

Abstract
Data quality in global metabolomics is of great importance for biomarker discovery and system biology studies. However, comprehensive metrics and methods to evaluate and compare the data quality of global metabolomics data sets are lacking. In this work, we combine newly developed metrics, along with well-known measures, to comprehensively and quantitatively characterize the data quality across two similar liquid chromatography coupled with mass spectrometry (LC–MS) platforms, with the goal of providing an efficient and improved ability to evaluate the data quality in global metabolite profiling experiments. A pooled human serum sample was run 50 times on two high-resolution LC-QTOF-MS platforms to provide profile and centroid MS data. These data were processed using Progenesis QI software and then analyzed using five important data quality measures, including retention time drift, the number of compounds detected, missing values, and MS reproducibility (2 measures). The detected compounds were fit to a γ distribution versus compound abundance, which was normalized to allow comparison of different platforms. To evaluate missing values, characteristic curves were obtained by plotting the compound detection percentage versus extraction frequency. To characterize reproducibility, the accumulative coefficient of variation (CV) versus the percentage of total compounds detected and intraclass correlation coefficient (ICC) versus compound abundance were investigated. Key findings include significantly better performance using profile mode data compared to centroid mode as well quantitatively better performance from the newer, higher resolution instrument. A summary table of results gives a snapshot of the experimental results and provides a template to evaluate the global metabolite profiling workflow. In total, these measures give a good overall view of data quality in global profiling and allow comparisons of data acquisition strategies and platforms as well as optimization of parameters.
Graphical Abstract
INTRODUCTION
Global metabolite profiling aims to measure comprehensively the small endogenous and exogenous metabolites detectable in biological samples that include cells, tissues, biofluids, and many others.1–3 Global profiling plays an essential role in metabolomics for disease biomarker discovery,4–6 altered pathway identification,7–9 and the potential for improved treatments and precision medicine10,11 among its many applications. The accurate global analysis of biological samples requires the maximization of the number of detected and eventually annotated metabolites along with reliable and reproducible detection of compounds. Liquid chromatography coupled with mass spectrometry (LC–MS) is currently the dominant technique for global metabolite profiling and has increasingly been applied in the field. The high sensitivity and high resolution provided by LC–MS produce rich metabolome information that can be extracted12 with improving data quality over the past two decades. Although significant progress has been made toward improving metabolome coverage, global profiling by LC–MS is still challenged by signal variability arising from a variety of experimental issues, and it generates large, complex data sets that can hinder interpretation. Because of the fluctuations that can affect an untargeted metabolomics study, especially by LC–MS, a set of measures of quantitative assessment should be applied to ensure that the obtained data are of high analytical quality. Good data quality in global metabolite profiling is crucial to maximize the available metabolic information and help minimize the misinterpretation of biological results.13–16 The main parameters of data quality have been evaluated in terms of metabolite coverage, missing values, and reproducibility.14
The number of metabolites that can be detected in a global metabolomic experiment is a key issue,17 and is reflected by how many metabolites can be reliably extracted from the raw data into a data set and subsequently identified.18 Insufficient detection limits the metabolome coverage and therefore, hypothesis development by, for example, missing potential biomarkers or insufficiently describing metabolic pathways and changes therein. However, no single detection technique, including LC–MS, can achieve full coverage of the entire metabolome, and different instrument platforms have a range of performance.19 For this reason, for example, multiple instrument platforms were used to describe the human serum metabolome initially as containing an estimated 4651 metabolites using a combination of NMR, gas chromatography–MS (GC–MS), and LC–MS.20
A missing value21 exists when a compound cannot be extracted from the data for one sample but can be extracted from the others. While some missing values across a data set are anticipated, such as in human samples that may or may not contain drug- or food-related metabolites, problematic missing values can result from the interferences of chemical noise or ion suppression (in the case of LC–MS) as well as impurities, hardware instabilities, detection limits, and software algorithms.14,22 Missing values, of which there are generally three types (completely, not completely, or not at random), limit the complete and accurate extraction of compound information in an experiment.23 Furthermore, statistical analyses in metabolomics presume missing value-free data sets. To solve this problem, the currently dominant solution is imputation.24–27 While imputation facilitates statistical data analysis,28 the imputed values are deductive rather than original and can negatively influence discoveries and explorations in metabolomics.29 Some approaches have directly neglected missing values; however, this approach comes with the cost of reduced compound coverage.
Good reproducibility is also an essential component of data quality and a critical goal in global metabolite profiling by LC–MS. Good reproducibility ensures that the measured peak abundances are sufficiently accurate to reflect the relevant biological differences between samples.30,31 Reproducibility is usually evaluated using the coefficient of variation (CV)32 between repetitions of identical samples, for example, quality control (QC) samples. In current practice, compounds with CV > 20–30% are typically filtered out of a metabolomic data set. Correlation methods, such as the Pearson correlation and intraclass correlation (ICC),33 have also been used to help characterize data quality in metabolomics.34
Various strategies for technical improvements in metabolomics have been aimed at raising data quality, and efforts are being made to drive improvements across the metabolomics community.15,16 For example, sample preparation methods to reduce the effects of sample matrices and maximize the sensitivity during detection are in essence attempts to improve data quality, as are methods developed to separate isomers, reduce ion suppression, and improve instrument performance. Efforts are also being made to develop and distribute new QC reference materials.16 Advanced mass spectrometers such as high-resolution time-of-flight (TOF) and Orbitrap instruments are increasingly being adopted to improve data quality in global metabolite profiling. A growing number of software platforms, including Agilent Profinder, Progenesis QI, XCMS,35 MZmine 2,36 and others, have been developed with enhanced algorithms for data preprocessing of raw data to optimize real peak detection, minimize missing values, and increase statistical significance. Efforts focused on data or batch normalization can be highly effective for improving data quality of large data sets.37 Nevertheless, there are major gaps in the efforts to improve data quality for global metabolite profiling. For example, it is not well known to what extent data quality can be improved by tuning one or more parameters in a complex LC–MS system, which comprises the LC–MS method, instrument, software, and data-processing parameters. It is also challenging to review data quality systematically and quantitatively without efficient tools. There is a lack of standardized metrics and measures focused on characterizing data quality that would enable comparisons among two or more conditions and the extent to which different factors change data quality in a quantitative fashion.
To address these issues, we describe our initial efforts to provide a reasonably comprehensive set of metrics (including both old and new measures) for the systematic characterization of data quality for global metabolite profiling. In particular, and as an example, we employed strategies to enable a systematic comparison across two platforms and two data formats that have different conditions and parameters. In this work, two platforms (Agilent 6545 and 6520 Q-TOF-MS systems) and two data formats (profile and centroid) were analyzed to demonstrate the approach, which revealed a number of observed differences in data quality. In particular, the difference between the profile and centroid data, which is often neglected in the literature, was found to make a very significant difference in data quality using Progenesis QI software. We chose a set of five metrics that provide a reasonably comprehensive view of data quality that are relatively easy to understand and interpret. In addition to standard metrics of data quality (retention time reproducibility, measured compound numbers, missing values, CV), new metrics were created, such as fitting a γ distribution to compound coverage, evaluating a characteristic point in the missing-value analysis, and plotting ICC versus compound abundance. Together with the more classic metrics, data quality was systematically characterized in a simple to use reporting format, and discoveries are reported. The results, along with the software scripts that are freely available, and should provide researchers with better, easy to use tools for evaluating the data quality of their global metabolite profiling experiments and analysis methods. These efforts will hopefully spur additional data quality metrics and, ultimately, the development of a consensus set of methods to evaluate and report data quality for global metabolomics data sets.
EXPERIMENTAL PROCEDURES
Chemicals.
Acetonitrile (ACN), methanol, and acetic acid were purchased from Thermo Fisher (Fair Lawn, NJ). Ammonium acetate was purchased from Sigma-Aldrich (St. Louis, MO). DI water (18.2 MΩ·cm at 25 °C) was produced using a MilliporeSigma water purification system (Model Synergy, Burlington, MA).
Sample Preparation.
To prepare the identical QC samples for analysis, approximately 2.5 mL of frozen commercial pooled human serum (Innovative Research, Novi, MI) was thawed at 4 °C, vortexed, and aliquoted into 50 μL of portions in 2 mL Eppendorf vials. Every 50 μL of the portion was mixed with 250 μL of cold methanol and vortexed to precipitate proteins.38 After 20 min of incubation at −20 °C, these mixtures were centrifuged at 20 800g for 10 min at 4 °C. The supernatants were transferred into clean 2.0 mL Eppendorf vials and then dried in an Eppendorf Vacufuge (Brinkmann Instruments, Westbury, NY). The residue in each Eppendorf vial was reconstituted in 50 μL of H2O/ACN (2:3 v/v), vortexed, and centrifuged at 20 800g for 10 min at 4 °C. The supernatants in all Eppendorf vials were pooled into a 5 mL Eppendorf vial, vortexed, and centrifuged at 5000g for 10 min at 4 °C to further remove any solid residue. The resultant supernatant was aliquoted into 50 μL of portions in 1.5 mL Eppendorf vials and stored at −80 °C. Prior to LC–MS analysis, eight aliquots were diluted to 200 μL each with H2O/ACN (2:3 v/v), pooled into a 2 mL LC vial, vortexed, and placed in the autosampler for LC injection and analysis.
High-Performance Liquid Chromatography-Electrospray Ionization (HPLC-ESI)-MS Experiments.
The HPLC-ESI-MS measurements were carried out using an Agilent 6545 Q-TOF-MS coupled to an Agilent 1290 Infinity LC pump, and an Agilent 6520 Q-TOF-MS coupled to an Agilent 1260 Infinity LC system (Agilent Technologies, Santa Clara, CA). The separation was performed using a Waters XBridge BEH Amide column (15 cm × 2.1 mm, 2.5 μm). The mobile phase consisted of (A) H2O/ACN (95:5, v/v) with 5 mM ammonium acetate and 0.1% acetic acid, and (B) H2O/ACN (5:95, v/v), 5 mM ammonium acetate, and 0.1% acetic acid. Gradient elution was performed as follows: 100% mobile phase B for 1.5 min, 100–78% B from 1.5 to 6.0 min, 78–50% B from 6.0 to 9.0 min, 50% B from 9.0 to 15.0 min, restoration to 100% B from 15.0 to 17.0 min, and continued 100% B from 17.0 to 30.0 to equilibrate the LC column (see Figure 1). The flow rate was 0.3 mL/min, the injection volume was 5 μL, followed by an H2O/ACN (5:95, v/v) needle wash for 10 s, and the column temperature was 35 °C. The ESI conditions were as follows: electrospray ion-source ESI Agilent Jet Stream Technology in positive ionization mode; voltage 3.8 kV; desolvation temperature 325 °C; cone flow 20 L/h; desolvation gas flow 600 L/h; nebulizer pressure 45 psi, N2 drying gas; MS scan rate of 1.03 spectra/s across the range m/z 60–1000. Data were acquired using MassHunter Data Acquisition Workstation v. B.06.01.6157 software (Agilent Technologies).
Figure 1.
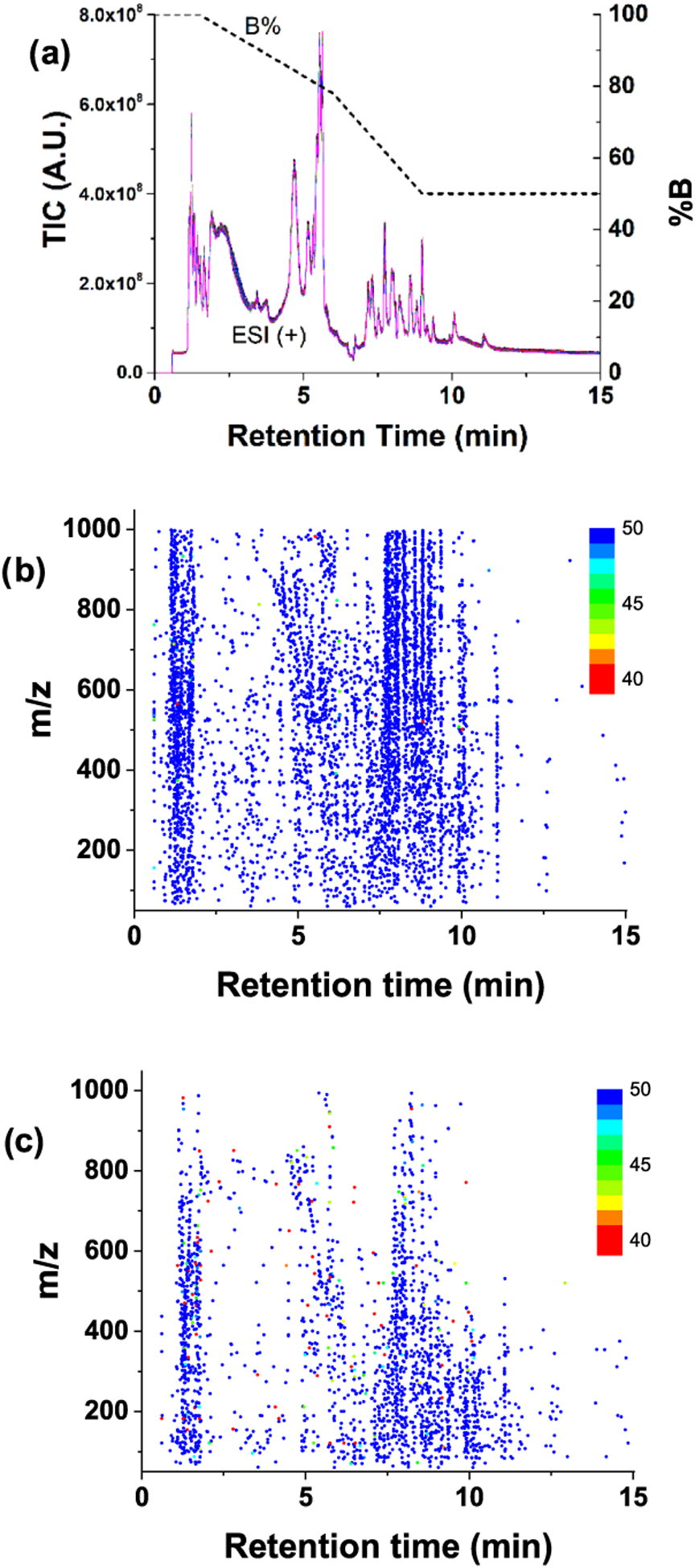
(a) Total ion count (TIC) chromatograms of 50 QC repetitions. Profile data and centroid data were processed, and the extracted compounds are shown in (b) and (c), respectively. Every compound was defined as having at least two ions, and a peak width of ≥3 s, as described in the main text. The color gradient indicates the detection frequency of compounds. Compounds detected in less than 40 samples (80%) were also marked as 40. Note: for plotting a, nonvarying signals observed in the blank were subtracted to reduce the offset.
Data Acquisition.
The same pooled serum sample was injected 50 times into both HPLC-ESI-Q-TOF systems using the same experimental parameters as much as possible. The data sets were stored in profile and centroid formats (biformat stored data). The conversion from the profile to the centroid format was performed with a threshold of 0.1% or 200 counts, whichever was higher, for the Agilent 6520 Q-TOF data. The same procedure was set to 0.01% or 100 counts for the Agilent 6545 Q-TOF instrument. MS resolution calculated from the data showed that the resolution for the 6545 instrument (R ~ 10 000–20 000) was roughly double that of the 6520 instrument (R ~ 5 000–10 000).
Software and Data Processing.
Progenesis QI (Version 2.2.5826.42898) from Nonlinear Dynamics (Durham, NC), was used to process the raw data (see Figure S1 for a description of the workflow). Centroid data files were extracted from the biformat raw data set using ProteoWizard msConvert freeware (http://proteowizard.sourceforge.net/)39 using the vendor (Agilent) algorithm. Profile raw data and converted LC–MS centroid data were imported into Progenesis QI for alignment, peak picking, and annotation. For the centroid data, median resolution values of 15 000 and 7500, were applied to the data from the Agilent 6545 and 6520 instruments, respectively. Data-processing parameters were set identically as much as possible to make the results comparable. In particular, the analysis was restricted to the 0.6–15 min retention time window and m/z of 60–1000. Default parameters for peak picking (automatic thresholds, minimum peak width = 3 s) and alignment were applied. Single-ion compounds were removed to reduce noise and false discovery as follows: after grouping the coeluting ions for compound identification and quantitation, every compound was defined by having two or more ions associated with the chromatographic peak. Only ions with a charge state of 1 were considered, and the analysis was limited to nine ion species, which included: [M + H]+, [M + Na]+, [M + K]+, [M + NH4]+, [M + H − H2O]+, [2M + H]+, [2M + Na]+, [2M + K]+, [2M + NH4]+, and [Misotope + H]+. Compounds with adduct ions were defined using neutral masses, and compounds with isotope ions only were defined using [M + H]+. Spectral matching was performed using the Human Metabolome Database (HMDB).40 The m/z accuracy tolerance was initially set to 10 ppm, which was a conservative approach taken for the older instrument; however, a 5 ppm tolerance was also evaluated to compare identified compound numbers.
Fitting Compound Number versus Abundance.
To better evaluate the metabolome coverage, the number of compounds detected was plotted versus the compound abundance to more easily visualize the nature and origin of missing values. Considering the challenge of comparing signals and compound numbers across different platforms and intensity ranges, we chose to calibrate the signal abundance at the point where the distribution of the detected number of compounds was maximal. To reduce skew, all abundances were first log10 transformed, which resulted in roughly normal-shaped distributions of the compound number versus ion count, which were fit using γ functions to take into account the slight skew of the data to higher abundances.
A histogram (h) of 100 equally spaced bins between the minimum and maximum values of the log-transformed abundance (x) was calculated. A γ distribution function, γ, was estimated using the Matlab function histfit to fit the histogram h as follows,
| (1) |
where A is the amplitude, α is the shape parameter, β is the scale parameter, and Γ is the γ function that has the formula . The mean μ and the standard deviation σ of the distribution can be calculated as μ = α/β and σ = α/β2, respectively.
Based on the fit, the log-transformed abundance was normalized by setting μ = 0 and similarly shifting h by the same amount. The area difference (i.e., the sum of non-overlapping areas) between the histogram h and γ distribution curve γ can also be used as a metric to evaluate data quality.
Missing Values.
To evaluate missing values, the compound number was plotted versus the detection frequency. These two dimensions were also normalized to 1, so that the coordinates of a characteristic point could be determined at either 0 or 20% missing. We chose this format for easy visualization, as it resembled a reflected receiver operating curve. The numbers of detected compounds and compounds with no missing value were also plotted versus the abundance. Furthermore, a three-dimensional (3-D) plot using the CV, abundance, and the percentage of compounds with missing values was developed.
CV and ICC.
The compounds were sorted by their median abundances (across the 50 QC samples), and then divided into 20 segments, each with 5% of the compounds. The accumulated compounds (from 0 to 100%) in 5% increments were used to calculate ICC. ICC provides an excellent metric to describe interobservational concordance, and, in particular, can detect changes in measurement values due to shifts and scaling effects better than the Pearson correlation. To compute the ICC, we used a two-way random, single-score approach defined as ICC (A,1) according to McGraw and Wong.33 The 20 accumulated percentages of compounds were plotted against the 20 corresponding ICCs. In addition, a 3-D plot using the ICC, CV, and the percentage of compounds was developed, in which the compounds were sorted by the CVs and divided into 20 segments, each with 0.05 of the CV. Then, the ICC and the corresponding compound numbers of the segments were calculated.
RESULTS AND DISCUSSION
Retention Time Drift and Compound Extraction versus Data Formats.
Figure 1a shows total ion count (TIC) chromatograms from the 50 samples acquired in profile mode on the Agilent 6545 instrument, and demonstrates the high reproducibility of the LC–MS experiments. Retention time drift was small, with values of −0.32 ± 0.29 and 0.04 ± 0.22 s for the 6520 and 6545 instruments, respectively. Figure 1b shows the m/z versus retention time for compounds extracted from the 50 sample data set collected on the Agilent 6545 in profile mode (6545(P)). As aforementioned, each compound was determined by grouping isotope and/or adduct ions to prevent improper large compound numbers and to reduce interference from chemical noise, as well as ungrouped isotope and/or adduct ion peaks. Filtering these single-ion features also contributes to the reduction in missing values and improves reproducibility, as is discussed below. The detection frequency of the compounds is marked in color (see Figure 1b). Some compounds could not be extracted from all 50 data, which resulted in missing values. Similarly, Figure 1c shows the m/z versus retention time of compounds extracted from the 50 sample data set collected on the Agilent 6545 in centroid mode (6545(C)). Comparing Figure 1b,c, the profile data resulted in significantly more compounds and fewer missing values than the centroid data. This may be due, in part, to the fact that Progenesis QI only accepts a single-resolution value for processing centroid data, which could reduce the number of compounds detected at higher m/z. At least for the analysis using Progenesis QI software, the more complete information provided by profile data is important to better filter background noise, and leads to cleaner data with more accurate peak detection and ultimately results in larger numbers of detected metabolite peaks.
Compound Numbers.
The evaluation of detected compounds primarily concerns how many compounds are extracted and identified. Analysis of the 6545(P), 6545(C), 6520(P), and 6520(C) data sets resulted in 5213, 2233, 1850, and 1230 detected compounds, respectively, as shown in Figure 2a. The Progenesis QI algorithm aligns and then stacks and adds the 50 m/z versus retention time data to define the compounds’ ion patterns, which increases the S/N compared to a single sample data. As a result, more compound ion patterns can be determined and extracted compared to those from the individual sample data. In addition, profile data provided many more compounds than centroid data, as discussed above. Not surprisingly, data from the 6520 instrument provided fewer compounds than the newer 6545 instrument, limited by its lower resolution and S/N ratio. Spectral matching to the HMDB library indicated that the highest number of matches was obtained using the 6545 instrument using profile mode data. The use of a tolerance of 5 ppm was also investigated and showed a similar trend across instrument platforms and acquisition modes, though the number of matches was reduced by ~20 to 30% in each case. A large number of detected and identified compounds increases data quality and improves the opportunity to measure the metabolome more comprehensively and thereby make insightful and novel metabolomics hypotheses.
Figure 2.
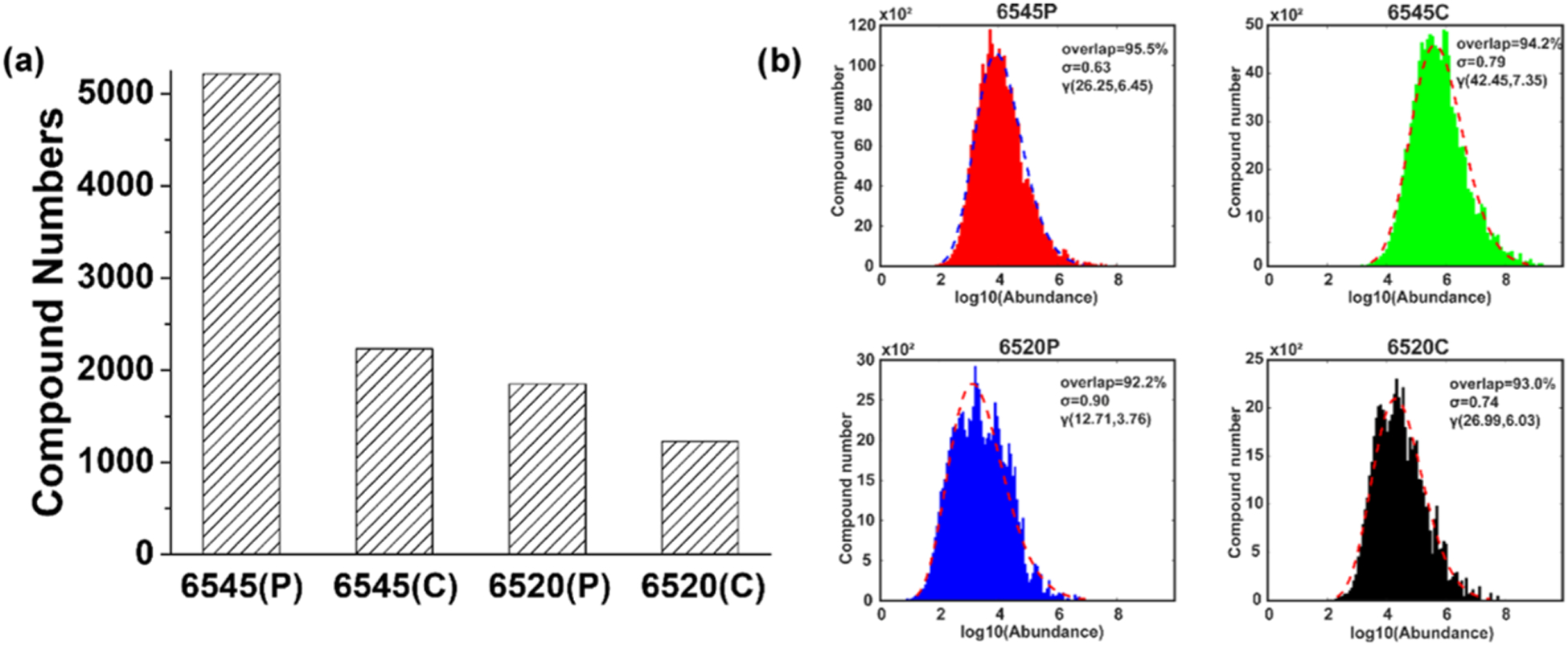
(a) Numbers of compounds extracted by Progenesis QI from the two different instrument platforms, 6545 and 6520, and either profile (P) or centroid (C) mode. (b) Compound numbers versus log10(abundance) and γ distribution γ(α, β) fits for the four types of data. The percentage of the overlapped area between the histogram and γ distributions is shown in each of the plots along with the standard deviation σ and the two parameters α and β.
Evaluation of the compound detection distribution provides additional and useful information on how the platform and software perform with respect to metabolite coverage as well as the nature of missing values. As shown in Figure 2b, the coverage results were fitted with a γ distribution. The area difference between the actual distribution and the fit provided a good overall means to characterize the data quality. The four approaches had area overlaps ranging from 92.2 to 95.5%, with the 6545 instrument showing a better fit and smaller σ, resulting from a more concentrated distribution, compared to the 6520 instrument. The higher resolution and higher S/N ratio of the 6545 instrument allowed data extraction of more compounds in the middle and low abundance ranges.
Missing Values and Characteristic Points.
Missing values induce problems such as unreliable compound identification and reporting, as well as biased statistical analysis. For example, compounds that cannot be reproducibly detected make them difficult to compare between samples and across studies, such that potentially key biomarkers are omitted. In LC–MS global metabolomic analysis, in which the data span a large dynamic range and data extraction typically depends on threshold levels, compound data can be absent for several reasons.22,23 Across a set of samples, some compounds are detected above the peak extraction thresholds in some samples but below the thresholds in others. Even if the samples are identical, LC–MS experiments include a number of steps from LC injection to MS detection and they are often not absolutely reproducible over time. Furthermore, impurities and random noise can produce undesirable or even false compounds in the final data set, which are absent from other samples and result in missing values. In addition, data extraction algorithms or software for data processing can fail to report some compounds from some data, resulting in missing values.23
As seen in Figure 3, the contrasting performance of each data set can be seen by plotting the compound number and detection frequency on a percentage basis. In many metabolomic applications, a data filter is often used to reduce the number of missing values. For example, compounds with more than 20% missing values are often eliminated from the data set prior to the imputation of the rest of the missing values, followed by statistical analysis. With a 20% missing-value cutoff chosen as a typical value, the percentages of compounds detected could be compared at a set of “characteristic points” that are easy to understand and report. These points were 99.88, 99.35, 97.54, and 92.35% for the 6545(P), 6520(P), 6545(C), and 6520(C), respectively. Additionally, as seen in the figure, there were 98.56, 96.81, 90.37, and 77.14% of the compounds detected in all of the samples for the 6545(P), 6520(P), 6545(C), and 6520(C), respectively. The area under the curve (AUC) is also provided, ranging from 0.957 to 0.999. A clear advantage for profile format data can be seen in this figure. Profile data provide complete information that is helpful to accurately determine compounds and facilitate the integration of ions peaks, resulting in fewer missing values compared to centroid data. The newer, 6545 instrument also performed better for both formats. The higher resolution and sensitivity provided by 6545 likely facilitated the accurate determination of the compounds and integration to suppress missing values.
Figure 3.
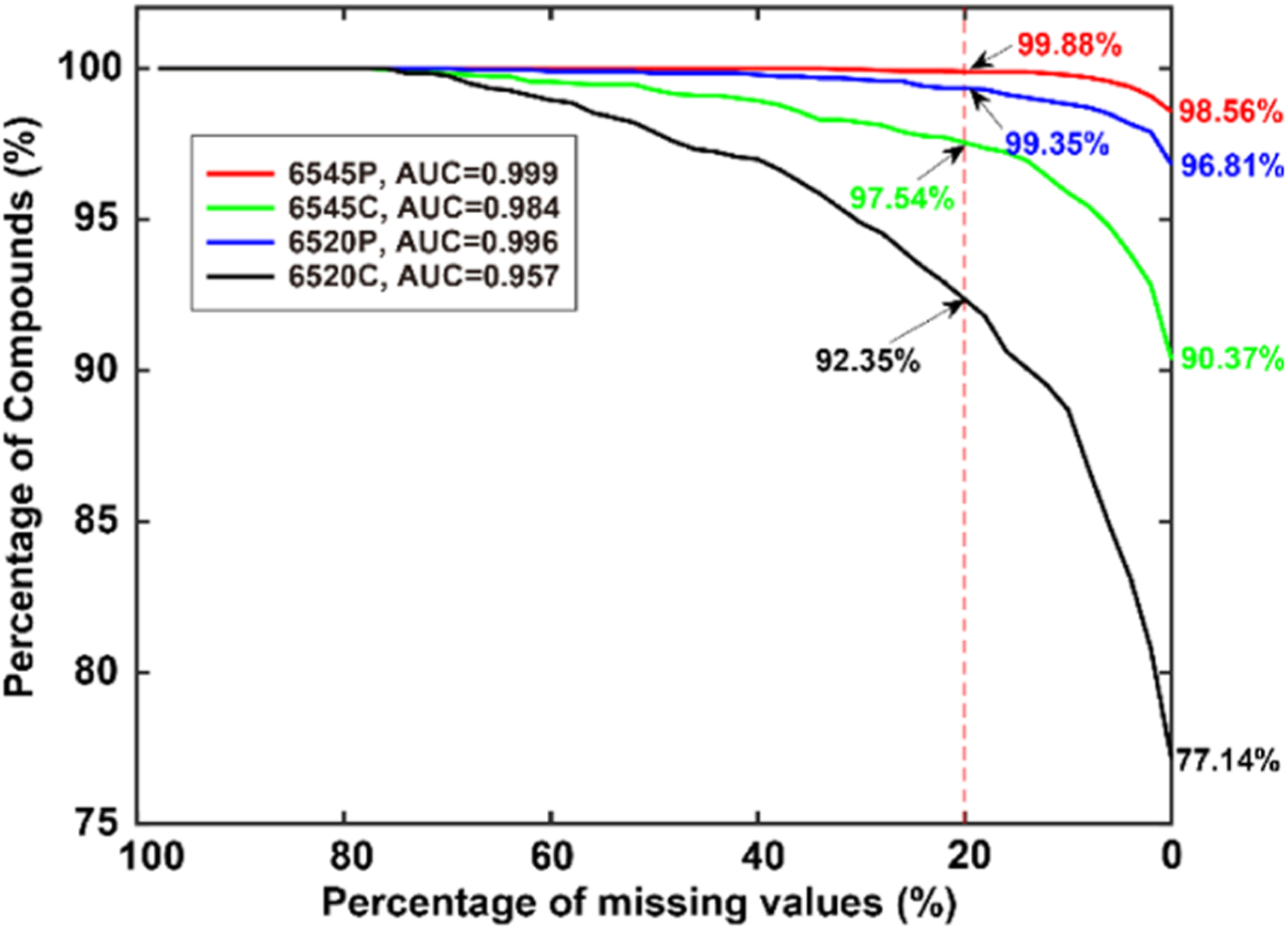
Missing values are visualized by plotting the percentage of compounds detected versus the percentage of missing values. As shown in the figure, 98.56% of the compounds were detected in all samples by the 6545(P) platform, while 99.88% of the compounds were detected with a missing rate of up to 20%. Poorer performance was seen, especially for the centroid data sets. Area under the curve (AUC) values are also provided.
As shown in Figure S2, individual curves for each of the instruments and data formats are shown, in which plots are shown for the number of compounds detected and compounds found in all samples (missing value free) versus the normalized intensity. These data provide additional information on the nature of the missing values, which are more prevalent for the lower concentration species and vary across the instrument platforms, recapitulating the overall trends seen in Figure 3. These results indicate how an advanced instrument or data format benefits the detection of missing value-free compounds.
CV and CV versus Compound Abundance.
To evaluate the reproducibility, CV versus compound abundance across the 50 samples was calculated. Only compounds with no missing values were considered. Figure 4a shows the accumulated percentage of compounds versus CVs measured for all platforms. It was found that the 6545(P) data had less than 10% of compounds with CV > 0.25, reflecting precise detection and data extraction. The 6545(C) and 6520(P) data showed higher compound percentages with CV > 0.25, while the 6520(C) had >50% compounds with CV > 0.25. This large CV reduces the reliability of statistical analysis considerably. Impressively, the median CV for the 6545(P) was ~5%, and ~70% of compounds had CV < 10%. A steep curve like the one shown in Figure 4a for the 6545(P) data represents high data quality, whereas using centroid data results in almost a twofold increase in CV.
Figure 4.
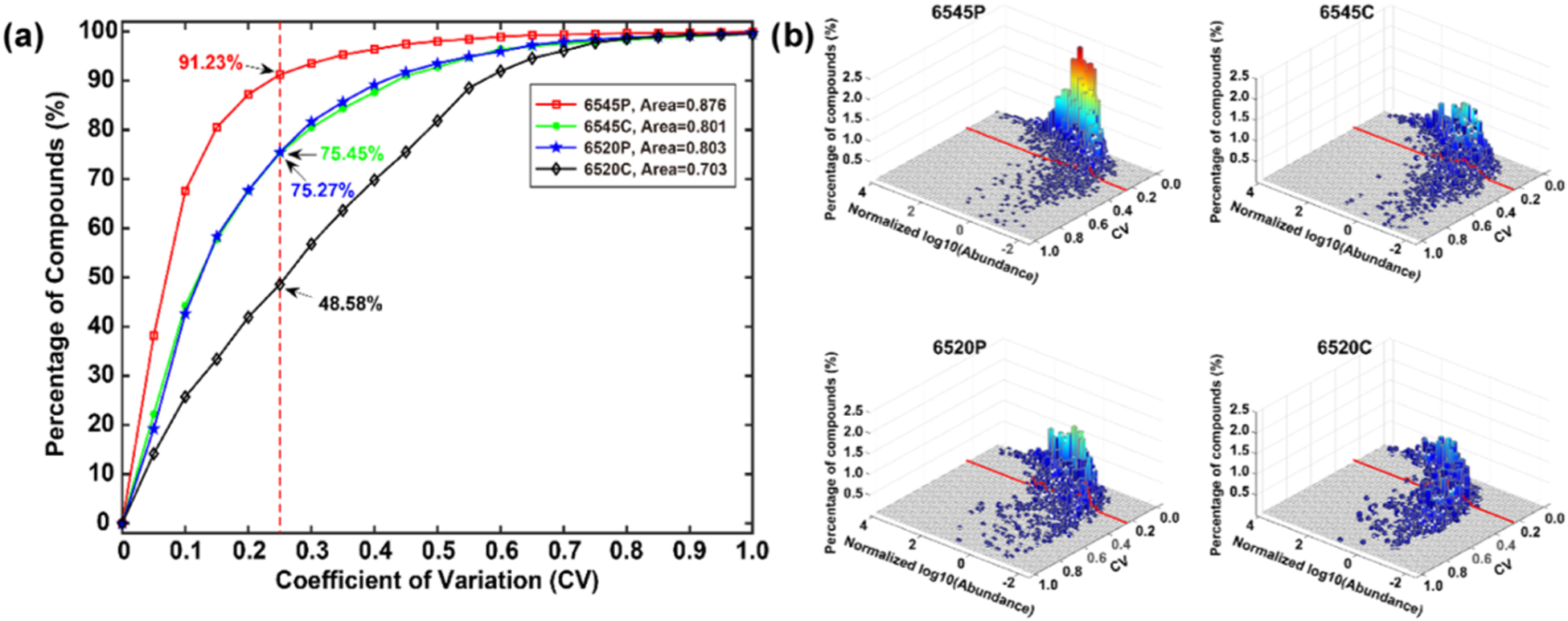
(a) CVs of the missing value-free compounds in the 50 samples versus the accumulated percentage of compounds. (b) CVs of the missing value-free compounds in the 50 samples set versus the normalized log10 (abundance) and percentage of compounds.
The relationship between the CV and signal intensity, log10(ion abundance), was also evaluated as a 3-D plot, as shown in Figure 4b, which could be a useful tool for parameter tuning or instrument comparisons. Not surprisingly, compounds with large abundance had small CV. When abundance decreased, the CV distribution started to extend to larger CV values, which was caused by insufficient S/N. However, the distributions were different in the four systems. For example, the 6545(P) data contained >90% of compounds with CV < 0.25, corresponding to the accumulated result in Figure 4a. The cross-section of the distribution had a median log abundance of approximately −1 at 0.25 CV. In contrast, the 6520(C) data contained <50% of compounds with CV < 0.25 and a median of ~0. High data quality is evident when a large percentage of the distribution has a small CV and when CV increases only at a relatively small abundance levels.
ICC versus Compound Abundance and ICC versus CV.
The reproducibility was also evaluated using correlation as a metric. We chose to use ICC because it is more discriminatory than the Pearson correlation, as it can detect changes such as drifts and scaling effects that the Pearson correlation cannot.41 In each data set, compounds were grouped into 20 fractions based on their log abundance. The cumulative correlation was calculated such that when the abundance reached the maximum, the correlation was calculated using all data in each data set. ICC was used to evaluate the data in an accumulated manner. Figure 5a shows that low abundance compounds had low correlation because of their low reproducibility arising from the influence of noise and other compounds, similar to the results and discussion discussed immediately above regarding CV. When the abundance range increased, the involvement of compounds with larger abundances improved the ICC and reproducibility for two reasons. First, large abundance compounds tend to have improved reproducibility due to their high S/N ratios. Second, mathematically, a large abundance compound influences the correlation more than a small abundance compound does. The results for the four data sets show that profile data exhibited much stronger correlation than centroid data. For example, at log10(abundance) = 0, the ICC was larger than 0.75, indicating excellent correlation; however, the 6520(C) and 6545(C) data sets had ICCs of only 0.31 and 0.43, respectively. With larger abundance compounds, the profile data produced an ICC of almost 1.0 near log10(abundance) = 2; however, the centroid data only reached a maximum of ~0.7. With the same data format, data from the 6545 platform performed slightly better than data from the 6520 platform, which again shows how an advanced instrument benefits data quality due to higher resolution and higher S/N. In the plot of ICC versus abundance, a steep curve and a high starting ICC represent high data quality. We also calculated the Pearson correlation coefficient (PCC) as a function of log10(abundance) and these data are shown in Figure S3. The results are similar to the ICC data, though less discriminatory.
Figure 5.
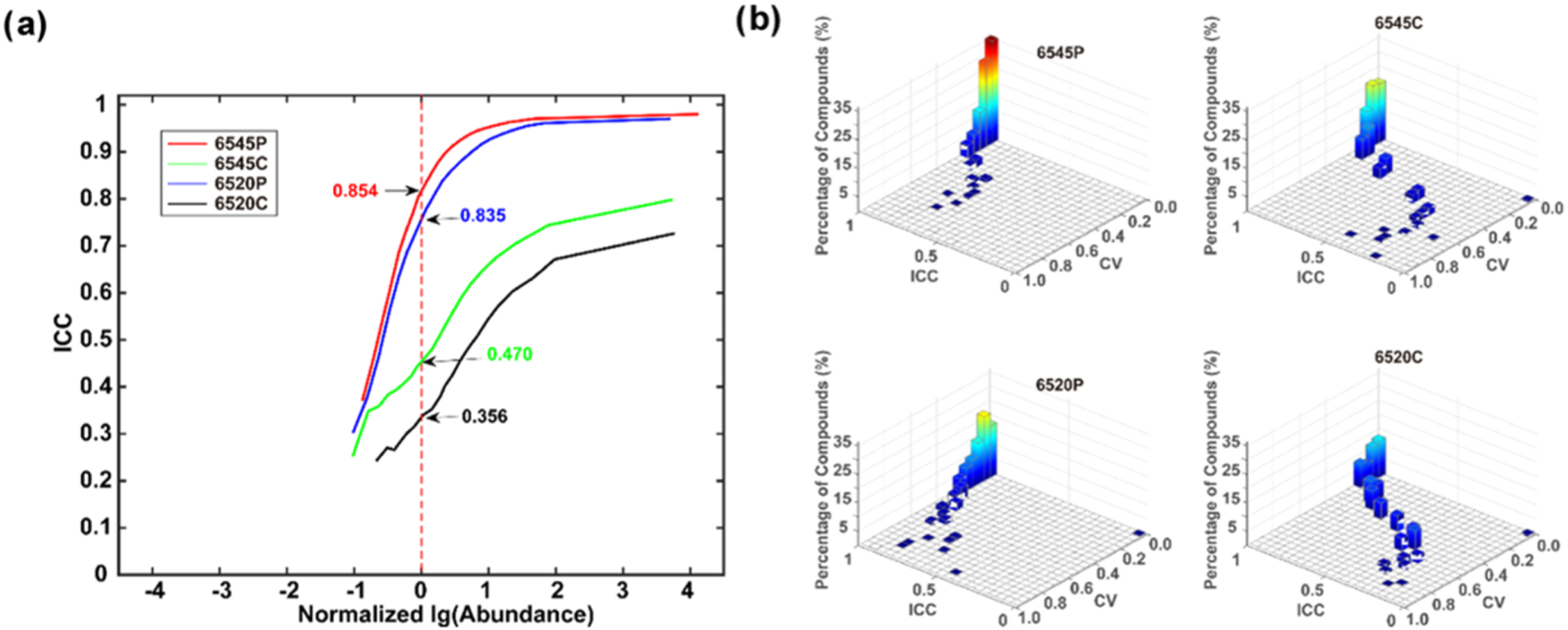
(a) ICC values versus percentages of compounds sorted by abundance for the 50 sample data set. The graph is normalized by setting the log10(abundance) = 0, where the number of detected metabolites is maximum. (b) 3-D plot of ICC versus CV and % compounds divided into 20 fractions along each axis.
Finally, the ICC was plotted versus CV to further explore reproducibility (see Figure 5b). When the CV was very small, for example, from 0 to 0.05, all systems had ICC > 0.95. When the CV increased, the ICC decreased but with different rates. The centroid data showed a decrease in ICC that was much faster than that for the profile data regardless of the instrument, indicating that the process of generating centroid and binned data likely cause the difference in CV. Furthermore, while the 6545(P) and 6520(P) data sets showed similar patterns, the 6545(P) had a higher percentage of compounds in a coordinate close to (CV = 0, ICC = 1), reflecting the positive contributions of high resolution and high sensitivity from the more advanced instrument. The 6545(C) and 6520(C) results show a similar phenomenon. These results indicate that the advanced instrument and profile format considerably reduced fluctuations in compound abundance. The more advanced instrument provided higher S/N and resolution, reducing the influence of noise and other ion species to enhance the reproducibility, especially at a lower signal intensity.
While a sample size of 50 QCs was chosen to mimic the effects seen in a global metabolomic study of moderate size (e.g., ~500 biological samples), we also analyzed the results for a smaller data set of five QC samples, and these data are shown in Figures S4–S9. Overall similar results were obtained with some notable findings. A somewhat smaller number of detected compounds was observed (see Figure S4), likely due to the lower S/N of the peaks in the Progenesis QI algorithm resulting from adding fewer spectra. Missing-value numbers were improved (see Figures S5 and S6) because there are fewer samples over which to detect the missing peaks, and the shorter time for acquisition of five samples allowed for less instrument drift. The CVs, ICCs, and Pearson correlations (Figures S7–S9) also improved for all data sets, although the 3-D distributions were quite similar when compared to the 50 QC data.
Consolidated Evaluation of Data Quality and Software.
The most important measures of data quality have been collected into a simple table for the 50 QC data (see Table 1) that provides a relatively comprehensive description of data quality for the different instruments and data formats evaluated here. We believe that this type of information can be useful for locally evaluating instrument performance, performing parameter and protocol optimization, for software development, and also for providing a useful summary in publications using global metabolomics data. All Progenesis QI-processed data sets and Matlab algorithms for assessing data quality, providing output plots, and producing the summary table can be downloaded from the Northwest Metabolomics Research Center website (http://nwmetabolomics.org/) and github at (https://github.com/jydong2018/metabolomics/). The raw and processed data can also be found at the Metabolomics Workbench,42 (https://www.metabolomicsworkbench.org), where it has been assigned Project ID PR000996. The data can be accessed directly via the Project DOI: 10.21228/M8Z692.
Table 1.
| quality measure | 6520 (C)a | 6520 (P) | 6545 (C) | 6545 (P) |
|---|---|---|---|---|
| retention time reproducibility (mean, SD, sec) | −0.32 ± 0.29 | 0.04 ± 0.22 | ||
| compounds detected (2 ions)b | 1230 | 1850 | 2233 | 5213 |
| percentage of compounds with less than 20% missing values | 92.35% | 99.35% | 97.54% | 99.88% |
| CV (percent of compounds with less than 25% CV) | 48.58% | 75.27% | 75.45% | 91.23% |
| ICC at most probable abundancec | 0.356 | 0.835 | 0.470 | 0.854 |
Notes: (C) = centroid mode; (P) = profile mode.
Compounds are defined as having at least two coeluting ions (see text for details).
Most probable abundance is normalized by setting log10(abundance) = 0, where the number of detected compounds is maximum.
CONCLUSIONS
High-quality data is a critical requirement in global metabolomics, and improved metrics are needed to describe the actual data quality of individual experiments, both for instrument parameter optimization and reporting purposes. In this work, we have combined known quality metrics and developed new metrics with the goal of providing a reasonably comprehensive set of overall measures of data quality for global metabolomics that are easy to understand and interpret. In the process of evaluating the compound numbers, missing values, and reproducibility in LC–MS global profiling of aqueous metabolites, the influence on data quality from different instruments and data formats was quantitatively demonstrated. This work shows, through the example of global metabolite profiling of human serum, that current LC–MS instrumentation and software can provide very good data quality. Not surprisingly, our analysis quantitatively demonstrated that an advanced instrument with high resolution and high S/N ratio detected more compounds, reduced missing values, and maintained a low CV. Somewhat surprisingly, the use of profile data or centroid data provides very different results due to the algorithms involved in producing and analyzing centroid data. This result may be due to unique features of the Progenesis QI software, and configurations of the resolution at multiple m/z instead of a single m/z might improve the performance of centroid data. Furthermore, the sample preparation and the LC–MS setup could also be adjusted to improve data quality.
For a particular application, and considering the many influencing factors that include different samples, instruments, parameters, and software, it is difficult at present to define absolute values or thresholds to evaluate data quality. The use of standard reference materials, such as the NIST 1950 or other QC samples,17 would be helpful to derive benchmarks for data quality measures for particular sample types and facilitate comparisons across laboratories. Nevertheless, the metrics and measurements developed in this work are suited for quantitative characterizations of data quality for applications such as testing the same samples on multiple instruments, tuning voltages in an instrument, tuning software parameters, and developing algorithms. As the quality of a data set depends on the compound numbers, missing values, and reproducibility, trade-offs could be necessary depending on the application. For the future, we anticipate that as such metrics become available for a variety of sample types and instruments, a consensus could be developed for minimal values and reporting standards. We believe that the adaptation of a consensus set of comprehensive metrics by the metabolomics community would be very beneficial for a number of purposes described above. The current efforts will hopefully spur the development of additional data quality metrics and especially a consensus on methods to evaluate and report data quality for global metabolomic data sets.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Qiang Fei in the College of Chemistry at Jilin University, China, for help in programming, and Yanan Yang and Donghui Yi at Agilent for providing raw data from the 6545 instrument. We also thank Dr. Fausto Carnevale Neto for his suggestions for improving our paper. We acknowledge the financial support from the NIH (P30CA015704, P30DK035816, and R01GM131491), Agilent and the University of Washington, and the National Natural Science Foundation of China (Grant No. 81871445). Metabolomics Workbench is supported by the NIH grant U2C-DK119886.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c01493.
Flow chart to process the raw LC–MS data (Figure S1), detected compounds and missing values versus compound abundance (Figure S2), the Pearson correlation coefficient versus compound abundance (Figure S3), missing-value performance for the reduced number of samples (Figure S4), detected compounds and missing values versus compound abundance for five QCs (Figure S5), the percentage of compounds versus CV (Figure S6), the 3-D plot of same versus log10(abundance) for five QCs, ICC values versus the percentage of compounds, the 3-D plot of same versus CV for five QCs (Figure S7), detected compounds and missing values versus compound abundance for five QCs (Figure S8), and the Pearson correlation coefficient versus compound abundance for five QCs (Figure S9) (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.analchem.0c01493
The authors declare no competing financial interest.
Contributor Information
Xinyu Zhang, Northwest Metabolomics Research Center, Department of Anesthesiology and Pain Medicine, University of Washington, Seattle, Washington 98109, United States.
Jiyang Dong, Department of Electronic Science, Xiamen University, Xiamen 361005, China.
Daniel Raftery, Northwest Metabolomics Research Center, Department of Anesthesiology and Pain Medicine, University of Washington, Seattle, Washington 98109, United States.
REFERENCES
- (1).Patti GJ; Yanes O; Siuzdak G Nat. Rev. Mol. Cell Biol 2012, 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Theodoridis GA; Gika HG; Want EJ; Wilson ID Anal. Chim. Acta 2012, 711, 7–16. [DOI] [PubMed] [Google Scholar]
- (3).Goodacre R; Vaidyanathan S; Dunn WB; Harrigan GG; Kell DB Trends Biotechnol. 2004, 22, 245–252. [DOI] [PubMed] [Google Scholar]
- (4).Sreekumar A; Poisson LM; Rajendiran TM; Khan AP; Cao Q; Yu J; Laxman B; Mehra R; Lonigro RJ; Li Y; et al. Nature 2009, 457, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- (5).Wang ZN; Klipfell E; Bennett BJ; Koeth R; Levison BS; Dugar B; Feldstein AE; Britt EB; Fu X; Chung YM; et al. Nature 2011, 472, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wikoff WR; Hanash S; DeFelice B; Miyamoto S; Barnett M; Zhao Y; Goodman G; Feng Z; Gandara D; Fiehn O; Taguchi A J. Clin. Oncol 2015, 33, 3880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Trushina E; Dutta T; Persson X-MT; Mielke MM; Petersen RC PLoS One 2013, 8, No. e63644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sperber H; Mathieu J; Wang Y; Ferreccio A; Hesson J; Xu Z; Fischer KA; Devi A; Detraux D; Gu H; Battle SL; Showalter M; Valensis C; Bielas JH; Ericson NG; Margaretha L; Robitaille AM; Margineantu D; Fiehn O; Hockenbery D; Blau CA; Raftery D; Margolin A; Hawkins RD; Moon RT; Ware CB; Ruohola-Baker H Nat. Cell Biol 2015, 17, 1523–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yanes O; Clark J; Wong DM; Patti GJ; Sanchez-Ruiz A; Benton HP; Trauger SA; Desponts C; Ding S; Siuzdak G Nat. Chem. Biol 2010, 6, 411–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Rohle D; Popovici-Miller J; Palaskas N; Turcan S; Grommes C; Campos C; Tsoi J; Clark O; Oldrini B; Komisopoulou E; et al. Science 2013, 340, 626–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wishart DS Nat. Rev. Drug Discovery 2016, 15, 473–484. [DOI] [PubMed] [Google Scholar]
- (12).Tugizimana F; Steenkamp PA; Piater LA; Dubery IA Metabolites 2016, 6, No. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gika HG; Macpherson E; Theodoridis GA; Wilson ID J. Chromatogr. B 2008, 871, 299–305. [DOI] [PubMed] [Google Scholar]
- (14).Engskog MKR; Haglöf J; Arvidsson T; Pettersson C Metabolomics 2016, 12, No. 114. [Google Scholar]
- (15).Broadhurst D; Goodacre R; Reinke SN; Kuligowski J; Wilson ID; Lewis MR; Dunn WB Metabolomics 2018, 14, No. 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Beger RD; Dunn WB; Bandukwala A; Bethan B; Broadhurst D; Clish CB; Dasari S; Derr L; Evans A; Fischer S; et al. Metabolomics 2019, 15, No. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Vuckovic D Anal. Bioanal. Chem 2012, 403, 1523–1548. [DOI] [PubMed] [Google Scholar]
- (18).Creek DJ; Dunn WB; Fiehn O; Griffin JL; Hall RD; Lei Z; Mistrik R; Neumann S; Schymanski EL; Sumner LW; et al. Metabolomics 2014, 10, 350–353. [Google Scholar]
- (19).Martin J-C; Maillot M; Mazerolles G; Verdu A; Lyan B; Migné C; Defoort C; Canlet C; Junot C; Guillou C; et al. Metabolomics 2015, 11, 807–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Psychogios N; Hau DD; Peng J; Guo AC; Mandal R; Bouatra S; Sinelnikov I; Krishnamurthy R; Eisner R; Gautam B; et al. PLoS One 2011, 6, No. e16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Bijlsma S; Bobeldijk I; Verheij ER; Ramaker R; Kochhar S; Macdonald IA; van Ommen B; Smilde AK Anal. Chem 2006, 78, 567–574. [DOI] [PubMed] [Google Scholar]
- (22).Di Guida R; Engel J; Allwood JW; Weber RJM; Jones MR; Sommer U; Viant MR; Dunn WB Metabolomics 2016, 12, No. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wei R; Wang J; Su M; Jia E; Chen S; Chen T; Ni Y Sci. Rep 2018, 8, No. 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Hrydziuszko O; Viant MR Metabolomics 2012, 8, 161–174. [Google Scholar]
- (25).Xia J; Wishart DS Nat. Protoc 2011, 6, 743–760. [DOI] [PubMed] [Google Scholar]
- (26).Castillo S; Gopalacharyulu P; Yetukuri L; Oresič M Chemom. Intell. Lab. Syst 2011, 108, 23–32. [Google Scholar]
- (27).Kokla M; Virtanen J; Kolehmainen M; Paananen J; Hanhineva K BMC Bioinf. 2019, 20, No. 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Little RJA; Rubin DB Statistical Analysis with Missing Data, 11–19; John Wiley & Sons, 2002. [Google Scholar]
- (29).Considine EC Metabolites 2019, 9, No. 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dunn WB; Broadhurst D; Begley P; Zelena E; Francis-McIntyre S; Anderson N; Brown M; Knowles JD; Halsall A; Haselden JN; et al. Nat. Protoc 2011, 6, 1060–1083. [DOI] [PubMed] [Google Scholar]
- (31).Gika HG; Zisi C; Theodoridis G; Wilson ID J. Chromatogr. B 2016, 1008, 15–25. [DOI] [PubMed] [Google Scholar]
- (32).Vorkas PA; Isaac G; Anwar MA; Davies AH; Want EJ; Nicholson JK; Holmes E Anal. Chem 2015, 87, 4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).McGraw KO; Wong SP Psychol. Methods 1996, 1, 30–46. [Google Scholar]
- (34).Sampson JN; Boca SM; Shu XO; Stolzenberg-Solomon RZ; Matthews CE; Hsing AW; Tan YT; Ji BT; Chow WH; Cai QY; et al. Cancer Epidemiol. Biomarkers Prev 2013, 22, 631–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G Anal. Chem 2006, 78, 779–787. [DOI] [PubMed] [Google Scholar]
- (36).Pluskal T; Castillo S; Villar-Briones A; Orešič M BMC Bioinf. 2010, 11, No. 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Fan SL; Kind T; Cajka T; Hazen SL; Tang WHW; Kaddurah-Daouk R; Irvin MR; Arnett DK; Barupal DK; Fiehn O Anal. Chem 2019, 91, 3590–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Gu H; Zhang P; Zhu J; Raftery D Anal. Chem 2015, 87, 12355–12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Chambers MC; MacLean B; Burke R; Amode D; Ruderman DL; Neumann S; Gatto L; Fischer B; Pratt B; Egertson J; et al. Nat. Biotechnol 2012, 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wishart DS; Feunang YD; Marcus A; Gua AC; Liang K; Vázquez-Fresno R; Sajed T; Johnson D; Li C; Karu N; et al. Nucleic Acids Res. 2018, 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Intraclass Correlation. https://en.wikipedia.org/wiki/Intraclass_correlation.
- (42).Sud M; Fahy E; Cotter D; Azam K; Vadivelu I; Burant C; Edison A; Fiehn O; Higashi R; Nair KS; Sumner S; Subramaniam S Nucleic Acids Res. 2016, 44, D463–D470. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.