

Abstract
Objective
To determine whether the polygenic risk score (PRS) derived from MEGASTROKE is associated with ischemic stroke (IS) and its subtypes in an independent tertiary health care system and to identify the PRS derived from gene sets of known biological pathways associated with IS.
Methods
Controls (n = 19,806/7,484, age ≥69/79 years) and cases (n = 1,184/951 for discovery/replication) of acute IS with European ancestry and clinical risk factors were identified by leveraging the Geisinger Electronic Health Record and chart review confirmation. All Geisinger MyCode patients with age ≥69/79 years and without any stroke-related diagnostic codes were included as low risk control. Genetic heritability and genetic correlation between Geisinger and MEGASTROKE (EUR) were calculated using the summary statistics of the genome-wide association study by linkage disequilibrium score regression. All PRS for any stroke (AS), any ischemic stroke (AIS), large artery stroke (LAS), cardioembolic stroke (CES), and small vessel stroke (SVS) were constructed by PRSice-2.
Results
A moderate heritability (10%–20%) for Geisinger sample as well as the genetic correlation between MEGASTROKE and the Geisinger cohort was identified. Variation of all 5 PRS significantly explained some of the phenotypic variations of Geisinger IS, and the R2 increased by raising the cutoff for the age of controls. PRSLAS, PRSCES, and PRSSVS derived from low-frequency common variants provided the best fit for modeling (R2 = 0.015 for PRSLAS). Gene sets analyses highlighted the association of PRS with Gene Ontology terms (vascular endothelial growth factor, amyloid precursor protein, and atherosclerosis). The PRSLAS, PRSCES, and PRSSVS explained the most variance of the corresponding subtypes of Geisinger IS suggesting shared etiologies and corroborated Geisinger TOAST subtyping.
Conclusions
We provide the first evidence that PRSs derived from MEGASTROKE have value in identifying shared etiologies and determining stroke subtypes.
Genome-wide association studies (GWAS) on ischemic stroke (IS) and its etiologic subtypes have been conducted for a decade, and some common variants or genes have been identified. These genetic variants/genes are mostly subtype specific, and their biological relevance to the etiology of stroke needs to be investigated.1 Meta/mega-analyses of GWAS, led by the MEGASTROKE consortium, have identified more stroke risk loci, but their effect sizes are quite small.2 In most diseases with a polygenic etiology, genome-wide significant markers explain a small proportion of the heritability of complex traits. However, converging evidence supports that a considerable proportion of phenotypic variation can be explained by the ensemble of individual markers not achieving that level of significance. Polygenic risk scores (PRSs) have been used to establish a common genetic basis for related disorders, irrelevant of single markers with a significant association or not, and to identify high- or low-risk individuals by a risk stratification for the purpose of personalized management.3
Pioneer studies have shown that a genetic risk score (GRS) derived from multiple loci has a limited power to predict IS4,5 or its subtype.6 The PRS from genome-wide loci has shown to be superior to multilocus GRS in the prediction of IS in a Japanese population despite a small training and testing samples.7 Through a risk stratification by PRS derived from MEGASTROKE summary statistics, a recent study has shown that the risk of incident stroke from the UK Biobank (UKB) cohort is 35% higher among those at the top third of PRS, and this association is independent of lifestyle factors.8 Genetic overlaps between stroke risk, early neurologic changes, and some of the cardiovascular risk factors (diabetes and hypertension) have been identified.9 Because IS is a multifactorial complex disease and the overall risk is determined by an interplay between genetic and environmental factors, a metaGRS has been developed through a machine learning (ML) approach to integrate multiple sets of GWAS summary statistics on stroke or its modifiable clinical risk factors such as hypertension, type 2 diabetes (T2D), dyslipidemia, body mass index (BMI), and coronary artery disease (CAD).10 Although the hazard ratio of this metaGRS for IS doubles that of previous GRS in the UKB cohort, for individuals with high metaGRS achieving currently recommended risk factor levels, this metaGRS approach remains insufficient to manage risk.10
PRS derived from stroke subtypes may augment the predictive power for patients with a similar etiology. The conventional classification methods stratify stroke subtypes into 5 major categories.11,12 PRS for atrial fibrillation (AFib) can significantly explain cardioembolic stroke (CES) risk, independent of other clinical risk factors.13
The purpose of this study is to estimate the heritability and genetic correlation between Geisinger and MEGASTROKE data sets and to determine the association of this MEGASTROKE-based PRS with IS and its subtypes in the Geisinger sample, which has similar inclusion criteria and the same subtype classified by MEGASTROKE. PRS derived from gene sets of known biological pathways will be evaluated to determine their association with a known or novel etiology of IS through a 2-step design using discovery and replication data sets. The shared etiology between MEGASTROKE and Geisinger TOAST subtypes through polygenic risk modeling will be explored.
Method
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Geisinger institutional review board. As an independent European cohort, Geisinger IS cases were obtained from the local Get With The Guidelines stroke registry and characterized by manual chart review,14 whereas controls were identified by leveraging the Geisinger electronic health record (EHR). The strategy of data analysis and sample sizes was illustrated (figure e-1, links.lww.com/NXG/A383).
The Geisinger MyCode Community Health Initiative cohort (n = 92,455) is a health system–based population,15 and it is also a geographically defined cohort that represents the patients who visit Geisinger clinics from the East and Central Pennsylvania. The study cohort was based on participants from the Geisinger's MyCode Community Health Initiative phase I and phase II16,17 consisting of 1,184 acute IS patients as the cases for discovery with validated European ancestry (EUR) and MRI data for the confirmation of diagnosis. These participants have consented to research using the deidentified genetic data and the corresponding EHRs.16,17 We also identified additional 941 IS patients through EHR with validated European ancestry and the corresponding genetic data as the cases for replication. Both discovery and replication cases were part of the stroke registry and had a primary hospital discharge diagnosis of IS and a brain MRI during the same encounter to confirm the diagnosis. Therefore, the positive predictive value for IS through this EHR process was 100%, and no coding bias was observed. Unlike the cases for discovery, the TOAST subtypes for replication cases were not determined.
Data were collected from January 1, 2007, through December 31, 2018, and analyzed from September 13, 2019, to January 31, 2020. Control subjects had no diagnosis codes, indicating IS from the International Classification of Diseases (ICD), Ninth or Tenth Revision. The diagnostic codes used for the identification of the study cohort are: (ICD-9) 431.X, 432.9, 433.X, and 434.X; and (ICD-10) 161.X, 162.9, and 163.X. As none of controls identified were overlapped with cases included in the stroke registry, the negative predictive value for IS by the EHR process was 100%. The mean (SD) age of controls having index age ≥79 (n = 7,484) or ≥69 (n = 19,806) years was 84.67 (3.21) or 77.98 (6.01) years. The age cutoffs of 69 and 79 for controls were based on mean age at onset for cases which is 59 for our cohort. As this design follows younger cases vs older controls, we expect to have 50% of controls having index age of 10 years or 20 years older than the onset age of cases. Covariates, including age and sex, were extracted from the EHR. Age was ascertained at the time of the index hospital admission.
Definition of Ischemic Stroke Subtypes and Extraction of Clinical Variables
Patient demographics, clinical information, and outcome measures were collected based on the neurologic examination and corresponding neuroimaging.14 ISs (age at onset >18 years) were classified according to the published TOAST criteria18 by individual chart review. For the subtype analyses, we excluded patients with a recurrent stroke of different TOAST subtypes and piHAT <0.2 to avoid the relatedness within each TOAST subtype. In the end, 218 patients with CES, 33 strokes of other determined etiology (DETERMINED), 277 large artery strokes (LAS), 200 small vessel strokes (SVS), and 225 strokes of undetermined etiology (UNDETERMINED) were included in the study. ASL was a synthesized TOAST subtype that represents a combination of Acute SVS (n = 79) and LAS (n = 124).
The diagnosis of clinical risk factors was based on structured data captured in the EHR. CAD ascertainment was based on a composite of myocardial infarction or coronary revascularization. The ICD-9 codes for myocardial infarction include 410.X, 411.X, 412.X, 413.X, or 414.X or ICD-10 codes of I20.X, I21.X, I22.X, I23.X, I24.X, or I25.X in hospitalization records. Coronary revascularization was assessed based on an OPCS-4 coded procedure for coronary artery bypass grafting (K40.1-40.4, K41.1-41.4, or K45.1-45.5) or coronary angioplasty with or without stenting (K49.1-49.2, K49.8-49.9, K50.2, K75.1-75.4, or K75.8-75.9). AFib ascertainment was based on the self-report of AFib, atrial flutter, or cardioversion in an interview with a trained nurse. The ICD-9 code of 427.3 or ICD-10 code of I48.X in hospitalization records or a history of a percutaneous ablation or cardioversion based on the OPCS-4 coded procedure (K57.1, K62.1, K62.2, K62.3, or K62.4). Type 2 diabetes ascertainment was based on self-report in an interview with a trained nurse or ICD-10 codes of E11.X and E13.X and ICD-9 codes of 249.5 and 250.X in hospitalization records. Smoking status and alcohol use were based on self-report in an interview with a trained nurse. Based on the summary statistics in table 1, the demographic and the frequency of clinical risk factors were comparable with some previously reported cohorts.19–22 This is a quality control (QC) step to avoid significant coding bias for comorbidities.
Table 1.
Characteristics of Ischemic Stroke Patients and Controls in the Geisinger Cohort

Genotyping, Imputation, and QC
Samples were genotyped using Infinium OmniExpress Exome array (Illumina) and GSA-24v1-0 array (Illumina) for phase I and II, respectively. Genotypes for both cohorts were imputed to HRC.r1-1 (Haplotype Reference Consortium reference panel, version r1.1) EUR reference genome (GRCh37 build) separately using Michigan Imputation Server, which used Eagle v2.3 and Minimac4 as the phasing and imputation algorithm, respectively.
Samples with the genotyping rate below 95% were excluded. Single nucleotide polymorphisms (SNPs) with an imputation info score of <0.7, minor allele frequency (MAF) <1%, and significant deviation (p < 10−4) from Hardy-Weinberg equilibrium (HWE) were removed. A pruned set of SNPs (608,437) was generated from high-quality genotyped SNPs (MAF >0.05, HWE p > 0.0001, LD pruned with r2 between SNPs <0.2) to calculate the kinship relatedness matrix by SAIGE.23 A total of 6,213,823 SNPs from the merged phase I and II sample were included in the analysis. Those SNPs with a significant difference in MAF between phase I (n = 57,118) and II (n = 28,462) samples (p < 5 × 10−5) were removed from the summary statistics. Principal component analysis (PCA) by a fast PCA approximation embedded in PLINK2 (cog-genomics.org/plink/2.0/) using 1000GENOME phase III (2014 version) as the reference genome indicated that all the selected cases and controls were of EUR (figure e-2, links.lww.com/NXG/A383).
Individual SNP Association Tests for IS
SAIGE,23 a linear mix model, which built a kinship matrix to account for the cryptic relatedness, was adopted to test genetic association and used saddle point approximation to calibrate the distribution of score test statistics while accounting for the imbalance of the case-control ratio. Because of the negative selection against effect alleles associated with stroke, the enrichment of noneffect alleles or protective alleles would be expected in older and nonstroke individuals. In a sensitivity analysis of subgroups of controls, the GWAS was conducted in a case-control design by considering all Geisinger MyCode patients with age >69 years or >79 years and without any stroke-related ICD-9 or ICD-10 codes as low-risk control, for a purpose of improving the discovery power of GWAS in late-onset diseases.24 The covariates such as sex and 5 major PCs were included in all the primary and secondary analyses. Index age was not included as a covariate because it led to a genome-wide deflation of the test statistics.
Functional Annotation
Open Targets Genetics (OpenTargets.org) and GWAS catalog (ebi.ac.uk/gwas/) were queried for top-associated SNPs to evaluate their functional impact and pleiotropy if any. The pathway-specific gene network was illustrated by string-db (string-db.org/).
Heritability (h2) and Genetic Correlation (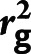 ) Calculations
) Calculations
We used the summary statistics from the GWAS to calculate adjust wide spacing disequilibrium score regression (LDSC25). The LD scores were estimated from an external reference sample, 1000GENOME with EUR. Only well-genotyped and imputed HapMap 3 SNPs (“w_hm3.snplist”) with the number of SNPs (877960/879085/878219/876828/878222 for any ischemic stroke (AIS)/any stroke (AS)/CES/LAS/SVS) were considered for the calculation of the genetic variance and covariance. To test for evidence of shared etiology between the base and target trait, we applied LDSC25,26 to quantify the extent of shared genetic contributions to IS between MEGASTROKE2 and Geisinger data sets at a genome-wide level. This shared etiology could be due to so-called horizontal pleiotropy (separate direct effects) or vertical pleiotropy (downstream effect).27 Effect allele for the Consortium GWAS2 was downloaded from MEGASTROKE.org for EUR. Based on an assumption of the expected chi-square statistic of a variant linearly correlated with LD score bin under a polygenic model,25 the stronger correlation could only be achieved when both data sets shared the same ancestry and when the LD score estimated for both data sets was obtained from the reference with the same ancestry. This procedure would help prevent bias of the estimates and decrease the standard error of LDSC estimate for genetic correlation.26
PRS Construction by PRSice-2 and Predictive Power for IS or TOAST Subtypes
We followed a tutorial28 when we constructed PRS, power analysis,29 and interpret the result. PRSice-227 is a p-value selection threshold approach. Removing SNPs with a low genotyping rate (geno >0.95), MAF <0.01; imputation “info score” >0.7; and individuals with a low genotyping rate (mind >0.9). PLINK2 was used for QC; effect allele for the Consortium GWAS2 was downloaded from MEGASTROKE.org for the European ancestry, and the genomic coordinates (hg19 version) of dbSNP were collected from UCSC Genome Browser (genome.ucsc.edu/). Shared markers with the same genomic coordinate and variant type between MEGASTROKE and Geisinger data were extracted. This is an autosomal-only analysis. No sample was overlapped between Geisinger and MEGASTROKE. Therefore, we assumed no substantial inflation of the association between the PRS and trait tested in the target data.
We initially set p-value thresholds at 0.001, 0.05, 0.01, 0.1, 0.2, 0.3, 0.4, 0.5, and 1. Because of the criticism of clumping by selecting an arbitrarily chosen correlation threshold for the removal of SNPs in LD, we tested the predictive power under the threshold ranging from 0.05 to 0.8. An informed LD clumping using the following PLINK command: —clump-p1 1, –clump-kb 1,000, —clump-r2 0.05 to 0.8 was conducted. After an evaluation of the trade-off by including less or more SNPs in the polygenic modeling by tuning the correlation threshold, we chose r2 = 0.1 because this level for LD pruning showed no systematic overestimation or underestimation of the PRS modeling. A total of 196,995 (AIS), 197,033 (AS), 197,529 (CES), 197,532 (LAS), and 197,549 (SVS) variants were retained for the 5 MEGASTROKE summary statistics. On average, around 25% of variants were kept with MAF < 0.025. PRSs were derived from MEGASTROKE by PRSice-2 with 10,000 permutation tests. The results were derived from testing over a range of p-value thresholds for base SNPs and also included the thresholding that gave the best predictive performance (best fit).
We also removed related individuals in the Geisinger sample with paired PI_HAT ≥0.2 and maintained the maximum number of cases. We ended up with 1,167 cases and 17,271 controls.
We selected the default mode of the PRS algorithm, PRSavg, which was calculated by the number of observed effective allele for each variant multiplied by the corresponding effect size derived from the MEGASTROKE, divided by the number of alleles included in the PRS from that individual, and finally sum of all from that individual. This approach would help prevent biased PRS toward more or less genetic markers included in the calculation for that individual. Nagelkerke pseudo-R2 (R2) and significance for the nonzero regression coefficient for PRS were calculated by PRSice-2 with clumping and thresholding (“P + T”). The IS was regressed on the PRSavg, including sex and first 5 PCs as covariates, to calculate the variance explained by PRSice-2. In line with previous PRS studies8,10,13 on STROKE, we also performed a z-score transformation, “PRSz-score = (PRSavg−mean(PRSavg))/SD(PRSavg),” of this raw PRSavg, and the logistic regression of TOAST subtypes was conducted. The odds ratios (ORs) with 95% confidence interval (95% CI) were calculated by R ‘glm’ with a nature log transformation.
We conducted a sensitivity analysis to determine whether this predictive power (R2 and significance for the nonzero regression coefficient for PRS) can be improved by raising the cutoff for the age of controls. The same number of controls as to the corresponding controls ≤69 or ≤79 by a random selection from controls ≤59 was simulated to determine whether this augmented predictive power, if any, was largely due to natural selection in aged nonstroke individuals but not due to the changed case:control ratio (table e-1, links.lww.com/NXG/A383).30
Gene Set–Based PRS Analyses
A two-step design was considered by randomly splitting 19,806 controls (age ≥ 69 years) into discovery (n = 1,184/10,983 for case/control) and replication (n = 951/8,823 for case/control) data sets to maintain the same case-control ratio (0.108). Those 951 IS cases were identified by Geisinger EHR but not included in the initial GWAS. We further removed related individuals (piHAT ≥0.20) from the discovery and replication data sets and ended up with 1,076/10,107 for case/control in discovery data set and 941/8,145 for case/control in replication data set. By comparing the result originated from all samples with the result derived from nonrelated individuals, any inflated R2 and p value would be determined.
The global genes were selected as a universal background for gene sets analyses, and the mapping file, “Homo_sapiens.GRCh37.87.gtf”, was downloaded from ftp://ftp.ensembl.org/pub/grch37/release-90/gtf/ho…
PRSavg derived from gene sets defined by the Gene Ontology Biological Process was calculated to test their association with IS under 2 MAF thresholds (MAF <0.025 or <1), which represents low-frequency common variants or all variants accordingly. A total of 7,350 Gene Ontology pathways of Biological Process and their related genes were defined by Molecular Signatures Database (“msigdb_v7.0_GMTs/c5.bp.v7.0.symbols.gmt” from gsea-msigdb.org/gsea/msigdb/index.jsp). The sex and 5 major PCs were included as covariates in the logistic regression model for each data set. A raw competitive p value, which indicated the level of enrichment, was calculated. The meta-analysis of summary statistics of discovery and replication data sets was conducted by metal with weighted effect size (coefficient) estimates using the inverse of the corresponding standard errors. Sample overlap correction was not performed because of no overlapping samples between discovery and replication samples. We reported the top 5 pathways that were significantly enriched from the gene-set analysis for PRS derived from each summary statistics stratified by 2 levels of MAF after the meta-analysis. All the p values presented as raw and only p value with −log10(p) ≥ 5.17 (−log10(0.05/7,350)) survived multiple testing for Bonferroni correction.
Data Availability
The summary statistics of our GWAS may be shared with third party on execution of data sharing agreement for reasonable requests.
Results
Characterization of Geisinger Ischemic Stroke and Controls
Demographics and clinical characteristics of IS cohorts (cases for discovery and replication) and controls were summarized in table 1 stratified by age for controls. There was a significant increase in the proportion of subjects having clinical risk factors for stroke in the case group vs control group (p < 0.01). These risk factors included smoking ever, CAD, AFib, diabetes mellitus, dyslipidemia, and history of hypertension. Other anthropometric factors such as BMI showed significant differences between cases and controls (p < 0.01).
No Genome-Wide Significant Association Identified by the GWAS
We first performed the GWAS using a linear mixed model. Manhattan and QQ plots for the GWAS results of control ≥69 years and ≥79 years were shown in figure e-3B, links.lww.com/NXG/A383, as mirrored toward each other. The top SNPs with p < 5 × 10−5 were listed in table e-2. No variants passed the genome-wide significant threshold (p < 5 × 10−8) of association for IS. Among the top variants with p < 5 × 10−5 (suggestive significance) in GWAS79yrs, all had decreased frequency of risk alleles for IS (p = 1.90 × 10−5, paired t test, increased effect size (β), and improved significance in association with IS than those in GWAS69yrs). The top loci were also associated with increased risk for various stroke-related (sub)phenotypes.
Heritability (h2) and Genetic Correlation (rg)
We present observed and liability scale h2, assuming that sample and population prevalence of Geisinger IS were 0.01 and 0.01 for control ≥69 years and 0.05 and 0.01 for control ≥79 years. A moderate heritability (∼10%) for Geisinger samples was identified (figure 1A). The estimates of explained heritability were increased for both observed and liability scale h2 when the cutoff for the index age of controls was raised to 79 years.
Figure 1. Estimated Heritability of Geisinger Ischemic Stroke and the Genetic Correlation Between Geisinger Sample and MEGASTROKE Sample.

The chip-based heritability (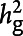 , A) and genetic correlation (rg, B) were calculated by LDSC using genotyped HapMap 3 SNPs (hm3). Both the observed scale and the liability scale h2, later of which was adjusted by the sample prevalence and population prevalence, were presented in the y-axis, including error bars for the estimates. We assumed a trait prevalence of 1% for all phenotypes and tested the robustness of heritability (
, A) and genetic correlation (rg, B) were calculated by LDSC using genotyped HapMap 3 SNPs (hm3). Both the observed scale and the liability scale h2, later of which was adjusted by the sample prevalence and population prevalence, were presented in the y-axis, including error bars for the estimates. We assumed a trait prevalence of 1% for all phenotypes and tested the robustness of heritability (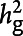 ) under 2 levels of controls. AIS = any ischemic stroke; AS = any stroke; CES = cardioembolic stroke; LAS = large artery stroke; LDSC = linkage disequilibrium score regression; SNP = single nucleotide polymorphism; SVS = small vessel stroke.
) under 2 levels of controls. AIS = any ischemic stroke; AS = any stroke; CES = cardioembolic stroke; LAS = large artery stroke; LDSC = linkage disequilibrium score regression; SNP = single nucleotide polymorphism; SVS = small vessel stroke.
The estimated genetic correlations of IS between each of the pairs of cohorts were shown in figure 1B. A significant genetic correlation between MEGASTROKE and Geisinger summary statistics (≥69 or ≥79) was observed with the most significant correlation for AS with rg = 0.586 or 0.545 and p = 2 × 10−4 or 2 × 10−8. Although there was no significant improvement of rg when the cutoff for the index age of controls was raised from 69 to 79 years, the significance of the correlation was improved with a smaller variance of rg across all pairs. Although there were similar numbers of well-characterized LAS, SVS, and CES in this Geisinger cohort, the highest rg was observed in LAS subtype with rg = 0.452 or 0.403 and p = 2 × 10−3 or 4.88 × 10−5, whereas the lowest rg observed in CES subtype (rg = 0.257 or 0.193 and p = 0.021 or 0.019).
Predictive Power of PRS for IS
The sample size and index age (mean and SD) stratified by sex were summarized in table e-1, links.lww.com/NXG/A383. The increased predictive power (R2 and p) of PRS by raising the cutoff for controls from 59 to 79 years old was independent of the case:control ratio and across all levels of thresholding p values observed, indicating that older low-risk control was, at least partially, driving the PRS's association with IS. The augmented predictive power was observed in all but the PRS derived from MEGASTROKE CES (red dots compared with pink dots or blue dots compared with light blue dots in figure 2A).
Figure 2. Sensitivity Analysis to Show the Predictive Power of PRS.

We conducted a sensitivity analysis to determine whether this predictive power (R2 and significance for the nonzero regression coefficient for PRS) can be improved by raising the cutoff for the age of controls. (A) We simulate the same number of controls as to the corresponding controls ≥69 or ≥79 by a random selection from controls ≥59 to determine this augmented predictive power, if any, was largely due to natural selection in aged nonstroke individuals but not due to the change in the case:control ratio. This improved predictive power was independent of the prevalence of the disease or case:control ratio as shown by this dot plot. (B) The association between PRSz-score derived from 5 summary statistics of MEGASTROKE and ischemic stroke was tested by logistic regression (phenotype ∼ PRSz-score + sex + PC1-5.). The PRS was calculated by PRSice-2 using the average score (avg) equation (default) from the best-fit modeling. The raw PRSavg was z score transformed into PRSz-score to compare the odds ratios across the analyses. Odds ratios (ORs) (y-axis) and significant levels (dot size) were calculated by the R glm. (C) The association of PRSz-score derived from the summary statistics of MEGASTROKE AIS with ischemic stroke and its major clinical risk factors were tested by the same logistic regression and visualized by the forest plot. AIS = any ischemic stroke; AS = any stroke; CES = cardioembolic stroke; LAS = large artery stroke; PRS = polygenic risk score; SVS = small vessel stroke.
Variation of all 5 PRSavg could significantly explain some of the phenotypic variations of Geisinger IS with the PRSLAS explained most of the phenotypic variance (R2 = 0.006, p = 7.2 × 10−12) for the best-fit model as compared to the PRS derived from other summary statistics (R2 = 0.003, p = 1.12 × 10−6 for PRSSVS; R2 = 0.004, p = 1.46 × 10−7 for PRSCES; R2 = 0.004, p = 3.04 × 10−8 for PRSAS; and R2 = 0.004, p = 1.87 × 10−7 for PRSAIS) (figure e-4A, links.lww.com/NXG/A383). We also conducted a restricted analysis by including samples with paired HAT score (piHAT) <0.2. As shown in figure e-4B, we still obtained a similar level of R2 (0.007 for PRSLAS; 0.003 for PRSSVS; 0.004 for PRSCES; 0.005 for PRSAS; and 0.004 for PRSAIS) and p value (1.69 × 10−11 for PRSLAS; 2.00 × 10−6 for PRSSVS; 2.22 × 10−7 for PRSCES; 1.31 × 10−8 for PRSAS; and 1.77 × 10−7 for PRSAIS) for the best-fit model.
The PRSz-score derived from MEGASTROKE LAS showed the strongest association with Geisinger IS phenotype (OR = 1.244, per 1−SD increase in the PRS, p = 1.57 × 10−11, 95% CI [1.168–1.326]) for controls ≥69, when compared with the PRSz-score derived from other MEGASTROKE summary statistics (figure 2B for the best-fit modeling). The association between PRS (i.e., PRSAIS) and other risk factors in the full model was accessed. This PRSAIS was significantly associated with hypertension, diabetes, CAD, and other comorbidities, but not with lifestyle changes captured in EHR such as drinking and smoking cigarettes (figure 2C).
Low-Frequency Variants Contributed More to the Association Between PRS and IS
Because the loci for cardiovascular diseases were significantly enriched for lifetime reproductive success by natural selection31 and identified that IS subtype-specific loci were more likely to be low MAF,32 we proposed that the genetic variants with lower MAF may contribute more to the phenotypic variation. When we partitioned the variants by MAF ≤0.01, 0.05, 0.1, 0.2, or to all, PRSLAS, PRSCES, and PRSSVS derived from low-frequency common variants (0.01 < MAF < 0.05) provided the best-fit modeling for Geisinger IS (see figure e-5A, links.lww.com/NXG/A383), suggesting that low-frequency common variants when taken together could have more contribution to the risk for IS subtypes. This result was confirmed by using the replication cases (n = 951) vs the same controls (n = 19,806) (figure e-5B, links.lww.com/NXG/A383). R2 for PRSLAS in discovery and replication data sets was 0.015 and 0.016 for MAF < 0.025 as compared to 0.006 and 0.007 for MAF < 1, respectively; R2 for PRSSVS was 0.011 and 0.012 for MAF < 0.025 as compared to 0.004 and 0.005 for MAF < 1, respectively. We selected p < 0.025 as 1 of 2 bar levels in the gene-set analysis to identify the enriched pathways associated with the potentially shared etiology.
PRS Derived From the Subsets of SNPs Associated With IS
We explored the top 5 pathways for the gene sets analyses stratified by 2 bar levels (MAF < 0.025 or < 1) (figure 3) after the meta-analysis of the summary statistics from discovery and replication data sets (figure 4). The summary statistics of the entire analysis were available in table e-3, links.lww.com/NXG/A384, and e-4, links.lww.com/NXG/A385. By comparing the results from nonrelated individuals (figure 4) with the result from the original data sets without removing related individuals (figure e-6A, links.lww.com/NXG/A383), we only observed a slightly inflated p value for the top gene sets when selecting all SNPs (MAF < 1) but not selecting SNPs with MAF < 0.025 in both discovery and replication data sets (figure e-7). This suggested that those pathway-related low-frequency common variants were not shared by related individuals with IS.
Figure 3. PRS Derived From Lower MAF Variants Provided the Best-Fit Modeling for the Ischemic Stroke.

Nonrelated individuals (piHAT ≤0.20) from the discovery and replication data sets with a random split of control samples were included in the association analysis. PRS derived from genetic variants with relatively lower MAF provided the best-fit modeling for the ischemic stroke (red dots) when PRS was constructed based on the summary statistics of TOAST subtypes such as LAS, SVS, and CES as compared to PRS constructed based on the summary statistics of AS or AIS. Both discovery data set and replication data set showed the same profile. The size of the dots represents the R2, a measure of the proportion of the variance explained by the model. The y-axis represents the significance of the model fit. The total number of variants included in the analysis under two MAF thresholds was also listed on the top. AIS = any ischemic stroke; AS = any stroke; CES = cardioembolic stroke; LAS = large artery stroke; MAF = minor allele frequency; PRS = polygenic risk score; SNP = single nucleotide polymorphism; SVS = small vessel stroke.
Figure 4. Gene Sets Analyses Illustrated the Top Five Pathways Enriched for Ischemic Stroke (Controls With Index Age ≥69 years) After Meta-analysis of Discovery Data Set (n = 1,076/10,107) and Replication Data set (n = 941/8,145) When the PRS Was Constructed Based on Each of the Five Summary Statistics of MEGASTROKE.

The sex and 5 major PCs were included as covariates in the logistic regression model for each data set. The meta-analysis was conducted by metal with weighted effect size (coefficient) estimates using the inverse of the corresponding standard errors. Sample overlap correction was not performed because of no overlapping samples between discovery and replication samples. The global genes were selected as a universal background for gene sets analyses, and the mapping file was “Homo_sapiens.GRCh37.87.gtf.” PRSs derived from gene sets defined by the Gene Ontology Biological Process were calculated to test their association with an ischemic stroke under 2 MAF thresholds (MAF < 0.025 or < 1), which represents low-frequency common variants or all variants accordingly. Seven thousand three hundred forty-nine pathways and their related gene sets were defined by Molecular Signatures Database (“msigdb_v7.0_GMTs/c5.bp.v7.0.symbols.gmt”). Exploration of the top 5 pathways enriched from PRS gene sets analyses after the meta-analysis of discovery and replication data sets using each summary statistics from MEGASTROKE to construct PRS under 2 levels of MAF thresholding (y-axis). The red or blue bar represents the ischemic stroke has a negative or positive association with the corresponding biological process according to the direction of the coefficient, respectively. All the p values in the x-axis were raw but survived multiple testing for Bonferroni correction as −log10(p) ≥ 5.17 (−log10(0.05/7,349)). AIS = any ischemic stroke; AS = any stroke; CES = cardioembolic stroke; LAS = large artery stroke; MAF = minor allele frequency; SVS = small vessel stroke; VEGF = vascular endothelial growth factor.
For all SNPs with various MAF, gene sets related to GO negative regulation of (RNA) biosynthetic process, the downstream terms of the metabolic process, were ranked to the top using PRSLAS and PRSCES. Top gene set related to apoptosis was enriched with a positive correlation (blue bar) with stroke using PRSLAS; gene sets related to embryonic heart tube development was one of the top pathways with a positive correlation with stroke using PRSSVS.
For genetic variants with MAF < 0.025, we observed the top pathways with a negative association (red bar) of stroke including the vascular endothelial growth factor signaling pathway and APP signaling pathway when using PRSAIS and PRSAS for all Geisinger IS. Genes related to the regulation of macrophage-derived foam cell differentiation and lipid complex assembly enriched in IS patients using PRSSVS, suggesting atherosclerosis in the pathogenesis of IS. Gene sets related to ion/transmembrane transport were the top pathways with a positive association with IS using PRSCES.
PRS Derived From the MEGASTROKE Subtypes Was Associated More With the Corresponding TOAST Subtypes of Geisinger Patients
Only cases from the discovery data set were subtyped by the TOAST criteria. As expected, PRSLAS (R2 = 0.004; p = 7.31 × 10−4), PRSCES (R2 = 0.005; p = 4.47 × 10−4), and PRSSVS (R2 = 0.003; p = 0.003) explained the most variance of the corresponding subtypes of Geisinger IS than PRS derived from other MEGASTROKE data sets (larger and warmer dots for the significant level and Nagelkerke pseudo-R2, respectively, see the arrow in figure 5 using base p < 0.1). For the synthesized group, ASL, with much more LAS cases (n = 124) than SVS cases (n = 79), the predictive power was the highest by PRSsvs (R2 = 0.007; p = 8.23 × 10−5). PRSCES could differentiate LAS from CES or SVS from CES (yellow arrows). TOAST Determined subtypes of Geisinger can be equally explained by PRSLAS and PRSSVS despite not reaching a significant level due to the small sample size (n = 33). Furthermore, none of the PRS could significantly explain the phenotypic variation of Geisinger Undetermined subtype.
Figure 5. The PRS Derived From MEGASTROKE Subtypes Was Mostly Associated With the Corresponding Geisinger TOAST Subtypes.

The dot plot demonstrated the association of PRS derived from MEGASTROKE on Geisinger TOAST subtypes when using base p < 0.1 as an example. The association between PRS and stroke subphenotypes was tested by logistic regression (phenotype ∼ PRSavg + sex + PC1–5). PRS derived from the MEGASTROKE consortium (y-axis) was calculated by PRSice-2 to determine their association with the TOAST subtypes of Geisinger ischemic stroke patients (x-axis). Nagelkerke pseudo-R2 (color of dots) and significant levels (size of dots) were calculated by PRSice-2 with clumping and thresholding (here using SNPs with base p < 0.1 as an example). *The significance of the association survived Bonferroni correction given 30 paralleled testing (punadjusted < 0.0017). We excluded any cases with a recurrent stroke of different TOAST subtypes. AIS = any ischemic stroke; AS = any stroke; CES = cardioembolic stroke (TOAST); DETERMINED = stroke of other determined etiology (TOAST); LAS = large artery stroke (TOAST); SNP = single nucleotide polymorphism; SVS = small vessel stroke (TOAST); UNDETERMINED = stroke of undetermined etiology (TOAST); ASL was a synthesized TOAST subtype that represents a combination of Acute SVS (n = 79) and LAS (n = 124).
Discussion
Among the top variants with p < 5 × 10−5 (suggestive significance) from GWAS79yrs, all had decreased frequency of risk alleles for IS, increased effect size (β or OR), and improved significance in association with IS than those from GWAS69yrs, suggesting that the protective alleles were enriched in the older nonstroke population. If a genetic variant was associated with fitness, selection would drive 1 allele to low frequency.33 The latter was the case even for traits without any obvious connection to fitness. Through an inquiry into the PheWAS summary statistics of stroke-related phenotypes obtained from UKB, we were able to identify that the risk alleles from the top loci were also associated with increased risk for various stroke-related (sub)phenotypes (i.e., atherosclerosis) or risk factors (i.e., hypertension, high cholesterol level, and cardiac arrhythmia), suggesting the potential pleiotropy of these variants.
Although there was no significant improvement of rg when the cutoff for the index age of controls was raised from 69 to 79 years, the significance of the correlation was improved with a smaller variance of rg across all pairs, suggesting that this case:control design could yield a more homogenous control population with enriched protective alleles. The lowest rg was observed using the summary statistics from MEGASTROKE CES subtype (rg = 0.257 or 0.193, p = 0.021 or 0.019), and no improvement in the prediction power for PRS derived from this CES summary statistics in the sensitivity analysis suggested that these low-frequency common variants may not show enrichment for the protective allele in aged controls. The similar findings were also reported for rare variants previously implicated in alone or familial forms of Afib. Those rare variants were detected at low frequencies in a general population but were not associated with Afib through an age stratification of the control population.34
PRSs derived from MEGASTROKE GWAS data sets can explain only a small fraction of the variance in the target phenotypes, and this proportion can be improved by using common variants with lower MAF. The lack of predictive power of PRS could be due to the genetic and environmental heterogenicity of IS with various causal mechanisms. It is possible that the predictive power of PRS can be improved by using a subset of SNPs derived from disease-relevant pathways or functional annotated subset of SNPs.35 The predictive power (R2) was improved when using a subset of low MAF variants, many of which were eQTL for the annotated genes nearby (data not shown). This lack of predictive power could also be due to the polygenic modeling algorithms, and how to improve the signal-to-noise ratio by ML approaches is an ongoing topic. The performance of PRSs over the life course in several cardiometabolic diseases and neoplasms has been evaluated in a prospective setting, and their value when integrated with the known clinical risk factors and biomarkers has been revealed.36 The cumulative risk for CAD, T2D, and AFib was disproportionally increased after 40 years old when patients were stratified by categorical PRS from higher (>97.5%) to lower (<2.5%) scores. Because of the pleiotropy of genetic risk factors for both IS and chronic diseases included in this study, we expect IS will show a similar pattern. The cumulative disease rate for IS will be disproportionally higher in the top PRS category. The polygenic contribution to early onset was much higher than that to late onset in the same disease.36 Our retrospective study from the sensitivity analysis alternatively confirmed this disproportional increased PRS burden for IS using younger cases vs 3 tiers of older controls (from 59, 69, to 79).
PRS alone cannot replace the need to investigate the sources of emboli or thrombosis and make a personalized treatment accordingly. Whether patients having high PRS value for specific pathways may indicate the potential causal mechanism of IS is still unknown and requires further investigation to validate. It is unclear to what extent PRS contributes to early-onset vs late-onset IS. Although PRS alone has a small but significant improvement in the prediction of IS (area under the curve for receiver operating characteristics [AUC-ROC] = 0.596 with 95% CI [0.577–0.616] as compared to AUC-ROC = 0.554 with 95% CI [0.534–0.573] for the base model, p = 4.26 × 10−6) (Supplementary figure e8, links.lww.com/NXG/A383), the combination of PRS and clinical risk factors has not been extensively evaluated using different ML algorithms and PRS construction models. Whether PRS contributed to the outcome prediction of IS cases is to be determined. IS is a multifactorial complex disease. The prediction of IS and its subtypes always follows the multivariate regression/classification model. This study confirmed the value of PRS derived from MEGASTROKE as one of the features in ML-based prediction modeling.
Furthermore, our study suffered from limited power to test for gene set analyses in Geisinger subcohort with the same TOAST subtypes because of the small sample size of well-characterized subtype cohorts.
Finally, our study was based on EUR subjects, limiting its generalizability to larger and more diverse cohorts. Recent research has focused on the generalizability of polygenic scores to non-EUR populations. Because of differences in variant frequencies and LD patterns between populations with different ancestries, reduced predictive power in non-EUR samples is anticipated, particularly in Africans.37 How to improve the treatment of LD and variant frequencies when applying polygenic scoring derived from Europeans to cohorts of non-EUR is an emerging field. On the other hand, data resources for non-EUR are currently inadequate, resulting in the rationale for large-scale GWAS in diverse human populations. Having realized the full and equitable potential of PRS, we should promote genetic studies on underserved populations.38
As a part of a multiple-level genetic association study, PRS constructed based on multiple loci from genes of curated biological pathways does not confer the biological information at the single-gene level. Thus, this PRS-based association study cannot directly convey the message of functional impact of genes on the definitive phenotype. Given all hypothesis (biological pathways) being considered, this study is trying to prioritize the top pathways from which the PRS could stratify IS cases into categorical high or low score for certain pathways based on different causal mechanisms through a biotyping approach. Clinically, we are often uncertain of the underlying stroke etiology. If nonroutine features such as PRS are able to aid in subtyping, the number of cryptogenic strokes could be potentially reduced. Thus, PRS, complemented with other clinical features of the patients, could facilitate more targeted secondary stroke prevention.
The future study will focus on (1) how individual genetic risk or polygenic risk extracted from curated biological pathways affects stroke outcome, such as recurrence and mortality and (2) how these identified subsets of genetic variants are correlated with some clinical subtypes of IS, particularly when the natural selection (negative or positive) may have more impact on the fitness in early-onset stroke rather than in late-onset stroke.
We provide the first evidence that PRSs derived from MEGASTROKE have value in identifying shared etiologies and determining the etiologic subtypes beyond TOAST in an independent cohort.
Acknowledgment
The authors thank Dr. Matthew T. Oetjens who provided critical review of the manuscript. The genetic data from Geisinger were made available through the collaboration with Regeneron Genetics Center (Author List and Contribution Statements at the Supplementary Material). V.A. has financial research support from the Defense Threat Reduction Agency (DTRA) grant no. HDTRA1-18-1-0008 subawarded to Geisinger and funds from the NIH grant no. R56HL116832 subawarded to Geisinger during the study period. R.Z. has financial research support from Bucknell University Initiative Program, Roche—Genentech Biotechnology Company, the Geisinger Health Plan Quality fund, and receives institutional support from the Geisinger Health System during the study period.
Glossary
- AFib
atrial fibrillation
- APP
amyloid precursor protein
- ASL
a synthesized TOAST subtype that represents a combination of Acute SVS (n = 79) and LAS (n = 124)
- AUC-ROC
area under the curve for receiver operating characteristics
- BMI
body mass index
- CAD
coronary artery disease
- CES
cardioembolic stroke
- CI
confidence interval
- DETERMINED
strokes of other determined etiology
- EHR
electronic health record
- EUR
European ancestry
- GO
Gene Ontology
- GWAS
genome-wide association study
- HWE
Hardy-Weinberg equilibrium
- ICD
International Classification of Diseases
- IS
ischemic stroke
- LAS
large artery stroke
- LDSC
linkage disequilibrium score regression
- MAF
minor allele frequency
- ML
machine learning
- OR
odds ratio
- PCA
principal component analysis
- PRS
polygenic risk score
- SNP
single nucleotide polymorphism
- SVS
small vessel stroke
- T2D
type 2 diabetes
- TOAST
trial of ORG 10172 in acute stroke treatment
- UNDETERMINED
strokes of undetermined etiology
Appendix 1. Authors
Appendix 2. Coinvestigators

Contributor Information
Jiang Li, Email: jli@geisinger.edu.
Durgesh P. Chaudhary, Email: dpchaudhary@geisinger.edu.
Ayesha Khan, Email: akhan2@geisinger.edu.
Christoph Griessenauer, Email: christoph.griessenauer@gmail.com.
David J. Carey, Email: djcarey@geisinger.edu.
Study Funding
No targeted funding reported.
Disclosure
All the authors report no disclosures relevant to the manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Neurology Working Group of the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium, the Stroke Genetics Network (SiGN), and the International Stroke Genetics Consortium (ISGC). Identification of additional risk loci for stroke and small vessel disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2016;15:695–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malik R, Chauhan G, Traylor M, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet 2018;50:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibrahim-Verbaas CA, Fornage M, Bis JC, et al. Predicting stroke through genetic risk functions: the CHARGE Risk Score Project. Stroke 2014;45:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malik R, Bevan S, Nalls MA, et al. Multilocus genetic risk score associates with ischemic stroke in case-control and prospective cohort studies. Stroke 2014;45:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tada H, Shiffman D, Smith JG, et al. Twelve-single nucleotide polymorphism genetic risk score identifies individuals at increased risk for future atrial fibrillation and stroke. Stroke 2014;45:2856–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hachiya T, Kamatani Y, Takahashi A, et al. Genetic predisposition to ischemic stroke: a polygenic risk score. Stroke 2017;48:253–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rutten-Jacobs LC, Larsson SC, Malik R, et al. Genetic risk, incident stroke, and the benefits of adhering to a healthy lifestyle: cohort study of 306 473 UK Biobank participants. BMJ 2018;363:k4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibanez L, Heitsch L, Dube U, et al. Overlap in the genetic architecture of stroke risk, early neurological changes, and cardiovascular risk factors. Stroke 2019;50:1339–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abraham G, Malik R, Yonova-Doing E, et al. Genomic risk score offers predictive performance comparable to clinical risk factors for ischaemic stroke. Nat Commun 2019;10:5819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radu RA, Terecoasa EO, Bajenaru OA, Tiu C. Etiologic classification of ischemic stroke: where do we stand? Clin Neurol Neurosurg 2017;159:93–106. [DOI] [PubMed] [Google Scholar]
- 12.Arsava EM, Helenius J, Avery R, et al. Assessment of the predictive validity of etiologic stroke classification. JAMA Neurol 2017;74:419–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pulit SL, Weng LC, McArdle PF, et al. Atrial fibrillation genetic risk differentiates cardioembolic stroke from other stroke subtypes. Neurol Genet 2018;4:e293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hendrix P, Sofoluke N, Adams MD, et al. Risk factors for acute ischemic stroke caused by anterior large vessel occlusion. Stroke 2019;50:1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oetjens MT, Kelly MA, Sturm AC, Martin CL, Ledbetter DH. Quantifying the polygenic contribution to variable expressivity in eleven rare genetic disorders. Nat Commun 2019;10:4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 2016;354:aaf6814. [DOI] [PubMed] [Google Scholar]
- 17.Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single U.S. health care system. Science 2016;354:aaf7000. [DOI] [PubMed] [Google Scholar]
- 18.Adams HP Jr, Bendixen BH, Kappelle LJ, et al. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in Acute Stroke Treatment. Stroke 1993;24:35–41. [DOI] [PubMed] [Google Scholar]
- 19.Howard G, Kissela BM, Kleindorfer DO, et al. Differences in the role of black race and stroke risk factors for first vs. recurrent stroke. Neurology 2016;86:637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhamoon MS, Sciacca RR, Rundek T, Sacco RL, Elkind MS. Recurrent stroke and cardiac risks after first ischemic stroke: the Northern Manhattan Study. Neurology 2006;66:641–646. [DOI] [PubMed] [Google Scholar]
- 21.Carandang R, Seshadri S, Beiser A, et al. Trends in incidence, lifetime risk, severity, and 30-day mortality of stroke over the past 50 years. JAMA 2006;296:2939–2946. [DOI] [PubMed] [Google Scholar]
- 22.Fang MC, Coca Perraillon M, Ghosh K, Cutler DM, Rosen AB. Trends in stroke rates, risk, and outcomes in the United States, 1988 to 2008. Am J Med 2014;127:608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou W, Nielsen JB, Fritsche LG, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet 2018;50:1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oliynyk RT. Evaluating the potential of younger cases and older controls cohorts to improve discovery power in genome-wide association studies of late-onset diseases. J Pers Med 2019;9:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulik-Sullivan BK, Loh PR, Finucane HK, et al. LD score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015;47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni G, Moser G, Schizophrenia Working Group of the Psychiatric Genomics Consortium, Wray NR, Lee SH. Estimation of genetic correlation via linkage disequilibrium score regression and genomic restricted maximum likelihood. Am J Hum Genet 2018;102:1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi SW, O'Reilly PF. PRSice-2: polygenic Risk Score Software for Biobank-Scale Data. Gigascience 2019;8:giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi SW, Mak TS, O'Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc 2020;15:2759–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet 2013;9:e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson G. On the utilization of polygenic risk scores for therapeutic targeting. PLoS Genet 2019;15:e1008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byars SG, Huang QQ, Gray LA, et al. Genetic loci associated with coronary artery disease harbor evidence of selection and antagonistic pleiotropy. PLoS Genet 2017;13:e1006328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malik R, Traylor M, Pulit SL, et al. Low-frequency and common genetic variation in ischemic stroke: the METASTROKE collaboration. Neurology 2016;86:1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wray NR, Yang J, Hayes BJ, Price AL, Goddard ME, Visscher PM. Pitfalls of predicting complex traits from SNPs. Nat Rev Genet 2013;14:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weeke P, Denny JC, Basterache L, et al. Examining rare and low-frequency genetic variants previously associated with lone or familial forms of atrial fibrillation in an electronic medical record system: a cautionary note. Circ Cardiovasc Genet 2015;8:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi J, Park JH, Duan J, et al. Winner's curse correction and variable thresholding improve performance of polygenic risk modeling based on genome-wide association study summary-level data. PLoS Genet 2016;12:e1006493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mars N, Koskela JT, Ripatti P, et al. Polygenic and clinical risk scores and their impact on age at onset and prediction of cardiometabolic diseases and common cancers. Nat Med 2020;26:549–557. [DOI] [PubMed] [Google Scholar]
- 37.Duncan L, Shen H, Gelaye B, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun 2019;10:3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 2019;51:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The summary statistics of our GWAS may be shared with third party on execution of data sharing agreement for reasonable requests.