

Abstract
Plaques of the amyloid beta (Aß) peptide are a pathological hallmark of Alzheimer’s disease (AD), the most common form of dementia. Mutations in Aß also cause familial forms of AD (fAD). Here, we use deep mutational scanning to quantify the effects of >14,000 mutations on the aggregation of Aß. The resulting genetic landscape reveals mechanistic insights into fibril nucleation, including the importance of charge and gatekeeper residues in the disordered region outside of the amyloid core in preventing nucleation. Strikingly, unlike computational predictors and previous measurements, the empirical nucleation scores accurately identify all known dominant fAD mutations in Aß, genetically validating that the mechanism of nucleation in a cell-based assay is likely to be very similar to the mechanism that causes the human disease. These results provide the first comprehensive atlas of how mutations alter the formation of any amyloid fibril and a resource for the interpretation of genetic variation in Aß.
Research organism: S. cerevisiae
eLife digest
Alzheimer’s disease is the most common form of dementia, affecting more than 50 million people worldwide. Despite more than 400 clinical trials, there are still no effective drugs that can prevent or treat the disease. A common target in Alzheimer’s disease trials is a small protein called amyloid beta. Amyloid beta proteins are ‘sticky’ molecules. In the brains of people with Alzheimer’s disease, they join to form first small aggregates and then long chains called fibrils, a process which is toxic to neurons.
Specific mutations in the gene for amyloid beta are known to cause rare, aggressive forms of Alzheimer’s disease that typically affect people in their fifties or sixties. But these are not the only mutations that can occur in amyloid beta. In principle, any part of the protein could undergo mutation. And given the size of the human population, it is likely that each of these mutations exists in someone alive today.
Seuma et al. reasoned that studying these mutations could help us understand the process by which amyloid beta forms new aggregates. Using an approach called deep mutational scanning, Seuma et al. mutated each point in the protein, one at a time. This produced more than 14,000 different versions of amyloid beta. Seuma et al. then measured how quickly these mutants were able to form aggregates by introducing them into yeast cells.
All the mutations known to cause early-onset Alzheimer’s disease accelerated amyloid beta aggregation in the yeast. But the results also revealed previously unknown properties that control how fast aggregation occurs. In addition, they highlighted a number of positions in the amyloid beta sequence that act as ‘gatekeepers’. In healthy brains, these gatekeepers prevent amyloid beta proteins from sticking together. When mutated, they drive the protein to form aggregates.
This comprehensive dataset will help researchers understand how proteins form toxic aggregates, which could in turn help them find ways to prevent this from happening. By providing an ‘atlas’ of all possible amyloid beta mutations, the dataset will also help clinicians interpret any new mutations they encounter in patients. By showing whether or not a mutation speeds up aggregation, the atlas will help clinicians predict whether that mutation increases the risk of Alzheimer’s disease.
Introduction
Amyloid plaques consisting of the amyloid beta (Aß) peptide are a pathological hallmark of Alzheimer’s disease (AD), the most common cause of dementia and a leading global cause of morbidity with very high societal and economic impact (Ballard et al., 2011; World Health Organization, 2012). Although most cases of AD are sporadic and of uncertain cause, rare familial forms of the disease also exist (Campion et al., 1999). These inherited forms of dementia typically have earlier onset and are caused by high penetrance mutations in multiple loci, including in the amyloid precursor protein (APP) gene, which encodes the protein from which Aß is derived by proteolytic cleavage (O'Brien and Wong, 2011). Several mutations in PSEN1 and PSEN2, the genes coding for the secretases performing sequential cleavage of APP, also lead to autosomal dominant forms of AD. The two most abundant isoforms of Aß generated upon cleavage are 42 and 40 amino acids (aa) in length, with the longer Aß peptide associated with increased aggregation in vitro and cellular toxicity (Meisl et al., 2014; Sandberg et al., 2010). The amyloid state is a thermodynamically low energy state but, both in vitro and in vivo, the spontaneous formation of amyloids is normally very slow because of the high kinetic barrier of fibril nucleation (Knowles et al., 2014). The process of nucleation generates oligomeric Aß species that have been hypothesized to be particularly toxic to cells and that then grow into fibrils (Michaels et al., 2020; Bolognesi et al., 2010; Cleary et al., 2005).
Fourteen different mutations in the Aß peptide have been reported to cause familial Alzheimer’s disease (fAD), with all but two having a dominant pattern of inheritance (Weggen and Beher, 2012; Van Cauwenberghe et al., 2016). However, it is not clear why these particular mutations cause fAD (Weggen and Beher, 2012; Van Cauwenberghe et al., 2016), and these 14 mutations represent only 3.7% of the possible 378 single nucleotide changes that can occur in Aß. As for nearly all disease genes, therefore, the molecular mechanism by which mutations cause the disease remains unclear and the vast majority of possible mutations in Aß are variants of uncertain significance (VUS). This makes the clinical interpretation of genetic variation in this locus a difficult challenge (Starita et al., 2017; Gelman et al., 2019). Moreover, given the human mutation rate and population size, it is likely that nearly all of these possible variants in Aß actually exist in at least one individual currently alive on the planet (Conrad et al., 2011). A comprehensive map of how all possible mutations affect the formation of Aß amyloids and how these changes relate to the human disease is therefore urgently needed.
More generally, amyloid fibrils are associated with many different human diseases (Knowles et al., 2014), but how mutations alter the propensity of proteins to aggregate into amyloid fibrils is not well understood and there has been no large-scale analysis of the effects of mutations on the formation of any amyloid fibril. Here, we address this shortcoming by quantifying the rate of fibril formation for >14,000 variants of Aß. This provides the first comprehensive map of how mutations alter the propensity of any protein to form amyloid fibrils. The resulting data provide numerous mechanistic insights into the process of Aß fibril nucleation. Moreover, they also accurately classify all the known dominant fAD mutations, validating the clinical relevance of a simple cell-based model and providing a comprehensive resource for the interpretation of clinical genetic data.
Results
Deep mutagenesis of Aß
To globally quantify the impact of mutations on the nucleation of Aß fibrils, we used an in vivo selection assay in which the nucleation of Aß is rate-limiting for the aggregation of a second amyloid, the yeast prion [PSI+] encoded by the sup35 gene (Chandramowlishwaran et al., 2018). Aggregation of Sup35p causes read-through of UGA stop codons, allowing growth-based selection using an auxotrophic marker containing a premature termination codon (Figure 1A and Figure 1—figure supplement 1A). We generated a library containing all possible single nucleotide variants of Aß42 fused to the nucleation (N) domain of Sup35p and quantified the effect of mutations on the rate of nucleation in triplicate by selection and deep sequencing (Faure et al., 2020; see Materials and methods). The selection assay was highly reproducible, with enrichment scores for aa substitutions strongly correlated between replicates (Figure 1B and Figure 1—figure supplement 1B).
Figure 1. Deep mutagenesis of amyloid beta (Aß) nucleation.
(A) In vivo Aß selection assay. Aß fused to the Sup35N domain seeds aggregation of endogenous Sup35p causing a read-through of a premature UGA in the Ade1-14 reporter, allowing the cells to grow in medium lacking adenine. (B) Correlation of nucleation scores for biological replicates 1 and 2 for single and double amino acid (aa) mutants. Pearson correlation coefficient and p-value are indicated (Figure 1—figure supplement 1B) n = 10,157 genotypes. (C) Correlation of nucleation scores with in vitro primary and secondary nucleation and elongation rate constants (Yang et al., 2018). Weighted Pearson correlation coefficient and p-value are indicated. (D) Nucleation scores as a function of principal component 1 (PC1) aa property changes (Bolognesi et al., 2019) for single and double aa mutants (n = 14,483 genotypes). Weighted Pearson correlation coefficient and p-value are indicated. (E) Solubility scores (Gray et al., 2019) as a function of PC1 changes (Bolognesi et al., 2019) for n = 895 single and double mutants. Pearson correlation coefficient and p-value are indicated.

Figure 1—figure supplement 1. Reproducibility of the assay and correlation with in vitro fibril nucleation.

In vivo nucleation scores are highly correlated with in vitro rates of amyloid nucleation
Comparing our in vivo enrichment scores to the qualitative effects of 16 mutations analysed in vitro across 10 previous publications validated the assay, with mutational effects matching the effects on in vitro nucleation previously reported for 14 Aß variants out of 16. (Supplementary file 1). Moreover, the in vivo scores correlate extremely well with the rate of nucleation of Aß variants in positions 21, 22, 23 (Yang et al., 2018; Törnquist et al., 2018; Figure 1C and Figure 1—figure supplement 1C). We henceforth refer to the in vivo enrichment scores as ‘nucleation scores’ (NS).
Two mechanisms of in vivo Aß aggregation
A prior deep mutational scan quantified the effects of mutations on the abundance of Aß fused to an enzymatic reporter (Gray et al., 2019). These ‘solubility scores’ do not predict the effects of mutations on Aß nucleation (Figure 1—figure supplement 1D). Previously we identified a principal component of aa properties (principal component 1 [PC1], related to changes in hydrophobicity) that predicts the aggregation and toxicity of the amyotrophic lateral sclerosis (ALS) protein TDP-43 when it is expressed in yeast (Bolognesi et al., 2019). PC1 is also not a good predictor of Aß nucleation (Figure 1D) but it does predict the previously reported changes in Aß solubility (Figure 1E), suggesting that Aß is aggregating by a similar process to TDP-43 in this alternative selection assay (Gray et al., 2019) but by a different mechanism in the nucleation selection.
Nucleation scores for 14,483 Aß variants
The distribution of mutational effects for Aß nucleation has a strong bias towards reduced nucleation, with 56% of single aa substitutions reducing nucleation but only 16% increasing it (Z-test, false discovery rate [FDR] = 0.1, Figure 2A). Moreover, mutations that decrease nucleation in our dataset typically have a larger effect than those that increase it, with many mutations reducing nucleation to the background rate observed for Aß variants containing premature termination codons (Figure 2A).
Figure 2. Modular organization of mutational effects in amyloid beta (Aß).
(A and B) Nucleation scores distribution for single (A) and double (B) amino acid (aa) mutants. n = 468 (missense), n = 31 (nonsense), n = 90 (synonymous) for singles, and n = 14,015 (missense) for doubles. Vertical dashed line indicates wild-type (WT) score (0). (C) Heatmap of nucleation scores for single aa mutants. The WT aa and position are indicated in the x-axis and the mutant aa is indicated on the y-axis, both coloured by aa class. Variants not present in the library are represented in white. Synonymous mutants are indicated with ‘*’ and familial Alzheimer’s disease (fAD) mutants with a box, coloured by fAD class. (D) Number of variants significantly increasing (blue) and decreasing (orange) nucleation at different false discovery rates (FDRs). Gatekeeper positions (D1, E3, D7, E11, L17, E22, and A42) are indicated on top of the corresponding bar and coloured on the basis of aa type. The N-terminal and C-terminal definitions are indicated on the x-axis. Gatekeeper positions are excluded from the N-terminal and C-terminal classes. (E) Aa position distributions for variants that increase (+), decrease (−), or have no effect on nucleation (WT-like) (FDR < 0.1). (F) Nucleation score distributions for the three clusters of positions defined on the basis of nucleation: Nt (2-26), Ct (27-41), and gatekeeper positions (clusters are mutually exclusive). Horizontal line indicates WT nucleation score (0). Nonsense (stop) mutants were only included in A and C. Boxplots represent median values and the lower and upper hinges correspond to the 25th and 75th percentiles, respectively. Whiskers extend from the hinge to the largest value no further than 1.5*IQR (interquartile range). Outliers are plotted individually or omitted when the boxplot is plotted together with individual data points or a violin plot.

Figure 2—figure supplement 1. Mutational effects in amyloid beta (Aß).

In addition to covering all aa changes obtainable through single nt mutations, our mutagenesis library was designed to contain a substantial fraction of double mutants. In total, we quantified the impact of 14,015 double aa variants of Aß. Double mutants were even more likely to reduce nucleation, with 63% decreasing and only 5.5% increasing nucleation (Z-test, FDR = 0.1; Figure 2B). Therefore, mutations more frequently decrease rather than increase Aß nucleation.
Aß has a modular mutational landscape
Inspecting the heatmap of mutational effects for aa changes at all positions in Aß reveals strong biases in the locations of mutations that increase and decrease nucleation (Figure 2C and D, and Figure 2—figure supplement 1A). Mutations that decrease nucleation are highly enriched in the C-terminus of Aß, whereas mutations that increase nucleation are enriched in the N-terminus (Figure 2E). Indeed, >84% of mutations in the C-terminus (residues 27-42) reduce nucleation and only 9.6% increase it (FDR = 0.1). In contrast, the effects of mutations are smaller (Figure 2F) and also more balanced in the first 26 aa of the peptide, with 38.6% decreasing and 20% increasing nucleation (FDR = 0.1).
These differences in the direction and strength of mutational effects between the N- and C-terminal regions of Aß suggest a modular organization of the peptide. This modularity is also reflected in the primary sequence of Aß, which has a hydrophobic C-terminus and a more polar and charged N-terminus (eight out of nine charged residues in Aß are found before residue 24 and the peptide consists entirely of hydrophobic residues from position 29) (Figures 2C and 3A). Consistent with this modular organization, mutations in the few hydrophobic residues in the N-terminus have effects that are more similar to mutations in polar residues in the N-terminus rather than in hydrophobic residues in the C-terminus. Similarly, mutations in the most C-terminal charged residue (K28) frequently strongly reduce nucleation, just as they do in the adjacent hydrophobic positions (Figure 3A).
Figure 3. Determinants of amyloid beta (Aß) nucleation.
(A) Effect of single aa mutants on nucleation for each Aß position. The wild-type (WT) aa and position are indicated on the x-axis and coloured on the basis of aa type. The horizontal line indicates the WT nucleation score (0). (B to D) Effect of each mutant aa on nucleation for the Ct (27-41) (B), the Nt (2-26) (C), and the negatively charged gatekeeper positions (D1, E3, D7, E11, and E22) (D). The three position clusters are mutually exclusive. Colour code indicates aa type. The horizontal line is set at the WT nucleation score (0). (E) Effect on nucleation for single aa mutations to proline, threonine, valine, and isoleucine. Mutations to other aa are indicated in grey. The horizontal line indicates WT nucleation score (0). Point size and shape indicate false discovery rate (FDR) and familial Alzheimer’s disease (fAD) class, respectively (see legend). (F) Nucleation scores as a function of hydrophobicity changes (Kyte and Doolittle, 1982) for single and double aa mutants in the Ct (27-41) cluster. Only double mutants with both mutations in the indicated position-range were used. Weighted Pearson correlation coefficient and p-value are indicated. (G) Nucleation score distributions arranged by the number of charged residues (y-axis) and the total net charge (x-axis) for single and double aa mutants in the full peptide (1-42). Only polar, charged, and glycine aa types were taken into account, for both WT and mutant residues. Colour gradient indicates the total number of charged residues. Numbers inside each cell indicate the number of positive and negative residues. The horizontal line indicates WT nucleation score (0). Boxplots represent median values and the lower and upper hinges correspond to the 25th and 75th percentiles, respectively. Whiskers extend from the hinge to the largest value no further than 1.5*IQR (interquartile range). Outliers are plotted individually or omitted when the boxplot is plotted together with individual data points or a violin plot.

Figure 3—figure supplement 1. Determinants of amyloid beta (Aß) nucleation.

Figure 3—figure supplement 2. Effect of mutations to each specific amino acid (aa) on amyloid beta (Aß) nucleation.

Gatekeeper residues act as anti-nucleators
Considering the entire Aß peptide, there are only seven positions in which mutations are not more likely to decrease rather than increase nucleation (FDR = 0.1; Figure 2D). Strikingly, these positions, which we refer to as ‘gatekeepers’ of nucleation (Rousseau et al., 2006; Pedersen et al., 2004), include five of the six negatively charged residues in Aß. The sixth gatekeeper is an unusual hydrophobic residue in the N-terminus, L17, where seven mutations increase nucleation and only one decreases it (FDR = 0.1; Figure 2D). The final aa of the peptide, A42, also has an unusual distribution of mutational effects that is different to the rest of the C-terminus, with four mutations increasing and three mutations decreasing nucleation (FDR = 0.1; Figure 2D).
Taken together, on the basis of mutational effects, we therefore distinguish the following mutually exclusive positions in Aß: the C-terminus (aa 27-41) where the majority of mutations strongly decrease nucleation, the N-terminus (aa 2-26) where mutations have smaller and more balanced effects, and seven gatekeeper residues (D1, E3, D7, E11, D22, L17, A42) where mutations frequently increase nucleation. We consider each of these classes below.
Mutations in the N- and C-terminal regions
Mutations in the C-terminus nearly all decrease nucleation (Figure 3A). This is consistent with the C-terminus forming part of the tightly packed amyloid core of all known structural polymorphs of both Aß42 (Colvin et al., 2016; Meier et al., 2017; Wälti et al., 2016; Xiao et al., 2015; Gremer et al., 2017; Lührs et al., 2005; Schmidt et al., 2015) and Aß40 (Kollmer et al., 2019; Lu et al., 2013; Qiang et al., 2012; Sgourakis et al., 2015; Paravastu et al., 2008; Schütz et al., 2015). Consistent with this, we quantified the nucleation of three C-terminal fragments of the peptide (aa 22-42, 24-42, 27-42) and found that they nucleate similarly or better than full length Aß (Figure 3—figure supplement 1C). Mutations to polar and charged residues in this region nearly all decrease nucleation, but so too do most changes to other hydrophobic residues (Figure 3B), suggesting specific side chain packing in this region is important for nucleation. The relative effects of different mutations are only partially captured by changes in hydrophobicity (Figure 3F; Pearson correlation coefficient, R = 0.45) and by predictors of aggregation potential (Figure 3—figure supplement 1A). Only a few mutations in this region increase nucleation: substitutions to isoleucine at positions 30, 34, and 39; mutations to valine at positions 29, 30, and 34; a change to threonine at position 30; changes to leucine and methionine at 36; and a mutation to phenylalanine at position 41 (FDR = 0.1).
Mutations in the N-terminus of Aß have a more balanced effect on nucleation, and these effects are not well predicted by either hydrophobicity or predictors of aggregation potential (Figure 3—figure supplement 1B,D and E). The effects of introducing particular aa are, however, biased, with the introduction of asparagine, isoleucine, and valine most likely to increase nucleation (Figure 3C and Figure 3—figure supplement 2). As at the C-terminus, the introduction of negative charged residues typically strongly reduces nucleation (Figure 3B and C). However, in contrast to what is observed in the C-terminus (Figure 3B), the effects of introducing positive charge are less severe (Figure 3C). Interestingly, the effects of mutations to proline, isoleucine, valine, and threonine in the N-terminus depend on the position in which they are made: mutations in the first 12 residues typically decrease nucleation, whereas mutations in the next four to nine residues increase nucleation (Figure 3E). The conformational rigidity of proline and the beta-branched side chains of isoleucine, valine, and threonine that disfavour helix formation suggest that disruption of a secondary structure in this region may favour nucleation. Interestingly, this same region was highlighted as the part of the peptide remaining most disordered across different states of the solution ensemble of Aß in molecular dynamics simulations, with the same region also making extensive long-range contacts in different states of the kinetic ensemble (Löhr et al., 2021).
The role of charge in limiting Aß nucleation
At five of six negatively charged positions in Aß, mutations frequently increase nucleation (Figures 2D and 3A). Moreover, the introduction of negative charge at other positions strongly decreases nucleation (Figure 3A), suggesting that negatively charged residues act as gatekeepers (Pedersen et al., 2004; Rousseau et al., 2006) to limit nucleation (Figure 3D and Figure 3—figure supplement 1D). In contrast, mutations in the three positively charged residues (R5, K16, K28) mostly decrease nucleation (Figure 2D). Mutating the negatively charged gatekeepers to the polar aa glutamine and asparagine, to positively charged residues (arginine and lysine), or to small side chains (glycine and alanine) increases nucleation (Figure 3D). Mutating the same positions to hydrophobic residues typically reduces nucleation (Figure 3D). This is consistent with a model in which the negative charge at these positions acts to limit nucleation, but that the overall polar and unstructured nature of the N-terminus must be maintained for effective nucleation.
To further investigate the role of charge in controlling Aß nucleation, we extended our analyses to the double mutants. Including double mutants allows the net charge of Aß to vary over a wider range and it also allows comparison of the nucleation of peptides with the same net charge but a different total number of charged residues (e.g., a net charge of −3 can result from a negative/positive aa composition of 6/3, as in wild-type Aß, or compositions of 7/4, 5/2, etc.). Considering all mutations between charged and polar residues or glycine reveals that, although reducing the net charge of the peptide from −3 progressively increases nucleation (Figure 3G), the total number of charged residues is also important: for a given net charge, nucleation is increased in peptides containing fewer charged residues of any sign (Figure 3G and Figure 3—figure supplement 1F and G). Thus, both the overall charge and the number of charged residues control the rate of Aß nucleation.
Hydrophobic gatekeeper residues
In addition to the five negatively charged gatekeeper residues, mutations most frequently increase nucleation of Aß in two specific hydrophobic residues: L17 and A42 (Figure 2C and D). At position 17, changes to polar, aromatic, and aliphatic aa all increase nucleation, as does the introduction of a positive charge and mutation to proline. Only a mutation to cysteine reduces nucleation (Figure 2C). This suggests a specific role for leucine at position 17 in limiting nucleation, perhaps as part of a nucleation-limiting secondary structure suggested by the mutational effects of proline, isoleucine, valine, and threonine in this region (Figure 3E).
Finally, the distribution of mutational effects at position 42 differs from that in the rest of the hydrophobic C-terminus of Aß, with mutations most often increasing nucleation (Figure 2D; FDR = 0.1). The mutations that increase nucleation are all to other aliphatic residues (Figures 2C and 3A). The distinction of position 42 is interesting because of the increased toxicity and aggregation propensity of Aß42 compared to the shorter Aß40 APP cleavage product (Meisl et al., 2014; Sandberg et al., 2010).
Nucleation scores accurately discriminate fAD mutations
To investigate how nucleation in the cell-based assay relates to the human disease, we considered all the mutations in Aß known to cause fAD. In total, there are 11 mutations in Aß reported to cause dominantly inherited fAD and one additional variant of unclear pathogenicity (H6R) (Janssen et al., 2003). These 12 known disease mutations are not well discriminated by commonly used computational variant effect predictors (Figure 4 and Figure 4—figure supplement 1A) or by computational predictors of protein aggregation and solubility (Figure 4 and Figure 4—figure supplement 1B). They are also poorly predicted by the previous deep mutational scan of Aß designed to quantify changes in protein solubility, suggesting the disease is unrelated to the biophysical process quantified in this assay (Gray et al., 2019; Figure 4—figure supplement 1C).
Figure 4. Amyloid beta (Aß) nucleation accurately discriminates dominant familial Alzheimer’s disease (fAD) variants.
Receiver operating characteristic (ROC) curves for 12 fAD mutants versus all other single aa mutants in the dataset. Area under the curve (AUC) values are indicated in the legend. Diagonal dashed line indicates the performance of a random classifier. The nucleation scores and categories for all fAD variants are reported in Supplementary file 1.
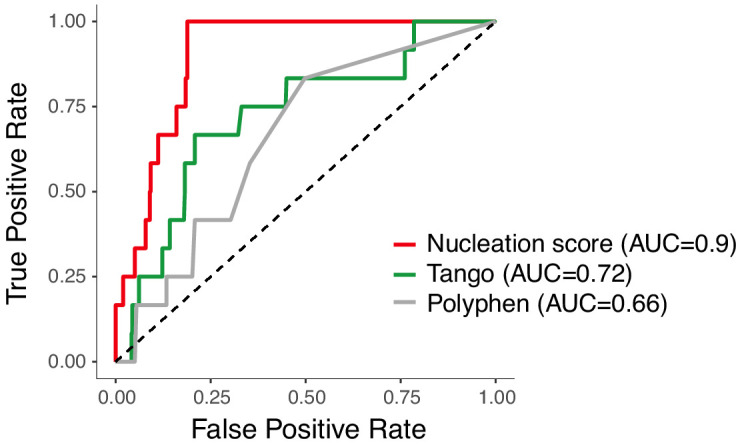
Figure 4—figure supplement 1. Discrimination of familial Alzheimer’s disease (fAD) variants by aggregation and variant effect predictors.

In contrast, the scores from our in vivo nucleation assay accurately classify the known dominant fAD mutations, with all 12 mutations increasing nucleation (Figure 4, area under the receiver operating characteristic curve, ROC−AUC = 0.9, two-tailed Z-test, p<2.2e-16). This suggests the biophysical events occurring in this simple cell-based assay are highly relevant to the development of the human disease.
Consistent with the overall mutational landscape, the known fAD mutations are also enriched in the N-terminus of Aß (Figure 2C). In some positions the known fAD mutations are the only mutation or one of only a few mutations that can increase nucleation. For example, based on our data, K16N is likely to be one of only two fAD mutations in position 16. However, in other positions, there are several additional variants that increase nucleation as much as the known fAD mutation. At position 11, for example, there are five mutations with a NS higher than the known E11K disease mutation (Figure 2C and D). Overall, our data suggest there are likely to be many additional dominant fAD mutations beyond the 12 that have been reported to date (Supplementary file 2).
In addition to the 12 known dominant fAD mutations, two additional variants in Aß have been suggested to act recessively to cause fAD (Di Fede et al., 2009; Tomiyama et al., 2008). One of these variants is a codon deletion (E22Δ) and is not present in our library. The other variant, A2V, does not have a dominant effect on nucleation in our assay (Figure 2C), consistent with a recessive pattern of inheritance and a different mechanism of action, such as reduced ß-cleavage and increased Aß42 generation, as previously proposed (Benilova et al., 2014). More generally, of the hundreds of aa changes possible in the peptide, our data prioritize 63 as candidate fAD variants (Supplementary file 2); 131 variants are likely to be benign, and 262 reduce Aß nucleation and so may even be protective. These include variants already reported in the gnomAD database of human genetic variation (Figure 4—figure supplement 1D). With the currently available data for patients carrying fAD mutations, we could not observe a correlation between NS and disease age-of-onset (Ryman et al., 2014; Figure 4—figure supplement 1E).
Discussion
Taken together, the data presented here provides the first large-scale analysis of how mutations promote and prevent the aggregation of an amyloid. The results reveal a modular organization for the impact of mutations on the nucleation of Aß. Moreover, they show that the rate of nucleation in a cell-based assay identifies all of the mutations in Aß that cause dominant fAD. The dataset therefore provides a useful resource for the future clinical interpretation of genetic variation in Aß.
A majority of mutations in the C-terminal core of Aß disrupt nucleation, consistent with specific hydrophobic contacts in this region being required for nucleation. In contrast, mutations that increase nucleation are enriched in the polar N-terminus with mutations in negatively charged gatekeeper residues and the L17 gatekeeper being particularly likely to accelerate aggregation. Indeed, decreasing both the net charge of the peptide and the total number of charged residues increases nucleation.
Little is known about the structure of Aß during fibril nucleation, but the results presented here are in general consistent with the nucleation transition state resembling the known mature fibril structures of Aß where the C-terminal region of the peptide is located in the amyloid core and the N-terminus is disordered and solvent exposed (Figure 5 and Figure 5—figure supplements 1 and 2). Although the N-terminus is not required for nucleation, it does affect the process when present and most mutations that accelerate nucleation are located in the N-terminus. Interestingly, the effects of mutations in residues immediately before position 17 suggest that the formation of a structural element in this region may interfere with nucleation.
Figure 5. Mutational landscape of the amyloid beta (Aß) amyloid fibril.
Average effect of mutations visualized on the cross-section of an Aß amyloid fibril (PDB accession 5KK3; Colvin et al., 2016). Nucleation gatekeeper residues and known familial Alzheimer’s disease (fAD) mutations positions are indicated by the wild-type (WT) aa identity on one of the two monomers; gatekeepers are indicated with blue dots and fAD are underlined. A single layer of the fibril is shown and the unstructured N-termini (aa 1-14) are shown with different random coil conformations for the two Aß monomers. See Figure 5—figure supplement 2 for alternative Aß42 amyloid polymorphs.

Figure 5—figure supplement 1. Modular organization of Aß42 and Aß40 polymorphs.

Figure 5—figure supplement 2. Modular organization of mutational effects and gatekeepers visualized on Aß42 polymorphs.

That accelerated nucleation is a common cause of fAD is also supported by the effects of mutations in APP outside of Aß and by the effects of mutations in PSEN1 and PSEN2. These mutations destabilise enzyme-substrate complexes, increasing the production of the longer Aß peptides that more effectively nucleates amyloid formation (Szaruga et al., 2017; Veugelen et al., 2016). In addition, Aß42 oligomers are hypothesised to be more toxic (Michaels et al., 2020; Bolognesi et al., 2010). It is possible that the effects of some of the mutations reported here on nucleation are also mediated by a change in the concentration of Aß rather than by an increase in a kinetic rate parameter. Some of the variants evaluated here may have additional effects, for example, altering cleavage of APP. Future work will be needed to test these hypotheses.
Comparing our results to the effects of mutations on Aß solubility quantified in a previous high-throughput analysis (Gray et al., 2019) provides evidence that, in the same type of cell (yeast), Aß can aggregate in at least two different ways. Moreover, the different performance of the two sets of scores from these datasets in classifying fAD mutations suggests that one of these aggregation processes (quantified by the nucleation assay employed here) is likely to be very similar to the aggregation that occurs in the human brain in fAD. The other pathway of aggregation (quantified by the solubility assay; Gray et al., 2019), however, is less obviously related to the human disease, because mutations that cause fAD do not consistently affect it. This second aggregation pathway is, at least to a large extent, driven by changes in hydrophobicity, similar to what we previously reported for the aggregation in yeast of the ALS protein, TDP-43 (Bolognesi et al., 2019).
More generally, our results highlight how the combination of deep mutational scanning and human genetics can be a general ‘genetic’ strategy to quantify the disease relevance of biological assays. Many in vitro and in vivo assays are proposed as ‘disease models’ in biomedical research with their relevance often justified by how ‘physiological’ the assays seem or how well phenotypes observed in the model match those observed in the human disease. The range of phenotypes that can be assessed and their similarity to the pathology of AD human brains are appealing features of many animal models of AD and many important insights have been derived – and will continue to be derived – from animal models (Sasaguri et al., 2017). However, there are applications where animal models cannot be realistically used, for example, for high-throughput compound screening for drug discovery and for testing hundreds or thousands of genetic variants of unknown significance. For these applications, in vitro or cell-based (Pimenova and Goate, 2020; Veugelen et al., 2016) assays are required and an important challenge is to evaluate the ‘disease relevance’ of different assays. Our study highlights an approach to achieve this, which is to use the complete set of known disease-causing mutations to quantify the ‘genetic agreement’ between an assay and a disease. Thus, although the yeast-based assay that we employed here might typically be dismissed as ‘non-physiological,’ ‘artificial,’ or ‘lacking many features important for a neurological disease,' unbiased massively parallel genetic analysis provides very strong evidence that it is reporting on biopysicall events that are extremely similar to – or the same as – those that cause the human disease. Indeed, one could argue that this simple system is now better validated as a model of fAD than many others, including animal models where the effects of only one or a few mutations (including control mutations) have ever been tested. Similarly strong agreement between mutational effects in a cellular assay and the set of mutations already known to cause a disease is observed for other diseases (Starita et al., 2017; Gelman et al., 2019), suggesting the generality of this approach.
We suggest therefore that the combination of deep mutational scanning and human genetics provides a general strategy to quantify the disease relevance of in vitro and cell-based assays. We encourage that deep mutagenesis should be employed early in discovery programmes to ‘genetically validate’ (or invalidate) the relevance of assays for particular diseases. The concordance between mutational effects in an assay and a disease is an unbiased metric that can be used to prioritize between different assays. Quantifying the ‘genetic agreement’ between an assay and a disease will help prevent time and resources being wasted on research that actually has little relevance to a disease.
Finally, the strikingly consistent effects of the dominant fAD mutations in our assay further strengthen the evidence that fAD is a ‘nucleation disease’ ultimately caused by an increased rate of amyloid nucleation (Aprile et al., 2017; Cohen et al., 2018; Knowles et al., 2009). This accelerated nucleation can be caused by the direct effects of mutations in Aß — such as those quantified here — or by changes in upstream factors (Szaruga et al., 2017). If this hypothesis is correct, then nucleation is the key bioph step to target to prevent or treat AD. We suggest that the ‘genetic validation’ of assays by mutational scanning and comparison to sets of known disease-causing mutations will be increasingly important in assay development and drug discovery pipelines.
Materials and methods
Plasmid library construction
The plasmid PCUP1-Sup35N-Aβ42 used in this study was a kind gift from the Chernoff lab (Chandramowlishwaran et al., 2018).
The Aβ coding sequence and two flanking regions of 52 bp and 72 bp, respectively, upstream and downstream of Aβ were amplified (primers MS_01 and MS_02, Supplementary file 3) by error-prone PCR (Mutazyme II DNA polymerase, Agilent). Thirty cycles of amplification and 0.01 ng of initial template were used to obtain a mutagenesis rate of 16 mutations/kb, according to the manufacturer’s protocol. The product was treated with DpnI (FastDigest, Thermo Scientific) for 2 hr and purified by column purification (MinElute PCR Purification Kit, Qiagen). The fragment was digested with EcoRI and XbaI restriction enzymes (FastDigest, Thermo Scientific) for 1 hr at 37°C and purified from a 2% agarose gel (QIAquick Gel Extraction Kit, Qiagen). In parallel, the PCUP1-Sup35N-Aβ42 plasmid was digested with the same restriction enzymes to remove the WT Aβ sequence, treated with alkaline phosphatase (FastAP, Thermo Scientific) for 1 hr at 37°C to dephosphorylate the 5’ ends, and purified from a 1% agarose gel (QIAquick Gel Extraction Kit, Qiagen).
Mutagenised Aβ was then ligated into the linearised plasmid in a 5:1 ratio (insert:vector) using a ligase treatment (T4, Thermo Scientific) overnight. The reaction was dialysed with a membrane filter (Merck Millipore) for 1 hr, concentrated 4x, and transformed in electrocompetent Escherichia coli cells (10-beta Electrocompetent, NEB). Cells were recovered in SOC medium and plated on LB with ampicillin. A total of 4.1 million transformants were estimated, ensuring that each variant of the library was represented more than 10 times; 50 ml of overnight E. coli culture was harvested to purify the Aβ plasmid library with a midi prep (Plasmid Midi Kit, Qiagen). The resulting library contained 29.9% of WT Aβ, 23.8% of sequences with 1 nt change, and 21.8% of sequences with 2 nt changes.
Large-scale yeast transformation
Saccharomyces cerevisiae [psi-pin-] (MATa ade1-14 his3 leu2-3,112 lys2 trp1 ura3-52) strain (also provided by the Chernoff lab) was used in all experiments in this study (Chandramowlishwaran et al., 2018).
Yeast cells were transformed with the Aβ plasmid library starting from an individual colony for each transformation tube. After an overnight pre-growth culture in YPDA medium at 30°C, cells were diluted to OD600 = 0.3 in 175 ml YPDA and incubated at 30°C 200 rpm for ~5 hr. When cells reached the exponential phase, they were harvested, washed with milliQ, and resuspended in sorbitol mixture (100 mM LiOAc, 10 mM Tris pH 8, 1 mM EDTA, 1M sorbitol). After a 30 min incubation at room temperature (RT), 5 µg of plasmid library and 175 μl of ssDNA (UltraPure, Thermo Scientific) were added to the cells. PEG mixture (100 mM LiOAc, 10 mM Tris pH 8, 1 mM EDTA pH 8, 40% PEG3350) was also added and cells were incubated for 30 min at RT and heat-shocked for 15 min at 42°C in a water bath. Cells were harvested, washed, resuspended in 350 ml recovery medium (YPD, sorbitol 0.5M, 70 mg/L adenine) and incubated for 1.5 hr at 30°C 200 rpm. After recovery, cells were resuspended in 350 ml -URA plasmid selection medium and allowed to grow for 50 hr. Transformation efficiency was calculated for each tube of transformation by plating an aliquote of cells in -URA plates. Between 1 and 2.5 million transformants per tube were obtained. Two days after transformation, the culture was diluted to OD600 = 0.02 in 1 l -URA medium and grown until the exponential phase. At this stage, cells were harvested and stored at −80°C in 25% glycerol.
Selection experiments
Three independent replicate selection experiments were performed. Tubes were thawed from the −80°C glycerol stocks and mixed proportionally to the number of transformants in a 1 l total -URA medium at OD600 = 0.05. A minimum of 3.7 million yeast transformants were used for each replicate to ensure the coverage of the full library and reaching therefore a 10x coverage of each variant.
Once the culture reached the exponential phase, cells were resuspended in 1 l protein inducing medium (-URA, 20% glucose, 100 µM Cu2SO4) at OD600 = 0.05. As a result, each variant was represented at least 100 times at this stage. After 24 hr the input pellets were collected by centrifuging 220 ml of cells and stored at −20°C for later DNA extraction (input pellets). In parallel, 18.5 million cells of the same culture underwent selection, with a starting coverage of at least 50 copies of each variant in the library. For selection, cells were plated on -ADE-URA selective medium in 145 cm2 plates (Nunc, Thermo Scientific) and let grow for 7 days at 30°C. Colonies were then scraped off the plates and recovered with PBS 1x to be centrifuged and stored at −20°C for later DNA extraction (output pellets).
For individual testing of specific variants, cells were plated on -URA (control) and -ADE-URA (selection) plates in three independent replicates. Individual growth was calculated as the percentage of colonies growing -ADE-URA relative to colonies growing in -URA.
DNA extraction
The input and output pellets (three replicates, six tubes in total) were thawed and resuspended in 2 ml extraction buffer (2% Triton-X, 1% SDS, 100 mM NaCl, 10 mM Tris pH 8, 1 mM EDTA pH 8), and underwent two cycles of freezing and thawing in an ethanol-dry ice bath (10 min) and at 62°C (10 min). Samples were then vortexed together with 1.5 ml of phenol:chloroform:isoamyl 25:24:1 and 1.5 g of glass beads (Sigma). The aqueous phase was recovered by centrifugation and mixed again with 1.5 ml phenol:chloroform:isoamyl 25:24:1. DNA precipitation was performed by adding 1:10 V of 3M NaOAc and 2.2 V of 100% cold ethanol to the aqueous phase and incubating the samples at −20°C for 1 hr. After a centrifugation step, pellets were dried overnight at RT.
Pellets were resuspended in 1 ml resuspension buffer (10 mM Tris pH 8, 1 mM EDTA pH 8) and treated with 7.5 μl RNase A (Thermo Scientific) for 30 min at 37°C. The DNA was finally purified using 75 μl of silica beads (QIAEX II Gel Extraction Kit, Qiagen), washed and eluted in 375 μl elution buffer.
DNA concentration in each sample was measured by quantitative PCR, using primers (MS_03 and MS_04, Supplementary file 3) that anneal to the origin of replication site of the plasmid at 58°C.
Sequencing library preparation
The library was prepared for high-throughput sequencing in two rounds of PCR (Q5 High-Fidelity DNA Polymerase, NEB). In PCR1, the Aβ region was amplified for 15 cycles at 68°C with frame-shifted primers (MS_05 to MS_18, Supplementary file 3) with homology to Illumina sequencing primers; 300 million of molecules were used for each input or output sample. The products of PCR1 were purified with an ExoSAP-IT treatment (Affymetrix) and a column purification step (QIAquick PCR Purification Kit) and then used as the template of PCR2. This PCR was run for 10 cycles at 62°C with Illumina indexed primers (MS_19 to MS_25, Supplementary file 2) specific for each sample (three inputs and three outputs). The six samples were then pooled together equimolarly. The final library sample was purified from a 2% agarose gel with silica beads (QIAEX II Gel Extraction Kit, Qiagen); 125 bp paired-end sequencing was run on an Illumina HiSeq2500 sequencer at the CRG Genomics Core Facility.
Data processing
FastQ files from paired-end sequencing of the Aß library before (‘input’) and after selection (‘output’) were processed using a custom pipeline (https://github.com/lehner-lab/DiMSum). DiMSum (Faure et al., 2020) is an R package that uses different sequencing processing tools such as FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (for quality assessment), Cutadapt (Martin, 2011) (for constant region trimming), and USEARCH (Edgar, 2010) (for paired-end read alignment). Sequences were trimmed at 5′ and 3′, allowing an error rate of 0.2 (i.e., read pairs were discarded if the constant regions contained more than 20% mismatches relative to the reference sequence). Sequences differing in length from the expected 126 bp or with a Phred base quality score below 30 were discarded. As a result of this processing, around 150 million total reads passed the filtering criteria.
At this stage, unique variants were aggregated and counted using Starcode (https://github.com/gui11aume/starcode). Variants containing indels and nonsynonymous variants with synonymous substitutions in other codons were excluded. The result is a table of variant counts which can be used for further analysis.
For downstream analysis, variants with less than 50 input reads in any of the replicates were excluded and only variants with a maximum of two aa mutations were used.
Nucleation scores and error estimates
On the basis of variant counts, the DiMSum pipeline (Faure et al., 2020; https://github.com/lehner-lab/DiMSum) was used to calculate nucleation scores (NS) and their error estimates. For each variant in each replicate NS was calculated as:
where for a specific variant and for Aß WT.
DiMSum models measurement error of NS by assuming that variants with similar counts in input and output samples have similar errors. Based on errors expected from Poisson-distributed count data, replicate-specific additive and multiplicative (one each for input and output samples) modifier terms are fit to best describe the observed variance of NS across all variants simultaneously.
After error calculation, NS were merged by using the error-weighted mean of each variant across replicates and centered using the error-weighted means frequency of synonymous substitutions arising from single nt changes. Merged NS and NS for each independent replicate, as well as their associated error estimates, are available in Supplementary file 4.
Nonsense (stop) mutants were excluded for the analysis except when indicated (Figure 2A and C and Figure 2—figure supplement 1A).
K-medoids clustering
We used K-medoids, or the partitioning around medoids algorithm, to cluster the matrix of single aa variant NS estimates by residue position with the number of clusters estimated by optimum average silhouette width, for values of K in [1,10]. The silhouette width is a measure of how similar each object (in this case residue position) is to its own cluster. In order to take into account uncertainty in NS estimates in the determination of the optimum number of clusters, we repeated this analysis after random resampling from the NS (error) distributions of each single aa variant (n = 100). Based on this clustering, we defined the N-terminus as aa 2-26 and the C-terminus as aa 27-41 (Figure 2—figure supplement 1B). Seven positions where as many (or more) single mutations increase as decrease nucleation were defined as ‘gatekeepers’ (D1, E3, D7, E11, L17, E22, A42) and excluded from the N- and C-terminus classes. Only those positions where most mutations are significantly different from WT (FDR = 0.1) were considered for the definition of gatekeepers.
Aa properties, aggregation, and variant effect predictors
Nucleation scores were correlated with aa properties and scores from aggregation, solubility, and variant effect prediction algorithms. Pearson correlations were weighted based on the error terms associated with the NS of each variant using the R package ‘weights.' The aa property features were retrieved from a curated collection of numerical indices representing various aa physicochemical and biochemical properties (http://www.genome.jp/aaindex/). We also used a principal component of these aa properties from a previous work (PC1; Bolognesi et al., 2019) that relates strongly to changes in hydrophobicity. For each variant (single and double aa mutants), the values of a specific aa property represent the difference between the mutant and the WT scores.
For the aggregation and solubility algorithms (Tango [Fernandez-Escamilla et al., 2004], Zyggregator [Tartaglia and Vendruscolo, 2008], CamSol [Sormanni et al., 2015], and Waltz [Oliveberg, 2010]), individual residue-level scores were summed to obtain a score per aa sequence. We then calculated the log value for each variant relative to the WT score (single and double aa mutants for Tango, Zyggregator, CamSol and single aa mutants for Waltz). For the variant effect predictors (Polyphen [Adzhubei et al., 2013] and CADD [Rentzsch et al., 2019]), we also calculated the log value for each variant (only single aa mutants) but in this case values were scaled relative to the lowest predicted score.
fAD, gnomAD, and Clinvar variants
The table of fAD mutations used in this study was taken from https://www.alzforum.org/mutations/app. Allele frequencies of APP variants were retrieved from gnomAD (Karczewski, 2020) (https://gnomad.broadinstitute.org/) and the clinical significance of variants was taken from their Clinvar (Landrum et al., 2014) classification (https://www.ncbi.nlm.nih.gov/clinvar).
ROC curves were built and AUC values were obtained using the ‘pROC’ R package.
PDB structures
The coordinates of the following PDB structures were used for Figure 5, Figure 5—figure supplements 1 and 2: 5OQV, 2NAO, 5KK3, 2BEG, 2MXU, 5AEF, 6SHS, 2LMN, 2LMP, 2LNQ, 2MVX, 2M4J, 2MPZ (Gremer et al., 2017; Colvin et al., 2016; Wälti et al., 2016; Lührs et al., 2005; Xiao et al., 2015; Schmidt et al., 2015; Kollmer et al., 2019; Lu et al., 2013; Qiang et al., 2012; Sgourakis et al., 2015; Schütz et al., 2015).
Acknowledgements
Work in the lab of BB is supported by the Spanish Ministry of Science, Innovation and Universities through the project RTI2018-101491-A-I00 (MICIU/FEDER), by the CERCA Program/Generalitat de Catalunya and by funding from the Agencia de Gestio d’Ajuts Universitaris i de Recerca (2019FI_B 01311) to MS Work in the lab of BL is supported by a European Research Council (ERC) Consolidator grant (616434), the Spanish Ministry of Science, Innovation and Universities (BFU2017-89488-P and SEV-2012–0208), the Bettencourt Schueller Foundation, Agencia de Gestio d’Ajuts Universitaris i de Recerca (AGAUR, 2017 SGR 1322.), and the CERCA Program/Generalitat de Catalunya. We acknowledge the support of the Spanish Ministry of Science and Innovation to the EMBL partnership and the Centro de Excelencia Severo Ochoa. We thank the Chernoff lab for kindly providing strains and plasmids and the CRG Genomics core facility for their assistance with sequencing.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Ben Lehner, Email: ben.lehner@crg.eu.
Benedetta Bolognesi, Email: bbolognesi@ibecbarcelona.eu.
Patrik Verstreken, KU Leuven, Belgium.
Huda Y Zoghbi, Texas Children's Hospital, United States.
Funding Information
This paper was supported by the following grants:
Ministerio de Ciencia e Innovación RTI2018-101491-A-I00 to Benedetta Bolognesi.
Ministerio de Ciencia e Innovación BFU2017-89488-P to Ben Lehner.
H2020 European Research Council 616434 to Ben Lehner.
Agència de Gestió d’Ajuts Universitaris i de Recerca SGR 1322 to Ben Lehner.
Agència de Gestió d’Ajuts Universitaris i de Recerca 2019FI_B 01311 to Mireia Seuma.
Fondation Bettencourt Schueller Prize to Ben Lehner.
Additional information
Competing interests
No competing interests declared.
Author contributions
Conceptualization, Formal analysis, Validation, Investigation, Visualization, Methodology, Writing - original draft, Writing - review and editing.
Software, Investigation, Visualization, Methodology.
Investigation.
Conceptualization, Supervision, Funding acquisition, Writing - original draft, Writing - review and editing.
Conceptualization, Supervision, Methodology, Funding acquisition, Writing - original draft, Writing - review and editing.
Additional files
For the same variants, the table also reports nucleation scores, as quantified in this study, and the qualitative agreement or disagreement with the previously published data.
For each mutation, the corresponding nucleation score (NS) is reported.
Read counts, nucleation scores, and associated error terms are reported for each Aß42 variant in each replicate. See sheet one for a deeper explanation of headers.
Data availability
Raw sequencing data and the processed data table (Supplementary file 4) have been deposited in NCBI's Gene Expression Omnibus (GEO) as record GSE151147. All code used for data analysis is available at https://github.com/BEBlab/abeta (copy archived at https://archive.softwareheritage.org/swh:1:rev:86e1e1be4ee6eb97c1c00b0bd53f98f4e4ea807f/).
The following dataset was generated:
Seuma M, Faure A, Badia M, Lehner B, Bolognesi B. 2020. The genetic landscape for amyloid beta fibril nucleation accurately discriminates familial Alzheimer's disease mutations. NCBI Gene Expression Omnibus. GSE151147
References
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen‐2. Current Protocols in Human Genetics. 2013;76:76. doi: 10.1002/0471142905.hg0720s76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aprile FA, Sormanni P, Perni M, Arosio P, Linse S, Knowles TPJ, Dobson CM, Vendruscolo M. Selective targeting of primary and secondary nucleation pathways in Aβ42 aggregation using a rational antibody scanning method. Science Advances. 2017;3:e1700488. doi: 10.1126/sciadv.1700488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. The Lancet. 2011;377:1019–1031. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- Benilova I, Gallardo R, Ungureanu A-A, Castillo Cano V, Snellinx A, Ramakers M, Bartic C, Rousseau F, Schymkowitz J, De Strooper B. The Alzheimer disease protective mutation A2T modulates kinetic and thermodynamic properties of Amyloid-β (Aβ) Aggregation. Journal of Biological Chemistry. 2014;289:30977–30989. doi: 10.1074/jbc.M114.599027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B, Kumita JR, Barros TP, Esbjorner EK, Luheshi LM, Crowther DC, Wilson MR, Dobson CM, Favrin G, Yerbury JJ. ANS binding reveals common features of cytotoxic amyloid species. ACS Chemical Biology. 2010;5:735–740. doi: 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- Bolognesi B, Faure AJ, Seuma M, Schmiedel JM, Tartaglia GG, Lehner B. The mutational landscape of a prion-like domain. Nature Communications. 2019;10:4162. doi: 10.1038/s41467-019-12101-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. The American Journal of Human Genetics. 1999;65:664–670. doi: 10.1086/302553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramowlishwaran P, Sun M, Casey KL, Romanyuk AV, Grizel AV, Sopova JV, Rubel AA, Nussbaum-Krammer C, Vorberg IM, Chernoff YO. Mammalian amyloidogenic proteins promote prion nucleation in yeast. Journal of Biological Chemistry. 2018;293:3436–3450. doi: 10.1074/jbc.M117.809004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nature Neuroscience. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Cohen SIA, Cukalevski R, Michaels TCT, Šarić A, Törnquist M, Vendruscolo M, Dobson CM, Buell AK, Knowles TPJ, Linse S. Distinct thermodynamic signatures of oligomer generation in the aggregation of the amyloid-β peptide. Nature Chemistry. 2018;10:523–531. doi: 10.1038/s41557-018-0023-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, Griffin RG. Atomic resolution structure of monomorphic aβ42 amyloid fibrils. Journal of the American Chemical Society. 2016;138:9663–9674. doi: 10.1021/jacs.6b05129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, Zilversmit M, Cartwright R, Rouleau GA, Daly M, Stone EA, Hurles ME, Awadalla P, 1000 Genomes Project Variation in Genome-Wide mutation rates within and between human families. Nature Genetics. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, Mazzoleni G, Merlin M, Giovagnoli AR, Prioni S, Erbetta A, Falcone C, Gobbi M, Colombo L, Bastone A, Beeg M, Manzoni C, Francescucci B, Spagnoli A, Cantù L, Del Favero E, Levy E, Salmona M, Tagliavini F. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science. 2009;323:1473–1477. doi: 10.1126/science.1168979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Faure AJ, Schmiedel JM, Baeza-Centurion P, Lehner B. DiMSum: an error model and pipeline for analyzing deep mutational scanning data and diagnosing common experimental pathologies. Genome Biology. 2020;21:207. doi: 10.1186/s13059-020-02091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nature Biotechnology. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Gelman H, Dines JN, Berg J, Berger AH, Brnich S, Hisama FM, James RG, Rubin AF, Shendure J, Shirts B, Fowler DM, Starita LM, Brotman Baty Institute Mutational Scanning Working Group Recommendations for the collection and use of multiplexed functional data for clinical variant interpretation. Genome Medicine. 2019;11:85. doi: 10.1186/s13073-019-0698-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray VE, Sitko K, Kameni FZN, Williamson M, Stephany JJ, Hasle N, Fowler DM. Elucidating the molecular determinants of aβ aggregation with deep mutational scanning. G3: Genes, Genomes, Genetics. 2019;9:3683–3689. doi: 10.1534/g3.119.400535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RBG, Tusche M. Fibril structure of Amyloid-ß(1-42) by cryoelectron microscopy. Science. 2017;9:eaao2825. doi: 10.1126/science.aao2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen JC, Beck JA, Campbell TA, Dickinson A, Fox NC, Harvey RJ, Houlden H, Rossor MN, Collinge J. Early onset familial Alzheimer's disease: Mutation frequency in 31 families. Neurology. 2003;60:235–239. doi: 10.1212/01.WNL.0000042088.22694.E3. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ. The mutational constraint spectrum quantified from variation in 141,456 humans. bioRxiv. 2020 doi: 10.1101/531210. [DOI] [PMC free article] [PubMed]
- Knowles TP, Waudby CA, Devlin GL, Cohen SI, Aguzzi A, Vendruscolo M, Terentjev EM, Welland ME, Dobson CM. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nature Reviews Molecular Cell Biology. 2014;15:384–396. doi: 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, Schmidt M, Sigurdson CJ, Jucker M, Fändrich M. Cryo-EM structure and polymorphism of aβ amyloid fibrils purified from Alzheimer's brain tissue. Nature Communications. 2019;10:4760. doi: 10.1038/s41467-019-12683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Research. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löhr T, Kohlhoff K, Heller GT, Camilloni C, Vendruscolo M. A kinetic ensemble of the Alzheimer’s Aβ peptide. Nature Computational Science. 2021;1:71–78. doi: 10.1038/s43588-020-00003-w. [DOI] [PubMed] [Google Scholar]
- Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β-Amyloid fibrils in Alzheimer’s Disease Brain Tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Döbeli H, Schubert D, Riek R. 3d structure of Alzheimer's amyloid-beta(1-42) fibrils. PNAS. 2005;102:17342–17347. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- Meier BH, Riek R, Böckmann A. Emerging structural understanding of amyloid fibrils by Solid-State NMR. Trends in Biochemical Sciences. 2017;42:777–787. doi: 10.1016/j.tibs.2017.08.001. [DOI] [PubMed] [Google Scholar]
- Meisl G, Yang X, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SI, Dobson CM, Linse S, Knowles TP. Differences in nucleation behavior underlie the contrasting aggregation kinetics of the aβ40 and aβ42 peptides. PNAS. 2014;111:9384–9389. doi: 10.1073/pnas.1401564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels TCT, Šarić A, Curk S, Bernfur K, Arosio P, Meisl G, Dear AJ, Cohen SIA, Dobson CM, Vendruscolo M, Linse S, Knowles TPJ. Dynamics of oligomer populations formed during the aggregation of Alzheimer's Aβ42 peptide. Nature Chemistry. 2020;12:445–451. doi: 10.1038/s41557-020-0452-1. [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer's disease. Annual Review of Neuroscience. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveberg M. Waltz, an exciting new move in amyloid prediction. Nature Methods. 2010;7:187–188. doi: 10.1038/nmeth0310-187. [DOI] [PubMed] [Google Scholar]
- Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer's beta-amyloid fibrils. PNAS. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen JS, Christensen G, Otzen DE. Modulation of S6 fibrillation by unfolding rates and gatekeeper residues. Journal of Molecular Biology. 2004;341:575–588. doi: 10.1016/j.jmb.2004.06.020. [DOI] [PubMed] [Google Scholar]
- Pimenova AA, Goate AM. Novel presenilin 1 and 2 double knock-out cell line for in vitro validation of PSEN1 and PSEN2 mutations. Neurobiology of Disease. 2020;138:104785. doi: 10.1016/j.nbd.2020.104785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang W, Yau WM, Luo Y, Mattson MP, Tycko R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. PNAS. 2012;109:4443–4448. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research. 2019;47:D886–D894. doi: 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Serrano L, Schymkowitz JW. How evolutionary pressure against protein aggregation shaped chaperone specificity. Journal of Molecular Biology. 2006;355:1037–1047. doi: 10.1016/j.jmb.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, Goate A, Frommelt P, Ghetti B, Langbaum JBS, Lopera F, Martins R, Masters CL, Mayeux RP, McDade E, Moreno S, Reiman EM, Ringman JM, Salloway S, Schofield PR, Sperling R, Tariot PN, Xiong C, Morris JC, Bateman RJ, And the Dominantly Inherited Alzheimer Network Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology. 2014;83:253–260. doi: 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg A, Luheshi LM, Söllvander S, Pereira de Barros T, Macao B, Knowles TP, Biverstål H, Lendel C, Ekholm-Petterson F, Dubnovitsky A, Lannfelt L, Dobson CM, Härd T. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. PNAS. 2010;107:15595–15600. doi: 10.1073/pnas.1001740107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H, Nilsson P, Hashimoto S, Nagata K, Saito T, De Strooper B, Hardy J, Vassar R, Winblad B, Saido TC. APP mouse models for Alzheimer's disease preclinical studies. The EMBO Journal. 2017;36:2473–2487. doi: 10.15252/embj.201797397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Rohou A, Lasker K, Yadav JK, Schiene-Fischer C, Fändrich M, Grigorieff N. Peptide dimer structure in an Aβ(1-42) fibril visualized with cryo-EM. PNAS. 2015;112:11858–11863. doi: 10.1073/pnas.1503455112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, Güntert P, Böckmann A, Glockshuber R, Meier BH. Atomic-resolution three-dimensional structure of amyloid β fibrils bearing the Osaka mutation. Angewandte Chemie International Edition. 2015;54:331–335. doi: 10.1002/anie.201408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgourakis NG, Yau WM, Qiang W. Modeling an in-register, parallel "iowa" aβ fibril structure using solid-state NMR data from labeled samples with rosetta. Structure. 2015;23:216–227. doi: 10.1016/j.str.2014.10.022. [DOI] [PubMed] [Google Scholar]
- Sormanni P, Aprile FA, Vendruscolo M. The CamSol method of rational design of protein mutants with enhanced solubility. Journal of Molecular Biology. 2015;427:478–490. doi: 10.1016/j.jmb.2014.09.026. [DOI] [PubMed] [Google Scholar]
- Starita LM, Ahituv N, Dunham MJ, Kitzman JO, Roth FP, Seelig G, Shendure J, Fowler DM. Variant interpretation: functional assays to the rescue. The American Journal of Human Genetics. 2017;101:315–325. doi: 10.1016/j.ajhg.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017;170:443–456. doi: 10.1016/j.cell.2017.07.004. [DOI] [PubMed] [Google Scholar]
- Tartaglia GG, Vendruscolo M. The zyggregator method for predicting protein aggregation propensities. Chemical Society Reviews. 2008;37:1395–1401. doi: 10.1039/b706784b. [DOI] [PubMed] [Google Scholar]
- Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Annals of Neurology. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- Törnquist M, Michaels TCT, Sanagavarapu K, Yang X, Meisl G, Cohen SIA, Knowles TPJ, Linse S. Secondary nucleation in amyloid formation. Chemical Communications. 2018;54:8667–8684. doi: 10.1039/C8CC02204F. [DOI] [PubMed] [Google Scholar]
- Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genetics in Medicine. 2016;18:421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veugelen S, Saito T, Saido TC, Chávez-Gutiérrez L, De Strooper B. Familial Alzheimer's disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron. 2016;90:410–416. doi: 10.1016/j.neuron.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Güntert P, Meier BH, Riek R. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. PNAS. 2016;113:E4976–E4984. doi: 10.1073/pnas.1600749113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weggen S, Beher D. Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer's disease. Alzheimer's Research & Therapy. 2012;4:9. doi: 10.1186/alzrt107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization . WHO Dementia: A Public Health Priority. WHO; 2012. https://www.who.int/mental_health/publications/dementia_report_2012/en/ [Google Scholar]
- Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nature Structural & Molecular Biology. 2015;22:499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Meisl G, Frohm B, Thulin E, Knowles TPJ, Linse S. On the role of sidechain size and charge in the aggregation of Aβ42 with familial mutations. PNAS. 2018;115:E5849–E5858. doi: 10.1073/pnas.1803539115. [DOI] [PMC free article] [PubMed] [Google Scholar]