

Abstract
The targeting of natural tolerogenic liver sinusoidal endothelial cells (LSEC) by nanoparticles (NPs), decorated with a stabilin receptor ligand, is capable of generating regulatory T-cells (Tregs), which can suppress antigen-specific immune responses, including to ovalbumin (OVA), a possible food allergen. In this regard, we have previously demonstrated that OVA-encapsulating PLGA nanoparticles eliminate allergic airway inflammation in OVA-sensitized mice, prophylactically and therapeutically. A competing approach is a nanocarrier platform that incorporates pharmaceutical agents interfering in mTOR (rapamycin) or NF-κB (curcumin) pathways, with the ability to induce a tolerogenic state in non-targeted antigen-presenting cells (APC) system-wide. First, we compared OVA-encapsulating, LSEC-targeting tolerogenic nanoparticles (TNPs) with non-targeted NPs incorporating curcumin and rapamycin (Rapa) in a murine eosinophilic airway inflammation model, which is Treg sensitive. This demonstrated roughly similar tolerogenic effects on allergic airway inflammation by stabilin-targeting NPOVA vs. non-targeted NPs delivering OVA plus Rapa. Reduction in eosinophilic inflammation and TH2-mediated immune responses in the lung were accompanied by increased Foxp3+ Treg recruitment and TGF-β production in both platforms. Since OVA incorporates IgE-binding as well as non-IgE binding epitopes, the next experiment explored the possibility of obtaining immune tolerance by non-anaphylactic T-cell epitopes. This was accomplished by incorporating OVA323-339 and OVA257-264 epitopes in liver-targeting NP to assess the prophylactic and therapeutic impact on allergic inflammation in transgenic OT-II mice. Importantly, we demonstrated that the MHC-II binding (former) but not the MHC-I binding (latter) epitope interfered in allergic airway inflammation, improving TNPOVA efficacy. The epitope-specific effect was transduced by TGF-β producing Tregs. In the final phase of experimentation, we used an OVA-induced anaphylaxis model to demonstrate that targeted delivery of OVA and its MHC-II epitope could significantly suppress the anaphylaxis symptom score, mast cell release, and the late-phase inflammatory response. In summary, these results demonstrate comparable efficacy of LSEC-targeting vs. pharmaceutical PLGA nanoparticles, as well as the ability of T-cell epitopes to achieve similar response outcomes as the intact allergens.
Keywords: immune tolerance, nanoparticles, LSECs, pharmacological regulator, T-cell epitope
Graphical Abstract
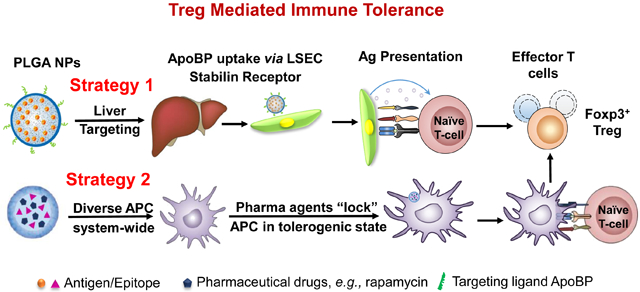
The induction of effective and durable antigen-specific immune tolerance has become a major objective for the long-term treatment of allergy and autoimmune diseases.1-3 This strategy avoids the immune suppressive effects of anti-inflammatory drugs, immunomodulatory agents, and monoclonal antibodies, which could enhance susceptibility to opportunistic infections or interfere in immune surveillance for cancer.4-6 A number of important recent advances have demonstrated the promise of using antigen-specific immune tolerance to alleviate or eliminate overactive immunity in the setting of autoimmune disease (e.g., rheumatoid arthritis, type I diabetes, experimental autoimmune encephalomyelitis) or life-threatening allergic disorders (e.g., food allergy, anaphylaxis, asthma).6-9 With this illumination comes the recognition of the powerful role of regulatory T-cells (Treg) in controlling antigen-specific tolerance.1,1, 10, 11 Against this backdrop, the introduction of multifunctional nanoparticles offers the advantage to improve engagement of the pathways leading to Treg generation based on the ability to target antigen-presenting cells (APC), which play a key role in engaging complementary and competing arms of the immune system in responding to foreign and self-antigens.2, 5, 12, 13 This awareness has sparked the development of a number of tolerogenic nanoparticle (TNP) platforms that leverage the unique properties of nanomaterials to modify the outcome of the immune response to allergens and autoimmune antigens, including to immunogenic epitopes displayed by these proteins.13-22
Among the emerging nanoparticle platforms, there are currently two major approaches for achieving antigen-specific immune modulation by an impact on cognate immunity and APC function (Fig. 1A). The first approach is to directly interfere in the function or elimination of antigen-specific, autoreactive T-cells by perturbing tolerogenic pathways intrinsic to T- and B-lymphocytes.18, 19, 23-25 This includes nanocarriers that use antigens or specific epitopes to induce anergy and/or apoptosis of cognate autoreactive T- and/or B-cells. One example includes SIGLEC-engaging tolerance-inducing antigenic liposomes (STALs), which use the display of an antigen plus a CD22 glycan ligand to induce apoptosis in mouse and human B-cells.26, 27 This allows targeting of a selective repertoire of antigens among the diversity of immunogens that are responsible for allergic and autoimmune disorders.13 The second approach seeks to induce dominant T- and B-cell tolerance by targeting APC that expand or reprogram T-cell and/or B-cell effectors into disease-suppressing regulatory cells (Fig. 1A).2, 5 The fundamental advantage of this approach is the sustained activation and expansion of regulatory cells, capable of comprehensive interference in the recruitment and activation pathways that allow adaptive and innate immune cells to participate in disease promotion.28, 29 This approach includes delivering antigens to harness natural tolerogenic APCs or to employ pharmaceutical agents for inducing a tolerogenic state in APCs.5, 17, 20, 30-34 Our preference is to target TNPs to the liver, which specializes in immune tolerance and prevention of inflammatory responses to a high load of microbial and foreign antigens being released into the portal circulation from the gastrointestinal tract.1, 35, 36 In particular, our approach has been to target liver sinusoidal endothelial cells (LSECs), which capture and present foreign antigens via the MHC-II complex, as well as releasing transforming growth factor-β (TGF-β) and IL-10 to direct the differentiation of naïve T-cells into Tregs (Fig. 1C).17, 37-39
Figure1.



(A) Schematic showing two main potential strategies for inducing immune tolerance. (A) Strategy 1 depicts a liver-targeting nanoparticle for allergen delivery to LSEC in the liver, where allergen processing and presentation to naïve T-cells induce antigen-specific Foxp3+ Tregs, which are recruited to the site of allergic inflammation in the lung. Strategy 2 shows nanoparticles with/without allergen encapsulation to be co-loaded with pharmaceutical agents that are capable of locking non-targeted APCs, which are distributed system-wide into a tolerogenic state. (B) Schematic showing the PLGA nanoparticle synthesis process, including carrier cargo loading (allergen, epitopes, pharmaceutical agents), using a w/o/w double emulsion method combined with solvent removal. The lower panel describes the ApoBP peptide surface attachment onto the particle surface by a NAEM spacer, using a two-step conjugation process to link the ApoBP cysteine group to the NAEM maleimide group.39 (C) The schematic in the left panel shows a working model for liver-targeting tolerogenic nanoparticles. Particles in the ~200 nm size range and attached ApoBP ligand deliver the antigens are epitopes to LSECs in the liver through endocytic uptake. Antigen processing and presentation to naïve T-cells are capable of generating Foxp3+ Tregs, which are recruited to the site of pathology, where they exert their immunosuppressive effects.39 The middle panel shows representative ex vivo IVIS images of the explanted hearts, livers, spleens, lungs, and kidneys collected from animals (24 h after injecting with 500 μg decorated or non-decorated NPs, containing 25 μg Dylight680-labeled OVA (n=4) in an independent experiment to confirm our previous findings.39 The detailed methodology was included in the supporting information. The right panel shows the use of confocal microscopy that reflects the intrahepatic distribution of free and encapsulated OVA. The red and green fluorescence colors represent Dylight680-labeled OVA and isolectin B4 stained LSECs, respectively.39 Quantification of the colocalization between OVA and LSECs was carried out using the calculation of Pearson’s correlation coefficient. Data are expressed as the mean ± SEM.
Another approach is to use nanoparticles loaded with pharmacological agents such as rapamycin, curcumin, and quercetin to promote the development of tolerogenic APC.16, 21, 22, 40 Noteworthy, these particles do not make use of targeting ligands, operating on the presumed basis that systemic dissemination will reach the relevant APC that are engaged in the immune response of interest.16 Rapamycin (Rapa), a natural product from Streptomyces hygroscopicus, binds to the FK506-binding protein to form a complex that allosterically inhibits the mTOR pathway.41, 42 This drug has potent immunosuppressive activity and is capable of interfering in allograft rejection as well as the ability to increase the generation of Foxp3-expressing Tregs as well as expanding inducible Treg populations.43-45 Kishimoto et al. have shown that Rapa-encapsulating PLGA nanoparticles are capable of inducing antigen-specific immune tolerance that can be achieved through the co-administration or co-encapsulation of a variety of antigenic proteins or representative peptides, without the need for liver targeting.16 Similarly, it has been demonstrated that the polyphenol, curcumin, can be used to induce immune tolerance by acting as an inhibitor of the NF-κB pathway, which is critical to APC function.46, 47 In this regard, it has been shown that liposomal co-delivery of antigens with various NF-κB inhibitors, including curcumin, is capable of suppressing inflammatory arthritis in an antigen-specific manner.21 However, despite the proven effectiveness of nanoparticles incorporating pharmaceutical agents, the nature of the non-targeted interaction with APCs is uncertain and no formal comparison has been made to the liver-targeting platform. Such a comparison would be helpful to assess the robustness of the platforms, as well as the possibility that Treg generated in the liver may differ in terms of their immune suppressive effects from the suppressor cells generated by pharmaceutical agents in other locations.
With regards to the treatment of life-threatening allergic disorders, an important consideration in developing tolerogenic platforms to achieve Treg-induced immune tolerance is the use of immunomodulatory T-cell epitopes instead of encapsulating the whole allergen.48, 49 This is advantageous from a manufacturing perspective as well as the ability to avoid peptide sequences that can trigger IgE-dependent hypersensitivity responses by crosslinking FcεR1 receptors on mast cells and basophils.48, 49 T-cell epitopes are discreet, linear peptides of short lengths (~10-20 amino acids) that can be presented to the T-cell antigen receptors (TCR) on CD4+ T-cells by MHC-II molecules, as a prelude to Treg development.49-51 It has been demonstrated in clinical trials that T-cell epitopes can be used for safe immunotherapy that avoids the induction of life-threatening or anaphylactic responses.52, 53 However, despite the utility of epitopes, there are downsides, including shorter half-lives, poor solubility, rapid in Vivo dilution, and poor bioavailability of the peptides.54 These pitfalls provide good justification for encapsulated delivery of T-cell epitopes to LSECs, which after endocytosis of the particles, are transported to the MHC II complex and the APC surface. There is no guarantee, however, that peptide sequences identified by epitope mapping are as effective for generating Treg immune responses as in the setting of using epitopes to generate helper T-cell responses.
In this communication, we compared the tolerogenic efficacy of LSEC-targeting TNPOVA nanoparticles to PLGA particles that deliver pharmaceutical agents (rapamycin and curcumin), with and without OVA co-encapsulation. We demonstrate that while targeting of TNPOVA to LSEC stabilin receptors is slightly less efficient in suppressing allergic airway inflammation compared to Rapa-encapsulating nanoparticles, that curcumin delivery is ineffective. In addition to TNPs delivering OVA, we also compared the prophylactic and therapeutic effects of encapsulated T-cell epitopes, which are presented by MHC-I and MHC-II complexes on LSECs in allergic inflammation in an OT-II mouse model as well as OVA-induced anaphylaxis model in C57Bl/6 mice. Our data demonstrate the success of the liver targeting nanoparticles in alleviating allergic inflammation with comparable success as rapamycin-delivering nanoparticles, as well as the effectiveness of T-cell epitopes on allergic inflammation and anaphylaxis. These results extend the use of the liver targeting platform for applications that can also be performed with T-cell epitopes, including for effective prevention of anaphylaxis.
Results and Discussion
Fabrication and characterization of PLGA NPs
Since the first goal of our study is to compare liver-targeting problem NPOVA with particles incorporating pharmaceutical agents, our synthesis procedure aimed to generate particles with comparable physicochemical characteristics (Fig. 1B). LSEC-targeting PLGA nanoparticles were constructed using a double emulsion technique (Fig. 1B, upper panel), previously described by us.39 OVA was passively encapsulated in the polymer matrix to achieve a loading capacity of 59 μg protein per mg PLGA. These carriers were constructed, either as bare particles (NPOVA) or particles in which the ApoB-100 peptide sequence (RLYRKRGLK, containing a GGC tag) was covalently attached to the particle surface (NPOVA/ApoBP) with a NAEM spacer (Figure 1B, lower panel).39 The coupling density of the peptide was 5 molar % compared to PLGA weight (Table 1). While Apo-B100, the major protein constituent of low-density lipoprotein (LDL), plays a role in LDL binding to LDL receptors.55, 56 LDL is also cleared from the blood by LSECs, which rely on the use of stabilin receptors (particularly stabilin-2) to mediate LDL uptake and degradation.57, 58 We have previously described the efficacy of ApoBP for LSEC targeting to the liver by binding to stabilin receptors, which are expressed on the LSEC surface and cellular interior, but not on Kupffer cells.59 The schematic in Fig. 1 C explains the mechanism of ApoBP targeting to clathrin-coated pits on LSECs, where peptide-mediated receptor binding leads to endocytosis, intracellular transport, and antigen presentation to naïve T-cells.39 Fig. 1 C also shows in Vivo IVIS imaging and confocal microscopy data to substantiate LSEC targeting effects in the liver; this will be discussed below.1
Table 1.
Comparison of different NP formulations for hydrodynamic size, PDI, zeta potential, loading capacity, and cargo encapsulation efficiency.
| Nanoparticle | Hydrodynamic size (nm) |
PDI | Zeta potential (mV) |
Loading capacity (μg/mg NPs) |
Encapsulation efficiency (%) |
OVA content (μg/mg NPs) |
|---|---|---|---|---|---|---|
| Empty NP | 231.2 ± 2.17 | 0.096 | −42.55 ± 2.99 | NA | NA | NA |
| NPOVA | 246.5 ± 3.01 | 0.105 | −44.37 ± 2.81 | NA | NA | 51.61 ± 2.32 |
| NPApoBP | 250.9 ± 3.81 | 0.112 | −11.44 ± 4.02 | 11.65 ± 0.75 (ApoBP ligand) |
5.1 (mol% of PLGA) |
NA |
| NPOVA/ApoBP | 270.8 ± 4.96 | 0.113 | −4.56 ± 2.25 | 12.96 ± 0.77 (ApoBP ligand) |
5.3 (mol% of PLGA) |
50.12 ± 2.18 |
| NPCurcumin | 251.2 ± 3.27 | 0.108 | −35.77 ± 3.52 | 66.12 ± 3.70 | 38.94 ± 2.14 | NA |
| NPOVA-Curcumin | 255.7 ± 4.44 | 0.129 | −38.42 ± 2.95 | 64.52 ± 2.69 | 37.06 ± 3.62 | 50.66 ± 2.68 |
| NPRapa | 246.3 ± 5.47 | 0.120 | −36.28 ± 3.41 | 23.44 ± 3.56 | 33.66 ± 5.24 | NA |
| NPOVA-Rapa | 252.5 ± 3.88 | 0.139 | −39.38 ± 2.99 | 22.31 ± 6.58 | 31.77 ± 3.94 | 46.88 ± 2.76 |
In contrast to the synthesis of targeted particles, the TNPs incorporating the pharmaceutical agents, curcumin and rapamycin, were constructed by passive encapsulation to achieve loading capacities of ~65 μg and ~22 μg per mg particle, respectively (Table 1). These particles were constructed to deliver the pharmaceutical ingredients independently (NPcurcumin or NP Rapa) of the antigen, as well as in combination with OVA (50 μg/mg) by particles, designated NPOVA-curcumin or NPOVA-Rapa. All the fabrication processes were optimized to allow the creation of particles with uniform sizes ~230-250 nm and a PDI ≤ 0.1. SEM images reveal that all the particles were roughly of similar size, spherical shape, and smooth surfaces (Fig. 2). The assessment of zeta potential showed that most particles displayed a negative surface charge (around −40 mV), except for ApoBP-conjugated particles, where the peptide attachment decreased the zeta potential to −4.56 mV (Table 1). Table 1 also shows the encapsulation efficacy of curcumin and or Rapa were ~38 wt% and ~32 wt%, respectively. An additional important characteristic is the release efficiency of antigens by TNPs, which was assessed by suspending TNPOVA in 2 mL PBS (10 mM, pH 7.4) for different time durations at 37 °C, using gentle shaking. This demonstrated a slow release of 40% of the antigen over two weeks (Fig. S3). In addition, the size of NPs stored in deionized water at 4 °C was assayed periodically, demonstrating good long-term stability (Fig. S3). The NPs also exhibited slow release of curcumin and rapamycin (Fig. S3).
Figure 2.
SEM pictures to show the morphology of PLGA nanoparticles prepared by encapsulation of pharmacological regulators, with or without the co-delivery of OVA. The morphology of liver-targeting particles is shown in Fig. S1.
LSECs represent a major tolerogenic APC subset, which occupies a large collective surface area in the liver and are capable of generating antigen-specific Tregs in Vivo.37 To assess the impact of TNPs on cytokine production by tissue culture LSECs, TGF-β, and IL-10 levels in the supernatant were measured after cellular exposure to 100 μg/mL particles for 24 h. All particles triggered TGF-β production, with NPOVA/ApoBP and NPOVA-Rapa showing the most significant increases (Fig. S1). TGF-β plays an important role in Treg generation as well as execution of their tolerogenic effects at the site of pathology.60, 61 Similarly, all nanoparticles except NPOVA, induced a significant increase in IL-10 production. IL-10 is an important anti-inflammatory cytokine that also potentiates Treg differentiation.62, 63 In this assay, the effect of NPOVA-Rapa was more robust than NPOVA/ApoBP.
Assessment of the biodistribution of ApoBP-decorated versus non-decorated particles
The tolerogenic particles were injected IV to deliver 25 μg OVA in 500 μg NPs per animal (n=4), before sacrifice after 24 h. Major organs such as the liver, spleen, heart, lung, and kidney were harvested and mounted in a Petri dish for IVIS imaging.39 The particle-encapsulated OVA accumulated in the liver, with lower retention in the lung and spleen (Fig. 1C, middle panel). Little or no distribution was observed in the kidney and heart. The ApoBP-decorated particles were sequestered in the liver with higher abundance than non-decorated NPOVA (Fig. 1C, middle panel).39 To visualize the intrahepatic distribution of the labeled NPs, isolectin B4 immunostaining was used to locate LSECs in liver tissue sections. Confocal microscopy showed that, compared to DyLight-labeled NPOVA, the co-localization of ApoBP-coated particles with isolectin-stained endothelial cells could be seen to increase (Fig. 1C, right panel).39 This was expressed as a co-localization index between the labeled particles and the isolectin B4-stained LSECs, demonstrating that the index values increased from 0.22±0.087 to 0.61±0. 131. These data corroborate our previous findings with NPOVA/ApoBP.39
Differential tolerogenic effects of TNPs on allergic airway inflammation in an OVA-sensitization model
We have previously demonstrated that NPOVA/ApoBP can be used to target LSECs in Vivo, with the ability to generate Tregs, capable of suppressing allergic inflammation in the lung.39 In order to determine how the effect of this delivery system compare with the effects of Rapa (NPRapa) and curcumin (NPcurcumin) delivering nanoparticles without or including OVA (i.e., NPOVA-curcumin or NPOVA-Rapa), a prophylactic TNP administration protocol was established before performing animal sensitization and challenge (Fig. 3A). C57BL/6 mice received intravenous particle injection via the tail vein, using a dose of 500 μg of NPs containing 25 μg OVA. Particles to deliver curcumin and Rapa were injected at doses of 32 and 10 μg, respectively, in each animal. Pretreatment was performed on days 0 and 7, followed by intraperitoneal (IP) sensitization with 10 μg OVA on days 14 and 21, before inhalation challenge with 10 mg/mL aerosolized OVA (for 20 min) on days 35 to 37. Animals were sacrificed on day 40 to allow blood withdrawal and harvesting of organs and BAL fluid for the analyses described below.
Figure 3.


TNP pretreatment interferes in OVA-induced antibody responses in a murine sensitization model. (A) Outline of the experimental animal protocol. Six to eight week old C57/BL6 mice received IV injection of NPOVA to deliver 25 μg OVA in 500 μg particles per animal on days 0 and 7. The animals were subsequently sensitized with two doses of OVA (10 μg/mouse) IP on days 14 and 21, before being exposed to aerosolized OVA inhalation (10 mg/mL) for 20 min on days 35-37. Animals were sacrificed for tissue harvesting and collection of BALF on day 40. The treatment groups (n=6) in the experiment included: (i) a control group without NP pretreatment, sensitization or challenge; (ii) no pretreatment before sensitization and challenge, or pretreatment with :(iii) NPOVA, (iv) NPOVA/ApoBP, (v)NPCurcumin+free OVA, (vi) NPOVA-Curcumin, (vii) NPRapa+free OVA, (viii) NPOVA-Rapa before sensitization and challenge. (B) Serum anti-OVA IgE, IgG1, and IgG2a antibody titers, as determined by ELISA. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by a Tukey’s test).
The serum was used to assess OVA-specific IgE, IgG1, and IgG2a antibody titers. Measurement of IgE titers, as a reflection of IL-4/CD40-mediated Ig kappa B-cell class switching,64 demonstrated that while all NPs decreased IgE production, the effect of ApoBP-decorated particles or particles incorporating Rapa and curcumin were more potent than NPOVA (Fig. 3B). IgG1 levels, another hallmark of IL-4 induced TH2 immunity, were also suppressed by all nanoparticles, with the most robust effect being assigned to NPOVA/ApoBP, NPOVA-Rapa, and NP Rapa + free OVA (Fig. 3B). In contrast, IgG2a levels, a marker of TH1-promoted Ig class switching, did not show any increase in response to OVA sensitization and challenge.
The TNP impact on antibody responses to OVA was also compared to the treatment impact on TH2 cytokines (IL-4, IL-5), IFN-γ (TH1 cytokine), and tolerogenic cytokines (IL-10 and TGF-β) in the lung (Fig. 4). This was accomplished by measuring cytokine levels in the bronchoalveolar lavage fluid (BALF) by ELISA. In addition to a role in Ig class switching, IL-4 is responsible for TH2 differentiation, expression of vascular cell adhesion molecules, eosinophil recruitment, and mucus secretion in inflamed airways.65, 66 Prophylactic treatment with nanoparticles demonstrated a reduction of IL-4 levels by all therapies, with the best effect attributable to NPOVA/ApoBP and NPOVA-Rapa administration, i.e., a reduction from 243 to 76 and 33 pg/mL, respectively (p<0.001) (Fig. 4A). The same trend was seen for IL-5, which plays a key role in generating eosinophilic airway inflammation (Fig. 4A).67, 68 IL-5 levels demonstrated a decrease from 47 pg/mL in untreated animals to 15, 14, and 11 pg/mL for animals treated with NPOVA/ApoBP, NPRapa plus free OVA, and NPOVA-Rapa, respectively. As expected, there was no change in IFN-γ production (Fig. 4A), which agrees with the IgG2a results. In contrast to decreased production of TH2 cytokines, there was a steep increase in TGF-β production in the BALF in response to TNP injection, with the highest levels being obtained in animals treated with NPOVA-Rapa, NPOVA-Rapa, NPRapa plus free OVA and NPOVA/ApoBP (Fig. 4B). In this regard, the response to the Rapa-delivering particles was higher (p<0.05) than the response to NPOVA/ApoBP. Despite obtaining an in Vitro IL-10 response for TNP-exposed LSECs, prophylactic treatment with the particles had minimal effects on IL-10 release in the BALF (Fig. 4B).
Figure 4.



Impact of TNP pretreatment on cytokine levels. Lung tissue and BALF from the experiment in Fig. 3A were used for the following analysis: (A) TH2 cytokine (IL-4 and IL-5) levels in the BALF, determined by ELISA. (B) TGF-β, INF-γ, and IL-10 levels in the BALF, determined by ELISA. (C) Differential eosinophil, neutrophil, and macrophage cell counts on BALF. BAL was performed using 1 mL of PBS lavage from each animal, for cytospinning and counting on slides. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by a Tukey’s test).
Differential cell counting of the BALF demonstrated robust eosinophil recruitment to the lung in response to OVA sensitization and challenge. This amounted to a total of 7,450,000 eosinophils appearing in the BALF from each animal Fig. 4C). While pretreatment with most NPs could be seen to reduce the eosinophil counts, NPOVA/ApoBP, NPOVA-Rapa, and NPRapa plus free OVA exhibited the strongest inhibitory effects (Fig. 4B). Again, the already significant response to NPOVA-Rapa was superseded by the response to NPOVA/ApoBP (p<0.05). Roughly similar trends were seen for the impact on neutrophil and macrophage recruitment, with a tendency for NPOVA-Rapa to be slightly more effective than NPOVA/ApoBP, although not statistically significant.
Histological examination of the lung tissue confirmed that OVA sensitization and challenge were accompanied by significant eosinophilic inflammation, particularly concerning smaller airways, alveolar ducts, alveoli, and small blood vessels in the lung (Fig. 5A). While some reduction in pulmonary inflammation was seen in response to all particles, treatment with NPOVA/ApoBP and NPOVA-Rapa to a large extent eliminated evidence of inflammation.
Figure 5.



Histological results showing that TNP pretreatment reduces allergic airway inflammation in the experiment described in Fig. 4A. (A) Representative lung tissue sections for formalin fixing and H&E staining. The scale bars represent 200 μm. (B) Foxp3+ T-cell recruitment to the lung during IHC analysis of tissues from the experiment. The scale bar represents 100 μm. Image-Pro Plus 6.0 software was used to detect cell nuclei and to calculate % cells, under 10x magnification. (C) A total of 12 independent fields were counted for each experimental group. The histogram on the right shows the Foxp3+ cell count for each group. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by a Tukey’s test).
In order to determine whether Foxp3+ cells are involved in the immune tolerizing effects of the particles, immunohistochemistry (IHC) staining was performed to discern Foxp3 expression in the harvested lung tissues. This demonstrated a significant increase in the number of Foxp3+ cells in the lungs of animals treated with NPOVA/ApoBP, NPOVA-Rapa, and NPRapa plus free OVA, compared to untreated animals (p<0.001) (Fig. 5B-C). Noteworthy, these cells were predominantly localized at pulmonary inflammation sites (Fig. 5B). NPs loaded with curcumin also increased the number of Foxp3+ Tregs, although not as prominent as the aforementioned groups. Quantification of Treg numbers showed that the response to NpOVA-Rapa was significantly higher than NPOVA/ApoBP (Fig. 5C).
The encapsulation of an MHC-II binding T-cell OVA epitope interferes in allergic airway inflammation in an OT-II mouse model
While treatment with nanoparticles encapsulating intact OVA was successful in alleviating allergic inflammation, an important consideration for TNP treatment in the clinic would be avoidance of IgE binding epitopes that could trigger an anaphylactic response before tolerization to whole allergen. An important development in allergen tolerance has been the substitution of whole allergens with immune-modulatory T-cell epitopes, capable of inducing tolerogenic effects without the danger of triggering mast cell by included IgE epitopes.49, 69 With regards to OVA, two epitope sequences, OVA257-264 (a.k.a. OT-I peptide) and OVA323-339 (OT-II peptide), have been described that are recognized by the Vα2/Vβ5.1-TCR of transgenic animals in the context of MHC-I and MHC-II molecules in OT-I and OT-II mouse models, respectively.70-74 To date these animal models have mostly been used to assess immune activation in the context of these MHC gene products, but not tolerization. Thus, to determine if the transgenic TCR expressed on CD4+ T-cells in OT-II mice can engage OVA323-339 in the context of the I-Ab gene product for generating Tregs that interfere with allergic airway inflammation, we used the protocol shown in Fig. S4 to perform the experimentation (Fig. 6 and 7). The effect of NPs on OT-I mice could not be assessed since this model does not allow the generation of allergic immune responses (Fig. S7). For the OT-II experiment, we compared the effect of NPOVA/ApoBP with NPOT-II/ApoBP and NPOT-I/ApoBP. These nanoparticles were synthesized as described in Fig. 1, except that OT-I and OT-II peptides were used for encapsulation in particles decorated with ApoB peptide (Fig. 6A). This yielded particles of ~270 nm, with uniform size, PDI ~0.1, epitope content of ~8 μg peptide per mg particle, and ApoBP coupling density of 5.3 mol% (Table 2 and Fig. S1).
Figure 6.




Pretreatment using epitope-encapsulating nanoparticles to assess the impact on OVA-induced serological responses and cytokine production in a transgenic OT-II murine model. Figure S4 outlines the experimental animal protocol. (A) Serum anti-OVA IgE and IgG1 antibody titers were determined by ELISA. (B) TH2 cytokine (IL-4, IL-5, and IL-13) levels in the BALF, determined by ELISA. (C) Differential eosinophil, neutrophil, and macrophage cell counts on BALF. (D) TGF-β, IL-10 and INF-γ levels in the BALF, determined by ELISA. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by a Tukey’s test).
Figure 7.


Impact of with epitope-encapsulating nanoparticles loaded with T-cell epitopes on allergic airway inflammation and Treg generation. This is from the same experiment as in Fig. 6. (A) Representative lung histology, as determined by H&E staining, scale bars represent 200 μm. (B) IHC for Foxp3+ T-cell recruitment to the lung. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by a Tukey’s test).
Table 2.
Characterization of liver-targeting TNPs encapsulating T-cell epitopes to assess hydrodynamic size, PDI, zeta potential, OVA content, and ApoBP attachment efficiency.
| Nanoparticle | Hydrodynamic size (nm) |
PDI | Zeta potential (mV) |
OVA/epitope content (μg/mg NPs) |
ApoBP Ligand loading efficiency (%) |
|---|---|---|---|---|---|
| NPOVA/ApoBP | 270.8 ± 4.96 | 0.113 | −4.56 ± 2.25 | 50.12 ± 2.18 | 5.3 (mol% of PLGA) |
| NPOT-II/ApoBP | 273.8 ± 2.35 | 0.118 | −5.59 ± 4.29 | 8.46 ± 2.02 | |
| NPOT-I/ApoBP | 267.9 ± 6.07 | 0.101 | −6.39 ± 4.18 | 8.99 ± 1.49 |
Prophylactic particle treatment was performed by IV injection on days 0 and 7, followed by IP OVA sensitization and inhalation challenge, before animal sacrifice on day 40 (Fig. S4). Assessment of OVA-specific IgE titers demonstrated that NPOVA/ApoBP and NPOT-II/ApoBP pretreatment could significantly decrease antibody titers, with the OT-II peptide significantly more effective (p-value) than the whole protein (Fig. 6A). In contrast, NPOT-I/ApoBP had no effect. The same trend was also seen during the assessment of OVA-specific IgG1 levels (Fig. 6A) as well as measuring TH2 cytokine levels (IL-4, IL-5, and IL-13) in the BALF (Fig. 6B). Performance of differential cell counts also confirmed that NPOT-II/ApoBP could significantly suppress the eosinophil, neutrophil, and macrophage numbers in the BALF. NPOVA/ApoBP was as effective as the OT-II peptide for impacting eosinophils, but with lesser effects on neutrophils and macrophages. Assessment of TGF-β levels in the BALF demonstrated that while NPOT-II/ApoBP treatment was associated with a significant increase of this tolerogenic cytokine, the response was much less robust during encapsulation OVA and comparatively weak for NPOT-I/ApoBP (Fig. 6C), H&E staining showed severe inflammation in the untreated control and the NPOT-I/ApoBP group, while the lungs of animals treated with NPOVA/ApoBP and NPOT-II/ApoBP were essentially devoid of eosinophilic inflammation (Fig. 7A). Moreover, IHC staining for Foxp3+ showed significant increases for all the particles, with NPOT-II/ApoBP inducing a robust effect compared to lesser (but still highly significant) response to NPOVA/ApoBP. The response to the encapsulated OT-I peptide was weak (Fig. 7B).
We also assessed the therapeutic impact of the particles on already-sensitized OT-II mice. Following IP sensitization on days 0 and 7, the mice were treated with IV particle injection on days 14 and 21 followed by inhalation challenge on three consecutive days (Fig. S5A). The OVA-specific IgE titer was significantly reduced by NPOT-II/ApoBP treatment (Fig. 8A) while NPOT-I/ApoBP exerted little effect. Histological analysis of lung tissue showed that, very similar to NPOT-II/ApoBP pretreatment, post-sensitization treatment could effectively reduce the eosinophilic inflammation, with modest reduction by NPOVA/ApoBP (Fig. 8B). Mice receiving NPOT-I/ApoBP did not show a significant effect. Differential accounting of eosinophils, neutrophils, and macrophages, as well as quantification of IL-5, confirmed the suppression of allergic inflammation by NPOT-II/ApoBP, which was slightly more effective than NPOVA/ApoBP (Fig. S5B-C). However, NPOT-II/ApoBP was more effective than NPOVA/ApoBP in elevating TGF-β levels in the BALF (Fig. S5C).
Figure 8.


Post-treatment effect of epitope-encapsulating nanoparticles on OVA-induced serological responses and allergic airway inflammation. Figure S5A outlines the experimental protocol. (A) Serum anti-OVA IgE level antibody titer was determined by ELISA. (B) Representative lung histology, as determined by H&E staining, scale bars represent 200 μm. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by a Tukey’s test).
TNP encapsulation of OVA and the OT-II epitope confers protective effects in a murine anaphylaxis model
In order to study a disease model that is more relevant to a systemic allergic event in humans, we also investigated the tolerogenic effects of the NPs on an adjuvant-free OVA sensitization model, where OVA challenge triggers mast cell release with the potential to induce anaphylaxis.75 This protocol involves intraperitoneal OVA sensitization once a week for 6 weeks, followed by intraperitoneal OVA challenge two weeks after the last sensitization. The ApoBP-conjugated TNPs containing encapsulated OVA or T-cell epitopes were IV injected before sensitization on two occasions, one week apart (Fig. 9A). Challenged mice, without pretreatment, developed robust anaphylactic manifestations within 30 min, maintaining an anaphylaxis score of 3 for 60 min, before a return to baseline by 120 min (Fig. 9B). This response was also accompanied by hypothermia. However, animals receiving NPOT-II/ApoBP and NPOVA/ApoBP showed little or no manifestations such as scratching or rubbing the nose and head, facial puffiness, pilar erecti, increased respiratory rate, labored respiration, and cyanosis (Fig. 9B). We also assessed the levels of mouse mast cell protease-1 (mMCPT-1) release to the serum as determined by a commercially available ELISA kit (Fig. 9C). This showed a significant increase in sensitized and exposed animals, with significant response reduction in animals treated with NPOT-II/ApoBP and NPOVA/ApoBP. Although there was some response reduction in mice receiving NPOT-I/ApoBP, the magnitude of the effect was significantly less than OVA or OT-II delivery. Consistent with Dr. Galli’s adjuvant-free model, we did not observe a significant increase in OVA-specific IgE levels (Fig. 9D). Instead, OVA-specific IgG, IgG1, and IgG2b levels were elevated by the allergen sensitization and challenge (Fig.9D). NPOT-II/ApoBP and NPOVA/ApoBP significantly reduced IgG (p<0.05) and IgG1 (p<0.01) levels, while NPOT-I/ApoBP reduced the IgG2b titer, similar to NPOT-II/ApoBP (p<0.05) (Fig.9D). In humans, an immediate hypersensitivity response can also be followed by a delayed inflammatory phase, which was assessed in mice by obtaining peritoneal lavage fluid that was analyzed for IL-4, IL-5, and TGF-β levels. This demonstrated a reduction of IL-4 and IL-5 production in response to NPOT-II/ApoBP and NPOVA/ApoBP pretreatment, which differed significantly from a lesser response by NPOT-I/ApoBP (Fig.9E). In addition, we also observed significant increases in TGF-β levels in animals treated with NPOT-II/ApoBP and NPOVA/ApoBP compared to treatment with other groups (Fig. 9E). The lesser responses to the OT-I peptide may reflect an unexplained contribution of an MHC-I mediated immune response to peritoneal sensitization. All considered, these data also show a robust tolerogenic effect of OVA and OT-II delivery by the conjugated TNP platform.
Figure 9.





Pretreatment using epitope-encapsulated tolerogenic nanoparticles to assess the impact on OVA-induced anaphylaxis responses, serological responses, and late phase cytokine production in an OVA anaphylaxis murine model. (A) Outline of the experimental animal protocol. Six to eight weeks old C57BL/6 mice received IV particle injections to deliver 25 μg OVA or 4 μg of OT-II or OT-I epitopes in 500 μg particles per mouse on weeks 0 and 1. The animals were subsequently sensitized by six doses of OVA (10 μg/mouse) IP over 7 weeks, before exposure to OVA challenge by IP injection (500 μg/mouse) on week 9. Animals were monitored for anaphylaxis scoring, as shown at the bottom of panel B. The treatment groups (n=6) in the experiment included: (i) a control group without NP pretreatment, sensitization or challenge; (ii) no pretreatment before sensitization and challenge; pretreatment with (iii) NPOVA/ApoBP, (iv) NPOT-II/ApoBP (v) NPOT-I/ ApoBP before sensitization and challenge. (B) Anaphylaxis scoring. (C) Serum mMCPT-1 level determined by ELISA. (D) Serum anti-OVA IgG, IgG1, and IgG2b antibody titers were determined by ELISA. (E) TH2 cytokine (IL-4 and IL-5) and TGF-β levels in the peritoneal lavage fluid, determined by ELISA. Data are expressed as the mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA followed by a Tukey’s test).
Discussion
In this paper, two main strategies to induce antigen-specific immune tolerance were compared by constructing LSEC-targeting TNPs and TNPs loaded with immunomodulators. For LSEC-targeting TNPs, ApoB peptide was conjugated to the OVA-loaded PLGA nanoparticles. In contrast, we also constructed curcumin and rapamycin nanoparticles without a surface ligand for comparing the effect to the liver-targeting platform, with and without OVA co-encapsulation. Tissue culture studies showed that both particle types could enhance the production of regulatory cytokines from LSECs, with NPOVA/ApoBP and NP Rapa-OVA performing the best. Animal experimentation in an OVA-induced allergic airway inflammation model also confirmed that NPOVA/ApoBP and NPRapa-OVA were the most effective TNPs for reducing TH2 cytokine responses (IL-4 and IL-13), eosinophil recruitment, and airway inflammation. The response was more robust with Rapa-OVA. Another assessment compared the efficacy of MHC-I and MHC-II binding OVA epitopes to the intact protein in a transgenic OT-II animal model, in which the use of OVA sensitization and inhalation challenge demonstrated that NPOT-II/ApoBP was even more effective than the whole protein in reducing the allergic airway inflammation through TGF-β production and Foxp3+ recruitment. This response reduction could be achieved by prophylactic as well as therapeutic interventions. We also demonstrate that delivery of OVA and the OT-II peptide is effective in reducing anaphylactic responses and mast cell release in a peritoneal sensitization and challenge anaphylaxis model. These results significantly extend the use of our liver-targeting TNP platform for additional design and applications to intervene in systemic allergic disorders.
The liver is considered as a natural tolerogenic organ based on its physiological function of filtering food and microbial antigens from the portal circulation, with a predilection for staging an anti-inflammatory innate immune response.76 LSECs represent the major tolerogenic APC type in the liver, which represents 15-20% of all hepatic cells with a combined surface area of ~200m2.37 Our liver-targeting PLGA platform was constructed to deliver OVA and OVA peptides to LSECs through surface attachment of ApoBP, which interacts with stabilin 1 and 2 scavenger receptors on the endothelial cell surface.59. This peptide sequence has also been used by other investigators to decorate nanoparticles and liposomes,77, 78 in addition to the use of poly(maleic anhydride-alt-1-octadecene to coat superparamagnetic iron oxide nanoparticles for LSEC targeting.17 The surface ligand conjugation promotes particle uptake via clathrin-coated pits, which allow antigen processing and presentation by MHC-II gene translation products.59 This leads to the activation of CD4+ T-cells that are required for the development of peripheral Tregs, with the involvement of TGF-β. In contrast, Rapa-loaded particles are capable of exerting tolerogenic effects, without the need for liver targeting. This effect is ascribed to therapeutic nanoparticles inducing antigen-specific immune tolerance without targeting a specific APC subset or even the necessity to incorporate the antigen in the tolerogenic particle.5, 16 For instance, Kishimoto et al. have shown that Rapa-encapsulating PLGA particles are capable of inducing durable antigen-specific immune tolerance when co-administered with encapsulated or free proteins or peptide antigens.16 The general assumption is that the systemic biodistribution of the Rapa particles, capable of inducing a tolerogenic state in unspecified APCs, can achieve a therapeutically beneficial effect. It is important to clarify, however, that our own NPRapa and NPRapa-OVA nanoparticles were constructed to achieve sizes of 230-250 nm, which generally favors biodistribution to the liver. We have previously demonstrated that the liver biodistribution of non-decorated NPOVA is capable of exerting a tolerogenic effect through sequestration by Kupffer or phagocytosing APCs, which also exhibit significant immune suppressive effects, although not as robust as LSECs.39 This notion is corroborated by the finding that non-decorated NPOVA could exert significant tolerogenic effects in our current experimentation, e.g., as demonstrated in Figures 3A, 4A, 4C, and 5. Thus, it is possible that Rapa particles in the 200 nm size range, may also distribute to liver APCs, where Rapa could act to reprogram APC activity as well as to contribute to Treg expansion.79, 80 In this regard, Rapa has been shown to promote Foxp3 expression, as well as being able to expand peripheral Treg proliferation 79, 81 Future studies will address the possibility that LSEC-targeting of pharmaceutical nanoparticles could enhance their tolerogenic potential.
Our study compared the effect of Rapa- versus curcumin-encapsulating NPs, demonstrating that the tolerogenic effects of NPRapa was considerably more efficacious than this NPCurcumin. These differences can be explained as follows: (i) Rapa inhibits the mTOR pathway that controls many different aspects of innate and cognate immunity to foreign and self-antigens;41, 43 (ii) Rapa directly promotes Treg expansion and differentiation in diseases such as rheumatic diseases;42 (iii) Rapa interferes in the immunostimulatory effects of APCs, allowing naïve CD4+ T-cells to differentiate into antigen-specific Foxp3+ Tregs;43 (iv) Rapa-encapsulating particles are capable of disrupting B-cell activation and differentiation, germinal center formation, antibody production and anaphylaxis.40, 82, 83 Although curcumin is also capable of inducing immune tolerance as a result of broad antioxidant effects and ability to interfere in the NF-κB pathway,84, 85 it lacks the antigen specificity and efficacy of Rapa, as demonstrated by our experimentation.
A potential shortcoming of the TNP platform for the delivery of whole protein allergens is the presence of linear and conformational IgE-binding epitopes that harbor the potential to induce mast cell degranulation.86, 87 This leads to the safety concern that systemic release of the allergen may trigger an anaphylactic response during the early phase of tolerization. Thus, it is necessary to consider developing NPs incorporating allergen sequences that promote Treg development by MHC-II presentation to naïve T cells. For example, T-cell peptide vaccines developed by Circassia Pharmaceuticals, have entered a phase 3 clinical trial for cat allergy and a phase 2b trial for house dust mite allergy.88 Another company, Aravax has shown the safety and efficacy of an intradermal vaccine for peanut allergy, comprised of a mixture of synthetic peanut protein epitopes.89 In a similar fashion, we demonstrate that the use of an MHC-II interactive, non-IgE binding OVA epitope is capable of tolerogenic effects that are comparable or even more effective than the whole protein (Figures 6-9 and Figures S4-S5). In contrast, an epitope sequence presented by an MHC-I gene product (OT-I peptide) exhibited lesser or no tolerogenic effects and was not accompanied by robust Treg generation or TGF-β production in the lung. Another type of epitope is regulatory T-cell epitopes, identified and characterized by De Groot et al.14, 15 These workers have demonstrated how “Tregitopes” can be used in different murine autoimmune models to suppress antigen-specific immune responses by Tregs.14, 15 These epitopes could be tested in future studies.
Our study was extended to examine the effect of liver-targeting TNPs on an animal anaphylaxis model, which is more directly related to a clinical response outcome premised on mast cell triggering.75 Anaphylaxis, as an acute life-threatening systemic disorder, exhibits a lifetime prevalence of 0.05% to 2.0% in developed countries.90, 91 Our results show that the prophylactic administration of TNPs incorporating OVA and the OT-II peptide could significantly suppress the anaphylactic score, mMCPT-1 levels and late-phase inflammatory response. This result is of potential relevance to the development of TNP platforms for the treatment of food allergies, including peanut-induced anaphylaxis. Thus, TNPs could reduce the need for daily maintenance therapy, therefore minimizing the risk of an adverse reaction to the oral peanut allergen intake. The ability to develop a TNP therapy with immunomodulatory T-cell epitopes also holds advantages for technology scale-up, which could also be applied to epitope delivery for the treatment of autoimmune diseases.
Finally, it is also worth mentioning the use of combination therapy with TNPs, including the possibility that liver-targeting TNPs may be combined with the NPs delivering pharmaceutical agents to achieve synergistic tolerogenic effects. This is exemplified by the use of nanoparticles to encapsulate an immunodominant epitope of the autoantigen, MOG (amino acid residues 35-55) plus IL-10 for prophylactic and therapeutic intervention in a chronic progressive experimental allergic encephalitis model.92 A recent study described the use of hybrid particles encapsulating TGF-β surface protein constructs such as: (i) MHC-I and -II multimers, presenting myelin basic protein peptides to autoreactive T-cells; (ii) anti-Fas mAb plus a recombinant PD-L1-Fc construct for apoptosis induction in autoreactive T-cells; or (iii) CD47-Fc for inhibiting NP sequestration, prolonging in Vivo half-life.93 We envisage the construction of a number of hybrid platforms that combine LSEC targeting, pharmaceutical enhancers of APC tolerogenic activity, enhancement of Treg generation and stability of Foxp3 expression, as well as T-cell epitopes for the custom design of TNPs that can be used for a range of antigen-specific immune disorders characterized by overactive immune function.
Conclusion
In summary, we compared the effectiveness of two major types of TNPs, liver-targeting PLGA nanoparticles and nanoparticles loaded with pharmaceutical agents. Cellular studies demonstrated the effective induction of regulatory and tolerogenic cytokines by LSEC-targeting and rapamycin-loaded nanoparticles, with roughly equivalent potencies. In animal studies, prophylactic treatment by LSEC-targeting and Rapa-encapsulating particles suppressed allergic airway inflammation by increasing Treg presence and TGF-β production in the lung. In addition, we compared the effectiveness of the LSEC-targeting particles containing T-cell epitopes and whole protein. NPs loaded with OT-II T-cell epitopes showed higher tolerogenic efficacy than NPs loaded with whole OVA, either prophylactically or during therapeutic administration to a transgenic OT-II mice model. The tolerogenic effects were also confirmed in the murine anaphylaxis model, where the whole protein and OT-II peptide suppressed the anaphylaxis score, mast cells release, and the late-phase inflammatory response. All considered, strategies to combine LSEC-targeting, pharmaceutical agents (e.g., rapamycin), and T-cell epitopes may enhance tolerogenic effects for treatment of systemic allergic as well as autoimmune disorders.
Materials and Methods
Reagents
A poly(D,L-lactide-co-glycolide) formulation, comprised of a 50:50 mix of lactide to glycolide co-polymers in molecular weight range of 38,000-54000, inclusive of ~5kDa PEG, was purchased from Sigma (St Louis, MO). The model antigen, ovalbumin (OVA), was bought from Sigma. OT-II peptide, i.e. OVA323-339, with the sequence of ISQAVHAAHAEINEAGR, and OT-I peptide, i.e. OVA257-264, with the sequence SIINFEKL, were purchased from InvivoGen (San Diego, CA). Analytical grade curcumin, dichloromethane, sodium cholate, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC), N-hydroxysuccinimide (NHS), and N-(2-Aminoethyl) maleimide (NAEM spacer) were obtained from Sigma. Rapamycin was purchased from LC Laboratories (Woburn, MA). The ApoB peptide RLYRKRGLK (ApoBP), along with GCC tag, was synthesized by Biomatik (Cambridge, Ontario, Canada). SV40-immortalized murine liver sinusoidal endothelial cells (LSECs), cell growth medium, and flasks were purchased from Applied Biological Materials (Vancouver, Canada). The ELISA kits for the measurement of cytokines, including TGF- β, IL-10, IL-4, IL-5, and IL-13, were purchased from R&D (Minneapolis, MN). The horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibodies for serological titration of IgG2a (A-10685) and IgE (PA1-84764) were purchased from Invitrogen (Waltham, MA). An antibody to assess IgG1 levels (ab97240) was from Abcam (Cambridge, MA). The 3,3',5,5'-tetramethylbenzidine (TMB) substrate kit was purchased from BD Biosciences (San Jose, CA).
Fabrication and characterization of LSEC-targeting NPs incorporating pharmaceutical agents and antigen epitopes
Pristine PLGA NPs without a surface ligand were synthesized using a double-emulsion method (w/o/w), combined with solvent evaporation, as previously described by us.39 Another particle was synthesized by conjugating the ApoB peptide (RLYRKRGLK, containing a GGC tag) to the particle surface, using a two-step reaction, that makes use of an N-(2-aminoethyl) maleimide (NAEM) spacer. Particles incorporating pharmaceutical agents were also fabricated as above, with the inclusion of synthesis parameters to optimize drug encapsulation. Briefly, 220 mg PLGA and pharmaceutical agents (2.2 mg of curcumin or rapamycin) were co-dissolved in 10 mL of DCM. 30 mg of an OVA solution (1 mL) was incrementally added into the organic solution, and then emulsified using ultrasonication for 1 min with a 4s on/4s off pulse at an intensity of 30% (Branson 450). The water/oil (w/o) mixture was poured into 45 mL of 1% sodium cholate solution and sonicated for 2 min, using the same parameters, and then added into 45 mL of 0.5% sodium cholate solution. The double emulsion (w/o/w) was stirred overnight for DCM evaporation. The mixture was centrifuged and washed in DI water (10,000 g, 10 min) to remove the non-encapsulated payloads, before suspension in DI water. For the epitope encapsulation, the fabrication method was adjusted slightly. Briefly, 1 mg of epitope solution (1 mL) was added into the PLGA solution (200 mg in 12 mL of DCM) and then sonicated for 40 s with a pulse of 4/4s on/off under 30 w. The primary emulsion was poured into 30 mL of 1% cholate solution and sonicated for 2 min, using the same parameters. The sonicate was poured into 35 mL of 0.5% cholate solution and stirred overnight for DCM removal. The purification process was the same as mentioned above.
The size and surface charge of the purified NPs were characterized using dynamic light scattering. The surface morphology was visualized by scanning electron microscopy. The microBCA assay and the nanodrop method were used to detect the loading capacity of OVA and its T-cell epitopes and the conjugation efficiency of the peptide ligand. Before use, the endotoxin level was measured and clarified by a chromogenic LAL assay.
Determination of NP-induced cytokine production by LSECs
LSECs were exposed to nanoparticles incorporating pharmacological inhibitors for 24 h. Supernatants were collected to determine the production of tolerogenic cytokines, including TGF-β, IL-4, IL-10, through the use of ELISA (R&D).
Use of LSEC-targeting NPs incorporating pharmaceutical agents and antigenic peptides to induce epitope-specific tolerance in a murine allergic airway disease model
OVA sensitization and inhalation challenge were used to test the efficacy of the nanoparticles in a well-established allergic airway disease model in C57BL/6 mice. The sensitization involved intraperitoneal injection of 0.5 mg/kg OVA on days 14 and 21, followed by inhalation challenge using aerosolized OVA (10 mg/mL) for 20 min. OVA nebulization was performed using a Schuco 2000 nebulizer (Allied Health Care Products, St. Louis, MO) for allergen delivery at a flow rate of 6 L/min in a nebulizer cup. The effect of the NPs encapsulating OVA and pharmacological inhibitors was assessed in 6 to 8-week-old female C57/BL6 mice, while the effect of the particles loaded with T-cell epitopes was assessed in transgenic OT-II animals (Jackson Laboratory, Bar Harbor, ME). OT-II mice express a transgenic T-cell antigen receptor that, with the assistance of CD4, recognizes OVA323-339 peptide, presented by the murine MHC-II molecule, I-Ab. For the pretreatment protocol, the particles were IV administered on days 0 and 7, before animal sensitization and allergen challenge, as described above (Fig. S4). For the post-treatment protocol, OT-II mice were sensitized by IP injection using the same OVA dose on days 0 and 7, before IV injection of the particles on days 14 and 21, using the same dose as before. The animal subsequently received aerosolized OVA inhalation from days 35- 37 (Fig. S5A). Animals were sacrificed on day 40, followed by the collection of BALF (1 mM EDTA in PBS) and lung tissues for histological and immunohistochemistry analysis. Animal care was based on principles established by the National Society for Medical Research (USA), with animal protocol approved by the Division of Laboratory Animals Medicine at UCLA.
Use of LSEC-targeting NPs for antigen-specific tolerance in a murine anaphylaxis model
We made use of an adjuvant-free OVA anaphylaxis model as described by Galli et al.75 Six to eight week old C57BL/6 mice received IV particle injections to deliver 25 μg OVA or 4 μg of either the OT-II or OT-I epitopes, delivered at a particle dose of 500 μg to each animal on weeks 0 and 1. The animals were subsequently sensitized by six doses of OVA (10 μg/mouse) IP on weeks 2, 3, 4, 5, 6, and 7, before exposure to OVA challenge by IP injection (500 μg/mouse) on week 9 (Galli paper). Animals were subsequently monitored to assess the anaphylaxis score (Fig.9A). Two days after the challenge, mice were sacrificed for tissue harvesting and collection of peritoneal lavage fluid. The treatment groups (n=6) in the experiment included: (i) a control group without NP pretreatment, sensitization or challenge; (ii) no pretreatment before sensitization and challenge; pretreatment with (iii) NPOVA/ApoBP, (iv) NPOT-II/ApoBP, (v) NPOT-I/ApoBP before sensitization and challenge. After the challenge, anaphylaxis scores and body temperatures were monitored by three independent people. The scoring criteria were: 0 = no symptoms; 1 = scratching and rubbing of the nose and head; 2 = puffiness around the eyes and mouth, diarrhea, pilar erecti, reduced activity, and/or decreased activity with increased respiratory rate; 3= wheezing, labored respiration, and cyanosis around the mouth and the tail; 4 = no activity after prodding or tremor and convulsion; 5 = death. The peritoneal inflammation and serum collection were performed two days after the challenge.
Assessment of differential cell counts and assessment of cytokines
Following animal sacrifice, bronchoalveolar lavage fluid (BALF) was cytospun onto glass slides for fixing and staining with Hema3 solutions I and II (Fisher Healthcare, Waltham, MA). Differential cell counts were performed under a Fisherbrand microscope. The cell-free BAL supernatants from allergic airway animal studies as well as the cell-free peritoneal lavage supernatants from the anaphylaxis model studies were used for the quantification of IL-4, IL-5, TGF-β, IL-10, and IFN-γ by ELISA (R&D), as per the manufacturers’ instructions.
Determination of antigen-specific antibody titers and mouse mast cell protease-1 (mMCPT-1) levels in serum
Blood was collected at sacrifice and the mouse serum separated at 4500g, 5min. Antigen-specific antibody titers were determined by ELISA, as previously described. Briefly, the plates were coated with OVA (2 μg per well) in citrate-buffered saline buffer (0.05 M CBS, pH 9.6) overnight at 4 °C. After washing using PBST (0.01 M PBS containing 0.05% [m/v] Tween 20, pH 7.4), PBS containing 10% FBS was used to block the plates for 2 hours. 100 μL of serum, serially diluted in PBS plus 1% FBS, was added into plates for 2-hour at 37 °C. 100 μL of a diluted suspension of HRP-conjugated goat anti-mouse secondary antibodies, recognizing IgE, IgG1, or IgG2a was added to the plates for 2 hours at 37 °C, before the addition of 50 μL TMB substrate for 30 min. The reaction was stopped by the addition of 50 μL sulfuric acid (2 M). The plates were read using a SpectraMax M5 microplate reader to record the optical density at a wavelength of 450 nm (OD450). Antibody titers were expressed as the highest dilution (titer) resulting in a doubling of OD values, compared to sera from non-treated animals. Levels of mMCPT-1 in serum were measured with an mMCPT-1enzyme-linked immunosorbent assay kit, following the manufacturer’s instructions.
Hematoxylin-eosin (H&E) staining and immunohistochemistry (IHC) analysis
Lung tissues were collected from sacrificed animals, before fixing in formalin and dehydration in 50% ethanol. Tissue sectioning was performed to generate 4 μm thick sections, placed on glass slides, before H&E and immunohistochemistry staining in the Translational Pathology Core Laboratory (TPCL) at UCLA, as previously described. Slides were scanned in an Aperio AT Turbo digital pathology scanner (Leica Biosystems) at 10× magnification
Statistical Analysis
Statistical analysis was performed on GraphPad Prism 7 software (GraphPad Software, La Jolla, CA), using one-way ANOVA or the Student t test to determine the level of significance. The results were expressed as mean ± SEM of at least three independent experiments. Statistical significance thresholds were set at *p < 0.05; **p < 0.01; ***p < 0.001.
Supplementary Material
Acknowledgment
Research reported in this publication was supported, in part, by the National Institute of Environmental Health Sciences of the National Institutes of Health under Award Number (U01 ES027237). The authors thank the CNSI Advanced Light Microscopy/Spectroscopy and Electron Imaging Center for NanoMachines Core Facilities, the Flow Cytometry Core Facility of Jonsson Comprehensive Cancer Center, and the Translational Pathology Core Laboratory (TPCL) Research Facility at UCLA.
Footnotes
Competing financial interests: Andre E. Nel is co-founder and equity holder in Westwood Biosciences Inc and Nammi Therapeutics. The remaining authors declare no conflict of interest.
Supporting Information: The Supporting Information is available free of charge at http://pubs.acs.org. Additional figures, tables, and results, e.g., cytokine production profile, SEM graph, stability of NPs and the in Vitro antigen and drug release profile, and experimental animal protocols, as described in the text.
References
- 1.Carballido JM; Santamaria P, Taming Autoimmunity: Translating Antigen-Specific Approaches to Induce Immune Tolerance. J. Exp. Med 2019, 216, 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serra P; Santamaria P, Antigen-Specific Therapeutic Approaches for Autoimmunity. Nat. Biotechnol 2019, 37, 238–251. [DOI] [PubMed] [Google Scholar]
- 3.Shakya AK; Nandakumar KS, Antigen-Specific Tolerization and Targeted Delivery as Therapeutic Strategies for Autoimmune Diseases. Trends Biotechnol. 2018, 36, 686–699. [DOI] [PubMed] [Google Scholar]
- 4.Ben-Akiva E; Est Witte S; Meyer RA; Rhodes KR; Green JJ, Polymeric Micro- and Nanoparticles for Immune Modulation. Biomater. Sci 2019, 7, 14–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kishimoto TK; Maldonado RA, Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front. Immunol 2018, 9, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pozsgay J; Szekanecz Z; Sármay G, Antigen-Specific Immunotherapies in Rheumatic Diseases. Nat. Rev. Rheumatol 2017, 13, 525–537. [DOI] [PubMed] [Google Scholar]
- 7.Stabler CL; Li Y; Stewart JM; Keselowsky BG, Engineering Immunomodulatory Biomaterials for Type 1 Diabetes. Nat. Rev. Mater 2019, 4, 429–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vickery BP; Scurlock AM; Jones SM; Burks AW, Mechanisms of Immune Tolerance Relevant to Food Allergy. J. Allergy Clin. Immunol 2011, 127, 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabatos-Peyton CA; Verhagen J; Wraith DC, Antigen-Specific Immunotherapy of Autoimmune and Allergic Diseases. Curr. Opin. Immunol 2010, 22, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakaguchi S; Yamaguchi T; Nomura T; Ono M, Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [DOI] [PubMed] [Google Scholar]
- 11.Bacher P; Scheffold A, The Effect of Regulatory T Cells on Tolerance to Airborne Allergens and Allergen Immunotherapy. J. Allergy Clin. Immunol 2018, 142, 1697–1709. [DOI] [PubMed] [Google Scholar]
- 12.Jonuleit H; Bopp T; Becker C, Treg Cells as Potential Cellular Targets for Functionalized Nanoparticles in Cancer Therapy. Nanomedicine 2016, 11, 2699–2709. [DOI] [PubMed] [Google Scholar]
- 13.Serra P; Santamaria P, Nanoparticle-Based Approaches to Immune Tolerance for the Treatment of Autoimmune Diseases. Eur. J. Immunol 2018, 48, 751–756. [DOI] [PubMed] [Google Scholar]
- 14.De Groot AS; Moise L; McMurry JA; Wambre E; Van Overtvelt L; Moingeon P; Scott DW; Martin W, Activation of Natural Regulatory T Cells by IgG Fc-Derived Peptide “Tregitopes”. Blood 2008, 112, 3303–3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cousens LP; Najafian N; Mingozzi F; Elyaman W; Mazer B; Moise L; Messitt TJ; Su Y; Sayegh M; High K; Khoury SJ; Scott DW; De Groot AS, in Vitro and in Vivo Studies of IgG-Derived Treg Epitopes (Tregitopes): A Promising New Tool for Tolerance Induction and Treatment of Autoimmunity. J. Clin. Immunol 2013, 33, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maldonado RA; LaMothe RA; Ferrari JD; Zhang A-H; Rossi RJ; Kolte PN; Griset AP; O’Neil C; Altreuter DH; Browning E; Johnston L; Farokhzad OC; Langer R; Scott DW; von Andrian UH; Kishimoto TK, Polymeric Synthetic Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E156–E165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carambia A; Freund B; Schwinge D; Bruns OT; Salmen SC; Ittrich H; Reimer R; Heine M; Huber S; Waurisch C; Eychmüller A; Wraith DC; Korn T; Nielsen P; Weller H; Schramm C; Luth S; Lohse AW; Heeren J; Herkel J, Nanoparticle-Based Autoantigen Delivery to Treg-Inducing Liver Sinusoidal Endothelial Cells Enables Control of Autoimmunity in Mice. J. Hepatol 2015, 62, 1349–1356. [DOI] [PubMed] [Google Scholar]
- 18.Yeste A; Takenaka MC; Mascanfroni ID; Nadeau M; Kenison JE; Patel B; Tukpah A-M; Babon JAB; DeNicola M; Kent SC; Pozo D; Quintana FJ, Tolerogenic Nanoparticles Inhibit T Cell-Mediated Autoimmunity through SOCS2. Sci. Signaling 2016, 9, 61–74. [DOI] [PubMed] [Google Scholar]
- 19.Duong BH; Tian H; Ota T; Completo G; Han S; Vela JL; Ota M; Kubitz M; Bovin N; Paulson JC; Nemazee D, Decoration of T-Independent Antigen with Ligands for CD22 and Siglec-G Can Suppress Immunity and Induce B Cell Tolerance in Vivo. J. Exp. Med 2009, 207, 173–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clemente-Casares X; Blanco J; Ambalavanan P; Yamanouchi J; Singha S; Fandos C; Tsai S; Wang J; Garabatos N; Izquierdo C; Agrawal S; Keough MB; Yong VW; James E; Moore A; Yang Y; Stratmann T; Serra P; Santamaria P, Expanding Antigen-Specific Regulatory Networks to Treat Autoimmunity. Nature 2016, 530, 434–440. [DOI] [PubMed] [Google Scholar]
- 21.Capini C; Jaturanpinyo M; Chang H-I; Mutalik S; McNally A; Street S; Steptoe R; O'Sullivan B; Davies N; Thomas R, Antigen-Specific Suppression of Inflammatory Arthritis Using Liposomes. J. Immunol 2009, 182, 3556–3565. [DOI] [PubMed] [Google Scholar]
- 22.LaMothe RA; Kolte PN; Vo T; Ferrari JD; Gelsinger TC; Wong J; Chan VT; Ahmed S; Srinivasan A; Deitemeyer P; Maldonado RA; Kishimoto TK, Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis. Front. Immunol 2018, 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson B; Park BJ; Verdaguer J; Amrani A; Santamaria P, Prevalent CD8+ T Cell Response against One Peptide/MHC Complex in Autoimmune Diabetes. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 9311–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groux H; O'Garra A; Bigler M; Rouleau M; Antonenko S; de Vries JE; Roncarolo MG, A CD4+ T Cell Subset Inhibits Antigen-Specific T-Cell Responses and Prevents Colitis. Nature 1997, 389, 737–742. [DOI] [PubMed] [Google Scholar]
- 25.Lieberman SM; Evans AM; Han B; Takaki T; Vinnitskaya Y; Caldwell JA; Serreze DV; Shabanowitz J; Hunt DF; Nathenson SG; Santamaria P; DiLorenzo TP, Identification of the B Cell Antigen Targeted by a Prevalent Population of Pathogenic CD8+ T Cells in Autoimmune Diabetes. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 8384–8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Macauley MS; Paulson JC, Siglecs Induce Tolerance to Cell Surface Antigens by Bim-Dependent Deletion of the Antigen-Reactive B Cells. J. Immunol 2014, 193, 4312–4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macauley MS; Pfrengle F; Rademacher C; Nycholat CM; Gale AJ; von Drygalski A; Paulson JC, Antigenic Liposomes Displaying CD22 Ligands Induce Antigen-Specific B Cell Apoptosis. J. Clin. Invest 2013, 123, 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sojka DK; Huang Y-H; Fowell DJ, Mechanisms of Regulatory T-Cell Suppression - A Diverse Arsenal for A Moving Target. Immunology 2008, 124, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corthay A, How Do Regulatory T Cells Work? Scand. J. Immunol 2009, 70, 326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tsai S; Shameli A; Yamanouchi J; Clemente-Casares X; Wang J; Serra P; Yang Y; Medarova Z; Moore A; Santamaria P, Reversal of Autoimmunity by Boosting Memory-Like Autoregulatory T Cells. Immunity 2010, 32, 568–580. [DOI] [PubMed] [Google Scholar]
- 31.Kuo R; Saito E; Miller SD; Shea LD, Peptide-Conjugated Nanoparticles Reduce Positive Co-Stimulatory Expression and T Cell Activity to Induce Tolerance. Mol. Ther 2017, 25, 1676–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider JL; Balu-Iyer SV, Phosphatidylserine Converts Immunogenic Recombinant Human Acid Alpha-Glucosidase to a Tolerogenic Form in a Mouse Model of Pompe Disease. J. Pharm. Sci 2016, 105, 3097–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yeste A; Nadeau M; Burns EJ; Weiner HL; Quintana FJ, Nanoparticle-Mediated Codelivery of Myelin Antigen and a Tolerogenic Small Molecule Suppresses Experimental Autoimmune Encephalomyelitis. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 11270–11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen C; He Y; Cheng K; Zhang D; Miao S; Zhang A; Meng F; Miao F; Zhang J, Killer Artificial Antigen-Presenting Cells Deplete Alloantigen-Specific T Cells in a Murine Model of Alloskin Transplantation. Immunol. Lett 2011, 138, 144–155. [DOI] [PubMed] [Google Scholar]
- 35.Tiegs G; Lohse AW, Immune Tolerance: What Is Unique About the Liver. J. Autoimmun 2010, 34, 1–6. [DOI] [PubMed] [Google Scholar]
- 36.Doherty DG, Immunity, Tolerance and Autoimmunity in the Liver: A Comprehensive Review. J. Autoimmun 2016, 66, 60–75. [DOI] [PubMed] [Google Scholar]
- 37.Knolle PA; Wohlleber D, Immunological Functions of Liver Sinusoidal Endothelial Cells. Cell. Mol. Immunol 2016, 13, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crispe IN, Liver Antigen-Presenting Cells. J. Hepatol 2011, 54, 357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Q; Wang X; Liu X; Kumar S; Gochman G; Ji Y; Liao Y-P; Chang CH; Situ W; Lu J; Jiang J; Mei K-C; Meng H; Xia T; Nel AE, Use of Polymeric Nanoparticle Platform Targeting the Liver to Induce Treg-Mediated Antigen-Specific Immune Tolerance in a Pulmonary Allergen Sensitization Model. ACS Nano 2019, 13, 4778–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang A-H; Rossi RJ; Yoon J; Wang H; Scott DW, Tolerogenic Nanoparticles to Induce Immunologic Tolerance: Prevention and Reversal of FVIII Inhibitor Formation. Cell. Immunol 2016, 301, 74–81. [DOI] [PubMed] [Google Scholar]
- 41.Thomson AW; Turnquist HR; Raimondi G, Immunoregulatory Functions of mTOR Inhibition. Nat. Rev. Immunol 2009, 9, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perl A, Activation of mTOR (Mechanistic Target of Rapamycin) in Rheumatic Diseases. Nat. Rev. Rheumatol 2016, 12, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turnquist HR; Raimondi G; Zahorchak AF; Fischer RT; Wang Z; Thomson AW, Rapamycin-Conditioned Dendritic Cells Are Poor Stimulators of Allogeneic CD4+ T Cells, but Enrich for Antigen-Specific Foxp3+ T Regulatory Cells and Promote Organ Transplant Tolerance. J. Immunol 2007, 178, 7018–7031. [DOI] [PubMed] [Google Scholar]
- 44.Stallone G; Infante B; Di Lorenzo A; Rascio F; Zaza G; Grandaliano G, mTOR Inhibitors Effects on Regulatory T Cells and on Dendritic Cells. J. Transl. Med 2016, 14, 152–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Passerini L; Barzaghi F; Curto R; Sartirana C; Barera G; Tucci F; Albarello L; Mariani A; Testoni PA; Bazzigaluppi E; Bosi E; Lampasona V; Neth O; Zama D; Hoenig M; Schulz A; Seidel MG; Rabbone I; Olek S; Roncarolo MG; Cicalese MP; Aiuti A; Bacchetta R, Treatment with Rapamycin Can Restore Regulatory T-Cell Function in IPEX Patients. J. Allergy Clin. Immunol 2020, 145, 1262–1271. [DOI] [PubMed] [Google Scholar]
- 46.Kim G-Y; Kim K-H; Lee S-H; Yoon M-S; Lee H-J; Moon D-O; Lee C-M; Ahn S-C; Park YC; Park Y-M, Curcumin Inhibits Immunostimulatory Function of Dendritic Cells: MAPKs and Translocation of NF-κB as Potential Targets. J. Immunol 2005, 174, 8116–8124. [DOI] [PubMed] [Google Scholar]
- 47.Olivera A; Moore TW; Hu F; Brown AP; Sun A; Liotta DC; Snyder JP; Yoon Y; Shim H; Marcus AI; Miller AH; Pace TWW, Inhibition of the NF-κB Signaling Pathway by the Curcumin Analog, 3,5-Bis(2-Pyridinylmethylidene)-4-Piperidone (Ef31): Anti-Inflammatory and Anti-Cancer Properties. Int. Immunopharmacol 2012, 12, 368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prickett SR; Rolland JM; O'Hehir RE, Immunoregulatory T Cell Epitope Peptides: The New Frontier in Allergy Therapy. Clin. Exp. Allergy 2015, 45, 1015–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Hehir RE; Prickett SR; Rolland JM, T Cell Epitope Peptide Therapy for Allergic Diseases. Curr. Allergy Asthma Rep 2016, 16, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akdis CA; Blaser K, Bypassing IgE and Targeting T Cells for Specific Immunotherapy of Allergy. Trends Immunol. 2001, 22, 175–178. [DOI] [PubMed] [Google Scholar]
- 51.Pentier J; Sewell A; Miles J, Advances in T-Cell Epitope Engineering. Front. Immunol 2013, 4, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hafner RP; Salapatek A; Patel D; Larché M; Laidler P, Validation of Peptide Immunotherapy as a New Approach in the Treatment of Allergic Rhinoconjunctivitis: The Clinical Benefits of Treatment with Amb a 1 Derived T Cell Epitopes. J. Allergy Clin. Immunol 2012, 129, 368–369.22051697 [Google Scholar]
- 53.Couroux P; Patel D; Armstrong K; Larché M; Hafner RP, Fel D 1-Derived Synthetic Peptide Immuno-Regulatory Epitopes Show a Long-Term Treatment Effect in Cat Allergic Subjects. Clin. Exp. Allergy 2015, 45, 974–981. [DOI] [PubMed] [Google Scholar]
- 54.Wen H; Jung H; Li X, Drug Delivery Approaches in Addressing Clinical Pharmacology-Related Issues: Opportunities and Challenges. AAPS J. 2015, 17, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Olsson U; Camejo G; Bondjers G, Binding of a Synthetic Apolipoprotein B-100 Peptide and Peptide Analogues to Chondroitin 6-Sulfate: Effects of the Lipid Environment. Biochemistry 1993, 32, 1858–1865. [DOI] [PubMed] [Google Scholar]
- 56.Olsson U; Camejo G; Hurt-Camejo E; Elfsber K; Wiklund O; Bondjers G, Possible Functional Interactions of Apolipoprotein B-100 Segments That Associate with Cell Proteoglycans and the Apob/E Receptor. Arterioscler., Thromb., Vasc. Biol 1997, 17, 149–155. [DOI] [PubMed] [Google Scholar]
- 57.Li R; Oteiza A; Sørensen KK; McCourt P; Olsen R; Smedsrød B; Svistounov D, Role of Liver Sinusoidal Endothelial Cells and Stabilins in Elimination of Oxidized Low-Density Lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol 2011, 300, G71–G81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sørensen KK; McCourt P; Berg T; Crossley C; Le Couteur D; Wake K; Smedsrød B, The Scavenger Endothelial Cell: A New Player in Homeostasis and Immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol 2012, 303, R1217–1230. [DOI] [PubMed] [Google Scholar]
- 59.Sørensen KK; Simon-Santamaria J; McCuskey RS; Smedsrød B, Liver Sinusoidal Endothelial Cells. Compr. Physiol 2015, 5, 1751–1774. [DOI] [PubMed] [Google Scholar]
- 60.Wan YY; Flavell RA, 'Yin-Yang' Functions of Transforming Growth Factor-Beta and T Regulatory Cells in Immune Regulation. Immunol. Rev 2007, 220, 199–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Letterio JJ; Roberts AB, Regulation of Immune Responses by TGF-Beta. Annu. Rev. Immunol 1998, 16, 137–161. [DOI] [PubMed] [Google Scholar]
- 62.Ng THS; Britton G; Hill E; Verhagen J; Burton B; Wraith D, Regulation of Adaptive Immunity; the Role of Interleukin-10. Front. Immunol 2013, 4, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Couper KN; Blount DG; Riley EM, IL-10: The Master Regulator of Immunity to Infection. J. Immunol 2008, 180, 5771–5777. [DOI] [PubMed] [Google Scholar]
- 64.Bacharier LB; Geha RS, Molecular Mechanisms of IgE Regulation. J. Allergy Clin. Immunol 2000, 105, S547–S558. [DOI] [PubMed] [Google Scholar]
- 65.Swain SL; Weinberg AD; English M; Huston G, IL-4 Directs the Development of Th2-Like Helper Effectors. J. Immunol 1990, 145, 3796–3806. [PubMed] [Google Scholar]
- 66.Seder RA; Paul WE; Davis MM; Fazekas de St Groth B, The Presence of Interleukin 4 During in Vitro Priming Determines the Lymphokine-Producing Potential of CD4+ T Cells from T Cell Receptor Transgenic Mice. J. Exp. Med 1992, 176, 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kouro T; Takatsu K, IL-5- and Eosinophil-Mediated Inflammation: From Discovery to Therapy. Int. Immunol 2009, 21, 1303–1309. [DOI] [PubMed] [Google Scholar]
- 68.Huston DP; Huston MM; Dickason RR; Martinez-Moczygemba M, Interleukin-5, A Therapeutic Target in Allergic Inflammation. Trans. Am. Clin. Climatol. Assoc 2000, 111, 46–59. [PMC free article] [PubMed] [Google Scholar]
- 69.Akdis M; Akdis CA, Therapeutic Manipulation of Immune Tolerance in Allergic Disease. Nat. Rev. Drug Discover 2009, 8, 645–660. [DOI] [PubMed] [Google Scholar]
- 70.Rötzschke O; Falk K; Stevanović S; Jung G; Walden P; Rammensee HG, Exact Prediction of a Natural T Cell Epitope. Eur. J. Immunol 1991, 21, 2891–2894. [DOI] [PubMed] [Google Scholar]
- 71.McFarland BJ; Sant AJ; Lybrand TP; Beeson C, Ovalbumin(323-339) Peptide Binds to the Major Histocompatibility Complex Class II I-A(D) Protein Using Two Functionally Distinct Registers. Biochemistry 1999, 38, 16663–16670. [DOI] [PubMed] [Google Scholar]
- 72.Johnsen G; Elsayed S, Antigenic and Allergenic Determinants of Ovalbumin--III. MHC Ia-Binding Peptide (OA 323-339) Interacts with Human and Rabbit Specific Antibodies. Mol. Immunol. 1990, 27, 821–827. [DOI] [PubMed] [Google Scholar]
- 73.Barnden MJ; Allison J; Heath WR; Carbone FR, Defective TCR Expression in Transgenic Mice Constructed Using cDNA-Based Alpha- and Beta-Chain Genes under the Control of Heterologous Regulatory Elements. Immunol. Cell Biol 1998, 76, 34–40. [DOI] [PubMed] [Google Scholar]
- 74.Hogquist KA; Jameson SC; Heath WR; Howard JL; Bevan MJ; Carbone FR, T Cell Receptor Antagonist Peptides Induce Positive Selection. Cell 1994, 76, 17–27. [DOI] [PubMed] [Google Scholar]
- 75.Balbino B; Sibilano R; Starkl P; Marichal T; Gaudenzio N; Karasuyama H; Bruhns P; Tsai M; Reber LL; Galli SJ, Pathways of Immediate Hypothermia and Leukocyte Infiltration in an Adjuvant-Free Mouse Model of Anaphylaxis. J. Allergy Clin. Immunol 2017, 139, 584–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horst AK; Neumann K; Diehl L; Tiegs G, Modulation of Liver Tolerance by Conventional and Nonconventional Antigen-Presenting Cells and Regulatory Immune Cells. Cell. Mol. Immunol 2016, 13, 277–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yu X; Chen L; Liu J; Dai B; Xu G; Shen G; Luo Q; Zhang Z, Immune Modulation of Liver Sinusoidal Endothelial Cells by Melittin Nanoparticles Suppresses Liver Metastasis. Nat. Commun 2019, 10, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Akhter A; Hayashi Y; Sakurai Y; Ohga N; Hida K; Harashima H, A Liposomal Delivery System That Targets Liver Endothelial Cells Based on a New Peptide Motif Present in the Apob-100 Sequence. Int. J. Pharm 2013, 456, 195–201. [DOI] [PubMed] [Google Scholar]
- 79.Chapman NM; Chi H, mTOR Signaling, Tregs and Immune Modulation. Immunotherapy 2014, 6, 1295–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Furukawa A; Wisel SA; Tang Q, Impact of Immune-Modulatory Drugs on Regulatory T Cell. Transplantation 2016, 100, 2288–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Battaglia M; Stabilini A; Migliavacca B; Horejs-Hoeck J; Kaupper T; Roncarolo M-G, Rapamycin Promotes Expansion of Functional CD4+CD25+Foxp3+ Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients. J. Immunol 2006, 177, 8338–8347. [DOI] [PubMed] [Google Scholar]
- 82.Meliani A; Boisgerault F; Ronzitti G; Collaud F; Leborgne C; Kishimoto TK; Mingozzi F, Antigen-Specific Modulation of Capsid Immunogenicity with Tolerogenic Nanoparticles Results in Successful aAv Vector Readministration. Mol. Ther 2016, 24, 34–35.26316391 [Google Scholar]
- 83.Mazor R; King EM; Onda M; Cuburu N; Addissie S; Crown D; Liu X-F; Kishimoto TK; Pastan I, Tolerogenic Nanoparticles Restore the Antitumor Activity of Recombinant Immunotoxins by Mitigating Immunogenicity. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E733–E742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Catanzaro M; Corsini E; Rosini M; Racchi M; Lanni C, Immunomodulators Inspired by Nature: A Review on Curcumin and Echinacea. Molecules 2018, 23, 2778–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Panda AK; Chakraborty D; Sarkar I; Khan T; Sa G, New Insights into Therapeutic Activity and Anticancer Properties of Curcumin. J. Exp. Pharmacol 2017, 9, 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galli SJ; Tsai M, IgE and Mast Cells in Allergic Disease. Nat. Med 2012, 18, 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Handlogten MW; Kiziltepe T; Serezani AP; Kaplan MH; Bilgicer B, Inhibition of Weak-Affinity Epitope-IgE Interactions Prevents Mast Cell Degranulation. Nat. Chem. Biol 2013, 9, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pfaar O; Bonini S; Cardona V; Demoly P; Jakob T; Jutel M; Kleine-Tebbe J; Klimek L; Klysner S; Kopp MV; Kuna P; Larché M; Muraro A; Schmidt-Weber CB; Shamji MH; Simonsen K; Somoza C; Valovirta E; Zieglmayer P; Zuberbier T; et al. Perspectives in Allergen Immunotherapy: 2017 and Beyond. Allergy 2018, 73, 5–23. [DOI] [PubMed] [Google Scholar]
- 89.Hoffmann HJ; Valovirta E; Pfaar O; Moingeon P; Schmid JM; Skaarup SH; Cardell L-O; Simonsen K; Larché M; Durham SR; Sørensen P, Novel Approaches and Perspectives in Allergen Immunotherapy. Allergy 2017, 72, 1022–1034. [DOI] [PubMed] [Google Scholar]
- 90.Sampson HA; Muñoz-Furlong A; Campbell RL; Adkinson NF; Bock SA; Branum A; Brown SGA; Camargo CA; Cydulka R; Galli SJ; Gidudu J; Gruchalla RS; Harlor AD; Hepner DL; Lewis LM; Lieberman PL; Metcalfe DD; O'Connor R; Muraro A; Rudman A; et al. Second Symposium on the Definition and Management of Anaphylaxis: Summary Report-Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network Symposium. J. Allergy Clin. Immunol 2006, 117, 391–397. [DOI] [PubMed] [Google Scholar]
- 91.Lieberman P; Camargo CA; Bohlke K; Jick H; Miller RL; Sheikh A; Simons FER, Epidemiology of Anaphylaxis: Findings of the American College of Allergy, Asthma and Immunology Epidemiology of Anaphylaxis Working Group. Ann. Allergy, Asthma, Immunol 2006, 97, 596–602. [DOI] [PubMed] [Google Scholar]
- 92.Cappellano G; Woldetsadik AD; Orilieri E; Shivakumar Y; Rizzi M; Carniato F; Gigliotti CL; Boggio E; Clemente N; Comi C; Dianzani C; Boldorini R; Chiocchetti A; Renò F; Dianzani U, Subcutaneous Inverse Vaccination with PLGA Particles Loaded with a MOG Peptide and IL-10 Decreases the Severity of Experimental Autoimmune Encephalomyelitis. Vaccine 2014, 32, 5681–5689. [DOI] [PubMed] [Google Scholar]
- 93.Pei W; Wan X; Shahzad KA; Zhang L; Song S; Jin X; Wang L; Zhao C; Shen C, Direct Modulation of Myelin-Autoreactive CD4+ and CD8+ T Cells in EAE Mice by a Tolerogenic Nanoparticle Co-Carrying Myelin Peptide-Loaded Major Histocompatibility Complexes, CD47 and Multiple Regulatory Molecules. Int. J. Nanomed 2018, 13, 3731–3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.