopathy. 19 , 20 , 22 However, it is currently unknown whether temporal partitioning of cardiac metabolism over the 24‐hours day is impaired during disease states (ie, the existence of “diurnal metabolic inflexibility”).
The purpose of the present study was to investigate whether diurnal metabolic inflexibility is observed in the heart during obesity. Accordingly, mice were fed a high fat diet for 20 weeks, followed by assessment of day‐night differences in cardiac glucose and fatty acid metabolism. Here, we report that despite profound impairment of 24‐hours rhythms in whole body substrate utilization and cardiac transcripts encoding for metabolism‐related proteins, day‐night differences in cardiac substrate oxidation were preserved. In contrast, day‐night differences in cardiac triglyceride synthesis were abolished during obesity, as were day‐night differences in cardiac high molecular weight lipid species (identified through lipidomics). Interestingly, restricting food intake to the 12‐hours dark (awake) period partially restored day‐night differences in cardiac triglyceride synthesis and lipidome in obese mice, in association with a partial reversal of adverse cardiac remodeling. Collectively, these studies highlight a lipid‐specific diurnal metabolic inflexibility of the heart during obesity, which can be partially restored by simply ensuring a lack of food intake during the usual sleep period.
2. MATERIALS AND METHODS
2.1. Mice
The present study utilized wild‐type C57B/6 mice (Jackson Labs). All experimental mice were male, and were housed at the Center for Comparative Medicine at the University of Alabama at Birmingham (UAB), under temperature‐, humidity‐, and light‐controlled conditions. A strict 12‐hour light/12‐hour dark cycle regime was enforced (lights on at 6AM; zeitgeber time [ZT] 0); the light/dark cycle was maintained throughout these studies, facilitating investigation of diurnal variations (as opposed to circadian rhythms). Mice were housed in either a standard micro‐isolator cage, or a CLAMS (Comprehensive Laboratory Animal Monitoring System, enabling control of food access, as well as whole body metabolic assessments, in a noninvasive automated fashion). All mice received water ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham.
2.2. Experimental protocol
Mice were allowed access to either a control diet (10% calories from fat, Research Diets, New Brunswick, NJ; catalog number D12450K) or a high fat diet (45% calories from fat, Research Diets, New Brunswick, NJ; catalog number D12451). For the first 17 weeks of the study, all mice were fed diets in an ad libitum fashion in standard micro‐isolator cages. At the end of week 17, all mice were transferred to a CLAMS, during which time they were maintained on the same diet that they had been consuming since initiation of the study. After a 1‐week period of acclimatization to the CLAMS (with continued ad libitum feeding), mice were divided into sub‐groups based on food access. More specifically, half of the mice had continuous 24‐hours access to food (ie, continued ad libitum feeding), while the other half had access to food only during the 12‐hours dark (active) period. In the latter case, food access was controlled by the CLAMS; each cage possesses feeders that open/close in a computer‐controlled fashion. Mice remained in the CLAMS for an additional 2 weeks, after which hearts were isolated for gravimetric, histologic, biochemical, and/or metabolic analyses.
2.3. Whole body behavioral and metabolic monitoring
Twenty‐four hour patterns of physical activity (beam breaks), food intake, and energy expenditure (indirect calorimetry) were measured in mice using a CLAMS during week 19 of the study, as described previously. 23 Lean and fat body masses were measured in a subset of mice at the end of the feeding study by quantitative magnetic resonance (QMR, Echo 3‐in‐1, Echo Medical System) at an established UAB core facility.
2.4. Histologic assessment
Cross sections from the medial heart were taken immediately upon removal of the heart, and fixed in formalin for 24‐hours. Wheat germ agglutinin (WGA) staining was utilized for measurement of myocyte cross‐sectional area; at least 45 myocytes were assessed per heart using Image‐J software (NIH), as described previously. 24 Picrosirius Red staining of collagen fibers was utilized for semi‐quantitative measurement of left ventricular fibrosis, using ImagePro Plus software (Media Cybernetics, Inc, Rockville, MD), as described previously. 25 Oil‐Red O staining of lipid was utilized to assess steatosis in the left ventricle, as described previously. 26 Briefly, ventricles were embedded in OCT embedding medium, cryosectioned (5 μm), stained with Oil‐Red O, and imaged (20× magnification with a Lecica DMRB microscope); all images were analyzed with Image Pro Plus software using color cube‐based selection criteria.
2.5. Echocardiographic analysis
Mouse echocardiography was performed under anesthesia with 1.5% isoflurane in 95% O2, using a VisualSonics VeVo 3100 Imaging System (VisualSonics, Toronto, Canada) at an established UAB core facility, and was analyzed by VisualSonics software. Body temperature was maintained at 37.0°C ± 0.5°C, while heart rate was sustained at 500 ± 100 beats per minute.
2.6. RNA sequencing
Transcriptomic analysis was performed through the use of RNA sequencing in the UAB Genomics Core facility. RNA was extracted from biventricular samples using standard procedures. 27 The quality of the RNA samples was tested using the Agilent BioAnalyzer and RNA with RIN values greater than 7.0 were used in downstream library preparation. The RNA was DNAse treated prior to library preparation. The RNA‐Sequencing libraries were generated using the NEBNext Ultra II RNA kit (NEB, Ipswich MA) following the manufacturer's protocol. The resulting libraries were sequenced on the Illumina NextSeq 500 (Illumina, Inc San Diego CA) using paired end 75bp sequencing reads, per standard methods.
2.7. Working mouse heart perfusions
Myocardial substrate utilization was measured ex vivo through isolated working mouse heart perfusions, as described previously. 19 , 20 , 28 , 29 All hearts were perfused in the working mode (nonrecirculating manner) for 40 minutes with a preload of 12.5 mm Hg and an afterload of 50 mm Hg. Standard Krebs‐Henseleit buffer was supplemented with 8 mM glucose, 0.4 mM oleate conjugated to 3% BSA (fraction V, fatty acid‐free; dialyzed), 10 µU/mL insulin (basal/fasting concentration), 2 mM β‐hydroxybutyrate, 0.2 mM acetoacetate, 0.05 mM L‐carnitine, and 0.13 mM glycerol. Metabolic fluxes were assessed through the use of distinct radiolabeled tracers: 1) [U‐14C]‐glucose (0.12 mCi/L; glycolysis, glucose oxidation, glycogen synthesis); and 2) [9,10‐3H]‐oleate (0.067 mCi/L; β‐oxidation, triglyceride synthesis). Measures of cardiac metabolism (eg, oxygen consumption) and function (eg, cardiac power) were monitored as described previously. 19 , 20 , 28 , 29 At the end of the perfusion period, hearts were snap‐frozen in liquid nitrogen and stored at −80°C prior to analysis. Data are presented as steady state values (ie, values during the last 10 min of the perfusion protocol). Heart perfusion conditions were chosen for consistency with a prior study describing the metabolic phenotype of the heart during fasting. 22
2.8. Quantitative RT‐PCR
Candidate gene expression analysis was performed by quantitative RT‐PCR, using previously described methods. 30 , 31 Specific Taqman assays were designed for each gene from mouse sequences available in GenBank. All quantitative RT‐PCR data are presented as fold change from an indicated control group.
2.9. Western blotting
Qualitative analysis of protein expression and phosphorylation status was performed via standard western blotting procedures, as described previously. 19 Briefly, 10‐30 µg protein lysate was separated on polyacrylamide gels and transferred to PVDF membranes. Membranes were probed for the following targets: p‐AktThr‐308 (Cell Signaling; 9275), Akt (Cell Signaling; 9272), and calsequestrin (Abcam; ab3516). A rabbit HRP‐conjugated secondary antibody (Cell Signaling; 7074) was used for chemiluminescent detection with Luminata Forte Western Blotting substrate (Millipore, WBLUF0100). In order to minimize the contribution that position on the gel might have on outcomes, samples were randomized on gels; samples were re‐ordered postimaging, only for the sake of illustration of representative images (note, all bands for representative images for an individual experiment were from the same gel).
2.10. Plasma non‐esterified fatty acids (NEFA) levels
Blood was collected at the time of euthanization, placed in EDTA‐containing tubes, and centrifuged at 3000 g for 10 minutes at 4°C. Resultant plasma was stored at −80°C prior to assessment of NEFA through use of a spectrophotometric‐based assay, as described previously. 13
2.11. Lipidomic analysis using liquid chromatography‐mass spectrometry (LC‐MS)
Lipid extraction and untargeted LC‐MS lipidomic analysis was performed as described previously, with slight modifications. 32 , 33 In brief, lipids were extracted from biventricular samples (between 10 and 30 mg) after spiking with six internal standards (lysophosphatidylcholine 13:0, phosphatidylcholine 19:0/19:0 and 14:0/14:0, phosphatidylethanolamine 17:0/17:0, phosphatidylglycerol 15:0/15:0, and phosphatidylserine 12:0/12:0; Avanti Polar Lipids Inc, Alabaster, USA). Samples (0.49‐1.52 μL) were injected into a 1290 Infinity HPLC coupled with a 6530 Accurate Mass Q‐TOF (Agilent Technologies Inc, Santa Clara, USA) via a dual electrospray ionization (ESI) source operated in the positive mode. Lipids were eluted on Zorbax Eclipse plus column (C18, 2.1 × 100 mm, 1.8 μm, Agilent Technologies Inc) over 83 minutes at 40°C using a gradient of solvent A (0.2% formic acid and 10 mM ammonium formate in water) and B (0.2% formic acid and 5 mM ammonium formate in methanol/acetonitrile/methyl tert‐butyl ether [MTBE], 55:35:10 [v/v/v]). MS data processing was achieved using Mass Hunter B.06.00 (Agilent Technologies Inc) and an in‐house bioinformatics script that we have developed and encoded in both Pearl and R languages to ensure optimal MS data alignment between chromatographic runs. In addition, frequency and adducts filters were applied, as well as signal intensity normalization (using cyclic loess algorithm) and imputation of missing values with k‐nearest neighbor on scaled data. This yields a data set listing MS features with their mass, retention time, and corrected signal intensity. For subsequent data mining, we used a targeted approach in which lipid features from this data set, including their fatty acids side chains, were annotated by alignment with our in‐house database which contains 498 lipids that were previously identified by MS/MS for the following selected lipid (sub)classes: (a) sphingolipids: ceramides (Cer) and spingomyelins (SM); (b) glycerolipids: diglycerides (DG) and triglycerides (TG); (c) glycerophospholipids: phosphatidylcholines (PC), phosphatidylethanolamines (PE), ether‐linked phosphatidylethanolamines (PE(O)), phosphatidylinositols PI); and (d) sterols: cholesteryl esters (CE).
2.12. Statistical analysis
Statistical analyses were performed using student t tests (for comparisons between only two experimental groups) and two‐way ANOVA, as described previously. 23 , 28 Briefly, analyses were performed on Prism statistical software to investigate the effects of diet and/or time‐of‐day, followed by Bonferroni post hoc analyses for pair‐wise comparisons (indicated in Figures). In all analyses, the null hypothesis of no model effects was rejected at P < .05. RNA‐Seq data from each experimental group were analyzed for rhythmicity in the R Programming Language with the MetaCycle Package. 34 Transcripts were considered to have a 24‐hours oscillation if they significantly fit a cosine curve (P < .05). Amplitude (peak‐to‐mesor difference) and phase (the zeitgeber time of the peak) were calculated within MetaCycle, and student t tests were employed to determining significant differences between feeding groups. Significant differences in mesor (daily average value) were calculated by DESeq2, 35 by calculating main effects of diet. Enrichr 36 was utilized for performing pathway analysis within the Reactome pathway database. 37 For the lipidomic analysis, the output text file containing the data set of annotated lipids with corrected signal intensity was imported into Mass Professional Pro software (version 12.6.1, Agilent Technologies Inc) and independent testing of each feature was achieved using an unpaired Student's t test, which was followed by a Storey's multiple testing correction whenever possible. We tested for lipids discriminating selected experimental diet groups based on the time‐of‐day using the following thresholds: (a) P‐value <.05 and (b) (Q‐value <.05 (corresponding to a False Discovery Rate of < 5%).
3. RESULTS
3.1. Whole body and cardiac alterations during chronic high fat feeding
We initially investigated the effects of 20‐wk ad libitum high fat feeding on whole body metabolic parameters. As expected, feeding mice a high fat diet led to accelerated body weight gain; statistically significant differences were observed between control and high fat diet fed mice within 2‐wks after initiation of the dietary intervention (Figure 1A). Assessment of body composition in a sub‐set of mice after 20‐wks revealed increased adiposity following high fat feeding (relative to control diet fed mice), with a trend for increased lean mass (Figure 1B). Assessment of energy balance parameters after 18‐wk of feeding revealed significantly increased daily energy expenditure in high fat fed mice, while physical activity and RER (respiratory exchange ratio) were both significantly lower; daily caloric intake did not differ between the two dietary groups (Table 1). 24‐hours patterns in caloric intake, physical activity, and energy expenditure did not differ between high fat and control diet fed mice (Figure 1C). However, 24‐hours oscillations in RER (an indicator of substrate utilization) were completely abolished in high fat fed mice (Figure 1C). Collectively, these observations indicate that 24‐hours rhythms in whole body substrate selection are disrupted in obese mice.
FIGURE 1.
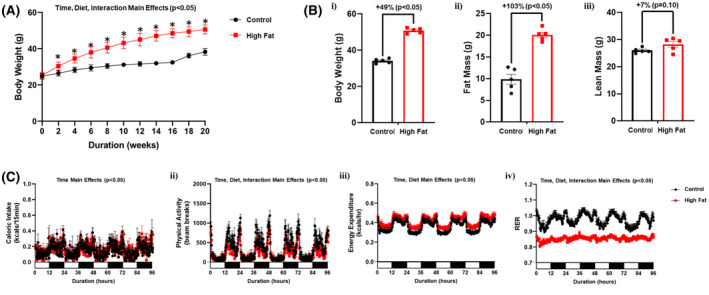
High fat feeding leads to whole body diurnal metabolic inflexibility. A, Body weight was assessed once every 2‐wk, in mice that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk (n = 20‐21). B, After 20‐wk of control or high fat diet feeding, body weight (i), fat mass (ii) and lean mass (iii) were assessed (n = 5). C, After 18‐wk of control or high fat diet feeding, 24‐h patterns in caloric intake (i), physical activity (ii), energy expenditure (iii), and respiratory exchange ratio (RER; iv) were assessed (n = 25‐27). Data are reported as mean ± SEM. Main effects for either time, diet, and/or interaction are reported at the top of the figure panels. *P < .05 for control (circle) vs high fat (square)
TABLE 1.
Caloric intake, physical activity, energy expenditure, and RER in ad libitum control and high fat diet fed mice
| Control diet Ad libitum | High fat diet Ad libitum | |
|---|---|---|
| 24‐h cumulative caloric intake (kcal/24‐h) | 14.4 ± 0.6 | 14.8 ± 0.4 |
| 24‐h cumulative physical activity (beam breaks; AU) | 32 703 ± 1856 | 24 257 ± 1353* |
| 24‐h average energy expenditure (kcals/h) | 0.355 ± 0.012 | 0.402 ± 0.018* |
| 24‐h average respiratory exchange ratio | 0.981 ± 0.008 | 0.856 ± 0.005* |
Caloric intake, physical activity, energy expenditure, and RER were monitored continuously using a CLAMS. Data are reported as mean ± SEM for 25‐27 mice per experimental group.
P < .05 for pair wise comparison between dietary groups.
The impact of chronic high fat feeding (20‐wk duration) on cardiac remodeling was next assessed at functional, gravimetric, and histologic levels. The majority of echocardiographic parameters were not significantly different between control and high fat diet fed mice, including preservation of ejection fraction and fractional shortening (Table 2). However, both stroke volume and left ventricular posterior wall thickness during systole were significantly increased in high fat fed mice (compared to controls; Table 2). Next, hearts were isolated from control and high fat fed mice. The biventricular weight to tibia length (BVW/TL) ratio was significantly increased in high fat fed mice (Figure 2A). Histologic analysis revealed that both cardiomyocyte area and cardiac fibrosis were significantly increased in high fat fed mice (Figure 2B,C). Moreover, oil red O staining (a marker of lipid deposition) was significantly increased in hearts of high fat fed mice (Figure 2D). Collectively, these observations reveal that chronic high fat feeding resulted in cardiac hypertrophy, fibrosis, and steatosis, in the presence of preserved ejection fraction.
TABLE 2.
Echocardiographic parameters in ad libitum control and high fat diet fed mice
| Control diet Ad libitum | High fat diet Ad libitum | |
|---|---|---|
| End systolic volume (uL) | 21.9 ± 5.2 | 22.0 ± 3.5 |
| End diastolic volume (uL) | 54.8 ± 6.8 | 69.2 ± 6.2 |
| Stroke volume (uL) | 32.9 ± 2.1 | 47.3 ± 2.7* |
| Ejection fraction (%) | 63.1 ± 7.2 | 69.1 ± 2.4 |
| Fractional shortening (%) | 35.3 ± 6.7 | 38.4 ± 1.8 |
| Left ventricular posterior wall thickness, systole (mm) | 1.29 ± 0.07 | 1.54 ± 0.07* |
| Left ventricular posterior wall thickness, diastole (mm) | 0.91 ± 0.06 | 1.32 ± 0.29 |
| Left ventricular inner diameter, systole (mm) | 2.36 ± 0.32 | 2.16 ± 0.34 |
| Left ventricular inner diameter, diastole (mm) | 3.58 ± 0.20 | 3.97 ± 0.15 |
| Intra‐ventricular septum thickness, systole (mm) | 1.44 ± 0.16 | 1.66 ± 0.13 |
| Intra‐ventricular septum thickness, diastole (mm) | 1.04 ± 0.10 | 1.11 ± 0.11 |
Data are reported as mean ± SEM for five mice per experimental group.
P < .05 for pair wise comparison between dietary groups.
FIGURE 2.
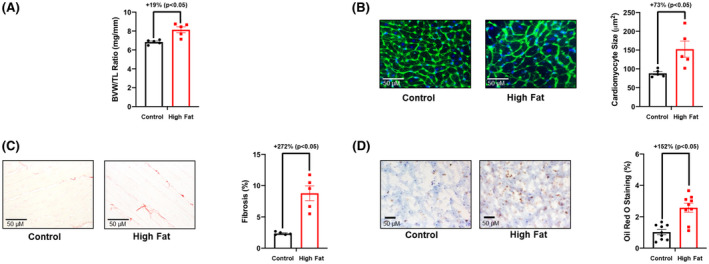
Adverse remodeling markers are increased in hearts of high fat fed mice. A, Biventricular weight to tibia length (BVW/TL) ratio was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk (n = 5). B, Cardiomyocyte size was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk (n = 5). C, Cardiac fibrosis was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk (n = 5). D, Cardiac steatosis was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk (n = 5). Data are reported as mean ± SEM
3.2. High fat diet‐induced obesity dramatically effects 24‐hours rhythms in cardiac metabolic genes
To gain insight regarding whether 24‐hours rhythms are altered in the heart during chronic high fat feeding, an unbiased transcriptomic approach was taken initially. Accordingly, hearts were isolated from control and high fat diet fed mice at 4‐hours intervals over the 24‐hours day. RNAseq was next performed, followed by MetaCycle analysis, for identification of transcripts undergoing 24‐hours oscillations. This analysis revealed that of the 14,575 genes expressed in the heart, 2472 genes exhibit significant 24‐hours oscillations in control hearts. In contrast, hearts isolated from high fat fed mice have 1833 genes with significant 24‐h oscillations. Interestingly, only 1064 genes oscillated in hearts from both control and high fat fed mice (Figure 3A and Table S1). Accordingly, 1408 genes are uniquely rhythmic in control hearts, while 769 genes are uniquely rhythmic in hearts of high fat fed mice (Figure 3A and Tables S2 and S3). Moreover, 375 and 179 genes are uniquely rhythmic in control and high fat fed mouse hearts (respectively) with a peak‐to‐trough ratio greater than 2 (Figure 3B). Bioinformatic analyses revealed that multiple metabolic processes were enriched in genes uniquely oscillating in control and high fat fed mouse hearts (Figure 3C). Example genes encoding for metabolism‐related proteins that were commonly rhythmic in both feeding groups (eg, nampt; encoding for nicotinamide phosphoribosyltransferase), uniquely rhythmic in control hearts (eg, accα; encoding for acetyl‐CoA carboxylase α), or uniquely rhythmic in high fat fed mouse hearts (eg, pdk4; encoding for pyruvate dehydrogenase kinase 4) are shown in Figure 3D. Next, MetaCycle and DESeq2 were utilized to identify differences in amplitude, phase, and/or mesor for those genes that commonly oscillate in hearts from both control and high fat fed mice; this analysis revealed a total of 509 differentially rhythmic transcripts between the two feeding groups (Figure 3E and Table S4). Interestingly, there appear to be a lower number of genes exhibiting peak expression levels at the light‐to‐dark and dark‐to‐light transitions in hearts from high fat fed mice (relative to controls; Figure 3F). Collectively, these data reveal that chronic high fat feeding dramatically alters temporal regulation of the cardiac transcriptome, particularly for metabolism‐related genes.
FIGURE 3.
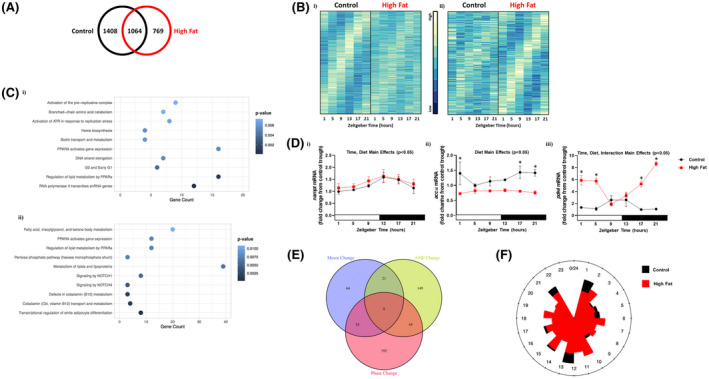
Bioinformatic analysis of RNAseq data reveals altered diurnal landscape of the cardiac transcriptome in obese mice. Hearts were isolated from mice (at ZT1, ZT5, ZT9, ZT13, ZT17, and ZT21) that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk, followed by RNAseq analysis (n = 4). A, Venn diagram indicating numbers of rhythmic transcripts in hearts of control and/or high fat fed mice. B, Heat map illustrating the transcripts that oscillate (with a peak‐to‐trough ratio greater than 2) uniquely in hearts of control (i) or high fat fed (ii) mice. C, Top 10 enriched pathways for transcripts that oscillate uniquely in hearts of control (i) or high fat fed (ii) mice. D, Examples of metabolism‐related transcripts that oscillate in hearts of both control and high fat fed mice (I; nampt), only in control mice (ii; accα), or only in high fat fed mice (iii; pdk4). E, Venn diagram indicating number of rhythmic transcripts with significant phase, mesor, and/or amplitude (AMP) changes in hearts of high fat fed mice (relative to controls). F, Phase relationship of rhythmic transcripts with significant differences between control and high fat fed mouse hearts; distance from point of origin represents number of transcripts, while direction from origin (ie, angle) represents phase
3.3. Differential influence of high fat feeding on day‐night differences in cardiac metabolism
Given that 24‐hours rhythms in both whole body substrate utilization (ie, RER; Figure 1C) and the cardiac transcriptome (particularly metabolism‐related genes; Figure 3) are dramatically attenuated/altered during chronic high fat feeding, we hypothesized that day‐night differences in cardiac metabolism would be similarly impacted (ie, diurnal metabolic inflexibility). Accordingly, glucose and oleate metabolism were studied in working mouse hearts isolated from control and high fat diet fed mice at ZT5 and ZT17 (times at which day‐night differences in cardiac metabolism have been reported previously 19 , 20 ). Hearts from control fed mice exhibited significant differences in cardiac glucose oxidation and triglyceride synthesis when assessed during the day (ZT5) versus the night (ZT17), with trends for day‐night differences in cardiac oleate oxidation, net lactate release, and [14C]‐labeled lactate release (Figure 4A). In contrast, glycogen synthesis did not exhibit a day‐night difference in control hearts (Figure 4A). In high fat fed mice, significant day‐night differences were observed for cardiac glucose oxidation and net lactate release, with trends for day‐night differences in oleate oxidation, [14C]‐labeled lactate release, and glycogen synthesis (Figure 4B). In contrast, triglyceride synthesis did not exhibit a day‐night difference in hearts isolated from high fat fed mice (Figure 4B). Importantly, no day‐night differences were observed in functional parameters in hearts isolated from either control or high fat fed mice (Table 3). Collectively, these data suggest a preservation of day‐night differences in cardiac oxidative metabolism in high fat fed mice, whereas diurnal regulation of triglyceride synthesis is selectively impaired.
FIGURE 4.
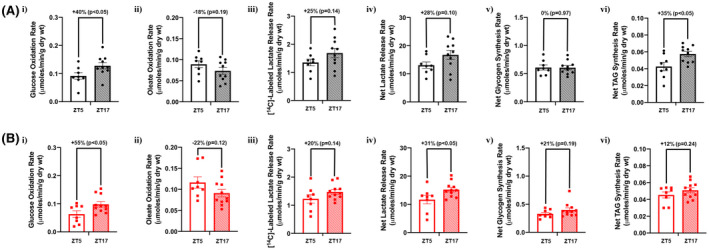
Selective diurnal metabolic inflexibility of cardiac triglyceride synthesis in obese mice. Hearts were isolated from mice (at ZT5 and ZT17) that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk, followed by assessment of cardiac metabolism ex vivo. A, Rates of cardiac glucose oxidation (i), fatty acid oxidation (ii), [14C]‐labeled lactate release (iii), net lactate release (iv), net glycogen synthesis (v), and net triglyceride synthesis (vi) in control mice (n = 8‐10). B, Rates of cardiac glucose oxidation (i), fatty acid oxidation (ii), [14C]‐labeled lactate release (iii), net lactate release (iv), net glycogen synthesis (v), and net triglyceride synthesis (vi) in high fat fed mice (n = 8‐11)
TABLE 3.
Functional parameters for ex vivo perfused hearts isolated from ad libitum control and high fat diet fed mice
| Control diet Ad libitum | High fat diet Ad libitum | |||
|---|---|---|---|---|
| ZT5 | ZT17 | ZT5 | ZT17 | |
| Cardiac power (CP; mW) | 1.19 ± 0.05 | 1.16 ± 0.06 | 1.15 ± 0.05 | 1.21 ± 0.08 |
| Heart rate (BPM) | 291 ± 15 | 304 ± 10 | 288 ± 9 | 285 ± 9 |
| Developed pressure (mm Hg) | 29.5 ± 3.2 | 31.4 ± 2.3 | 30.3 ± 2.9 | 33.1 ± 1.5 |
| Rate pressure product (BPM × mm Hg) | 8272 ± 656 | 9480 ± 629 | 8653 ± 695 | 9417 ± 497 |
| Oxygen consumption (MVO2; umoles/min/g dry wt) | 7.10 ± 0.68 | 7.56 ± 0.97 | 8.71 ± 1.16 | 7.81 ± 0.69 |
| Cardiac efficiency (CP/MVO2) | 0.18 ± 0.02 | 0.17 ± 0.02 | 0.15 ± 0.02 | 0.16 ± 0.01 |
Data are reported as mean ± SEM for 8‐11 mice per experimental group.
3.4. Day‐night differences in cardiac lipid species are abolished by chronic high fat feeding
Given that day‐night differences in cardiac triglyceride synthesis were selectively impaired in high fat fed mice, we next investigated potential contributing factors that are intrinsic (cardiac gene expression and signaling) and extrinsic (plasma NEFA) to the myocardium. Diurnal rhythms in cd36 (fatty acid translocase; involved in fatty acid uptake into the cell), mcd (malonyl‐CoA decarboxylase; involved in controlling malonyl‐CoA levels in the cell, a key allosteric regulator of fatty acid entry into the mitochondrial matrix), lcad (long chain acyl‐CoA dehydrogenase; a fatty acid β‐oxidation enzyme), and lipe (hormone sensitive lipase, involved in triglyceride turnover) were investigated. In control hearts, expression of all these genes appeared to increase near the light‐to‐dark phase transition (Figure 5A). Cosinor analysis revealed that three out of the four genes fit a cosine curve in control hearts (namely cd36, lcad, and lipe); none of these genes fit a cosine curve in hearts of obese mice. In addition, all four lipid metabolism genes are chronically elevated in hearts of obese mice (ie, diet main effect). The phosphorylation status of a key insulin signaling component (Akt) was next investigated, due to established regulation of triglyceride turnover by insulin, as well as altered myocardial insulin signaling during obesity. Significant day (ZT5) versus night (ZT17) differences in the phosphorylation status of Akt were not observed in hearts of either control or high fat fed mice (Figure 5B). Plasma NEFA levels exhibited a significant diurnal variation in control mice, but not high fat fed mice (as assessed by Cosinor analysis; Figure 5C). Moreover, plasma NEFA levels were chronically elevated in high fat fed mice (ie, diet main effect). Collectively, these data suggest that diurnal variations in factors that are intrinsic (ie, metabolic gene expression) and extrinsic (ie, plasma NEFA levels) to the heart, which are known to influence cardiac triglyceride turnover, are altered during obesity.
FIGURE 5.
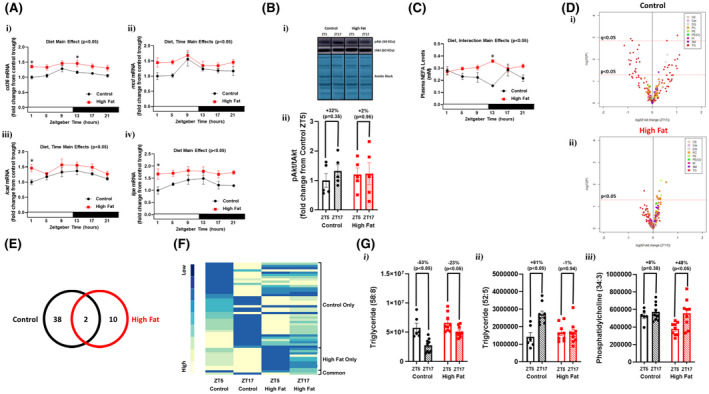
Diminished day‐night differences in the cardiac lipidome of obese mice. Hearts and plasma were isolated from mice (at ZT1, ZT5, ZT9, ZT13, ZT17, and ZT21) that were fed either a control or high fat diet in an ad libitum fashion for 20‐wk (n = 5‐9). A, Diurnal variations in cardiac cd36 (i), mcd (ii), lcad (iii), and lipe (iv) mRNA levels in control and high fat fed mice. B, Day (ZT5) versus night (ZT17) differences in the ratio of phosphorylated Akt to total Akt (ie, pAkt/tAkt) in hearts of control and high fat fed mice. C, Diurnal variations in plasma NEFA levels in control and high fat fed mice. D, Volcano plots indicating day‐night (ZT5‐ZT17) differences in the cardiac lipidome of control (i) and high fat fed (ii) mice; the x axis corresponds to fold changes (FC) of MS signal intensity values for all annotated lipids at ZT17 versus ZT5 (log2), the y axis corresponds to P‐values (‐log 10); the horizontal dotted lines indicates the cut‐off for P < .05 and Q < 0.05, as indicated; color indicates distinct lipid species classification abbreviated: CE (cholesterol ester), Cer (ceramide), DG (diglyceride), PC (phosphatidylcholine), PE (phosphatidylethanolamine), PE(O) (ether‐linked PE), PI (phosphatidylinositol), SM (sphingomyelin), and TG (triglyceride). E, Venn diagram indicating numbers of lipid species with day‐night differences in hearts of control and/or high fat fed mice (based on P < .05). F, Heat map illustrating lipid species in hearts of control and/or high fat fed mice with day‐night differences in levels (based on P < .05); sub‐grouping is based on whether day‐night differences were observed in control mice only, high fat fed mice only, or common in both groups. G, Examples of lipid species with day‐night differences in hearts of both control and high fat fed mice (i), only in control mice (ii), or only in high fat fed mice (iii)
To determine the extent to which diurnal regulation of cardiac lipid metabolism was impacted, a lipidomic approach was next taken. Of the 1039 MS features detected and included in the final data set, 208 lipid species were annotated for the selected (sub)classes, namely 27 sphingolipids (1 ceramide and 26 sphingomyelins), 93 glycerolipids (6 diglycerides and 87 triglycerides), 86 glycerophospholipids (70 phosphatidylcholines, 8 phosphatidylethanolamines, 3 ether‐linked phosphatidylethanolamine, and 5 phosphatidylinositols), and 2 sterols (namely cholesterol esters). From these 208 lipid species, 40 exhibited day‐night differences in control hearts using a P‐value <.05, which decreased to 5 using a Q‐value <.05 (Figure 5A‐B). Of the 40 lipid species exhibiting day‐night differences in control hearts, 36 were triglycerides, 3 were diglycerides, and 1 was a sphingomyelin (Figure 5D and Table S5). In high fat fed mice, day‐night differences were observed for only 12 cardiac lipid species using a P‐value <.05, but none of them passed the criteria Q‐value <.05 (Figure 5D‐E). Of the 12 lipid species exhibiting day‐night differences in hearts of high fat fed mice, 10 were phospholipids, 1 was a triglyceride, and 1 was a diglyceride (Figure 5D and Table S6). Comparison of lipid species exhibiting day‐night differences in hearts from control versus high fat diet fed mice revealed that only 2 lipid species are common to both (Figure 5E); importantly, day‐night differences in these common lipid species were attenuated in hearts from high fat fed mice (Figure 5F and Tables S5 and S6). Example lipid species that were commonly rhythmic in both feeding groups (eg, triglyceride 58:8), uniquely rhythmic in control hearts (eg, triglyceride 52:5), or uniquely rhythmic in high fat fed mouse hearts (eg, phosphatidylcholine 34:3) are shown in Figure 5G. Collectively, these data reveal a dramatic attenuation in day‐night differences in the cardiac lipidome during chronic high fat feeding.
3.5. Time‐of‐day‐restricted feeding partially reverses adverse remodeling of the heart during obesity
We have previously reported that restricting a high fat diet to the dark (active) period prevents high fat diet‐induced cardiac dysfunction in the mouse. 29 However, whether such an intervention can reverse adverse remodeling of the heart in an obese mouse (ie, treatment strategy) is unknown. As such, mice were fed control or high fat diets for 18‐wks in an ad libitum fashion, followed by 2‐wks of restricting the same diet only to the 12‐hours dark (active) period. Body weight of high fat fed mice remained significantly increased following the two week time‐of‐day‐restricted feeding period, compared to control fed mice (Figure 6A,B). Similarly, adiposity remained increased in high fat fed mice at the end of the 2‐wk time‐of‐day‐restricted feeding period (Figure 6B). Placement of mice in metabolic cages during time‐of‐day‐restricted feeding revealed no significant differences in total daily caloric intake between control and high fat diet fed mice (Table 4). During this time‐of‐day‐restricted feeding period, daily energy expenditure remained significantly increased in high fat fed mice, while physical activity and RER remained significantly lower (Table 4). 24‐hours patterns in caloric intake and physical activity did not differ between high fat and control diet fed mice during time‐of‐day‐restricted feeding (Figure 6C). The amplitude of day‐night differences in energy expenditure was also comparable between control and high fat diet fed mice (Figure 6C). Importantly, 24‐hours oscillations in RER were observed in both control and high fat fed mice during time‐of‐day restricted feeding (although the amplitude was lower in high fat fed mice; Figure 6C). Collectively, these observations suggest that whole body metabolic rhythms are partially restored in obese mice through time‐of‐day‐restricted feeding.
FIGURE 6.
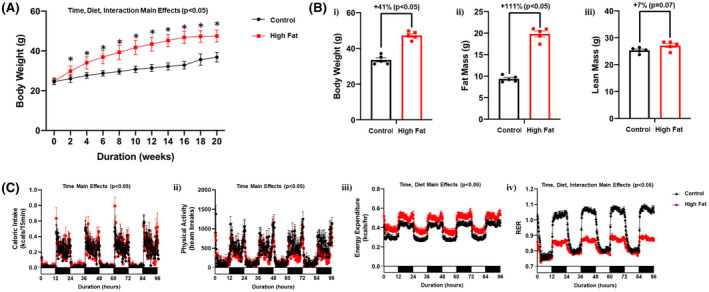
Time‐of‐day‐restricted feeding partially restores whole body diurnal metabolic flexibility in obese mice. A, Body weight was assessed once every 2‐wk, in mice that were fed either a control or high fat diet in an ad libitum fashion for 18‐wk, followed by food access only during the 12‐h dark (active) phase for 2‐wk (n = 19‐20). B, After 18‐wk of control or high fat diet feeding in an ad libitum fashion plus 2‐wk dark‐phase restricted feeding, body weight (i), fat mass (ii) and lean mass (iii) were assessed (n = 5). C, 24‐h patterns in caloric intake (i), physical activity (ii), energy expenditure (iii), and respiratory exchange ratio (RER; iv) were assessed during the first week of the dark‐phase restricted feeding protocol (n = 20‐21). Data are reported as mean ± SEM. Main effects for either time, diet, and/or interaction are reported at the top of the figure panels. *P < .05 for control (circle) vs high fat (square)
TABLE 4.
Caloric intake, physical activity, energy expenditure, and RER in time‐of‐day‐restricted control and high fat diet fed mice
| Control diet Ad libitum | High fat diet Ad libitum | |
|---|---|---|
| 24‐h cumulative caloric intake (kcal/24‐h) | 13.4 ± 0.4 | 13.9 ± 0.9 |
| 24‐h cumulative physical activity (beam breaks; AU) | 36 345 ± 2131 | 28 252 ± 1422* |
| 24‐h average energy expenditure (kcals/h) | 0.352 ± 0.019 | 0.429 ± 0.028* |
| 24‐h average respiratory exchange ratio | 0.944 ± 0.006 | 0.836 ± 0.003* |
Caloric intake, physical activity, energy expenditure, and RER were monitored continuously using a CLAMS. Data are reported as mean ± SEM for 20‐21 mice per experimental group.
P < .05 for pair wise comparison between dietary groups.
The impact of time‐of‐day‐restricted feeding for 2 weeks on cardiac remodeling was next assessed at functional, gravimetric, and histologic levels. No echocardiographic parameters were significantly different between control and high fat diet fed mice after the 2‐wk time‐of‐day‐restricted feeding period (Table 5). Similarly, the BVW/TL ratio and cardiomyocyte size was not significantly different between the two experimental groups (although a trend was observed for both parameters to be increased in high fat fed mice; Figure 7A,B). Although cardiac fibrosis remained significantly increased in high fat fed mice (Figure 7B) following 2‐wk time‐of‐day‐restricted feeding, cardiac steatosis was not different between the two feeding groups (Figure 7C,D). Collectively, these observations reveal that restricting food intake to the active period for only 2 weeks partially normalizes adverse cardiac remodeling in obese mice.
TABLE 5.
Echocardiographic parameters in time‐of‐day‐restricted control and high fat diet fed mice
| Control diet time‐restricted | High fat diet time‐restricted | |
|---|---|---|
| End systolic volume (uL) | 27.5 ± 4.8 | 26.3 ± 5.5 |
| End diastolic volume (uL) | 59.9 ± 4.3 | 64.6 ± 6.9 |
| Stroke volume (uL) | 32.4 ± 2.3 | 38.4 ± 3.1 |
| Ejection fraction (%) | 55.2 ± 5.5 | 60.7 ± 5.2 |
| Fractional shortening (%) | 28.6 ± 3.7 | 32.5 ± 3.6 |
| Left ventricular posterior wall thickness, systole (mm) | 1.23 ± 0.07 | 1.32 ± 0.06 |
| Left ventricular posterior wall thickness, diastole (mm) | 0.93 ± 0.06 | 1.02 ± 0.08 |
| Left ventricular inner diameter, systole (mm) | 2.68 ± 0.20 | 2.62 ± 0.23 |
| Left ventricular inner diameter, diastole (mm) | 3.74 ± 0.11 | 3.85 ± 0.17 |
| Intra‐ventricular septum thickness, systole (mm) | 1.35 ± 0.04 | 1.72 ± 0.22 |
| Intra‐ventricular septum thickness, diastole (mm) | 1.03 ± 0.03 | 1.15 ± 0.06 |
Data are reported as mean ± SEM for five mice per experimental group.
FIGURE 7.
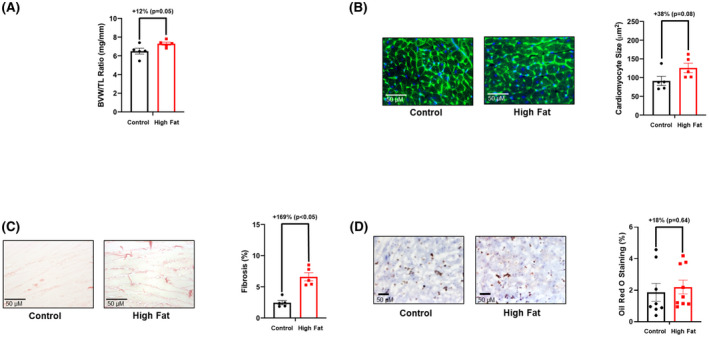
Time‐of‐day‐restricted feeding partially reverses adverse remodeling markers in hearts of high fat fed mice. A, Biventricular weight to tibia length (BVW/TL) ratio was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 18‐wk, followed by food access only during the 12‐h dark (active) phase for 2‐wk (n = 5). B, Cardiomyocyte size was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 18‐wk, followed by food access only during the 12‐h dark (active) phase for 2‐wk (n = 5). C, Cardiac fibrosis was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 18‐wk, followed by food access only during the 12‐h dark (active) phase for 2‐wk (n = 5). D, Cardiac steatosis was determined in mice that were fed either a control or high fat diet in an ad libitum fashion for 18‐wk, followed by food access only during the 12‐h dark (active) phase for 2‐wk (n = 5). Data are reported as mean ± SEM
3.6. Time‐of‐day‐restricted feeding selectively augments day‐night differences in cardiac lipid metabolism during obesity
We next tested the hypothesis that time‐of‐day‐restricted feeding augments day‐night differences in cardiac metabolism (similar to that observed for whole body metabolism; Figure 6C). Contrary to this hypothesis, no significant day‐night differences were observed in cardiac glucose oxidation, oleate oxidation, [14C]‐labeled lactate release, net lactate release, or net glycogen synthesis when mice were subjected to time‐of‐day‐restricted feeding (neither control nor high fat fed mice; Figure 8A,B). However, significant day‐night differences were observed for triglyceride synthesis in hearts isolated from both control and high fat fed mice following 2‐wk time‐of‐day‐restricted feeding (Figure 8A,B). Importantly, no day‐night differences were observed in functional parameters in hearts isolated from either control or high fat fed mice (Table 6). Lipidomic analysis was next performed, revealing augmented day‐night differences in cardiac lipid species following time‐of‐day‐restricted feeding (Figure 8C). Using a q‐value, 47 cardiac lipid species exhibited significant day‐night differences in control fed mice following 2‐wk time‐of‐day‐restricted feeding; 42 were triglycerides, 2 were diglycerides, 1 was a cholesterol ester, and 1 was a ceramide (Figure 8C and Table S7). Similarly, 15 cardiac lipid species exhibited significant day‐night differences in high fat fed mice following 2‐wk time‐of‐day‐restricted feeding (using a q‐value); 13 were triglycerides, 1 was a phospholipid, and 1 was a ceramide (Figure 8C and Table S8). Interestingly, prominent day‐night differences are observed in both carbon chain length and degree of unsaturation of fatty acyl species of triglycerides in the heart following 2‐wk time‐of‐day‐restricted feeding; both parameters are lower during the dark (active) period (Figure 8D). Collectively, these observations suggest that restricting feeding to the active period selectively augments day‐night differences in cardiac lipid metabolism.
FIGURE 8.
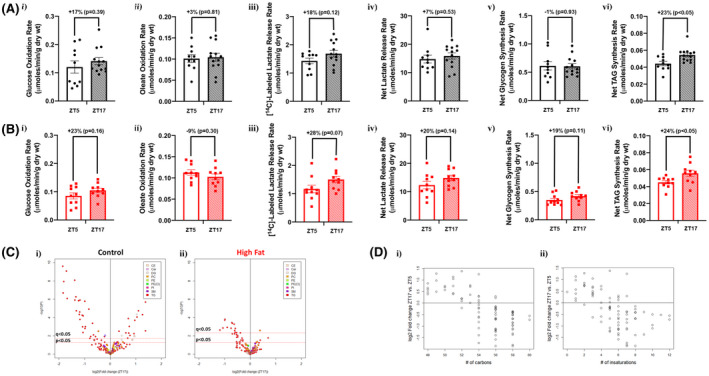
Time‐of‐day‐restricted feeding partially restores diurnal metabolic flexibility of cardiac nonoxidative lipid metabolism in obese mice. Hearts were isolated from mice (at ZT5 and ZT17) that were fed either a control or high fat diet in an ad libitum fashion for 18‐wk, followed by food access only during the 12‐h dark (active) phase for 2‐wk. A, Rates of cardiac glucose oxidation (i), fatty acid oxidation (ii), [14C]‐labeled lactate release (iii), net lactate release (iv), net glycogen synthesis (v), and net triglyceride synthesis (vi) in control diet fed mice (n = 10‐13). B, Rates of cardiac glucose oxidation (i), fatty acid oxidation (ii), [14C]‐labeled lactate release (iii), net lactate release (iv), net glycogen synthesis (v), and net triglyceride synthesis (vi) in high fat diet fed mice (n = 10). C, Volcano plots indicating day‐night differences in the cardiac lipidome of control (i) and high fat fed (ii) mice; color indicates distinct lipid species classification (n = 8‐9). D, Relationship between MS signal intensity values for individual annotated triglycerides in (i) control and (ii) high fat fed mice expressed as fold changes between ZT17 and ZT5 (log2) with carbon chain length (i) and degree of unsaturation (ii) of cardiac lipid species (n = 8‐9)
TABLE 6.
Functional parameters for ex vivo perfused hearts isolated from time‐of‐day‐restricted control and high fat diet fed mice
| Control diet time‐restricted | High fat diet time‐restricted | |||
|---|---|---|---|---|
| ZT5 | ZT17 | ZT5 | ZT17 | |
| Cardiac power (CP; mW) | 1.32 ± 0.08 | 1.36 ± 0.08 | 1.24 ± 0.06 | 1.19 ± 0.07 |
| Heart rate (BPM) | 307 ± 13 | 314 ± 10 | 293 ± 15 | 297 ± 17 |
| Developed Pressure (mm Hg) | 26.7 ± 2.2 | 31.6 ± 1.9 | 29.8 ± 2.3 | 30.7 ± 2.0 |
| Rate pressure product (BPM × mm Hg) | 8107 ± 655 | 9816 ± 534 | 8728 ± 770 | 8940 ± 569 |
| Oxygen consumption (MVO2; umoles/min/g dry wt) | 8.02 ± 1.22 | 9.45 ± 0.83 | 7.86 ± 0.92 | 7.60 ± 0.88 |
| Cardiac efficiency (CP/MVO2) | 0.19 ± 0.02 | 0.15 ± 0.01 | 0.17 ± 0.02 | 0.17 ± 0.02 |
Data are reported as mean ± SEM for 10‐13 mice per experimental group.
4. DISCUSSION
The purpose of the present study was to investigate whether diurnal metabolic inflexibility occurs in the heart during obesity. Chronic high fat feeding‐induced obesity (ie, increased body weight and adiposity; Figure 1A,B) and abolished diurnal rhythms in whole body substrate selection (ie, loss of RER 24‐hours oscillations; Figure 1C). RNAseq analysis revealed dramatic alterations in the diurnal landscape of the cardiac transcriptome during obesity; many differentially oscillating genes in hearts of obese mice have established metabolic functions (Figure 3). However, subsequent investigation of cardiac substrate oxidation surprisingly revealed a persistence of day‐night differences in glucose oxidation in obese mice (Figure 4). In contrast, day‐night differences in cardiac triglyceride synthesis were aberrant in obese mice, which was associated with a dramatic dysregulation of day‐night fluctuations in the cardiac lipidome (Figures 4 and 5). Next, food availability was restricted to the 12‐hours dark (awake) period, in an attempt to normalize day‐night differences in cardiac lipid metabolism during obesity. This intervention partially restored 24‐hours oscillations in whole body substrate selection in obese mice (Figure 6C). Similarly, time‐of‐day‐restricted feeding augmented day‐night differences in cardiac triglyceride synthesis and lipidome (Figure 8). This was associated with a partial reversal of adverse cardiac remodeling in obese mice after 2‐wks (Figures 2 and 7). Collectively, these data reveal selective diurnal metabolic inflexibility for cardiac lipid metabolism during obesity, which is partially restored by modulating the timing of food intake.
The healthy myocardium exhibits profound metabolic plasticity. Perturbations in workload, substrate availability, and multiple neurohumoral factors are typically met with appropriate shifts in cardiac metabolic fluxes, thus, ensuring adequate contractile performance, both in the short‐ and long‐ term. In contrast, cardiac metabolic inflexibility (defined as an inability of metabolic processes to shift appropriately in response to physiologic stimuli) has been reported during various disease states. 15 Examples include chronic augmentation of cardiac glucose oxidation, fatty acid oxidation, and glycolysis during pressure overload, obesity/diabetes, and heart failure, respectively. 11 , 14 , 16 , 38 , 39 In the case of obesity, evidence supports the concept of impaired cardiac energetic reserve contributing toward contractile dysfunction (particularly in response to increased workload). 40 However, the extent to which myocardial substrate selection is inflexible during obesity is less clear. Although the heart increases fatty acid oxidation rates during diet‐induced obesity, isolated perfused hearts from control and high fat fed mice exhibit comparable changes in substrate switching when acutely challenged with either high workloads or increased fatty acid availability. 20 , 41 Similarly, evidence exists suggesting that the heart can also switch between substrates during diabetes. 42 In contrast, hearts of obese Zucker rats (a genetic model of obesity) fail to increase fatty acid oxidation in response to fasting 43 ; the latter may be secondary to intrinsic myocardial metabolic inflexibility in this particular model and/or a differential neurohumoral response to fasting (ie, extracardiac contribution). The possibility, therefore, remains that the heart during obesity retains some level of substrate selection flexibility (albeit in the presence of mitochondrial dysfunction and impaired energetic reserve).
Workload, substrate availability, and neurohumoral factors fluctuate over the course of the day, in association with sleep‐wake and fasting‐feeding cycles. 44 , 45 It is, therefore, not surprising that cardiac metabolism is temporally partitioned over a 24‐hours period. 17 In general, the heart increases oxidative metabolism during the active period, promotes nutrient storage toward the end of the active period, while turnover of cellular constituents appears to be augmented during the sleep period. 17 It has been hypothesized that disruption of cardiac metabolism temporal partitioning may contribute toward cardiac dysfunction development. 17 For example, failure to increase cardiac oxidative metabolism upon awakening would limit energetic reserve in the face of increased workload, while failure to replace damaged cellular constituents during the sleep period would be anticipated to precipitate cardiomyocyte dysfunction. To date, no studies have investigated directly whether 24‐hours rhythms in cardiac metabolism are altered in a disease state. Only indirect evidence currently exists, primarily at the transcriptional level. For example, diurnal rhythms in distinct transcripts encoding for metabolism‐related proteins are altered in the heart during pressure overload and diabetes, as well as following a myocardial infarction. 46 , 47 , 48 However, given that gene expression cannot always predict metabolic flux accurately, 49 there is a need to better understand whether diurnal metabolic inflexibility occurs during cardiac disease states.
Here, we hypothesized that obesity would attenuate day‐night differences in cardiac glucose and fatty acid metabolism (the primary substrates utilized by the heart for oxidative metabolism). Consistent with prior studies, 19 , 23 , 28 both whole body (evidenced by a rise in RER) and cardiac glucose oxidation increase during the dark (awake) period when mice are fed a control (normal) diet (Figures 1C and 4A). Conversely, whole body (evidenced by a decline in RER) and cardiac fatty acid oxidation increase during the light (sleep) period (although this did not reach statistical significant for cardiac fatty acid oxidation; Figures 1C and 4A). High fat feeding‐induced obesity abolished 24‐hours rhythms in whole body substrate selection (Figure 1C). However, contrary to our hypothesis, day‐night differences in both cardiac glucose and fatty acid oxidation were essentially identical between control and high fat fed mice (ie, diurnal metabolic flexibility is preserved for cardiac substrate oxidation during obesity; Figure 4A,B). This is despite a dramatic perturbation of the diurnal landscape of the cardiac transcriptome during obesity (Figure 3). More specifically, only 22% of the transcripts that oscillate in control hearts are unaffected by chronic high fat feeding. Interestingly, these unaffected genes include core circadian clock components (bmal1, clock, npas2, nr1d1, nr1d2, per2, per3, and cry2). Circadian clocks are cell autonomous molecular mechanisms composed of a series of transcriptional positive and negative feedback loops, which temporally orchestrate biological processes, thus, ensuring that they occur at an appropriate time of the day. 50 Prior studies have reported that genetic disruption of the cardiomyocyte circadian clock in mice abolishes 24‐hours rhythms in cardiac metabolism (despite normal sleep‐wake and fasting‐feeding cycles). 19 , 20 , 28 This includes cardiac glucose metabolism, in part through regulation of glucose uptake 19 ; interestingly, diurnal variations in both scl2a1 and slc2a4 (encoding for GLUT1 and GLUT4, respectively) mRNA levels persist in hearts of high fat fed mice (unpublished observations). These observations raise the possibility that the cardiomyocyte circadian clock orchestrates persistent day‐night differences in cardiac oxidative metabolism during obesity.
In contrast to oxidative metabolism, day‐night differences in nonoxidative lipid metabolism were notably impacted in the heart during obesity. More specifically, day‐night differences in cardiac triglyceride synthesis were markedly attenuated in high fat fed mice (Figure 4A,B). Moreover, targeted lipidomics revealed that of the 40 lipid species exhibiting day‐night differences in control hearts, only 2 exhibit day‐night differences in hearts from high fat fed mice (both of which have attenuated diurnal patterns compared to controls; Figure 5D‐G). In addition, 10 lipid species exhibit day‐night differences uniquely in hearts of obese mice. These profound alterations in the diurnal lipidome of the heart during obesity were associated with a large number of differentially rhythmic transcripts in hearts of obese mice, with known functions in fatty acid and lipid metabolism. The latter include agpat3 (1‐acylglycerol‐3‐phosphate O‐acyltransferase 3), acot1 (acyl‐coA thioesterase 1), sgpl1 (sphingosine‐1‐phosphate lyase 1), and fabp4 (fatty acid binding protein 4) (Table S3), as well as cd36, lcad, and lipe (Figure 5A). In contrast, mRNAs known to encode for specific key regulators of triglyceride metabolism (srebf1, mlxipl, nr1h3, pnpla2, and abhd5, encoding for SREBP1, CHREBP, LXRα, ATGL, and CGI58, respectively) were not significantly rhythmic in hearts of control and/or high fat fed mice (Tables S1‐S3). Interestingly, diurnal variations in plasma NEFA levels were also markedly disrupted in high fat fed mice (Figure 5C). Unfortunately, plasma triglyceride and glucose were not assessed in the current study; diurnal variations in these circulating substrates have been reported previously to be altered during prolonged high fat feeding. 51 , 52 Collectively, these data suggest that during obesity, cardiac nonoxidative lipid metabolism displays diurnal metabolic inflexibility, potentially due to changes in factors that are intrinsic (eg, gene expression) and extrinsic (eg, neurohumoral milieu) to the myocardium.
Multiple studies suggest that the time of day at which nutrients are consumed influences the development of cardiometabolic diseases. 53 In preclinical models, preventing high fat diet consumption during the sleep period protects against development of adiposity, dyslipidemia, hyperinsulinemia, and glucose intolerance. 54 Conversely, enforcing high fat diet consumption during the sleep period augments these cardiometabolic parameters. 54 In terms of cardiac function, we have previously reported that removal of food during the sleep period prevented chronic (16‐wk) high fat feeding‐induced contractile dysfunction in mice. 29 Subsequently, we observed that a daily 4‐hours high fat meal at the end of the active period‐induced cardiac dysfunction in mice (compared to the same 4‐hours meal at the beginning of the active period). 55 Moreover, acutely challenging the heart with lipid during the sleep period attenuated both cardiac contractility and efficiency; the same lipid challenge was without effect when performed during the active period. 56 Collectively, these studies suggest that lipids exert detrimental effects on the heart at the end of the active period and/or during the sleep period. However, whether this knowledge can be utilized for an effective treatment (as opposed to preventative) strategy is less clear. In addition, although several studies report whole body metabolic responses to time‐of‐day‐restricted feeding, little is known regarding its influence on cardiac metabolism. Here, we report that preventing food intake during the light (sleep) phase augments day‐night differences in both cardiac triglyceride synthesis and lipidome in obese mice (Figure 8). In contrast, this intervention blunted day‐night differences in substrate oxidation (in both control and high fat fed mice; Figure 8A,B). Importantly, 2‐wk of time‐of‐day restricted feeding partially normalized cardiac mass, wall thickness, cardiomyocyte size, and cardiac steatosis in obese mice, with an attenuation of cardiac fibrosis (Figures 2 and 7). These data reveal that changing the time‐of‐day at which food is consumed, as opposed to macronutrient quantity/quality, can elicit cardiovascular benefit during obesity. Future studies are required to determine the precise mechanisms by which time‐of‐day‐restricted feeding confers cardiovascular benefit.
In summary, the current study reveals a persistence of diurnal metabolic flexibility for cardiac substrate oxidation during obesity. In contrast, nonoxidative lipid metabolism develops diurnal metabolic inflexibility in the heart during obesity. We also report that restricting food intake to the 12‐hours dark (active) period in obese mice partially restores day‐night difference in cardiac lipid metabolism, as well as partially reverses adverse cardiac remodeling. Given that prior studies have reported that skipping breakfast and late night snacking increases risk of heart disease, our findings are consistent with the concept that calorically dense meals should be consumed toward the beginning of the day, in order to promote heart health. 53 , 57 , 58
CONFLICT OF INTEREST
The authors have stated explicitly that there are no conflicts of interest in connection with this article.
AUTHOR CONTRIBUTIONS
A. Diwan, S. Frank, C. Des Rosiers, and M.E. Young designed research; S. Mia, R. Sonkar, M.N. Latimer, I.F. Robillard, and M.E. Young performed research; R. Sonkar, L. Williams, M.N. Latimer, I.F. Robillard, and M.E. Young analyzed data; R. Sonkar, C. Des Rosiers, and M.E. Young wrote the manuscript.
Supporting information
DataSet S1
DataSet S2
DataSet S3
DataSet S4
DataSet S5
DataSet S6
DataSet S7
DataSet S8
ACKNOWLEDGMENTS
This work was supported by the National Heart, Lung, and Blood Institute (HL123574 and HL142216), and benefited from an infrastructure grant supported by the Canadian Foundation for Innovation and by the Montreal Heart Foundation. We would like to thank Maximiliano Grenett and Stephanie Reed for technical assistance.
Mia S, Sonkar R, Williams L, et al. Impact of obesity on day‐night differences in cardiac metabolism. The FASEB Journal. 2021;35:e21298. 10.1096/fj.202001706RR
REFERENCES
- 1. Bhupathiraju SN, Hu FB. Epidemiology of obesity and diabetes and their cardiovascular complications. Circ Res. 2016;118:1723‐1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Poirier P, Giles TD, Bray GA, et al. Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation. 2006;113:898‐918. [DOI] [PubMed] [Google Scholar]
- 3. GBD 2016 Causes of Death Collaborators . Global, regional, and national age‐sex specific mortality for 264 causes of death, 1980–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1151‐1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics‐‐2015 update: a report from the American Heart Association. Circulation. 2015;131:e29‐322. [DOI] [PubMed] [Google Scholar]
- 5. Brown CD, Higgins M, Donato KA, et al. Body mass index and the prevalence of hypertension and dyslipidemia. Obes Res. 2000;8:605‐619. [DOI] [PubMed] [Google Scholar]
- 6. Kenchaiah S, Evans JC, Levy D, et al. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305‐313. [DOI] [PubMed] [Google Scholar]
- 7. Hubert HB, Feinleib M, McNamara PM, Castelli WP. Obesity as an independent risk factor for cardiovascular disease: a 26‐year follow‐up of participants in the Framingham Heart Study. Circulation. 1983;67:968‐977. [DOI] [PubMed] [Google Scholar]
- 8. Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251‐259. [DOI] [PubMed] [Google Scholar]
- 9. Ebong IA, Goff DC Jr, Rodriguez CJ, Chen H, Bertoni AG. Mechanisms of heart failure in obesity. Obes Res Clin Pract. 2014;8:e540‐e548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. 2008;88:389‐419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Allard M, Schonekess B, Henning S, English D, Lopaschuk G. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. 1994;267:H742‐H750. [DOI] [PubMed] [Google Scholar]
- 12. Goodwin G, Taylor C, Taegtmeyer H. Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem. 1998;273:29530‐29539. [DOI] [PubMed] [Google Scholar]
- 13. Brewer RA, Collins HE, Berry RD, et al. Temporal partitioning of adaptive responses of the murine heart to fasting. Life Sci. 2018;197:30‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207‐258. [DOI] [PubMed] [Google Scholar]
- 15. Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci. 2004;1015:202‐213. [DOI] [PubMed] [Google Scholar]
- 16. Young M, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation. 2002;105:1861‐1870. [DOI] [PubMed] [Google Scholar]
- 17. Young ME. Temporal partitioning of cardiac metabolism by the cardiomyocyte circadian clock. Exp Physiol. 2016;101:1035‐1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Degaute JP, van de Borne P, Linkowski P, Van Cauter E. Quantitative analysis of the 24‐hour blood pressure and heart rate patterns in young men. Hypertension. 1991;18:199‐210. [DOI] [PubMed] [Google Scholar]
- 19. Durgan DJ, Pat BM, Laczy B, et al. O‐GlcNAcylation, novel post‐translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606‐44619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsai JY, Kienesberger PC, Pulinilkunnil T, et al. Direct regulation of myocardial triglyceride metabolism by the cardiomyocyte circadian clock. J Biol Chem. 2010;285:2918‐2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McGinnis GR, Tang Y, Brewer RA, et al. Genetic disruption of the cardiomyocyte circadian clock differentially influences insulin‐mediated processes in the heart. J Mol Cell Cardiol. 2017;110:80‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Young ME, Brewer RA, Peliciari‐Garcia RA, et al. Cardiomyocyte‐specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms. 2014;29:257‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bray MS, Ratcliffe WF, Grenett MH, Brewer RA, Gamble KL, Young ME. Quantitative analysis of light‐phase restricted feeding reveals metabolic dyssynchrony in mice. Int J Obes (Lond). 2013;37:843‐852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ingle KA, Kain V, Goel M, Prabhu SD, Young ME, Halade GV. Cardiomyocyte‐specific Bmal1 deletion in mice triggers diastolic dysfunction, extracellular matrix response, and impaired resolution of inflammation. Am J Physiol Heart Circ Physiol. 2015;309:H1827‐H1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Durgan DJ, Tsai JY, Grenett MH, et al. Evidence suggesting that the cardiomyocyte circadian clock modulates responsiveness of the heart to hypertrophic stimuli in mice. Chronobiol Int. 2011;28:187‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goodpaster BH, Theriault R, Watkins SC, Kelley DE. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metab Clin Exp. 2000;49:467‐472. [DOI] [PubMed] [Google Scholar]
- 27. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. Anal Biochem. 1987;162:156‐159. [DOI] [PubMed] [Google Scholar]
- 28. Bray M, Shaw C, Moore M, et al. Disruption of the circadian clock within the cardiomyocyte influences myocardial contractile function; metabolism; and gene expression. Am J Physiol Heart Circ Physiol. 2008;294:H1036‐H1047. [DOI] [PubMed] [Google Scholar]
- 29. Tsai JY, Villegas‐Montoya C, Boland BB, et al. Influence of dark phase restricted high fat feeding on myocardial adaptation in mice. J Mol Cell Cardiol. 2013;55:147‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gibson UE, Heid CA, Williams PM. A novel method for real time quantitative RT‐PCR. Genome Res. 1996;6:995‐1001. [DOI] [PubMed] [Google Scholar]
- 31. Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6:986‐994. [DOI] [PubMed] [Google Scholar]
- 32. Forest A, Ruiz M, Bouchard B, et al. Comprehensive and reproducible untargeted lipidomic workflow using LC‐QTOF validated for human plasma analysis. J Proteome Res. 2018;17:3657‐3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ruiz M, Cuillerier A, Daneault C, et al. Lipidomics unveils lipid dyshomeostasis and low circulating plasmalogens as biomarkers in a monogenic mitochondrial disorder. JCI insight. 2019;4:e123231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu G, Anafi RC, Hughes ME, Kornacker K, Hogenesch JB. MetaCycle: an integrated R package to evaluate periodicity in large scale data. Bioinformatics. 2016;32:3351‐3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90‐W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fabregat A, Jupe S, Matthews L, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46:D649‐D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140‐1151. [DOI] [PubMed] [Google Scholar]
- 39. Wende AR, Brahma MK, McGinnis GR, Young ME. Metabolic origins of heart failure. JACC Basic Transl Sci. 2017;2:297‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luptak I, Sverdlov AL, Panagia M, et al. Decreased ATP production and myocardial contractile reserve in metabolic heart disease. J Mol Cell Cardiol. 2018;116:106‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet‐induced obesity. Circulation. 2009;119:2818‐2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Larsen TS, Aasum E. Metabolic (in)flexibility of the diabetic heart. Cardiovasc Drugs Ther. 2008;22:91‐95. [DOI] [PubMed] [Google Scholar]
- 43. Young M, Guthrie P, Razeghi P, et al. Impaired long chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51:2587‐2595. [DOI] [PubMed] [Google Scholar]
- 44. Martino TA, Young ME. Influence of the cardiomyocyte circadian clock on cardiac physiology and pathophysiology. J Biol Rhythms. 2015;30:183‐205. [DOI] [PubMed] [Google Scholar]
- 45. Gamble KL, Berry R, Frank SJ, Young ME. Circadian clock control of endocrine factors. Nat Rev Endocrinol. 2014;10:466‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Young M, Wilson C, Razeghi P, Guthrie P, Taegtmeyer H. Alterations of the circadian clock in the heart by streptozotocin‐induced diabetes. J Mol Cell Cardiol. 2002;34:223‐231. [DOI] [PubMed] [Google Scholar]
- 47. Kung T, Egbejimi O, Cui J, et al. Rapid attenuation of circadian clock gene oscillations in the rat heart following ischemia‐reperfusion. J Mol Cell Cardiol. 2007;43:744‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Young M, Razeghi P, Cedars A, Guthrie P, Taegtmeyer H. Intrinsic diurnal variations in cardiac metabolism and contractile function. Circ Res. 2001;89:1199‐1208. [DOI] [PubMed] [Google Scholar]
- 49. Taegtmeyer H, Young ME, Lopaschuk GD, et al. Assessing cardiac metabolism: a scientific statement from the American Heart Association. Circ Res. 2016;118:1659‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kohsaka A, Laposky AD, Ramsey KM, et al. High‐fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007;6:414‐421. [DOI] [PubMed] [Google Scholar]
- 52. De Gasquet P, Griglio S, Pequignot‐Planche E, Malewiak MI. Diurnal changes in plasma and liver lipids and lipoprotein lipase activity in heart and adipose tissue in rats fed a high and low fat diet. J Nutr. 1977;107:199‐212. [DOI] [PubMed] [Google Scholar]
- 53. St‐Onge MP, Ard J, Baskin ML, et al. Meal timing and frequency: implications for cardiovascular disease prevention: a scientific statement from the American Heart Association. Circulation. 2017;135:e96‐e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bray MS, Tsai JY, Villegas‐Montoya C, et al. Time‐of‐day‐dependent dietary fat consumption influences multiple cardiometabolic syndrome parameters in mice. Int J Obes (Lond). 2010;34:1589‐1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peliciari‐Garcia RA, Goel M, Aristorenas JA, et al. Altered myocardial metabolic adaptation to increased fatty acid availability in cardiomyocyte‐specific CLOCK mutant mice. Biochim Biophys Acta. 2016;1860:1579‐1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Durgan D, Moore M, Ha N, et al. Circadian rhythms in myocardial metabolism and contractile function: influence of workload and oleate. Am J Physiol Heart Circ Physiol. 2007;293:H2385‐H2393. [DOI] [PubMed] [Google Scholar]
- 57. Cahill LE, Chiuve SE, Mekary RA, et al. Prospective study of breakfast eating and incident coronary heart disease in a cohort of male US health professionals. Circulation. 2013;128:337‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rong S, Snetselaar LG, Xu G, et al. Association of Skipping Breakfast With Cardiovascular and All‐Cause Mortality. J Am Coll Cardiol. 2019;73:2025‐2032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DataSet S1
DataSet S2
DataSet S3
DataSet S4
DataSet S5
DataSet S6
DataSet S7
DataSet S8