

Abstract
Objective
We describe a third patient with brain small vessel disease 3 (BSVD3), being the first with a homozygous essential splice site variant in the COLGALT1 gene, with a more severe phenotype than the 2 children reported earlier.
Methods
Analysis of whole exome sequencing (WES) data of the child and parents was performed. We validated the missplicing of the homozygous variant using reverse transcription PCR and Sanger sequencing of the mRNA in a lymphocyte culture.
Results
The patient presented antenatally with porencephaly on ultrasound and MRI. Postnatally, he showed a severe developmental delay, refractory epilepsy, spastic quadriplegia, and a progressive hydrocephalus. WES revealed a homozygous canonical splice site variant NM_024656.3:c.625-2A>C. PCR and Sanger sequencing of the mRNA demonstrated that 2 cryptic splice sites are activated, causing a frameshift in the major transcript and in-frame deletion in a minor transcript.
Conclusions
We report a third patient with biallelic pathogenic variants in COLGALT1, confirming the role of this gene in autosomal recessive BSVD3.
Brain small vessel disease 1 (BSVD1, OMIM#175780) and 2 (BSVD2, OMIM#614483) are autosomal dominant syndromes associated with, respectively, COL4A1 and COL4A2 variants.1,2 These variants can cause a broad spectrum of cerebrovascular, renal, ocular, cardiac, and muscular abnormalities.3–5 BSVD is caused by a defect in the α1α1α2 trimer made from the proteins Col4a1 and Col4a2 that forms type IV collagen. Because of inappropriate 3D-construction, there is an instability of the basal membrane in arteries, creating a high vulnerability to cerebrovascular incidents.6 This leads to the characterizing symptoms of BSVD: porencephalic cysts, periventricular leukoencephalopathy, lacunar infarcts, and cerebral microbleeds and calcifications.1
Variants in the COLGALT1 gene cause brain small vessel disease 3 (BSVD3, OMIM #618360). The COLGALT1 gene encodes the collagen beta galactosyltransferase 1 (ColGalT1) protein. ColGalT1 initiates glycosylation of CoL4a1 (and perhaps Col4a2), an important step in the formation of the triple helix of collagen IV. Miyatake et al.7 described 2 patients with a compound heterozygous variant in the COLGALT1 gene and showed that reduced ColGalT1 activity results in decreased CoL4a1 protein, thereby resulting in reduced secretion of type IV collagen.
Here, we describe a third patient with BSVD3 and the first with a homozygous variant in the COLGALT1 gene.
Methods
Identification of Variants Using Whole Exome Sequencing
Whole exome sequencing (WES) was performed as previously described by Neveling et al.8 Briefly, capture of exons was performed using a SureSelectXT Human All Exon 50 MbKit V5 (Agilent, Santa Clara, CA). Sequencing was performed using a Hiseq 4000 (Illumina, San Diego, CA). Read mapping and variant calling were conducted using BWA (bio-bwa.sourceforge.net/index.shtml) and GATK (software.broadinstitute.org/gatk/; Broad Institute, Cambridge, MA), respectively.
Assessment of Pathogenicity
Lymphocytes were Epstein-Barr virus (EBV) transformed and cultured in the presence or absence of cycloheximide to block translation and, consequently, possible nonsense-mediated decay (NMD). Total RNA was isolated from these cells using QIAamp RNA Blood Mini Kit (QIAGEN, Hilden, Germany) and subjected to reverse-transcriptase PCR using cDNA iScript cDNA Synthesis Kit (Bio-rad Laboratories, Hercules, CA). Subsequently, the copy DNA was subjected to Sanger sequencing using the following primers Forward: 5′-TGGCTACGGACCACAACATG-3′ (in exon 2) and Reverse: 5′-CCGCCTCAGGTTGATCATGA-3′ (exon 8).
Data Availability
Further clinical and genetic data that are deidentified are available on request.
Results
Case Presentation
Antenatally, ventriculomegaly was seen on a regular 20 weeks ultrasound. Prenatal MRI at 21 weeks of gestation showed a cortical mantle defect, periventricular hemorrhage, and ventriculomegaly (figure 1, A and B). The nonconsanguineous parents had no family history of neurologic disease.
Figure 1. Prenatal MRI.
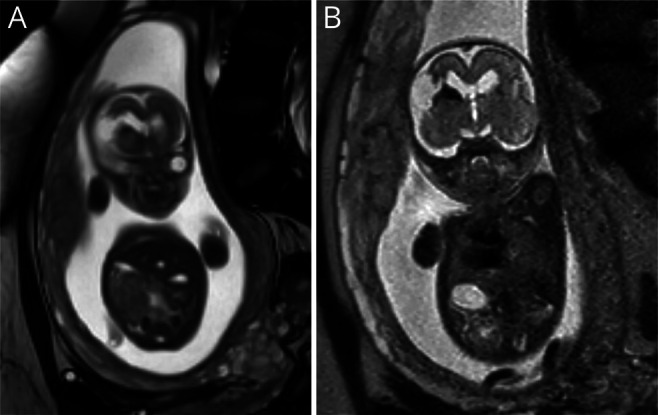
Fetal MRI at 21 weeks of gestational age. (A) T2-weighted coronal MRI showing defect of the frontal cortical mantle. (B) Coronal MRI showing the same defect with hypointensities, indicating hemorrhage.
A boy of 2,642 g was born after an uncomplicated birth at full term. Clinical examination postnatally showed microcephaly (−3.12 SD) and jitteriness. The ophthalmologist described a megalocornea of the right eye, strabismus, and bilateral pale optic nerves.
The postnatal cerebral MRI scan (figure 2, A–F) revealed porencephalic cysts, multiple white matter abnormalities, and elaborate hemorrhages (figure 2D), which had been progressive compared with the antenatal MRI. A COL4A1/COL4A2-related disorder was suspected; however, Sanger sequencing of the COL4A1 and COL4A2 genes was negative.
Figure 2. MRI of the Brain.

MRI of 1 day after birth (A–F) and at 10 months (G and H). Postnatal MRI shows porencephalic cysts, bleeding, and ischemic changes. (A) T2. (B) Fluid-attenuated inversion recovery weighted images. (C) T1. (D) Fast field echo showing extensive hypointensive artifacts indicating hemorrhage. (E and F) Diffusion weighted imaging with hyperintensities (E) and corresponding hypointensities apparent diffusion coefficient (F), suggestive of recent ischemia. MRI at 10 months shows extreme hydrocephalus and loss of white matter (G). After receiving a ventriculoperitoneal drain (H), major loss of brain parenchyma is even more obvious; moreover, pericerebral hygromas are now present (white arrows).
At the age of 6.5 months, infantile spasms (West syndrome) was diagnosed and prednisolone was started. Zonisamide and later nitrazepam was added because of ongoing seizures. Furthermore, he showed a spastic tetraplegy with axial hypotonia.
At the age of 10 months, he presented with vomiting, a bulging fontanel, and an increase in the head circumference. A repeated MRI revealed no signs of new hemorrhages, but progressive dilatation of the lateral ventricles and the porencephalic cyst (figure 1, G and H). After a ventriculoperitoneal shunt was placed, hydrocephaly was controlled, but he continued to suffer from intractable epilepsy, spastic tetraplagia, and severe psychomotor retardation. Some weeks before his fourth birthday, he suddenly developed an irregular breathing pattern, low saturation, and hypotension and died.
Identification of COLGALT1 Variants and Assessment of Pathogenicity
Using WES, a homozygous essential splice site variant c.625-2A>C (NM_024656.3) was detected in intron 4 of the COLGALT1 gene. This variant was not present in the Genome Aggregation Database (gnomad.broadinstitute.org) control population.
To prove that the splice site variant was causing a splicing defect, mRN92A samples were obtained from an EBV-transformed lymphocyte and a fibroblast culture taken from the patient, which were treated with cycloheximide or left untreated. The mRNA samples, as well as those of controls, were subjected to reverse-transcriptase and COLGALT1 specific PCR. In the control samples, a single band of ∼820 bp was seen on agarose gels, whereas all samples of the patient showed 2 distinct bands of ∼800 and ∼850 bp (not shown). The intensities of the ∼850 bp bands increased on cycloheximide treatment, suggesting that the larger product was indeed prone to NMD. Subsequent Sanger sequencing of a cycloheximide treated sample (figure 3) revealed that the larger product corresponded to a retention of 34 bases from intron 4, r.624_625ins[625-34_625-1; 625-2A>C], predicting the frameshift p.(Gly209fs), which results in a premature termination codon after 60 amino acids that is prone to NMD. The smaller transcript revealed a deletion of the first 12 bases of exon 5, corresponding to an in-frame deletion of 4 amino acids, r.625_636del; p(Gly209_Lys212del), not prone to NMD. Both aberrant transcripts exist because of the use of cryptic splice sites that were not visibly used in cycloheximide-treated control cells (figure 3A). The effect of the minor transcript, coding for a ColGalT1 protein with deletion of 4 well-conserved amino acids, has not been tested. This could well be acting as wildtype, or as loss of function. However, because the larger transcript is also the major transcript (the sequence of the higher peaks in figure 3B correspond to the larger transcript) and leads to a frame shift, the c.625-2A>C splice site variant undoubtedly results in markedly decreased CoLGaLT1 protein levels and function.
Figure 3. The c.625-2A>C Variant in COLGALT1 Results in 2 Aberrant Transcripts.

Reverse-transcriptase PCR and subsequent Sanger sequencing of Epstein-Barr virus–transformed cycloheximide-treated lymphocytes of a control (A) and the patient (B) resulted in normal splicing of intron 4 from the control and aberrant splicing for the patient. The 625-2A>C variant (indicated) resulted in the use of 2 cryptic splice sites (splicing positions indicated by arrow heads), 1 in intron 4, and 1 in exon 5. Thus, 2 populations of transcripts were detected in different quantities in the patient (compare peak heights). The major transcript contains 34 bases of intron 4 (retention) starting from position c.625-34 (indicated). The minor transcript lost 12 bases of exon 5, fusing c.624 to c.637 (indicated). Exon sizes are not to scale. Exon 4 (Ex4), intron 4 (indicated), exon 5 (Ex5), and exon 5 missing 12 bases (ΔEx5) are indicated by boxes, and partial sequences of these gene components are shown below the chromatograms and in the schematics.
Discussion
We describe a boy with a severe form of congenital porencephaly, currently the third patient in the world with BSVD3. Pathogenic variants in COLGALT1 are extremely rare. A recent study screened more than 100,000 persons from 7 ethnic groups, showing no variants in 6 of the 7 ethnic groups; in the other group, a heterozygous pathogenic variant with a frequency of 0.12/1,000 was found.9 Our patient has a similar phenotype compared with the 2 children described earlier,7 although he seems more severely affected. The MRI changes with porencephalic cysts, hemorrhages, leukoencephalopathy, and ischemia confirmed the clinical phenotype.10 A history of porencephalic cysts, cerebral small vessel disease, and eye disease is also described in patients with variants in the COL4A1 and COL4A2 genes.1,2
The Col4a1 and Col4a2 peptides undergo post-translational modifications in the endoplasmic reticulum forming the α1α1α2 trimer. The translational modification of the CoL4a1 and CoL4a2 nascent peptides is an extensive process and plays a crucial role in the stabilization, secretion, and function of collagen IV. One of the important steps in this process is glycosylation. This is where CoLGalT1, a galactosyltransferase that initiates glycosylation of collagens, may play its crucial role. Although it was not proven, the decrease in Col4A1 protein levels in the absence of CoLGalT1 (or the presence of ColGalT1 mutants) suggests that ColGalT1 is involved in the glycosylation of CoL4a1 (and perhaps Col4a2).7 Further research into the glycosylation of collagen IV by ColGalT1 and the effects of glycosylation in its stability and trafficking is needed to establish this mechanism.
In conclusion, we report a severe case of congenital porencephaly caused by a homozygous COLGALT1 variant on and explain how this splice site variant lead to aberrant splicing.
Glossary
- BSVD
brain small vessel disease
- ColGalT1
collagen beta galactosyltransferase 1
- EBV
Epstein-Barr virus
- NMD
nonsense-mediated decay
- WES
whole exome sequencing
Appendix. Authors
Contributor Information
Mariel W.A. Teunissen, Email: mwavanduijn@gmail.com.
Erik-Jan Kamsteeg, Email: erik-jan.kamsteeg@radboudumc.nl.
Suzanne C.E.H. Sallevelt, Email: suzanne.sallevelt@mumc.nl.
Maartje Pennings, Email: maartje.pennings@radboudumc.nl.
Noel J.C. Bauer, Email: n.bauer@mumc.nl.
R. Jeroen Vermeulen, Email: jeroen.vermeulen@mumc.nl.
Study Funding
The authors report no targeted funding.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Meuwissen ME, Halley DJ, Smit LS, et al. The expanding phenotype of COL4A1 and COL4A2 mutations: clinical data on 13 newly identified families and a review of the literature. Genet Med 2015;17:843–853. [DOI] [PubMed] [Google Scholar]
- 2.Verbeek E, Meuwissen ME, Verheijen FW, et al. COL4A2 mutation associated with familial porencephaly and small-vessel disease. Eur J Hum Genet 2012;20:844–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Favor J, Gloeckner CJ, Janik D, et al. Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics 2007;175:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sibon I, Coupry I, Menegon P, et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol 2007;62:177–184. [DOI] [PubMed] [Google Scholar]
- 5.Plaisier E, Gribouval O, Alamowitch S, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med 2007;357:2687–2695. [DOI] [PubMed] [Google Scholar]
- 6.Kuo DS, Labelle-Dumais C, Gould DB. COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum Mol Genet 2012;21:R97–R110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyatake S, Schneeberger S, Koyama N, et al. Biallelic COLGALT1 variants are associated with cerebral small vessel disease. Ann Neurol 2018;84:843–853. [DOI] [PubMed] [Google Scholar]
- 8.Neveling K, Feenstra I, Gilissen C, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat 2013;34:1721–1726. [DOI] [PubMed] [Google Scholar]
- 9.Grami N, Chong M, Lali R, et al. Global assessment of Mendelian stroke genetic prevalence in 101 635 individuals from 7 ethnic groups. Stroke 2020;51:1290–1293. [DOI] [PubMed] [Google Scholar]
- 10.Vahedi K, Boukobza M, Massin P, Gould DB, Tournier-Lasserve E, Bousser MG. Clinical and brain MRI follow-up study of a family with COL4A1 mutation. Neurology 2007;69:1564–1568. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Further clinical and genetic data that are deidentified are available on request.